
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly selective Ag foam gas diffusion electrodes for CO2 electroreduction by pulsed hydrogen bubble templation†
Hendrik
Hoffmann
 *a,
Maximilian
Kutter
a,
Jens
Osiewacz
b,
Melanie-Cornelia
Paulisch-Rinke
c,
Steffen
Lechner
a,
Barbara
Ellendorff
b,
Annika
Hilgert
a,
Ingo
Manke
c,
Thomas
Turek
b and
Christina
Roth
a
*a,
Maximilian
Kutter
a,
Jens
Osiewacz
b,
Melanie-Cornelia
Paulisch-Rinke
c,
Steffen
Lechner
a,
Barbara
Ellendorff
b,
Annika
Hilgert
a,
Ingo
Manke
c,
Thomas
Turek
b and
Christina
Roth
a
aElectrochemical Process Engineering, Universität Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany. E-mail: hendrik.hoffmann@uni-bayreuth.de
bInstitute of Chemical and Electrochemical Process Engineering, Clausthal University of Technology, Leibnizstrasse 17, 38678 Clausthal-Zellerfeld, Germany
cHelmholtz-Zentrum Berlin für Materialien und Energie, Department Electrochemical Energy Storage, Hahn-Meitner-Platz 1, 14109 Berlin, Germany
First published on 9th October 2023
Abstract
The electrochemical reduction of carbon dioxide to valuable fossil-free products opens up a way to close the carbon cycle, if based solely on renewable energy sources. Making the process industrially viable, however, needs high CO2 conversion rates, efficient electrodes, and high selectivity for desired products. To reach this goal, highly catalytically active porous electrodes with maximized surface areas are required. We combined pulsed electrochemical deposition of the Ag foam catalyst with ionomer infiltration of the electrode to produce Ag-based gas diffusion electrodes (GDEs) in a facile and fast production process. Using the dynamic hydrogen bubble templation method (DHBT), we utilized the parasitic hydrogen evolution reaction (HER) to aid the solvent free structuring of the 3D catalyst network and directly manufacture a GDE. Different deposition parameters and in particular pulse-to-pause ratios increased the amount of deposited catalyst and successfully reduced the overpotential during CO2RR operation. To inhibit electrode flooding and decrease CO2 mass transport limitations during CO2RR, we further infiltrated the electrode with a suitable perfluorosulfonic acid ionomer. SEM and EDS analyses showed a homogeneous Ag/F distribution along the cross section of the electrodes. These electrodes catalyzed the conversion of CO2 to CO at industrially viable current densities of 500 mA cm−2 with an unprecedented faradaic efficiency up to 76% in 1 M KHCO3.
Broader contextA new direct fabrication method for polymer-based Ag-DHBT-GDEs was developed, combining pulsed electrochemical deposition of Ag catalysts with ionomer infiltration of a sputtered Ag-GDL. The sputtering process prepared the insulating polymer fabric by depositing a conductive thin layer. This allowed for the electrochemical deposition of Ag catalysts, resulting in a conductive GDL that maintained its porous structure essential for gas transport during CO2 electrolysis. The DHBT method utilized the parasitic HER to structure the 3D catalyst network and directly fabricate a future benchmark GDE. While galvanostatic deposition (DC) produced Ag foam structures, they did not show improved performance compared to sputtered Ag electrodes. To address this, a pulsed DHBT process (PC) was introduced, resulting in efficient Ag deposition with high mass loading and ECSA values. Linear sweep voltammetry tests demonstrated improved performance for PC-DHBT electrodes compared to DC-DHBT-GDEs at industrially relevant current densities. To further enhance CO2 electrolysis, the Ag foam GDE was infiltrated with a perfluorosulfonic acid ionomer. This prevented flooding and improved CO2 mass transport. SEM and EDS analysis confirmed a homogeneous Ag/F distribution. The infiltrated electrode enabled CO2 electrolysis at higher current densities while maintaining its CO selectivity. Overall, this chapter presents a novel approach for fabricating polymer-based Ag-DHBT-GDEs using a combination of sputtering, electrochemical deposition and ionomer infiltration. The developed GDE allows for efficient CO2 electrolysis with unprecedented performance and selectivity. |
1. Introduction
Electrochemical carbon dioxide reduction reaction (CO2RR) can contribute decisively to overcoming the challenge of rising greenhouse gas emissions on our planet. Combining renewable energy sources like wind and solar power with the CO2RR, the carbon cycle can be closed and fossil-free chemicals like syngas (CO + H2), formic and acetic acid, or hydrocarbons, such as ethylene, can be produced, depending on the catalyst material applied.1 Ag-based catalysts are very promising in the field of CO2RR, since they show high selectivities towards the reduction of CO2 to CO with current efficiencies close to 100%. A highly efficient reduction process achieving high conversion rates is also indispensable to make the concept industrially viable. Towards this goal, highly catalytically active porous electrodes with large surface areas are required. All of the above requirements are met by 3D porous metal foam electrodes, that provide a free-standing structure, a connected hierarchical pore system with increased surface area and high mass transfer coefficients.2,3 An efficient, fast and solvent-free electrochemical deposition method to produce highly porous 3D structured foam electrodes is the dynamic hydrogen bubble templation (DHBT).4–6 This technique takes advantage of the parasitic hydrogen evolution reaction (HER) by which metal foam electrodes can be produced within seconds in a very controlled fashion. Metal ions are reduced and electrochemically deposited by applying sufficiently high overpotentials, while simultaneously the hydrogen bubbles originating from the HER detach from the substrate and work as a negative template for the formation of macro-porous layers and nanoscale interconnecting foam walls.7–10Most of the electrodes fabricated by DHBT are tested in H-cell configurations and evaluated for CO2RR in aqueous electrolyte, saturated with CO2.4–6,11–19 Here, the successful design of the DHBT foam electrodes and its effects on the CO2RR performance can be studied very effectively. A significant drawback in aqueous-fed systems is the limited CO2 solubility, resulting in long diffusion pathways of CO2 within the bulk electrolyte. Consequently, CO2 electrolysis cannot be performed at industrially relevant conditions in aqueous solution in H-cell configurations due to the rapid depletion of CO2 within the porous electrodes. Hence, lab scale testing is limited to maximum current densities of approx. 35 mA cm−2, which is far below the commercially relevant current densities (≥200 mA cm−2).20–22 Therefore, the transfer of these foam electrodes towards GDEs is necessary to take the next development step towards industrial application in order to approach a higher technology readiness level. One way to bypass such limitations is the utilization of gas diffusion electrodes (GDEs) in flow cell configurations and thereby avoiding the solvation limit of CO2 and decreasing diffusion lengths to approx. 50 nm.22 By intense contact and thus creating a three-phase boundary between the liquid phase, the gas phase and the catalytically active material, the mass transport is enhanced and industrially-required current densities can be accomplished.23–26 GDEs based on conductive materials are very prominent in the field of CO2RR, where carbon felts or metal meshes function as current collector and gas diffusion layers (GDL).27 Non-conductive GDLs based on polytetrafluoroethylene (PTFE) have become popular only recently and provide higher flooding resistance and achieve more stable conditions with improved CO2RR activity.23,28–32
On top of the GDLs, a coating of the active catalytic layer needs to be airbrushed,33 doctor-bladed,34,35 sputtered32,36,37 or electrochemically deposited38–40 to achieve a porous 3D composite structure. Luo et al.41 reported a manufacturing strategy to enhance the CO2RR activity by electrochemical deposition of a porous Zn catalyst on a Cu substrate and turning the metal structure into a GDE by fine-tuning with hydrophobic parts. Zelocualtecatl Montiel et al.33 deposited Bi foam structures electrochemically on a Cu substrate, which were removed by ultrasonication in an alcoholic solvent, before the Bi nanoparticle containing ink was airbrushed on a carbon GDL to manufacture the final GDE. These examples show that the DHBT approach can be implemented to manufacture a GDE in a facile and fast way. Yet, the goal of directly applying the electrochemical DHBT process for Ag catalysts on a polymer substrate and upscaling it to GDE processing has not been reached. This work addresses the development of a fast manufacturing routine for Ag-based GDE, which catalyze the CO2RR towards CO up to industrial current densities. Based on a non-conductive polymer filter material as gas diffusion layer, we implemented a thin conductive Ag layer on its surface by sputtering technique. By pulsed electrodeposition above the limiting current density a porous Ag foam was deposited on top of the porous sputter layer, forming a highly porous and active 3D structured electrode. Ultimately, the infiltration with a perfluorinated ionomer led to a GDE which enabled CO2RR towards CO at current densities up to 500 mA cm−2 in KHCO3 electrolyte with unprecedented high selectivity.
2. Experimental section
Preparation of Ag foam GDEs
All Ag foam GDEs were produced by the DHBT method at room temperature, using a two-electrode setup. To enable electrodeposition, a thin electrically conductive Ag layer (thickness = 120 nm) was galvanostatically sputtered (Balzers Union SCD 004) on top of a porous substrate, which consisted of a polymeric bilayer fibre fabric (polytetrafluoroethylene, PTFE and polypropylene, PP, Sterlitech Aspire Laminated ePTFE membrane QP952). The Ag-sputtered fibre fabrics were used as working electrodes (WE) and placed in a glass beaker, filled with an acidic aqueous electrolyte (0.02 M Ag2SO4 + 1.5 M H2SO4) together with a platinum foil (0.1 mm thickness, 99.995%, Goodfellow) which served as counter electrode (CE). For DHBT synthesis direct current (DC) and pulsed current (PC) deposition was applied using a potentiostat (Gamry Ref 3000, Gamry Instruments, USA). Different charge densities were used (20, 30, 40 and 50 A s cm−2) and the pulse-to-pause ratios were varied (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 2:1, 1:1, 1:2, 1:4), while the pause time was kept constant at 10 ms. To maintain a constant geometric area (Ageo) of 5.8 cm2, the sputtered substrates were mounted into an in-house built holder. During deposition, the distance between WE and CE was maintained at 8 mm. After deposition the GDEs were removed from the electrolytic bath immediately, dip washed in 0.1 M KHCO3 to neutralize residual sulphuric acid, before they were dip washed with ultrapure water and dried in air.
:0, 2:1, 1:1, 1:2, 1:4), while the pause time was kept constant at 10 ms. To maintain a constant geometric area (Ageo) of 5.8 cm2, the sputtered substrates were mounted into an in-house built holder. During deposition, the distance between WE and CE was maintained at 8 mm. After deposition the GDEs were removed from the electrolytic bath immediately, dip washed in 0.1 M KHCO3 to neutralize residual sulphuric acid, before they were dip washed with ultrapure water and dried in air.
Hydrophobic treatment of the DHBT-GDEs
A hydrophobic treatment of the GDEs was carried out by dropcasting a perfluorosulfonic acid (PFSA) suspension onto the electrodes. The suspension contained 1-propanol, water and 12–18 wt% (7–11 mg) of a short side chain ionomer with an equivalent weight of 800 gmol−1 (3 M-800EW, 3 M, USA). For the dropcasting process a planar position of the GDE was necessary to guarantee a uniform distribution of the ionomer suspension. After the dropcasting process the GDEs were dried in air for 24 h.Electrode performance and product analysis
Performance tests and CO2RR experiments of the GDEs with a geometric surface area of 3.14 cm2 were performed in three-electrode set-ups at room temperature in the same custom-built gas-tight polymer flow-cell (SI-2, ESI†) in two testing facilities (Bayreuth and Clausthal). In Bayreuth testing was performed using a potentiostat (Gamry Reference 3000, Gamry Instruments, USA). While testing, Ag-based GDEs, a platinum mesh (Ageo = 3.14 cm2, 99.95%, Biologic) and a HydroFlex® (Gaskatel GmbH, Germany) reversible hydrogen electrode (RHE) functioned as working electrode (WE), counter electrode (CE) and reference electrode (RE), respectively. Electrolyte containing compartments were separated by a Selemion anion exchange membrane (AGC Chemicals Europe, England). The gas compartment of the cell was continuously supplied with CO2 (≥99.995%, Air Liquide, France), at a flow rate of 20 N mL min−1, using a calibrated mass flow controller (Bronkhorst, Netherlands), while a backpressure of 30 mbar was maintained by a backpressure controller (Bronkhorst, Netherlands). The electrolyte utilized for the CO2RR (1 M KHCO3 solution) was prepared of powder (>99.7% KHCO3, Carl Roth, Germany) and ultrapure water (Barnstead GenPure Pro, Thermo Scientific, USA) respectively. The electrolyte was supplied and pumped using peristaltic pumps (DOSAFlex, Dosatronic GmbH, Germany) with a constant pump rate of 3 L h−1. Linear sweep voltammetry measurements (LSV) were accomplished applying the current interrupt (CI) mode to correct for the inner ohmic resistance (iR). The potential range was scanned with a constant rate of 10 mV s−1 up to −1.5 V vs. RHE. CO2RR chronopotentiometric measurements (CP) lasted up to 100 hours. For the CO2RR, different current densities (10, 30, 50, 100, 200, 300, 400 and 500 mA cm−2) were tested. The gas outlet of the cell was in-line connected to a gas chromatograph (GC) (GC-2014, Shimadzu, Japan) to detect and quantify the gaseous products of the CO2RR. The product gases H2, CO, and CO2 were analyzed every 5 min of electrolysis and separated in HayeSep capillary columns connected in series (HayeSep Q + HayeSep R, 80/100 mesh, 2 + 2 m × 1/8 in.). The GC was equipped with a thermal conductivity detector (TCD) for the detection of H2. A flame ionization detector (FID) was used to detect CO (in the form of CH4 after passing through a methanation unit before passing the FID, carrier gas = Ar 5.0).In Clausthal, the 1 M KHCO3 solution prepared from KHCO3 (>99 wt%, Carl Roth) was cycled through the catholyte and anolyte chambers at a flow rate of 480 mL min−1 each in isolated cycles. The electrolyte chambers were separated by an anion exchange membrane (fumasep® FAA-3-130-PK, fumatech). CO2 (99.995%, Linde) was supplied to the gas chamber at a flow rate of 50 N mL min−1. The experiments were conducted at ambient temperature (25 °C), with a catholyte pressure of 1000 mbar and a gas pressure of 1030 mbar, corresponding to a 30 mbar differential pressure over the GDE. Measurements were performed using an electrochemical workstation (Zahner ZenniumPro, Zahner GmbH) in a three-electrode configuration, with the GDE as WE, a nickel mesh as CE and a RHE (HydroFlex, Gaskatel) as reference. Each current density was applied galvanostatically for 30 min, starting from lowest to highest. At the 15 min and 30 min mark for each current density, gas products from gas and electrolyte chamber were analyzed using gas chromatography (Trace1310, ThermoFischer) in a two channel setup with column stripper (Hayesep Q and 5 Å molecular sieve), equipped with two TCDs.
The WE potential was continuously measured against the reference electrode and averaged for each current density after steady-state was reached. iR compensation was conducted for each point of measurement using the high frequency response method (HFR) to accurately account for the changing ohmic resistance caused by bubble formation.42,43 For this, an alternating current with an amplitude of 20 mA and a frequency of 15 kHz was superimposed over the direct current. The impedance response at this frequency was determined beforehand by impedance spectroscopy to be free of capacitive and inductive features at the measured current densities for the given setup. The faradaic efficiencies (FE) of the specific products i were determined by dividing their partial currents Ii by the total current It using eqn (1), where xi, zi, F, and ṅ are the volume fractions of the detected gaseous product, the number of electrons involved for a given reduction product, the Faraday constant, and the molar flow rate, respectively.
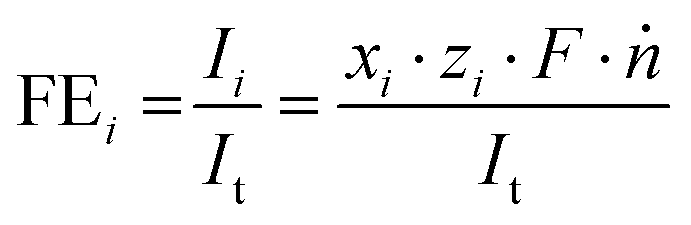 | (1) |
Electrochemical surface area
The electrochemical surface area (ECSA) of the GDEs was measured in alkaline electrolyte (1 M KHCO3), in a three-electrode set-up, using an in-house built H-cell. After conditioning the electrodes at the starting potential of 0.07 V vs. RHE for 30 s, the cyclic voltammograms (CVs) were recorded in the non-faradaic region between 0.03–0.07 V vs. RHE. The electrochemical double layer capacitance (CDL) was calculated according to eqn (2), where I was the average value of the charging current from the anodic and cathodic sweep, and V, the scan rate used to calculate the ECSA from eqn (3). Ten different scan rates were applied (10 to 100 mV s−1) and the step size was set to 2 mV. The double layer capacitance of the reference (Cref = 1.5 × 10−4 F ± 7.6 × 10−8 F) was determined experimentally from multiple CVs at ten different scan rates with the protocol described above, using a Ag sputtered polymer substrate, as described in the section about the preparation of the Ag foam GDEs.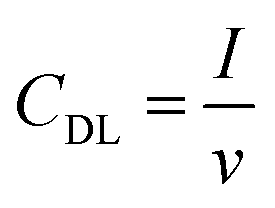 | (2) |
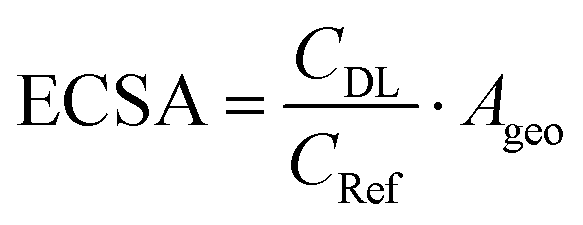 | (3) |
Gravimetric measurements
A high precision balance (Kern ABT 220-5DM, resolution of 0.01 mg) was used to weigh the Ag catalysts before and after electrodeposition and the corresponding mass differences (Δm) were calculated with respect to the total masses of the sputtered substrate.Contact angle measurements
To evaluate the hydrophobic properties of the GDEs, contact angle measurements were conducted using an aqueous 1 M KHCO3 solution. A drop of liquid (10 μl) was dispensed onto the centre of the tested electrode, using the dispensing option of the SCA 20 software (DataPhysics Instruments, Germany).Microstructure analysis
High resolution images were recorded using the secondary electron detector (SED) of a scanning electron microscope (SEM) (Zeiss Ultra plus, Carl Zeiss, Germany). To analyse the cross-section of electrodes at high resolution a Zeiss Crossbeam 340 Gallium-Focused Ion Beam-SEM (FIB-SEM) was applied. Pieces of 5 mm × 10 mm were cut out from the centre of the electrode with a scalpel and were mounted on a sample holder using a carbon pad. For the microstructural investigation, the sample surfaces and the cutting surfaces were analysed. FIB cuts were performed by applying an acceleration voltage of 3 keV and a current of 30 nA. Images were taken at an acceleration voltage of 1 keV. For chemical analyses a Bruker XFlash 6–100 energy-dispersive X-ray spectroscopy (EDS) detector was used. For these measurements an acceleration voltage of 5 keV was applied.X-ray diffraction
The Ag phase in the samples was analyzed by X-ray diffraction (Philips X’Pert-MPD PW 3040/00) in Bragg–Brentano geometry using unfiltered CuKα radiation (PW3373/00 Cu LFF at 40 kV and 40 mA). The 10 mm × 20 mm samples were placed on a sample holder steel disc and the sample height was adjusted using a dial gauge (Käfer GmbH, Germany). Diffractograms were recorded with a proportional point detector (WP3011) at a step size of 0.02° 2θ and a scan rate of 0.12° 2θ min−1 between 10° and 90° 2θ. Cu Kα2 reflections were removed post measurement with Highscore software.Physisorption measurements
The IUPAC recommendations for gas physisorption were taken into account to conduct the gas physisorption measurements.44 Therefore, the measurements were performed with Krypton as adsorptive gas at 77.15 K. Krypton physisorption data were collected in the p/p0 range of 0.09–0.2 (fixed p0: 0.35 kPa) by using an Anton Paar QuantaTec ASiQ-MP-MP-AG setup. The data were evaluated via Brunauer–Emmett–Teller (BET) method.45 Prior to the measurements the samples were cut in pieces and degassed at 120 °C for 12 h.Capillary flow porometry
Flow-through pores and bubble point pressure were determined using capillary flow porometry (Porometer 3Gzh, Quantachrome). A high-wetting liquid, with contact angle of zero, (Porofil, Quantachrome) was applied to the probe of 18 mm diameter, which was adjusted in the device. Afterwards, the fluid was driven out of the pores with a pressure gradient. The resulting air flow was detected on top of the probe. The bubble point pressure was determined at a flow rate of 0.1 L min−1. The flow-through pore distribution was finally determined by comparing the air flow through the wet and dry sample.3. Results and discussion
Modification of the polymer substrate for electrodeposition
To turn the insulating polymer fabric into a suitable substrate for electrodeposition, it was necessary to apply a conductive layer that retains its porous character to enable gas permeability in the GDE application. Fig. 1a shows the substrate that consists of a bipolymeric fibre fabric, made of a thin PTFE fibre layer (thickness ∼ 20 μm) on top of a larger PET fibre layer (thickness ∼ 105 μm), functioning as backing layer to increase its mechanical stability. Fig. 1b shows the thin Ag layer, which was sputtered on top of the polymer fibres and served as the contact layer for the later GDE structure. | ||
| Fig. 1 Analysis of electrode microstructure by scanning electron microscopy. (a) Cross-section of a pristine polymer fibre fabric and (b) top-down view of the polymer substrate, which was sputtered with Ag for 360 s. | ||
We chose Ag as contact layer material to avoid any influence or contamination by an additional metal on the CO2RR. Next to the electrical conductivity, which enables high-quality electrodeposition and high CO2RR activity at the later stages, another requirement for an efficient GDE is the facile and even supply of CO2 gas to the final catalyst layer. Therefore, the Ag sputter layer needed to be as thick as possible for electrical conductivity without losing its porosity as caused by a completely closed Ag surface layer on top of the PTFE fibres. The optimum parameters for the sputtered layer are shown in Table 1.
| Parameter | Value |
|---|---|
| Time, t [s] | 360–420 |
| Current, I [A] | 0.016 |
| Δm [mg] | 1.09 ± 0.12 |
| Specific mass, mspec [mg cm−2] | 0.13 |
| Thickness, d [nm] | 123.68 ± 13.95 |
Preparation and characterization of the DHBT microstructure
The sputtered Ag/polymer fibre substrate was used as WE in the electrochemical DHBT process as described in the experimental section above. Fig. 2a shows a graphical representation of the DHBT process, which makes use of the HER taking place on the conductive substrate, with its bubbles as the negative template responsible for the final porous foam structure. SEM images in Fig. 2b show the characteristic DHBT foam structure, consisting of microporous craters formed by hydrogen bubbles and the nanoporous foam walls consisting of Ag nano needles. An EDS analysis of the cross-section of a GDE after DHBT can be seen in the ESI†(SI-5), where the porous Ag foam forms the top layer of the GDE, with the hydrophobic PTFE layer underneath. This PTFE coating should prevent the electrolyte from entering the pores and prevent electrolyte flooding during CO2RR operation. Fig. 2c shows the XRD patterns of the stainless steel sample holder, the polymer substrate and the two Ag samples. Five small reflections can be attributed to the sample holder itself. XRD measurement of the polymer substrate revealed a decrease in intensity of the background signal. Additionally, amorphous reflections of the semi-crystalline structure of the PET (reflex at 22° and 26° 2θ) and the sharp crystalline peak of the PTFE and PET at 18° 2θ can be detected.46,47 Adding a very thin Ag sputter layer of approx. 130 nm on top results in five small reflections at 38°, 44°, 64°, 77° and 81° 2θ, representing the (111), (200), (220), (311) and (222) plane of the sputtered Ag. The XRD pattern of the electrodeposited Ag foam shows an increase in intensity of all characteristic fcc Ag reflections, accompanied by a decrease in intensity of the crystalline PTFE and amorphous PET reflections, indicating the successful preparation of a ready-to-use metallic GDE.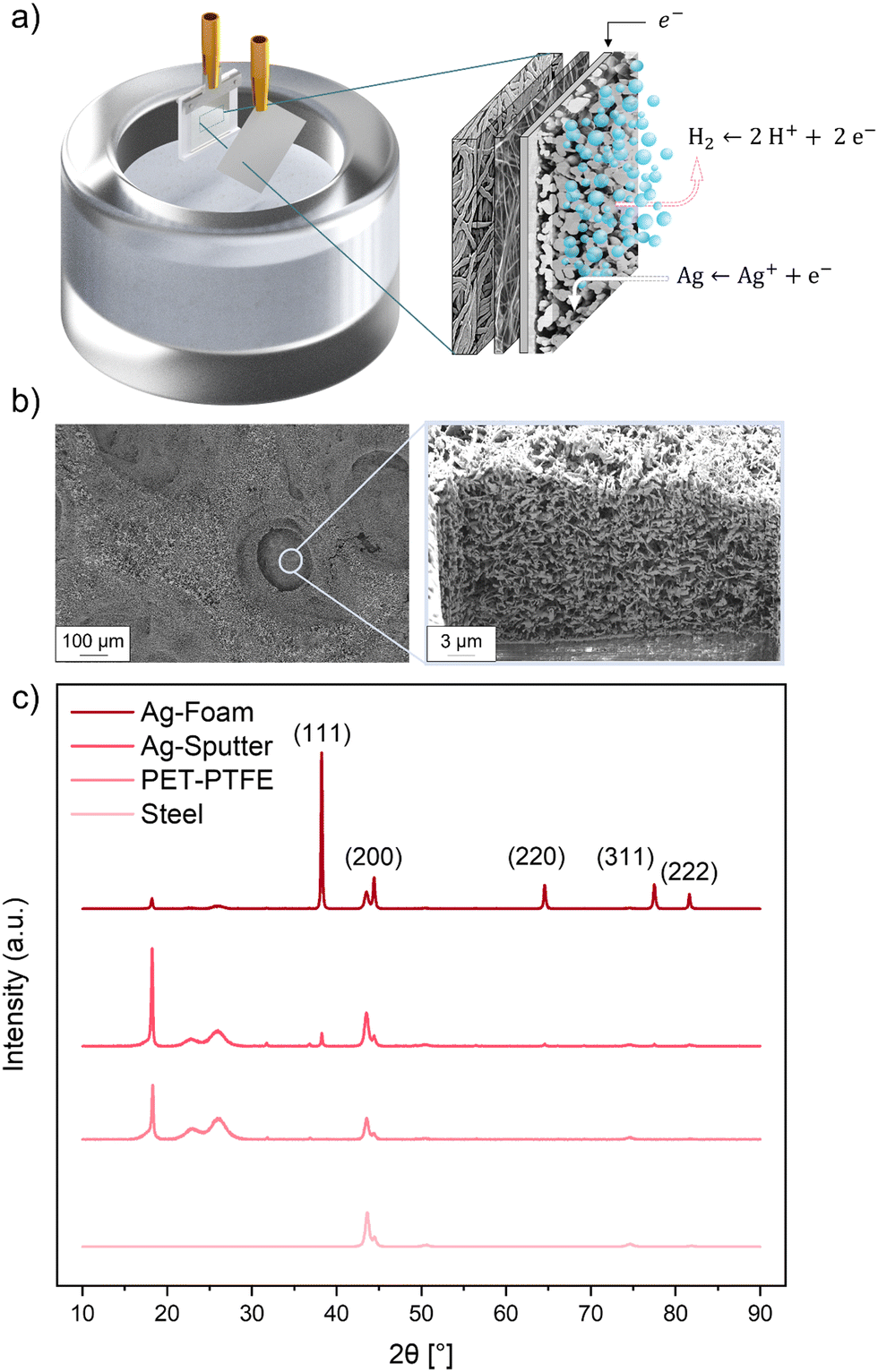 | ||
| Fig. 2 Analysis of the dynamic hydrogen bubble templation process. (a) Graphical representation of the working principle of the DHBT method, (b) SEM images of the Ag foam pores by HER occurring during DHBT method and high resolution image of a Ag pore with agglomerated Ag nanoparticles on top of the porous sputter layer, (c) XRD pattern of the reference steel plate (ICDD: 04-006-1881), PTFE-PET substrate (ICDD: 00-054-1595, 00-050-2275), the Ag sputter layer (ICDD: 04-001-2617) and the Ag DHBT foam (04-006-1881). | ||
Parameter selection for dynamic hydrogen bubble templation
To analyse the successful implementation of the DHBT process, the electrodeposited Ag mass was measured and the Ag deposition Efficiency (DEAg) was calculated (eqn (S1), ESI†). Fig. 3a demonstrates an increase in deposited Ag mass with increasing charge densities under galvanostatic deposition mode. By DC deposition, the highest Ag catalyst loading with ∼26 mg Ag at 50 A s cm−2 can be reached. For each electrode, the DEAg stays approximately on the same level for every deposition charge density (SI-1, ESI†). This result indicates that the increase in Ag loading is a consequence of increasing deposition time during DC deposition rather than of increased coulombic efficiency during the DHBT process. Based on the measurements of the double-layer capacitance, the calculated ECSA also increased with increasing deposition time under DC mode, compared to the sputtered electrode (300 s) (Fig. 3a). Similar results have been shown and reported for model electrodes for H-cell applications in an earlier study.48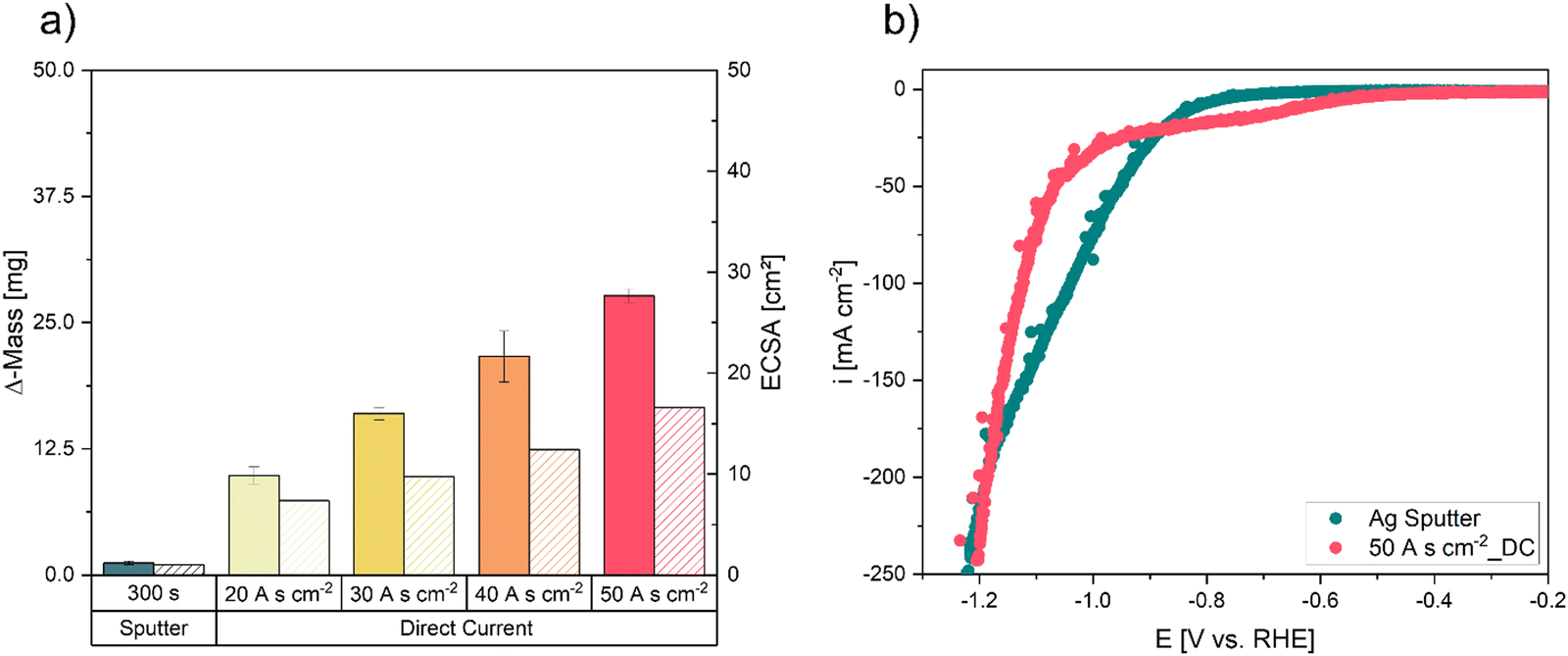 | ||
| Fig. 3 Characterization of the Ag-sputtered electrode compared to the DHBT electrodes manufactured in DC mode. (a) Deposited Ag mass and ECSA values of Ag-sputtered electrode and DHBT foam electrodes with increasing charge densities during DC mode, (b) linear sweep voltammetry curves for the Ag sputter electrode and the DC-50 A s cm−2 electrode to test their electrochemical performance (IR corrected potentials by applied CI method). | ||
The sputtered electrode (sputter time = 300 s) and the DC electrode with the highest ECSA and Δm, respectively (DC-50 A s cm−2), were tested and compared for their electrochemical performance in testing conditions identical to the CO2RR (Fig. 3b). In comparison to the sputtered electrode, the DC-50 A s cm−2 electrode exhibits slightly elevated current densities within the potential range of −0.6 to 1.0 V vs. RHE. This most certainly represents the onset potential for CO2RR at approximately −0.65 V vs. RHE, equating to an approximate current density of −10 mA cm−2. Below −1.0 V vs. RHE, the current densities of both electrodes increase steeply, with the sputtered electrode showing slightly higher values. The desirable value of 200 mA cm−2 can be reached for both electrodes at the tested potential of −1.2 V vs. RHE. Consequently, the intricate foam structure does not appear to lead to beneficial effects and higher performance.
As the DHBT electrode shows similar behaviour in the LSV sweep compared to the sputtered GDE at the target value of 200 mA cm−2, we focused on reducing the electrode overpotential by increasing the catalyst loading and hence, the electrode performance. A similar strategy of reduced overpotentials to increase the electrodes flooding resistance by inhibiting electrowetting, has been reported by Yang et al.49,50 Hence, we went from DC to PC electrodeposition, to realize a higher metal deposition efficiency by limiting HER during electrodeposition.51 By interrupting the direct current a quasi-stationary state of the reduction products during electrodeposition is obtained, while the depletion of the metal ions close to the surface area can be reduced and the competing HER should be limited. Nikolić et al.52 demonstrated that inhibiting electrolyte stirring by less pronounced HER bubble formation resulted in the formation of dendrites and small metal agglomerates, due to decreasing pulse-to-pause ratios in PC deposition. This effect should result in an increased surface area which we intended for optimized activity.
Fig. 4 illustrates the results of deposited Ag mass, the DEAg and the resulting total ECSA values as well as the specific ECSA in dependence of the applied pulse-to-pause ratios. Higher charge densities as well as lower pulse-to-pause ratios show an increase in Ag mass loadings (Fig. 4a). Electrodes deposited with the lowest pulse-to-pause ratio (1:4) show the highest increase in Ag loading, which results in the highest DEAg (Fig. 4b). Interestingly, the DEAg stayed on a similar level for pulse-to-pause ratios of 2:1 and 1:1 but increased at 1:2 and 1:4 with increasing deposition time. The reason for this might be the reduced cell potential with increasing deposition time (SI-3, ESI†). This effect is more pronounced at lower pulse-to-pause ratios, as the deposition times here are higher, when comparing the same charge density. These results indicate that the Ag loading can be significantly increased by higher deposition charges and shorter current pulses, so that Ag+ ions have enough time to diffuse from the bulk electrolyte to the electrode surface, where the electrochemical reduction takes place to form the delicate Ag foam structures.
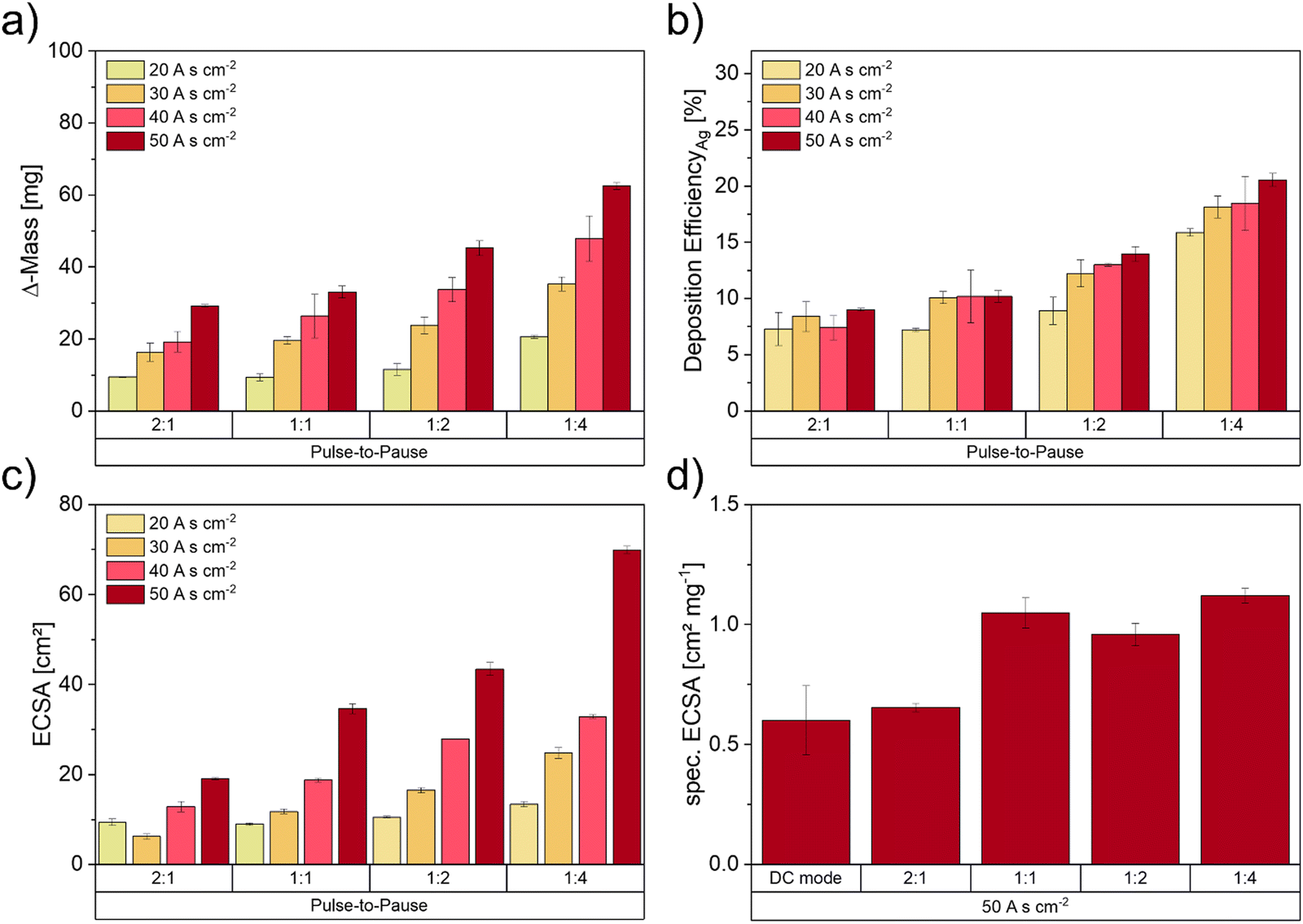 | ||
| Fig. 4 Electrochemically deposited masses (a) and the calculated FE of the DHBT in dependence of the varied pulse-to-pause ratios (b), calculated ECSA values by measurement of the double layer capacity and specific ECSA (d) of the deposited samples with 50 A s cm−2 in dependence of the pulse-to-pause ratios. | ||
Fig. 4c shows the calculated ECSA values based on the measurements of the double-layer capacitance. As the total ECSA values increased with increasing deposition times, electrodes deposited with 50 A s cm−2 show the highest values at every deposition condition. Except for one sample (30 A s cm−2_2:1), where the low ECSA value could be explained by a detached foam structure, we can report an increasing trend of ECSA by decreasing pulse-to-pause ratios for every deposition time. As the increase in deposited mass requires a higher volume and the decrease in pulse-to-pause ratios results in a decrease in agglomerate size, we therefore assume that the total value of ECSA is directly linked to the Ag mass deposited after the successful DHBT process (SI-4, ESI†). Including the error for deposited masses of 7.2% for 20 A s cm−2 and 3.4% for 50 A s cm−2, the correlation between the deposited mass and the ECSA is as high as 0.94. Thus, we can assume that the correlation is linear and almost ideal.
Since the analysis of the specific ECSA (Fig. 4d) shows an increase for decreasing pulse times, the mass change can not be the only affecting component. The reason for the rise in specific ECSA could be found in the microstructure of the foam, which may change with the pulse duration due to the necessity for the system to build new crystallisation nuclei at the start of each pulse. A quantitative analysis of Ag particles can be done by FIB tomography, which has been proven to be a successful characterisation method for Ag foam model electrodes in an earlier study.48
To analyze the electrochemical performance of the electrodes, LSV measurements in flow cell mode were performed (Fig. 5a). The various specimen achieve higher current densities and hence, a higher performance of the electrodes with decreasing pulse-to-pause ratios. A comparison of all tested electrodes reveals, that the overpotential was reduced by approximately 200 mV by the electrode deposited with the lowest pulse-to-pause ratio (50 A s cm−2_1:4). Since PC_2:0 and PC_1:4 electrodes show large differences in performance and 2:1, 1:1 and 1:2 samples show almost similar behaviour despite significant Ag mass differences of 29 mg, 33 mg and 45 mg, the mass of the deposited catalyst is not the only reason for the increase in performance. Also, the foam morphology has a large influence and depends on the pulse-to-pause ratio. Since DC electrodes were deposited at higher current densities with a more pronounced formation of larger foam craters and larger Ag agglomerates, it can be assumed that this macroporosity is not desired, as it showed poor electrode performance. Hence, a more compact and densely structured nanoporous foam, which is deposited at lower pulse-to-pause ratios appears to be more suitable for the CO2RR. This result is in good agreement with a previous study, where model electrodes deposited at higher current densities resulted in higher catalytically active morphologies by a more compact and dense foam structure, which showed increased CO2RR performance.48
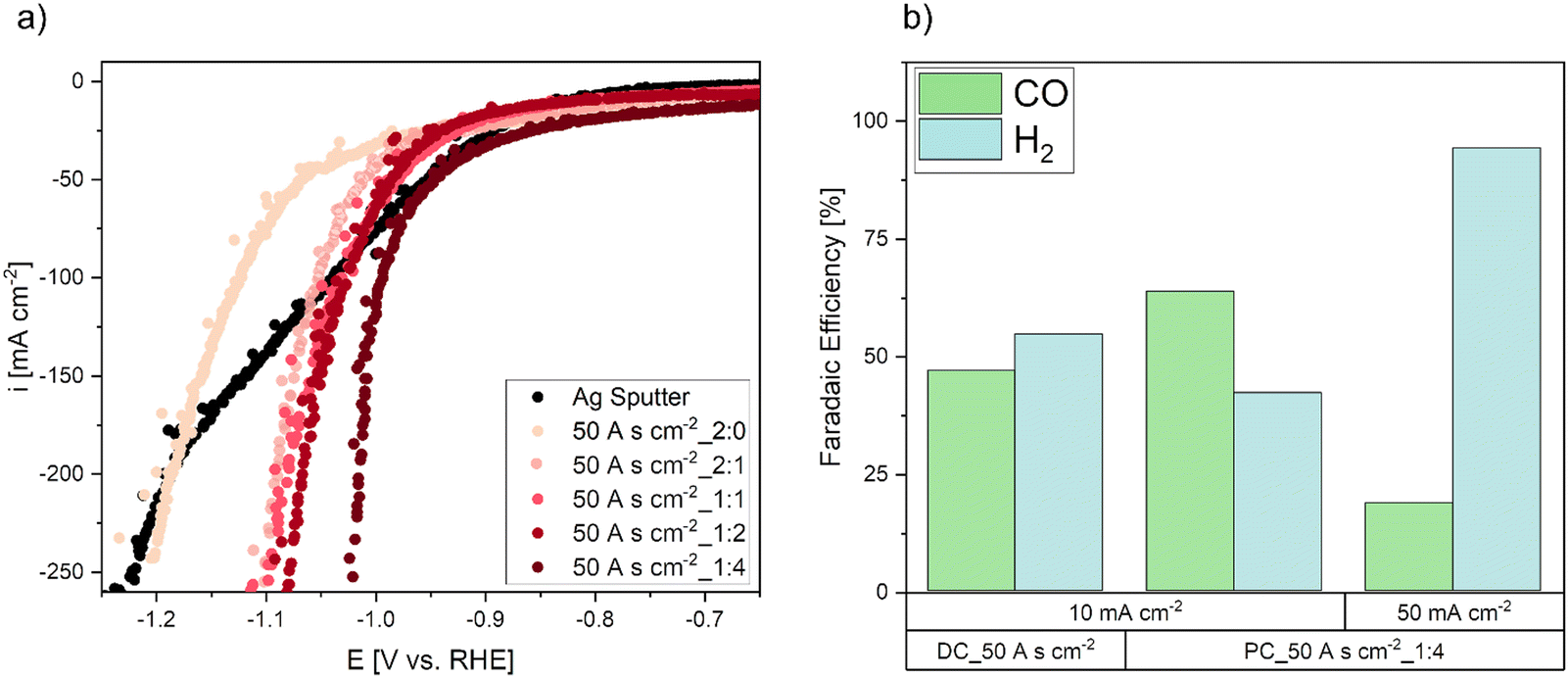 | ||
| Fig. 5 Electrochemical performance of the electrodeposited electrodes compared to the sputtered electrode. (a) Linear sweep voltammetry measurements for GDEs deposited with a charge density of 50 A s cm−2, (b) faradaic efficiency of the detected CO2RR products after 1 h of operation (b). | ||
As the CO2RR performance of the electrodes cannot be evaluated by LSV scans alone, due to parallel CO2RR and HER, galvanostatic operation at various current densities for 1 h was tested. The results of the galvanostatic CO2RR experiments are shown in Fig. 5b. At relatively low current densities of 10 mA cm−2, the DC electrode already shows higher HER selectivity than the PC_50A s cm−2_1:4 electrode. Increasing the galvanostatic operation from 10 to 50 mA cm−2 shifted the selectivity towards HER even for the PC electrode. This behaviour can be explained by a hydrophilic Ag catalyst layer and therefore significant mass transport limitations for CO2. As the electrode consists of pure Ag, the surface of the electrode is completely flooded with electrolyte and the reacting CO2 gas requires long pathways to be transported to the three-phase boundary regions.
Without hydrophobic components to reduce diffusion lengths in the GDE, the mass transport limitations are reached quickly by increasing current densities. As a result, the CO2RR selectivity shifts towards the undesired HER. Therefore, an additional hydrophobic component is needed to improve the performance of the GDE structure.
Perfluorinated ionomer infiltration for higher performance
Several approaches have been reported in the literature over the last few years on how to manage GDE flooding by the electrolyte and control electrode wettability.50 Especially incorporating different fluorinated and non-fluorinated polymers, such as PTFE, PFSA (e.g. Nafion) or PSMIM (e.g. Sustanion), can establish a suitable hydrophobic microenvironment.29,31,35,53,54 This results in decreased electrode flooding and promotes gas transport in the catalyst layer, which in turn will lead to improved current densities during the CO2RR.23,50,55 Furthermore, functional groups containing ionomers, such as PFSA with its sulfonic acid group, can not only provide hydrophobic regions (PTFE backbone), but also water pathways inside the ionomer structure within the GDE during CO2RR operation. This beneficial acidic microenvironment results in a higher ion transport, which is sufficient to sustain a proton supply for the complex multistep CO2 reduction reaction.23,56–58 These ionomer properties can be significantly improved by the choice of the ionomer type, the dispersion solvents or drying parameters and will have a strong influence on viscosity, distribution in the catalyst layer, structural conformation, water uptake and therefore ion conductivity.55 García de Arquer et al.23 designed and implemented a catalyst:ionomer bulk heterojunction (CIBH) based on a long-side-chain PFSA into the Ag-GDEs and reported a CO2RR partial current density as high as 400 mA cm−2.To enable the formation of favorable connected three-phase boundaries within the GDE structure, we implemented an infiltration process of a short side-chain perfluorosulfonic acid ionomer (3M-800EW). The beneficial effect of its low equivalent weight and the short-side chain results in a higher proton conductivity than long-side-chain ionomers used in previous studies.55,59 Studies by Lees et al.58 showed that an optimized ionomer loading is needed for the electrodes to perform at a high FECO. With increasing ionomer loading the FECO decreases and promotes the HER due to blockage of the pores by the ionomer. Consequently, we studied the influence of different ionomer loadings in our Ag foam GDE on the hydrophobic behaviour of the surface and the resulting ECSA. A graphical representation of the dropcasting infiltration process is shown in Fig. 6a.
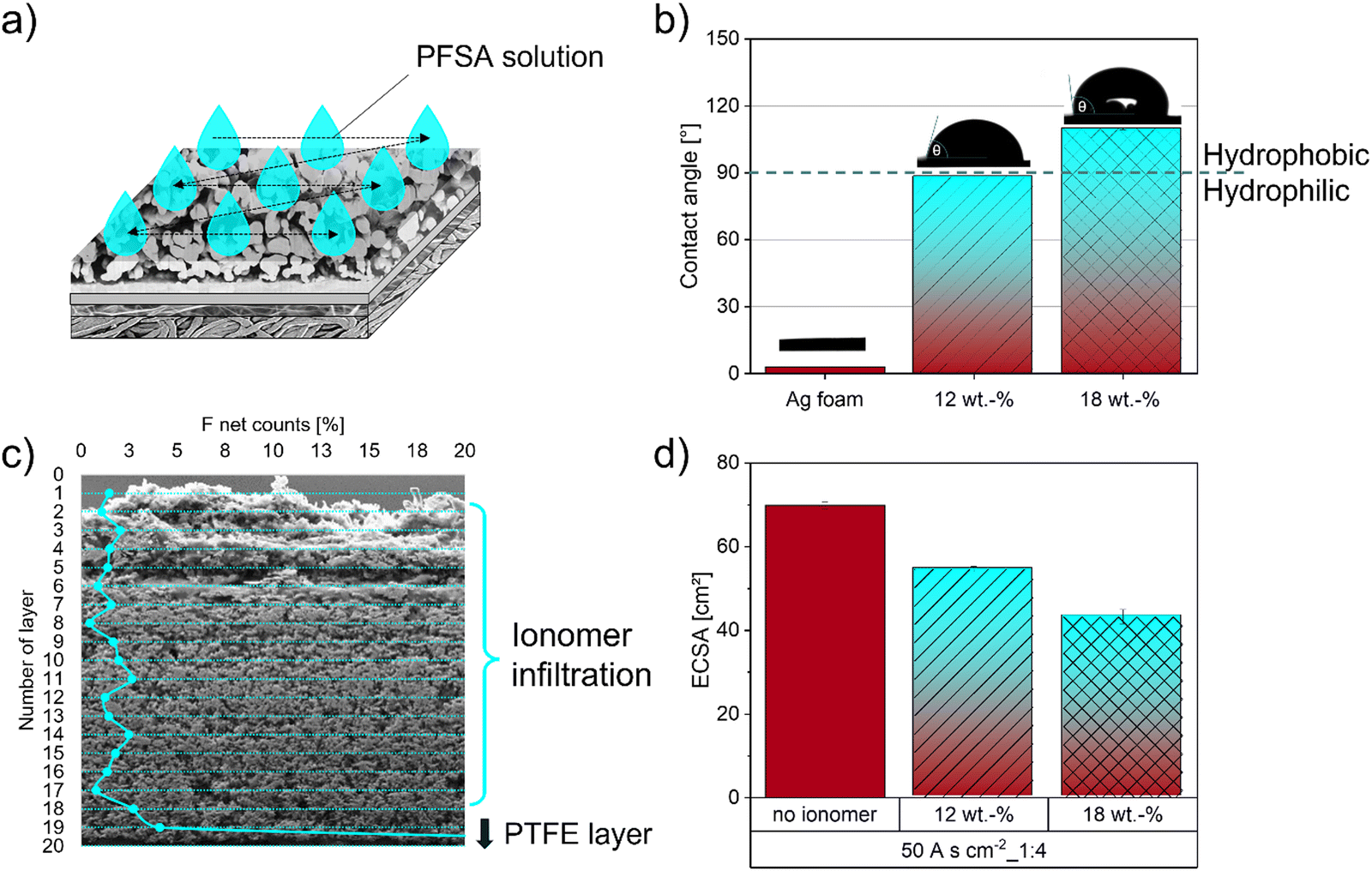 | ||
| Fig. 6 Graphical representation of the PFSA infiltration and its influence on the Ag electrode. (a) Infiltration routine of the electrodes, (b) surface contact angle of the Ag foam electrodes before and after infiltration with different PFSA contents, (c) F distribution along the thickness of the GDE and (d) effect of different weight percentage of perfluorinated polymer on the ECSA of the GDE. | ||
To study the influence of different ionomer concentrations on the hydrophobic behaviour of the electrode surface, the resulting contact angles were measured and presented in Fig. 6b. The contact angle increases by increasing ionomer concentration from approximately 3° of the pure Ag foam to 89° and 110° for 12 wt% and 18 wt% PFSA, respectively, which implies an increase in hydrophobicity of the surface. To investigate the quality of the infiltration process within the GDE we performed line scans on cross-sections of the GDEs. The results in Fig. 6c present an almost uniform F distribution across the thickness of the Ag foam on top of the PTFE layer of the polymer substrate. The increase in F signal at the bottom of the electrode is related to the PTFE fibre layer itself (SI-5, ESI†). To estimate the influence of the perfluorinated ionomer on the active catalytic sites we conducted ECSA measurements. The results in Fig. 6d clearly indicate that the ECSA is decreased with higher ionomer concentration as expected.50,60
To monitor possible changes in morphology regarding catalyst surface area, pore volume and pore diameter during the different processing steps, the BET method and capillary flow porometry were used, yielding the results shown in Fig. 7. By sputtering a thin layer of Ag on top of the polymer substrate, the surface area and the pore volume increased from 0.69 m2 g−1 to 0.85 m2 g−1 and from 34.5 cm3g−1 to 37.6 cm3g−1, respectively. Electrodeposition of the porous Ag foam only showed a minor increase of the surface area as well as pore volume, which was as expected according to earlier results.48 The infiltration of the Ag foam by the ionomer closed almost 50% of the pore volume compared to the starting substrate, which resulted in a BET surface area of 0.35 m2 g−1 and a pore volume of 20.8 cm3 g−1.
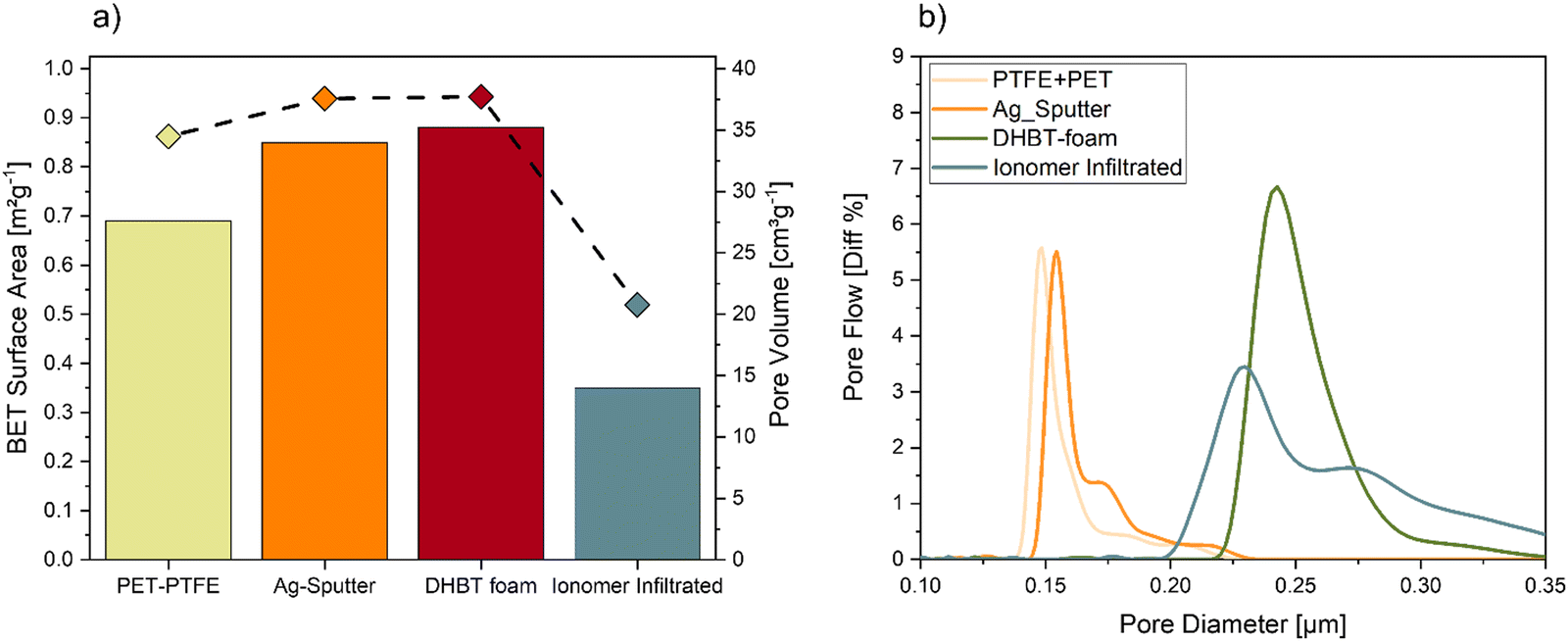 | ||
| Fig. 7 Characterization of the microstructure at each processing step during GDE manufacturing. (a) Calculated BET surface area by and measured pore volume by capillary flow porometry, (b) distribution of pore diameters. | ||
To compare our manufactured GDE to a commercially available, state of the art GDE, an Ag based Covestro GDE61 was tested up to current densities of 300 mA cm−2 (Fig. 8a) in the same electrochemical cell set-up. After showing the highest FECO of 100% at 30 mA cm−2, the FE decreased consistently with increasing current densities at galvanostatic operation and CO2RR shifted towards HER. At current densities of 300 mA cm−2 the commercial GDE showed a FECO of 55%. During testing, the working potential at the GDE linearly increased with increasing current densities.
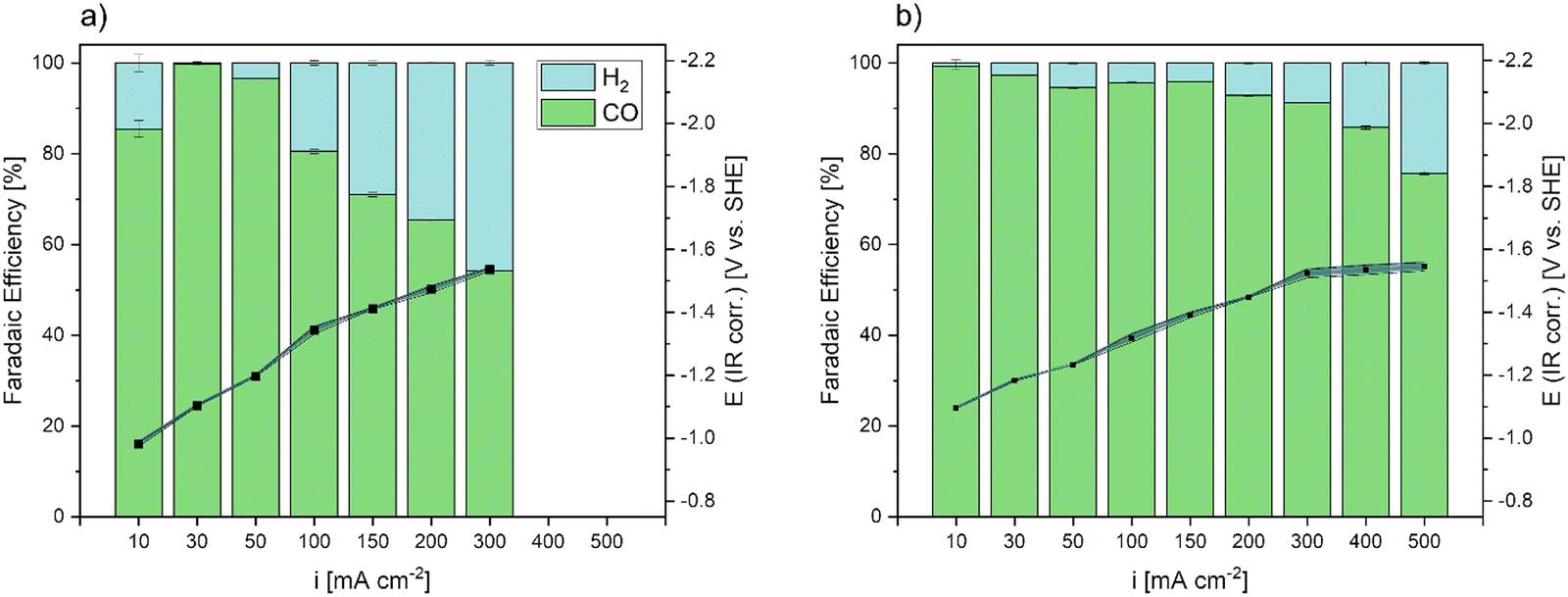 | ||
| Fig. 8 Normalized faradaic efficiencies of CO and H2 after galvanostatic operation at current densities up to 500 mA cm−2 and the recorded WE potential after correction for the inner resistance. (a) CO2RR results of a commercial Covestro GDE and (b) CO2RR results of the Ag DHBT GDE. Each curve represents the mean of two replicate measurements with the respective standard deviations indicated by the shaded areas. | ||
The Ag DHBT GDE with the most pronounced hydrophobic behaviour (18 wt.-%) was tested in galvanostatic CO2RR at each current density in comparison (Fig. 8b). Here, it needs to be mentioned that unlike other groups that tested similar electrode arrangements,31 only traces of C1+ products were produced, as we confirmed by NMR analysis (SI-7a, ESI†). Since it has been reported that Ag electrodes successfully convert CO2 to products other than CO during electrochemical reduction, the reason for this may be the flow cell design and the use of an anion exchange membrane. A use of the latter can cause diffusion of carbonate (CO32−) and formate ions to the anolyte compartment, which results in the electrochemical oxidation of the products.62 As the main aim in this study was the optimization of GDEs for CO production, investigation of tuning product selectivity by different cell-designs and ion exchange membranes is part of ongoing work.
The galvanostatic CO2RR performance of the Ag DHBT GDE was investigated, revealing excellent CO selectivity. At a current density of 10 mA cm−2, the FECO reached 100%, and it remained above 93% within the range of 30 mA cm−2 to 200 mA cm−2, while exhibiting negligible selectivity for the competing HER. Further increasing the applied current densities up to 300 mA cm−2, our fabricated Ag DHBT GDE exhibited a remarkable FECO of 91%, showcasing a performance improvement of 65% compared to the commercial GDE, especially at industrially relevant current densities. Even at higher current densities of 500 mA cm−2, the FECO remained above 76%, indicating the prevailance of CO2RR throughout the tested range. Notably, the WE potentials increased for both electrodes with increasing current densities, exhibiting similar values. While Ag electrodes are generally known for efficient CO2 conversion to CO,32,36,63 our GDE demonstrates exceptionally high CO selectivity at elevated current densities (500 mA cm−2) in a 1 M KHCO3 electrolyte. This performance surpasses current literature results and establishes our GDE as a future benchmark electrode32,36,63–67 (Table S2, ESI†).
We believe that the CO conversion performance of our Ag DHBT GDE can be achieved by the interplay of controlled pore sizes, amount of high surface area catalyst and the well distributed phase-separated PFSA. The hydrophobic property plays an important role in preventing the electrode from flooding, whereas the hydrophilic part can control the transport and availability of protons at the catalyst surface. The high number of catalytically active sites, given by the Ag nanostructured GDE reduces the energy barrier of the initial electron transfer step and consequently, improves the stabilization of the important COOHads intermediate.68 As already proposed in previous studies on the CO2RR mechanisms,57,69,70 the rate-limiting factor in this reaction is the combined proton and electron donation step (PEDS). We assume that the SO3− group of PFSA can provide protons for the reaction quickly and efficiently, resulting in a fast conversion rate from COOHads to COads. In addition to that, SO3− groups can have a decisive effect on OH− inhibition, generating higher pH values, which are required for the successful suppression of the HER, as already stated by Berlinguette et al.58 and Bell et al.53,54 We propose that the interplay of the COOHads stabilizing nanostructured Ag morphology, the fast PEDS by the highly proton conductive PFSA and the increase of local pH by OH− species provided by the fast cathodic reactions (CO2RR/HER), are responsible for FECO of more than 90% at current densities up to 300 mA cm−2.
To investigate the stability of the Ag DHBT GDE under conditions relevant to industrial applications, we performed galvanostatic long-term measurements at 200 mA cm−2 for a duration of up to 100 h (Fig. 9). Initially, during the initial 42 h, the GDE exhibited remarkably stable operating potentials with minor shifts of approximately 50 mV. In this phase of CO2RR, we posit the existence of short diffusion pathways, facilitating the reaction of solvated CO2 within the two-phase boundary at the catalytically active Ag particles, with subsequent rapid desorption of the formed CO molecules.
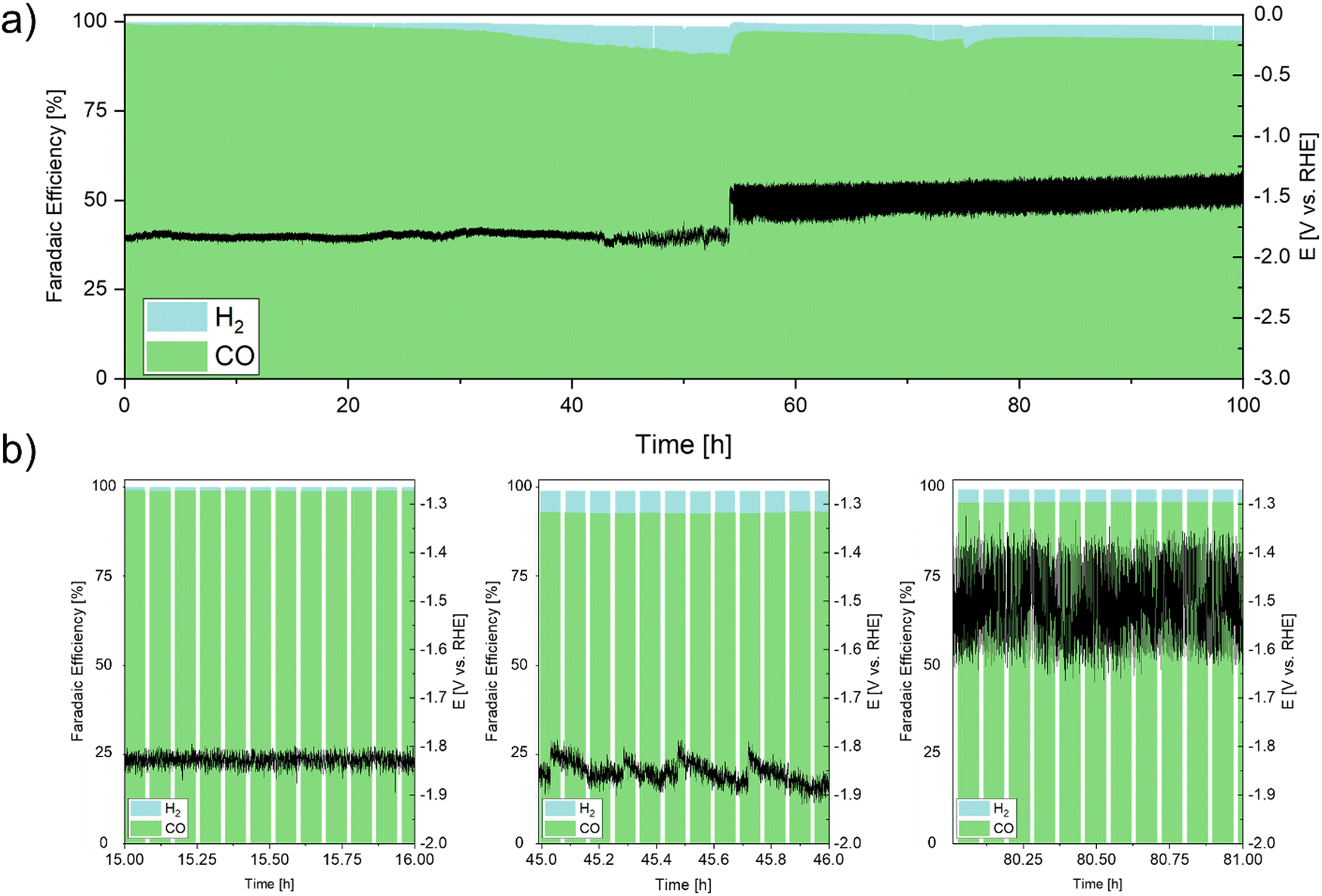 | ||
| Fig. 9 Long-term stability tests of the DHBT-GDE in galvanostatic mode at 200 mA cm−2 for 100 h. (a) WE potential without compensation and normalized FE of the only detected products over 100 h, (b) WE potential and FE of 1 h of operation each, during three different operating stages. | ||
Between the 43rd and 54th h of operation, the formation of salt precipitates was initiated on the backside of the GDE (PET layer). This led to an initial obstruction of CO2 pathways toward the catalyst layer within the GDE and the gas-outlet of the flow cell. Consequently, there was a gradual transition from flow-by to flow-through mode, causing CO2 to traverse the GDE towards the electrolyte side. This transition manifested as a progressively intensifying “saw-tooth” pattern in the potential curve, which became more pronounced over extended operation periods. Simultaneously, an increase in hydrogen evolution reaction (HER) selectivity occurred as protons were electrochemically reduced.71,72 During the shift from flow-by to flow-through mode, the working potential of the electrode experienced an alteration of around 100 mV. Subsequent to 54 h of operation, the electrode functioned entirely in CO2 flow-through mode, resulting in a potential shift of about 200 mV.
The severe changes in overpotential during the course of the experiment can be attributed to the shift from flow-by to flow-through mode, triggered by the accumulation of salt precipitates obstructing the CO2 outlet in the electrochemical cell. The restriction on CO2 gas exiting through its designated outlet prompted its permeation through the porous GDE. This transition not only enhanced mass transport by directly traversing the porous Ag foam layer, but also led to an augmented concentration of CO2 within the electrolyte due to its dissolution. The HER rate was reduced and maintained below 5%, contributing to an average FECO of 96% over the 100 h duration.
Post-mortem analysis of the microstructure after electrochemical long-term CO2RR by FIB-SEM analysis revealed only minor changes in needle shape of the foam walls, but an overall change in the foam morphology can be excluded. FIB-SEM-EDS analysis detected potassium within the structure, which can be attributed to certain amounts of precipitated salt crystals after 100 h of operation. The ionomer appears to be long-term stable as the F signal is homogeneously detected across the cross-section of the electrode (SI-9, SI-11 and SI-12, ESI†). The potassium detection through FIB analysis, as illustrated in SI-12 (ESI†), serves as strong evidence supporting the presence of electrolyte within the porous structure of the GDE during its operation. While this outcome implies that electrolyte has infiltrated throughout the entire thickness of the electrode's porous structure, the notably high FECO value suggests the continued existence of unobstructed pathways for gas diffusion. Consequently, it is reasonable to infer that there is a partial saturation of the GDE with electrolyte during operation, aligning with the desirable state of having a wetted GDE, without it being flooded.73 Analysis of the electrolyte compartments by 19F-NMR have shown, that no degradation of the PFSA occurs during the long-term electrochemical CO2RR experiment highlighting the stability of the system at industrially relevant conditions (SI-7b, ESI†). The low solubility limits of carbonate and bicarbonate species can lead to salt precipitation by crystallization on the surface of the GDL due to shielding effects that limit the evaporation of water between isolated salt crystals and their porous and hygroscopic nature, which pumps the electrolyte through its structure via capillary forces. Consequently, gas diffusion pathways in the electrode pores are blocked by precipitated salts, which consume CO2 and form bicarbonates through reactions with the carbonate deposits.50 Jeanty et al.74 demonstrated the importance of preventing the gas chamber from flooding and how the degree of turbulence and the amount of gas movement in the gas chamber can be improved, leading to optimised long-term stability of CO2 electrolysis by avoiding salt deposits.
4. Conclusions
We demonstrated a simple and rapid fabrication process for Ag-based GDEs by combining pulsed DHBT of Ag catalysts with ionomer infiltration. Before DHBT, a sputtering process was necessary to transform the insulating bipolymeric fibre fabric into a conductive substrate. The sputtered layer met the requirements for electrodeposition, leading to a conductive GDL that could withstand deposition currents up to 500 mA cm−2 without losing its porous character, which was essential for gas transport and distribution in the Ag foam catalyst structure during CO2RR.Galvanostatic deposition of Ag foams showed no advantage in electrode performance over sputtered Ag electrodes. Therefore, we switched from direct current DHBT to pulsed current DHBT and varied the pulse-to-pause ratios. The study revealed an efficient Ag deposition process resulting in high Ag mass deposition and high ECSA values while maintaining the porous foam structure by decreasing pulsing times. Linear sweep voltammetry tests showed a performance enhancement of about 200 mV at industrially viable current densities of PC DHBT electrodes compared to DC DHBT electrodes, resulting in slightly higher CO2RR selectivity at galvanostatic CO2RR operation. Increasing the operating current density required further electrode development, as HER became predominant for pure Ag foam electrodes at intermediate current densities. The incorporation of perfluorinated PFSA ionomer into the Ag foam GDE enabled efficient CO2RR operation at high current densities of up to 500 mA cm−2, while preserving a remarkable faradaic selectivity of 76% for CO production. This notable accomplishment positions the infiltrated electrode as a benchmark when compared to previous studies (Table S2, ESI†). In order to study the electrolyte distribution and the formation of the three-phase-boundary within the electrodes, operando analysis by synchrotron imaging should become the next step to gain more insights into electrolyte intrusion into the porous structure under operating conditions.72,75 These studies can help to improve electrode design by confining the pore structure during the manufacturing process and resulting in even better CO2RR performance.
Future work will incorporate the study of high current density deposition on the structure-performance relationship of the DHBT-GDE and the effect on the long term stability due to prevention of salt precipitations at the backside of the electrode, which needs to be improved to enhance the lifetime of the GDE. Additionally, we intend to present an extensive 4D depiction of electrolyte distribution. This will contribute to cultivating a profound comprehension of the exact localization of the three-phase boundary within porous Ag GDEs during CO2RR operations, thereby enhancing our understanding.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Conceptualization, C. R., T. T. and I. M.; methodology, H. H. and S. L.; validation, H. H. and M. K.; formal analysis, M. K., and H. H.; investigation, H. H., M. K, J. O., S. L., B. E. and A. H.; resources, C. R., T. T. and I. M.; data curation, H. H., M. P.-R.; writing – original draft preparation, H. H., M. K.; writing – review & editing, H. H. and C. R.; visualization, H. H. and M. P.-R.; supervision, C. R.; project administration, C. R. and H. H.; funding acquisition, C. R., T. T. and I. M.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors would like to thank the German Research Foundation (DFG) in the framework of the research unit “Multiscale analysis of complex three-phase systems: Oxygen and CO2 reduction at gas-diffusion electrodes in aqueous electrolyte” (FOR 2397; research grants RO 2454/16-1, RO 2454/16-2, TU 89/13-1, TU 89/13-2, MA 5039/3-1, MA 5039/3-2) for the financial support of this study. The authors would like to thank Mark Muggli (3M Advanced Materials Division, Dyneon GmbH) for provision of materials, Ruben Jäger (Humboldt-Universität zu Berlin) for assistance in NMR analysis, Tilo Held (University of Bayreuth) for electrochemical data treatment and Holger Kropf (Helmholtz-Zentrum Berlin) for his assistance with FIB analyses.References
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media, Electrochim. Acta, 1994, 39, 1833–1839, DOI:10.1016/0013-4686(94)85172-7.
- L. F. Arenas, C. Ponce de León and F. C. Walsh, Three-dimensional porous metal electrodes: Fabrication, characterisation and use, Curr. Opin. Electrochem., 2019, 16, 1–9, DOI:10.1016/j.coelec.2019.02.002.
- R. Du, et al., Engineering Self-Supported Noble Metal Foams Toward Electrocatalysis and Beyond, Adv. Energy Mater., 2020, 10, 1901945, DOI:10.1002/aenm.201901945.
- K. Klingan, et al., Reactivity Determinants in Electrodeposited Cu Foams for Electrochemical CO2 Reduction, ChemSusChem, 2018, 11, 3449–3459, DOI:10.1002/cssc.201801582.
- J. Darayen, et al., Porous Electrodeposited Cu as a Potential Electrode for Electrochemical Reduction Reactions of CO2, Appl. Sci., 2021, 11, 11104, DOI:10.3390/app112311104.
- T. Kottakkat, et al., Electrodeposited AgCu Foam Catalysts for Enhanced Reduction of CO2 to CO, ACS Appl. Mater. Interfaces, 2019, 11, 14734–14744, DOI:10.1021/acsami.8b22071.
- S. Cherevko, X. Xing and C.-H. Chung, Electrodeposition of three-dimensional porous silver foams, Electrochem. Commun., 2010, 12, 467–470, DOI:10.1016/j.elecom.2010.01.021.
- S. Vesztergom, et al., Hydrogen Bubble Templated Metal Foams as Efficient Catalysts of CO2 Electroreduction, ChemCatChem, 2021, 13, 1039–1058, DOI:10.1002/cctc.202001145.
- J. Wang, H. Shao, S. Ren, A. Hu and M. Li, Fabrication of porous Ni-Co catalytic electrode with high performance in hydrogen evolution reaction, Appl. Surf. Sci., 2021, 539, 148045, DOI:10.1016/j.apsusc.2020.148045.
- B. J. Plowman, L. A. Jones and S. K. Bhargava, Building with bubbles: the formation of high surface area honeycomb-like films via hydrogen bubble templated electrodeposition, Chem. Commun., 2015, 51, 4331–4346, 10.1039/c4cc06638c.
- Y. Yu, et al., Comparative Study between Pristine Ag and Ag Foam for Electrochemical Synthesis of Syngas with Carbon Dioxide and Water, Catalysts, 2019, 9, 57, DOI:10.3390/catal9010057.
- Q. Li, et al., Tuning Sn-Cu Catalysis for Electrochemical Reduction of CO2 on Partially Reduced Oxides SnOx-CuOx-Modified Cu Electrodes, Catalysts, 2019, 9, 476, DOI:10.3390/catal9050476.
- A. Dutta, C. E. Morstein, M. Rahaman, A. Cedeño López and P. Broekmann, Beyond Copper in CO2 Electrolysis: Effective Hydrocarbon Production on Silver-Nanofoam Catalysts, ACS Catal., 2018, 8, 8357–8368, DOI:10.1021/acscatal.8b01738.
- S. Sen, D. Liu and G. T. R. Palmore, Electrochemical Reduction of CO2 at Copper Nanofoams, ACS Catal., 2014, 4, 3091–3095, DOI:10.1021/cs500522g.
- D. Du, et al., Achieving Both High Selectivity and Current Density for CO2 Reduction to Formate on Nanoporous Tin Foam Electrocatalysts, ChemistrySelect, 2016, 1, 1711–1715, DOI:10.1002/slct.201600451.
- C. Chen, et al., Sharp Cu@Sn nanocones on Cu foam for highly selective and efficient electrochemical reduction of CO2 to formate, J. Mater. Chem. A, 2018, 6, 19621–19630, 10.1039/C8TA06826G.
- M. Rahaman, K. Kiran, I. Zelocualtecatl Montiel, A. Dutta and P. Broekmann, Suppression of the Hydrogen Evolution Reaction Is the Key: Selective Electrosynthesis of Formate from CO2 over Porous In55Cu45 Catalysts, ACS Appl. Mater. Interfaces, 2021, 13, 35677–35688, DOI:10.1021/acsami.1c07829.
- A. Dutta, et al., A Tandem (Bi2O3 → Bi met) Catalyst for Highly Efficient ec-CO2 Conversion into Formate: Operando Raman Spectroscopic Evidence for a Reaction Pathway Change, ACS Catal., 2021, 11, 4988–5003, DOI:10.1021/acscatal.0c05317.
- H. Mehrabi, et al., Electrochemical Control of the Morphology and Functional Properties of Hierarchically Structured, Dendritic Cu Surfaces, Energy Technol., 2023, 2201124, DOI:10.1002/ente.202201124.
- S. Hernandez-Aldave and E. Andreoli, Fundamentals of Gas Diffusion Electrodes and Electrolysers for Carbon Dioxide Utilisation: Challenges and Opportunities, Catalysts, 2020, 10, 713, DOI:10.3390/catal10060713.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Fundamentals and Challenges of Electrochemical CO2 Reduction Using Two-Dimensional Materials, Chem, 2017, 3, 560–587, DOI:10.1016/j.chempr.2017.09.009.
- T. Burdyny and W. A. Smith, CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions, Energy Environ. Sci., 2019, 12, 1442–1453, 10.1039/C8EE03134G.
- F. P. García de Arquer, et al., CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2, Science, 2020, 367, 661–666, DOI:10.1126/science.aay4217.
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Gas diffusion electrodes and membranes for CO2 reduction electrolysers, Nat. Rev. Mater., 2022, 7, 55–64, DOI:10.1038/s41578-021-00356-2.
- B. J. Etzold, et al., Understanding the activity transport nexus in water and CO2 electrolysis: State of the art, challenges and perspectives, Chem. Eng. J., 2021, 424, 130501, DOI:10.1016/j.cej.2021.130501.
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, Gas-Diffusion Electrodes for Carbon Dioxide Reduction: A New Paradigm, ACS Energy Lett., 2019, 4, 317–324, DOI:10.1021/acsenergylett.8b02035.
- V. M. Schmidt, Elektrochemische Verfahrenstechnik. Grundlagen, Reaktionstechnik, Prozeßoptimierung, Wiley-VCH, Weinheim, 2004 Search PubMed.
- Y. Xu, et al., Self-Cleaning CO2 Reduction Systems: Unsteady Electrochemical Forcing Enables Stability, ACS Energy Lett., 2021, 6, 809–815, DOI:10.1021/acsenergylett.0c02401.
- C.-T. Dinh, et al., CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface, Science, 2018, 360, 783–787, DOI:10.1126/science.aas9100.
- Y. Jännsch, M. Hämmerle, E. Simon, M. Fleischer and R. Moos, Contributions of Pulsed Operation Along with Proper Choice of the Substrate for Stabilizing the Catalyst Performance in Electrochemical Reduction of CO2 Toward Ethylene in Gas Diffusion Electrode Based Flow Cell Reactors, Energy Technol., 2022, 10, 2200046, DOI:10.1002/ente.202200046.
- C.-T. Dinh, F. P. García de Arquer, D. Sinton and E. H. Sargent, High Rate, Selective, and Stable Electroreduction of CO2 to CO in Basic and Neutral Media, ACS Energy Lett., 2018, 3, 2835–2840, DOI:10.1021/acsenergylett.8b01734.
- A. Senocrate, et al., Importance of Substrate Pore Size and Wetting Behavior in Gas Diffusion Electrodes for CO2 Reduction, ACS Appl. Energy Mater., 2022, 5, 14504–14512, DOI:10.1021/acsaem.2c03054.
- I. Zelocualtecatl Montiel, et al., CO2 Conversion at High Current Densities: Stabilization of Bi(III) Containing Electrocatalysts under CO2 Gas Flow Conditions, ACS Catal., 2022, 12(17), 10872–10886, DOI:10.1021/acscatal.2c02549.
- G. Liu, D. McLaughlin, S. Thiele and C. Pham, Correlating Catalyst Ink Design and Catalyst Layer Fabrication with Electrochemical CO2 Reduction Performance, Chemical Engineering Journal, 2023, 460 DOI:10.1016/j.cej.2023.141757.
- K. Junge Puring, et al., Electrochemical CO2 Reduction: Tailoring Catalyst Layers in Gas Diffusion Electrodes, Adv. Sustainable Syst., 2021, 5, 2000088, DOI:10.1002/adsu.202000088.
- N. B. D. Monti, et al., Facile Fabrication of Ag Electrodes for CO2-to-CO Conversion with Near-Unity Selectivity and High Mass Activity, ACS Appl. Energy Mater., 2022, 5, 14779–14788, DOI:10.1021/acsaem.2c02143.
- F. Huq, et al., Influence of the PTFE Membrane Thickness on the CO2 Electroreduction Performance of Sputtered Cu-PTFE Gas Diffusion Electrodes, ChemElectroChem, 2021, 9(1) DOI:10.1002/celc.202101279.
- S. Oh, et al., Fabrication of Large Area Ag Gas Diffusion Electrode via Electrodeposition for Electrochemical CO2 Reduction, Coatings, 2020, 10, 341, DOI:10.3390/coatings10040341.
- Y. S. Ham, et al., Proton-exchange membrane CO2 electrolyzer for CO production using Ag catalyst directly electrodeposited onto gas diffusion layer, J. Power Sources, 2019, 437, 226898, DOI:10.1016/j.jpowsour.2019.226898.
- J. Zhang, W. Luo and A. Züttel, Self-supported copper-based gas diffusion electrodes for CO2 electrochemical reduction, J. Mater. Chem. A, 2019, 7, 26285–26292, 10.1039/C9TA06736A.
- W. Luo, J. Zhang, M. Li and A. Züttel, Boosting CO Production in Electrocatalytic CO2 Reduction on Highly Porous Zn Catalysts, ACS Catal., 2019, 9, 3783–3791, DOI:10.1021/acscatal.8b05109.
- A. R. Heenan, J. Hamonnet and A. T. Marshall, Why Careful iR Compensation and Reporting of Electrode Potentials Are Critical for the CO2 Reduction Reaction, ACS Energy Lett., 2022, 7, 2357–2361, DOI:10.1021/acsenergylett.2c00800.
- W. Zheng, iR Compensation for Electrocatalysis Studies: Considerations and Recommendations, ACS Energy Lett., 2023, 8, 1952–1958, DOI:10.1021/acsenergylett.3c00366.
- M. Thommes, et al., Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report, Pure Appl. Chem., 2015, 87, 1051–1069, DOI:10.1515/pac-2014-1117.
- S. Brunauer, P. H. Emmett and E. Teller, Adsorption of Gases in Multimolecular Layers, J. Am. Chem. Soc., 1938, 60, 309–319, DOI:10.1021/ja01269a023.
- L. Chi, et al., Novel g-C3N4/TiO2/PAA/PTFE ultrafiltration membrane enabling enhanced antifouling and exceptional visible-light photocatalytic self-cleaning, Catal. Today, 2019, 335, 527–537, DOI:10.1016/j.cattod.2019.02.027.
- S. Fernández, A. Martínez-Steele, J. J. Gandía and F. B. Naranjo, Radio frequency sputter deposition of high-quality conductive and transparent ZnO:Al films on polymer substrates for thin film solar cells applications, Thin Solid Films, 2009, 517, 3152–3156, DOI:10.1016/j.tsf.2008.11.097.
- H. Hoffmann, et al., Multi-scale morphology characterization of hierarchically porous silver foam electrodes for electrochemical CO2 reduction, Commun. Chem., 2023, 6 DOI:10.1038/s42004-023-00847-z.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, Role of the Carbon-Based Gas Diffusion Layer on Flooding in a Gas Diffusion Electrode Cell for Electrochemical CO2 Reduction, ACS Energy Lett., 2021, 6, 33–40, DOI:10.1021/acsenergylett.0c02184.
- M. Li, et al., The role of electrode wettability in electrochemical reduction of carbon dioxide, J. Mater. Chem. A, 2021, 9, 19369–19409, 10.1039/d1ta03636j.
- N. Kanani Galvanotechnik. Grundlagen, Verfahren und Praxis einer Schlüsseltechnologie. 3rd ed. Hanser, München, 2020 Search PubMed.
- N. D. Nikolić and G. Branković, Effect of parameters of square-wave pulsating current on copper electrodeposition in the hydrogen co-deposition range, Electrochem. Commun., 2010, 12, 740–744, DOI:10.1016/j.elecom.2010.03.021.
- J. C. Bui, et al., Engineering Catalyst-Electrolyte Microenvironments to Optimize the Activity and Selectivity for the Electrochemical Reduction of CO2 on Cu and Ag, Acc. Chem. Res., 2022, 55, 484–494, DOI:10.1021/acs.accounts.1c00650.
- C. Kim, et al., Tailored catalyst microenvironments for CO2 electroreduction to multicarbon products on copper using bilayer ionomer coatings, Nat. Energy, 2021, 6, 1026–1034, DOI:10.1038/s41560-021-00920-8.
- A. Kusoglu and A. Z. Weber, New Insights into Perfluorinated Sulfonic-Acid Ionomers, Chem. Rev., 2017, 117, 987–1104, DOI:10.1021/acs.chemrev.6b00159.
- Y. Xue, Y. Guo, H. Cui and Z. Zhou, Catalyst Design for Electrochemical Reduction of CO2 to Multicarbon Products, Small methods, 2021, 5, e2100736, DOI:10.1002/smtd.202100736.
- J. Rosen, et al., Mechanistic Insights into the Electrochemical Reduction of CO2 to CO on Nanostructured Ag Surfaces, ACS Catal., 2015, 5, 4293–4299, DOI:10.1021/acscatal.5b00840.
- E. W. Lees, et al., Linking gas diffusion electrode composition to CO2 reduction in a flow cell, J. Mater. Chem. A, 2020, 8, 19493–19501, 10.1039/d0ta03570j.
- G. A. Giffin, G. M. Haugen, S. J. Hamrock and V. Di Noto, Interplay between structure and relaxations in perfluorosulfonic acid proton conducting membranes, J. Am. Chem. Soc., 2013, 135, 822–834, DOI:10.1021/ja3099799.
- L. de Sousa, C. Harmoko, N. Benes and G. Mul, Optimizing the Ink Formulation for Preparation of Cu-Based Gas Diffusion Electrodes Yielding Ethylene in Electroreduction of CO2, ACS ES&T Engg, 2021, 1, 1649–1658, DOI:10.1021/acsestengg.1c00228.
- A. Bulan, Method of manufacturing gas diffusion electrodes, 2006 Search PubMed.
- G. O. Larrazábal, et al., Analysis of Mass Flows and Membrane Cross-over in CO2 Reduction at High Current Densities in an MEA-Type Electrolyzer, ACS Appl. Mater. Interfaces, 2019, 11, 41281–41288, DOI:10.1021/acsami.9b13081.
- M. Sassenburg, et al., Characterizing CO2 Reduction Catalysts on Gas Diffusion Electrodes: Comparing Activity, Selectivity, and Stability of Transition Metal Catalysts, ACS Appl. Energy Mater., 2022, 5, 5983–5994, DOI:10.1021/acsaem.2c00160.
- S. Verma, X. Lu, S. Ma, R. I. Masel and P. J. A. Kenis, The effect of electrolyte composition on the electroreduction of CO2 to CO on Ag based gas diffusion electrodes, Phys. Chem. Chem. Phys., 2016, 18, 7075–7084, 10.1039/c5cp05665a.
- S. C. Abeyweera, J. Yu, J. P. Perdew, Q. Yan and Y. Sun, Hierarchically 3D Porous Ag Nanostructures Derived from Silver Benzenethiolate Nanoboxes: Enabling CO2 Reduction with a Near-Unity Selectivity and Mass-Specific Current Density over 500 A/g, Nano Lett., 2020, 20, 2806–2811, DOI:10.1021/acs.nanolett.0c00518.
- S. Liu, et al., Shape-Dependent Electrocatalytic Reduction of CO2 to CO on Triangular Silver Nanoplates, J. Am. Chem. Soc., 2017, 139, 2160–2163, DOI:10.1021/jacs.6b12103.
- C. Kim, et al., Achieving Selective and Efficient Electrocatalytic Activity for CO2 Reduction Using Immobilized Silver Nanoparticles, J. Am. Chem. Soc., 2015, 137, 13844–13850, DOI:10.1021/jacs.5b06568.
- M. Ma, B. J. Trześniewski, J. Xie and W. A. Smith, Selective and Efficient Reduction of Carbon Dioxide to Carbon Monoxide on Oxide-Derived Nanostructured Silver Electrocatalysts, Angew. Chem., 2016, 55, 9748–9752, DOI:10.1002/anie.201604654.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, Catalysts and Reaction Pathways for the Electrochemical Reduction of Carbon Dioxide, J. Phys. Chem. Lett., 2015, 6, 4073–4082, DOI:10.1021/acs.jpclett.5b01559.
- H. A. Hansen, J. B. Varley, A. A. Peterson and J. K. Nørskov, Understanding Trends in the Electrocatalytic Activity of Metals and Enzymes for CO2 Reduction to CO, J. Phys. Chem. Lett., 2013, 4, 388–392, DOI:10.1021/jz3021155.
- C. Lee, et al., Bubble Formation in the Electrolyte Triggers Voltage Instability in CO2 Electrolyzers, iScience, 2020, 23, 101094, DOI:10.1016/j.isci.2020.101094.
- H. Hoffmann, et al., Development of a Modular Operando Cell for X-ray Imaging of Strongly Absorbing Silver-Based Gas Diffusion Electrodes, J. Electrochem. Soc., 2022, 169, 44508, DOI:10.1149/1945-7111/ac6220.
- L.-C. Weng, A. T. Bell and A. Z. Weber, Modeling gas-diffusion electrodes for CO2 reduction, Phys. Chem. Chem. Phys., 2018, 20, 16973–16984, 10.1039/c8cp01319e.
- P. Jeanty, et al., Upscaling and continuous operation of electrochemical CO2 to CO conversion in aqueous solutions on silver gas diffusion electrodes, J. CO2 Util., 2018, 24, 454–462, DOI:10.1016/j.jcou.2018.01.011.
- M. C. Paulisch, et al., Operando Synchrotron Imaging of Electrolyte Distribution in Silver-Based Gas Diffusion Electrodes During Oxygen Reduction Reaction in Highly Alkaline Media, ACS Appl. Energy Mater., 2021, 4, 7497–7503, DOI:10.1021/acsaem.1c01524.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00220a |
| This journal is © The Royal Society of Chemistry 2024 |