
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances and perspectives on intercalation layered compounds part 1: design and applications in the field of energy
Chiara
Bisio
 *ab,
Jocelyne
Brendlé
*c,
Sébastien
Cahen
d,
Yongjun
Feng
e,
Seong-Ju
Hwang
f,
Klara
Melanova
g,
Morena
Nocchetti
*h,
Dermot
O'Hare
i,
Pierre
Rabu
j and
Fabrice
Leroux
*k
*ab,
Jocelyne
Brendlé
*c,
Sébastien
Cahen
d,
Yongjun
Feng
e,
Seong-Ju
Hwang
f,
Klara
Melanova
g,
Morena
Nocchetti
*h,
Dermot
O'Hare
i,
Pierre
Rabu
j and
Fabrice
Leroux
*k
aDipartimento di Scienze e Innovazione Tecnologica, Università del Piemonte Orientale, Viale T. Michel 11, 15121 Alessandria, AL, Italy. E-mail: chiara.bisio@uniupo.it
bCNR-SCITEC Istituto di Scienze e Tecnologie Chimiche “Giulio Natta”, Via C. Golgi 19, 20133 Milano, MI, Italy
cInstitut de Science des Matériaux de Mulhouse CNRS UMR 7361, Université de Haute-Alsace, Université de Strasbourg, 3b rue Alfred Werner, 68093 Mulhouse CEDEX, France. E-mail: jocelyne.brendle@uha.fr
dInstitut Jean Lamour – UMR 7198 CNRS-Université de Lorraine, Groupe Matériaux Carbonés, Campus ARTEM – 2 Allée André Guinier, BP 50840, F54011, NancyCedex, Francia
eState Key Laboratory of Chemical Resource Engineering, Beijing Engineering Center for Hierarchical Catalysts, Beijing University of Chemical Technology, No. 15 Beisanhuan East Road, Beijing, 100029, China
fDepartment of Materials Science and Engineering, College of Engineering, Yonsei University, Seoul 03722, Republic of Korea
gCenter of Materials and Nanotechnologies, Faculty of Chemical Technology, University of Pardubice, Studentská 95, 532 10 Pardubice, Czech Republic
hDepartment of Pharmaceutical Sciences, University of Perugia, Via del Liceo 1, 06123 Perugia, Italy. E-mail: morena.nocchetti@unipg.it
iChemistry Research Laboratory, University of Oxford Department of Chemistry, 12 Mansfield Road, Oxford, OX1 3TA, UK
jInstitut de Physique et Chimie des Matériaux de Strasbourg, CNRS – Université de Strasbourg, UMR7504, 23 rue du Loess, BP43, 67034 Strasbourg cedex 2, France
kInstitut de Chimie de Clermont-Ferrand, Université Clermont Auvergne, UMR CNRS 6296, Clermont Auvergne INP, 24 av Blaise Pascal, BP 80026, 63171 Aubière cedex, France. E-mail: fabrice.leroux@uca.fr
First published on 11th July 2024
Abstract
Herein, initially, we present a general overview of the global financial support for chemistry devoted to materials science, specifically intercalation layered compounds (ILCs). Subsequently, the strategies to synthesise these host structures and the corresponding guest–host hybrid assemblies are exemplified on the basis of some families of materials, including pillared clays (PILCs), porous clay heterostructures (PCHs), zirconium phosphate (ZrP), layered double hydroxides (LDHs), graphite intercalation compounds (GICs), graphene-based materials, and MXenes. Additionally, a non-exhaustive survey on their possible application in the field of energy through electrochemical storage, mostly as electrode materials but also as electrolyte additives, is presented, including lithium technologies based on lithium ion batteries (LIBs), and beyond LiBs with a focus on possible alternatives such XIBs (X = Na (NIB), K (KIB), Al (AIB), Zn (ZIB), and Cl (CIB)), reversible Mg batteries (RMBs), dual-ion batteries (DIBs), Zn-air and Zn-sulphur batteries and supercapacitors as well as their relevance in other fields related to (opto)electronics. This selective panorama should help readers better understand the reason why ILCs are expected to meet the challenge of tomorrow as electrode materials.
1. Introduction
Today, science is required more than ever to improve our daily life, and specifically, the science of materials must address new challenges concerning major human issues, including breakthrough solutions in the fields of medicine, energy storage and transport, agriculture and the environment. Furthermore, this must be achieved using approaches complying with sustainable development, and if possible, the circular economy. These ever-increasing requirements are causing certain technologies to be reconsidered in terms of their carbon footprint, the reduction of fossil fuels, the rarefaction of elements and their balance from the mine to their end of life, which is the so-called life cycle assessment combined with sustainable development during their period of use. Thus, as scientists, we must consider these criteria because we are creating the materials of tomorrow.There is an urgent need for fundamental research to feed its successes into current technologies by crossing the famous technology readiness levels (TRLs) as quickly as possible from proof of concept to prototype manufacturing. Because of their reactivity, layered materials and, more generally, intercalation compounds are of great interest in many fields such as catalysis, photo-physical processes, electronics, energy, drug delivery, biomaterials, coatings, composites as polymer fillers, and water remediation. Funding agencies contributing the most in the domain of intercalation compounds (according to about 3600 articles) are the National Science Foundation of China (NSFC) holding about 18% of the total contribution, the European Commission holding 5.5%; the United States Department of Energy (DOE) holding 5.5%; the National Science Foundation (NSF) holding 4.9%; the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, holding 4.1%; Japan Society for the Promotion of Science with 3.1%; the German Research Foundation (DFG) with 2.9%; Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPQ) with 2.6% and Coordenacao de Aperfeicoamento de Pessoal de Nivel Superior (CAPES) with 2.2%.
Here, our aim is to unambiguously show the advantages, limitations and possible disadvantages of intercalation compounds in addressing scientific and technological challenges to the greatest extent. Among the intercalation layered compounds (ILCs), the family of graphene, transition-metal dichalcogenides, layered double hydroxides, layered phosphates, smectite clays, layered silicates, perovskite-type materials, zeolites and mesoporous materials are still investigated and we want to give the most useful information. To achieve this, a non-exhaustive state of the art is compiled and organised into two parts.
This present part covers:
- Design and synthesis strategies (e.g. modelling and prediction by increasingly fine simulation, process design, new synthetic methods, sustainable and green synthesis, scaled-up processes, hierarchical assemblies, core–shell structures, additive manufacturing, spray, electrospinning, nanosheets, and superlattice-like heterostructures).
- Energy (e.g. new-generation solar cells, Li-ion batteries, and H2 storage), supercapacitors (energy grid saving, lighting and emission/shaping in polymers).
Alternatively, the second part is devoted to:
- Catalysis (e.g. photo-, bio-, electro-catalysis, water electrolysis, H2 production, fine chemicals, separations, recycling, and efficiency).
- Environment (CO2 capture, soil remediation, and plant feeding).
- Health (drug delivery/release, patch, bone regeneration, and biomaterials).
- Engineering coating/composites as polymer fillers/cements/textiles (anti-oxidation, anti-corrosion, and fire retardant).
This should highlight some expectations regarding future developments and suitable applications of intercalation compounds.
2. Design and synthesis strategies
2.1. Where do we come from and where are we going in the case of intercalation compounds?
Exchange reactions, and more generally involving intercalation chemistry are often considered as the possibility of adapting the host structure to specific properties. Two-dimensional (2D) materials have been recently reviewed5–7 including layered transition metal chalcogenides.8,9
Ion-exchange materials are well-described in the literature, and their members usually belong to a family, as exemplified by the anionic exchangers such as layered double hydroxides (LDHs) and cationic exchangers such as cationic clays, alkali transition metal (bronze) oxide or metal(IV) phosphates. In some specific cases, opposite charge ions or neutral species can be incorporated in a host structure. In the case of the former, cation exchange reactions are reported for hydrotalcite by revisiting sulphate and alkaline cations both interleaved, as found for the shigaite mineral.10
An additional step involves the use of the spatial confinement provided by a two-dimensional network. A recent review highlighted the interest in using 2D materials as nano-reactors for the confined synthesis of large variety of nanostructured materials showing promising for electrocatalytic applications.11
In the case of cationic clays, ion exchange reactions lead to a wide family of modified 2D materials. Clay-based catalysts can be prepared by ion-exchange reactions.12 In acid-activated clays, the intercalation of protons between the interlayer spaces of clay is carried out by treating the solids in acid solution. Consequently, slight modification of the silicate layer (often related to a partial lamellae amorphization) and textural properties is achieved.13
However, appropriate modification with different functional groups is an interesting approach to reduce the destructive effects of acid activation and increase the interface properties for a wide range of applications.14
The introduction of organic species and/or polymers in the interlayer space of clays results in the modification of their hydrophilic character. Alkylammonium and alkyl-phosphonium cations are the main surfactants used in the intercalation process.
Anionic surfactants have been also used for clay modification.15 Non-ionic surfactants display better properties than other surfactants because they are thermally and chemically stable, preserve the exchangeable inorganic cations and are normally non-toxic.16 Gemini surfactants have been also used for the modification of clays.17,18
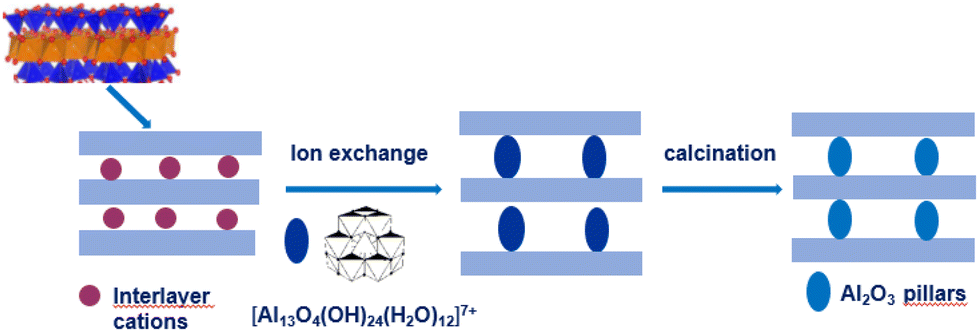 | ||
| Fig. 1 Preparation of Al2O3 pillared clays. | ||
Porous clay heterostructures (PCHs) are a class of interesting materials that can be obtained from cationic clays and are characterised by a combination of microporous and mesoporous structures, surface acidity and cation exchange capacity.22 The method used to prepare PCHs is the intercalation of cationic surfactants by long-chain alkylammonium into the interlayer space of smectite clay minerals, followed by the addition of neutral amine co-surfactant molecules. Subsequently, a silica precursor (e.g. tetraethyl orthosilicate (TEOS)) is added. The 3D framework of the mesoporous silica structure between the intermediate layers of clay minerals is obtained by calcination.22,23
The so-called “organoclay colloidal route” (Fig. 2) is a novel method to prepare porous architectures from alkylammonium-exchanged smectite clays. These nanostructures can be then exploited to host metal–oxide nanoparticles, which can be either already preformed or formed through an in situ process.24
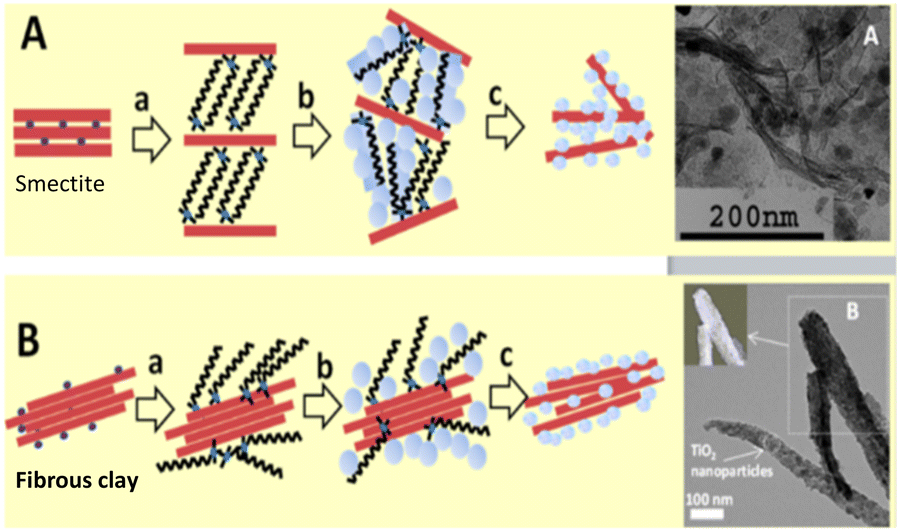 | ||
| Fig. 2 Synthesis of smectites (A) of fibrous clays and (B) semiconductor nanoarchitectures via the three-step “organoclay colloidal route”: (a) formation of an organoclay due to the replacement of the interlayer cations by alkylammonium ions, (b) treatment with metal oxide precursors and (c) calcination to obtain nanoarchitectures containing the semiconductor, reproduced with permission from ref. 24. Copyright 2019, Beilstein-Institut. | ||
 | ||
| Fig. 3 Preparation of graphene derivatives, graphene oxide and reduced graphene oxide. | ||
GIC to form graphene can also be prepared by multi-step processes using chemical intercalation, where no electrochemical cell is used, and thus no co-intercalated solvent molecule is involved. Firstly, the direct intercalation of potassium is realised by chemical vapor transport (CVT) to form the golden-brown KC8 GIC, which is then dispersed in THF. Finally, the solvent is washed/substituted with water; nevertheless, the graphene material remains dispersed in the solution and a dry material is not obtained.28 Redox intercalation into graphite can lead to the formation of graphene oxide using the well-known Hummers’ method, which can be modified to decrease the cost and free toxic gases by using other oxidative reagents and solvents.29
The combination of electrochemical intercalation and oxidation/exfoliation is also feasible to produce exfoliated graphite oxide, which can be subsequently treated to form reduced graphite oxide (RGO) with few functional groups,30 and all the processes leading to the formation of graphene materials have been reviewed.31
Given that it is not possible to intercalate graphene because it is a monolayer, several works attempted intercalation under graphene, meaning between epitaxial graphene and its substrate. In most cases, the intercalation process is performed under UHV (ultra-high vacuum) by exposing graphene to molecular species.32
It should be noted that a bottom-up strategy instead of the top-down approach has been developed in the last decade, especially for the preparation of N-doped graphene 2D materials, i.e. graphitic C3N4, a metal-free 2D material with remarkable photocatalytic properties. The experimental procedure involves sequential molecule self-assembly, alcohol molecules intercalation, thermal-induced exfoliation and polycondensation process, leading to porous few-layer g-C3N4. Claimed as a simple bottom-up method by authors, several steps are necessary considering the re-stacking due to reinforced π–π interactions.33 Other ways for the synthesis of these materials have also been exhaustively reviewed.34
Exfoliation is facilitated in the case of materials having low charge density or, in other words, with cohesive electrostatic forces that are not too strong. However, when the charge density is high, some possibilities remain, as exemplified for the positively charged LDH sheets.35 In this review, the exfoliation methods and mechanisms of exfoliation are classified according to liquid exfoliation, organic molecule-driven intercalation, and mechanical shearing or stirring as well as the 2D associated filler loading to form diverse polymer nanocomposites where the interfacial interactions are the key for efficient exfoliation. However, this usually requires a large amount of solvents. Also, the relatively small quantity of exfoliated materials per unit volume strongly limits their application, and thus often remains a laboratory feat with no real technology transfer possible. Another point of attention is that exfoliation leading to the total exposure of the inter-lamellar species is not desirable in many cases. The de-cohesion of the structural stack leads to the possible migration of guest species.
The exfoliation of cationic smectites (e.g. montmorillonite) can be achieved in both polar and non-polar solvents.36
In an aqueous environment, the interlayer-exchangeable cations are surrounded by water molecules, thus increasing the interlayer distance. In a diluted suspension, clays can naturally dissociate into particles, which can be then partly or completely exfoliated. These processes strongly depend on the concentration of the clay suspension and the type of cations present in the interlayer space. In this case, organic modification is needed for exfoliation in nonpolar media, leading to the transformation of the hydrophilic surface into a hydrophobic one and an increase in the interlayer space of clays. The exfoliation degree is influenced by several factors such as the nature of the organic species, and degree of organic modification and polarity and chemical nature of the solvent.36
2.2. For better particle size control
Alongside exfoliation, the controlled synthesis of small particles is also strongly desirable. To achieve this, the bottom-up preparation of ultrathin LDHs assisted by a reactor is a promising strategy. Previously, Duan and co-workers developed the separate nucleation and aging step (SNAS) method using a commercial colloidal mill reactor to facilitate the scaled-up preparation of nanostructured LDHs.37 This method utilises forced micro-mixing technology to rapidly blend the metal salt solution with the alkali solution in the gap between the rotors. Nucleation of the particles occurs within seconds, resulting in the formation of LDH nanosheets that possess a small and uniform particle size ranging from 60 to 80 nm.38 Therefore, attempting to add a laminar inhibitor in the reactor-assisted synthesis process of LDHs may potentially inhibit the growth of LDHs in the c-axis direction, resulting in the formation of ultra-thin LDHs. Formamide, a stable lamellar inhibitor, can be adsorbed on the LDH layer and suppress the growth of LDHs in the Z-axis direction. Monolayer LDHs with a thickness of ∼1 nm were obtained by modifying the industry colloid mill reactor through the addition of a small amount of layer growth inhibitor formamide using SNAS method.39Another way that is also complicated to implement involves the use of the so-called topochemical reactions. This was nicely illustrated by the recent heterostructures obtained from the ingress of Cu(I) into vacancies of the layered crystalline structure of an Fe7S8 hexagonal nanoplate.40 The topochemical reaction is also reported for anions. Known for decades for mobile oxygen anions, the topochemical reaction has been reported for chalcogen anions and proceeds through a de-intercalation reaction from stable pristine materials, as exemplified by layered cobalt chalcogenides, leading to new metastable compounds41 or oxy-chalcogenide, where the insertion of copper occurs via the chalcogen dimer redox reaction.42 In the case of the latter, it was illustrated with the de-intercalation/re-intercalation of sulphur in the layered oxy-chalcogenide La2O2S2, where structure prediction and electron microscopy helped to understand its topochemical soft chemistry reaction.43
Importantly, topochemical insertion and desorption reactions are applied to mobile ions in the prospect of next-generation energy storage mechanisms. Fluoride anion (de)intercalation was reported in the Ruddlesden–Popper-type perovskite LaSrMnO4 by substituting an oxygen atom in the apical sites with fluorine, yielding oxyfluorides of interest as the cathode.44 Similar de-fluorination was reported for Sr2TiO3F2 as the anode in fluoride-ion battery.45
In other cases, the topochemical reaction proceeds via the dissolution of a pristine host structure, followed by nucleation and growth rather than by transient solid-state intercalation. Thus, the filling of the sites that are vacant, and therefore available, does not take place by migration into the structure but through a dissolution-reprecipitation front, as elucidated with the transformation of gibbsite into LiAl-LDH by multinuclear NMR46 or the transformation from layered single hydroxide (LSH) in LDHs using a solid-state kinetic model.47
2.3. Exemplifying zirconium phosphate (ZrP)
Zirconium phosphate (ZrP) is an interesting cationic exchanger, where several chemical methods, such as refluxing, hydrothermal, sol–gel, microemulsion and microwave methods, can be used to synthesise amorphous or crystalline phases of ZrP.48 All the above-mentioned synthetic strategies involve the addition of phosphate to a solution of zirconium(IV) salt, often in the presence of complexing agents such as HF,49 oxalic acid,50 and formamide,51 resulting in the precipitation of amorphous or crystalline ZrP. The choice of preparation method and reaction conditions strongly influences not only the degree of crystallinity, but also the shape and size of the particles. Presently, chemists are trying to develop more environmentally friendly methods, which avoid the use of excess acids and complexing agents. Several solid-state methods using various alkali metal phosphates have been developed. Flower like α-ZrP was obtained by grinding ZrOCl2·8H2O together with Na3PO4, followed by hydrothermal treatment.52 Bevara et al. synthesised highly crystalline K2Zr(PO4)2 by heating pressed pellets containing ZrO2 and KPO3 at 750 °C.53 The minimal liquid approach, in which reagents are mixed in their as-obtained state and concentration, was used for the synthesis α-ZrP.54 ZrOCl2·8H2O was mixed with concentrated orthophosphoric acid (P/Zr = 2 or 3) and heated at 25–120 °C for 24 h. Larger crystals were obtained at a higher P/Zr ratio and the crystallinity of the sample increased with an increase in the reaction temperature. When a small amount of fluoride ions was added to the reaction mixture, the highly crystalline product was obtained and the its morphology changed from platelets to rod-shape particles with an increase in the F/Zr ratio. Highly crystalline γ-ZrP could be obtained using NaH2PO4 instead of orthophosphoric acid.55 Similarly, α-Zr(NH4PO4)2·2H2O could be prepared using ZrOCl2·8H2O, (NH4)2HPO4 and a small amount of Na.56Another direction in the development of methods for the synthesis of ZrP is the preparation of materials with properties tailored to the specific application, especially ZrP with a high specific surface and suitable porosity for ion-exchange and catalytic applications. The so-called template method using surfactants, which can be cationic (e.g. cetyltrimethylammonium57 and tetradecyltrimethylammonium58 bromide), anionic (sodium dodecylsulfate59) or neutral (e.g. Brij5660 Pluronic P123,61 and Triton-X62) to precipitate ZrP, followed by calcination seems to be very useful. Tian and coworkers synthesised mesoporous ZrP using yeast cells as a template.63 Another way to synthesise mesoporous ZrP is via the calcination of zirconium phosphonates.64 An interesting paper, describing the preparation of ZrP with different morphologies, including rod-like (minimalistic method), spherical (surfactant templating) and cube-like (sol–gel), was published in 2020.65 Highly porous monolithic zirconium phosphate was prepared via sol–gel synthesis in the presence of poly(ethylene oxide) and poly(acrylamide), followed by supercritical drying.66a,b In this case, the presence of both polymers was necessary to form a macroscopic structure with the macropore size controlled by changing the ratio between the polymers and orthophosphoric acid.
The growing interest in nanotechnologies is leading to the development of new methods for the synthesis of nanoparticles with a controlled shape and size. Hajipour and coworkers prepared hexagonal α-ZrP nanoparticles using poly(vinyl alcohol) or polyvinylpyrrolidone in water solution.67 Hexagonal nanocrystalline ZrP particles were formed by the addition of H3PO4 to an alcoholic solution of zirconium propionate.68 The dry samples possessed an α-ZrP structure with intercalated alcohol. By heating this sample to 120 °C, a new phase with 3D structure consisting of cube-like nanoparticles was obtained.69 This phase was called τ′-Zr(HPO4)2 due to its structural similarity with the already known τ-ZrP. Recently, a microwave-assisted hydrothermal method was used for the synthesis of hexagonal ZrP nanodiscs.70
Single-layer α-ZrP nanosheets can also be obtained using the so-called “top-down” method, i.e., by exfoliation of already synthesised ZrP using various chemicals and solvents. The methods for the exfoliation of ZrP were reviewed recently.71 A widely used amine for the exfoliation of α-ZrP is propylamine or methylamine.72 Recently, the exfoliation of α-ZrP using allylamine was described.73 Alkanolamines can also serve as an exfoliating agent and the minimum amount necessary for exfoliation is less than that of monoamine.74 Other agents widely used for the exfoliation of α-ZrP are tetraalkylammonium hydroxides.75 Aqueous exfoliation is the most common, but the dispersion of nanosheets in an organic solvent is desirable for many applications. In this case, the replacement of water with the desired organic solvent can be realised after exfoliation through multiple cycles of centrifuge-rinsing or by heating in the case of high-boiling point organic solvents. Exfoliation can be also achieved by the intercalation of excessively large amines76 such as poly(ether)amines with a molecular weight of about thousand g mol−1, or intercalation of large amines followed by the intercalation of polymers.77 However, the main drawback associated with the use of poly(ether)amine is its difficult removal from the medium, but an exfoliation state can be achieved in organic solvents. For instance, nanosized α-ZrP prepared in methanol78 could be directly exfoliated in methanol.79
2.4. Welcome to a new family: MXenes
Although intercalation compounds have been known for decades or centuries, some of them are revisited and others have been synthesised more recently such as carbon nanotubes as 1D-compounds and metal oxides frameworks (MOF) as 3D-compounds, in the early and late 1990s, respectively, while MXenes as 2D-compounds emerged in the 2010s.A little more than a decade ago, the latter family of intercalation compounds, MXenes, emerged with the synthesis of Ti3C2 from the leaching of Al from Ti3AlC2 in an aqueous HF solution. Since then, these materials have been widely studied due to their excellent properties, which are related to their 2D structure, adjustable bandgap and richness of their surface chemistry, among others, as highlighted in the highly cited review.78
These 2D intercalation compounds composed of transition metal carbides, carbo-nitrides and nitrides are of interest for interlayer engineering and the associated host–guest intercalation chemistry,79 and in particular for electrochemical energy storage applications mostly as electrode materials in supercapacitors and rechargeable batteries due to their high electrical conductivity, allowing a higher power density.80
Unit blocks obtained from topochemical reactions or from MXenes can form heterostructures such as Ti3C2Tx/C/MoO2 microspheres and 2D/2D/0D heterojunctions to fine tune the electric/dielectric properties for wave absorption and shielding.81
Despite the high potential of MXenes, the use of HF in the etching process poses a serious problem for their large-scale manufacture and the production of high-quality crystals. Therefore, alternative synthesis pathways have been explored such as deposition by chemical vapour and plasma-enhanced pulsed laser.82 MXene-based core–shell nanocomposites have also been investigated using core–shell MXene functionalised with carboxylate groups interacting with PEI/PAA in contact with gold nanoparticles for application in catalysis.83
3. Energy
3.1. The interest in ILCs as seen by machine learning methods
Given that layered structures can be kept apart either by van der Waals or not, their interlayer space can accommodate relatively cumbersome guest species. In the literature, this has been well demonstrated in organic–inorganic hybrid materials, where the layered structure is used as a host to protect, deliver or prevent its cargo from migration, as shown in the companion article (part 2). In the field of energy, this tunability in the interlayer domain size is of interest for new technology beyond LiB, adopting larger size cations or anions, given that the diffusion of the guest in and out of the host is not limited by the size of the vacancies such as in tunnel or canal void-based open-structures. A recent article reviewed the reason why some 2D-materials including transition metal oxides, chalcogenides, carbides and nitrides perform efficiently as positive electrode materials in a large range of batteries (AIBs and dual batteries).84 Another review highlighted the importance of selecting guest species from an interlayer engineering point of view to improve selective ion separation membranes and the ion storage performance in battery.85Indeed, intercalation layered materials with their inherent versatile open-structure are considered a solution for the challenging design of new energy concepts. Performing computational science using first-principles calculations on more than 9000 compounds as well as machine learning, a recent study helped to better understand the reason for the stability or instability of layered compounds upon intercalation and de-intercalation processes.86 Other machine learning methods provide insights into 2D-materials, developing a binding energy and structural accommodation-based classification model to screen anode materials for next-generation batteries,87 considering their 2D topology and inherent heterostructure with grain boundaries, which act as the surface adsorption sites of Na+ cations for the same application in NIBs88 or to find a “universal design strategy” for cathode materials that should be air stable such as O3 layered oxides.89 This predictive understanding of the stability toward host–guest possibilities using first-principles calculations is often applied to energy storage applications to better understand the mechanisms and predict virtual structures to synthesise or modulate the host–guest interaction by changing the nature of the ligands for a specific demand regarding the reversibility of the electrochemical intercalation process.90
Among the intercalation compounds, 2D materials present an open-structure for fast ion diffusion and charge transport that is suitable for electrochemical energy storage and electro-catalysis, as recently reviewed,91 and their role as a filler in polymer composite electrolytes (PCE), as either active ion conductive or passive non-ion conductive. These inorganic platelets include graphene oxide, boron nitride, transition metal chalcogenides, phosphorene, MXenes and 2D layered clay minerals, such as layered double hydroxides and silicates (vide infra).92 This review emphasises the benefit of using 2D materials to enhance the PCE in lithium and post-lithium batteries. Another review reported GO/LDH and GO/MXenes for different types of energy storage such as LiS, NIB, KIB and supercapacitors, underlining the importance of combining materials to create an interconnected network of 2D/2D materials, thus preventing the agglomeration of the layers, and thus promoting the ion mass transport.93
3.2. Li ion batteries: LiB
Presently, although the field of lithium-ion batteries is now very mature in terms of electrode materials, and this technology has been transferred and commercialised,94 significant advances are still being made in intercalation chemistry to identify possible “beyond Li battery” technologies given that its limits can now be exceeded by others and less affected by the rarefaction of its elements.At the anode, due to its superior performance including high practical specific capacity provided (360 mA h g−1vs. theoretical 372 mA h g−1), low operation potential (0.1 V vs. Li+/Li), and reversible capacity, graphite remains the best anode material in the Li-ion battery technology (LIB). Lithium intercalation in graphite is possibly the most famous example of intercalation reaction involved in energy applications and has been (once again) recently reviewed. Considering graphite as the anode in LIB, its ambient temperature performance, particle size and morphology as well as electrode engineering are impact parameters still posing a challenge.95,96
The advantages and drawbacks of using synthetic versus natural graphite are also under debate, as well as the lithium diffusion coefficient in the carbon matrix depending on the stage GIC for high-rate applications. In LIB, lithium intercalation into graphite occurs together with the formation of a solid electrolyte interphase (SEI), whose stability is a key point in aging and safety issues. As a perspective, recycling processes constitute a part of the future of graphite intercalated by lithium given that its recovery is a promising route to circumvent mining of natural graphite, which is becoming a critical resource.97 The staging intercalation of lithium into graphite accompanied with the Daumas-Hérold model is well known (Fig. 4), but some planar structural mechanisms remain controversial in the recent literature.98
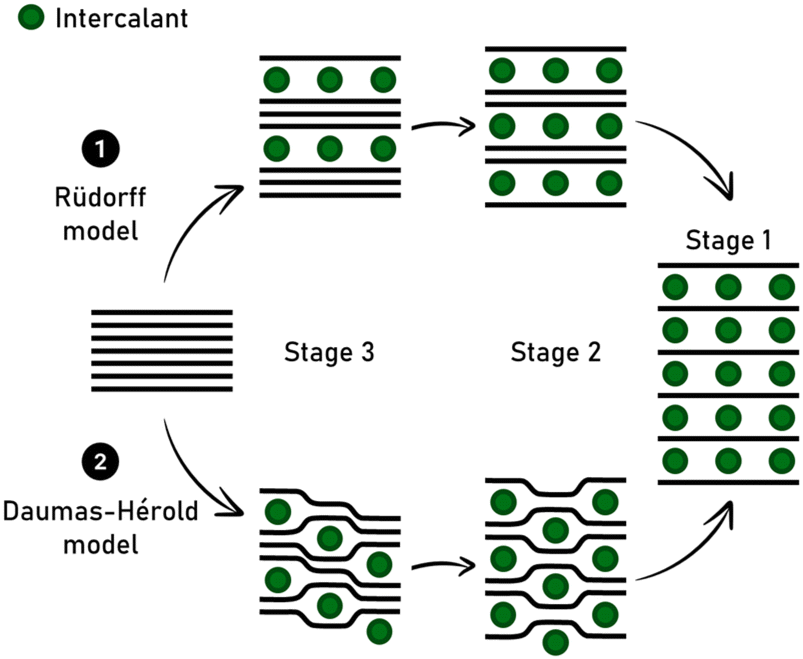 | ||
| Fig. 4 Schematic representation of the Rüdorff model versus the Daumas-Hérold model, considering the flexible feature of graphene sheets. This model describes the continuous transformation from third to second stage GIC. | ||
Thus, the staging phenomena still need to be fully elucidated and appear more complex than expected and complementary operando techniques and ex situ analyses should be explored, as pointed out in the recent study by Komaba and co-workers concerning K-ion batteries.99 In this technology, graphite remains a promising material and intercalation reactions need to be investigated further (see following section).
At the nanoscale, outstanding properties have been predicted for graphene as a promising material for electrochemical energy storage applications. However, single-layer graphene cannot be intercalated, and considering LIB, the most promising result can be obtained by the adsorption of Li+ on both surfaces of its layer to form Li2C6 with a theoretical capacity of 744 mA h g−1. However, the defects formed during the synthesis of graphene and its very high surface area blemish this ideal picture and make graphene-based cells unfeasible for alkali-ion technology.100 Chemical doping of graphite, graphene or few-layers graphene is also envisaged to be a feasible strategy to improve the affinity between alkali ions and the carbon matrix in the corresponding batteries.101
The interest in ILCs can be observed in the case of fast-charging, as reported for Li-based anode materials using vanadium-based oxide.102 The conductive open network is based on the inherent heterostructure formed by the stacking of two vacancy-ordered types of sheets interleaved by either Na+ or Mg2+ cations, leading to a large spacing, which facilitates the diffusion process, while enhancing the electronic conductivity in the vicinity of the vanadium redox sites through a rapid multi-electron reaction, acting as a pseudocapacitive redox surface in the inner open-structure.
Clays are also used to prepare electrode materials for rechargeable metal-ion batteries. One reported strategy is their use to prepare high-capacity Si anodes, given that in view of up-scaling their production, it is more convenient to extract silicon from earth-abundant or marine abundant (pelagic clays), eco-friendly and low-cost sources rather than from synthetic silica. Starting from natural halloysite, Zhou et al. (2016)103 prepared interconnected Si nanoparticles through the selective acid etching of halloysite, enabling the removal of the alumina sheets, followed by magnesiothermic reduction at 700 °C with the assistance of NaCl processes. Due to its small particle size (in the range of 20–50 nm) and porous structure, this new Si anode exhibited a satisfactory performance as an anode for LIB with a specific capacity of 800 mA h g−1 at a current density of 1 C after 1000 cycles. Magnesiothermic reduction was also applied, starting from montmorillonite,104 talc,105,106 LAPONITE®,107 vermiculite,108 attapulgite109 and pelagic clays.110 In the case of talc, by fine tuning the preparative methods, a mixture of Si and SiO2 particles having sizes going from tens to hundreds nanometres were formed.105 The addition of a carbon layer on these particles imparts electronic conductivity and improved durability towards stresses associated with the volume change of Si/SiOx during cycling. Similarly, pelagic clays and sucrose were used as the source of silicon and carbon, respectively, to form a carbon-coated porous silicon anode demonstrating a performance with a specific capacities of 540 mA g−1 under the current density of 0.5 A g−1 after 120 cycles.110
Si nanosheets possessing hierarchical porous structures were also obtained from vermiculite through a mild (300 °C) and low-cost strategy involving chemical delamination, acidification and low-temperature aluminothermic reduction in a eutectic molten system.111 The formation of Si nanosheets was explained by the self-templating role of vermiculite assisted by subsequent mild reduction reaction. High lithium-storage properties were obtained with a reversible capacity as high as 1269 mA h g−1 at 1.0 A g−1 after 300 cycles and excellent rate capability with desirable capacities of 1314 mA h g−1 at 4.0 A g−1. Porous silicon/carbon composite nanosheets were also fabricated by reducing the carbon-coated expanded vermiculite with metallic Al in molten salts.112 Under these conditions, the layered structure of vermiculite was retained with a thickness of less than 50 nm and the formed carbon nanolayer served as a diffusion barrier and mechanical support for the growth of mesoporous silicon nanosheets. Consequently, the obtained anode displayed remarkable electrochemical performances, with a reversible capacity of 1837 mA h g−1 at 4 A g−1 and 71.5% retention of the initial capacity after 500 cycles.
Attapulgite and polyacrylonitrile (PAN) were also used to form nano-sized attapulgite-based aerogels. It was demonstrated that the carbonised AT-based aerogels used as active anode materials of LIBs exhibited the average discharge capacity of 534.6 mA h g−1 at 100 mA g−1 after 50 cycles.113
A relationship between the type of clay and the morphology of the resulting Si was established by Chen et al.104 Attapulgite led to the formation of 0D nanoparticles, whereas Si derived from halloysite showed a 3D interconnected framework and 2D nano Si stemming from montmorillonite and vermiculite, indicating that in this case, the original morphology was maintained.
Beside using clay-derived silicon and silica anodes, iron oxide-carbon-clay nanocomposites are also good candidates for use in LIBs, as demonstrated by Alonso-Domínguez et al.114 Using an environmentally friendly process involving sepiolite bentonite, oxyhydroxide-type phase, FeOOH, sodium alginate and glucose, composites having high capacity values of ∼2500 mA h g−1 after 30 cycles at 1 A g−1 and capacity retention close to 92% were obtained. Sepiolite-based composites display outstanding cycling stability, as interpreted in terms of the optimal interaction between iron oxide nanoparticles and sepiolite surface through hydrogen bonds, which is related to the particular structural and compositional features of sepiolite.
At the cathode, based on the common alkali-rich layered cathode material Naz[LixMny]O2, close comparisons between similar compositions have identified the case of the first charge hysteresis with O2− oxidised, and then cleaved in discharge.115 The authors scrutinised the honeycomb superstructure arising from the local ordering of lithium and transition metal ions during first cycle and noted that the presence of this superstructure prevented the migration of manganese ions, and therefore the loss of energy density.
Presently, the original approaches are still being developed such as misfit layer heterostructure SnS/TiS2 studied as a superior anode material for LIB and based on a conversion-alloy mechanism upon lithiation/delithiation.116
However, overall, the current LiB technology is stalled because the capacities at the cathode, and also at the anode are limited.
3.3. Li–S batteries
Compared to the traditional carbon cathode, Li–S batteries have attracted increasing attention and are considered as one of the most promising next-generation energy storage systems owing to their remarkably high energy density (2600 W h kg−1) and earth abundance and large theoretical specific capacity of sulfur (1675 mA h g−1). In this case, clays are excellent candidates to circumvent the drawbacks of sulfur cathodes.117,118 Indeed, their interlayer space can accommodate the volume of expansion of sulfur cathodes, the dissolution and shuttle effect of polysulfides can be suppressed by the confinement of the sulfur cathode in different clays (montmorillonite and halloysite) and clay-sulfur cathodes enable the free transportation of lithium, resulting in efficient and straightforward ion diffusion.119 For example, a Li-montmorillonite-sulfur cathode prepared by a simple process including the incorporation of 80 wt% sulfur powder demonstrated a high capacity and nearly sulfur loading-independent cell performance.118 Sulphur infiltrated in vermiculite led to cathodes demonstrating an outstanding rate capacity and cycling stability.120 In this case, it was concluded that due to the cations present at the surface of vermiculite, polysulfides anions (Sn2−) can be adsorbed, thus preventing them from dissolution; moreover, it was assumed that the excess surface charge is most probably compensated by excess Li+ in the space charge zones, which is beneficial for charge transfer and local conductivity. A comparison between the performance of this cathode and regular carbon–sulfur based cathodes revealed that a promising rate capability and much better cycling stability are obtained with vermiculite-sulfur cathodes, showing the potential of clays as hosts for sulfur and the formation of high-performance cathodes for lithium-chalcogen batteries. A conductive polymer coating on LDH provides a suitable volume to reserve the lithium polysulfides (LiPSs), thus increasing the kinetics of sulfur-based redox reactions.121 Polypyrrole associated with LDH platelets improved the electrode performance by preventing the shuttling of LiPSs. Similarly, it was found that LDH cations act as sulphur hosts, and in particular NiFe by interacting with LiPSs, resulting in a better performance in the LiS battery.122Beyond intercalation, conversion is also possible for a large number of electrode materials, which can have very large theoretical capacities. Conversion-type materials in particular transition metal fluorides and sulphides have been reported as high-energy density anodes and cathodes.123
3.4. Beyond LiB technology using ILCs
New trends are also emerging to find breakthrough solutions with expanded concepts of intercalation chemistry pushing materials to their limits to have sufficiently open-structures. In addition, the following discussion is intended to be non-exhaustive, and thus includes solutions to compensate for the scarcity of the elements that make up lithium batteries (LIBs) such as cobalt, nickel, copper (as the current collector) and lithium. Even graphite is becoming a critical material. These batteries include Na- (NIBs), K- (KIBs), and aqueous Zn- (ZIBs) ion batteries.Graphite can also be employed as the anode in emerging K-ion battery technology using liquid organic electrolyte at room temperature but it shows a moderate rate capability and relatively fast capacity fading, which are related to the dramatic volume expansion after intercalation into the van der Waals galleries (interplanar distance increases from 335 pm for pristine graphite to 535 pm for KC8 1st stage GIC). Thus, it is necessary to transpose the knowledge on LIB and NIB technologies to KIB technologies, but developments are mandatory concerning the nature and structure of the anodic carbon materials, the involved intercalation mechanisms, and the composition of the electrolyte.130
We believe that new emerging materials or revisited well-known compositions should satisfy some of the requirements to go beyond LIB technology. For instance, the building block approach is a fascinating method to vary the stoichiometric combination of redox metals, as recently illustrated by the 2D non-van der Waals layered material Li2MP2S6.131
To improve the inherent low-rate capacity and poor cycling stability of 2D ternary metal phosphorus, defects are introduced in the MnPS3 layers, and the presence of interleaved species such as methylammonium Me4N+, tetraethylammonium Et4N+ or pyridine PyH+ cations yields the hybrid layered compound: Mn1−xPS3y2x (y = Et4N+, Me4N+, and pyH+), respectively.132
The Na+ energy storage mechanism of MnPS3 can be described as follows (according to the XRD phase identification in Fig. 5):

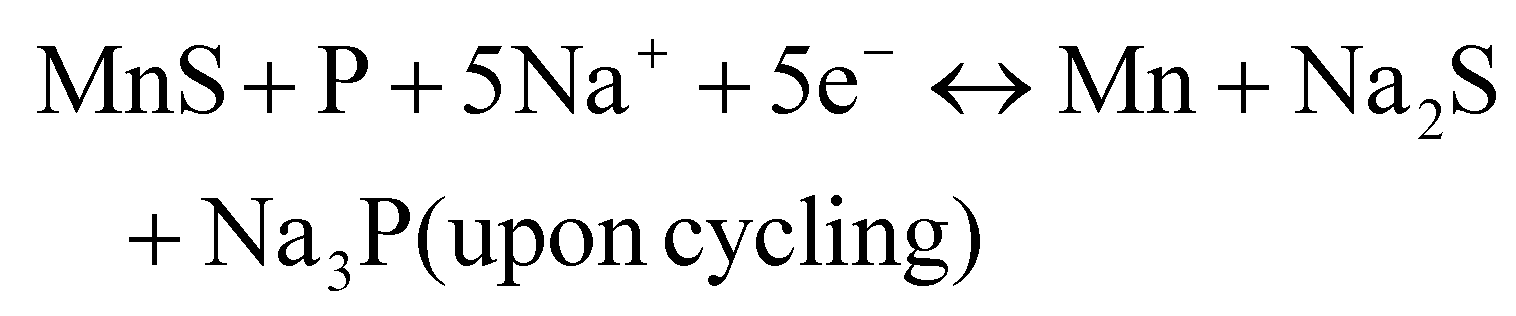
 | ||
| Fig. 5 (a) Initial charge–discharge curve of the Mn1−xPS3(Me4N+)2x electrode at 0.05 A g−1 and (b) corresponding ex situ XRD patterns at different voltage stages. Reproduced with permission from ref. 132. Copyright (2024), Elsevier. | ||
Intralayer defect vacancy engineering by CVD has been found to create electronic conductivity and an expandable spacing suitable for the rapid diffusion and mitigation of the Na+ cations, as well as material volume expansion upon intercalation and deintercalation when used as electrode materials in NIBs. For example, it was reported that the Mn1−xPS3y2x hybrid composite electrodes exhibited a higher specific capacity and rate performance than the MnPS3 electrode (Table 1) and interestingly Mn1−xPS3(Me4N+)2x with a larger interlayer distance in the series exhibited a higher specific capacity due to the better access of the electrolyte.
| ICLs | Guest | Battery or SC | Capacity | Retention (%) (N cycles) | Ref. |
|---|---|---|---|---|---|
| a (VOH) = Hydrated vanadium oxide. b PA/HA = Propylamine/hexylamine. c ABS = azobenzene sulfonic acid. d EDA = ethylenediamine. e PEDOT:PSS = poly(3,4-ethylenedioxythiophene) polystyrene sulfonate; and. f NH4+ = rechargeable aqueous ammonium-ions battery. | |||||
| TiSe2 | PA/HAb | NIB | 417 mA h g−1 (@1 A g−1) | 95 (2000) | 135 |
| MoSe2 | Ethylene glycol | NIB | 326 mA h g−1 (@5 A g−1) | 87 (200) | 134 |
| MnPS3 | Me4N+ | NIB | 890 mA h g−1 (@0.1 A g−1) | 94 (700) | 132 |
| KVOPO4 | K+ | NIB | 120 mA h g−1 | 88 (150) | 133 |
| MXene V2C | ABSc | KIB (anode) & Dual (SC) | 174 mA h g−1 | 80 (900) | 142 |
| MXene V2C | — | 121 mA h g−1 (@0.05 A g−1) | 45 (900) | ||
| Graphite | Na+/ether-based molecule | NIB | 110 mA h g−1 (@0.1 A g−1) | >99% (2500) | 125 |
| Graphite | K + -solvent | KIB | 130 | ||
| Graphite | — | AIB (cathode) AlCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :urea :urea |
73 mA h g−1 (@0.1 A g−1) | 160 | |
| (VOH)a | Mo6+ | AZIB | 430 mA h g−1 (@0.1 A g−1) | 95 (1000) | 168 |
| Birnessite δ-MnO2 | Na+, Cu2+ | AZIB | 576 mA h g−1 | 147 | |
| LDH NiCo | Glucose | Alkaline Zn | 224 mA h g−1 | 86 (2000) | 151 |
| LDH CoFe | Cl− | ACIB | 190 mA h g−1 (@0.2 A g−1) | 66 (200) | 179 |
| LDH Ni5Ti | Cl− | CIB | 129 mA h g−1 (@1 A g−1) | 100 (1000) | 180 |
| LDH Mn3Al | CO32− | NH4+f |
184 mA h g−1 (@0.1 A g−1) | 182 | |
| MXene Ti3C2Tx | EDAd | SC (H2SO4) | 486 F g−1 | 89.7 (10000) |
190 |
| 1313 F cm−3 | |||||
| MXene Ti3C2Tx | PEDOT/PSSe | SC (H2SO4) | 1065 F cm−3 | 191 | |
| LDH NiCo | Acetate | SC | 1032 F g−1 | 187 | |
| NiCo | PO43− | SC | 87 mA h g−1 | 92 (10000) |
188 |
| 2070 F g−1 | |||||
Due to the high redox potential of V5+/V4+, vanadium-based materials have been considered as electrode materials in NIBs. Interestingly the authors focused on vanadium-based phosphate AVOPO4 and used KVOPO4, which is commonly employed in KIBs but not in NIBs.133 The small amount of K+ remaining after the exchange of K+ for Na+ in the first cycle was found to have a “pillar” effect, thus facilitating the diffusion kinetics and reversible storage of Na+ cations, and finally completely outperforming NaVOPO4 (Table 1).
In the strategy that interleaved species may be more active, exhibiting a better performance than the pristine compounds, some studies reported the intercalation of ethylene glycol in 1T/2H-MoSe2, leading to a 1T phase percentage as high as 80%.134 Ethylene glycol was used as a sacrificial guest to produce an interstratified conductive layer, thus helping in electronically connecting all the materials as well as preventing the restacking of MoSe2 upon intercalation/deintercalation cycling. When used as the anode in NIBs, the performance of the 1T/2H-MoSe2@C composite hybrid layered material was appealing in terms of energy and capacity retention (Table 1).
Two subsequent intercalations between TiSe2 induced the delamination of the platelets in well exposed nanosheets, which exhibited a low diffusion barrier towards Na+ cation diffusion.135 Subsequently, propylamine, followed by hexamine insertion led to the spontaneous exfoliation of the layered structure. A rapid electrochemical reaction in NIB was observed due to the fact that the redox active sites were well exposed, combining the efficient inner-wettability of the electrolyte with the stability of the structure. All these factors resulted in a high specific capacity, which was associated with the low polarisation (Table 1). According to the TEM, ex situ XPS and CV characterisation, the mechanism of both the sodiation and desodiation processes was found to proceed in two steps of intercalation and conversion, as follows (V vs. Na+/Na):
| TiSe2 + xe− + xNa+ → NaxTiSe2 (1.62 V) |
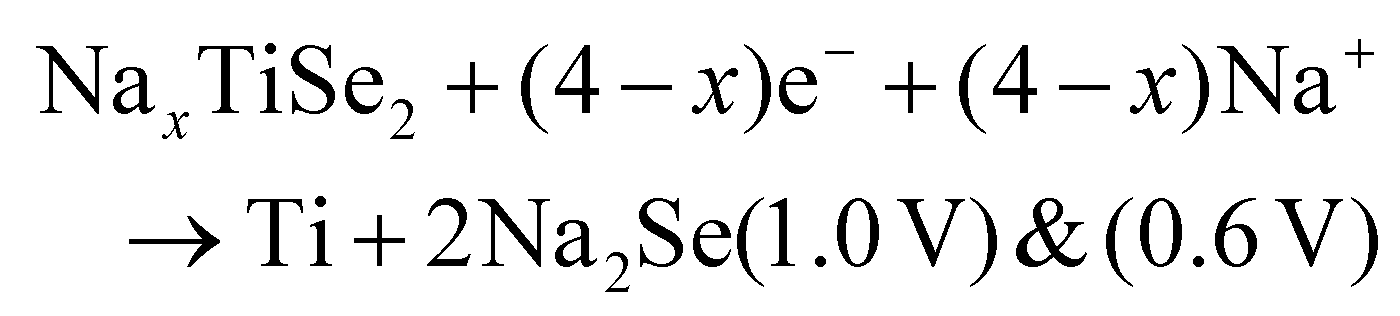
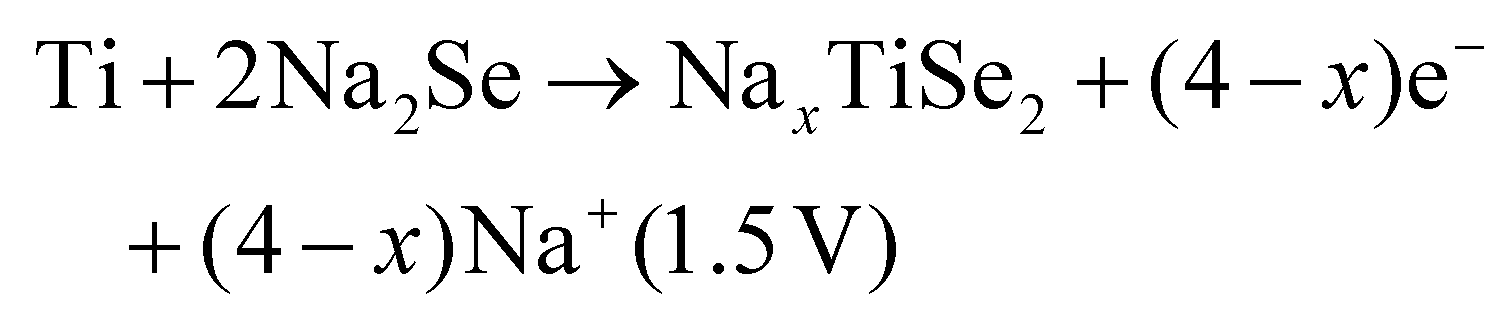
| NaxTiSe2 → TiSe2 + xe− + xNa+ (1.9 V) |
According to the HRTEM analysis, the (400) lattice fringes were indexed to Na2Se, which is lamellar.
Topochemical reaction may affect the insertion of big-size mobile ions in KIBs. Indeed, in addition to the interlayer space, the intralayer domain is rich in terms of substitution for many families of layered structures, thus leading to an almost infinite possible combination. This versatility is largely reported in the literature for LiNixCoyMnzO2, which is known as the NMC layered structure, using some Al3+ cation substitution to stabilise the structure. A recent example reported the use of KxCr0.75Ti0.25O2 instead of KxCrO2 as the cathode in KIBs.136 This topochemical exchange reaction usually stabilizes the pristine layered structure upon charge/discharge cycling and this example shows that layered structures can be diversely modified for application in new technology.
Another approach is to interstratify sheets of layered structures. Recently, the use of the unique intergrowth of the same composition but presenting different stacking sequences was reported, resulting in the formation of a biphasic P2/P3 of K0.7Mn0.67Ni0.33O2 composite.137 This composite showed a high energy density and capacity retention when used as the cathode in KIBs.
Clay-based silicon and silica anodes have also attracted attention in recent years for applications in NIBs and KIBs but it was shown that they are not suitable candidates for these applications.138 More interesting materials are composites prepared by wrapping halloysite with a conductive a layer of polypyrrole (PPy) via in situ polymerisation. Consequently, a cathode material that could maintain a capacity of 126 mA h g−1 after 280 cycles at a current density of 200 mA g−1 was obtained.139 Sodium-montmorillonite was also used as a sodium–ion–conductor interface in a sodium-based composite anode, which was prepared by the mechanical mixing of sodium and sodium montmorillonite. In particular, the use of sodium montmorillonite provided sodium diffusion channels, reduced the nucleation potential of sodium and reduced the generation of sodium dendrites during repeated Na plating and stripping processes. Consequently, an excellent cycling performance and favourable rate capability in both carbonate and ether electrolytes were obtained.140
In NIBs, new intercalation compounds have also been considered such as MXene Ti3C2Tx exhibiting reversible Na+ intercalation/deintercalation in a nonaqueous electrolyte and good capacity retention upon cycling.141
Grafting azobenzene sulfonic acid (ASA) onto V2C MXene induced the expansion of the interlayer space, which is beneficial to enhance the specific capacity and rate capability in KIBs.142
The better performance of ASA V2C compared to V2C was explained based on the hopping site for K+ cations at the sulfonate anion and an extra storage site at the azobenzene unit, in addition to the response of the V2C layers (Fig. 6).
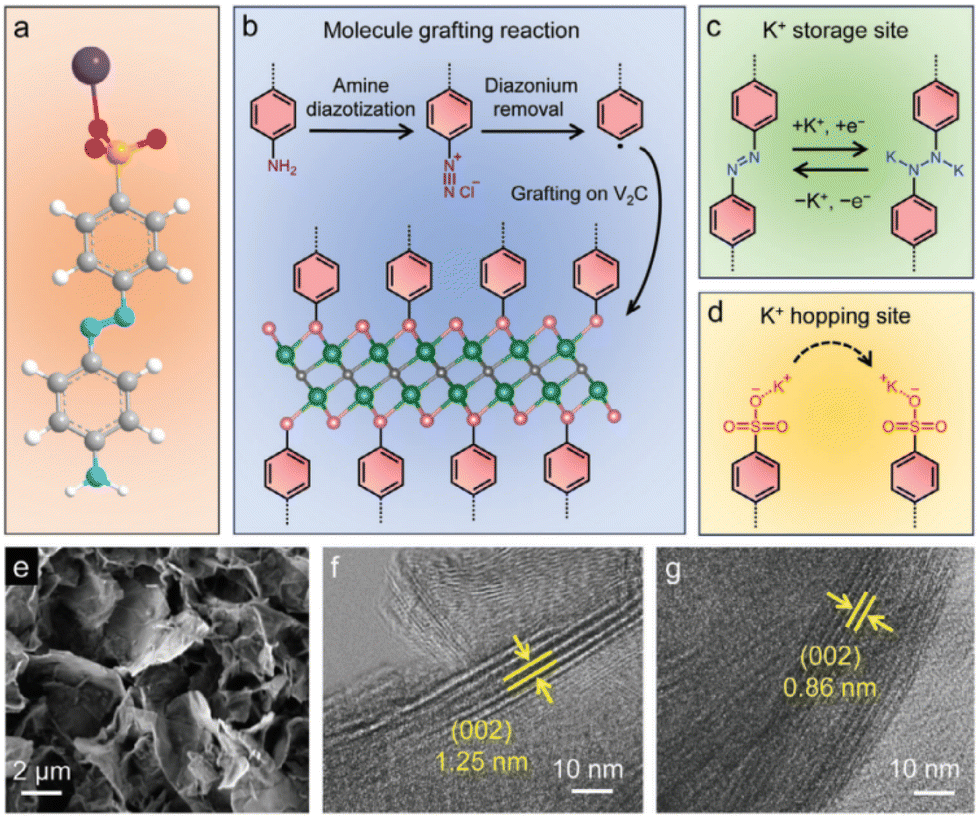 | ||
| Fig. 6 Schematic illustration showing (a) 4-aminoazobenzene-4′-sulfonic acid sodium salt (white sphere: H; grey sphere: C; blue sphere: N; orange sphere: S; red sphere: O; and purple sphere: Na), (b) molecule grafting reaction (black sphere: C; green sphere: V; and pink sphere: terminals of V2C MXene), (c) azobenzene unit as the extra K+-storage site, and (d) sulfonate anion as the K+-hopping site. (e) Scanning electron microscopy image of ASA-V2C nanoflakes. HR-TEM images of (f) ASA-V2C film and (g) pristine V2C film. Reproduced with permission from ref. 142. Copyright (2024), Wiley-VCH GmbH. | ||
It is also demonstrated by synchrotron operando XRD that the organic moieties alleviated the structural distortion upon K+ intercalation and de-intercalation, which was strongly visible for V2C but not ASA V2C.
Similarly, using δ-MnO2 as the cathode in AZIBs, birnessite manganese oxide is co-intercalated by sodium and copper.147 Their co-intercalation has been found to boost the electrochemical performances due to the activation effect of Cu2+ on the Mn2+/Mn4+ redox pair on the surface, and Na+ cations stabilise the host structure during the two-electron transfer reaction occurring in AZIBs.
The intercalation of inorganic redox centres has been reported for vanadium oxides and their potential use in aqueous zinc-ion batteries, AZIBs.148 In this study, Mo3V15O35·nH2O nanosheets were obtained via a one-step hydrothermal approach using ammonium salt of two transition metals, i.e., NH4VO3 and (NH4)6Mo7O24·4H2O, leading to the presence of V5+/V4+ and Mo6+/Mo4+ oxidation states, respectively, according to XPS (Fig. 7). The intercalation of Mo6+/Mo4+ was found to enhance the electrochemical response when used as the cathode in AZIBs due to the additional redox reactions involving Mo4+/Mo5+/Mo6+ to promote the diffusion due to the expanded layer spacing as well as to stabilise the capacity upon insertion and de-insertion of hydrated Zn2+ cation cycling.
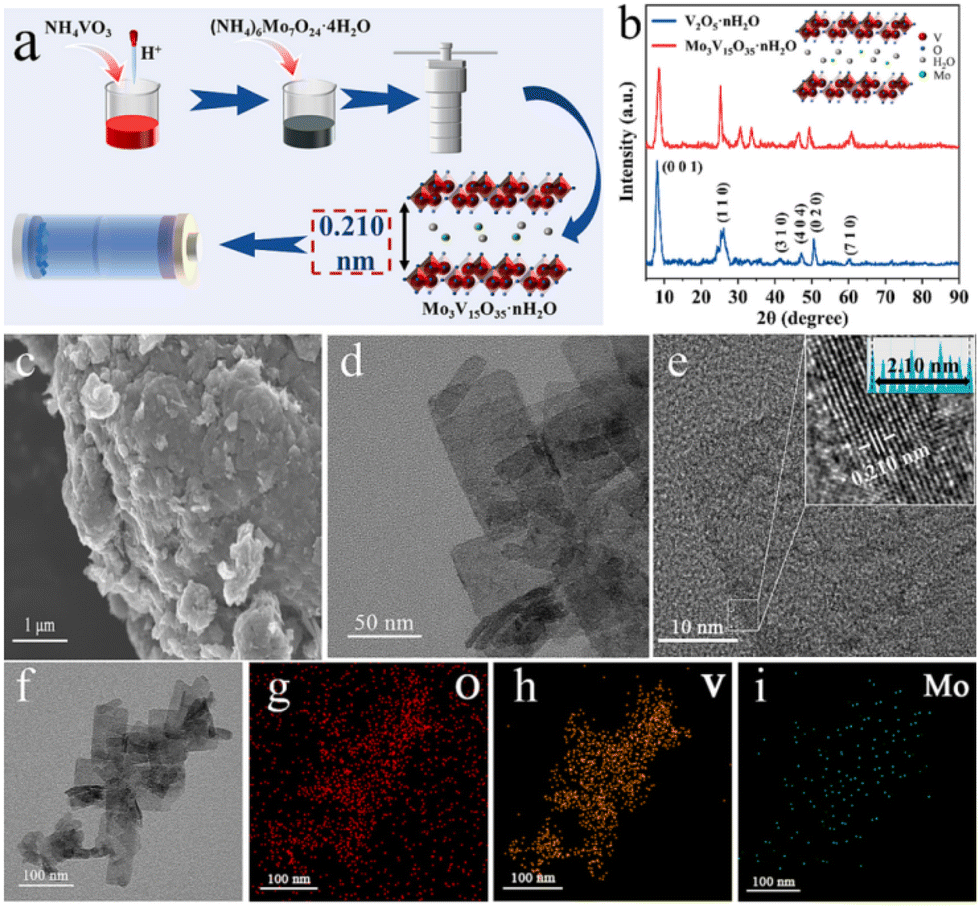 | ||
| Fig. 7 (a) Schematic of the sample preparation of Mo3V15O35·nH2O. (b) XRD patterns of V2O5·nH2O and Mo3V15O35·nH2O. (c) SEM image, d and (e) TEM and HRTEM images, and (f–i) TEM elemental mapping images of Mo3V15O35·nH2O. Reproduced with permission from ref. 148. Copyright (2024), Elsevier. | ||
Bi-(multi) phasic compounds exhibit phase boundaries of interest for the interfacial adsorption-insertion mechanism, as predicted and experimentally observed for a biphasic vanadate in aqueous medium with the adsorption and insertion of Zn2+ and H+, leading to stable cycling capacities.145 For example, the modification of zinc anodes using ball-milled clay containing Ti3C2Tx to build a three-dimensional (3D) framework on the surface of zinc foil was reported by Li et al.149 One interesting feature in this process is that electrostatic adsorption of functional groups increases the transport dynamics of the Zn2+ ions, while the 2D diffusion of Zn2+ ions in the plane is constrained by the confinement effect of Ti3C2Tx, which uniformly nucleates the Zn2+ ions and restricts dendrite growth. Consequently, a stable long cycle was achieved (2000 h, 0.5 mA h cm−2). One additional interest of this preparative method is the protecting effect of the ball-milled clay toward zinc self-corrosion and electrochemical corrosion in the electrolytes. LDH nanosheets decorated on carbon improved the energy efficiency of an alkaline zinc–iron flow battery due to the presence of a mesoporous texture. This originates from the in situ grown hydroxyl-covered LDH platelets, making the adsorption and ion mass transport easier in the zinc-based battery.150 Another article reported the use of glucose-pillared Ni–Co LDH as a cathode material, providing a high specific capacity and stability upon cycling in alkaline zinc batteries.151 To suppress the growth of dendrites at the Zn anode, a chitosan gel was coated on its surface, while partially sulfidated NiCoFe LDH platelets that were efficiently exfoliated were combined with reduced graphite oxide to be used a cathode, resulting in an outstanding specific power.152 Interestingly, sulfur mismatch substitution of NiFe LDH when deposited on nitrogen-doped graphene led to electron transfer between the sulfur anion and Fe3+, producing Fe4+ species, which enabled a high power and energy density to be achieved in a Zn-air battery, together with long cycle stability.153 The LDH crystalline structure presenting metal-sulfur (especially the Cu–S) bonds by subtle modulation of the copper and sulfur interaction led to large specific capacity in a zinc battery.154
Reversible Mg batteries (RMBs) have been evaluated given that they may provide a high volumetric capacity and avoid the growth of dendrites at the metal anode. However, a limitation of this technology is finding open-structures that are sufficiently efficient and stable against the insertion of Mg2+ ions.155 An article reviewed the most representative 1D, 2D and 3D intercalation compounds. Another article compared the multivalent-ion batteries based on Mg2+, Zn2+, and Ca2+ ions intercalated in a disordered bi-dimensional compound, and the authors presented defective CaTiO3-based perovskites of interest for rechargeable Ca-based batteries.156
This is nicely exemplified by the use of α-MoO3 as the cathode in AIBs in the air-stable Al(ClO4)3·9H2O/succinonitrile hydrated eutectic electrolyte (ASHEE) system.157 The layered structure was found to be stable upon cycling and the in situ combination of XAS and XRD demonstrated the average valence state between Mo6+ and Mo4+/Mo5+, which was associated with the smooth variation of the local bond distance.
The insertion of the trivalent Al3+ ions in AIBs was also presented in cation-deficient anatase TiO2, where the vacant sites were used for intercalation.158
Intercalation occurs in graphite used as the cathodic host material and AlCl4− ions as the intercalated species, involving acceptor-type GIC. Given that the intercalated/deintercalated species in the graphitic cathode are different from that involved in the simultaneous anodic process (deposition of Al atoms/ions from the Al-foil), there is no “rocking-chair” mechanism in this technology. This implies the formulation of very specific ionic liquid electrolytes with the drawback of corrosion phenomena versus materials.159
The intercalation of Al3+ in an Al battery was also reported with graphite as the cathode and AlCl3/urea as a cheap ionic liquid electrolyte.160 Second-stage GIC was observed during charging and a high coulombic efficiency and cycling stability were recorded at a high current density.
Carbon electrodes were reviewed based on graphite intercalation compounds (GIC) for ion batteries.161 An additional review focused on theoretical calculations to better understand their band structure and structural change and stability against Li ions diffusion.162 A critical issue was reported for carbon-based materials and composites in KIBs in non-aqueous electrolyte.163 Another study reported that graphite has a poor performance, while soft carbon appears to be of interest upon cycling.164
Another study reported interest in the insertion of chloride anions into Fe-based LDH to produce some type of green rust, which helped to electrochemically convert Fe(OH)2 to FeOOH, as tested in rechargeable alkaline iron batteries.165 This iron redox improved the cycling stability, according to a combination of characterisation techniques including operando X-ray diffraction and molecular dynamics simulations. Through the phase evolution identified by XRD (Fig. 8), this elegant redox system involves some type of cascade reduction as follows:
| Fe(OH)2 + OH− → FeOOH + H2O + e− |
| 3Fe(OH)2 + Cl− → [Fe2+2Fe3+(HO−)6]+[Cl]− + e− |
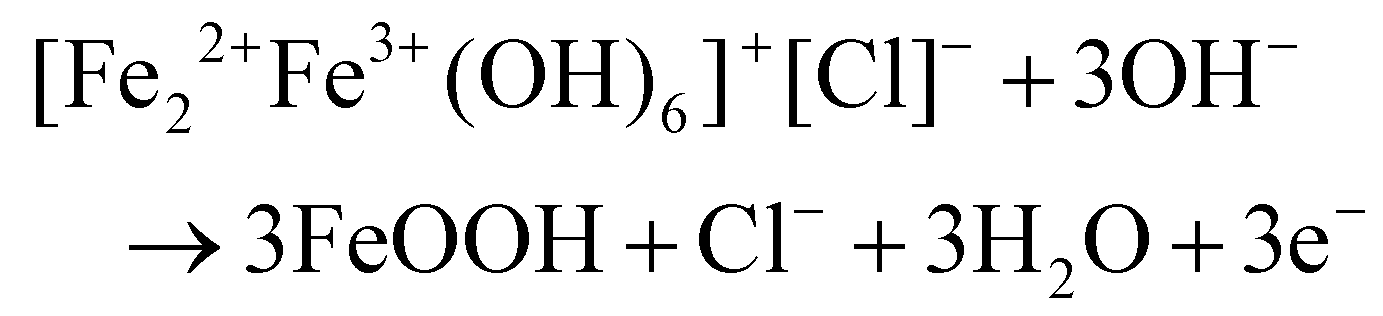
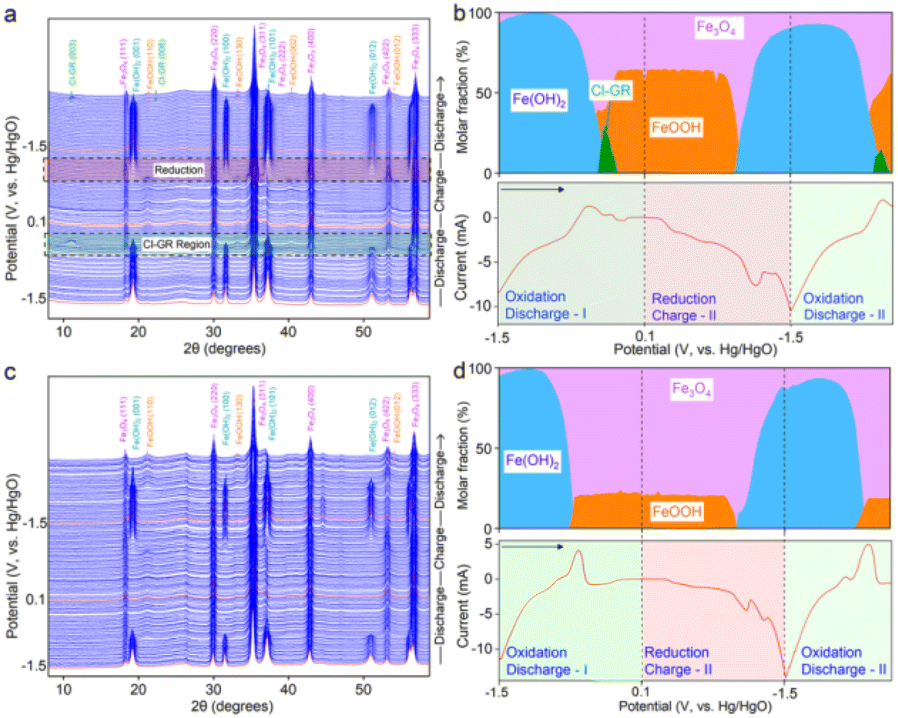 | ||
| Fig. 8 (a and c) Waterfall plots and (b and d) phase ratio analyses of iron oxides in (a and b) NaOH/NaCl and (c and d) NaOH electrolytes using operando XRD measurements together with cyclic voltammetry at a scan rate of 0.5 mV s−1. The selected regions in (a) show the phase transitions during the Cl− insertion (discharge) and Fe(OH)2 formation (charge). They highlight the XRD patterns depicting the Cl-GR formation when iron oxides were discharged from −0.42 to −0.18 V and the formation of Fe(OH)2 when iron oxides were charged from −1.02 to −1.5 V, respectively. The X-ray wavelength was corrected to Cu Kα source (λ = 0.15406 nm). Arrows in (b) and (d) indicate the voltage scan direction. Reproduced with permission from ref. 165. Copyright (2023), the American Chemical Society. | ||
Indeed, the green rust intermediate avoids the direct reaction predicted by Pourbaix diagrams, as follows:
| 3Fe(OH)2 + 2OH− → Fe3O4 + 4H2O + 2e− |
Dual-ion batteries (DIBs), emerging energy storage systems, use the properties of graphite to insert both anions and cations, thus acting as unique materials associated with the electrochemical redox reactions at both electrodes.166 Recently, two reviews summarised the advantages (high energy density and low cost) and disadvantages (cycling stability and electrolyte decomposition) of DIBs operating at a working potential over 4.5 V vs. Li/Li+ as well as some promising strategies.
The modification of the interlayer distance between graphene planes is also a method to explore and to improve intercalation, especially in sodium-ion batteries. Besides alkali metal ion GIC, other covalent or ionic intercalation compounds can be envisaged to develop battery technologies such as fluorinated graphite, FeCl3-GIC, and anion intercalated graphite. The possibility of developing high-voltage dual-ion batteries with both the formation of cationic GIC and anionic GIC as the anode and cathode, respectively, is also under development. The use of advanced electrolytes including ionic liquids is necessary for subsequent development to withstand larger operating voltage.167,168
Similar to “small changes may have big effects”, the interlayer distance in a Ruddlesden–Popper perovskite oxide, La1.2Sr1.8Mn2O7, which is known to be suitable for fluoride-ion intercalation/deintercalation reactions, was optimized as a function of the substitution x in La2-2xSr1+2xMn2O7.176 The authors observed the optimized expansion of the interlayer distance x = 0.35, which also corresponds to the composition in the series presenting the highest capacity and rate performance as the cathode in FIBs. The authors explained the good performance in the high regime by the fact that a larger spacing suppresses the coulombic repulsion between fluoride-ions and oxide ions.
Other halide-ion batteries have also been studied as possible post-lithium technology. Chloride-ion batteries (CIB) using LDHs as cathode have also been reported.177 Computational screening gives some insight into the optimized cation LDH composition for anion insertion electrochemistry. This family of compounds has also been studied for the fabrication of flexible supercapacitors and alkali metal (Li, Na, K) ion batteries.178 CoFe LDHs crosslinked with carbon nanotubes (CNT) were studied as anodes in an aqueous CIB.179 The redox reaction is based on the valence changes in the transition metals and both LDH cations contribute to the capacity by repetitive oxidation and reduction redox reactions through a topochemical mechanism occurring within the platelets, while CNTs supply the electronic conductivity. This opens their possible application in aqueous desalination technology. Another article reported an unsual Ni5Ti-Cl LDH composition for CIB, giving rise to efficient retention of the reversible capacity upon cycling.180
Another family of layered structure is considered to fulfil the requirement as stable anode materials in CIBs, namely, layered perovskite oxychlorides A2TMO3Cl where A is a strong alkaline metal that can interact with Cl− ions and T is a transition metal for the redox reaction.181 The authors selected (A, T) = (Ca, Co) and (Ba, Rh) in these compounds, which maintained their layered structure during the Cl− (de)intercalation reaction process.
Another forefront research in new energy technology concerns rechargeable aqueous batteries based on ammonium ions. A proof of concept study was carried out on a rocking-chair battery using 3,4,9,10-perylenetetracarboxylic di-imide at the anode and MnnAl (n = 2 or 3)-based LDH as the cathode.182 The full battery was found to deliver an energy density as high as 45.8 W h kg−1, which was mostly explained by the rapid amorphization of the LDH framework, thus facilitating the mass transport and storage of NH4+ ions.
3.5. High-power batteries and supercapacitors
On the border between efficient energy density and power density, rapid battery-like intercalation may find a place, the so-called pseudocapacitive storage, which should be described by capacitance in mA h g−1 rather than F g−1 given that it originates from the response of rapid redox active sites and not only the surface process. In this field, layered structures have attracted significant attention such as all types of 2D framework possessing redox sites, once the latter are well exposed due to the large surface area of the high aspect ratio platelets and/or the presence of highly diffusive pathways, thus connecting most of the active mass of the open-structure.183 However, only a few materials can compete with the layered structure of metal oxide frameworks (MOF), which possess a very large tunnel-based structure, under the condition that the connectivity between transition metals and their ligands is stable during the rapid redox reaction.A recent article reported the use of hybrid LDH materials as both electrodes in an aqueous battery-type device.184 Their feasibility was demonstrated using two electroactive species interleaved into the LDH host structure, i.e., riboflavin phosphate on one side (negative electrode) and ferrocene carboxylate (FCm) (positive electrode) on the other side, which are then placed face to face. In this set-up, the complete cell cycled up to 2.2 V vs. Ag/AgCl in sodium acetate aqueous electrolyte.
A controlled morphology is known to be important to improve the electrochemical performance, as exemplified by copper hydroxide nitrate nanostructures.185 This was exemplified via the simple hydrolysis of a salt and oxide or precipitation with alkaline solution, ultrasonication, microwave, and urea at a high temperature or under reflux. As shown in Fig. 9, the crystal aspect ratio Lz/Lxy reporting the thickness to lateral size reflects the extent of the structural coherence length along the stacking direction (Lz) and in the plane of the hydroxide layers (Lxy), respectively. This shape anisotropy may be controlled by the chemical preparation and it was found to result in different crystalline domains, i.e., Lz/Lxy < 1 for platelet-like shape or Lz/Lxy > 1 for rod-like particles, thus forming a landscape that can be divided into three large zones (Fig. 9), which affect the electrochemical activity. In terms of electrochemical activity, some nanostructured submicron particles exhibited a capacity of up to 197 mA h g−1, corresponding to nearly 90% of the theoretical capacity for a two-electron redox process.
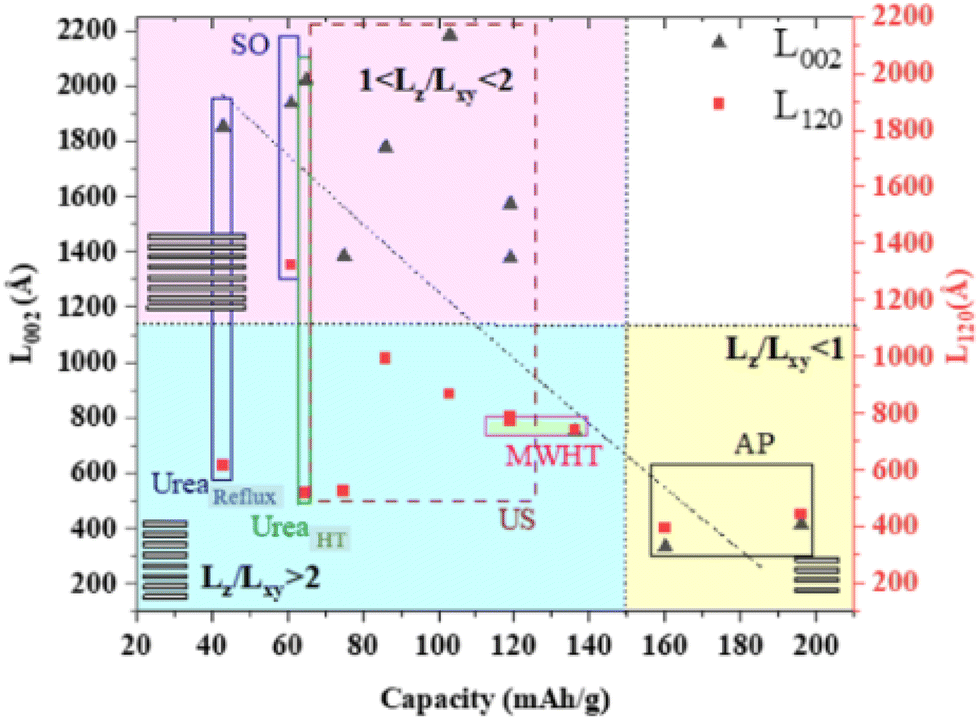 | ||
| Fig. 9 Correlation between the capacities obtained from the CV of the first cycle and structural coherence lengths Lz and/LxY obtained from the XRD analysis for the Cu2(OH)3(NO3) series of samples with US: ultra-sonification, AP: alkaline precipitation, MWHT: microwave high-temperature, and SO: salt and oxide method. Reproduced with permission from ref. 185. Copyright (2023), the American Chemical Society. | ||
Similarly, an intercalation compound of copper sulfide184 was used as both the positive and negative electrode material in a complete aqueous battery system but its performance still need to be optimised.186 Interestingly, in terms of the solid-state kinetic approach, copper sulfide was prepared by a pseudomorphic transformation from layered copper hydroxide salt (LSH) (Cu2(OH)3NO3) precursor using amine digestion or microwave process. The activation energy to convert LSH into CuS is dependent on the transformation process, while a more open-structure (larger anions inside the interlayer LSH) was found to increase the entropy term, thus more prompt to react and to be transformed into CuS.
It is well known that energy and power are the main characteristics of electrochemical storage systems. After these preliminary studies of power batteries using aqueous electrolyte electrode materials, other systems can provide even more power but often at the expense of energy (unless there is rapid redox reaction but then these are power batteries, as previously discussed), which are often named supercapacitors; however, their mechanism is still based on redox rapid reactions. As mentioned above, the capacitance is often reported in F g−1 but it should be expressed in mA h g−1 given that it is a faradaic process involving a rapid electron transfer and not strictly a capacitive process related to the adsorption of mobile ions in the diffuse layer.
Limited by scarcity of the chemical elements Ir or Ru, some model materials considered as super-capacitor electrodes have been abandoned in favour of ILCs. For instance, but rather counterintuitively, acetate inserted into an NiCo-based LDH open-structure led to a reduced interlayer spacing, which was found to enhance the structural stability, while providing a high specific capacitance at a high current density.187 Scrutinised by AC impedance, the intercalation of acetate anions was found to lower the internal electrode material resistance, thus improving the rate performance by better ion conductivity. When placed as the positive electrode and assembled in an asymmetric supercapacitor, the cycling was found to be stable (Table 1).
In the same LDH cation composition but electrodeposited, the intercalation of PO43− was found to significantly increase the access to and the quantity of the active redox sites.188 Consequently, the hybrid composite when used as a cathode in an asymmetric supercapacitor showed exceptional cycle stability.
Other ILCs have been reported such as layered metal chalcogenide nanocrystals189 and Ti3C2Tx MXene structure when intercalated by ethylenediamine (EDA)190 or assembled with polymer.191 The EDA-Ti3C2Tx intercalation compound was mostly mesoporous and exhibited low charge transfer resistances in H2SO4 aqueous electrolytes, which is associated with a large specific capacitance and good retention upon cycling. In the Ti3C2Tx/PEDOT:PSS hybrid film, PEDOT polymer chains pillared MXene flakes to prevent their aggregation or self-restacking as well as acted as a conductive bridge, accelerating the electrochemical reaction through multidimensional electronic transport channels. The outstanding volumetric power density of the films is of interest for flexible and wearable electronic devices.190 When mixed with LDH, MXenes exhibit high capacity as electrode materials for supercapacitors.192
In high-power-ion hybrid Na-ion full cell capacitors, an MXene Ti2C negative electrode associated with alluaudite Na2Fe2(SO4)3 as the positive electrode delivered a relatively high voltage of 2.4 V corresponding to 90 mA h g−1 at 1.0 A g−1 (based on the weight of the negative electrode).193
Attention was also paid to clays having different structures such as nanotubes (halloysite) and 2D nanosheets (montmorillonite) as electrode materials for supercapacitors.194–198 However, montmorillonites face limitations due to their low electronic conductivity and high electrical resistance.199 Alternatively, halloysite, with its unique tubular structure, shows promise by providing channels for electrolyte ions, thereby enhancing the specific capacitance.199,200 However, it suffers from low conductivity and a small active specific surface.201 In this case, acid activation has emerged as a suitable method to address these weaknesses by increasing the number of active sites, specific surface area, and porosity through inner-wall dissolution.202
Various strategies have been proposed to address the issue of low conductivity in clay-based materials, including the formation of composites starting from conductive polymers such as polyaniline (PANI)203 and combination of montmorillonite or halloysite with carbon-based materials such as porous carbon, carbon multiwall nanotubes, and reduced graphene oxide.197 Montmorillonite has also been used as a template for the growth of nickel telluride nanostructures204 and cobalt boride@clay mineral hybrid composites have been developed.205 Furthermore, layer assemblies of NiMnLDHs and/or poly(3,4-ethylenedioxythiophene) (PEDOT) on perforated halloysite nanotube templates have been achieved through the growth and polymerisation method.206 A relevant approach involves the formation of core–shell heterostructures with hydrangea shapes using LDH@polypyrrole as counterpart materials of clay minerals for high-performance hybrid supercapacitors.207
The investigation of the electrochemical properties of halloysite-polyaniline (PANI) composites formed via the in situ polymerisation of aniline demonstrated that the composites prepared using halloysite etched with hydrochloric acid at 0 °C exhibited the highest specific capacitance of about 710 F g−1 at a current density of 1 A g−1, with good cycling stability, retaining around 77% of its specific capacitance after 300 charge–discharge cycles.208 MMT/PANI/Co3O4 prepared through a two-step process achieved a specific capacitance of 234 F g−1 with a capacitance retention of approximately 97% after 1000 cycles.203 Clay-conducting poly-o-toluidine hybrid nanocomposites are also promising compounds for supercapacitor applications.209
Combining clays and carbon-based materials is also widely performed to improve the poor conductivity of montmorillonite and halloysite. For example, montmorillonite reduced graphite oxide (rGO) nanocomposites exhibited a high specific capacitance and high cycle stability.210 Using a non-liquid crystal spinning method followed by chemical reduction to produce rGO/clay fibers, it was possible to achieve a high conductivity and capacitance.211 The comparison between polypyrrole-coated carbon nanotubes and Cloisite 30B®/polypyrrole-carbon nanotube composites demonstrated that the addition of Cloisite 30B® led to an enhanced electro-active surface and higher specific capacitance.212 rGO/montmorillonite/polyvinylcarbazole (PVK) nanocomposites showed an increased specific capacitance compared to pristine rGO/polyvinylcarbazole, which was attributed to the dopant effect of montmorillonite and PVK.213 The inhibitory effect of montmorillonite nanosheets on the restacking of graphene sheets and enhancement in the electrochemical performance were also proven for 3D reduced graphene oxide/montmorillonite aerogels.214 A robust strategy to form halloysite/carbon composites starting from halloysite and formaldehyde-free phenol-furfural resin synthesised from phenol and sugar-based furfural was also proposed.215 However, a challenge encountered is finding the best condition enabling the offset of the poor conductivity of halloysite with a renewable carbon precursor having a higher biomass content. Halloysite was also used to meet the demand for flexible and wearable supercapacitors, and in this frame, a novel chemical crosslinking of cellulose/ionic liquid/halloysite nanotube dispersions was formed.216 Nacre-inspired HPA-rGO nanocomposite films having high electrical conductivity and higher specific capacitance compared with other graphene-based films were successfully prepared.217 N-doped graphene quantum dots deposited on Fe3O4-halloysites were also formed,218 and the synergetic effects of the different compounds enabled the formation of high-performance materials, where Fe3O4 increased the energy density and Fe3O4-halloysite shortened the diffusion path for electrons and electrolyte ions to the graphene quantum dots and absorbed the mechanical stress during cycling, while also providing excess sites for charge storage. It was also reported that a novel hybrid material based on halloysite, NiCo2S4 (NCS), and carbon achieved a maximum specific capacity of 544 mA h g−1 at a scan rate of 5 mV mA h g−1.219 In the field of CeO2-clay, it was concluded that its high capacitance is mainly attributed to the increase in specific surface area and improved wettability, with the unique interlayer structure of clay minerals contributing to the cycle stability. A comparison of the literature data on various composites suggests that the performance of CeO2-clay composites is superior, highlighting their potential.220
3.6. Use of clays in dye-sensitised solar cells (DSSCs)
In the field of energy, clays can also serve as nanofillers for dye-sensitised solar cell (DSSC, now also known as Gratzel cells) electrolytes, as recently reviewed.221–223 The electrolytes in DSSCs are responsible, not only for the regeneration of the dye impregnated on mesoporous TiO2, but also for charge transport between the photoanode and the counter electrode (CE) of the solar cell.222 Attempts to replace liquid and gel electrolytes were governed by different facts such as their lack of stability with time at the required temperature owing to the leakage and/or evaporation of their organic solvent, leading to the need for hermetic sealing. Thus, to overcome these problems, polymer-based electrolytes have been introduced for application in DSSCs but despite their advantages over liquid or gel electrolytes, their power conversion efficiency is lower than that of liquid and gel-based electrolytes given that the movement of ions is hindered by their polymeric matrices.Accordingly, the use of nanofillers as multi-functional additives, especially clays is one way to solve this problem and clays play key roles in the properties of electrolytes and the performance of DSSCs.
2D-materials have been found to exceed 3D-materials in terms of the pre-intercalation of guest species to ensure structural stability and/or provide an additional property such as water repellence of interest when the materials are used in solar cells. This was nicely illustrated by the 2D perovskite analogues used in solar cells, the so-called perovskite solar cells (PSCs), where a hydrophobic organic bifunctional spacer with 1,4-phenylenedimethanammonium (PDMA) containing the Dion–Jacobson 2D perovskite material (PDMA)FA2Pb3I10 was used224 (Fig. 10). The hybrid layered materials when assembled in a solar cell and exposed to humidity were found to be moisture resistant without noticeable degradation, presenting a suitable efficiency of 7%. Similarly, diammonium cations were intercalated in a PbI2 layered structure in a 2D/3D perovskite heterojunction.225 The intercalation helped to orientate the 2D nanoplate acting as a template during the growth of the perovskite layer. It was also observed that a reduction in defects and better carrier extraction led to an efficient power conversion efficiency, and thus the authors concluded that the pre-intercalation strategy is appealing to address both efficiency and stability challenges in PSCs.
 | ||
| Fig. 10 Perovskite films with BDMA pre-intercalation and photovoltaic performance of PSCs with steady-state efficiency under continuous illumination at the maximum power point for the control and BDMA PSC. Reproduced with permission from ref. 225. Copyright (2024), Elsevier. | ||
Due to their beneficial properties, clays have been widely used as nanofillers for electrolytes in dye-sensitised solar cells (DSSCs).226 Non-modified clays such as saponite and LAPONITE®, as well as grafted and organically modified clays such as montmorillonite and talc, have been utilised.227,230,228,229 These clays are incorporated in various polymers or polymer mixtures, such as poly(n-isopropyl acrylamide) (PNIPAAm) and iodide poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF-HFP), using conventional methods such as in situ polymerisation or solution/melt intercalation.192,194 For instance, the addition of 5 wt% montmorillonite to PVDF-HFP electrolyte significantly improved the diffusivity, conductivity, and power conversion efficiency in DSSCs.231 The coupling of substituted Zn-porphyrins with PMMA and nanoclay-based gel electrolytes in DSSC devices enabled a PCE of around 1.55% to be achieved.232 This innovative approach demonstrates potential for the development of stable and efficient gel-state electrolytes for DSSCs.
3.7. Hydrogen storage layered materials
The quest for efficient and sustainable hydrogen storage materials has led to the exploration of various options, including clay minerals.233 One solution is underground hydrogen storage,234 where clays, being ubiquitous secondary minerals in natural underground settings, play a significant role. Studies have delved into the contribution of clays in geochemical and reaction path modeling of hydrogen storage in sedimentary rocks.235 Additionally, research has focused on the wettability of clay surfaces by hydrogen in the presence of brine, which influences the injectivities, withdrawal rates, storage capacity, and containment security.236 Recent investigations have explored the impact of pore structure parameters on hydrogen adsorption under different conditions, revealing that specific surface area and micropore volume positively affect hydrogen adsorption, while average pore width has a negative effect.237Several studies have focused on solid-state hydrogen storage for electrochemical applications, where montmorillonite has shown promise. Modification techniques such as acid-activation, ion-exchange, and grafting have been explored to enhance the hydrogen adsorption capabilities.238,239 For instance, metal-organo-clays (MOC) synthesised by the in situ formation of metal nanoparticles in montmorillonite-supported polyol dendrimers demonstrated improved hydrogen affinity.240 Copper-loaded organo-montmorillonite was prepared, showing enhanced hydrogen affinity due to the reduced nanoparticle mobility and aggregation, favoring the physical condensation of hydrogen.241 Ca2Mn3O8/CaMn3O6/montmorillonite composites, achieving increased hydrogen storage capacity due to spillover, redox, and physisorption mechanisms, are also good candidates.242 Halloysite has also been explored for hydrogen storage, with acid-treated halloysite and hexagonal boron nitride composites exhibiting enhanced hydrogen storage capacity due to their increased number of adsorption sites.243 Additionally, a composite electrode based on halloysite nanotube-Li0.9Ni0.5Co0.5O2−x/LiFeO2 nanostructures showed high electrochemical hydrogen storage.244 The optimisation of the key parameters governing the formation of composites has led to hydrogen storage capacities six times higher than that of pristine halloysite. Clay minerals have been integrated into carbon-based materials to develop clay-carbon composites for hydrogen storage. Combining clay minerals with carbon materials such as activated carbon, graphene, and carbon nanotubes has shown potential for enhancing the hydrogen adsorption capabilities.245,246
However, despite the progress, challenges remain, including developing scalable synthesis methods, exploring new composite formulations, and assessing the practical feasibility of clay-based hydrogen storage systems.
3.8. Magnetism/optoelectronics/tools for characterisation (equipment, analytical, etc.)
In nanoelectronics, intercalation into graphene (i.e. intercalation of species between a monoatomic graphene layer and its substrate) is an efficient functionalisation method to significantly modify its band structure. Numerous studied elements have been already checked versus epitaxial graphene obtained by CVT or SiC evaporation. Attention has been paid to alkali and alkaline-earth lanthanide metals, some transition metals and even noble metals such as gold, as highlighted in a recent review.247 These authors also presented an original experimental study investigating the intercalation mechanism, electronic properties and superconductivity. Employing a deposition-annealing two-step process, electronic decorrelation of graphene from its growth substrate is possible, enhancing its electronic properties and modifying its band structure. Depending on the nature of the metallic element, two different intercalation mechanisms have been proposed. For elements with low affinity versus graphite (d-block metals for instance), a diffusion mechanism through graphene defects is evidenced, with the intercalated species located between the buffer layer and true graphene. Alternatively, considering s-block or f-block elements whose affinity versus graphite is higher, a possible diffusion through carbon layer is proposed.Since the discovery of superconducting properties in CaC6 with a critical temperature of 11.5 K,248 the highest for this class of materials, the physical properties of donor-type GIC have been deeply (re)investigated. Especially, interest has been focused on alkaline-earth-based GIC and the nature of their superconductivity is commonly explained by the BCS theory and electron–phonon coupling, which can be easily studied in these materials. Although beryllium still has not been intercalated in graphite, successful bulk intercalation reactions have been performed with Ca, Sr and even Ba.249 In the latter case, the increase in the interlayer distance together with the decrease in the charge transfer upon intercalation led to a very low Tc of only 65 mK.249
To understand the superconducting properties of intercalated graphite, it is interesting to focus on the nanoscale. As claimed before, intercalation into graphene (one-atomic-layer-honeycomb-network) is impossible but the experimental study intercalated bilayer of few-layers graphene can be realised and brings information on the evolution of coupling versus dimensionality.250
Superconducting properties can also be tuned at the nanoscale by twisting, heterostructure elaboration or interlayer separation control to adapt the electronic structure.251
The optimised synthesis of misfit compounds containing BiSe, Bi2Se3, and MoSe2 constituent layers led to the formation of (BiSe)0.97(Bi2Se3)1.26(BiSe)0.97(MoSe2) containing metallic 1T-MoSe2, showing charge transfer and low resistivity with a dependence on the metallic temperature.252 Upon Cu topological intercalation into the parent insulator heterostructure compound (PbSe)5(Bi2Se3)6, a superconducting state was reached for Cux(PbSe)5(Bi2Se3)6 (CPSBS with x = 1.47).253
Another heterostructure based on Bi2S3/AgBiS2 prepared by a cation exchange reaction from the binary parent compound Bi2S3 showed a high light absorption coefficient due the enhancement in its photoinduced properties.254
A two-dimensional transition metal nitride Ti2N (MXene) obtained from the selective etching of Al from the ternary layered transition metal nitride Ti2AIN exhibited surface-enhanced Raman scattering (SERS) activity.255
Intercalation chemistry combined with exfoliation is known to be a versatile strategy to modulate heterostructures and their electronic structure. This was exemplified with δ-Sr0.50V2O5 and its intercalation-driven exfoliation to 2D nanosheets.256
Concerning tools for the characterisation of intercalated graphite and carbon-based materials, numerous operando techniques (diffraction, microscopy, and spectroscopy257) have been developed to understand their behaviour upon alkali ion intercalation in the corresponding technology,258 NIBs259 and KIB.260,261
A diacetylene monomer layered crystal structure hosting organic amines through intercalation was found to polymerize, leading to polydiacetylene with amine, which structure presented some tunable stimuli (mechano)-responsive colour-change properties.262 Specifically, the colour change was tuned based on the nature of the intercalated amines.
4. Summary of the latest developments
The comparison of the performance of layered structure in terms of capacity and its associated retention with time, voltage window, polarisation and hysteresis, energy and power densities is usually a difficult task given that the experimental conditions including the mass of active material, the relative amount of electronic carbon black, nature of the electrolyte and salt, and cycling regime may be different among studies. Therefore, a selection of the most recent articles is given in Table 1, reporting the best performances for some lamellar materials to streamline “beyond Li” future technologies. In this case, the energy and power densities are not reported given that the data in the literature are not complete and often refer to complete electrochemical devices, making a comparison among electrode materials difficult.Table 1 summarizes the benefits of 2D materials for some beyond LIB electrochemical systems. The possibility in substituting intra-layered redox metals is not unusual given that many inorganic compounds present a solid solution. However, some 2D compounds present the possibility of having multiple substitutions such as LDH, resulting in the presence of diverse redox metals.263 These versatile 2D templates may be of interest to increase the electronic delocalization, i.e. the electronic conductivity to ease the intercalation and deintercalation in some of these beyond LIB systems and optimize the energy density, which is not only available for intercalation but also conversion-type mechanisms, where the volume change should be minimised.
The expansion of the interlayer spacing is of high value for 2D systems given that it permits big-size ions to migrate in and out of the structure compared to other rigid open-frameworks formed by tunnels. Indeed, even a small change in the spacing value may have a significant effect on the long-term performance, as illustrated by KVOPO4 in NIBs instead of NaVOPO4133 or for the solid solution La2−2xSr1+2xMn2O7 tested in FIBs.176 Evidently this is more pronounced with a large expansion such as that observed for acetate in NiCo LDH for SC application,187 and even more when the interleaved species is redox-active such as V2C MXene pre-intercalated with azobenzene sulfonic acid tested in KIB.132
5. Recycling ILCs
Recycling spent lithium-ion batteries is scarcely reported but should increase to respond to the strong requirement regarding sustainability and life cycle assessment of used materials. In one study,264 the authors reported the fluorination process of spent cathode materials (LCO and NMC) converted to CoF2 and mixed transition metal fluorides, which could further deliver energy.A study reported the use of water molecule intercalation to regenerate the de-lithiated cathode material NCM (LiNi0.55Co0.15Mn0.3O2) via mild calcination.265 This intercalation led to a phase transformation induced by the lattice expansion along the stacking direction, which was nicely used to form a renewed electrode material after soft hydrothermal treatment. As another perspective, although graphite recycling remains of low interest economically, it will become mandatory regarding sustainable policies, and thus recent works in this field are also emerging in the literature.266–269
6. Remarks and outlook
In this review, we aimed to present some up-to-date insights on the synthesis of ILCs and the associated elaborate experimental protocols to modify, functionalize, and decorate these materials, making them suitable to be mixed with other constituents such as polymers or compounds to form a larger-scale architecture, and also to endow them with specific properties. We can distinguish the two main approaches, which are the bottom-up and top-down approaches. The first is related to synthesis in micellar medium, where the nucleation is favoured over the phenomenon of crystal growth, and the second is exfoliation such as for clays, α-ZrP and GIC to form graphene. Other methods make it possible to obtain hybrid materials.Thus, clay modification has been described through organo-modification, and more specifically in the case of cationic clay organoclay, the colloidal route. Another interesting treatment is the intercalation-exfoliation reaction (Hummers-based interaction reaction), which is somehow reminiscent of the reductive intercalative polymerisation (RIP)270 or of the topochemical oxidative reaction (TOR).271
The use of ICs in the field of electrochemical energy storage gives rise to abundant literature given that ILCs meet almost all the criteria, where the first is linked to the diffusion of species and the second is linked to their electronic conductivity, which can be improved. A recent summary was present on technologies linked to lithium-ion batteries (LIB) and those after LiB, the so-called beyond LIB such as XIB (X = Na (NIB), K (KIB), Al (AIB), Zn (ZIB), Cl (CIB)), and dual-ion batteries (DIB) as well as other possible systems such as reversible Mg batteries (RMB), Zn-air, and Zn-sulphur, where ILCs feature adaptable compositions that can often accommodate redox centres for electrochemical reactions occurring at potentials conducive to their use as either an anode or cathode of a complete system. The advantages of ILCs for these beyond LIB technologies are briefly summarised in Table 2.
| Intralayer redox metal substitution | Interlayer spacing | Interleaved redox species | Exfoliation and interstratification |
|---|---|---|---|
| Tune potential discharge and charge | Insertion and desertion of large size ions | Pillared 2D stacking | To form composites with other compounds |
| Energy density↗ | Electrolyte wettability | No structural collapse | Adaptable response |
| Hysteresis↘ | Diffusion↗ | Number of cycles↗ | Dual system |
| Conversion reaction | Power density↗ | Additional capacity performance | Energy & power↗ |
| Volume change limitation | Performance↗ | ||
| XIB and SC mostly concerned: | |||
| All | KIB, AIB, ZIB | KIB, CIB | All |
This was classically illustrated by the positive materials used in LIBs. However, surprisingly even the least expected ILCs in terms of redox reaction for electrochemical storage systems have been considered or in part to form composite electrode materials. This is the case in particular for clays with conjugated polymers to limit the stress caused by changes in volume under electrochemical grinding or to form templates for carbonization to obtain carbon with a high specific surface area for supercapacitor applications.
Finally, it is obvious that the field of energy for intercalation compounds has witnessed significant advancements and holds great promise for addressing the challenges associated with energy storage. Several perspectives emerge for the future of energy storage based on ILCs, as follows:
- the development of new synthesis methods with a focus on the development of sustainable and environmentally friendly solutions (with low CO2 footprint),
- given that these versatile materials are scalable nowadays, to target a high technology readiness level (TRL), avoiding the valley of death, which limits many new proposals resulting from research but to transfer those that are innovative but also pragmatic in economic and sustainable development terms and no longer simple easy alternatives, and
- to develop new recycling strategies to minimize the environmental impact of intercalation-based energy storage technologies, and integration of renewable energy sources into the grid as compounds that can help to address their intermittency.
ILCs may be viewed as a chemical toolbox that can tackle critical features for energy storage and future challenges, while also balancing sustainability and scalability simultaneously, as well as ensuring a greener future. This explains the reason why many ILCs revisited in the case of beyond LIB systems.
We are convinced that ILCs will address the related challenges due to their versatility in terms of chemical composition, structuring, shaping as well as being ion exchange materials with the possibility to expand their interlayer space to increase the diffusion process and be organo-modified with active redox guests. The unstacking through exfoliation results in the formation of more exposed layers, i.e., redox centres, as well as the possibility of being interstratified when restacked with other layers to form heterostructures or to be simply mixed with other materials to form complex architectures of interest.
Data availability
Manuscript Ms. Ref. No.: DT-PER-03-2024-000755 entitled “Recent Advances and Perspectives on Intercalation Compounds Part 1: Design and applications in the field of energy”, authored by Chiara Bisio, Jocelyne Brendlé, Sébastien Cahen, Yongjun Feng, Seong-Ju Hwang, Morena Nocchetti, Dermot O'Hare, Pierre Rabu, Klara Melanova, Fabrice Leroux.No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
All co-authors as well as all colleagues from the board of the International Symposium on Intercalation Compounds (ISIC) are warmly acknowledged.References
- F. Chang, I. Tezsevin, J. W. de Rijk, J. D. Meeldijk, J. P. Hofmann, S. Er, P. Ngene and P. E. de Jongh, Nat. Catal., 2022, 5(3), 222–230 CrossRef CAS.
- S. Cahen, L. Speyer, P. Lagrange and C. Herold, Eur. J. Inorg. Chem., 2019, 45, 4798–4806 CrossRef.
- A. Lyo, H. Ogino, S. Ishio and H. Eisaki, Adv. Mater., 2023, 35(15), 2209964 Search PubMed.
- A. Lyo, H. Fujihisa, Y. Gotoh, S. Ishida, H. Eisaki, H. Ogino and K. Kawashima, Carbon, 2023, 215, 118381–118398 CrossRef.
- M. Laipan, L. Xiang, J. Yu, B. R. Martin, R. Zhu, J. Zhu, H. He, A. Clearfield and L. Sun, Prog. Mater. Sci., 2020, 109, 100631 CrossRef CAS.
- C. Xu, M. Zhang, X. Yin, Q. Gao, S. Jiang, J. Cheng, X. Kong, B. Liu and H.-Q. Peng, J. Mater. Chem. A, 2023, 11, 18502–18529 RSC.
- Y. C. Lin, R. Torsi, R. Younas, C. L. Hinkle, A. F. Rigosi, H. M. Hill, K. Zhang, S. Huang, C. E. Shuck, C. Chen, Y. H. Lin, D. Maldonado-Lopez, J. L. Mendoza-Cortes, J. Ferrie, S. Kar, N. Nayir, S. Ragjabpour, A. C. T. Van Duin, X. Liu, D. Jariwala, J. Jang, J. Shi, W. Mortelmans, R. Jaramillo, J. M. J. Lopes, R. Engel-herbert, A. Trofe, T. Ignatova, S. H. Lee, Z. Mao, L. Damian, Y. Wang, J. L. Knappenberger Jr, Z. Wang, S. Law, G. Bepete, Y. Wang, D. Zhou, J.-X. Lin, M. S. Scheurer, J. Li, P. Wang, G. Yu, S. Wu, D. Akinwande, J. M. Redwing, M. Terones and J. A. Robinson, ACS Nano, 2023, 17(11), 9694–9747 CrossRef CAS PubMed.
- Z. Guo, F. Sun and W. Yuan, Cryst. Growth Des., 2017, 17(4), 2238–2253 CrossRef CAS.
- K. B. Ibrahim, T. A. Shifa, S. Zorzi, M. G. Sendeku, E. Moretti and A. Vomiero, Prog. Mater. Sci., 2024, 144, 101287–101315 CrossRef.
- A. R. Sotiles, L. M. Baika, M. T. Grassi and F. Wypych, J. Am. Chem. Soc., 2019, 141, 531–540 CrossRef CAS PubMed.
- Z. Li, X. Zhang, H. Cheng, J. Liu, M. Shao, M. Wei, D. G. Evans, H. Zhang and X. Duan, Adv. Energy Mater., 2020, 10, 1900486 CrossRef CAS.
- S. Yahya, S. K. M. Wahab and F. W. Harun, Renewable Energy, 2020, 157, 164–172 CrossRef CAS.
- S. Barakan and V. Aghazadeh, Environ. Sci. Pollut. Res. Int., 2021, 28, 2572–2599 CrossRef CAS PubMed.
- H. Han, M. K. Rafiq, T. Zhou, R. Xu, O. Mašek and X. Li, J. Hazard. Mater., 2019, 369, 780–796 CrossRef CAS PubMed.
- S. Akbulut, Z. N. Kurt and S. Arasan, Earth Sci. Res. J., 2012, 16, 95–101 Search PubMed.
- S. Taymouri and J. Varshosaz, Adv. Biomed. Res., 2016, 5, 48 CrossRef PubMed.
- G. Xue, M. Gao, Z. Gu, Z. Luo and Z. Hu, Chem. Eng. J., 2013, 218, 223–231 CrossRef CAS.
- R. Wei, Y. Mo, D. Fu, H. Liu and B. Xu, Molecules, 2023, 28, 2021–2037 CrossRef CAS PubMed.
- J. M. Thomas and W. J. Thomas, Principles and Practice of Heterogeneous Catalysis, 2nd edn, 2015 Search PubMed.
- T. Mishra and D. K. Mahato, J. Environ. Chem. Eng., 2016, 4, 1224–1230 CrossRef CAS.
- F. Tomul, Appl. Clay Sci., 2016, 120, 121–134 CrossRef CAS.
- (a) L. Chmielarz, A. Kowalczyk, M. Skoczek, M. Rutkowska, B. Gil, P. Natkański, M. Radko, M. Motak, R. Dębek and J. Ryczkowski, Appl. Clay Sci., 2018, 160, 116–125 CrossRef CAS.
- M. Munoz, M. Greber, K. B. Tayed, C. Lamonier and C. I. Carole, Green Process. Synth., 2023, 12, 20230026–20230038 CrossRef CAS.
- E. Ruiz-Hitzky, P. Aranda, M. Akkari, N. Khaorapapong and M. Ogawa, Beilstein J. Nanotechnol., 2019, 10, 1140–1156 CrossRef CAS PubMed.
- R. Ma and T. Sasaki, Acc. Chem. Res., 2015, 48(1), 136–143 CrossRef CAS PubMed.
- (a) M. Khan, M. N. Tahir, S. F. Adil, H. U. Khan, M. R. H. Siddiqui, A. A. Al-Warthan and W. Tremel, J. Mater. Chem. A, 2015, 3, 18753–18808 RSC; (b) M. Yi and Z. Shen, J. Mater. Chem. A, 2015, 3(22), 11700–11715 RSC.
- S. E. Lowe, G. P. Simon and Y. L. Zhong, Curr. Opin. Colloid Interface Sci., 2015, 20(5–6), 329–338 Search PubMed.
- G. Bepete, F. Hof, K. Huang, K. Kampioti, E. Anglaret, C. Drummond and A. Penicaud, Phys. Status Solidi RRL, 2016, 10(12), 895–899 CrossRef CAS.
- N. I. Zaaba, K. L. Foo, U. Hashim, S. J. Tan, W. W. Liu and C. H. Voon, Procedia Eng., 2017, 184, 469–477 CrossRef CAS.
- J. Cao, P. He, M. A. Mohammed, X. Zhao, R. J. Young, B. Derby, I. A. Kinloch and R. A. W. Dryfe, J. Am. Chem. Soc., 2017, 139(48), 17446–17456 CrossRef CAS PubMed.
- C. Backes, A. M. Abdelkader, C. Alonso, A. Andrieux-Ledier, R. Arenal, J. Azpeitia, N. Balakrishnan, L. Banszerus, J. Barjon and R. Bartali, 2D Mater., 2020, 7, 022001 CrossRef CAS.
- E. Grånäs, T. Gerber, U. A. Schröder, K. Schulte, J. N. Andersen, T. Michel and J. Knudsen, Surf. Sci., 2016, 651, 57–61 CrossRef.
- Y. Xiao, G. Tian, W. Li, Y. Xie, B. Jiang, C. Tian, D. Zhao and H. Fu, J. Am. Chem. Soc., 2019, 141(6), 2508–2515 CrossRef CAS PubMed.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116(12), 7159–7329 CrossRef CAS PubMed.
- N. Mao, C. H. Zhou, D. S. Tong, W. H. Yu and C. X. C. Lin, Appl. Clay Sci., 2017, 144, 60–78 CrossRef CAS.
- T. T. Zhu, C. H. Zhou, F. B. Kabwe, Q. Q. Wu, C. S. Li and J. R. Zhang, Appl. Clay Sci., 2019, 169, 48–66 CrossRef CAS.
- Y. Zhao, F. Li, R. Zhang, D. G. Evans and X. Duan, Chem. Mater., 2002, 14, 4286–4291 CrossRef CAS.
- S. Guo, D. G. Evans, D. Li and X. Duan, AIChE J., 2009, 55(8), 2024–2034 CrossRef CAS.
- S. Bai, T. Li, H. Wang, L. Tan, Y. Zhao and Y.-F Song, Chem. Eng. J., 2021, 419, 129390 CrossRef CAS.
- D. Barman, S. Ghosh, S. Paul, B. Dalal and S. K. De, Chem. Mater., 2018, 30(16), 5550–5560 CrossRef CAS.
- X. Zhou, B. Wilfong, H. Vivanco, J. Paglione, C. M. Brown and E. E. Rodriguez, J. Am. Chem. Soc., 2016, 138(50), 16432–16442 CrossRef CAS PubMed.
- S. Sasaki, D. Driss, E. Grange, J.-Y. Mevellec, M. T. Caldes, C. Guillot-Deudon, S. Cadars, B. Corraze, E. Janod, S. Jobic and L. Cario, Angew. Chem., Int. Ed., 2018, 57(41), 13618–13623 CrossRef CAS PubMed.
- S. Sasaki, M. T. Caldes, C. Guillot-Deudon, I. Braems, G. Steciuk, L. Palatinus, E. Gautron, G. Frapper, E. Janod, B. Corraze, S. Jobic and L. Cario, Nat. Commun., 2021, 12(1), 3605 CrossRef CAS PubMed.
- Y. Wang, K. Yamamoto, Y. Tsujimoto, T. Matsunaga, D. Zhang, Z. Cao, K. Nakanishi, T. Uchiyama, T. Watanabe, T. Takami, H. Miki, H. Iba, K. Maeda, H. Kageyama and Y. Uchimoto, Chem. Mater., 2022, 34(2), 609–616 CrossRef CAS.
- K. Wissel, S. Dasgupta, A. Benes, R. Schoch, M. Bauer, R. Witte, A. D. Fortes, E. Erdem, J. Rohrer and O. Clemens, J. Mater. Chem. A, 2018, 6(44), 22013–22026 RSC.
- T. R. Graham, J. Z. Hu, X. Zhang, M. Dembowski, N. R. Jaegers, C. Wan, M. Bowden, A. S. Lipton, A. R. Felmy, S. B. Clark, K. M. Rosso and C. I. Pearce, Inorg. Chem., 2019, 58(18), 12385–12394 CrossRef CAS PubMed.
- T. Stimpfling, A. Langry, H. Hintze-Bruening and F. Leroux, J. Colloid Interface Sci., 2016, 462, 260–271 CrossRef CAS PubMed.
- (a) A. Bashir, S. Ahad, L. A. Malik, A. Qureashi, T. Manzoor, G. N. Dar and A. H. Pandith, Ind. Eng. Chem. Res., 2020, 59(52), 22353–22397 CrossRef CAS; (b) M. Pica, A. Donnadio and M. Casciola, Coord. Chem. Rev., 2018, 374, 218–235 CrossRef CAS; (c) H. P. Xiao and S. H. Liu, Mater. Des., 2018, 155, 19–35 CrossRef CAS.
- G. Alberti and E. Torraca, J. Inorg. Nucl. Chem., 1968, 30, 317–318 CrossRef CAS.
- D. Capitani, M. Casciola, A. Donnadio and R. Vivani, Inorg. Chem., 2010, 49(20), 9409–9415 CrossRef CAS PubMed.
- J. F. Yu, H. Ding, J. Lampron, B. R. Martin, A. Clearfield and L. Y. Sun, Inorg. Chem., 2020, 59(2), 1204–1210 CrossRef CAS PubMed.
- W. J. Mu, Q. H. Yu, R. Zhang, X. L. Li, R. Hu, Y. He, H. Y. Wei, Y. Jian and Y. C. Yang, J. Mater. Chem. A, 2017, 5(46), 24388–24395 RSC.
- S. Bevara, P. Giri, S. J. Patwe, S. N. Achary, R. K. Mishra, A. Kumar, A. K. Sinha, C. P. Kaushi and A. K. Tyagi, J. Environ. Chem. Eng., 2018, 6(2), 2248–2261 CrossRef CAS.
- Y. Cheng, X. D. Wang, S. Jaenicke and G. K. Chuah, ChemSusChem, 2017, 10(16), 3235–3242 CrossRef CAS PubMed.
- Y. Cheng, X. D. Wang, S. Jaenicke and K. Chuah, Inorg. Chem., 2018, 57(8), 4370–4378 CrossRef CAS PubMed.
- Y. Cheng, H. W. Zhang, J. A. Jaenicke, E. C. P. Tan and G. K. Chuah, ACS Sustainable Chem. Eng., 2019, 7(1), 895–904 CrossRef CAS.
- (a) U. Ciesla, S. Schacht, G. D. Stucky, K. K. Unger and F. Schuth, Angew. Chem., Int. Ed. Engl., 1996, 35(5), 541–543 CrossRef CAS; (b) J. Jimenez-Jimenez, P. Maireles-Torres, P. Olivera-Pastor, E. Rodriguez-Castellon, A. Jimenez-Lopez, D. J. Jones and J. Roziere, Adv. Mater., 1998, 10(10), 812–815 CrossRef CAS.
- A. Tarafdar, A. B. Panda, N. C. Pradhan and P. Pramanik, Microporous Mesoporous Mater., 2006, 95(1–3), 360–365 CrossRef CAS.
- Y. Sun, P. Afanasiev, M. Vrinat and G. Coudurier, J. Mater. Chem., 2000, 10(10), 2320–2324 RSC.
- Z. Y. Yuan, T. Z. Ren, A. Azioune, J. J. Pireaux and B. L. Su, Catal. Today, 2005, 105(3–4), 647–654 CrossRef CAS.
- K. Saravanan, K. S. Park, S. Jeon and J. W. Bae, ACS Omega, 2018, 3(1), 808–820 CrossRef CAS PubMed.
- R. Chakraborty, K. Bhattacharaya and P. Chattopadhyay, Appl. Radiat. Isot., 2014, 85, 34–38 CrossRef CAS PubMed.
- X. Y. Tian, W. He, J. J. Cui, X. D. Zhang, W. J. Zhou, S. P. Yan, X. N. Sun, X. X. Han, S. S. Han and Y. Z. Yue, J. Colloid Interface Sci., 2010, 343(1), 344–349 CrossRef CAS PubMed.
- G. Alberti, M. Casciola, F. Marmottini and R. Vivani, J. Porous Mater., 1999, 6(4), 299–305 CrossRef CAS.
- M. V. Ramos-Garces, J. Sanchez, K. La Luz-Rivera, D. E. Del Toro-Pedrosa, T. F. Jaramillo and J. L. Colon, Dalton Trans., 2020, 49(12), 3892–3900 RSC.
- (a) Y. Zhu, K. Kanamori, N. Moitra, K. Kadono, S. Ohi, N. Shimobayashi and K. Nakanishi, Microporous Mesoporous Mater., 2016, 225, 122–127 CrossRef CAS; (b) Y. Zhu, T. Shimizu, T. Kitajima, K. Morisato, N. Moitra, N. Brun, T. Kiyomura, K. Kanamori, K. Takeda, H. Kurata, M. Tafu and K. Nakanishi, New J. Chem., 2015, 39(4), 2444–2450 RSC.
- A. R. Hajipour and H. Karimi, Mater. Lett., 2014, 116, 356–358 CrossRef CAS.
- M. Pica, A. Donnadio, D. Capitani, R. Vivani, E. Tronia and M. Casciola, Inorg. Chem., 2011, 50(22), 11623–11630 CrossRef CAS PubMed.
- M. Pica, R. Vivani, A. Donnadio, E. Troni, S. Fop and M. Casciola, Inorg. Chem., 2015, 54(18), 9146–9153 CrossRef CAS PubMed.
- (a) Y. H. Yu, Y. P. Chen, M. X. Zeng and Z. D. Cheng, Mater. Lett., 2016, 163, 158–161 CrossRef CAS; (b) X. Y. Li, G. D. Ding, B. L. Thompson, L. D. Hao, D. A. Deming, Z. M. Heiden and Q. Zhang, ACS Appl. Mater. Interfaces, 2020, 12(27), 30670–30679 CrossRef PubMed; (c) D. L. Huang, H. F. Xu, B. S. Jacob, R. Ma, S. Yuan, L. C. Zhang, M. S. Mannan and Z. D. Cheng, J. Loss Prev. Process Ind., 2020, 66, 104183 CrossRef.
- H. Ding, A. Ahmed, K. Y. Shen and L. Y. Sun, Aggregate, 2022, e174 CrossRef CAS.
- (a) G. Alberti, M. Casciola and U. Costantino, J. Colloid Interface Sci., 1985, 107(1), 256–263 CrossRef CAS; (b) R. M. Tindwa, D. K. Ellis, G. Z. Peng and A. Clearfield, J. Chem. Soc., Faraday Trans., 1985, 81, 545–552 RSC; (c) G. Alberti and J. Marmottini, J. Colloid Interface Sci., 1993, 157, 513–515 CrossRef CAS.
- F. C. A. Ding, H. Ding, Z. Q. Shen, L. Qian, J. Ouyang, S. S. Zeng, T. A. P. Seery, J. Li, G. Z. Wu, S. E. Chavez, A. T. Smith, L. Liu, Y. Li and L. Y. Sun, Langmuir, 2021, 37(25), 7760–7770 CrossRef CAS PubMed.
- (a) M. Pica, A. Donnadio, V. Bianchi, S. Fop and M. Casciola, Carbohydr. Polym., 2013, 97(1), 210–216 CrossRef CAS PubMed; (b) X. Lin, D. Schmelter, S. Imanian and H. Hintze-Bruening, Sci. Rep., 2019, 9, 16389–16404 CrossRef PubMed; (c) H. W. Huang, M. L. Li, Y. Q. Tian, Y. H. Xie, X. X. Sheng, X. Jian and X. Y. Zhang, “Exfoliation and functionalization of alpha-zirconium phosphate in one pot for waterborne epoxy coatings with enhanced anticorrosion performance”, Prog. Org. Coat., 2020, 138, 105390 CrossRef CAS.
- H. Ding, S. T. Khan, K. N. Aguirre, R. S. Camarda, J. B. Gafney, A. Clearfield and L. Y. Sun, Inorg. Chem., 2020, 59(11), 7822–7829 CrossRef CAS PubMed.
- (a) L. Y. Sun, W. J. Boo, H. J. Sue and A. Clearfield, New J. Chem., 2007, 31(1), 39–43 RSC; (b) M. Wong, R. Ishige, K. L. White, P. Li, D. Kim, R. Krishnamoorti, R. Gunther, T. Higuchi, H. J. Jinnai, A. Takahara, R. Nishimura and H. J. Sue, Nat. Commun., 2014, 5, 3589 CrossRef PubMed.
- H. J. Sue, K. T. Gam, N. Bestaoui, N. Spurr and A. Clearfield, Chem. Mater., 2004, 16(2), 242–249 CrossRef CAS.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2(2), 16098 CrossRef CAS.
- H. Chen, H. Ma and C. Li, ACS Nano, 2021, 15(10), 15502–15537 CrossRef CAS PubMed.
- P. A. Shinde, A. M. Patil, S. Lee, E. Jung and S. C. J. Jun, J. Mater. Chem. A, 2022, 10(3), 1105–1149 RSC.
- Z. Wu, Z. Yang, C. Jin, Y. Zhao and R. Che, ACS Appl. Mater. Interfaces, 2021, 13(4), 5866–5876 CrossRef CAS PubMed.
- L. Verger, C. Xu, V. Natu, H. M. Cheng, W. Ren and M. W. Barsoum, Curr. Opin. Solid State Mater. Sci., 2019, 23(3), 149–163 CrossRef CAS.
- K. Li, T. Jiao, R. Xing, G. Zou, Q. Zhao, J. Zhou, L. Zhang and Q. Peng, Green Energy Environ., 2018, 3(2), 147–155 CrossRef.
- Y. T. He, X. X. Shen and Y. H. Zhang, ACS Appl. Nano Mater., 2024 DOI:10.1021/acsanm.3c06242.
- J. L. Yang, Y. Zhang, Y. Z. Ge, S. Tang, J. Li, H. Zhang, X. D. Shi, Z. T. Wang and X. L. Tian, Adv. Mater., 2024, 11141 Search PubMed.
- N. Kawaguchi, K. Shibata and T. Mizoguchi, ACS Phys. Chem. Au, 2024, 4(3), 281–291 CrossRef CAS PubMed.
- S. Parida, A. Mishra, Q. Yang, A. Dobley, C. B. Carter and A. M. Dongare, J. Mater. Sci., 2024, 59(3), 828–846 CrossRef.
- J. L. Liang, C. B. Wei, D. X. Huo and H. Li, J. Energy Storage, 2024, 85, 111044 CrossRef.
- H. L. Li, J. Y. Wang, S. Xu, A. Y. Chen, H. Y. Lu, Y. Jin, S. H. Guo and J. Zhu, Adv. Mater., 2024, 3073 CAS.
- F. Li, R. Hu, Z. Y. Huang, S. W. Luo, H. Qiao, J. X. Zhong and X. Qi, Appl. Mater. Today, 2024, 36, 102069 CrossRef.
- B. Yang, A. G. Tamirat, D. Bin, Y. Yao, H. Lu and Y. Xia, Adv. Funct. Mater., 2021, 31(52), 2104543 CrossRef CAS.
- V. Vijayakumar, M. Ghosh, K. Asokan, S. B. Sukumaran, S. Kurungot, J. Mindemark, D. Brandell, M. Winter and J. R. Nair, Adv. Energy Mater., 2023, 13(15), 202203326 Search PubMed.
- H. Chaudhuri and Y.-S. Yun, J. Power Sources, 2023, 564, 232870 CrossRef CAS.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- W. Zhao, C. Zhao, H. Wu, L. Li and C. Zhang, J. Energy Storage, 2024, 81, 110409 CrossRef.
- Y. Liu, H. Shi and Z. S. Wu, Energy Environ. Sci., 2023, 16, 4834–4871 RSC.
- J. Asenbauer, T. Eisenmann, M. Kuenzel, A. Kazzazi, Z. Chen and D. Bresser, Sustainable Energy Fuels, 2020, 4(11), 5387–5416 RSC.
- M. Druee, M. Seyring and M. Rettenmayr, J. Power Sources, 2017, 353, 58–66 CrossRef CAS.
- H. Onuma, K. Kubota, S. Muratsubaki, W. Ota, M. Shishkin, H. Sato, K. Yamashita, S. Yasuno and S. Komaba, J. Mater. Chem. A, 2021, 9(18), 11187–11200 RSC.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14(3), 271–279 CrossRef CAS PubMed.
- K. Share, A. P. Cohn, R. Carter, B. Rogers and C. L. Pint, ACS Nano, 2016, 10(10), 9738–9744 CrossRef CAS PubMed.
- Z. Chen, Q. M. Wang, S. Bai, X. Wang, W. L. Lin and Y. N. Zhang, J. Colloid Interface Sci., 2024, 659, 463–473 CrossRef CAS PubMed.
- X. Zhou, L. Wu, J. Wang, J. Tang, L. Xi and B. Wang, J. Power Sources, 2016, 324, 33–40 CrossRef CAS.
- Q. Chen, S. Liu, R. Zhu, D. Wu, H. Fu, J. Zhu and H. He, J. Power Sources, 2018, 405, 61–69 CrossRef CAS.
- J. Ryu, D. Hong, S. Choi and S. Park, ACS Nano, 2016, 10, 2843–2851 CrossRef CAS PubMed.
- Y.-K. Park, M. Boyer, G. S. Hwang and J.-W. Lee, J. Electroanal. Chem., 2019, 833, 552–559 CrossRef CAS.
- K. Adpakpang, S. B. Patil, S. M. Oh, J.-H. Kang, M. Lacroix and S.-J. Wang, Electrochim. Acta, 2016, 204, 60–68 CrossRef CAS.
- X.-Y. Yue, A. Abulikemu, X.-L. Li, Q.-Q. Qiu, F. Wang, X.-J. Wu and Y.-N. Zhou, J. Power Sources, 2019, 410–411, 132–136 CrossRef CAS.
- L. Sun, T. Su, L. Xu and B. Du, Phys. Chem. Chem. Phys., 2016, 18, 1521–1525 RSC.
- P. Zhang, C. Zhao, W. Ning, S. Miao, L. Nan, Q. Gao and X. Shin, Colloids Surf., A, 2022, 642, 128605–129615 CrossRef CAS.
- L. Hou, B. Xing, W. Kang, H. Zeng, H. Guo, S. Cheng, G. Huang, C. Cao, Z. Chen and C. Zhang, Appl. Clay Sci., 2022, 218, 106418–106429 CrossRef CAS.
- X. Huang, D. Cen, R. Wei, H. Fan and Z. Bao, ACS Appl. Mater. Interfaces, 2019, 11, 26854–26862 CrossRef CAS PubMed.
- Y. Lan and D. Chen, J. Mater. Sci., 2018, 53, 2054–2064 CrossRef CAS.
- D. Alonso-Domínguez, M. P. Pico, I. Álvarez-Serrano and M. L. López, Nanomaterials, 2018, 8, 805–825 CrossRef PubMed.
- R. A. House, U. Maitra, M. A. Perez-Osorio, J. G. Lozano, L. Jin, J. W. Somerville, L. C. Duda, A. Nag, A. Walters, K.-J. Zhou, M. R. Roberts and P. G. Bruce, Nature, 2020, 577, 502 CrossRef CAS PubMed.
- Y.-E. Huang, W. Lin, C. Shi, L. Li, K. Fan, X.-Y. Huang, X. Wu and K.-Z. Du, J. Power Sources, 2021, 494, 229712 CrossRef CAS.
- X. Long, Z.-H. Luo, W.-H. Zhou, S.-K. Zhu, Y. Song, H. Li, C.-N. Geng, B. Shi, Z.-Y. Han, G.-M. Zhou, W. Lv and J.-J. Shao, Energy Storage Mater., 2022, 52, 120–129 CrossRef.
- W. Chen, T. Lei, X. Lv, Y. Hu, Y. Yan, Y. Jiao, W. He, Z. Li, C. Yan and J. Xiong, Adv. Mater., 2018, 30, 1804084–1804092 CrossRef PubMed.
- Y. Lan, Y. Liu, J. Li, D. Chen, G. He and I. P. Parkin, Adv. Sci., 2021, 8, 2004036–2004061 CrossRef CAS PubMed.
- F. Wu, H. Lv, S. Chen, S. Lorger, V. Srot, M. Oschatz, P. A. van Aken, X. Wu, J. Maier and Y. Yu, Adv. Funct. Mater., 2019, 29, 1902820–1902832 CrossRef.
- H. Dong, S. Qi, L. Wang, X. Chen, Y. Xiao, Y. Wang, B. Sun, G. Wang and S. Chen, Small, 2023, 19(30), 2300843 CrossRef CAS PubMed.
- Q. Liu, Y. Zhang, Y. Zhou, M. Wang, R. Li and W. Yue, J. Solid State Electrochem., 2023, 27(3), 797–807 CrossRef CAS.
- S.-H. Yu, X. Feng, N. Zhang, J. Seo and H. D. Abruna, Acc. Chem. Res., 2018, 51(2), 273–281 CrossRef CAS PubMed.
- B. Jache, J. O. Binder, T. Abe and P. Adelhelm, Phys. Chem. Chem. Phys., 2016, 18(21), 14299–14316 RSC.
- H. Kim, J. Hong, Y.-U. Park, J. Kim, I. Hwang and K. Kang, Adv. Funct. Mater., 2015, 25(4), 534–541 CrossRef CAS.
- Y. Li, Y. Lu, P. Adelhelm, M.-M. Titirici and Y.-S. Hu, Chem. Soc. Rev., 2019, 48(17), 4655–4687 RSC.
- H. Kim, J. Hong and K. Kang, Energy Environ. Sci., 2015, 8(10), 2963–2969 RSC.
- W. Luo, F. Shen and L. B. Hu, Acc. Chem. Res., 2016, 49(2), 231–240 CrossRef CAS PubMed.
- J. Sun, G. Zheng, H.-W. Lee, N. Liu, H. Wang, H. Yao, W. Yang and Y. Cui, Nano Lett., 2014, 14(8), 4573–4580 CrossRef CAS PubMed.
- Z. L. Jian, W. Luo and X. L. Ji, J. Am. Chem. Soc., 2015, 137(36), 11566–11569 CrossRef CAS PubMed.
- S. Sundaramoorthy, N. Gerasimchuk, K. Ghosh, S. P. Kelley and A. Choudhury, Chem. Mater., 2024, 36(8), 3574–3587 CrossRef CAS.
- L. S. Zhong, H. N. Chen, W. H. Xie, W. F. Jia, Y. H. Xiao, B. C. Cheng, L. X. Lin and S. J. Lei, Chem. Eng. J., 2024, 481, 148370 CrossRef CAS.
- R. Z. Wei, Y. Lu, W. J. Ren, Y. P. Han, A. P. V. L. Saroja, X. M. Xia, P. He, C. A. F. Nason, Z. X. Sun, J. A. Darr, J. Y. Luo, M. Zhou and Y. Xu, JPhys Energy, 2024, 6, 25022 CrossRef.
- L. Liu, B. X. Li, J. Q. Wang, H. F. Du, Z. Z. Du and W. Ai, Small, 2024, 9647 Search PubMed.
- L. H. Li, S. Lu, X. Y. Wang, J. L. Luo, H. Xu, H. Q. Gu, L. Tan, X. Du, Z. L. Niu, X. C. Zheng and D. Li, Energy Storage Mater., 2024, 65, 103131 CrossRef.
- W. Ko, S. Lee, H. Par, J. Kang, J. Ahn, Y. Lee, G. Oh, J. K. Yoo, J. Y. Hwang and J. Kim, Carbon Energy, 2024, 454 CrossRef.
- L. P. Duan, C. Y. Shao, J. Y. Liao, L. L. Song, Y. N. Zhang, R. K. Li, S. H. Guo, X. S. Zhou and H. S. Zhou, Angew. Chem. Inter. Ed., 2024, 63(17), 868 Search PubMed.
- Y. Lan, Y. Liu, J. Li, D. Chen, G. He and I. P. Parkin, Adv. Sci., 2021, 8, 2004036–2004061 CrossRef CAS PubMed.
- X. Cao, S. Yingjuan, Y. Sun, D. Xie, H. Li and M. Liu, Appl. Clay Sci., 2021, 213, 106265–106273 CrossRef CAS.
- C. Luo, H. Wang, Y. Qian, X. Shi, Z. Mao, G. Li, C. Yang, Y. Gong, A. Tang and H. Yang, J. Power Sources, 2002, 548, 232038–232047 CrossRef.
- S. Kajiyama, L. Szabova, K. Sodeyama, H. Iinuma, R. Morita, K. Gotoh, Y. Tateyama, M. Okubo and A. Yamada, ACS Nano, 2016, 10(3), 3334–3341 CrossRef CAS PubMed.
- D. Sabaghi, J. Polcak, H. Y. J. Yang, X. D. Li, A. Morag, D. Q. Li, A. S. Nia, S. H. Khosravi, T. Sikola, X. L. Feng and M. H. Yu, Adv. Energy Mater., 2024, 14(3), 2302961 CrossRef CAS.
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Chem. Rev., 2020, 120(14), 6358–6466 CrossRef CAS PubMed.
- M. Song, H. Tan, D. Chao and H. J. Fan, Adv. Funct. Mater., 2018, 28(41), 1802564 CrossRef.
- L. Shan, Y. Wang, S. Liang, B. Thang, Y. Yang, Z. Wang, N. Lu and J. Zhou, InfoMat, 2021, 3(9), 1028–1036 CrossRef CAS.
- A. A. Zhang, X. X. Yin, X. Zhang, J. J. Ba, J. P. Li, Y. J. Wei and Y. Z. Wang, ACS Appl. Energy Mater., 2024, 7(3), 1298–1305 CrossRef CAS.
- X. Gao, C. Shen, H. Dong, Y. H. Dai, P. Jiang, I. P. Parkin, H. B. Zhang, C. J. Carmalt and G. J. He, Energy Environ. Sci., 2024, 17, 2287–2297 RSC.
- G. F. Shi, P. Zhao, P. Gao, Y. Y. Xing and B. X. Shen, J. Energy Storage, 2024, 78, 110057 CrossRef.
- Y. M. Li, W. H. Li, W.-Y. Diao, F. Tao, X.-L. Wu and X.-Y. Zhang, ACS Appl. Mater. Interfaces, 2022, 14(20), 23558–23569 CrossRef CAS PubMed.
- P. Wang, K. Zhang, H. Li, J. Hu and M. Zheng, Small, 2023, 308791 Search PubMed.
- Q. Liu, P. Zhang, Z. Wang, S. Liu, X. Ren, K. Qian, X. Chen, J. Li, J. Yao, Y. Gan, L. Lv, H. Wan and H. Wang, J. Alloys Compd., 2023, 963, 171227 CrossRef CAS.
- H. Yousefzadeh, A. Noori, M. S. Rahmanifar, N. Hassani, M. Neek-Amal, M. F. El-Kady, A. Vinu, R. B. Kaner and M. F. Mousavi, Adv. Energy Mater., 2023, 13(41), 2302137 CrossRef CAS.
- X. Han, N. Li, J. S. Baik, P. Xiong, Y. Kang, Q. Liu, J. Y. Lee, C. S. Kim and H. S. Park, Adv. Funct. Mater., 2023, 33(11), 2212233 CrossRef CAS.
- L. Yang, J. Lu, E. Zhu, J. Zhang, X. Guan, B. Liu, P. Yin and G. Wang, Appl. Surf. Sci., 2024, 645, 158847 CrossRef CAS.
- M. Mao, T. Gao, S. Hou and C. Wang, Chem. Soc. Rev., 2018, 47(23), 8804–8841 RSC.
- S. Kang, K. G. Reeves, T. Koketsu, J. Ma, O. J. Borkiewicz, P. Strasser, A. Ponrouch and D. Dambournet, ACS Appl. Energy Mater., 2020, 3(9), 9143–9150 CrossRef CAS.
- F. H. Hsu, S. Y. Hsu, R. Subramani, T. C. Cheng, B. H. Chen, J. L. Chen, J. M. Chen and K. T. Lu, J. Energy Storage, 2024, 84A, 110693 CrossRef.
- T. Koketsu, J. Ma, B. J. Morgan, M. Body, C. Legein, W. Dachraoui, M. Giannini, A. Demortiere, M. Salanne, F. Dardoize, H. Groult, O. J. Borkiewicz, K. W. Chapman, P. Strasser and D. Dambournet, Nat. Mater., 2017, 16(11), 1142 CrossRef CAS PubMed.
- S. Heguri, N. Kawade, T. Fujisawa, A. Yamaguchi, A. Sumiyama, K. Tanigaki and M. Kobayashi, Phys. Rev. Lett., 2015, 114(24), 247201/1–247201/5 CrossRef CAS PubMed.
- M. Angell, C.-J. Pan, Y. Rong, C. Yuan, M.-C. Lin, B.-J. Hwang and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(5), 834–839 CrossRef CAS PubMed.
- J. Xu, Y. Dou, Z. Wei, J. Ma, Y. Deng, Y. Li, H. Liu and S. Dou, Adv. Sci., 2017, 4(10), 1700146 CrossRef PubMed.
- H. Zhang, Y. Yang, D. Ren, L. Wang and X. He, Energy Storage Mater., 2021, 36, 147–170 CrossRef.
- X. Wu, Y. Chen, Z. Xing, C. Lam, K. Wai, S.-S. Pang, W. Zhang and Z. Ju, Adv. Energy Mater., 2019, 9(21), 1900343 CrossRef.
- Z. Jian, W. Luo and X. Ji, J. Am. Chem. Soc., 2015, 137(36), 11566–11569 CrossRef CAS PubMed.
- S. N. Jagadeesan, G. D. Barbosa, F. Guo, L. Zhang, A. M. M. Abeykoon, G. Kwon, D. Olds, C. H. Turner and X. Teng, Chem. Mater., 2023, 35(16), 6517–6526 CrossRef CAS.
- Y. Sui, C. Liu, R. C. Masse, Z. G. Neale, M. Atif, M. AlSalhi and G. Cao, Energy Storage Mater., 2020, 25, 1–32 CrossRef.
- M. Liu, W. Zhang and W. Zheng, ChemSusChem, 2022, 16(4), e202201375 CrossRef PubMed.
- S. Xia, X. Wu, Z. Zhang, Y. Cui and W. Liu, Chem, 2019, 5(4), 753–785 CAS.
- P. G. Balkanloo, A. P. Marjani, F. Zanbili and M. Mahmoudian, Appl. Clay Sci., 2022, 228, 106632–106644 CrossRef.
- S. Chua, R. Fang, Z. Sun, M. Wu, Z. Gu, Y. Wang, J. N. Hart, N. Sharma, F. Li and D.-W. Wang, Chem., 2018, 24(69), 18180–18203 CrossRef CAS PubMed.
- Y. Ma, L. B. Li, L. G. X. Gao, X.-Y. Yang and Y. You, Electrochim. Acta, 2016, 187, 535–542 CrossRef CAS.
- R. Liu, B. Yuan, S. Zhong, J. Liu, L. Dong, Y. Ji, Y. Dong, C. Yang and W. He, Nano Sel., 2021, 2, 2308–2345 CrossRef CAS.
- N. Banitaba, D. Semnani, E. Heydari, B. Rezaei and A. A. Ensafi, J. Min., Met. Mater. Soc., 2019, 4537–4546 CrossRef.
- N. S. M. Johari, S. B. R. S. Adnan and N. Ahmad, Solid State Ionics, 2022, 377, 115882 CrossRef.
- K. Wissel, S. Dasgupta, A. Benes, R. Schoch, M. Bauer, R. Witte, A. D. Fortes, E. Erdem, J. Rohrer and O. Clemens, J. Mater. Chem. A, 2018, 6(44), 22013–22026 RSC.
- H. Miki, K. Yamamoto, C. Shuo, T. Matsunaga, M. Kumar, N. Thakur, Y. Sakaguchi, T. Watanabe, H. Iba, H. Kageyama and Y. Uchimoto, Solid State Ionics, 2024, 406, 116480 CrossRef CAS.
- Q. Yin, T. Wang, Z. Song, S. Yang, Y. Miao, Y. Wu, Y. Sui, J. Qi, Y. Li and D. Zhao, Chem. Eng. J., 2023, 459, 141545 CrossRef CAS.
- Q. Lin and L. Wang, J. Semicond., 2023, 44(4), 041601 CrossRef.
- Z. Wu, Y. Wu, Q. Yuan, J. Zhang, Y. Dou and J. Han, ACS Appl. Mater. Interfaces, 2023, 15(32), 38540–38549 CrossRef CAS PubMed.
- Z. Song, Q. Yin, S. Yang, Y. Miao, Y. Wu, Y.-Z. Li, Y. Ren, Y. Sui, J. Qi and J. Han, Small, 2023, 19(43), 2302896 CrossRef CAS PubMed.
- D. X. Wang, F. S. Zhang, J. L. Wang, X. Q. Shi, P. L. Gong, H. J. Liu, M. Q. Wu, Y. J. Wei and R. Q. Lian, J. Mater. Chem. A, 2024, 12(14), 8302–8310 RSC.
- Q. Liu, F. Ye, K. Guan, Y. Yang, H. Dong, Y. Wu, Z. Tang and L. Hu, Adv. Energy Mater., 2023, 13(5), 2202908 CrossRef CAS.
- S. J. Panchu, K. Raju and H. C. Swart, ChemElectroChem, 2024, e202300810 CrossRef.
- J. Sarmet, F. Leroux, C. Taviot-Guého, P. Gerlach, C. Douard, T. Brousse, G. Toussaint and P. Stevens, Molecules, 2023, 28, 1006–1020 CrossRef CAS PubMed.
- J. Sarmet, C. Taviot-Gueho, R. Thirouard, F. Leroux, C. Douard, I. Gaalich, T. Brousse, G. Toussaint and P. Stevens, Cryst. Growth Des., 2023, 23(4), 2634–2643 CrossRef CAS.
- J. Sarmet, F. Leroux, C. Taviot-Gueho, P. Gerlach, C. Douard, T. Brousse, G. Toussaint and P. Stevens, J. Solid State Chem., 2024, 332, 124592–124602 CrossRef CAS.
- Q. Q. Zhang, S. R. Wang, Y. L. Lan, J. P. Deng, M. Z. Fan, G. B. Du and W. G. Zhao, J. Colloid Interface Sci., 2024, 660, 597–607 CrossRef CAS PubMed.
- S. S. Cui, X. H. Ren, H. F. Yin, H. Q. Fan, C. Wang, M. C. Zhang, Y. Tang, H. D. Yuan and Y. L. Xin, J. Energy Storage, 2024, 85, 111092 CrossRef.
- G. A. Muller, J. B. Cook, H.-S. Kim, S. H. Tolbert and B. Dunn, Nano Lett., 2015, 15(3), 1911–1917 CrossRef CAS PubMed.
- P. Xu, H. Xiao, X. Liang, T. Zhang, F. Zhang, C. Liu, B. Lang and Q. Gao, Carbon, 2021, 173135–173144 Search PubMed.
- L. Li, N. Zhang, M. Zhang, X. Zhang and Z. Zhang, Dalton Trans., 2019, 48(5), 1747–1756 RSC.
- Z. Lin, X. Li, H. Zhang, B. B. Xu, P. Wasnik, H. Li, M. V. Singh, Y. Ma, T. Li and Z. Guo, Inorg. Chem. Front., 2023, 10(15), 4358–4392 RSC.
- X. Wang, S. Kajiyama, H. Iinuma, E. Hosono, S. Oro, I. Moriguchi, M. Okubo and A. Yamada, Nat. Commun., 2015, 6, 6544 CrossRef CAS PubMed.
- L. Zhang, W.-B. Zhang, S.-S. Chai, X.-W. Han, Q. Zhang, X. Bao, Y. W. Guo, X.-L. Zhang, X. Zhou, S.-B. Guo and X.-J. Ma, J. Electrochem. Soc., 2021, 168, 070558 CrossRef CAS.
- C. H. Chen, Y. Z. Ma and C. L. Wang, Sustainable Mater. Technol., 2019, 19, e00086 CrossRef CAS.
- S. Maiti, A. Pramanik, S. Chattopadhyay, G. De and S. Mahanty, J. Colloid Interface Sci., 2016, 464, 73 CrossRef CAS PubMed.
- Y. Lan, Y. Liu, J. Li, D. Chen, G. He and I. Parkin, Adv. Sci., 2021, 8, 2004036 CrossRef CAS PubMed.
- S. Ummartyoti, N. Bunnak and H. Manuspiya, Renewable Sustainable Energy Rev., 2016, 61, 466–472 CrossRef.
- A. Kausar, Polym. Technol. Eng., 2017, 57, 548–564 CrossRef.
- N. Pandi, S. H. Sonawane, A. K. Kola, U. K. Zore, P. H. Borse, S. B. Ambade and M. AshokKumar, Energy, Ecol. Environ., 2020, 1–13 Search PubMed.
- C. Cheng, W. Song, Q. Zhao and H. Zhang, Nanotechnol. Rev., 2020, 9(1), 323–344 CrossRef.
- D. Garcia-Garcia, J. Ferri, L. Ripoll, M. Hidalgo, J. López-Martínez and R. Balart, Appl. Surf. Sci., 2017, 422, 616–625 CrossRef CAS.
- W. Xu, B. Mu and A. Wang, New J. Chem., 2016, 40, 2687–2695 RSC.
- N. Y. W. Zaw, S. Jo, J. Park, N. Kitchamsetti, N. Jayababu and D. Kim, Appl. Clay Sci., 2022, 225, 106539–106545 CrossRef CAS.
- S. S. Chai, L. Zhang, W.-B. Zhang, X. Bao, Y.-W. Guo, X.-W. Han and J. Ma, Appl. Clay Sci., 2022, 218, 106426–106435 CrossRef CAS.
- J. Zhou, S. Dai, Y. Li, F. Han, Y. Yuan, J. Tang and W. Tang, Chem. Eng. J., 2018, 350, 835–843 CrossRef CAS.
- J. Jiang, X. Huang, R. Sun, X. Chen and S. Han, J. Colloid Interface Sci., 2023, 640, 662–679 CrossRef CAS PubMed.
- Y. Zhao, Y. Li, X. Quan and C. Li, Electrochim. Acta, 2019, 321, 134715 CrossRef CAS.
- R. Baby Suneetha, ECS Trans., 2022, 107(1), 4003 CrossRef.
- S. Zhang, Y. Yang, R. Xiao, M. Yu, Y. Zhang, X. Sun, L. Lu, X. Wu and Y. Chen, Appl. Clay Sci., 2021, 200, 105821–105830 CrossRef CAS.
- G. Chen, Y. Ai, I. T. Mugaanin, W. Ma, B. S. Hsiao, K. Hoi and M. Zhu, J. Power Sources, 2020, 450, 227637–227676 CrossRef CAS.
- R. Oraon, A. De Adhikari, S. K. Tiwari and G. C. Naya, Dalton Trans., 2016, 45, 9113–9126 RSC.
- M. Ates and S. Caliskan, Polym.-Plast. Technol. Mater., 2019, 58(14), 1481–1494 CAS.
- W. Ge, Q. Ma, Z. Ai, W. Wang, F. Jia and S. Song, Appl. Clay Sci., 2021, 206, 106022–106030 CrossRef CAS.
- X. Yang, X. Zeng, G. Han, D. Sui, X. Song and Y. Zhang, Nanomaterials, 2020, 10, 1703–1720 CrossRef CAS PubMed.
- S. Guo, K. Zhao, Z. Feng, Y. Hou, H. Li, J. Zhao, Y. Tian and H. Song, Appl. Surf. Sci., 2018, 455, 599–607 CrossRef CAS.
- C. Wu, T. Zhou, Y. Du, S. Dou, H. Zhang, L. Jiang and Q. Cheng, Nano Energy, 2019, 58, 517–527 CrossRef CAS.
- A. B. Ganganbiona, A. D. Chowdhury and R. Doong, Electrochim. Acta, 2017, 245, 912–923 CrossRef.
- S. K. Shinde, D. P. Dubal, H. M. Yadav, A. D. Jagadale, N. Maile, S. S. Karade, D.-S. Lee and D.-Yo. Kim, Ceram. Int., 2022, 48, 25020–25033 CrossRef CAS.
- S. S. Chai, W.-B. Zhang, J. L. Yang, L. Zhang, X.-W. Han, M. M. Theint and X.-J. Ma, J. Rare Earths, 2023, 41, 728–739 CrossRef CAS.
- S. Venkatesan and Y.-L. Lee, Coord. Chem. Rev., 2017, 353, 58–112 CrossRef CAS.
- H. Iftikhar, G. G. Sonai, S. G. Hashmi, A. F. Nogueira and P. D. Lund, Materials, 2019, 12, 1998–2066 CrossRef CAS PubMed.
- S. Ummartyoti, N. Bunnak and H. Manuspiya, Renewable Sustainable Energy Rev., 2016, 61, 466–472 CrossRef.
- Y. Li, J. V. Milic, A. Ummadisingu, J. Y. Seo, J. H. Im, H. S. Kim, Y. Liu, M. I. Dar, S. M. Zakeeruddin, P. Wang, A. Hagfeldt and M. Grätzel, Nano Lett., 2019, 19(1), 150–157 CrossRef CAS PubMed.
- F. Long, Y. H. Guo, L. G. Yuan, H. Yin, Y. R. Tao, Z. G. Jiang, S. M. Peng, B. Wu, K. Y. Yan, M. Liu, X. H. Lu, W. W. Meng, M. Z. Long and G. F. Zhou, Chem. Eng. J., 2024, 485, 149963 CrossRef CAS.
- S. Yun, J. N. Freitas, A. F. Nogueira, Y. Wang, S. Ahmad and Z.-S. Wang, Prog. Polym. Sci., 2016, 59, 1–40 CrossRef CAS.
- D. Costenaro, C. Bisio, F. Carniato, G. Gatti, F. Oswald, T. B. Meyer and L. Marchese, Sol. Energy Mater. Sol. Cells, 2013, 117, 9–14 CrossRef CAS.
- R. K. Gupta, H.-W. Rhee, I. Bedja, A. N. AlHazaa and A. Khan, J. Power Sources, 2021, 490, 229509 CrossRef CAS.
- C. W. Tu, K. Y. Liu, A. T. Chien, M. H. Yen, T. H. Weng, K. C. Ho and K. F. Lin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 47–53 CrossRef CAS.
- M. A. S. Andrade, A. F. Nogueira, K. Miettunen, A. Tiihonen, P. D. Lund and H. O. Pastore, J. Power Sources, 2016, 325, 161–170 CrossRef CAS.
- L.-H. Chen, S. Venkatesan, I.-P. Liu and Y. L. Lee, J. Oleo Sci., 2020, 69(6), 539–547 CrossRef CAS PubMed.
- N. Sangiorgi, A. Sangiorgi, A. Sanson, M. Licchelli, M. Orbelli and A. Biroli, Processes, 2023, 11, 463–475 CrossRef CAS.
- Z. Chen, K. O. Kirlikovali, K. B. Idrees, M. C. Wasson and K. Farha, Chem, 2022, 8, 693–716 CAS.
- S. Flesch, D. Pudlo, D. Albrecht, A. Jacob and F. Enzmann, Int. J. Hydrogen Energy, 2018, 43, 20822–20835 CrossRef CAS.
- C. Hemme and W. Van Berk, Appl. Sci., 2018, 8, 2282 CrossRef CAS.
- A. Al-Yaseri, D. Wolff-Boenisch, C. A. Fauziah and S. Iglauern, Int. J. Hydrogen Energy, 2021, 46, 34356–34361 CrossRef CAS.
- P. Ghosh, B. K. Prusty, P. Sandilya and V. Y. Turlapati, Energy Fuels, 2023, 37, 6757–6769 CrossRef CAS.
- V. A. Arus, S. Nousir, E. Sennour, T. C. Shiao, I. D. Nistor, R. Roy and A. Azzouz, Int. J. Hydrogen Energy, 2018, 43, 7964–7972 CrossRef CAS.
- B. E. Alver, Clay Miner., 2017, 52(1), 67–73 CrossRef CAS.
- A. Azzouz, S. Nousir, N. Bouazizi and R. Roy, TechConnect Briefs, 2014, 72–75 CAS.
- R. Sennour, T. C. Shiao, V. A. Arus, M. N. Tahir, N. Bouazizi, R. Roy and A. Azzouz, Phys. Chem. Chem. Phys., 2017, 19, 29333–29343 RSC.
- F. Samimi, M. Ghiyasiyan-Arani and M. Salavati-Niasari, Fuel, 2022, 320, 123933–123945 CrossRef CAS.
- R. N. Muthu, S. Rajashabala and R. Kannan, Renewable Energy, 2016, 90, 554–564 CrossRef CAS.
- R. Monsef and M. Salavati-Niasari, Fuel, 2023, 332, 126015–126031 CrossRef CAS.
- J. S. Boruah and D. Chowdhury, Minerals, 2023, 13, 26 CrossRef CAS.
- C. Ruiz-García, J. Pérez-Carvajal, A. Berenguer-Murcia, M. Darder, P. Aranda and E. Ruiz-Hitzky, J. Phys. Chem. Chem. Phys., 2013, 15, 18635–18641 RSC.
- L. Daukiya, M. N. Nair, M. Cranney, F. Vonau, S. Hajjar-Garreau, D. Aubel and L. Simon, Prog. Surf. Sci., 2019, 94(1), 1–20 CrossRef CAS.
- T. E. Weller, M. Ellerby, S. S. Saxena, R. P. Smith and N. T. Skipper, Nat. Phys., 2005, 1, 39 Search PubMed; N. Emery, C. Hérold, M. d'Astuto, V. Garcia, Ch. Bellin, J. F. Marêché, P. Lagrange and G. Loupias, Phys. Rev. Lett., 2005, 95, 087003 CrossRef CAS PubMed.
- I. El Hajj, L. Speyer, S. Cahen, L. Herbuvaux, P. Lagrange, G. Medjahdi and C. Herold, Carbon, 2022, 186, 431–436 CrossRef CAS.
- S. Ichinokura, K. Sugawara, A. Takayama, T. Takahashi and S. Hasegawa, ACS Nano, 2016, 10(2), 2761–2765 CrossRef CAS PubMed.
- M. Yankowitz, S. Chen, H. Polshyn, Y. Zhang, K. Watanabe, T. Taniguchi, D. Graf, A. F. Young and C. R. Dean, Science, 2019, 363(6431), 1059–1064 CrossRef CAS PubMed.
- M. A. Choffel, R. N. Gannon, F. Goehler, A. M. Miller, D. L. Medlin, T. Seyller and D. Johnson, Chem. Mater., 2021, 33(16), 6403–6411 CrossRef CAS.
- K. Nakayama, H. Kimizuka, Y. Tanaka, T. Sato, S. Souma, T. Takahashi, S. Sasaki, K. Segawa and Y. Ando, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92(10), 100508–100513 CrossRef.
- S. Paul, B. Dalal, R. Jana, A. Shit, A. Datta and S. Kumar De, J. Phys. Chem. C, 2020, 124(23), 12824–12833 CrossRef CAS.
- B. Soundiraraju and B. K. George, ACS Nano, 2017, 11(9), 8892–8900 CrossRef CAS PubMed.
- J. L. Andrews, L. R. De Jesus, T. M. Tolhurst, P. M. Marley, A. Moewes and S. Banerjee, Chem. Mater., 2017, 29(7), 3285–3294 CrossRef CAS.
- J. P. Pender, G. Jha and C. B. Mullins, ACS Nano, 2020, 14(2), 1243–1295 CrossRef CAS PubMed.
- T. Liu, L. Lin, X. Bi, L. Tian, K. Yang, J. Liu, M. Li, Z. Chen, J. Lu, M. Li, Z. Chen, J. Lu, K. Amine, K. Xu and F. Pan, Nat. Nanotechnol., 2019, 14(1), 50–56 CrossRef CAS PubMed.
- H. Kim, J. Hong and K. Kang, Energy Environ. Sci., 2015, 8(10), 2963–2969 RSC.
- X. Bie, K. Kubota, T. Hosaka, K. Chihara and S. Komaba, J. Mater. Chem. A, 2017, 5(9), 4325–4330 RSC.
- L. Fan, R. Ma, Q. Zhang, X. Jia and B. Lu, Angew. Chem., Int. Ed., 2019, 58(31), 10500–10505 CrossRef CAS PubMed.
- Y. Ishijima, H. Imai and Y. Oaki, Chem, 2017, 3(3), 509–521 CAS.
- A. Rouag, R. Porhiel, K. Lemoine, F. Leroux, J. M. Grenèche, D. Delbègue, C. Iojoiu and K. Guérin, Dalton Trans., 2024, 53(17), 7628 RSC.
- D. D. Zhu, Y. Su, J. Z. Chen, X. Z. Ou, X. D. Zhang, W. Xie, Y. Y. Zhou, Y. N. Guo, Q. S. Dai, P. Jia, J. T. Yan, L. Geng, B. Y. Guo, L. Q. Zhang, Y. F. Tang, Q. Huang and J. Y. Huang, Nanoresearch, 2023, 6289 Search PubMed.
- S. W. Liu, J. C. Yang, S. P. Hao, S. J. Jiang, X. H. Li, O. Dolotko, F. X. Wu, Y. J. Li and Z. J. He, Chem. Eng. J., 2024, 479, 147607 CrossRef CAS.
- S. Ghosh, M. Bhar, U. Bhattacharjee, K. P. Yalamanchili, S. Krishnamurthy and S. K. Martha, J. Mater. Chem. A, 2024, 12, 11362–11377 RSC.
- G. Liu, L. Ma, X. Xi and Z. Nie, Waste Manage., 2024, 178(15), 105–114 CrossRef CAS PubMed.
- Y. Gao, S. Zhang, S. Lin, Z. Li, Y. Chen and C. Wang, Environ. Res., 2024, 247, 118216 CrossRef CAS PubMed.
- S. Yang, Q. Gao, Y. Li, H. Cai, X. Li, G. Sun, S. Zhuang, Y. Tong, H. Luo and M. Lu, J. Energy Clim., 2024, 93, 24–31 CAS.
- M. G. Kanatzidis, L. M. Tonge, T. J. Marks, H. O. Marcy and C. R. Kannewurf, J. Am. Chem. Soc., 1987, 109(12), 3797–3799 CrossRef CAS.
- P. Vialat, C. Mousty, C. Taviot-Gueho, G. Renaudin, H. Martinez, J.-C. Dupin, E. Elkaim and F. Leroux, Adv. Funct. Mater., 2014, 24, 4831–4842 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |