
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A cobalt(II) coordination polymer-derived catalyst engineered via temperature-induced semi-reversible single-crystal-to-single-crystal (SCSC) dehydration for efficient liquid-phase epoxidation of olefins†
Siya T.
Hulushe
 *a,
Gareth M.
Watkins
a and
Setshaba D.
Khanye
b
*a,
Gareth M.
Watkins
a and
Setshaba D.
Khanye
b
aDepartment of Chemistry, Rhodes University, Makhanda 6139, South Africa. E-mail: hulushesiya@gmail.com
bDivision of Pharmaceutical Chemistry, Faculty of Pharmacy, Rhodes University, Makhanda 6139, South Africa
First published on 20th June 2024
Abstract
Single-crystal-to-single-crystal (SCSC) transformations provide more avenues for phase transitions, which have piqued great interest in crystal engineering. In this work, a 3D Co(II)-based coordination polymer (CP), {Co2(OH2)8(btec)}·4H2O (1), (where (btec)4− = 1,2,4,5-benzenetetracarboxylate) undergoes SCSC transition upon heating at 180 °C to afford an anhydrous phase [Co2(btec)] (1′). Room-temperature water-vapour induced semi-reversible SCSC transformation of 1′ involves condensation of two water molecules coordinating to the metal cluster, yielding a new framework [Co2(OH2)2(btec)] (2). These SCSC transitions were accomplished through a sequential bond breaking and new bond formation process which was accompanied by colour changes from orange (1) → violet (1′) → pink (2). All materials were structurally elucidated by single-crystal X-ray diffraction (SCXRD) and further established by various analytical techniques. According to SCXRD data, all the frameworks possess octahedral geometries around the cobalt(II) sphere. SCXRD studies further revealed that 1 is a polymeric architecture with a binodal 4-c sql topology while 1′ and 2 possess (3,6)-c kgd and (4,6)-c scu 3D nets, respectively. By virtue of multitopicity exhibited by the tetracarboxylate, the coordination number of the linker around the Co(II) sphere increased from four (in 1) to eight (in 1′) and then decreased to six (in 2). Most interestingly, permanent porosity could be observed for the dihydrate 2, originated from potential void space as substantiated by dinitrogen (N2) sorption isotherm. These porous frameworks were active catalysts for the aerobic epoxidation of the model substrate cyclohexene using molecular oxygen (O2) as the final oxidant in the presence of the sacrificial i-butyraldehyde (IBA) reductant. For using the dihydrous phase 2, cyclohexene and various other olefins were catalytically oxidised to their corresponding epoxides with up to 38.5% conversion and 99.0% selectivity. The catalyst 2 can be expediently recycled in four runs without significant loss of activity. This research demonstrates that a little innovation in the active-site-engineered organic–inorganic hybrid materials can significantly enhance the catalytic performance and selectivity of coordination polymer-derived heterogeneous catalysts.
1. Introduction
The catalytic epoxidation of olefins is a significant commercial process since epoxides are commonly used as crucial intermediates in chemical synthesis.1 There has recently been an upsurge of interest in studying the aerobic epoxidation of olefins in an attempt to develop more efficient and environmentally benign routes.2 The “Mukaiyama–Yamada” approach has been described to easily achieve liquid-phase aerobic epoxidation of olefins over various transition metal (TM) complexes under very mild conditions by utilizing ubiquitous dioxygen (O2) as an oxidant and aliphatic aldehyde (i.e., i-butyraldehyde; IBA) as a co-reagent.3 Although the use of IBA is undesirable, it is still worth investigating for its several advantages, including relatively mild operating conditions (low temperatures and ambient pressure),4 excellent epoxide selectivity, low cost, and the ecologically friendly nature of the oxidant.5 Generally, after years of investigation, this aerobic epoxidation reaction has been known to involve TM centres,6–9 such as copper, cobalt, and manganese, for higher efficiency of catalytic activities.9,10–14 However, aerobic epoxidation, particularly the unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, constitutes one of the most coveted but challenging issues.11 For instance, owing to the reductive nature of the double bond, many non-ideal oxidative products can be produced.
C bond, constitutes one of the most coveted but challenging issues.11 For instance, owing to the reductive nature of the double bond, many non-ideal oxidative products can be produced.
Preparation of high-value-added products without the use of organic solvents and with only O2 as the oxidizing reagent is an ever-ongoing goal for the development of green methods that are more efficient and selective while generating fewer toxic residues and by-products.15–17 Furthermore, greener technologies based on O2 and organic peroxide (R-OOH; R = alkyl chain), such as tert-butyl hydroperoxide (t-BuOOH) and hydrogen peroxide (H2O2), have gained popularity in recent years due to their environmental friendliness.18
A vast variety of coordination polymers (CPs) have been reported, with those based on M2 diad paddle-wheel-type [M2(COO)4] (M = Zn, Cu) secondary building units (SBUs) widely regarded as one of the most representative class.19 Carboxylic acid bridging ligands, in particular, coordinate with metal ions to form one-dimensional (1D) and two-dimensional (2D) layers, which are further supported by solvent(s) to generate three-dimensional (3D) architectures.7 One downside of CPs is that their open metal sites (OMSs) are occupied by these solvents, which significantly limits their effectiveness in potential applications,20 such as separation and gas storage, catalysis science, magnetism, luminescence, drug delivery, and so on.21–38 To address this issue, the terminal coordinated solvent molecules saturating the metal sites are evacuated via temperature-induced dehydration.39 External stimuli such as heat or mechanic force, on the other hand, could produce defects and damaged regions,40 which otherwise precludes the atomic level characterisation of OMSs via single-crystal X-ray diffraction (SCXRD). Therefore, creating OMSs through single-crystal-to-single-crystal (SCSC) transformation still remains a challenge.41 Furthermore, SCSC transformations pose a great challenge since the vast majority of the solid-state processes that begin with single crystals frequently result in polycrystalline products.42
Heterogeneous catalysis,43–46 for instance, will be undoubtedly favourable if one can identify the crystal structures of the active species and precisely pinpoint the catalytic centres with apparent coordination geometries.40,47 SCSC transformations permit the observation and understanding of how the positions of atoms and molecules change during a solid-state phase transition. Moreover, SCSC transformations guide the development of new catalysts and certainly aid in the understanding of the reaction mechanism.48 In addition, SCSC transformations often offer materials with peculiar structures that are likely distinct from those derived using traditional synthetic routes. Meanwhile, CPs have proven to be effective heterogeneous catalysts, primarily for liquid-phase reactions.49–56 Therefore, the synthesis of versatile and efficient CP catalysts, either through classical methods or solid-state transformation for organic oxidation, is significant and highly demanded.57 For example, a Co(II)-based coordination polymer {[Co2(2,2′-bipy)2(btec)]·H2O}n (2,2′-bipy = 2,2′-bipyridine; (btec)4− = 1,2,4,5-benzenetetracarboxylate) was discovered to be an efficient heterogeneous catalyst for selective oxidation of cyclohexene with O2, in which the conversion was 33.2% and the total selectivity of 2-cyclohexene-1-ol, 2-cyclohexene-1-one, and 2-cyclohexene-1-hydroperoxide was 90% under mild and solvent-free conditions.58 Interestingly, in situ SCSC dehydration transformation from a hydrous [Co3(OH)2(tpta)(H2O)4] (tpta = terphenyl-3,2′′,5′′,3′-tetracarboxylate) to an anhydrous [Co3(OH)2(tpta)] coordination network enhanced cyclohexene conversion (73.6%) and selectivity (64.9%) towards 2-cyclohexene-1-one.7 This temperature-induced SCSC transformation was shown not only to improve the conversion and selectivity, but also the turnover number (TON) from an 88 to 161.
Here, in this study, we constructed a 3D coordination polymer {Co2(OH2)8(btec)}·4H2O (1) under hydrothermal conditions. Moreover, we address the single-crystal structural characterisation of a microporous framework 1 that underwent a single-crystal-to-single-crystal (SCSC) phase transition into [Co2(btec)] (1′). Furthermore, the completely dehydrated framework 1′, in turn, has been shown to undergo a semi-reversible SCSC transformation upon hydration at room temperature (RT; 25 °C) to afford [Co2(OH2)2(btec)] (2). Our attention has been directed towards the advancement of catalysts based on Co(II)-containing CPs. This choice is not solely due to the well-established redox properties and exceptional oxygen transfer capabilities of cobaltous ions, but also because of their high spin state in the weak crystal field of carboxylates, large orbital moment through a sizeable spin–orbit coupling, relative abundance, and cost-effectiveness. Additionally, Co(II) coordination polymer-derived catalysts have been relatively less explored in the aerobic epoxidation of olefins. The tetratopic (btec)4− ligand was chosen since it can undergo deprotonation, completely or partially, and therefore, can coordinate to the metal ions in various coordination modes to yield a variety of interesting frameworks. In this contribution, we show that three different coordination modes to cobalt(II) are possible for a semi-rigid ligand containing four terminal carboxylic functional groups through dehydration and rehydration. Herein, we report epoxidation of olefins over networks 1, 1′, and 2 using environmentally benign oxidants either with or without IBA under mild reaction conditions. Network 2 was discovered to be a stable heterogeneous catalyst during the reaction process and could be reused at least four times without a significant decrease in catalytic activity, demonstrating its potential application in the liquid-phase epoxidation of olefins for the production of important fine chemicals. The CPs were further characterised by powder X-ray diffraction (PXRD), elemental analyses, infrared (IR) spectra, differential scanning calorimetry (DSC), thermogravimetric (TG) and, gas and moisture sorption isotherms. The aerobic epoxidation products were qualitatively identified by gas chromatograph (GC) analysis and nuclear magnetic resonance (NMR) spectroscopy, and quantitatively analysed using gas chromatographic-mass spectrometry (GC-MS). Although the hydrothermally prepared structure of 1′ is known, to the best of our knowledge, we are the first group to synthesise this framework through in situ SCSC dehydration, and explore it as a heterogeneous catalyst in the epoxidation of olefins.
2. Experimental section
2.1 General
All commercially available reagents were of analytical grade and used as received without further purification unless otherwise noted. Protea Chemicals (South Africa) and BM Scientific/Parow Industria (South Africa) supplied the solvents. Cobaltous acetate tetrahydrate (Co(OAc)2·4H2O, 99.0%), cobaltous nitrate hexahydrate (Co(NO3)2·6H2O, 98.0%), 1,2,4,5-benzenetetra-carboxylic acid (H4btec, 95.0%), tert-butylhydroperoxide solution (t-BuOOH, 70% aqueous), sodium hydroxide (NaOH, 99.5%), calcium dichloride (CaCl2, 99.99%), anhydrous magnesium sulphate (MgSO4, 99.5%), were all obtained from Sigma-Aldrich and Merck (South Africa). 1,2-Dichloroethane (1,2-DCE, 99.99%) was purchased from Fisher Chemical Co. Hydrogen peroxide (H2O2, 30%), chloroform (CHCl3, 99.8%), hexene (99.0%), and ethyl acetate (EtOAc, ≥99.5%) were obtained from Merck Chemical Company. N,N′-Dimethylformamide (DMF, ≥99.8%), methanol (MeOH, 99.9%), anhydrous ethanol (EtOH, 99.5%) were purchased from Acros Organics. i-Butyraldehyde (IBA, 98.0%) was obtained from the Alfa Aesar Co. Acetonitrile (MeCN, 99.0%) of high-performance liquid chromatography (HPLC) grade was purchased from Ranbaxy Fine Chemicals Ltd (India). The preparative hydrothermal method that allowed us to grow X-ray-quality crystals of 1 is described herein. For catalytic activity measurements, reagent grade cyclohexene (99.9%), 4-methylcyclohexene (99.0%), cyclopentene (96.0%), 4-methylcyclopentene (99.0%), cyclooctene (98.0%), limonene (98.0%), styrene (99.9%), and trans-stilbene (98.0%) were purchased from Sigma Aldrich Chemical Co. Dimethyl sulfoxide-d6 (DMSO-d6) and deuterated chloroform (CDCl3) were bought from Sigma-Aldrich (Germany) with >98% chemical purity. Cyclohexene were dried with anhydrous CaCl2, while 70% t-BuOOH was dried over anhydrous MgSO4 in an ice bath. Only 18 MΩ cm deionised water (Milli-Q Water Purification System) was utilised for the preparation of 1 and 2. Compounds 1, 1′, and 2 were synthesised as described below.2.2 Synthesis of {Co2(OH2)8(btec)}·4H2O (1)
Single-crystals suitable for X-ray structure determination were prepared by dissolving cobalt(II) acetate tetrahydrate (Co(OAc)2·4H2O; 125 mg, 0.50 mmol) and H4btec (63.5 mg, 0.25 mmol) individually in 10.0 mL of deionised water. Suitable aliquots of a freshly prepared 0.10 M NaOH were added drop-wise to the ligand solution to achieve pH = 4.55. The red and colourless solutions were mixed together, transferred to a Teflon-lined stainless-steel vessel, tightly sealed, and heated under autogenous pressure to 120 °C for 3 days. Orange prism-like crystals of 1 thus formed in the vessel were cooled to RT, isolated, washed with deionised water (5.00 mL × 3) and cold methanol (5.00 mL × 3), and air-dried for 24 h. 1 was the favoured product with a relatively lower pH value (pH ≤ 4.55). Yield: 63% (based on the ligand). Anal. calcd for C10H26O20Co2 (584.2 g mol−1): C, 20.56; H, 4.49; Co, 20.2%. Found: C, 20.62; H, 4.41; Co, 19.6%. IR (ATR; cm−1): 3329 (vs, br) ν(O–Hwater); 3013 (w) ν(C–Haliphatic); 1666 (s); 1549 (vs); 1604 (vs) νasym(COO−); 1437 (m); 1383 (vs) νsym(COO−); 1377 (w); 1269 (m); 989 (w); 948 (w); 860 (m); 809 (w); 796 (w); 746 (m); 657 (w).2.3 Synthesis of [Co2(btec)] (1′)
A selected crystal of 1 (approx. 0.10 mg) with 0.183 × 0.174 × 0.093 mm3 dimensions was employed for in situ high-temperature dehydration (at 180 °C for 60 min) in a single-crystal-to-single-crystal (SCSC) manner to give a violet prism of 1′, obtained in 100% yield. The dehydration experiment was performed to investigate the SCSC phase transition induced by the removal of both lattice and coordinated water molecules in 1. Anal. calcd for C10H2O8Co2 (368.0 g mol−1): C, 32.64; H, 0.55; Co, 32.0%. Found: C, 32.79; H, 0.47; Co, 31.8%. IR (ATR; cm−1): 3063 (w) ν(C–Haliphatic); 1572 (vs); 1559 (vs) νasym(COO−); 1429 (m); 1402 (sh); 1395 (vs) νsym(COO−); 1137 (w); 939 (m); 862 (m); 843 (m); 808 (s); 772 (s); 648 (w).2.4 Synthesis of [Co2(OH2)2(btec)] (2)
The purpose of this synthetic procedure was to determine whether the rehydration of 1′ is partially or completely reversible. The pink single crystal of 2 was synthesised by exposing the violet anhydrous phase of 1′ to water vapour in a closed vacuum desiccator (approx. 45.0% average relative humidity (RH) at 27 °C) for 30 days. Anal. calcd for C10H6O10Co2 (404.0 g mol−1): C, 29.73; H, 1.50; Co, 29.2%. Found: C, 29.81; H, 1.48; Co, 28.5%. IR (ATR; cm−1): 3284 (vs, br) ν(O–Hwater); 2995 (w) ν(C–Haliphatic); 1669 (s); 1553 (vs) νasym(COO−); 1445 (m); 1389 (s); 1461 (vs) νsym(COO−); 1143 (m); 995 (sh); 943 (m); 861 (m); 842 (m); 806 (s); 781 (s); 639 (w).2.5 Single-crystal X-ray crystallography
Single-crystals of 1, 1′, and 2 were selected individually under a polarised light microscope, immersed in Paratone oil (to prevent solvent loss and crystal degradation), mounted on a 75.0 μm diameter Kapton loop, and measured at 150 K under liquid nitrogen stream. Intensity data were collected using a Bruker D8 VENTURE with Photon III detector diffractometer,59,60 equipped with a graphite monochromator and a Mo Kα (λ = 0.71073 Å), with fine-focus sealed tube operated at 2.00 kW (50.0 kV, 40.0 mA). Crystals were centred by looking at a digital camera on the APEX-IV software. Data were collected using ϕ and ω scans of 0.50° per frame, and a full sphere of data was obtained. All reflections were emerged and integrated with the SAINT and XPREP software packages, respectively.61,62 Absorption corrections were applied using the multi-scan techniques TWINABS63/SADABS64 and the structures were solved by the direct methods package SHELXT65 and refined with the SHELXL66 algorithms incorporated into the program X-Seed.67 The final anisotropic full-matrix least-squares refinement was done on F2. The aromatic protons were placed in geometrically idealised positions (C–H = 0.94–0.96 Å) and constrained to ride on their parent atoms with Uiso(H) = 1.2Ueqv(C). SHELX constraints and restraints were used to model the structures. The non-hydrogen atoms were refined with anisotropic displacement parameters. The potential solvent accessible area or void space and symmetry checks were calculated using the PLATON program packages,68 which also confirmed the proposed space groups. Structural data for 1, 1′ and 2 have been deposited with the Cambridge Crystallographic Data Centre (CCDC deposition numbers 2288634–2288636).†It should be noted that network 1 was processed as a 2-component twin. All crystallographic data and structure refinement parameters of the frameworks are provided in Table 1. Bond distances and angles, as well as relevant hydrogen-bond contacts are detailed in Tables S1–S4; refer to the ESI.†
| Compounds | 1 | 1′ | 2 |
|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||∑|Fo|. b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w − (Fo2)2]}1/2, w = 1/[σ2(Fo)2 + (aP)2 + bP], where P = [Fo2 + 2Fc2]/3. | |||
| Formula | C10H26O20Co2 | C10H2O8Co2 | C10H6O10Co2 |
| Mol. wt. (g mol−1) | 584.2 | 368.0 | 404.0 |
| T (K) | 150.0(2) | 150.0(2) | 150.0(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal habit | Prism | Prism | Prism |
| Crystal colour | Orange | Violet | Pink |
| Crystal system Space group | Triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Monoclinic C2/m | Triclinic P |
| a (Å) | 10.7969(8) | 6.1151(7) | 3.3576(6) |
| b (Å) | 11.1079(8) | 17.4400(18) | 9.0091(16) |
| c (Å) | 11.4544(8) | 4.5477(4) | 9.5434(15) |
| α (deg) | 82.796(3) | 90.00 | 113.881(5) |
| β (deg) | 62.783(2) | 115.606(4) | 99.313(6) |
| γ (deg) | 62.288(3) | 90.00 | 93.915(6) |
| Vol. (Å3) | 1074.84(14) | 437.37(8) | 257.60(8) |
| Z, Z′ | 4, 2 | 4, ½ | 2, 1 |
| D calcd (g cm−3) | 1.805 | 2.794 | 2.604 |
| μ (mm−1) | 1.638 | 3.839 | 3.286 |
| F(000) | 600.0 | 360.0 | 200.0 |
| Crystal size (mm3) | 0.183 × 0.174 × 0.093 | 0.183 × 0.174 × 0.093 | 0.183 × 0.174 × 0.093 |
| hkl ranges | −13 ≤ h ≤ 13 | −7 ≤ h ≤ 7 | −4 ≤ h ≤ 4 |
| −13 ≤ h ≤ 13 | −20 ≤ h ≤ 20 | −10 ≤ h ≤ 10 | |
| −14 ≤ h ≤ 14 | −5 ≤ h ≤ 5 | −11 ≤ h ≤ 11 | |
| 2θ range (deg) | 4.024–52.908 | 7.75–50.22 | 4.772–50.738 |
| No. reflns. | 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 246 246 |
2281 | 11857 |
| Unique reflns. | 4428 | 397 | 927 |
| Abs. corr. | Multi-scan | Multi-scan | Multi-scan |
| Data/restrainsParameter | 4428/0399 | 394/050 | 927/0104 |
| R(int)/R(sigma) | 0.0925/0.0352 | 0.0307/0.0197 | 0.1079/0.0426 |
| GOF | 1.078 | 1.142 | 1.071 |
| R 1 [I > 2σ(I)]a wR2 [I > 2σ(I)]a | 0.0449 0.1042 | 0.0308 0.0692 | 0.06240.1275 |
| R 1 (all data)b wR2 (all data)b | 0.0741 0.1207 | 0.0369 0.0764 | 0.08960.1434 |
| Max./min. e− density (e Å−3) | 0.72/−0.62 | 0.28/−0.24 | 0.76/−0.62 |
2.6 Powder X-ray diffraction (PXRD)
PXRD data were recorded on a Bruker D8 ADVANCE diffractometer with copper radiation (Cu-Kα; λ = 1.5418) and a secondary monochromator, operated at 40.0 kV and 40.0 mA for a Cu-target tube and a graphite monochromator. A flat-plate configuration was utilised, and the typical data collection range was between 2θ range of 5.0–60.0°. Simulation of the PXRD plots were performed by single crystal data and the diffraction crystal module of Mercury 2020.2.0 program.692.7 Elemental and topology analyses
Elemental analyses (C, H, N) were carried out on an Elementar Analysensysteme varioMICRO V1.6.2 GmbH analysis system. Cobalt content analysis (prior and post catalytic runs) were performed on atomic absorption spectroscopy (AAS) utilizing a Varian 1275 spectrometer with a laminar acetylene burner. The structures were also subjected to a topological analysis employing program package Topos 4.0 incorporating TopCryst software.70 The Reticular Chemistry Structure Resource (RCSR) three-letter codes were utilised to designate the network topologies.702.8 IR spectroscopy
IR data were recorded on a PerkinElmer Spectrum 100 spectrometer with a KBr beam splitter and an attenuated total reflection (ATR) attachment. Mid-infrared spectra with frequency of 4000–650 cm−1 (note: br – broad, m – medium, sh – shoulder, s – strong, vs – very strong, and w – weak) were obtained by placing samples on a diamond/ZnSe crystal plate and using the force gauge between 100 and 149.2.9 Thermal analysis
Thermogravimetric (TG) data, using temperature ranging from 30 to 1000 °C, and with heating rate set at 10.0 °C min−1, was collected on a PerkinElmer TG-4000 (Pyris Version 4.01 software) instrument employing nitrogen gas (N2) purge at a ramp rate of 20.0 mL min−1. Crystalline samples (heated between 30–250 °C at a rate of 5.00 °C min−1) were analysed by differential scanning calorimetry (DSC) employing a TA DSC 2500 instrument with hermetically sealed TZero aluminium pans. The instruments were both calibrated using standard reference materials. Dehydration of the crystal was carried out on a DOF-30E/45E vacuum drying oven at atmospheric pressure.2.10 Adsorption properties
Gas adsorption isotherms were carried out on Micromeritics ASAP 2020 HD analyzer (Accelerated Surface Area and Porosimetry System), determined by the N2 adsorption–desorption, at 77 K. Samples to be analysed were degassed with helium gas at 120 °C on Flow Prep 060. Essentially, ca. 65.0 mg of activated samples were utilised for the gas adsorption–desorption measurements. Finally, the instrument was used with nitrogen as the analytical gas, and the determination was based on a 5-point method with 30 adsorption and 30 desorption points measured. The custom-built relative humidity (RH) vapour balance instrument was utilised to conduct dynamic vapour sorption (DVS) experiments. This gravimetric sorption apparatus consists of a small stainless-steel chamber equipped with a thermostat (at 25 °C) to maintain a constant sample temperature, as well as a flow controller to generate a controllable variable vapour pressure. The adsorbed mass was continuously measured in relation to the relative pressure. Both the microbalance and mass flow controller systems were controlled by custom-written software. This system exhibits a sensitivity of 0.1 μg and a dynamic range of 150 mg. With regards to relative humidity (RH), the system maintains an accuracy of ±3% across a range of 2–70%. To achieve equilibrium, a rate of change in mass per time unit (dm/dt) of 0.001% min−1 was established as the equilibration parameter. Water was employed to study the phase transition of a dehydrated sample.2.11 Gas chromatographic-mass spectrometry/gas chromatography (GC-MS/GC)
GC-MS/GC data were conducted on an Agilent 7000 Series Triple Quad with FID detector and a capillary column (Rxi – 5Sil MS, column length: 30.0 m; internal diameter: 0.25 mm). MS spectra were compared to those in the NIST library. A Shimadzu gas chromatograph Model GC-15A (Shimadzu, Kyoto, Japan) equipped with a flame-ionisation detector and a model GC-R4A Chromatopac integrating recorder was used for GC analyses. The column used was Teknokroma® (TRB-1) 30.0 m × 0.32 mm × 3.00 μm.2.12 Nuclear magnetic resonance (NMR)
NMR spectral data were recorded at room temperature utilizing a Bruker Avance III HD 400 MHz NMR spectrometer with tetramethylsilane (TMS) serving as an internal reference. The coupling constant (J) is expressed in Hertz (Hz). All chemical shift values are given in parts per million (ppm) in comparison to dimethyl sulfoxide-d6 (DMSO-d6; δH 2.50), and deuterated chloroform (CDCl3; δH 7.26).2.13 Catalytic oxidation of the cyclohexene substrate model
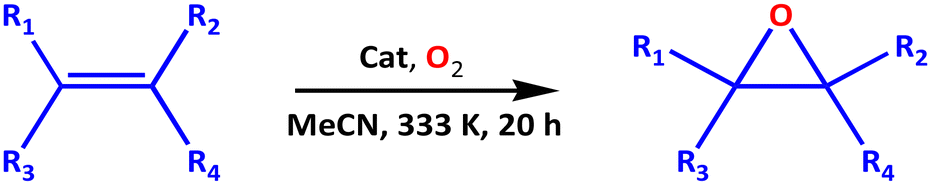
Caution: Solutions that contain high levels of t-BuOOH or hydrogen peroxide (H2O2) in the presence of a metal can be extremely hazardous, and it is crucial to handle these reactions with the necessary safety measures. Catalytic reactions were performed in a 50 mL three-necked Schlenk-flask equipped with a reflux condenser, magnetic stir bar, and oxygen gas supply. The flask was charged with 5.00 mmol of the corresponding olefin, co-reagent (183.0 μL, 2.00 mmol), and catalyst (2.00 mol%) before 10.0 mL of freshly distilled MeCN was slowly added. The reaction mixture was evacuated thoroughly to remove gas impurities and purged three times with oxygen. To commence the reaction, the mixture was stirred in an oil bath at 333 K under atmospheric pressure for 20 h while molecular oxygen was fed into the reactor vessel by bubbling at a constant flow rate (20.0 mL min−1), by means of an adjustable valve. Progression of the reaction was monitored via thin-layer chromatography (TLC). The reaction was quenched by adding 3.00 mL of ethyl acetate (EtOAc) which led to the precipitation of solid catalyst which was filtered off and air-dried at RT. Deionised water was utilised to wash the EtOAc layer which was then dried over anhydrous MgSO4, and the organic products were isolated upon removing the solvent under reduced pressure. The crude organic products were separated by the flash column chromatography on silica gel 70–260 mesh (cyclohexane/EtOAc, 5:1) into various fractions. GC-MS/GC was employed to analysed and quantified the products. Blank experiments were also carried out, confirming that no oxidation or almost no oxidation occurred unless the catalyst as well as co-reagent were present. For the control experiments, reactions were performed to evaluate the role of (a) solvent, (b) aerobic versus anaerobic environment, (c) oxygen flow rate, (d) catalyst loading, (e) reaction time, (f) reaction temperature, and (g) stirring speed. We may note that for avoiding vaporisation of cyclohexene and its products, all reactions were performed at temperatures not higher than 343 K. In the case of anaerobic oxidation using t-BuOOH, it is noteworthy to mention that the cyclohexene peroxides were quantitatively determined by means of an indirect method using triphenylphosphine (Ph3P).71
2.14 Recycling experiments
For the catalytic cycle, the catalyst 2 was separated from the reaction mixture by centrifugation at 1000 rpm for 5.00 min, washed with deionised water (5.00 mL × 3) and MeCN (5.00 mL × 3), dried at 353 K under vacuum overnight, which could be employed for the next run. The recycled CP catalyst was subsequently reused for the next epoxidation of the model substrate, keeping the same reaction conditions. The recovered catalysts were then characterised by IR spectra that displayed identical data to that of freshly prepared samples and powder X-ray diffraction (PXRD) patterns which verified crystallinity of the architectures was preserved.2.15 Hot filtration test
For a hot filtration test, the Co(II)-CPs were removed from the hot reaction mixture after 10 h by filtration while the reaction was still ongoing for another 10 h. The leaching of metal was carried out using the AAS instrument.3. Results and discussion
3.1 Design, synthesis and crystal structure of network 1
The hydrothermal reaction of Co(OAc)2·4H2O with (btec)4−, obtained by in situ deprotonation of H4btec, enabled us to isolate orange prisms of compound {Co2(OH2)8(btec)}·4H2O (1) in good yield (Scheme S1†). Single-crystal X-ray diffraction disclosed that 1 crystallised in the triclinic space group P. It is worth mentioning that the structure of 1 has been reported previously.72 The asymmetric units contain two identical crystallographically independent Co(II) centres (each located on an inversion centre), one-half of the fully deprotonated bridging (btec)4− anion, and two coordinated water molecules (lying in a general position) per cobaltous ion, and four free aqua molecules constitute the structure of 1. Fig. S1a, ESI† illustrates two identical asymmetric units of 1 with ellipsoids drawn at 50% probability. The dihedral angle between two phenyl rings of two adjacent (btec)4− molecules in the asymmetric unit is 4.19°, in agreement with the semi-rigid nature of this spacers. All the Co(II) species possess a {CoO6} octahedral stereochemistry defined by the oxygen atoms [O9W(i), O10W, and O11W(ii) and O12W in the first residue] of four water molecules [Co1–OW/Co2–OW bond distances of 2.029(3)–2.128(3) Å] occupying both the axial and equatorial sites and the donor atoms [O5, O5(iii), O8 and O8(iv) in the second residue] of two (btec)4− spacers with Co3–O/Co4–O bond lengths of 2.085(3)–2.123(3) Å only occupying the equatorial positions. All the bond distances mentioned are within the normal range,48,58 as indicated in Tables 2 and S1.†
| CP | Co1–O1 | Co1–O2 | Co1–O4 | Co2–O4 | Co1–O9W | Co1–O10W | Co2–O11W | Co2–O5W | Co2–O12W | Co1⋯Co1 | Co1(3)⋯Co2(4) | Co1(2)⋯Co4(3) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n/a = not applicable. | ||||||||||||
| 1 | 2.107(3) | n/a | n/a | 2.104(3) | 2.066(4) | 2.085(3) | 2.029(3) | n/a | 2.128(3) | n/a | 9.230(8) | 5.398(5) |
| 1′ | 2.024(3) | 2.079(3) | n/a | 2.194(3) | n/a | n/a | n/a | n/a | n/a | 3.358(4), 4.465(6), 4.547(7) | n/a | n/a |
| 2 | 1.991(6) | 2.088(5) | 2.172(6) | n/a | n/a | n/a | n/a | 2.087(8) | n/a | 3.357(6), 5.665(7) | n/a | n/a |
In more detail, the crystallographically independent cobalt(II) cations (Co1, Co2, Co3, and Co4) which are located on crystallographic inversion centres, are coordinated by two ligands through the oxygen atoms of their carboxylate groups (symmetry codes: (i) 1 − x, 2 − y, 1 − z; (ii) 2 − x, 1 − y, 2 − z; (iii) 1 − x, 1 − y, 2 − z; (iv) −x, 2 − y, 1 − z).
Carboxylates provide several possible coordination modes, viz. monodentate, bridging bidentate, bidentate, and tridentate, of the independent carboxylate groups (Scheme 1). Additionally, each of the carboxylate groups of the ligand assume a μ1–η1:η0syn-monodentate coordination mode (type-I, Scheme 1), and the entire linker acts as μ4-bridge connecting four Co(II)-ions. Upon a closer observation of the structure, the cis ∠O–Co–O angles are in the range of 86.11(12)°–93.89(12)°, and the trans ∠O–Co–O angles are 180.00° as shown in Table S1.† The structure of 1 consists of a 3D CP with open channels (Fig. 1a) where lattice water molecules are embedded. The crystallographically identified uncoordinated or guest water molecules are located outside and between the 2D sheets (Fig. 1b) while the coordinated water molecules protrude within the channels, resulting in the unit cell containing no residual solvent accessible voids. In the solid state, two sets of the distorted {CoO6} octahedron (Fig. 1c) are further linked alternatively by (btec)4− into parallel (4,4) square grid layers running in the (011) crystallographic plane. The layers in 1 are stacked along the a-direction following the ABAB sequence, leading to a 1D intersecting channel system and narrow open windows, with an approx. effective cross-section of ca. 11.5 Å × 11.3 Å (interatomic distances between opposite Co atoms of the grid void as shown in Fig. S2a†). The values of the intralayer Co⋯Co separation corresponding to the edges of two pairs of rectangles are 9.230(8) Å (Co1⋯Co2v/Co3⋯Co4; symmetry codes: (v) = x, y, 1 + z) and 5.398(5) Å (Co1⋯Co4vi/Co2⋯Co3; symmetry code: (vi) = x, y, z). Due to twelve molecules of lattice and terminal coordinated water molecules per formula unit, numerous hydrogen bonding interactions between these aqua molecules and oxygen atoms of (btec)4− ligands are found within the architecture of 1. Consequently, a 3D supramolecular framework of 1 is generated through extensive O–H⋯O hydrogen bond interactions (Fig. S2b, ESI and Table S4†). The distorted {CoO6} octahedron form zig-zag ladder-like chains linked by the (btec)4− ligand which act as 4-c (connected) nodes to furnish the 2D architecture with edge-transitive square lattice (sql) net (Fig. 1d).
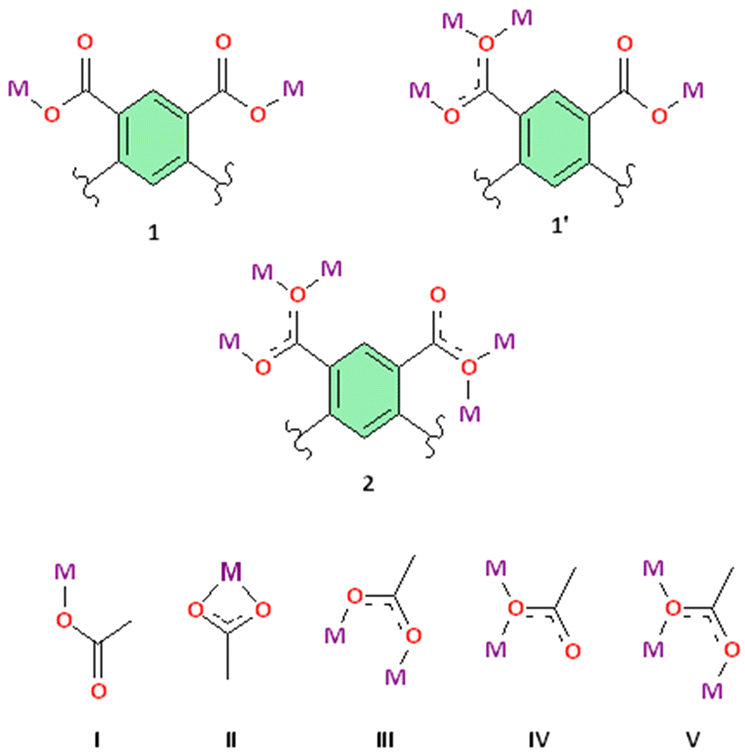 | ||
| Scheme 1 Coordination modes observed in 1, 1′, and 2 along with various types of non-bridging and bridging modes of carboxylate groups. | ||
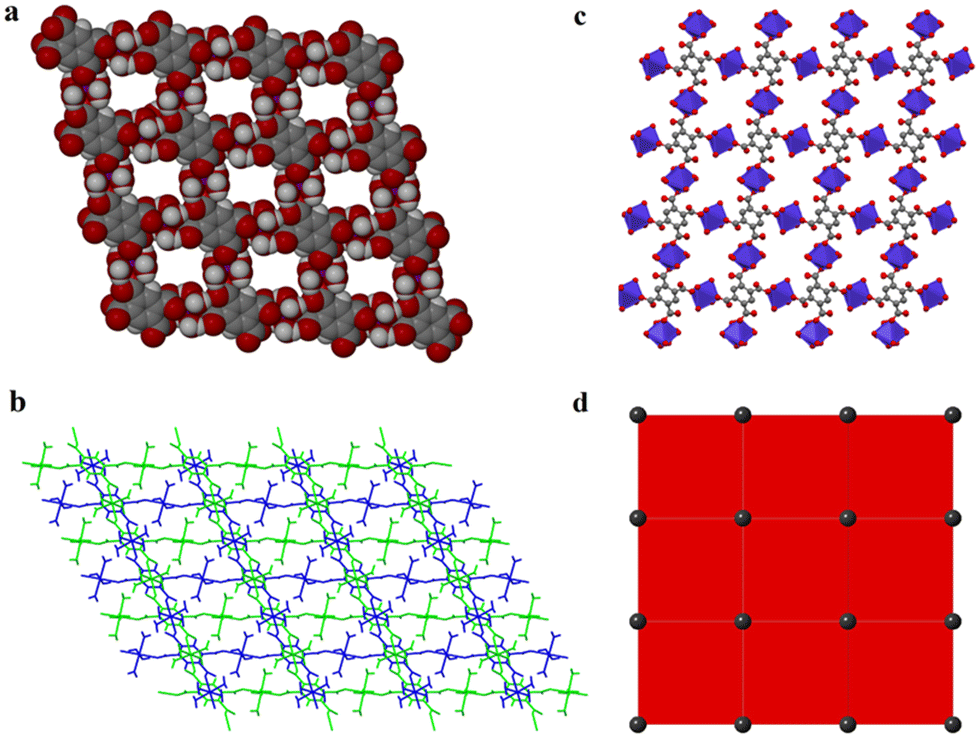 | ||
| Fig. 1 (a) A space-filling view of the microporous network 1 showing 2D channels along the crystallographic a-axis, in which the lattice water molecules are omitted for clarity. (b) Projection along the b-axis of the 3D crystal structure of 1 showing the coordinated water molecules jutting between the 2D sheets. (c) Polyhedral view of 1 showing the {CoO6}cluster running along the a-axis (colour code: Co indigo polyhedra; C grey; O red; H atoms are omitted for clarity). (d) View of the square lattice (sql) topology with a 4-c uninodal net. | ||
3.2 Synthesis, in situ SCSC transformation and crystal structure of 1′
Single-crystals of 1′, as violet platelets, were first reported by Kumagai et al., 2002.73 The group described a hydrothermal technique that produced 1′ after 6 days with a 60% yield. In contrast, our attempts to isolate 1′via conventional CP synthetic routes gave rise to either an amorphous or non-crystalline material. This compelled us to proceed with the temperature-induced SCSC dehydration of 1. Upon drying the crystal in vacuo at 180 °C for 60 min, 1 underwent a de-twinning phase transition to produce 1′ formulated as [Co2(btec)]. Our method is conveniently effective to shorten the reaction time and gives a 100% yield. Optical microscopic examination indicated that the colour of the crystals changed from orange to violet, suggesting the possible structural transformation from 1 to 1′. Additionally, the colour change signalled the loss of the water molecules coordinated to the Co(II)-based paddle-wheel moiety.This 3D anhydrous phase 1′ is determined to possess a monoclinic space group C2/m (Z = 4) with the Co(II)-ion occupying the 2-fold axis while the (btec)4− anion is lying on a mirror plane. Interestingly, a phase transition was seen during the structural transformation to give 1′ and, their lattice parameters underwent a significant variation, representing approx. 41.0% volumetric shrinkage (based on the initial volume of 1). In accordance with the single-crystal data, the crystal structure of 1′ involves a crystallographically asymmetric unit with one-half of cobalt(II) cation and one-quarter of the (btec)4− bridging ligand. The repeating structural motif is a dimer consisting of two edge-sharing symmetry-related cobaltous ions which are bridged by oxygen atoms of (btec)4− units (Fig. S1b, ESI†). The coordination environment around the metal centre in 1′ is a six-coordinate distorted octahedral geometry {CoO6} with four oxygen atoms of carboxylate groups belonging to two different ligands coordinating in a μ1–η1:η0 and μ3–η1:η2 (types-I and -V, Scheme 1) fashion. 1′ possesses the syn–anti/anti–anti carboxylate bridges.37,74 Upon closer inspection of the structure, all the water molecules in 1′, guest and coordinated, have been evacuated and the framework is now composed of four crystallographically unique cobalt centres and four unique (btec)4− secondary building units (SBUs). Moreover, the structure (Fig. 2a) is formed by 1D zig-zag chains of these edge-sharing dimers of octahedral Co(II)-ions, generating distinguishable 2-fold interpenetrated hexagonal grids running parallel to the ab-axis as illustrated in Fig. 2b. These networks are linked to one another through O–C–O bridges to form layers parallel to the ac-plane. The Co1 site are now coordinated by bridging and non-bridging carboxylate oxygen atoms (O1, O1(i), O2(ii), O2(iii), O2(iv) and O2(v)) with Co1–O distances ranging from 2.024(3) to 2.195(3) Å (Tables 2 and S2†). The cis and trans ∠O–Co1–O angles are in the range of 76.37(12)°–110.83(12)° and 159.07(13)–170.87(12)° (Table S2†), respectively. Each Co1 atom consist of Co⋯Co distances of 3.358(4)–4.547(7) Å and the ∠Co1(vi)–O2–Co1(iv) angles is 103.63(13)°, (symmetry codes: (i) 1 − x, y, 2 − z; (ii) x, y, 1 + z; (iii) 1 − x, y, 1 − z; (iv) 1/2 − x, 3/2 − y, 1 − z; (v) 1/2 + x, 3/2 − y, 1 + z; (vi) x, y, −1 + z). This anhydrous lattice possesses crystallographically hexagonal grids with ca. 7.1 Å × 7.0 Å (down c-axis) and 6.6 Å × 4.5 Å (down b-axis) dimensions.
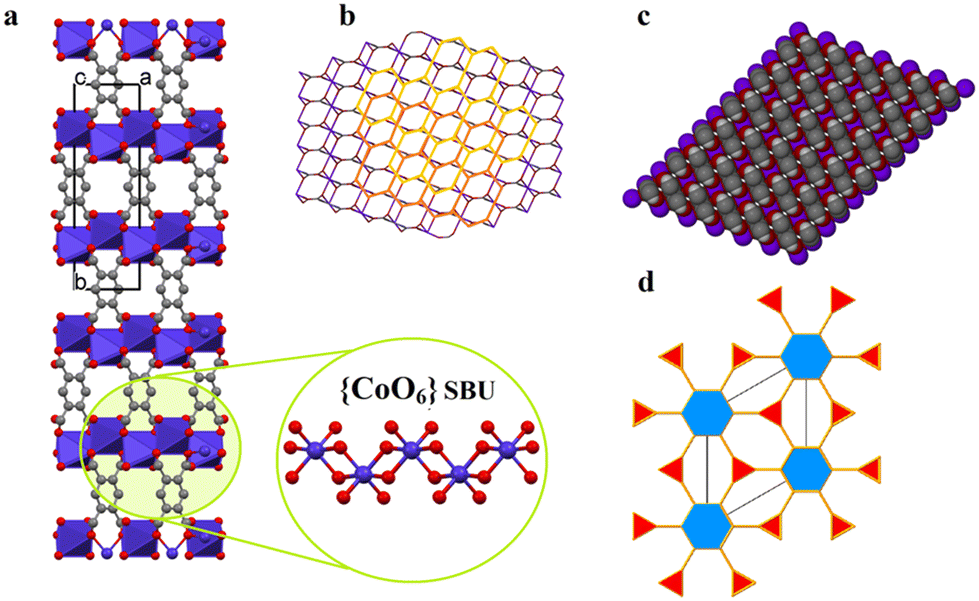 | ||
| Fig. 2 (a) Polyhedral view of 1′ showing the {CoO6} subunit running along the a-axis (colour code: Co indigo polyhedra; C grey; O red; H atoms are omitted for clarity). (b) 2-fold interpenetrated hexagonal grids of 1′ orientated along the crystallographic b-axis. (c) A space-filling view of the nanoporous network 1′ directed in the b-axis. (d) View of the Kagomé-dual (kgd) topology with a (3,6)-c binodal net. | ||
Moreover, this scaffold has deformed such that on half of the {Co2(COO)4} motif, the coordination sites that were vacated by the axial and equatorial coordinated H2O molecules are now occupied by four carboxylate oxygen atoms. However, the coordination sphere of the metal centres in the paddle-wheel core and the dimensionality of the framework is therefore unchanged. Essentially, 1′ exhibits no classical hydrogen bond interactions. Furthermore, SCSC dehydration influenced the aromatic edge-to-edge type π–π stacking interactions (Table S5†), resulting in the formation of nanoporous structural networks as displayed in Fig. 2c. The phenyl rings running parallel to each other (down the b-axis) are determinately separated after the conversion of 1 to 1′, and are 4.548(3) Å apart, displaying zero degrees angle between their planes. The framework can be described as a 2-nodal (3,6)-c Kagomé-dual (kgd) net topological type (Fig. 2d) with point (Schläfli) symbol {43}2{46}. Essentially, no solvent-accessible void space was found for this scaffold. It is important noting the distinction between the structures of 1 and 1′. Both frameworks possess the common [Co2(btec)] core, with 1 having twelve molecules of water per formula unit. Accordingly, the calculated densities of 1.81 g cm−3 for microporous 1 and 2.79 g cm−3 for nanoporous 1′ differ significantly, indicating that the latter has a more tightly packed network architecture than the former.
3.3 Semi-reversible SCSC hydration and crystal structure of 2
Remarkably, a hydrous compound having the formula [Co2(OH2)2(btec)] (designated as 2 herein) was generated when a small crystal of 1′, with dimensions: 0.183 × 0.174 × 0.093 mm3, was hand-picked and subjected to water vapour at 45% relative humidity (RH). Subsequently, a colour change was noticed from violet 1′ to pink 2 (rehydrated crystal) after 21 days. The transformation was completed after 30 days, as it was validated by X-ray diffraction (see discussion below). However, structural conversion from 1 to 2via1′ is, in fact, partially reversible. From this semi-reversible transformation, it is reasonably proposed that the dynamic SCSC transformation from 1′ to 2 can be attributed to water molecule attack on the Co(II) cations with the aid of the volatility of aqua molecules at RT. The colour change observed earlier is suggested to be caused by a reset of the Co coordination sphere illustrated by the construction of the Co–Owater bonds and departure of the Co–Ocarboxylate bonds without catastrophic breakdown of the crystal structure. Meanwhile, during this process, the coordination mode of the carboxylate groups is significantly changed, from multidentate (i.e., bridging and bidentate) to monodentate. Besides, the infeasible structural conversion from 2 to 1 (by a direct route or via1′) could be rationally ascribed to the compactness of these two crystal structures, in which framework 2, characterised by lower porosity, is much more stable than 1. It is worth pointing out that the semi-reversible water vapour induced SCSC transformation was monitored every twenty-four hours using single-crystal unit cell determination. Moreover, the SCSC semi-reversible transformation from dehydrated 1′ to hydrated 2 resulted in a slight increase in the unit cell as well as space group change (high-symmetry to low-symmetry). Additionally, the resolved crystal structure of 2 therefore confirmed the SCSC transformation. Structural analysis reveals that 2 crystallises in the triclinic space group P with an asymmetric unit consisting of two crystallographically independent Co(II) dications, one-half (btec)4−, and one coordinated water molecule. As depicted in Fig. S1c, ESI,† the Co1 ion is a six-coordinated octahedron surrounded by carboxylate oxygen atoms (O1, O1(i), O4(ii), O4(iii), O4(iv), and O4(v)) of four different (btec)4− ligands (symmetry codes: (i) −x, 2 − y, 1 − z; (ii) −1 + x, 1 + y, z; (iii) x, 1 + y, z; (iv) −x, 1 − y, 1 − z; (v) 1 − x, 1 − y, 1 − z). The two Co1 and Co2 atoms are connected by the four (btec)4− ligands, forming two distinct octahedra, {Co(μ2-O2)O4} and {Co(OH2)2O4} with equal Co1⋯Co1vi/Co2⋯Co2vii intermetallic distances of 3.357(6) Å (symmetry codes: (vi) 1 + x, y − 1, z; (vii) 1 + x, y, 1 + z). Additionally, the Co1 and Co2 atoms are separated by a distance of 5.665(7) Å. Moreover, six oxygen atoms of carboxylate groups belonging to four different ligands coordinating the Co1 sphere in a μ2-η2:η0 (type-IV, Scheme 1) bridging mode while the other four oxygen atoms of four different carboxylates coordinate the Co2 centre in μ3-η1:η2 (type-V, Scheme 1) manner with syn–anti/anti–anti configurations. The Co1 site also adopts an octahedral geometry, coordinated by two coordinated terminal water molecules (O5W, and O5W(v); building a trans-[Co(OH2)2]2+ cationic subunit), and four carboxylate oxygen atoms (O2, O2(vi), O2(vii) and O2(viii)), symmetry codes: (vi) 1 + x, y, z; (vii) −x, 1 − y, −z; (viii) 1 − x, 1 − y, −z. Therefore, 2 is composed of (btec)4− linkers and infinite chains of trans-corner sharing {CoO6} polyhedra cluster as shown in Fig. 3a. Notably, the coordinating aqua molecules are attached to the Co2 centre with two equal Co2–O5W bond lengths of 2.086(7) Å. Compound 2 presents Co1–O and Co2–O bond distances (1.991(6)–2.173(6) Å and 2.087(6)–2.185(6) Å, respectively) that are comparable to the length of a typical Co–O coordination bond.75 Thus, the bond lengths reported here are within normal statistical errors. The tables of bond lengths and angles are supplied in Tables 2 and S3.† Essentially, the cis ∠O–Co1–O and ∠O–Co2–O angles are in the range of 75.8(2)°–92.9(2)° and 76.4(2)°–92.4(2)°, respectively, while their trans angles vary from 103.6(2)°–180.00° and 104.2(2)°–180.0(3)°. As in framework 1′, the dihedral angle between the planar phenyl rings from two adjacent tetradeprotonated ligands unexpectedly remain 0.00° in 2. In addition, the πcentroid⋯πcentroid and C–H⋯πcentroid interactions (Table S5†) within layers of 2 are observed after rehydration. The metal-bound water molecules in the diaquacobalt(II) fragments act as donors (interconnecting along the c-axis) in hydrogen bond interactions involving the free oxygen atoms of the carboxyl groups as acceptors (Table S4†). This hydrous lattice possesses distorted rectangular channels with dimensions of ca. 9.0 Å × 9.0 Å (down a-axis). Surprisingly, two faces of the channel are formed by (btec)4− spacers with the benzene ring and the terminal coordinated water molecules of the trans-[Co(OH2)2]2+ species pointing towards the inner portion of the cavity. The structure of 2, compared to that of 1, display a more efficient packing owing to an increase in the number of crystallisation water tethered to Co(II)-atoms. These water molecules link the 2-fold interpenetrated square 2D layers through hydrogen bonds (Fig. S2c, ESI and Table S4†), contributing to the stabilisation of the 3D structure. Subsequently, the 2D layer and 3D framework 2 (Fig. 3b and c) are shown to possess a small potential void space. A topology approach was employed to gain a better understanding of the nature of this structure; each (btec)4− spacer acts as 4-fold connector and the trans-diaquacobalt(II) units serve as simple linear nodes. Thus, the entire framework can be described as an unusual binodal (4,6)-c square cube (scu) net topology (Fig. 3d) with space symbol {35}.
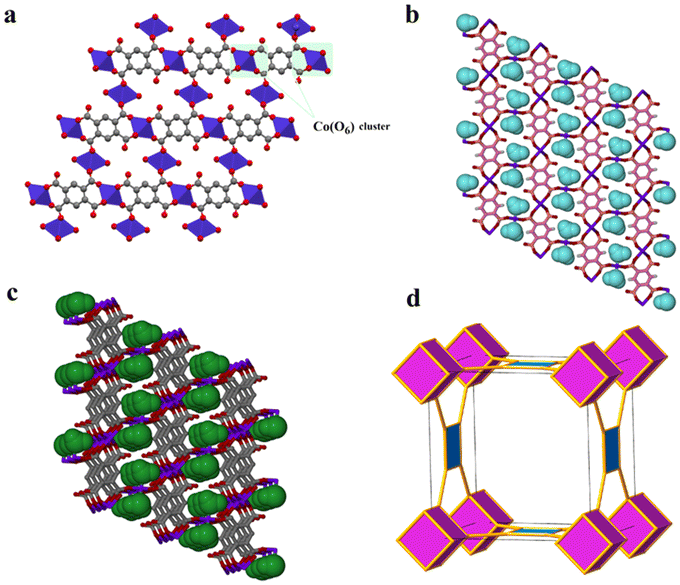 | ||
| Fig. 3 (a) Polyhedral view of 2 highlighting the repeating dinuclear {Co(μ2-O2)O4} motif (colour code: Co indigo polyhedra; C grey; O red; H atoms are omitted for clarity). (b) Perspective view of the 2D layer of 2 on the (011) plane, with the coordinated water molecules shown as light-blue calotte model. (c) The 3D architecture of 2 on the (011) plane, with the coordinated water molecules shown as green space-fill model. (d) Topological representation of a binodal (4,6)-c square cube (scu) net possessed by framework 2. | ||
3.4 Elemental analysis, infrared (IR) spectroscopy, thermochromism and polarised microscope
Validated by single-crystal data, the error of actual carbon and hydrogen contents in frameworks 1, 1′, and 2 was well under 0.5% while for cobalt was less than 1%. To further verify the in situ SCSC phase transitions via dehydration and rehydration, the frameworks 1′ and 2 were examined by IR spectroscopy and compared with the pristine framework 1. Therefore, we will simply attempt to rationalise specific observations and compare significant features of the three networks. One obvious difference between the three networks was the number of uncoordinated and coordinated water molecules. Consequently, the asymmetric vas(OH) and symmetric vs(OH) modes were assigned. In the high-frequency region, the vs(OH) was attributed at 3444 and 3284 cm−1 to 1 and 2, respectively, as this band appears absent in 1′. Also, small-to-medium bands appear for all three scaffolds approx. at 3079 cm−1 and was tentatively assigned as stretching bands of C–H functional group. In the present study, the (btec)4− anion in 1′ has a mirror bisecting the C–H bonds and a C2 plane perpendicular to it, while it contains only an inversion centre in both 1 and 2. The H–O–H bending modes are observed at ca. 1665 cm−1 in 1 and 2, and again was not observed in 1′. Additionally, the in-plane δ(OH) assigned at 1269 cm−1 in 1 and 1272 cm−1 in 2. Therefore, the obtained vibrational bands of the O–H groups clearly indicate the hydrous nature of 1 and 2, which supports the earlier discussed crystallographic data and further validating the stoichiometric calculations established from TG analysis (see later). In the mid-frequency region of Fig. S3,† the weak-to-medium vibrational bands observed at 1437 cm−1 for 1, 1429 cm−1 for 1′ and 1445 cm−1 for 2 were characteristic of asymmetric stretching of a carbonyl group. Moreover, the vibration mode at approx. 1495 cm−1 is ascribed to the aromatic ν(CC) and the free δ(OH) group in both hydrate forms.76 The asymmetric and symmetric stretching vibrations of the carboxylate group for the three CP solids were observed between 1550–1356 cm−1. Essentially, the frequencies of the asymmetric νas(COO) and symmetric νs(COO) stretches revealed the coordination modes existing in 1, 1′ and 2. This vibrational difference, [(Δv) = vas(COO) − vs(COO)], suggested monodentate, bidentate and bridging coordination modes exhibited by the frameworks. For 1 and 2, the Δv-value is ca. 201 (Δ > 200 cm−1), and 92 cm−1, showing monodentate and bidentate coordination modes, respectively. In contrast, the Δv-value is 164 cm−1 (150 < Δ < 200 cm−1) for the intermediate network 1′, indicating a bridging or mixed coordination mode typical of a paddle-wheel motif.77 In the low-frequency region, on the other hand, the out-of-plane C–H bending of the (btec)4− anion was observed in the frameworks at approx. 745 cm−1. All the spectra described above are characteristic of the geometry and coordination environment of a tetracarboxylate species.71 It is usual for a Co(II)-ion in octahedral coordination of weak ligands to have an orange and pink hue, as in 1 and 2, respectively.71
The orange crystals of 1 at RT (25 °C) change their colour to violet at ca. 180 °C to afford 1′. The crystals of 2, on the other hand, which are pink at RT, change to tint-blue at ca. 180 °C. Interestingly, compound 1′ has a strong violet to purple colour and exhibits dichroism under a polarizing microscope. Prismatic crystals of 1′ transmit dark blue in one polarisation and pale red in the other.
3.5 Sorption properties and powder X-ray diffraction (PXRD) studies
Nitrogen sorption experiments were carried out to evaluate the pore characteristics of compounds 1, 1′ and 2. To pre-treat the CPs, each fresh sample was soaked in MeOH solvent for 3 days before being heated under vacuum at 120 °C for 5 h to eliminate all solvent molecules, generating the activated frameworks. Based on the nitrogen adsorption/desorption isotherms in Fig. 4a–c, the porous frameworks 1, 1′ and 2 all possessed typical type-III (according to the IUPAC classification) curve with a Brunauer–Emmett–Teller (BET) surface area of 50.5, 94.4, and 86.4 m2 g−1 and total pore volume of 0.100, 0.148 and 0.123 cm3 g−1, respectively. The pore-size distribution revealed pore diameters of about 126.2, 79.2 and 80.4 Å, which is somewhat consistent with the crystal structure analysis.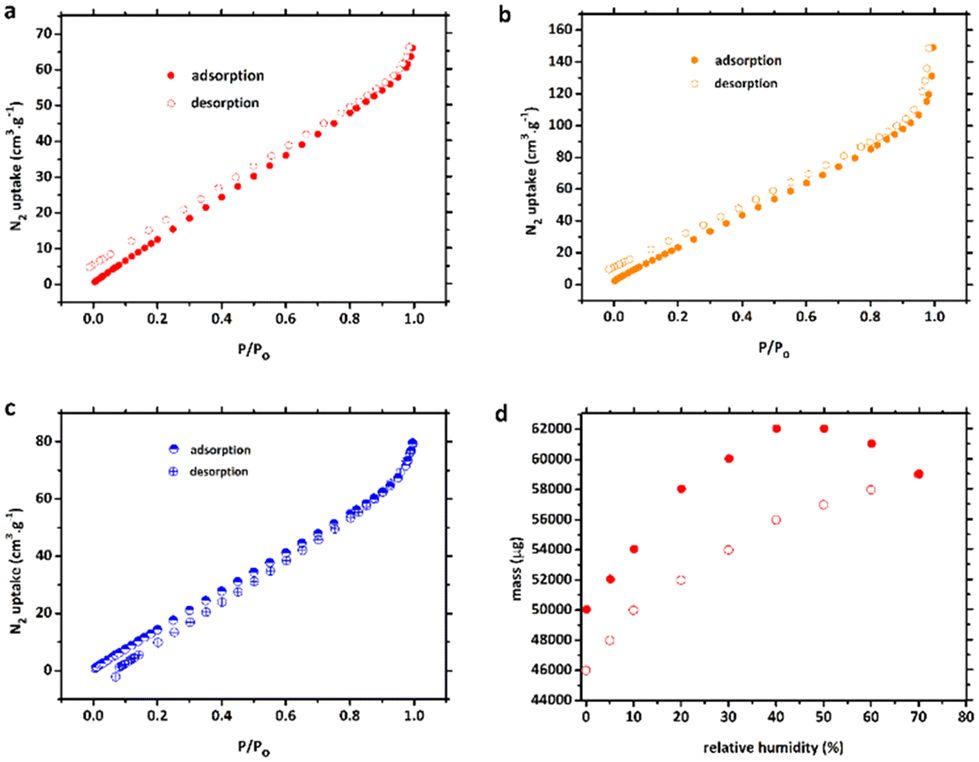 | ||
| Fig. 4 N2 adsorption isotherms for networks (a) 1, (b) 1′, and (c) 2 at 77 K. (d) Water vapour sorption profile of the dehydrated CP 1′ at 23 °C (296 K), with each data point only being allowed to equilibrate for 60 min. | ||
The adsorption and desorption processes of 1′ were very slow (Fig. 4d), resulting in none of the data points on the vapour sorption isotherms reaching equilibrium within the 60 min time frame for either process. Nonetheless, upon pausing for 60 min at 70% relative humidity (RH), the normalised mass of water adsorbed during the experiment amounted to 0.153 g g−1. It is noteworthy that the total quantity of water molecules adsorbed at 45% RH, as determined by single-crystal X-ray diffraction (SCXRD) data, was slightly lower (2.0 molecules per [Co2(btec)] unit) than that adsorbed at 70% RH (3.1 molecules per formula unit, based on the vapour sorption isotherm experiment). However, these results provide confirmation that 1′ exhibited semi-reversible water uptake at room temperature under both 45% and 70% RH conditions.
PXRD measurements were conducted to evaluate the phase purity and assess structural insight about the CP materials. As illustrated in Fig. 5, the bulk phase purity of the scaffolds could be confirmed by matching their powder X-ray diffraction (PXRD) patterns with those of crystallographically characterised samples. The PXRD phases of 1′ (shown as a red pattern in Fig. 5), measured at RT, exhibits some changes when compared to 1 (blue pattern) indicating some structural transformation. According to the PXRD patterns, sharp reflexes around 2θ = 8.73°, 9.03°, 11.96°, 13.37°, 16.50° in 1 are corresponded to reflections of (001), (010), (011), (011) and (211) planes, respectively. All of these lattice planes either disappear or slightly shift towards higher 2θ values compared to the 1′, owing to the elimination of guest and coordinated water molecules. Furthermore, a few Bragg peaks are significantly more apparent in the experimental PXRD diffractions, which are ascribed to minor preferred orientation effects. Although all of the diffractograms shown are typical for metal–organic systems, the experimental diffractogram of 2 (dark yellow in Fig. 5) obtained after rehydration illustrates significant change in the reflections appearing at 11.46°, 18.02°, and 19.03°. The related reflections represent the structural change, which was formed via a semi-reversible water-triggered SCSC transformation. Moreover, shifting these diffraction peaks to high angles suggests a possible unit cell reduction during framework 1′ rehydration.
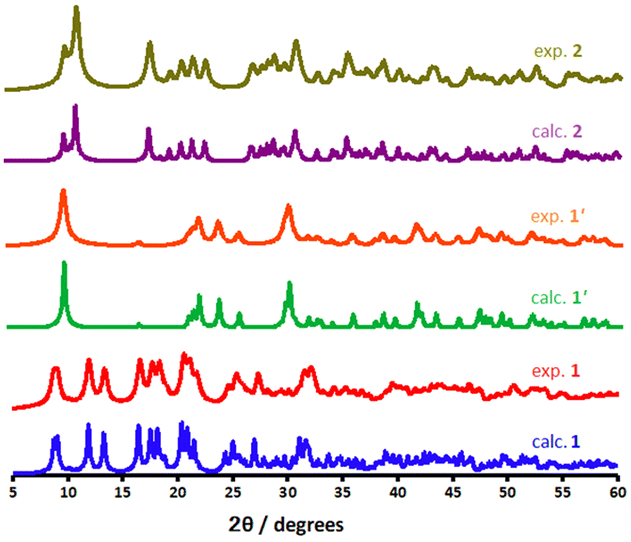 | ||
| Fig. 5 Calculated and experimental PXRD patterns for the as-synthesised 1, dehydrated phase 1′ and dihydrous form 2. | ||
3.6 Thermal analysis
Thermal investigation of the virgin framework 1 was performed using differential scanning calorimetry (DSC) at a rate of 5.00 K min−1. DSC measurements shown in Fig. S4† revealed two broad exothermic peaks for 1 at ca. 109.3 and 142.4 °C (steps 1 and 2, respectively), corresponding to a solid-to-solid phase transformation of complex 1 to 1′. Nevertheless, the cooling DSC experiment failed to exhibit any discernible peak, thereby suggesting the absolute irreversibility of the transformation in the presence of a N2 gas purge.Furthermore, the thermal stability of 1–2 was examined using thermogravimetric (TG) measurements in the temperature range 30–1000 °C under N2 atmosphere (Fig. S5a–c, ESI, ESI†). The thermogram of the compound 1 revealed a multi-step weight loss between 30 and 163.8 °C, corresponding to the loss of guest and coordinated water molecules, before the compound decayed at ca. 317.5 °C. The first mass loss of 9.81% (30–82.5 °C) based on the asymmetric unit accounts for twelve water molecules, which most likely corresponded to the four guest and six coordinated water molecules. The second mass loss of 2.57% (82.5–163.9 °C) corresponded to the loss of the last remaining two coordinated water molecules and the subsequent degradation of the entire framework at ca. 317.5 °C to generate the cobaltous oxide (CoO2) residue. Compounds 1′ is stable up to ca. 181.6 °C and then the whole framework starts losing residues to also form CoO2. Owing to the compactness and huge number of coordination bonds between anions and cations, the structure of 1′ decomposes at ca. 420.1 °C. It is noteworthy that the two bound aqua molecules in 2 can be entirely removed at approx. 120.2 °C and the water-free framework with open cobaltous sites is stable up to ca. 291.7 °C based on TG traces. Such thermally stable CPs with porous active-site-engineered sites could be employed as heterogeneous catalysts for higher-temperature organic transformation reactions.
3.7 Application of 1, 1′, and 2 as catalysts in olefins epoxidation
The activation of molecular oxygen (O2) is a critical step in biology, chemistry, and industry.29,78–82 To this end, attempts have also been made to heterogenise the molecular components in order to induce heterogeneous activation of O2.9,83 We selected epoxidation of various olefins because epoxides are important starting materials for a wide range of industrial and chemical applications.79,84 We realised that any of the three frameworks could act as a catalyst for the epoxidation of olefins using either O2 or any of the two peroxides (i.e., t-BuOOH and H2O2) as oxidizing agents. Scheme 2 depicts simplified chemical pathways for two proposed processes (i.e., allylic and epoxidation products). However, we conducted a series of control experiments to optimise the reaction conditions for improved catalytic efficiency and to avoid the formation of undesirable side product(s).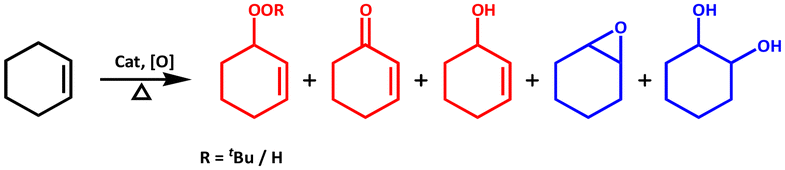 | ||
| Scheme 2 Major oxidation products of substrate cyclohexene; allylic (red) and epoxidation (blue) products. | ||
Of course, one of the preliminary experimental studies revealed that catalysts 1–2 are essential in olefin oxidation reaction, as without them and the sacrificial co-reagent, only traces of products were detected in most cases. Cyclohexene was chosen as the model substrate over catalyst 1 to examine the influence of solvent on the aerobic oxidation of olefins. In liquid-phase catalysis, the solvent is frequently a critical component influencing catalyst performance. Highly polar alkylamine solvents (e.g., MeCN and DMF) possess significant epoxidation activity.85
In this study, initial catalytic activity of 2.00 mol% catalyst 1 was tested using cyclohexene (5.00 mmol) in 10.0 mL MeCN media in the presence of O2 (flow rate of 10.0 mL min−1) at 333 K for 24 h. i-Butyraldehyde (IBA) was selected as an ideal reductant (or co-reagent) for this reaction system.86 We were able achieve 33.3% cyclohexene conversion and 95.1% selectivity to cyclohexene oxide (or cycloaliphatic epoxide) in MeCN (Table 3, entry 1) before screening alternative solvents such as DMF, CHCl3, MeOH or 1,2-DCE. When compared to other solvents, DMF exhibited the highest conversion (38.7%) and but lowest selectivity (79.0%; Table 3, entry 2).
a
| Entry | CP | Solvent | Conv.b (%) | Sel.c (%) | TONd |
|---|---|---|---|---|---|
| a Reaction conditions: cyclohexene (5.00 mmol), CP catalyst (2.00 mol%), co-reagent (IBA; 2.00 mmol), O2 (at 10.0 mL min−1), and solvent (10.0 mL) was stirred at 333 K for 24 h. b Determined by GC-MS/GC. c Epoxide yield/% conversion. d TON (turnover number) = moles of cyclohexene conversion per mole of the dinuclear Co(II) cluster (i.e., {Co(OH2)2O4}) of the catalyst. e Co(NO3)2·6H2O. The reaction vessel was degassed and operated under nitrogen (N2; flow rate at 10.0 mL min−1). f Argon (Ar; flow rate at 10.0 mL min−1). g Atmosphere. | |||||
| 1 | 1 | MeCN | 33.3 | 95.1 | 7.92 |
| 2 | 1 | DMF | 38.7 | 79.0 | 7.64 |
| 3 | 1 | CHCl3 | 31.2 | 87.6 | 6.83 |
| 4 | 1 | MeOH | 29.3 | 94.9 | 6.97 |
| 5 | 1 | 1,2-DCE | 32.3 | 90.6 | 7.32 |
| 6 | 1′ | MeCN | 26.6 | 78.7 | 5.23 |
| 7 | 2 | MeCN | 35.9 | 96.4 | 9.53 |
| 8 | Co(NO3)2e | MeCN | 35.5 | 86.3 | 7.75 |
| 9 | Blank | MeCN | 17.4 | 73.1 | 3.18 |
| 10f | 2 | MeCN | 23.7 | 77.4 | 4.59 |
| 11g | 2 | MeCN | 19.1 | 71.2 | 3.40 |
We did achieve ≤29.3% conversion of cyclohexene in CHCl3, MeOH and 1,2-DCE, but the selectivities to the epoxide were around 87.6, 94.9 and 90.6%, respectively (Table 3, entries 3–5). Cyclohexene conversion occurred in the following order: DMF > MeCN > 1,2-DCE > CHCl3 > MeOH, while cyclohexene oxide selectivity followed the order: MeCN > MeOH > 1,2-DCE > CHCl3 > DMF. Among these solvents, MeCN is the most active in catalysing cyclohexene, with the highest epoxide selectivity (95.1%) and higher cyclohexene conversion (33.3%) when 1 was employed (Fig. 6a). Considering the fairly high conversion and higher selectivity displayed by MeCN, this weak donor solvent was selected for further studies. This behaviour may be attributed to the high permittivity of MeCN.87 Besides, MeCN has been shown to be miscible with most aqueous oxidants for cyclohexene (sebaceous phase), ensuring a broad contact surface for efficient cyclohexene epoxidation.88 As illustrated in Table 3 (entry 6), when MeCN was used as the green solvent, 1′ demonstrated poor catalytic activity among the test catalysts under a molecular oxygen atmosphere. Among the catalysts assessed, 2 had the highest activity (35.9% conversion and 96.4% selectivity towards the epoxide; Table 3, entry 7). When Co(NO3)2·6H2O was employed as a homogeneous catalyst, a higher conversion (86.3%) was obtained, but the selectivity for epoxide was significantly decreased to 35.5% (Table 3, entry 8). Essentially, the lack of an accessible void in the unit cell implies that the open Co(II)-sites on the surfaces are responsible for the catalytic activity of 2. However, the aerobic epoxidation of the substrate in the presence of the co-reagent, using MeCN as a solvent, easily occurred even without the addition of 2, and 17.4% cyclohexene conversion was obtained with 73.1% epoxide selectivity (Table 3, entry 9; blank). Unexpectedly, the efficiency of the epoxidation reactions was dramatically lowered in inert conditions. Cyclohexene conversions of 23.7% and 19.1%, with respective epoxide selectivities of 77.4% and 71.2% (Table 3, entries 10 and 11), were significantly lower under flowing N2 and Ar compared to the reference experiment using molecular oxygen. These observations confirm the influence of O2 for the reaction mechanism (see later). When the flow rate of the dioxygen was halved (5.00 mL min−1) (Table 4, entry 1), the conversion was insignificantly reduced, and the selectivity towards the epoxide was slightly decreased from 96.4 to 95.2%. However, when the flow rate of O2 was doubled (20.0 mL min−1), cyclohexene conversion increased to 38.5%, with 99.0% selectivity for cyclohexene oxide (entry 2 in Table 4).
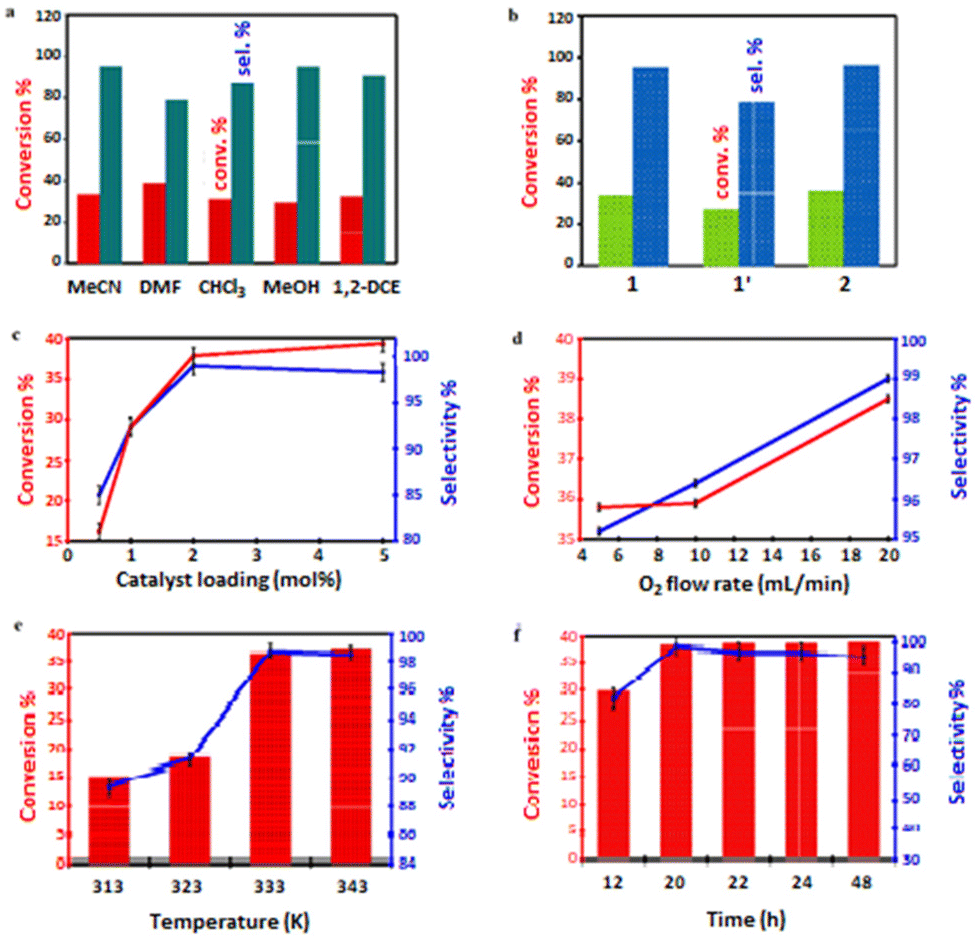 | ||
| Fig. 6 Effect of experimental controls on conversion (%) and selectivity (%) for the epoxidation of cyclohexene: (a) employing 1 as a catalyst in different solvents; looking at various variables such as (b) catalyst type, (c) catalyst loading (mol%), (d) O2 flow rate (mL min−1), (e) temperature (K), (f) time (h). | ||
a
| Entry | Oxidant | Conv.b (%) | Sel.b (%) |
|---|---|---|---|
| a Reaction conditions: cyclohexene (5.00 mmol), catalyst 2 (2.00 mol%), co-reagent (IBA; 2.00 mmol). b Conversion % and selectivity %. c O2 was purged at 5.00, and 20.0 mL min−1, respectively, t-BuOOH/H2O2 (10.0 mmol), and MeCN (10.0 mL) was stirred at 333 K for 20 h. d Conversion was determined by GC-MS/GC. e Selectivity was calculated as epoxide yield/% conversion. f In the absence of the co-reagent/catalyst. | |||
| 1c | O2 | 35.8 | 95.2 |
| 2c | O2 | 38.5 | 99.0 |
| 3d | t-BuOOH | 17.9 | 81.8 |
| 4e | H2O2 | 13.5 | 70.8 |
| 5f | t-BuOOH | 18.8 | 76.0 |
| 6f | H2O2 | 15.4 | 66.3 |
Because an insufficient amount of dioxygen could decrease the contact possibility of the reactant molecules, the optimal flow rate of 20.0 mL min−1 was therefore determined to be the most suitable quantity for further experiments. Then, in the presence and absence of the reductant, we investigated the influence of anaerobic conditions on the epoxidation of cyclohexene using t-BuOOH as the oxidant. Generally, tert-butyl-2-cyclohexenyl-1-peroxide is the main product of the catalytic epoxidation reaction detected when using Co(II)-containing CP as the catalyst.82,89 However, a different and intriguing catalytic result was obtained when 2 was utilised as the catalytic agent in cyclohexene epoxidation. In contrast to other reports about the lesser catalytic effect of Co-based CPs,7 the catalytic data presented here show that cyclohexene epoxidation in the presence of 2 was relatively fast, and allowed for multiple turnovers, whereas the extent of the reaction conducted in the absence of the catalyst under the same conditions was almost insignificant (17.4% conversion after 24 h; entry 9 in Table 3). Notably, 17.9% cyclohexene conversion was selective for tert-butyl-2-cyclohexenyl-1-peroxide (11.7%), 2-cyclohexen-1-ol (12.3%), and 2-cyclohexen-1-one (19.2%) as by-products, with cyclohexene oxide (38.6%) as the desired product after 24 h when 2 was used in the presence of IBA (Table 4, entry 3). There was no improvement without the co-reagent, neither in selectivity nor in the conversion of cyclohexene. Of course, the major product was tert-butyl-2-cyclohexenyl-1-peroxide. The presence of t-BuOOH and IBA clearly produced a free-radical-based cyclohexene oxidation, resulting in the selectivity of cyclohexene oxide, 2-cyclohexen-1-one, and 2-cyclohexen-1-ol, with tert-butyl-2-cyclohexenyl-1-peroxide showing the least selectivity. In addition to negligible amounts for other oxidation products, network 2 was predominantly selective towards the epoxide (76.0%; entry 5 in Table 4). Importantly, there was neither 2-cyclohexene-1-one nor 2-cyclohexene-1-ol (allylic products) detected in our catalytic method when the reaction was carried out in the absence of t-BuOOH. Regardless of whether catalyst 2 and reductant were present or absent, cyclohexene was able to oxidise to cyclohexene oxide with at least 13.5% conversion and up to 81.8% selectivity even under anaerobic reaction conditions (Table 4; entries 3–6).
Using 30% (wt) H2O2 instead of t-BuOOH, the catalytic reaction over catalyst 2 in the absence of IBA gave 13.5% conversion and 70.8% selectivity (Table 4, entry 4) for the epoxidation products (cyclohexene oxide and cyclohexane-1,2-diol) and 19.2% selectivity towards the allylic products. Typical epoxidation by-products such as 2,3-epoxy-cyclohexanone, phenol, and cyclohexene dimer (minor products) were not detected in our system. When t-BuOOH and H2O2 were each implemented in MeCN media without IBA or the catalyst (Table 4, entries 5 and 6), the former oxidant demonstrated significantly higher conversion and selectivity. Moreover, these results suggested that the catalyst, oxidant and reductant individually played a key role in the anaerobic oxidation of cyclohexene. However, the oxidant H2O2 partially dissolved the catalyst 2 after 24 h, and the metal content was 22.9% cobalt, as confirmed by AAS.
We also made efforts to identify the optimal loading of the catalyst in order to carry out the epoxidation reactions efficiently. In this case, 2 was opted for since it outperformed frameworks 1 and 1′ catalytically (Fig. 6b).
For this purpose, different catalyst loadings of 2 (i.e., 0.50, 1.00, and 5.00 mol%) were utilised for the aerobic oxidation of cyclohexene at 333 K (O2 bubbled at 20.0 mL min−1). After 24 h, cyclohexene conversion varied in the expected order: 0.50 mol% (16.2%) < 1.00 mol% (29.1%) < 2.00 mol% (38.5% as determined before), and <5.00 mol% (39.4%). Meanwhile, the corresponding selectivities for cyclohexene oxide were 85.0, 92.4, 99.0, and 98.3%, as shown in Fig. 6c. Because the results at 2.00 and 5.00 mol% catalyst loading were comparable, we chose to use only 2.00 mol% in an attempt to limit the quantity of the catalyst.
Furthermore, the effect of the oxygen concentration on the reaction system was investigated. Fig. 6d and Table 4 illustrate that the oxygen flow rate can influence the oxidative reaction. The conversion of the substrate and the selectivity for cyclohexene oxide both increased as the oxygen flow rate was raised.
To assess the optimal reaction temperature, cyclohexene was oxidised with 2.00 mol% of the catalyst 2 at RT, 313, 323, 333, and 343 K, as shown in Fig. 6e. This epoxidation procedure was carried out with O2 as the final oxidant, flowing at 20.0 mL min−1 for 24 h. It was discovered that higher temperatures were beneficial for enhancing the conversion of cyclohexene as well as selectivity. With the effect of temperature first tested at RT, the reaction barely occurred. When the reaction temperatures were separately elevated to 313 and 323 K, the selectivity for cyclohexene oxide improved to 89.4 and 91.2%, respectively, while cyclohexene conversion rose to 15.3 and 18.2%. Moreover, the epoxide was produced with high conversion and selectivity when the temperature was further raised to 333 K. This demonstrated that more deeply oxidised products were obtained at higher temperatures. However, higher temperature than 333 K resulted in a considerable production of undesirable products. The effect of time on the epoxidation reaction was also evaluated at 333 K in the 12–24 h range. As displayed in Fig. 6f, extending the reaction period from 12 to 20 h enhanced the conversion of the substrate from 29.8 to 35.1% and epoxide selectivity from 81.6 to 98.8%. However, the selectivity towards the epoxide dropped when the reaction time was further prolonged, which led to the formation of 2-cyclohexene-1-ol and 2-cyclohexene-1-one, with the latter showing a higher selectivity of 29.5%. A further increase in the reaction time to 24 h did not bring about higher conversion and more excellent selectivity.
Preliminary tests were conducted to determine the conditions under which the reaction was not controlled by external diffusion. Following that, four experiments were performed at stirring speeds of 350, 500, 750 and 900 rpm. There were no changes in the initial reaction rate, indicating that the reaction was not regulated by external diffusion at the stirring rate of 350 rpm. Therefore, the experiments have always been carried out at 350 rpm.
We also compared the activity and selectivity of 2 in the oxidation of cyclohexene to those of other cobalt-containing CP materials. It is worth emphasizing that the conversion and selectivity of the oxidation reaction catalysed by 2 are among the highest when compared to the reported Co(II)-CP catalysts (Table 5), considering the relatively mild reaction conditions presented herein.
| Entry | Catalyst/additive | Sel. (%) | Conv. (%) | Oxidant/solvent | Temp. (K) | Time (h) | Ref. |
|---|---|---|---|---|---|---|---|
| a [(CoCl3)(tptz)][(CoCl(H2O))]·¼H2O; tptz = 2,4,6-tris(2-pyridyl)-1,3,5-triazine; tpta = terphenyl-3,2′′,5′′,3′-tetracarboxylate; L = 4-((1-carboxy-2-(1H-imidazol-4-yl)ethylamino)methyl)-benzoate; ZIF = zeolitic imidazolate framework; IBA = i-butyraldehyde; 1,4-phda = 1,4-phenylenediacetate; 4,4′-bpa = 1,2-bis(4-pyridyl)ethane; H2-DHBDA = 2,5-dihydroxy-1,4-benzenediacetate; bpe = 1,2-bis(4-pyridyl)ethylene; 3-pmp = 3-pyridylmethylphosphonate; bdpb = 1,4-bis[(3,5-dimethyl)pyrazol-4-yl]benzene; DOBDC = 2,5-dihydroxyterephthalate; L-Glu = L-glutamate; TPPS = tetrasulfonatophenyl porphyrin; HL = 2-hydroxyquinoline-4-carboxylate; mef = mefenamate; HIm = imidazole; acet = acetylacetonate; 4,4′-bpy = 4,4′-bipyridine. | |||||||
| 1 | [Co3(OH)2(tpta)] | 22.1 | 73.6 | t-BuOOH | 343 | 7.0 | 7 |
| 2 | [Co(L)(H2O)2(H2O)] | 12.8 | 82.6 | t-BuOOH | 333 | 6.0 | 8 |
| 3 | Co-ZIF/IBA | 98.5 | 100 | O2/MeCN | 308 | 5.0 | 16 |
| 4 | [Co(1,4-phda)(4,4′-bpa)] | 10.0 | 68.0 | t-BuOOH/MeCN | 343 | 20 | 18 |
| 5 | [Co(H2-DHBDA)(bpe)] | 36.0 | 82.0 | H2O2/MeCN | 313 | 24 | 79 |
| 6 | 2 | 66.0 | 15.0 | H2O2/MeCN | 333 | 20 | Herein |
| 7 | [Co(3-pmp)]/IBA | 68.0 | 93.0 | O2/MeCN | 333 | 16 | 80 |
| 8 | [Co4O(bdpb)3] | 3.00 | 27.5 | t-BuOOH | 343 | 20 | 82 |
| 9 | 2 | 82.0 | 18.0 | t-BuOOH/MeCN | 333 | 20 | Herein |
| 10 | [Co2(DOBDC)(H2O)2]·8H2O | 0.50 | 10.5 | O2 | 353 | 15 | 90 |
| 11 | L-Glu-Co(II) | 93.2 | 82.5 | O2 | 343 | 24 | 91 |
| 12 | [CoTPPS]/IBA | 0.95 | 93.0 | O2 | 298 | 3.0 | 92 |
| 13 | 2/IBA | 99.0 | 38.5 | O2/MeCN | 333 | 20 | Herein |
| 14 | [Co(HL)2(H2O)4]/IBA | 69.0 | 82.0 | Air/MeCN | 333 | 6.0 | 93 |
| 15 | [Co(mef)2(HIm)2(MeOH)2] | 100 | 100 | t-BuOOH/1,2-DCE | 348 | 4.0 | 94 |
| 16 | [Co(acet)(4,4′-bpy)]/IBA | 88.0 | 65.3 | O2/MeCN | 298 | 14 | 95 |
| 17 | Co-CPa | 100 | 54.0 | t-BuOOH/MeCN | 353 | 8.0 | 96 |
3.8 Heterogeneous nature and recyclability
To validate the heterogeneous character of the catalytic process, 2 was removed from the hot solution by filtration after 10 h, exhibiting approx. 24.6% conversion of the model substrate. Fig. 7a shows that no further catalytic conversion occurred, demonstrating that cobalt ions (if any) leached from the catalytic species are not responsible for the observed activity. The filtrate was also analysed by AAS, which revealed that no free cobalt(II) ions were detected in the filtrate due to the metal concentration exceeding the instrument's lower detection limit. These results suggested that 2 was a genuinely heterogeneous catalyst. Network 2 could be easily recovered and reused in the additional oxidation three times with minimal efficiency loss, implying that the catalyst is efficient and recyclable. Moreover, the epoxide activity and selectivity after four cycles with the active catalytic species described in this work are roughly 37.0 (±1.5) and 97.0 (±2.0)% (Fig. 7b), respectively.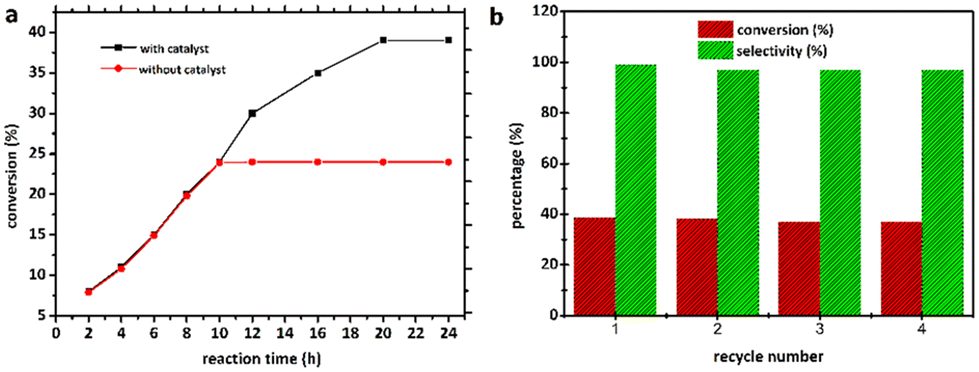 | ||
| Fig. 7 (a) Leaching test for catalytic epoxidation of cyclohexene catalysed by 2. (b) Conversion (%) and selectivity (%) for the epoxidation of cyclohexene over 2 for the fresh catalyst (1st) and for three successive reuses (2nd, 3rd, and 4th). | ||
IR examination (Fig. S6†) of a fresh and reused catalyst 2 (after being rinsed and vacuum dried at 100 °C for 60 min) revealed no significant differences, indicating that the CP remained in its initial crystalline state. After four catalytic cycles, the crystalline structure of catalyst 2 was therefore preserved (see Fig. 8). The strong resistance to degradation during the catalytic process correlates with its higher thermal stability of up to 260 °C.
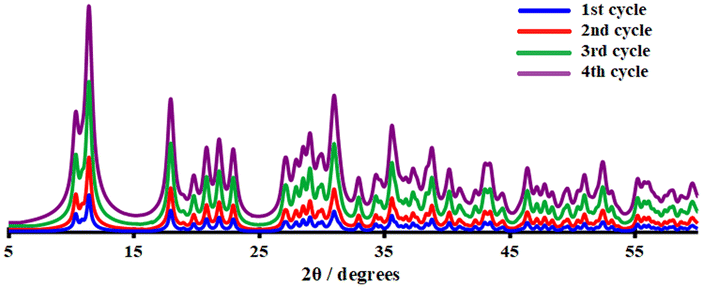 | ||
| Fig. 8 PXRD patterns of catalyst 2 after the first (blue), second (red), third (green), and fourth (purple) recycled runs. | ||
3.9 Substrate scope
Motivated by the excellent catalytic performance of 2 in the oxidation of cyclohexene, the scope of the substrates was widened to look into their general applicability. The preceding control and optimisation experiments enabled us to investigate the epoxidation reactions of diverse substrates, and the results are shown in Table 6. In a typical epoxidation reaction, olefin and reductant (5:2 ratio) were reacted over 2.00 mol% of catalyst 2 in 10.0 mL MeCN at 333 K, and the dioxygen was circulated through the reactor at 20.0 mL min−1 for 20 h. Using 2, it is obvious that the substrates can be smoothly and selectively transformed into their matching epoxides. As illustrated in Table 6 (entries 1–5), various cyclic olefins (4-methylcyclohexene, cyclopentene, 4-methylcyclopentene, and limonene) and terminal olefins (styrene and trans-stilbene; entries 6 and 7, respectively) converted between ca. 34.6% and 39.5% after 20 h and returned sizeable epoxide yields (approx. 32.2–39.0%). All the epoxidation reaction products were analysed by GC-MS/GC spectra (Fig. S7†) and essentially identified by 1H NMR data (see Fig. S8 in the ESI†). In earlier studies of the selective oxidation of cyclic olefins, such as cyclohexene or styrene, over a variety of catalysts with molecular oxygen or peroxides as the oxidizing agents,88 a radical reaction route was presumed in which the oxygen species (see mechanistic insights later) played an important role. In most cases, the quantity of the undesirable products was limited to small scales. Under identical reaction conditions, 4-methylcyclohexene presented 98.3% selectivity towards the epoxide.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Product | Time (h) | Conv. (%) | Sel. (%) | TONc | Yield (%) |
|
a Reaction conditions: catalyst (2.00 mol%), molar ratio of olefin to IBA (5:2), O2 (at 20.0 mL min−1), and MeCN (10.0 mL) was stirred at 333 K for 20 h.
b All values are based on GC-MS/GC spectra.
c TON (turnover number) = moles of olefin conversion per mole of the {Co(OH2)2O4} cluster of the catalyst.
|
|||||||
| 1 |
|
|
20 | 38.5 | 99.0 | 9.53 | 38.1 |
| 2 |
|
|
20 | 39.5 | 98.3 | 9.71 | 38.8 |
| 3 |
|
|
20 | 37.2 | 99.1 | 9.22 | 36.9 |
| 4 |
|
|
20 | 39.4 | 99.0 | 9.75 | 39.0 |
| 5 |
|
|
20 | 37.3 | 93.3 | 8.70 | 34.8 |
| 6 |

|
|
20 | 39.1 | 94.4 | 9.23 | 36.9 |
| 7 |
|
|
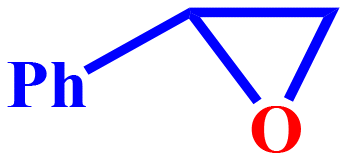
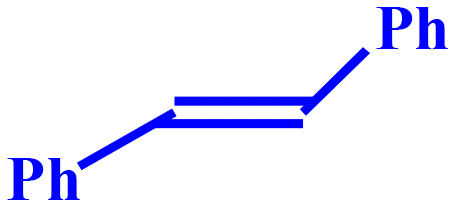
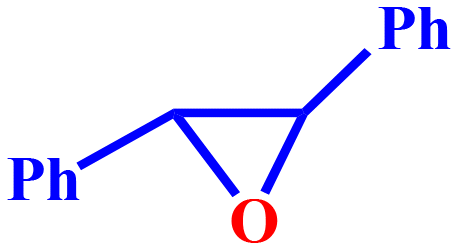
20 | 34.6 | 93.1 | 8.05 | 32.2 |
This is most likely due to the presence of a methyl group, which has a positive inductive (+I) effect and makes 4-methylcyclohexene a more electron-rich olefin. Cyclopentene displayed superior reactivity owing to smaller ring size and easy accessibility. Cyclohexene and 4-metylcyclopentene, on the other hand, underwent comparable transformations. The production of the epoxide component suggested the involvement of a radical mechanism in the epoxidation of cyclic olefins. Notably, the amount of epoxide produced by conformationally rigid molecules, such as limonene and trans-stilbene was negligibly small. Additionally, the conversion of styrene was more substantial than that of trans-stilbene. It could be due to the styrene-containing terminal alkene, which is the high activity position for epoxidation reaction.
3.10 Mechanism
Based on literature review and our experimental observations, we proposed a possible reaction mechanism.97 The Mukaiyama–Yamada epoxidation reaction3,97 (Scheme 3, route A) describes the mechanism for the transition metal-catalysed aerobic oxidation of olefins in the presence of aldehyde as a key co-reagent. In this work, aldehyde autoxidation serves as a sacrificial step in the catalytic process (step I in Fig. 9). The allylic pathway is more favourable without the aldehyde in the Co(II)-catalysed alkene oxidation reaction.71,90,98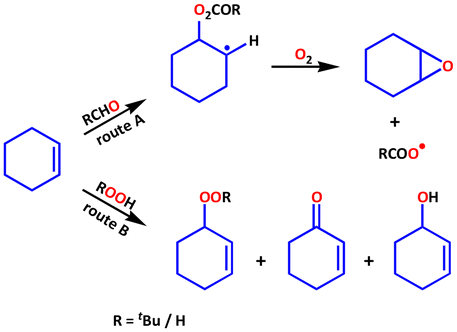 | ||
| Scheme 3 Schematic representation for the formation of the oxidation products of cyclohexene in the presence of both IBA and O2 and, when either t-BuOOH or H2O2 is used instead. | ||
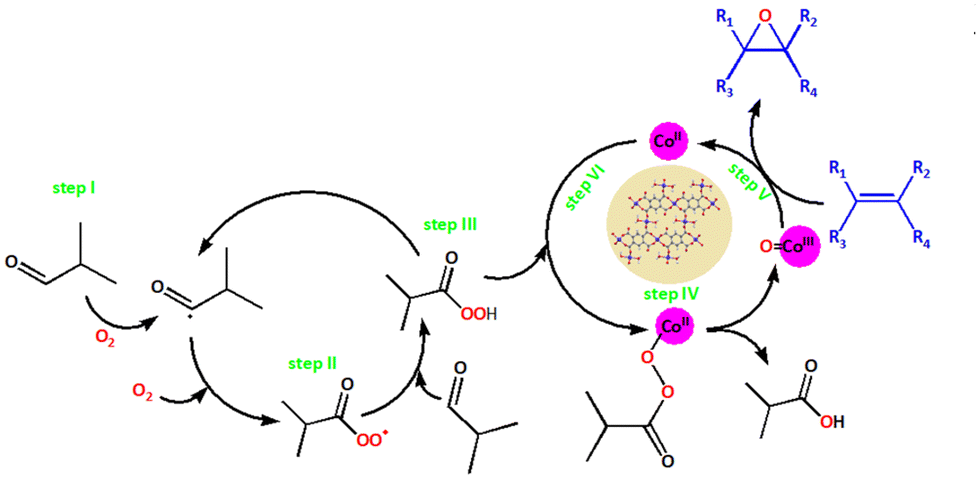 | ||
| Fig. 9 Possible reaction mechanism steps for the formation of peroxo species catalysed by the heterogeneous catalyst 2 in the epoxidation of olefins. {Co(OH2)2O4} active species is represented by CoII. | ||
The co-reactant IBA was transformed via an in situ dehydrogenation reaction into an acylperoxy radical (step II), which was the dominating oxidizing species and aided oxygen transfer to the substrate. Cyclohexene is an excellent substrate for determining whether the oxidizing species favours epoxidation or allylic oxidation.99 During our catalytic assays, the latter pathway was the most prevalent, indicating that the Co(II) active sites ({Co(OH2)2O4} cluster) mainly stabilised the acylperoxy radical. Afterwards, the fragile carbonyl radical disintegrated to form an acyl radical, which can then be oxidised to an acylperoxy radical. This acylperoxy radical species was less selective towards the formation of epoxide in the presence of peroxides and led to the formation of side products through the allylic oxidation pathway (Scheme 3, route B). However, the acylperoxy radical reacted with another IBA molecule to yield a peroxycarboxylic acid (step III), thereby producing another acyl radical for the next catalytic run. Finally, the peroxycarboxylic acid interacted with the Co(II)-CP to form a high-valent Co(III)-CP intermediate (step IV), which then underwent oxygen transfer with the substrate to give the epoxide product along with the formation of Co(II) species to continue the catalytic cycle (steps V and VI).
4. Conclusions
This work effectively demonstrates the capacity of solid-state transitions to produce a unique cobalt-CP that cannot be obtained through traditional solvent-mediated techniques.In this study, it has been observed that compound 1 undergoes a single-crystal-to-single-crystal (SCSC) transformation upon heating at 180 °C. This transformation led to the formation of an anhydrous phase denoted as 1′ ([Co2(btec)]; coloured violet). Furthermore, it has been noted that the presence of water vapour at room temperature could induce a semi-reversible SCSC transformation from 1′ to 2 ([Co2(OH2)2(btec)]; pink). SCSC inclusion of coordinated water molecules into a coordinatively saturated coordination polymer (CP) cluster at room temperature is a rare phenomenon. As an atmospheric water harvester, 1′ demonstrated an efficiency of 175 and 263 mL per kg per CPday at 45% and 70% relative humidity (RH), respectively. The CPs exhibited colorimetrically rewritable behaviour due to reversible structural transitions, enabling them to function as temperature or water probes that can be visually identified. Additionally, the catalytic oxidation of cyclohexene using molecular oxygen (O2) as the final oxidizing agent was systematically tested with these three cobalt-containing CPs, both in the presence and absence of i-butyraldehyde (IBA) reductant. It was found that tert-butylhydroperoxide (t-BuOOH) was a more effective oxidant compared to hydrogen peroxide (H2O2), which showed minimal activity, especially in the presence of 1′ catalyst. The absence of a catalyst allowed for the epoxidation of cyclohexene with t-BuOOH to occur. Overall catalytic activity per mole of catalyst was observed to follow the order of 2 > 1 > 1′. The presence of O2 and the addition of a sacrificial co-reagent accelerated the epoxidation reaction. However, O2 alone was not enough to achieve rates higher than the conversion in the absence of the catalyst. As time progressed, the conversion of cyclohexene increased until it reached almost complete conversion. The determination of the cyclohexene conversion using t-BuOOH over 1, 1′, 2 resulted in the deduction that the Co(II)-sites in 2 exhibit higher reactivity in generating radicals and facilitating radical-based oxidation compared to the metal-sites in 1 and 1′. This analysis substantiates the notion that the liquid-phase conversion of cyclohexene with O2 serves as an optimal and delicate test reaction for assessing the capacity and selectivity of novel solid oxidation catalysts, particularly those derived from cobalt-based CPs. The most effective catalyst, based on the {Co(OH2)2O4} cluster and the selectivity for cyclohexene epoxide, possessed a turnover number (TON) of 9.53. This indicated that the catalytic reaction was significantly enhanced by catalyst 2. Furthermore, the catalytic activity of 2 was evaluated in the epoxidation of various cyclic olefins (4-methylcyclohexene, cyclopentene, 4-methylcyclopentene, cyclooctene, and limonene) as well as terminal olefins (styrene and trans-stilbene). The results demonstrated that the newly developed scu-net CP 2 displayed remarkable activity in the epoxidation of the aforementioned alkenes under optimised conditions. This research work not only showcase the correlation between structure and property, but also offers valuable knowledge on the development and construction of a highly effective and selective heterogeneous catalyst for crucial catalysis applications, specifically in oxidative transformations. Moreover, these findings have stimulated our curiosity to explore the wide range of structures found in main-group metal-based CPs and the corresponding SCSC conversion processes. This exploration will facilitate the examination of the intricate interplay between the lattice molecules and the framework, ultimately opening the door for a deep understanding of their catalytic capabilities within these networks.
Author contributions
Siya T. Hulushe: conceptualisation, data curation and formal analysis, experimental investigation, methodology, validation, visualisation and the writing of the manuscript; Gareth M. Watkins: resources and supervision, reviewed and edited the manuscript; Setshaba D. Khanye: funding acquisition and resources, reviewed and edited the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors express their gratitude to Professor Rui Werner Maçedo Krause for his valuable assistance with GC-MS/GC data. We extend our appreciation to Dr Vincent J. Smith for granting us permission to utilise the in-house single-crystal X-ray diffraction facility. We acknowledge and are thankful for the assistance provided by Professor Edith Antunes (University of the Western Cape) in conducting BET adsorption/desorption experiments. Distinguished Professor Leonard J. Barbour and Dr Alexios I. Vicatos of the Department of Chemistry at Stellenbosch University are highly valued and acknowledged for performing dynamic vapour sorption (DVS) experiments. Furthermore, we would like to acknowledge the financial support received from the National Research Funding (NRF) of South Africa, the Atlantic Philantropies Scholarship for young academics, and the Rhodes University Research Council.References
- X. Song, W. Zhu, K. Li, J. Wang, H. Niu, H. Gao, W. Gao, W. Zhang, J. Yu and M. Jia, Catal. Today, 2015, 259, 59–65 CrossRef
.
- S. D. Bishopp, J. L. Scott and L. Torrente-Murciano, Green Chem., 2014, 16, 3281–3285 RSC
- T. Nagata, K. Imagawa, T. Yamada and T. Mukaiyama, Chem. Lett., 1994, 23, 1259–1262 CrossRef
- A. Savara, C. E. Chan-Thaw, I. Rossetti, A. Villa and P. Prati, ChemCatChem, 2014, 6, 3464–3473 CrossRef CAS
- X. Zhou and H. Ji, Chem. Eng. J., 2010, 156, 411–417 CrossRef CAS
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS
-
(a) T. Yan, Z. Li and W. Pan, Sol. Energy Mater. Sol. Cells, 2022, 240, 111645 CrossRef CAS
- F. Yu, X. Xiong, K. Huang, Y. Zhoua and B. Li, CrystEngComm, 2017, 19, 2126–2132 RSC
- G. Kumar, G. Kumar and R. Gupta, Inorg. Chem., 2015, 54, 2603–2615 CrossRef CAS PubMed
- T. Zhang, Y.-Q. Hu, T. Han, Y.-Q. Zhai and Y.-Z. Zheng, ACS Appl. Mater. Interfaces, 2018, 10, 15786–15792 CrossRef CAS PubMed
- M. B. Meder and L. H. Gade, Eur. J. Inorg. Chem., 2004, 2004, 2716–2722 CrossRef
- A. Corma, H. García and F. X. LIabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed
- B. Yu, S. M. Zhang and X. Wang, Angew. Chem., Int. Ed., 2021, 60, 17404–17409 CrossRef CAS PubMed
- M. J. Beier, W. Kleist, M. T. Wharmby, R. Kissner, B. Kimmerle, P. A. Wright, J.-D. Grunwaldt and A. Baiker, Chem. – Eur. J., 2012, 18, 887–898 CrossRef CAS PubMed
- P. Cancino, A. Vega, A. Santiago-Portillo, S. Navalon, M. P. Alvaro, P. Aguirre, E. Spodine and H. García, Catal. Sci. Technol., 2016, 6, 3727–3736 RSC
- A. P. Zhang, L. Q. Li, J. Li, Y. Zhang and S. Gao, Catal. Commun., 2011, 12, 1183–1187 CrossRef CAS
- W.-J. Cui, S.-M. Zhang, Y.-Y. Ma, Y. Wang, R.-X. Miao and Z.-G. Han, Inorg. Chem., 2022, 61, 9421–9432 CrossRef CAS PubMed
- J. Othong, J. Boonmak, J. Ha, S. Leelasubcharoen and S. Youngme, Cryst. Growth Des., 2017, 17, 1824–1835 CrossRef CAS
- X. Zhang, Y.-Z. Zhang, Y.-Q. Jin, L. Geng, D.-S. Zhang, H. Hu, T. Li, B. Wang and J.-R. Li, Inorg. Chem., 2020, 59, 11728–11735 CrossRef CAS PubMed
- Y. Hu, R. Abazari, S. Sanati, M. Nadafan, C. L. Carpenter-Warren, A. M. Z. Slawin, Y. Zhou and A. M. Kirillov, ACS Appl. Mater. Interfaces, 2023, 15, 37300–37311 CrossRef CAS PubMed
- A. Kobayashi, M. Fujii, Y. Shigeta, M. Yoshida and M. Kato, Inorg. Chem., 2019, 58, 4456–4464 CrossRef CAS PubMed
- S. Ahmed, A. Kumar and P. A. Mukherjee, Chem. Commun., 2023, 59, 3229–3232 RSC
- S. Ahmed, A. Kumar and P. A. Mukherjee, Chem. Mater., 2022, 34, 9656–9665 CrossRef CAS
- N. Hosono and T. Uemura, Acc. Chem. Res., 2021, 54, 3593–3603 CrossRef CAS PubMed
- X. Zhang, T. Kitao, A. Nishijima and T. Uemura, ACS Macro Lett., 2023, 12, 415–420 CrossRef CAS PubMed
- Y. Zhang, F. de Azambuja and T. N. Parac-Vogt, Catal. Sci. Technol., 2022, 12, 3190–3201 RSC
- M. Dosen, Y. Kawada, S. Shibata, K. Tsuge, Y. Sasaki, A. Kobayashi, M. Kato, S. Ishizaka and N. Kitamura, Inorg. Chem., 2019, 58, 8419–8431 CrossRef CAS PubMed
- S. Ghosh, S. Kamilya, T. Pramanik, A. Mohanty, M. Rouzières, R. Herchel, S. Mehta and A. Mondal, Dalton Trans., 2021, 50, 7725–7735 RSC
- S. Ghosh, S. Kamilya, T. Pramanik, M. Rouzières, R. Herchel, S. Mehta and A. Mondal, Inorg. Chem., 2020, 59, 13009–13013 CrossRef CAS PubMed
- M. Woellner, S. Hausdorf, N. Klein, P. Mueller, M. W. Smith and S. Kaskel, Adv. Mater., 2018, 30, 1704679 CrossRef PubMed
- I. A. Lázaro and R. S. Forgan, Coord. Chem. Rev., 2019, 380, 230–259 CrossRef
- X.-Y. Yao, Q. Wang, Q. Liu, M. Pang, X.-M. Du, B. Zhao, Y. Li and W.-J. Ruan, ACS Omega, 2020, 5, 712–717 CrossRef CAS PubMed
- R. A. Yang and M. L. Sarazen, Catal. Sci. Technol., 2021, 11, 5282–5296 RSC
- F.-Y. Xie, Q. Yang, J.-S. Wang, H.-Y. Yu, Y. Li and W.-J. Ruan, Dalton Trans., 2021, 50, 17228–17234 RSC
- V. Kumar, Q. Evrard, C. Leuvrey, M. Lenertz, Y. Garcia, P. Rabu and G. Rogez, Inorg. Chem., 2023, 62, 21101–21114 CrossRef CAS PubMed
- K. Leng, Y. Sun, X. Li, S. Sun and W. Xu, Cryst. Growth Des., 2016, 16, 1168–1171 CrossRef CAS
- O. Akintola, A. Buchholz, H. Görls and W. Plass, Eur. J. Inorg. Chem., 2021, 2021, 2266–2273 CrossRef CAS
- E. Echenique-Errandonea, J. M. Pérez, S. Rojas, J. Cepeda, J. M. Seco, I. Fernández and A. Rodríguez-Diéguez, Dalton Trans., 2021, 50, 11720–11724 RSC
-
(a) S. T. Hulushe, G. M. Watkins and S. D. Khanye, ACS Omega, 2024, 9, 7511–7528 CAS
- H. Yuan, N. Li, W. Fan, H. Cai and D. Zhao, Adv. Sci., 2021, 9, 2104374 CrossRef PubMed
-
(a) S. Hyun, L. Yang, D. Kim and O.-S. Jung, Dalton Trans., 2019, 48, 10927–10932 RSC
-
(a) R. Fernández de Luis, J. Orive, E. S. Larrea, M. K. Urtiaga and M. I. Arriortua, Cryst. Growth Des., 2014, 14, 658–670 CrossRef
-
(a) Z. Wu, Y. Li, C. Zhang, X. Huang, B. Peng and G. Wang, Chem Catal., 2022, 2, 1009–1045 CrossRef CAS
- F. de Azambuja, A. Loosen, D. Conic, M. van den Besselaar, J. N. Harvey and T. N. Parac-Vogt, ACS Catal., 2021, 11, 7647–7658 CrossRef CAS
- M. Mu, Y. Wang, Y. Qin, X. Yan, Y. Li and L. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 22856–22863 CrossRef CAS PubMed
-
(a) H.-Y. Li, S.-N. Zhao, S.-Q. Zang and J. Li, Chem. Soc. Rev., 2020, 49, 6364–6401 RSC
- W. M. Bloch, A. Burgun, C. J. Coghlan, R. Lee, M. L. Coote, C. J. Doonan and C. J. Sumby, Nat. Chem., 2014, 6, 906–912 CrossRef CAS PubMed
- D. Yan, Y. Chen, Y. Yang, Z. Guo and J. Guo, Inorg. Chem., 2022, 61, 1360–1367 CrossRef CAS PubMed
- Y. J. Wang, Y. Y. Zhou, H. G. Hao, M. Song, N. Zhang, S. Yao, J. H. Yan, Z. M. Zhang and T. B. Lu, Inorg. Chem., 2018, 57, 1342–1349 CrossRef CAS PubMed
- Z.-Y. Gu, J. Park, A. Raiff, Z. Wei and H.-C. Zhou, ChemCatChem, 2014, 6, 67–75 CrossRef CAS
- A. Dhakshinamoorthy, M. Opanasenko, J. Čejka and H. Garcia, Catal. Sci. Technol., 2013, 3, 2509–2540 RSC
- J. Gascon, A. Corma, F. Kapteijn and F. X. LIabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS
- M. Ranocchiari and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2011, 13, 6388–6396 RSC
- P. García-García, M. Muller and A. Corma, Chem. Sci., 2014, 5, 2979–2990 RSC
- E. Loukopoulos, A. Abdul-Sada, E. M. E. Viseux, I. N. Lykakis and G. E. Kostakis, Cryst. Growth Des., 2018, 18, 5638–5651 CrossRef CAS
- Y. Xiao, W. Guo, H. Chen, H. Li, X. Xu, P. Wu, Y. Shen, B. Zheng, F. Huo and W. D. Wei, Mater. Chem. Front., 2019, 3, 1580–1585 RSC
- M. R. Mian, U. Afrin, M. S. Fataftah, K. B. Idrees, T. Islamoglu, D. E. Freedman and O. K. Farha, Inorg. Chem., 2020, 59, 8444–8450 CrossRef CAS PubMed
- J. Hao, S. Li, L. Han, L. Cheng, Q. Suo, Y. Xiao, X. Jiao, X. Feng, W. Bai and X. Song, Inorg. Chim. Acta, 2014, 421, 246–254 CrossRef CAS
-
Bruker AXS Inc., APEX4 (v2021.4.0), Bruker AXS Inc., Madison, Wisconsin, USA, 2021 Search PubMed
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed
-
Bruker AXS Inc., SAINT (version 8.40B), Bruker AXS Inc., Madison, Wisconsin, USA, 2021 Search PubMed
-
G. M. Sheldrick, XPREP, Georg-August-Universität, Göttingen, Göttingen, Germany, 2015 Search PubMed
-
G. M. Sheldrick, TWINABS (version 2012/1), Georg-August-Universität, Göttingen, Göttingen, Germany, 2012 Search PubMed
-
Bruker AXS GmBH, SADABS, Karlsruhe (Germany), 2016 Search PubMed
-
Bruker AXS Inc., Bruker AXS Inc., Madison, Wisconsin, USA, 2018
-
G. M. Sheldrick, SHELXL–2016/6, Universität of Göttingen, Göttingen, 2016 Search PubMed
- L. J. Barbour, X-Seed, 4, J. Appl. Crystallogr., 2020, 53, 1141–1146 CrossRef CAS
-
(a) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS
- I. Luz, A. León, M. Boronat, F. X. LIabrés i Xamena and A. Corma, Catal. Sci. Technol., 2013, 3, 371–379 RSC
- O. Fabelo, J. Pasán, L. Canãdillas-Delgado, F. S. Delgado, A. Labrador, F. Lloret, M. Julve and C. Ruiz-Pérez, Cryst. Growth Des., 2008, 8, 3984–3992 CrossRef CAS
- H. Kumagai, C. J. Kepert and M. Kurmoo, Inorg. Chem., 2002, 41, 3410–3422 CrossRef CAS PubMed
- S. Komar, P. A. Mukherjee, M. G. B. Drew, J. Ribas and N. R. Chaudhuri, Inorg. Chem., 2003, 42, 2545–2552 CrossRef PubMed
- L. R. D′souza, N. N. Harmalkar, S. S. Harmalkar, S. B. Tayade and S. N. Dhuri, ACS Omega, 2022, 7, 5698–5712 CrossRef PubMed
- H. Shahroosvand, F. Nasouti, E. Mohajerani and A. Khabbazi, Opt. Mater., 2012, 35, 79–84 CrossRef CAS
- F. Fan, L. Zhao, Q. Zeng, L. Zhang, X. Zhang, T. Wang and Y. Fu, ACS Appl. Mater. Interfaces, 2023, 15, 37086–37092 CrossRef CAS PubMed
-
B. Meunier, Biomimetic Oxidations Catalysed by Transition Metal Complexes, Imperial College Press, London, 2000 Search PubMed
- R. Sen, D. Saha, M. Mal, P. Brandão and Z. Lin, Eur. J. Inorg. Chem., 2013, 5103–5109 CrossRef CAS
- R. Sen, D. Saha, M. Mal, P. Brandão, G. Rogez and Z. Lin, Eur. J. Inorg. Chem., 2013, 5020–5026 CrossRef CAS
- H. Fei, J. W. Shin, Y. S. Meng, M. Adelhardt, J. Sutter, K. Meyer and S. M. Cohen, J. Am. Chem. Soc., 2014, 136, 4965–4973 CrossRef CAS PubMed
- G. Kumar and S. K. Das, Inorg. Chem. Front., 2017, 4, 202–233 RSC
- Q.-H. Xia, H.-Q. Ge, C.-P. Ye, Z.-M. Liu and K.-X. Su, Chem. Rev., 2005, 105, 1603–1662 CrossRef CAS PubMed
- A. P. Woodham, G. Meijer and A. Fielicke, Angew. Chem., Int. Ed., 2012, 51, 4444–4447 CrossRef CAS PubMed
- Y. B. N. Tran and P. T. K. Nguyen, New J. Chem., 2021, 45, 2090–2102 RSC
- Y. Zheng, Q. Shen, Z. Li, X. Jing and C. Duan, Inorg. Chem., 2022, 61, 11156–11164 CrossRef CAS PubMed
- K. L. Walker, L. M. Dornan, R. N. Zare, R. M. Waymouth and M. J. Muldoon, J. Am. Chem. Soc., 2017, 139, 12495–12503 CrossRef CAS PubMed
- Q. Wang, X. Liang, R. Bi, Y. Liu, Y. He, J. Feng and D. Li, Dalton Trans., 2019, 48, 16402–16411 RSC
- G. Tuci, G. Giambastiani, S. Kwon, P. C. Stair, R. Q. Snurr and A. Rossin, ACS Catal., 2014, 4, 1032–1039 CrossRef CAS
- U. Junghans, C. Suttkus, J. Lincke, D. Lässig, H. Krautscheid and R. Gläser, Microporous Mesoporous Mater., 2015, 216, 151–160 CrossRef CAS
- L. Tabrizi, H. Chiniforoshan and P. Mcardle, J. Coord. Chem., 2015, 68, 980–992 CrossRef CAS
- E. Angelescu, O. D. Pavel, R. Ionescu, R. Bîrjega, M. Badea and R. Zăvoianu, J. Mol. Catal. A: Chem., 2012, 352, 21–30 CrossRef CAS
- M. Maghami, F. Farzaneh, J. Simpson and A. Moazeni, Polyhedron, 2014, 73, 22–29 CrossRef CAS
- T. Takai, T. Yamada and T. Mukaiyama, Chem. Lett., 1991, 20, 1499–1502 CrossRef
- D. W. Zhang, J. P. Dutasta, V. Dufaud, L. Guy and A. Martinez, ACS Catal., 2017, 7, 7340–7345 CrossRef CAS
- W. Nam, H. J. Kim, S. H. Kim, R. Y. N. Ho and J. S. Valentine, Inorg. Chem., 1996, 35, 1045–1049 CrossRef CAS PubMed
- Y. N. Wei, H. Li, F. Yue, Q. Xu, J. D. Wang and Y. Zhang, RSC Adv., 2016, 6, 107104–107108 RSC
- C. Shen, J. Ma, T. Zhang, S. Zhang, C. Zhang, H. Cheng, Y. Ge, L. Liu, Z. Tong and B. Zhang, Appl. Clay Sci., 2020, 187, 105478 CrossRef CAS
- B. N. Patra, P. Ghosh, N. Sepay, S. Gayen, S. Koner, P. Brandão, Z. Lin, R. Debnath, J. L. Pratihar, T. Maity and D. Mal, Appl. Organomet. Chem., 2022, 36, e6552 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Tables for crystallographic data, bond lengths and angles, packing structures (Tables S1–S5), Scheme S1, ortep views along with atom numbering schemes for compounds 1, 1′, and 2 (Fig. S1 and S2), IR spectra, DSC and TG curves, PXRD patterns (Fig. S3–S6) for 1, 1′, and 2, additional characterisation techniques (Fig. S7 and S8), which include GC-MS/GC analysis and 1H NMR spectra (PDF). CCDC 2288634, 2288635 and 2288636. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00739e |
| This journal is © The Royal Society of Chemistry 2024 |