
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable optical properties of isoreticular UiO-67 MOFs for photocatalysis: a theoretical study†
Manuel A.
Treto-Suárez
 *a,
Yoan
Hidalgo-Rosa
b,
Mario
Saavedra-Torres
c,
Bryan D.
Koivisto
d,
Karel
Mena Ulecia
e,
Dayán
Páez-Hernández
f,
Ximena
Zarate
g and
Eduardo
Schott
*h
*a,
Yoan
Hidalgo-Rosa
b,
Mario
Saavedra-Torres
c,
Bryan D.
Koivisto
d,
Karel
Mena Ulecia
e,
Dayán
Páez-Hernández
f,
Ximena
Zarate
g and
Eduardo
Schott
*h
aDepartamento de Física y Química, Facultad de Ingeniería, IDETECO, Universidad Autónoma de Chile, Av. Alemania 01090, 4810101-Temuco, Chile. E-mail: manuel.treto@uautonoma.cl
bCentro de Nanotecnología Aplicada, Facultad de Ciencias, Ingeniería y Tecnología, Universidad Mayor, Camino La Pirámide 5750, Huechuraba, Santiago, 8580745, Chile
cMillennium Nucleus in Catalytic Processes towards Sustainable Chemistry (CSC), Chile
dDepartment of Chemistry and Biology, Toronto Metropolitan University, Toronto, Ontario, Canada
eDepartamento de Ciencias Biológicas y Químicas, Facultad de Recursos Naturales, Universidad Católica de Temuco, Ave. Rudecindo Ortega 02950, Temuco, Chile
fDoctorado en Fisicoquímica Molecular, Center of Applied Nanosciences (CANS), Universidad Andres Bello, Ave. República #275, Santiago de Chile, Chile
gInstituto de Ciencias Aplicadas, Facultad de Ingeniería, Universidad Autónoma de Chile, Santiago, Chile
hDepartamento de Química Inorgánica, Facultad de Química y de Farmacia, Centro de Energía UC, Centro de Investigación en Nanotecnología y Materiales Avanzados CIEN-UC, Pontificia Universidad Católica de Chile, Avenida Vicuña Mackenna, 4860, Santiago, Chile. E-mail: edschott@uc.cl; maschotte@gmail.com
First published on 13th June 2024
Abstract
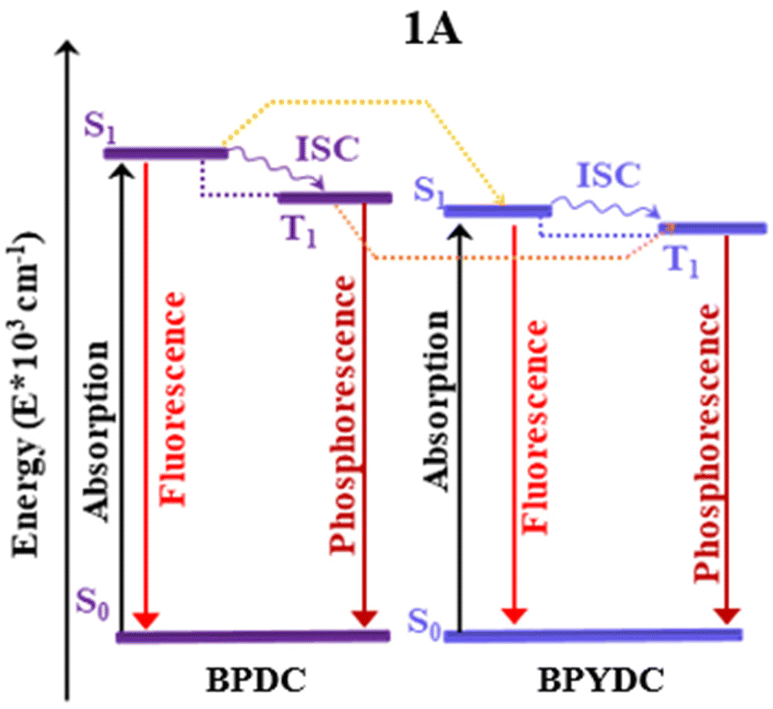
A theoretical study of the reported photocatalytic systems based on Zr-based MOF (UiO-67) with biphenyl-4,4′-dicarboxylic acid (bpdc) and 2,2′-bipyridine-5,5′-dicarboxylic acid (bpydc) as linkers was performed. Quantum chemical calculations were carried out to understand the optical properties of the materials and to facilitate the rational design of new UiO-67 derivatives with potentially improved features as photocatalysts under ambient conditions. Hence, the effect of the structural modifications on the optical properties was studied considering different designs based on the nature of the linkers: in 1 only the bpdc linker was considered, or the mixture 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 between bpdc and bpydc linkers (labeled as 1A). Also, substituents R, –NH2, and –SH, were included in the 1A MOF only over the bpdc linker (labeled as 1A-bpdc-R) and on both bpdc and bpydc linkers (labeled as 1A-R). Thus a family of six isoreticular UiO-67 derivatives was theoretically characterized using Density Functional Theory (DFT) calculations on the ground singlet (S0) and first excited states (singlet and triplet) using Time-Dependent Density Functional Theory (TD-DFT), multiconfigurational post-Hartree–Fock method via Complete Active Space Self-Consistent Field (CASSCF). In addition, the use of periodic DFT calculations suggest that the energy transfer (ET) channel between bpdc and bpydc linkers might generate more luminescence quenching of 1A when compare to 1. Besides, the results suggest that the 1A-R (R: –SH and NH2) can be used under ambient conditions; however, the ET exhibited by 1A, cannot take place in the same magnitude in these systems. These ET can favor the photocatalytic reduction of a potential metal ion, that can coordinate with the bpydc ligand, via LMCT transition. Consequently, the MOF might be photocatalytically active against molecules of interest (such as H2, N2, CO2, among others) with photo-reduced metal ions. These theoretical results serve as a useful tool to guide experimental efforts in the design of new photocatalytic MOF-based systems.
:1 between bpdc and bpydc linkers (labeled as 1A). Also, substituents R, –NH2, and –SH, were included in the 1A MOF only over the bpdc linker (labeled as 1A-bpdc-R) and on both bpdc and bpydc linkers (labeled as 1A-R). Thus a family of six isoreticular UiO-67 derivatives was theoretically characterized using Density Functional Theory (DFT) calculations on the ground singlet (S0) and first excited states (singlet and triplet) using Time-Dependent Density Functional Theory (TD-DFT), multiconfigurational post-Hartree–Fock method via Complete Active Space Self-Consistent Field (CASSCF). In addition, the use of periodic DFT calculations suggest that the energy transfer (ET) channel between bpdc and bpydc linkers might generate more luminescence quenching of 1A when compare to 1. Besides, the results suggest that the 1A-R (R: –SH and NH2) can be used under ambient conditions; however, the ET exhibited by 1A, cannot take place in the same magnitude in these systems. These ET can favor the photocatalytic reduction of a potential metal ion, that can coordinate with the bpydc ligand, via LMCT transition. Consequently, the MOF might be photocatalytically active against molecules of interest (such as H2, N2, CO2, among others) with photo-reduced metal ions. These theoretical results serve as a useful tool to guide experimental efforts in the design of new photocatalytic MOF-based systems.
Introduction
Metal–organic frameworks (MOFs) are a class of materials formed by metal ions (or metallic clusters) coordinated to organic ligands (linkers) to form various multi-dimensional structures. These structures are generated from the extension of the coordination compounds and their repetition in space in one, two, or three dimensions.1,2 These materials have structural diversity and can exhibit exceptional properties such as high specific surface areas, permanent porosity, readily accessible cavities, and well-dispersed active sites, among others.3–8 Their intrinsic hybrid organic–inorganic nature and the possibility to incorporate functional groups in the structures provide attractive photophysical properties that allow their applications as luminescence sensors9,10 and/or photocatalysts.6,8,11–14 Despite all these advantages, reported MOFs have been limited in several photocatalytic processes due to higher charge recombination, low yield under UV-light illumination, and limited applications under sunlight (visible light).15These photophysical properties depend on several mechanisms which are necessary to be fully understood to achieve improved properties. These mechanisms are based on energy transfer (ET)11,16 and charge transfer (CT)11,17 which compete with other radiative and nonradiative deactivation processes occurring from the excited states.18–21 The ET has been described by two possible mechanisms (i) the Dexter (DET) mechanism22 (which describes the electronic exchange due to orbital overlap, including short-range interactions) and (ii) the coulombic mechanism, known as Förster resonance energy transfer (FRET)23,24 (which considers long-range dipole–dipole interactions). Also, the CT mechanisms (or Photoinduced Electron Transfer (PET)) are responsible for the optical properties in many systems.10,21,25,26 In MOFs, this electron transfer mechanism can occur between the metals and the organic linkers, known as ligand-to-metal charge transfer (LMCT) or metal-to-ligand charge transfer (MLCT).11,17,19,27,28 The electron transfer might also occur between the organic linkers known as ligand-to-ligand charge transfer (LLCT mechanism). All the previously described processes can provide activation or deactivation pathways leading to a higher quantum yield or photocatalytic activity.11,17 For example, recently Santaclara, J. G. et al. reported13 two titanium-based MOFs with potential applications in photocatalysis for solar fuel generation. These MOFs displayed a remarkably long decay lifetime due to an LMCT transition. Our research group reported a theoretical study of an Eu-based MOF sensor that displayed luminescence quenching by aniline due to ET, using quantum chemical computations, to elucidate the detection principle of the turn-off luminescence mechanism.9
On the other hand, the structural modifications of MOFs have been a strategy to modulate the optical and photocatalytic properties of MOFs, by promoting broader light absorption in the visible spectra, improving the efficiency charge separation and the lifetime in the excited state, among others.11,14,15,29–31 For example, Hao Huang et al.15 recently reported the improvement of the visible-light-assisted photocatalytic N2 fixation on the MIL-125-Ti4+ MOF by ligands functionalization with withdrawing groups (–NH2, –OH, and –CH3). This result showed the potential of combining light-harvesting and catalytic components in MOFs for green photocatalytic processes.15 Another strategy is to employ linkers with broad light absorption in the visible spectrum and perform substitutions with metals to improve charge separation efficiency in the excited state.32,33 In this sense, Mehdi Rahimi-Nasrabad et al. reported a review of the progress over the last decade in the chemistry of UiO-67 MOF materials.34 The authors noted that the design of functional materials with preferred properties can be achieved by the inclusion of functional groups, molecules, and nanoparticles into the pores, linkers, and nodes. From this perspective, it is expected that shortly, UiO-67 as a crucial MOF will be a very wide field of research. Some works have been exploring how to improve the photocatalytic properties of UiO-67 by incorporating linkers as the 2,2′-bipyridine-5,5′-dicarboxylic acid (bpydc) for anchor various catalytic active metals or metal complexes.11,35–37 Thus, Yang An et al. reported11 the photocatalytic properties improvement of UiO-67 by incorporating Ce4+ coordinated to bipyridine dicarboxylate ligands. All these strategies be achieved through rational design due to the high tunability of MOFs’ composition (linkers and nodes) and their unique characteristics. For these reasons, future research should focus on the precise and controlled design to allow improved photocatalytic processes in MOFs.15,33,38,39 Therefore, a detailed understanding of the charge (or energy) transfer mechanisms nature in MOFs is crucial to designing the next generation of MOF-based materials via computational methodologies.40–44
Despite the advantages of MOFs, there is a lack of theoretical investigations into their photophysical properties.12,15 This is likely due to the complexity of studying photocatalysis having transition metals with partially filled f- or d-orbitals as the presence of these orbitals might result in near-degenerate electron configurations. Thus, these systems require rigorous computational simulations that can describe the electronic structure, because the wave function has a multiconfigurational character. In this sense, the Hartree–Fock (HF) and the Density Functional Theory (DFT) methods provide a good description of the system in their ground and excited states (to analyze the deactivation processes). Furthermore, for systems containing heavy metals, it is also necessary to apply post-Hartree–Fock methods. This theoretical level can make a correct description of the correlation in the systems.9,30,45 However, these methods are computationally expensive for large systems (such as MOFs) and are not available for periodic models.43 In the case of MOFs, an alternative strategy is to truncate the periodic structure in a representative fragment (node and linker), as highly localized electron states characterize the optical properties. At the same time, diffusion of the reactants through the pores is not usually the rate-limiting step.9,16,30,43,46 Performing this consideration, the precise MOF photophysical and chemical properties modeling is possible in excellent agreement with the experimental evidence.9,30,43 This strategy has allowed more precise modeling of local effects to explore different levels of theory beyond DFT and is effective at describing the excited states with multiconfigurational character.9,43 For example, our group accomplished a theoretical methodology to comprehend the photophysical mechanisms of light emission in different cases, such as the Eu–MOF9 ([Eu2(PmBC)4(NO3)2(H2O)6]) (PmBC = 4-(pyrimidin-5-yl)benzoate), the MOF [Zn2(OBA)4(byp)2] (byp = 4,4′-bipyridine; H2OBA = 4,4′-oxybis[benzoic acid])10 and three isostructural MIL125-Ti.44
Hence, quantum chemical calculations are a powerful tool to rationalize experimental observations and to predict the optical properties and the effect of structural modifications in a material. Thus, it is possible to modulate a material to absorb light in the visible range of the spectrum, and to reduce the charge recombination. In this regard, in this report, the optical properties of a reported photocatalytic system based on Zr-based MOF (UiO-67)11 are studied. The UiO-67 commonly is composed of a Zr cluster with biphenyl-4,4-dicarboxylic acid (bpdc) as a linker (named 1). Furthermore, in this work, some bpdc linkers are replaced with the dianion of 2,2′-bipyridine-5,5′-dicarboxylic acid (bpydc) (named as 1A). After obtaining 1A, this MOF has been used to form bpydc-metal complexes, thus generating complexes of Re, Ce, Eu, Ru, Rh, Pt, or Pd, among others.11,47–51 These alterations produce a modified UiO-67 MOF which demonstrated a higher photocatalytic hydrogen rate, CO2 reduction, water oxidation, or other catalytic activities.11,51–53 Yang An et al. proposed that due to the presence of organic linkers with different functionalities a new energy transfer might be active, giving access to new or improved properties.11 This possible ET from excited bpdc to bpydc might inhibit the recombination of the charge carriers and therefore improve the efficiency of the photocatalyst. These conclusions are based on emission lifetime and transient absorption measurements.11 However, these photophysical mechanisms have not been deeply studied so far. On the other hand, the UiO-67 systems show an intense absorption band at 365 nm, thus achieving a red shift in the absorption band to promote broader light absorption in the visible spectra without affecting the ET between bpdc and bpydc is a challenge. This red shift might be promoted via substitutions in the linker which could allow them to be used under sunlight (visible light) irradiation, possibly affecting their photocatalytic activity.15 Thus, further theoretical studies are needed to guide experimental efforts in the design of new MOF-based systems with better optical properties to be used in photocatalytic processes.
In this framework, reported herein, a theoretical protocol is accomplished for exploring electronic and photophysical properties, and the effect of the linker functionalization (using auxochrome groups such as –NH2 and SH) in based MOFs materials such as UiO-67 using quantum chemical calculation methods at a rigorous theoretical level, such as DFT and CASSCF/NEVPT2, as well as periodic DFT. These two substituent groups were selected as they have been shown to improve the absorption under sunlight and the photocatalytic properties in previous MOF-based systems.15,44,54,55 It was found that the proposed methodology allows us to understand the optical properties of the studied UiO-67 MOFs, suggesting that the ET between bpdc and bpydc linkers is possible. These results suggest that the modifications proposed in the linkers promote a broader light absorption in the visible range of the spectrum. These theoretical results will be a useful tool to guide experimental efforts in the design of new MOF-based systems.
Methodology and computational details
The exploration of MOFs often involves the utilization of finite structural models, an approach that has been widely documented in scientific literature.9,16,29,43,44,56 Despite the extended network structures characteristic of these systems, it has been observed that they frequently exhibit highly localized electronic states. This feature has made it feasible to apply finite structural models in the investigation of the electronic and photophysical properties of MOFs, with results that align well with experimental evidence.11 In the first step, the methodology implemented in this research revolves around the truncation of an extended structure of UiO-67 to a representative fragment called a cluster model or finite fragment. This approach is advantageous in the study of MOFs because it allows the treatment of a fragment representative of the entirety of the extended solid with methods and high-level theory as implemented in molecular modeling suites. The extended structure of UiO-67 is formed by connecting Zr6O4(OH)4 inorganic cluster metal with 4,4′-biphenyl-dicarboxylate (bpdc) as linker resulting in a cubic framework (labeled as 1). In this sense, the chosen finite fragment [Zr6O4(OH)4(COO)10(bpdc)(bpydc)] is composed of a cluster [Zr6O4(OH)4(COO)12], and bpdc and bpydc linkers, (labeled as 1A). The remaining ten linkers were truncated to formiate-capped (HCOO−), (see Fig. 1). The representative fragment was selected from crystallographic data reported for a UiO-67.57,58 Furthermore, to see and analyze the effect of the bpydc linker on the optical properties of UiO-67, the model considering only bpdc linkers was studied (labeled as 1). The proposed isoreticular structures (with –NH2 and –SH substituent) were studied considering two substituent schemes to 1A, (i) the substitutions were performed only at bpdc linker (labeled as 1A-bpdc-R) (see Fig. 2a) and (ii) the substitutions were performed at the bpydc and the bpdc linkers (labeled as 1A-R) (see Fig. 2b).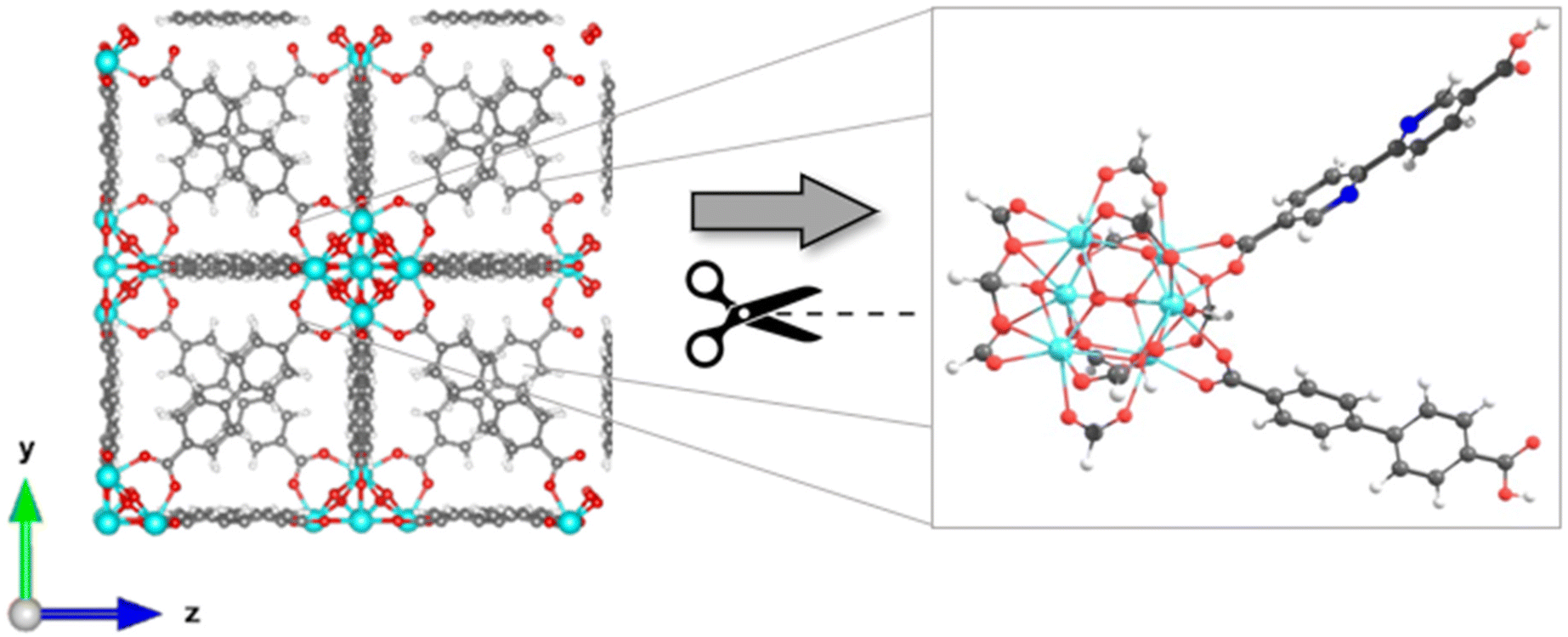 | ||
| Fig. 1 Schematic representation of the extended structure reported to UiO-67-bpdc-bpydc (1A) (left panel) and finite structure (node/linkers) used to study the optical properties (right panel). Atoms are denoted with dark gray (carbon), light gray (hydrogen), red (oxygen), darker blue (nitrogen), and blue (zirconium) spheres. | ||
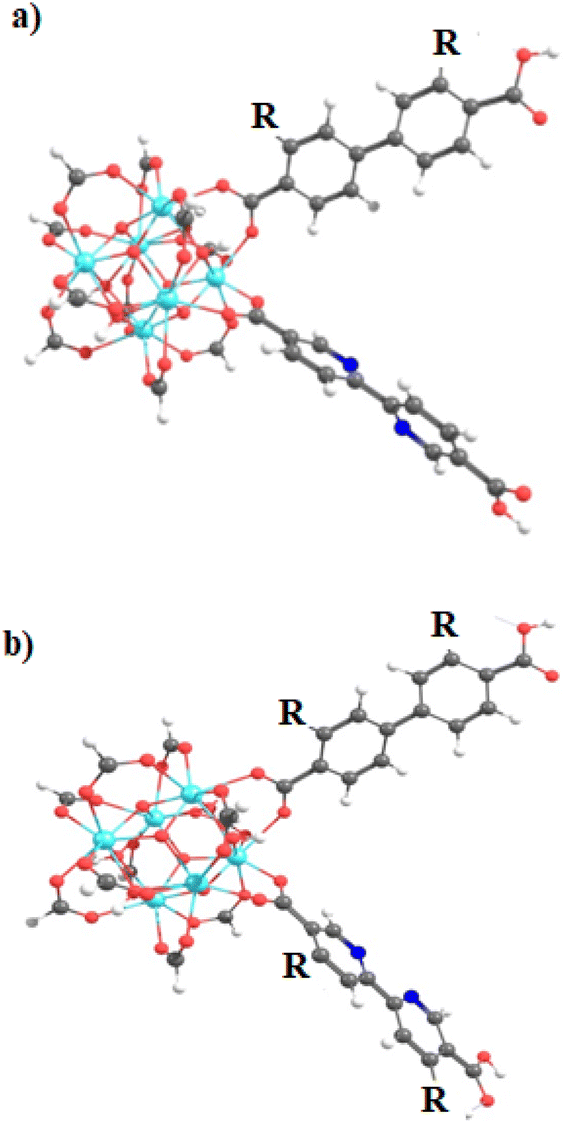 | ||
| Fig. 2 Schematic representation of the finite structure (Zr-node/linkers) with the substituents R (–H, –NH2, and –SH) in (a) the bpdc (1A-bpdc-R) and (b) both linkers (1A-R). Atoms are denoted with dark gray (carbon), light gray (hydrogen), red (oxygen), darker blue (nitrogen), and blue (zirconium). | ||
According to Kasha's rule,59,60 a molecule relaxes to the lowest vibrational level and the lowest electronic excitation state, and from there, can decay via radiative or non-radiative pathways to the S0 state. In this sense, the optical properties depend on the electronic structure in the ground (S0) but also on the first-excited states (singlet (S1) or triplet (T1)).61 Our group has explored similar behaviors in different MOF luminescence sensors.44,62 Those works evidenced that the optical properties cannot be appropriately described considering only the relative energies and electronic structure of the S0 state. Whereas, also the S1 state together with the kinetic parameters of the electron transfer and emission deactivation process.44,62 For these reasons, in the herein report, all studied systems were optimized in the S0, S1, and T1 states via DFT methods to describe the molecular structure, the absorption spectra (from the S0 state), the deactivation pathway (from S1 and T1 states), and the proposed photophysical mechanisms. The optimizations were performed using the valence triple-zeta with two sets of polarization functions (def2-TZVPP) basis set63 for all atoms and the Becke 3-parameter, Lee–Yang–Parr (B3LYP) hybrid functional.64 All local minima were studied through vibrational frequency analysis to ensure no negative eigenvalues in the Hessian matrix. On the other hand, the vertical excitations of UV-Vis absorption spectra (from optimized S0 state electronic structure) and deactivation process (from optimized S1 and T1 state electronic structures) were computed via the Time-Dependent Density Functional Theory (TD-DFT) method at the same theory level (def2-TZVPP/B3LYP) and also using the Coulomb Attenuating Method-Becke, 3-parameter, Lee–Yang–Parr (CAM-B3LYP) functional. The CAM-B3LYP functional was used to analyze the possible charge transfer transitions as this functional corrects the underestimation that the B3LYP functional has shown before.62,65–67 The kinetic parameters associated with the radiative deactivations (radiative rate (krd) and radiative lifetime (τrd) of the emission) were computed using the emission energy (Ei,j) and the transition dipole moment (μi,j) (eqn (1)).61,68 Furthermore, the reorganization energy (l) and the free-energy change (ΔG) due to the excitation process were also calculated via Marcus's theory68 to evaluate the changes in the excited states.
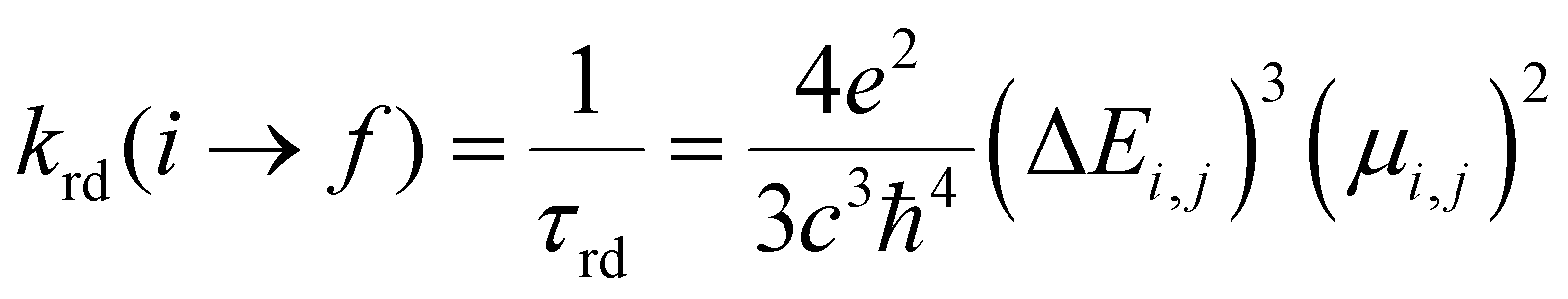 | (1) |
On the other hand, the wave functions that describe the S0, the S1, and the T1 states of the systems were also modeled via CASSCF methods45,69 in an active space of 8 electrons in 8 orbitals to obtain the first 20 singlets and 20 triplets. Additionally, the effect of electronic dynamic correlation was incorporated via the n-electron valence state perturbation theory (NEVPT2) strategy, to correct energy computations from the CASSCF. An active space that comprises ten electrons within ten orbitals was utilized to deduce the energy values of the S0, S1, and T1 electronic states. These calculations were accomplished to confirm the appropriate electronic configuration for the ET mechanism. All calculations were performed using ORCA 5 program package.70
The interactions between the linker (bpdc and bpydc) with the metal centers (node) were studied via the Morokuma–Ziegler decomposition scheme from optimized geometries in the S0 state using ADF 2020 package,71 to accomplish a quantitative description of the chemical interaction. The interaction energy (ΔEInt) can be determined and decomposed in the Pauli repulsion (ΔEPauli), electrostatic interaction (ΔEElec), dispersion energy (ΔEDis), and the orbital terms (ΔEOrb)72,73 using eqn (2). This last term, ΔEOrb, quantifies the orbital interaction and polarization effects due to the fragment interaction and can be deeply studied via the natural orbital of chemical valence (NOCV) methodology proposed by Mitoraj.71
| ΔEInt = ΔEPauli + ΔEElec + ΔEOrb + ΔEdis. | (2) |
Furthermore, periodic DFT calculations were performed on 1 and 1A systems. Thus, UiO-67 and UiO-67-bpydc cells were used to model 1 and 1A structures taking the crystal structure allocated in the Crystallography Open Database (COD) (ID codes on Table S9†) and modifying it agreeing with the herein studied model. Each unit cell was reduced by symmetry to a primitive cell, corresponding with a rhombohedral shape. Cell parameters were optimized for UiO67 (1), UiO67-bpydc, and 1A and the models of each unit cell model were optimized and compared with the experimental crystal data. All structures show good agreement with the experimental parameters (Table S9†). All calculations were modeled through a linear combination of atomic orbitals (LCAO) periodic approximations, employing density functional methods. The structural optimizations were modeled using a gradient generalized Perdew–Burke–Ernzerhof (PBE) functional74 sampling the first Brillouin zone with a 3 × 3 × 3 Monkhorst–Pack k-point scheme.75 Moreover, a 5 × 5 × 5 k-point grid scheme was employed to Projected Density of States calculations with a hybrid HSE06 functional. PseudoDojo family set of pseudopotentials and Grimme-D3 dispersion correction (DFT-D3)76,77 were included in all calculations. QuantumATK 2022.1278 was used for all ab initio calculations and Figures have been represented through VESTA software.79
Results and discussion
This section displays the theoretical results and subsequent discussion achieved to rationalize the experimental observations (optical properties and the photophysical mechanisms) in terms of electronic and molecular structures of the UiO-67 MOFs isoreticular photocatalysts. 1A MOF shows experimentally an absorption band at 365 nm and a radiative decay ∼400 nm (excited at 300 nm). This system displayed the lowest emission intensity compared to 1 due to the inclusion of the bpydc linker. It has been indicated that a new ET is produced.11 Hence, in this report, on other isoreticular structures (including –NH2 and –SH in the linkers as substituents) considering two substituent schemes (1A-bpdc-R and 1A-R, where R: H, –NH2 and –SH) were studied (see Fig. 2). Those substitutions aim to provoke a red shift in the absorption bands (365 nm) which could increase their capacity to work under sunlight (visible light) irradiation. The herein-shown theoretical results, agree with the previously reported experimental evidence and suggest that based on optical terms, the 1A-SH and 1A-NH2 could display a red shift in the absorption bands (visible light). However, in those two possible structures, the ET would not be possible, due to the spectral overlap between emission (from bpdc-R) and absorption (of bpydc-R) (which favor the ET mechanism) is not observed.Structural analyses
A structural analysis of the finite fragments was performed exploring the changes in some selected dihedral angles and bond distances in the S0 and the first excited states (S1 and T1 states). The structural optimizations were performed to get a minimum in the potential energy surface to reproduce the UV-Vis absorption spectra (from the optimized structure in the S0 state) and the emission (from the optimized structure in the S0, S1, and T1 state) via TD-DFT methods (Fig. 3 and S1 in ESI† show the optimized structures (node/linkers) in S0 and first excited states (S1 and T1), respectively). According to the selected dihedral angle values and bond distances (see Table S1 in the ESI†), the optimized structures do not show a large difference between them nor between the proposed isoreticular systems. Only the dihedral angles γ(C4–C5–C6–C7) in the bpdc linker and γ(N1–C4–C5–N2) in the bpydc linker display an increase of planarity (between the linkers rings) in the excited states which agree with the experimental emission of 1. However, reported 1A system displays a decrease in emission intensity after the bpydc incorporations. In this sense, the reproduction and deep analysis of the absorption and emission spectra are important factors in the understanding of the photophysical mechanisms.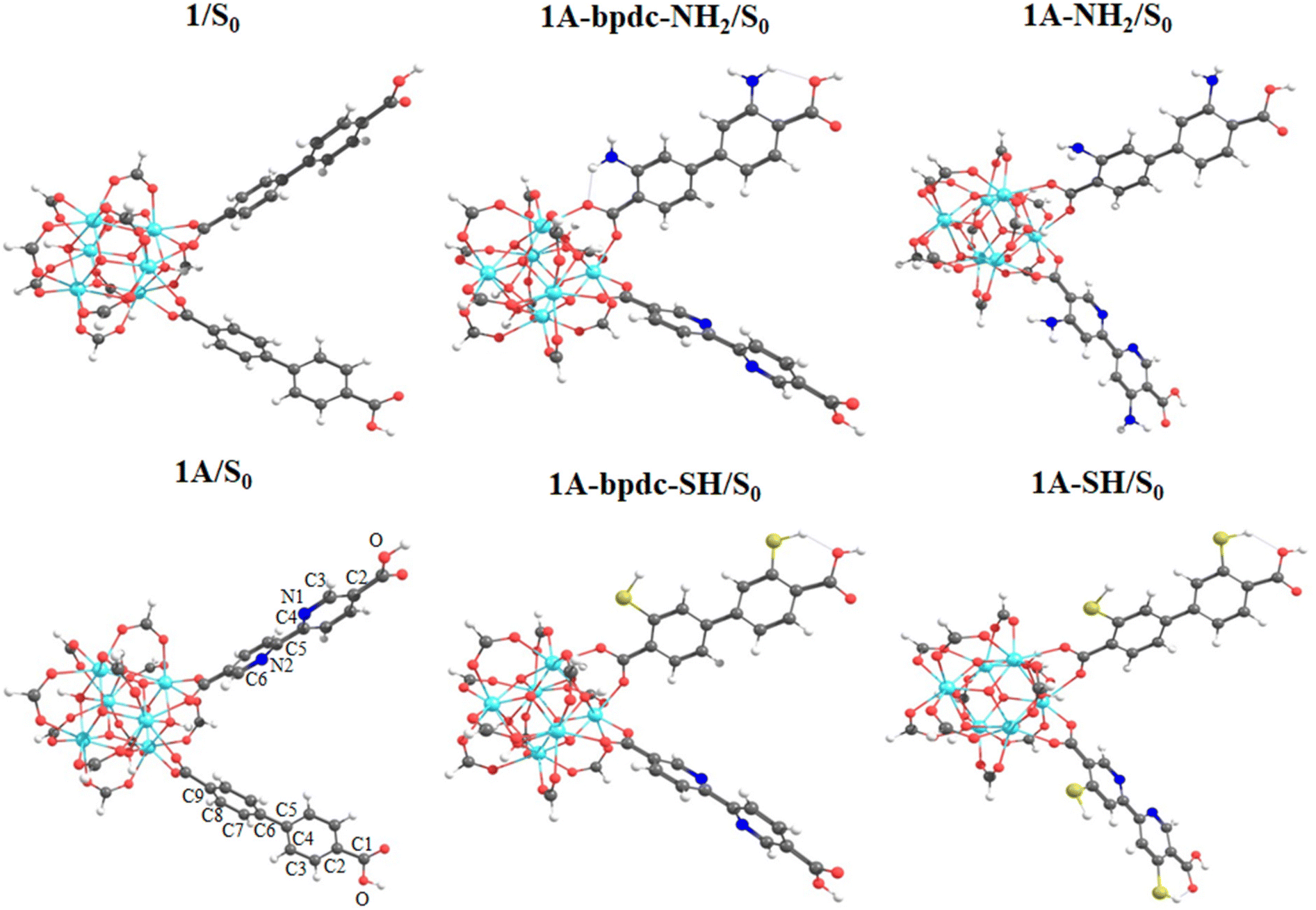 | ||
| Fig. 3 Optimized structures of the studied systems (1, 1A, 1A-bpdc-NH2, 1A-bpdc-SH, 1A-NH2 and 1A-SH) in the S0 state via DFT/B3LYP/Def2-TZPP theory level. Atoms are denoted with light green (titanium), gray (carbon), light gray (hydrogen), red (oxygen), darker blue (nitrogen), blue (zirconium), and yellow (sulfur) spheres. | ||
Absorption and excited state deactivation mechanisms via TDDFT
In this section, the computed vertical excitations of UV-Vis absorption (from S0 state) and emission (from S1 and T1 states) spectra are analyzed via the TD-DFT method to understand the optical properties of the selected fragments. Experimentally, system 1 shows an absorption band at 365 nm and a radiative decay at ∼400 nm when is excited at 300 nm. The intensity of this radiative decay decreases with the inclusion of a bpydc linker in the structure (1A), which was associated with a new energy transfer channel between bpdc and bpydc linkers.11 With this, we performed the theoretical study of the optical properties of these systems, and evaluated the functionalization effect of the linkers (with auxochrome groups such as –NH2, and SH) over the optical properties of these systems was also studied.Systems 1 and 1A
In 1, the most intense singlet → singlet absorption transitions correspond to π → π* transition localized on bpdc linkers (see MOs isosurface Fig. 4 and Table S2 in ESI†). In the case of reported 1A, these transitions are also localized over the linkers. The computed transitions (∼295 nm and 290 nm for 1 and 1A, respectively) agree with the previously reported experimental absorption band (365 nm). These transitions are consistent with HOMO (H) → LUMO (L) for 1 and H → L+1 1A (localized over the bpdc linker). These results suggest that the proposed finite structural model reproduces the optical properties of these systems. In terms of energy, the B3LYP functional underestimates by around 60 nm of the experimental value, while the CAM-B3LYP displays an underestimation of around 100 nm (see Tables S2 and S3 in ESI†). In the case of reported 1A, another transition (higher in energy) was computed which showed a contribution from the bpydc linker (H-1 to L). This result suggests that the PET process, from the lone pair of the nitrogen atoms, to partially occupied H-1, is thermodynamically possible over the bpydc linker. This mechanism is responsible for the optical properties in many systems causing the luminescence quenching.10,21,25,26 The experimental results suggest that the emission quenching of 1A, due to the inclusion of the bpydc linker in the structure, occurs by a new energy transfer (ET) between bpdc and bpydc linkers following the PET process.11 In this sense, the analysis of the emission spectra is crucial to understand the optical properties of these systems. The emission spectra were computed from the optimized structure on S1 and T1 states (see Tables S2–S4 in ESI†) via TDDFT methods to analyze the energy, the intensity of the radiative decay (based on oscillator strength), and the necessary overlap between absorption and emission spectra of the linkers. The overlap helps to probe that the ET between bpdc and bpydc can occur. For 1, the radiative decays (from S1) were computed at 339 nm (with B3LYP functional) and 313 nm (with CAM-B3LYP functional) displaying a π* → π character (see Fig. 4) with an underestimation of around 60 nm and 87 nm, respectively, according to the measured experimental emission wavelengths ∼398 nm.11 In the case of 1A, two emission bands were computed which are localized over bpydc and bpdc, at 605 nm and 318 nm with B3LYP (480 nm and 297 nm with CAM-B3LYP, see Table S3 in ESI†) in the S1 state, respectively. The emission localized over bpydc displayed π* → n character, while the band localized over bpdc linker, showed a π* → π (see MOs isosurface in Fig. 5). The B3LYP functional causes a larger energy underestimation than CAM-B3LYP for the π* → n transition. This capacity of CAM-B3LYP functional has been reported in several previous studies.62,65–67 On the other hand, based on the oscillator stranger (f), the emission centered over bpydc (f = 0.4 × 10−5) is less intense than the localized over bpdc linkers in 1A (f = 1.171) as in 1 (f = 1.278). This result agrees with the experimental evidence and suggests that the π* → n decay which involves bpydc can affect the emission intensity. Furthermore, the spectral overlap (between bpdc and bpydc linker) is verified by analyzing the emission (bpdc) and the absorption (bpydc) spectra (see Fig. 5 and S3†). This evidence suggests that the emission generated by bpdc can be absorbed by the bpydc linker, generating an energy transfer (ET) that produces a luminescence quenching of 1A (based on f value). On the other hand, the computed radiative deactivation processes display lifetime (τrad) within the value range of fluorescence, lower than 10−6 s (see Tables S2–S4†) for all systems. These results and the experimental evidence suggest that radiative deactivation occurs from the S1 state.80–82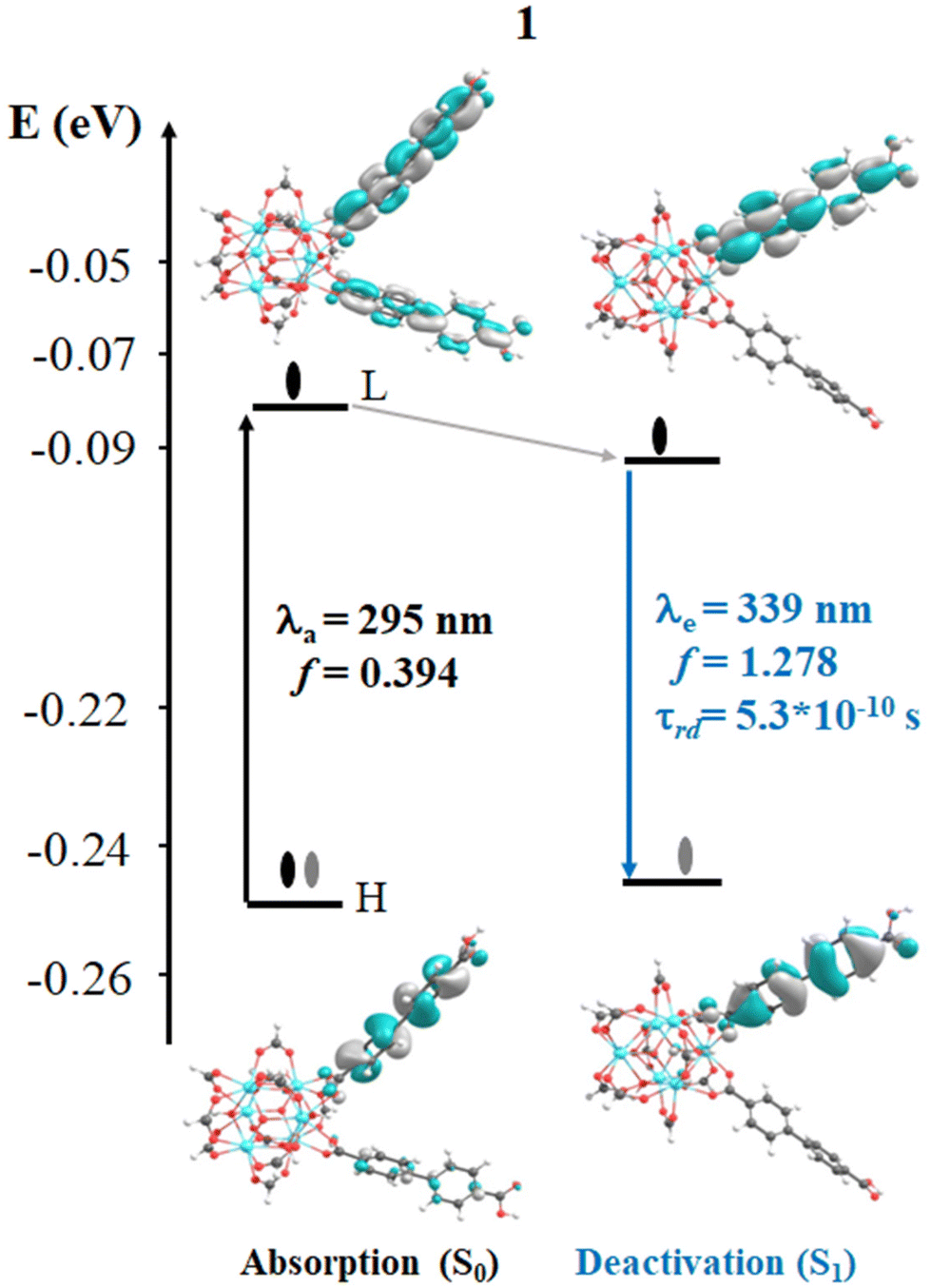 | ||
| Fig. 4 Schematic representation of the photophysical mechanism based on FMOs analysis on the S0 (absorption) and S1 states to 1 system. Where, λa and λe are the computed wavelength of absorption (black) and deactivation (blue), respectively; f is the oscillator strength; τrad is the radiative transition lifetime. Isosurface contour values are set as 0.03 with TDDFT/Def2-TZPP/B3LYP theory level. | ||
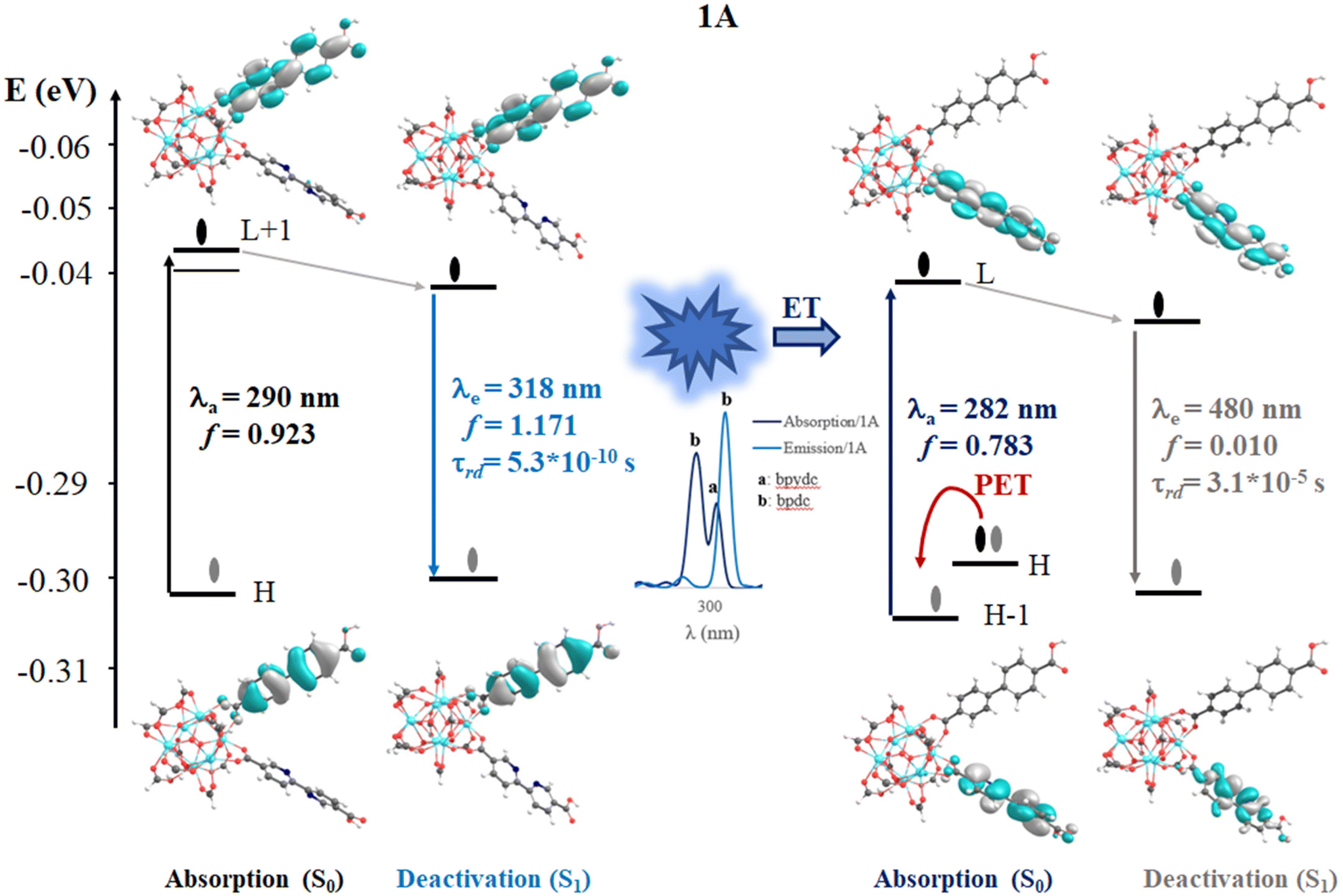 | ||
| Fig. 5 Schematic representation of the photophysical mechanism based on FMOs analysis on the S0 (absorption) and S1 states to 1A system. Where, λa and λe are the computed wavelength of absorption (black and dark blue) and deactivation (light blue and gray), respectively; f is the oscillator strength; τrad is the radiative transition lifetime and ET is energy transfer. The spectral overlap (emission bpdc/absorption at bpydc) is represented. Isosurface contour values are set as 0.03 with TDDFT/Def2-TZPP/B3LYP theory level. | ||
These theoretical results agree with the experimental reports11 in terms of energy, intensity, and the ET between bpdc and bpydc linkers. The difference in energy, between the computed and experimental values, is within the error range commonly reported for TD-DFT.19,83,84 These results suggest that the theoretical tools used to understand the optical properties in these MOFs-based systems are reasonable. Fig. 4 and 5 display the photophysical mechanism proposed for 1 and 1A.
Isoreticular systems
The computed vertical transitions for the S0 state show that the most intense singlet → singlet absorption transitions (see Table S2 in ESI†) correspond to an n → π* transition localized on the bpdc-R (R: –SH and –NH2) linkers (see MOs isosurface Fig. 6 and S4 in ESI†). The computed transitions (358 nm, 357 nm, 362 nm, and 378 nm for 1A-bpdc-NH2, 1A-bpdc-SH, 1A-NH2, and 1A-SH, respectively) are localized over the bpdc-R linker with a large contribution of the auxochrome groups. These wavelengths have a larger value than the observed for 1A and show an increase as the donor character of the R group gets bigger (the –SH shows the larger value). These changes in the absorption band energies are given by the stabilization of the molecular orbitals localized on the bpdc linker and the substituent groups (–NH2 and –SH) (see Table S2† and Fig. 6 and 7). This behavior suggests that these groups can cause a broadening in the light-harvesting capability of the studied MOF (∼70 and 130 nm for 1A-NH2, and 1A-SH, respectively). Although these results are promising for visible light irradiation applications, the photocatalytic properties of the MOFs also depend on the deactivation processes. In this sense, the analysis of the emission spectra is also essential to understanding the optical properties of these systems.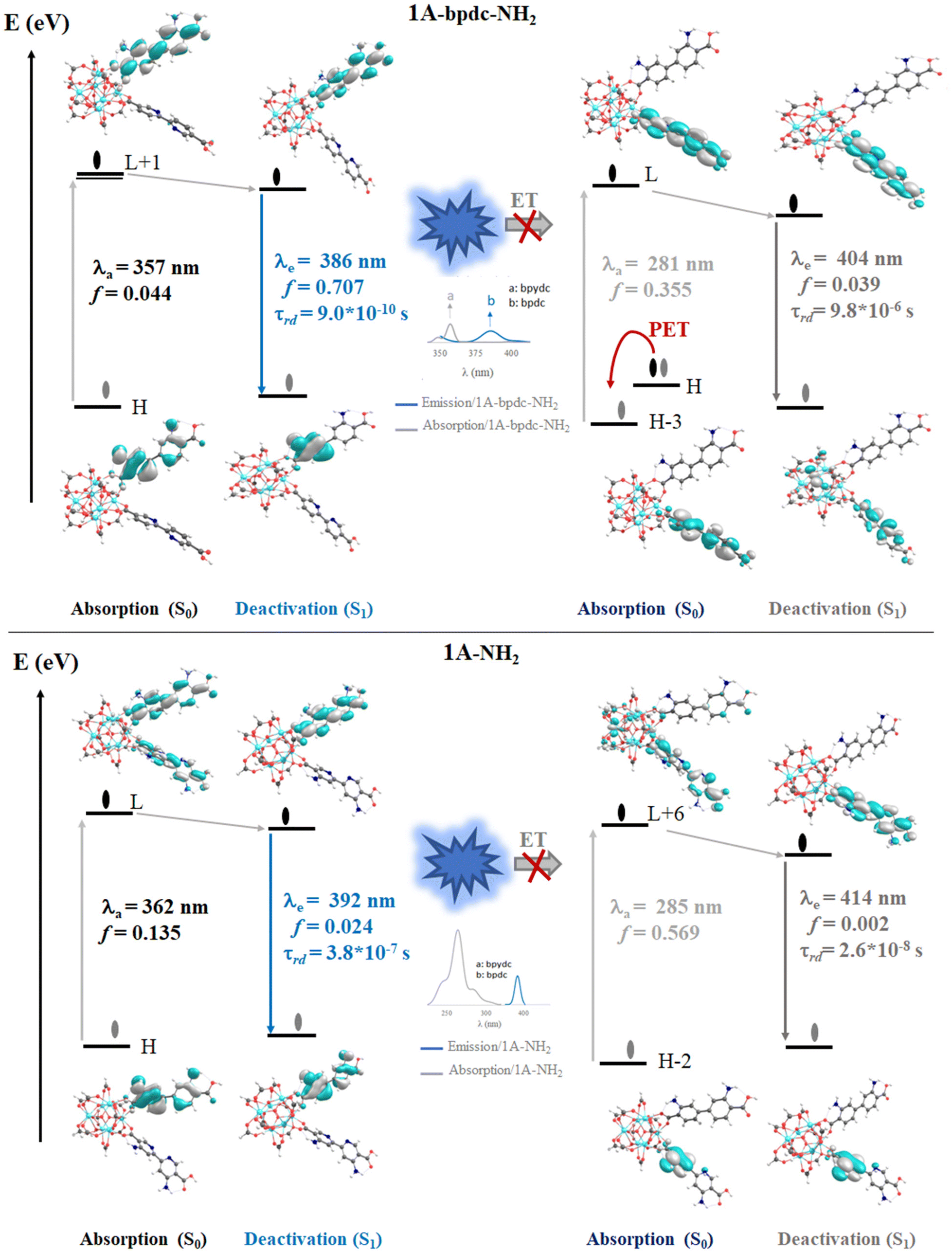 | ||
| Fig. 6 Schematic representation of the photophysical mechanism based on FMOs analysis on the S0 (absorption) and S1 states to 1A-bpdc-NH2 and 1A-NH2 systems. Where, λa and λe are the computed wavelength of absorption (gray) and deactivation (orange and dark gray), respectively; f is the oscillator strength; τrad is the radiative transition lifetime and ET is energy transfer. The spectral overlap (emission bpdc/absorption at bpydc) is represented. Isosurface contour values are set as 0.03 with TDDFT/Def2-TZPP/B3LYP theory level. | ||
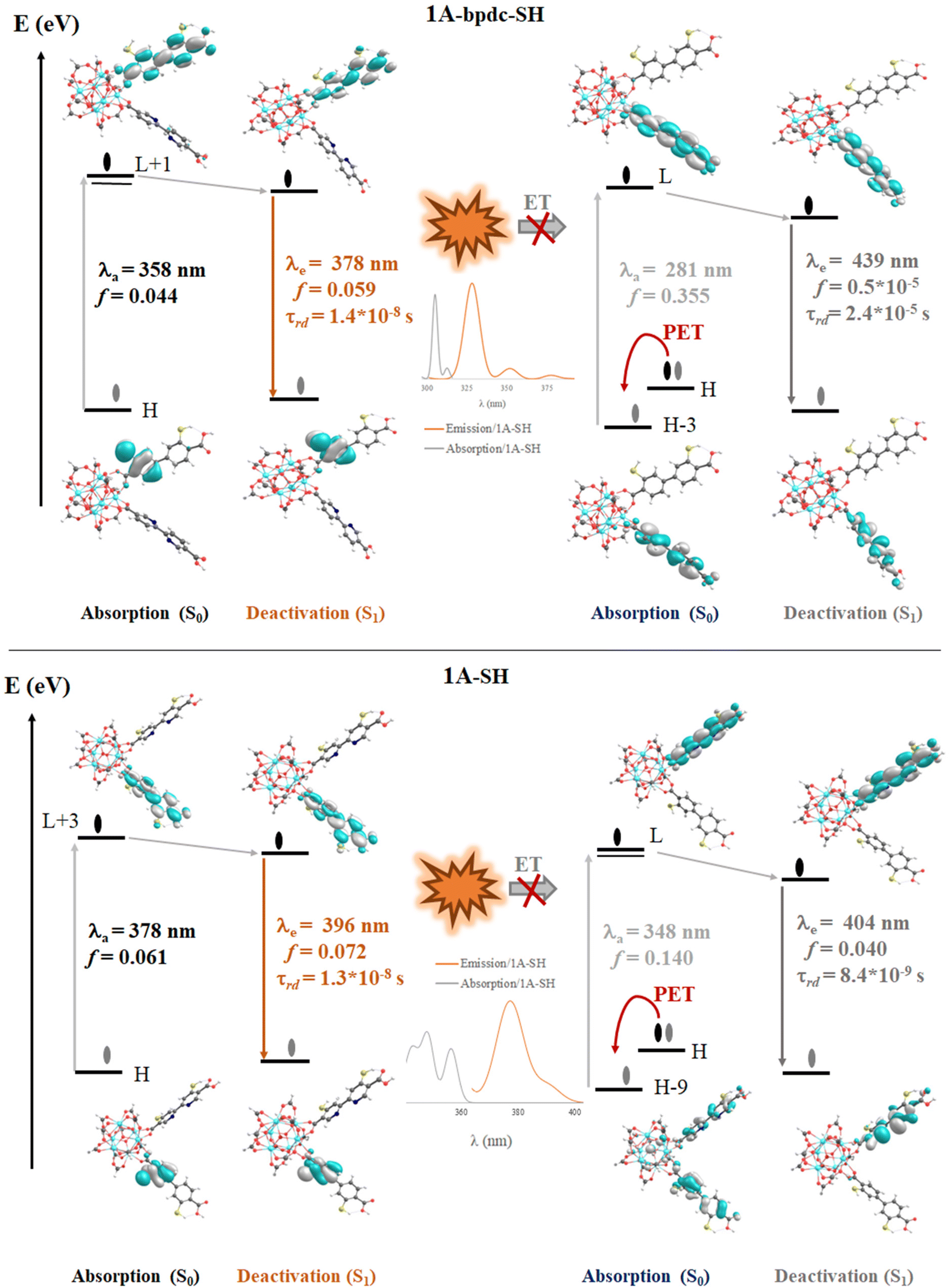 | ||
| Fig. 7 Schematic representation of the photophysical mechanism based on FMOs analysis on the S0 (absorption) and S1 states to 1A-bpdc-SH and 1A-SH system. Where, λa and λe are the computed wavelength of absorption (gray) and deactivation (orange and dark gray), respectively; f is the oscillator strength; τrad is the radiative transition lifetime and ET is energy transfer. The spectral overlap (emission bpdc/absorption at bpydc) is represented. Isosurface contour values are set as 0.03 with TDDFT/Def2-TZPP/B3LYP theory level. | ||
The emission spectra were computed from the optimized structure on the S1 and T1 states (see Tables S2–S4 in ESI†) via TDDFT methods. In the case of the isoreticular 1A-bpdc-R and 1A-R (R: –SH and –NH2) systems, the radiative decays (from S1) were computed at ∼380 nm and ∼400 nm (with B3LYP functional). The orbitals involved in the deactivation process for these MOFs are localized over bpdc-R and bpydc-R, respectively, with a π* → n character for both cases (see Fig. 6 and 7). The deactivation process that involves orbitals from bpdc-R is less intense than those for 1 and 1A (see Table S2†), which can be associated with an increase in the π* → n character of these transitions. Contrary, the deactivation processes involving the bpydc-R linker's orbitals result in a higher “emission” intensity relative to 1 and 1A. These results suggest the ET between the linkers displayed by 1A cannot take place in the same magnitude. The computed spectra confirm this theory, as the spectral overlap (between bpdc and bpydc linker) is not observed for 1A-bpdc-R and 1A-R (see Fig. 6 and 7), whereas this overlap was observed for 1A. These results suggest that the emission generated by bpdc-R cannot be absorbed by the bpydc-R, inhibiting the ET and therefore, producing a deactivation with higher emission intensity from the bpydc-R linker. For these systems, the computed radiative deactivation processes also show a τrad within the value range of fluorescence, lower than 10−6 s (see Tables S2–S4†). These results and the experimental evidence suggest that from the S1 states a radiative deactivation process dominates.80–82
Excited states deactivation mechanisms via CASSCF/NEVPT2 methods
The theoretical and experimental results suggest that the ET followed by the PET process can cause a low emission intensity for 1A. However, the thermodynamic possibility of the ET and PET processes is not sufficient to conclude that this emission takes place.61,62,85 In this sense, the Multireference Complete Active Space Self-Consistent Field (CASSCF)86,87 combined with second-order perturbation theory (NEVPT2)87 is a powerful computational method used to determine the existence of a state with the electronic configuration and energy position that validates the ET process. Despite that the fragmentation model used for TD-DFT calculation allows to reproduce the optical properties in good agreement with the experimental results, this model is still too big to perform multiconfigurational calculations. Considering that the most intense absorption transitions and excited state deactivation of 1A are centered in the linker, the fragmentation scheme proposed by Beltrán-Leiva M. J. et al. reported for lanthanide complexes88 and by our group, in a luminescent MOF-based systems9 was applied. In the experimental report, the authors studied the free linkers to validate that the emission process is centered on the bpydc.11 Hence, we used TD-DFT to reproduce this study for bpdc and bpydc linkers and to validate the fragmentation scheme proposed to compute the multiconfigurational calculations. The structural analysis displays the same change for the selected dihedral angles and bond distances in the S0, S1, and T1 states were then observed for 1A (see Table S5 and Fig. S5 in ESI†). These optimized structures do not display a large difference between S0 and S1 states. Similar to 1A, the dihedral angles γ(C4–C5–C6–C7) in the bpdc linker and γ(N1–C4–C5–N2) in the bpydc linker show an increase of planarity (between the aromatic rings) in the excited states (see Table S5 and Fig. S1 in ESI†). On the other hand, Tables S5 and S7 in ESI† show the most intense singlet → singlet absorption transitions and emission data, respectively, for the free bpdc and bpydc linkers (see Fig. S6†). These models display the same absorption transition and emission deactivation in terms the energy and intensity (based on the f value) as observed for 1A. Furthermore, the same thermodynamical possibility of PET for bpydc linker and the spectral overlap between emission (from bpdc) and absorption (bpydc) (which favor the ET mechanism) are reproduced in the free linkers (see Fig. S6 and Table S9†). These results also show, as a previous experimental report,11 that the optical properties of 1A agree with the free linkers in terms of the energy and intensity of transitions and, therefore, the most intense absorption transitions and excited state deactivation are localized over the linkers.The bpdc-R and bpydc-R were also studied to validate the proposed fragmentation schema to compute the multiconfigurational calculations. The optimized structures do not display a large difference between S0 and S1 states showing the same change to the selected dihedral angles and bond distances as observed for the MOFs model (see Table S5 and Fig. S7†). In the case of the computed transitions, the free linkers display the equivalent absorption transition and emission deactivation in terms of energy and intensity (based on f value) than observed for 1A-bpdc-R and 1A-R. In addition, the same thermodynamic possibility of PET to bpydc linker and the fact of the ET mechanism between the bpdc-R and bpydc-R are observed (see Fig. S8 and S9 in ESI†). Based on the experimental and theoretical evidence, we separated the models into two fragments. Those two fragments consist of the two linkers with unmodified structures obtained from the MOF models. This reduced model study was performed to accomplish the multiconfigurational calculations to determine the existence of a state with the electronic configuration and energy position that validate the ET between these linkers.
The CASSCF/NEVPT2 method is employed to elucidating the electronic states involved in the photophysical processes of 1A and 1A-SH. These systems present ligand-centered emission, which is characteristic of the MOFs based on closed-shell metal ions. Considering that the origin of the optical properties (absorption and emission) involves the photophysical process localized on the linker, the following procedure was carried out. The linker bpdc, bpydc, bpdc-R, and bpydc-R was cut from the system, i.e.1A and 1A-SH (optimized ground electronic state), and this unmodified geometry was used as starting geometries for the CASSCF/NEVPT2 calculations (see Table S9 in ESI†). The CAS(8,8)SCF/NEVPT2 calculations predict the S1 electronic state of the bpdc linker at 31085 cm−1, whereas the T1 electronic state appears at 28242 cm−1. Therefore, the T1 electronic state is localized at 2843 cm−1 (0.35 eV) lower than the S1 electronic state. For the bpydc linker, the S1 electronic state appears at 27205 cm−1 which is close to the position of the T1 electronic state (26867 cm−1). Based on the linker's singlet and triplet energy states in 1A, it is considered that an ET from bpdc (donor) to bpydc (acceptor) is possible (See Fig. 8). The energy of the S1 electronic state in the bpdc linker is located at 3880 cm−1 (0.48 eV) above the S1 electronic state in the bpydc linker. The energy gap between a T1 state of bpdc and the T1 state of bpydc is 1375 cm−1 (0.17 eV). Thus, the bpdc linkers can favor the population of the bpydc linker's S1 states, via its high energy excited electronic states (S1 and T1). It should be emphasized that besides the energy gap, the spin–orbit coupling (SOC) provokes a combination of states with different multiplicities.89 Thus, a non-radiative mechanism might be followed to fill the luminescent state of the 1A system. To sum up, these findings corroborate the suppression of 1A emission experimentally observed, resulting from an ET from the bpdc linker to the bpydc linker, also agreeing with our results derived from the DFT approach. On the other hand, the S1 and T1 states of bpdc-SH appear at 6671 and 6974 cm−1 lower than the corresponding first excited states (S1 and T1) of the bpydc-SH linker. Hence, the ET path from bpdc-SH to bpydc-SH is not allowed due to the –SH functional group inclusion, resulting in the turn-off of the emission. This result agrees with the TD-DFT calculations that predict a dark excited state (low emission intensity) for the 1A-SH, according to the predicted oscillator strength.
 | ||
| Fig. 8 Energy diagrams for the most probable energy transfer pathways in 1A. ISC, intersystem crossing. | ||
Morokuma–Ziegler energy decomposition analysis
The Energy decomposition analysis (EDA) was used to accomplish a description of the linkers and Zirconium (node) interactions. According to ΔEint, the metal-linker interaction is equivalent for bpdc-R and bpydc-R linkers (R: H and NH2). However, the ΔEint values decrease with the donor character increase of the –SH group (see Table S10 in ESI†). The ΔEElect represents ∼70% of the total stabilization energy (see Table S10†) which suggests that the electrostatic interaction (between –COO− group and Zr) plays the most important role, compared to the orbital interaction (∼30%). On the other hand, according to the contours of the NOCV deformation density (ρ), the charge flow in the interaction is established between the carboxylate group and the Zirconium atoms (node) (indicated by red to blue in Fig. 9). The charge flow from the bpydc-R is larger than in bpdc-R linkers, increasing the rings’ contribution (see ρ in Fig. 9). These results suggest that the donor character of the R group might increase the charge flow from bpydc linkers. This suggests an improvement in the photocatalytic reduction of a potential metal ion that can coordinate with this ligand (Re, Ce, Eu, Ru, Rh, Pt, or Pd11,47–50) (via LMCT). Consequently, the MOF might be photocatalytically active towards gases of interest (such as H2, N2, among others) with these potential photo-reduced metal ions.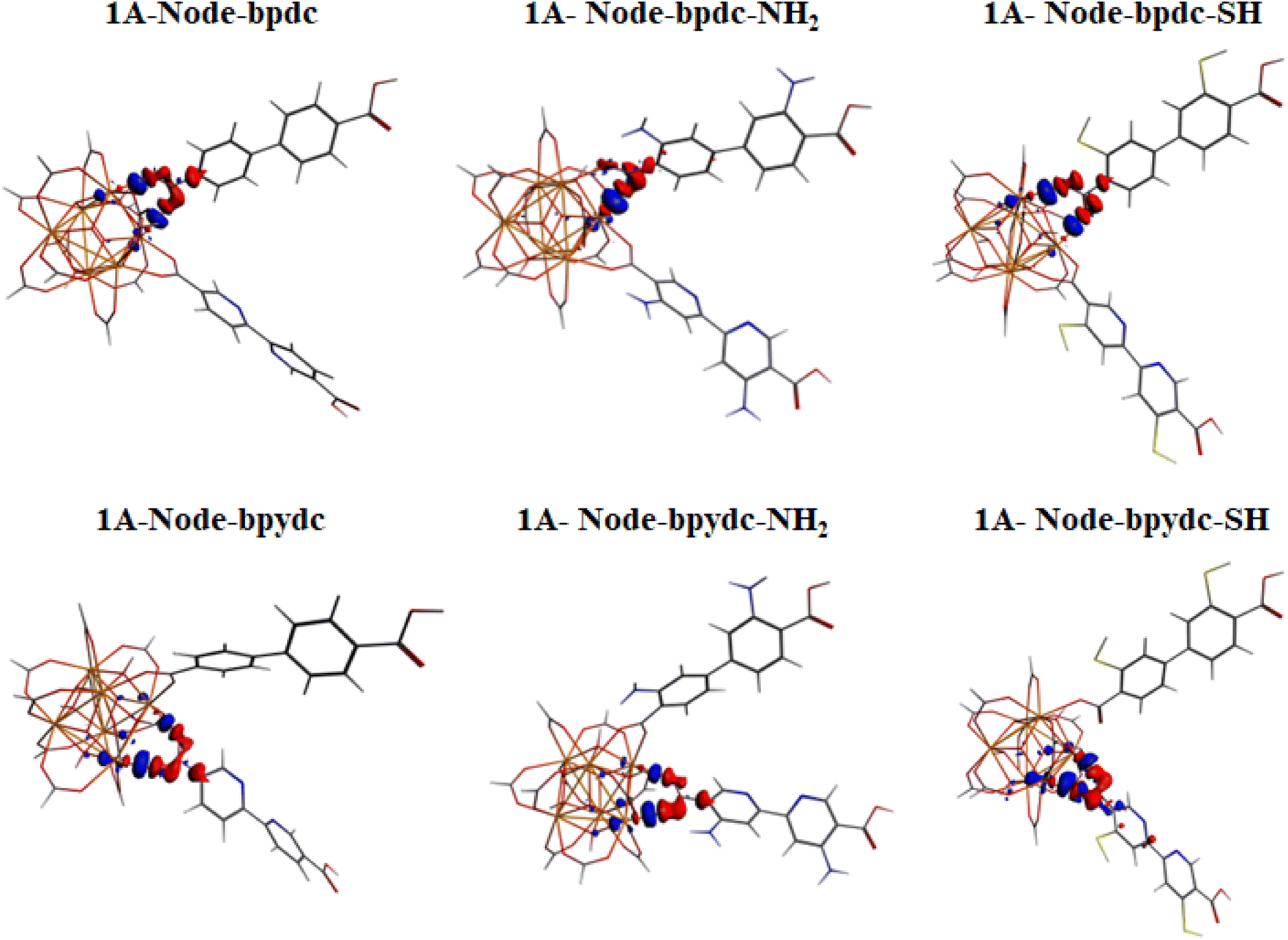 | ||
| Fig. 9 Contours of the NOCV deformation density (ρ). Isosurface contour values of the NOCV deformation density (ρ) are set as 0.002. | ||
Periodic modelling
Periodic calculations of 1 and 1A were performed including electronic band gap and projected densities of states (PDOS), which revealed information about the composition of the electron states. According to Table S11,† the band gap values of 1A show the minor value of band gap (3.31 eV), followed by the 1 (3.42 eV) and UiO-67-bpydc (3.54 eV). All structures showed a good band gap agreement with the experimental parameters (see Table S11 in ESI†). The PDOS by fragments (Fig. S10†) shows that linkers have a main role around the Fermi level, with states between −1.5 to −2 eV, and 1.5 to 2 eV for the conduction and valence states, respectively. The Zr6 node appears with an increased states population on ranges of −5 to −3 and 3 to 5, far of the Fermi level. Also, it can be noted a general improvement when the bpydc is incorporated into UiO-67, i.e. a slight band gap reduction, in agreement with optical properties and transition results described previously on TD-DFT analysis section, particularly increasing the number of states on the valence/conduction region when is compared against UiO-67 (particularly between −1.5 and 3 eV, for UiO-67-BPYDC). For 1A that have a mix of bpydc and bpdc, shows a slightly reduction on the band gap, with bpydc on the valence band and bpydc on the conduction band, showing that the mix is good enough to reduce the gap. However, these values would be affected by the substituent(s) groups and position, homogeneous distribution of bpydc and bpdc of linkers, among other factors. The PDOS by elements (Fig. S12†) show that carbon atoms with a larger number of occupied and unoccupied states are placed near to Fermi level, followed by nitrogen. Both atom types at same energy levels for 1A and UiO-67-bpdc. While the effect of zirconium and hydrogen atoms are not relevant regarding the levels around the Fermi level, the largest population given by carbon atoms, relates that the aromatic states plays a main role on adsorption and emission processes. So, it can be expected that incorporating substituents in the bpdc and bpydc linkers positions has a relevant effect on the optic properties of the materials.Conclusions
The computational protocol used in this work allowed us to appreciate the optical properties and substituent effects (R: –NH2 and –SH) of four isoreticular Zr-based MOF (UiO-67) with the bpdc and bpydc as the linkers (1A). This protocol was able to reproduce the optical properties of the reported 1A. These results suggest that the emission quenching of 1A occurs by a new ET between bpdc and bpydc linkers following the PET process (from lone pair of the nitrogen atoms (localized in HOMO) to partially occupied H-1). This electronic rearrangement generates a luminescence quenching in 1A. On the other hand, the substitutions in the linkers (with R: –NH2 and –SH) can generate a broadened light-harvesting of the 1A which increases as the donor character of the R group increases. These changes in the absorption band energies are given by the stabilization of the molecular orbital localized on the bpdc linker with the substituents –NH2 and –SH. The deactivations from bpdc-R linkers are less intense than 1 and 1A due to an increase in the π* → n character while the bpydc-R linker displays an increase in the intensity relative to 1 and 1A. These results suggest that the new isoreticular systems could have application under ambient conditions. However, the ET exhibited by 1A, cannot take place in the same magnitude in the proposed systems (1A-bpdc-R and 1A-R) which was confirmed via CASSCF calculation. According to EDA and the NOCV deformation density, the metal-linker interaction is comparable for all studied systems suggesting that the electrostatic interaction plays the most important role, compared to the other terms of the interaction energy. The charge flow in the interaction is established from the carboxylate group to Zr atoms (node) increasing with the donor character of the R group which might improve photocatalytic reduction if catalytic-metal ions coordinate at the bpydc ligands (via LMCT). Consequently, the photocatalytic activation of the interest in these potential photo-reduced metal ions. These theoretical results serve as a useful guide in the experimental design of new photocatalytic systems based on MOFs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by ANID, FONDECYT/Postdoctoral ANID 3210271 and 3230141, ANID/FONDAP/1523A0006; FONDECYT 1241917, FONDECYT 1231194 and the Anillos de Ciencia y Tecnología ACT210057. This research was enabled in part by support provided by Graham (https://docs.alliancecan.ca/wiki/Graham) and the Digital Research Alliance of Canada (alliancecan.ca).References
- S. R. Batten, N. R. Champness, X. M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O’Keeffe, M. P. Suh and J. Reedijk, Terminology of metal–organic frameworks and coordination polymers (IUPAC Recommendations), Pure Appl. Chem., 2013, 85(8), 1715–1724 CrossRef CAS.
- O. Ola and M. M. Maroto-Valer, Review of material design and reactor engineering on TiO2 photocatalysis for CO2 reduction, J. Photochem. Photobiol., C, 2015, 24, 16–42 CrossRef CAS.
- Y. Zhang, J. Guo, L. Shi, Y. Zhu, K. Hou, Y. Zheng and Z. Tang, Tunable chiral metal organic frameworks toward visible light-driven asymmetric catalysis, Sci. Adv., 2017, 3, 1–8 Search PubMed.
- S. Zhang, L. Li, S. Zhao, Z. Sun, M. Hong and J. Luo, Hierarchical metal-organic framework nanoflowers for effective CO2 transformation driven by visible light, J. Mater. Chem. A, 2015, 3, 15764–15768 RSC.
- H. Bin Wu and X. W. Lou, Metal-organic frameworks and their derived materials for electrochemical energy storage and conversion: Promises and challenges, Sci. Adv., 2017, 3, 1–17 Search PubMed.
- A. Dhakshinamoorthy, Z. Li, S. Yang and H. Garcia, Metal–organic framework heterojunctions for photocatalysis, Chem. Soc. Rev., 2024, 53, 3002–3035 RSC.
- A. Dhakshinamoorthy, A. M. Asiri and H. García, Metal–Organic Frameworks as Multifunctional Solid Catalysts, Trends Chem., 2020, 2, 454–466 CrossRef CAS.
- A. Dhakshinamoorthy, S. Navalón, A. Primo and H. García, Selective Gas–Phase Hydrogenation of CO2 to Methanol Catalysed by Metal–Organic Frameworks, Angew. Chem., 2024, 136, e202311241 CrossRef.
- Y. Hidalgo-Rosa, M. A. Treto-Suárez, E. Schott, X. Zarate and D. Páez-Hernández, Sensing mechanism elucidation of a europium(III) metal organic framework selective to aniline: A theoretical insight by means of multiconfigurational calculations, J. Comput. Chem., 2020, 41, 1956–1964 CrossRef CAS PubMed.
- Y. Hidalgo-Rosa, M. A. Treto-Suárez, E. Schott, X. Zarate and D. Páez-Hernández, Sensing mechanism elucidation of a chemosensor based on a metal-organic framework selective to explosive aromatic compounds, Int. J. Quantum Chem., 2020, 120, 1–11 CrossRef.
- Y. An, Y. Liu, H. Bian, Z. Wang, P. Wang, Z. Zheng, Y. Dai, M. H. Whangbo and B. Huang, Improving the photocatalytic hydrogen evolution of UiO-67 by incorporating Ce4+-coordinated bipyridinedicarboxylate ligands, Sci. Bull., 2019, 64, 1502–1509 CrossRef CAS PubMed.
- G. Li, F. Li, J. Liu and C. Fan, Fe-based MOFs for photocatalytic N2 reduction: Key role of transition metal iron in nitrogen activation, J. Solid State Chem., 2020, 285, 121245 CrossRef CAS.
- J. G. Santaclara, M. A. Nasalevich, S. Castellanos, W. H. Evers, F. C. M. Spoor, K. Rock, L. D. A. Siebbeles, F. Kapteijn, F. Grozema, A. Houtepen, J. Gascon, J. Hunger and M. A. Van Der Veen, Organic Linker Defines the Excited-State Decay of Photocatalytic MIL-125(Ti)-Type Materials, ChemSusChem, 2016, 9, 388–395 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, A. M. Asiri and H. García, Metal–Organic Framework (MOF) Compounds: Photocatalysts for Redox Reactions and Solar Fuel Production, Angew. Chem., Int. Ed., 2016, 55, 5414–5445 CrossRef CAS PubMed.
- H. Huang, X. S. Wang, D. Philo, F. Ichihara, H. Song, Y. Li, D. Li, T. Qiu, S. Wang and J. Ye, Toward visible-light-assisted photocatalytic nitrogen fixation: A titanium metal organic framework with functionalized ligands, Appl. Catal., B, 2020, 267, 118686 CrossRef CAS.
- Z. H. Yan, M. H. Du, J. Liu, S. Jin, C. Wang, G. L. Zhuang, X. J. Kong, L. S. Long and L. S. Zheng, Photo-generated dinuclear {Eu(II)}2 active sites for selective CO2 reduction in a photosensitizing metal-organic framework, Nat. Commun., 2018, 9, 1–9 CrossRef PubMed.
- X. P. Wu, L. Gagliardi and D. G. Truhlar, Cerium Metal-Organic Framework for Photocatalysis, J. Am. Chem. Soc., 2018, 140, 7904–7912 CrossRef CAS PubMed.
- D. Escudero, Revising Intramolecular Photoinduced Electron Transfer (PET) from First Principles, Acc. Chem. Res., 2016, 49, 1816–1824 CrossRef CAS PubMed.
- M. A. Treto-Suárez, Y. Hidalgo-Rosa, E. Schott, X. Zarate and D. Páez-Hernández, Understanding the Selective-Sensing Mechanism of Al3+ Cation by a Chemical Sensor Based on Schiff Base: A Theoretical Approach, J. Phys. Chem. A, 2019, 123, 6970–6977 CrossRef PubMed.
- M. López-Espejel, A. Gómez-Treviño, B. M. Muñoz-Flores, M. A. Treto-Suarez, E. Schott, D. Páez-Hernández, X. Zarate and V. M. Jiménez-Pérez, Organotin Schiff bases as halofluorochromic dyes: green synthesis, chemio-photophysical characterization, DFT, and their fluorescent bioimaging in vitro, J. Mater. Chem. B, 2021, 9, 7698–7712 RSC.
- M. A. Treto-Suárez, Y. Hidalgo-Rosa, E. Schott, D. Páez-Hernández and X. Zarate, Fluorescence turn-on and turn-off mechanisms of a dual-selective chemosensor of Bi3+ and pH changes: Insights from a theoretical perspective, Dyes Pigm., 2021, 185, 108934 CrossRef.
- D. L. Dexter, A theory of sensitized luminescence in solids, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- M. Zhu, Y. Zhou, L. Yang, L. Li, D. Qi, M. Bai, Y. Chen, H. Du and Y. Bian, Synergistic coupling of fluorescent ‘turn-Off’ with spectral overlap modulated FRET for ratiometric Ag+ sensor, Inorg. Chem., 2014, 53, 12186–12190 CrossRef CAS PubMed.
- D. B. Syed Arshad Hussain, D. Dey, S. Chakraborty, J. Saha, A. D. Roy, S. Chakraborty and P. Debnath, Fluorescence Resonance Energy Transfer (FRET) sensor, J. Spectrosc. Dyn., 2015, 4, 1–13 Search PubMed.
- K. R. Leight, B. E. Esarey, A. E. Murray and J. J. Reczek, Predictable tuning of absorption properties in modular aromatic donor-acceptor liquid crystals, Chem. Mater., 2012, 24, 3318–3328 CrossRef CAS.
- A. Taherpour, M. Jamshidi, O. Rezaei and A. R. Belverdi, Photoinduced electron transfer process on emission spectrum of N,N′-bis(salicylidene)-1,2-phenylenediamine as a Mg2+ cation chemosensor: A first principle DFT and TDDFT study, J. Mol. Struct., 2018, 1161, 339–344 CrossRef CAS.
- J. Wang, A. S. Cherevan, C. Hannecart, S. Naghdi, S. P. Nandan, T. Gupta and D. Eder, Ti-based MOFs: New insights on the impact of ligand composition and hole scavengers on stability, charge separation and photocatalytic hydrogen evolution, Appl. Catal., B, 2021, 283, 119626 CrossRef CAS.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Catalysis and photocatalysis by metal organic frameworks, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- Y. Hidalgo-Rosa, J. Santoyo-Flores, M. A. Treto-Suárez, E. Schott, D. Páez-Hernández and X. Zarate, Tuning the sensitization pathway T1→ 5DJ in Eu-based MOF through modification of the antenna ligand. A theoretical approach via multiconfigurational quantum calculations, J. Lumin., 2023, 260, 119896 CrossRef CAS.
- Y. Hidalgo-Rosa, M. Saavedra-Torres, B. D. Koivisto, M. A. Treto-Suárez, D. Páez-Hernández, X. Zarate and E. Schott, Rare-earth-based metal–organic frameworks with improved visible-light-harvesting properties: a quantum chemistry study, J. Mater. Sci., 2023, 58, 8862–8877 CrossRef CAS.
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro, B. Ferrer and H. García, Metal–Organic Frameworks as Photocatalysts for Solar-Driven Overall Water Splitting, Chem. Rev., 2023, 123, 445–490 CrossRef PubMed.
- A. Hastings, D. Ray, W. Jeong, L. Gagliardi, O. K. Farha and A. Hixon, Advancement of Actinide Metal-Organic Framework Chemistry via Synthesis of Pu-UiO-66, J. Am. Chem. Soc., 2020, 142, 9363–9371 CrossRef CAS PubMed.
- M. R. Mian, L. R. Redfern, S. M. Pratik, D. Ray, J. Liu, K. B. Idrees, T. Islamoglu, L. Gagliardi and O. K. Farha, Precise Control of Cu Nanoparticle Size and Catalytic Activity through Pore Templating in Zr Metal–Organic Frameworks, Chem. Mater., 2020, 32, 3078–3086 CrossRef CAS.
- A. H. Vahabi, F. Norouzi, E. Sheibani and M. Rahimi-Nasrabadi, Functionalized Zr-UiO-67 metal-organic frameworks: Structural landscape and application, Coord. Chem. Rev., 2021, 445, 214050 CrossRef CAS.
- S. Yang, B. Pattengale, S. Lee and J. Huang, Real-Time Visualization of Active Species in a Single-Site Metal–Organic Framework Photocatalyst, ACS Energy Lett., 2018, 3, 532–539 CrossRef CAS.
- K. M. Batoo, E. Ali, S. A. Hussein, S. Chandra, A. S. Abdulwahid, S. H. Kareem, M. F. Ijaz, A. A. Omran, M. K. Abid and A. Alawadi, UiO-67 metal-organic frameworks with dual amino/iodo functionalization, and mixed Zr/Ce clusters: Highly selective and efficient photocatalyst for CO2 transformation to methanol, J. Mol. Struct., 2024, 1312, 138492 CrossRef CAS.
- K. M. Choi, D. Kim, B. Rungtaweevoranit, C. A. Trickett, J. T. D. Barmanbek, A. S. Alshammari, P. Yang and O. M. Yaghi, Plasmon-Enhanced Photocatalytic CO2 Conversion within Metal–Organic Frameworks under Visible Light, J. Am. Chem. Soc., 2017, 139, 356–362 CrossRef CAS PubMed.
- Y. Zhang, F. Mao, L. Wang, H. Yuan, P. F. Liu and H. G. Yang, Recent Advances in Photocatalysis over Metal–Organic Frameworks–Based Materials, Sol. RRL, 2020, 4, 1900438 CrossRef CAS.
- D. Yang, S. O. Odoh, J. Borycz, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, Tuning Zr6 Metal-Organic Framework (MOF) Nodes as Catalyst Supports: Site Densities and Electron-Donor Properties Influence Molecular Iridium Complexes as Ethylene Conversion Catalysts, ACS Catal., 2016, 6, 235–247 CrossRef CAS.
- I. Garagounis, A. Vourros, D. Stoukides, D. Dasopoulos and M. Stoukides, Electrochemical synthesis of ammonia: Recent efforts and future outlook, Membranes, 2019, 9, 1–17 CrossRef PubMed.
- C. Huang, J. Dong, W. Sun, Z. Xue, J. Ma, L. Zheng, C. Liu, X. Li, K. Zhou, X. Qiao, Q. Song, W. Ma, L. Zhang, Z. Lin and T. Wang, Coordination mode engineering in stacked-nanosheet metal–organic frameworks to enhance catalytic reactivity and structural robustness, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed.
- X. Y. Xie, P. Xiao, W. H. Fang, G. Cui and W. Thiel, Probing Photocatalytic Nitrogen Reduction to Ammonia with Water on the Rutile TiO2 (110) Surface by First-Principles Calculations, ACS Catal., 2019, 9, 9178–9187 CrossRef CAS.
- V. Bernales, M. A. Ortuño, D. G. Truhlar, C. J. Cramer and L. Gagliardi, Computational design of functionalized metal-organic framework nodes for catalysis, ACS Cent. Sci., 2018, 4, 5–19 CrossRef CAS PubMed.
- M. A. Treto-Suárez, Y. Hidalgo-Rosa, K. Mena Ulecia, D. Paez-Hernández, B. D. Koivisto, X. Zarate and E. Schott, Tunning the optical properties of a photocatalytic metal–organic framework by means of molecular modelling, New J. Chem., 2023, 47, 3430–3444 RSC.
- M. J. Beltrán-Leiva, E. Solis-Céspedes and D. Páez-Hernández, The role of the excited state dynamic of the antenna ligand in the lanthanide sensitization mechanism, Dalton Trans., 2020, 49, 7444–7450 RSC.
- S. R. Zhang, D. Y. Du, J. S. Qin, S. J. Bao, S. L. Li, W. W. He, Y. Q. Lan, P. Shen and Z. M. Su, A Microporous Anionic Metal–Organic Framework for Sensing Luminescence of Lanthanide(III) Ions and Selective Absorption of Dyes by Ionic Exchange, Chem. – Eur. J., 2014, 20, 3589 CrossRef CAS PubMed.
- X. Deng, J. Albero, L. Xu, H. García and Z. Li, Construction of a Stable Ru–Re Hybrid System Based on Multifunctional MOF-253 for Efficient Photocatalytic CO2 Reduction, Inorg. Chem., 2018, 57, 8276–8286 CrossRef CAS PubMed.
- M. F. M. B. Chambers, X. Wang, N. Elgrishi, C. H. Hendon, A. Walsh, J. Bonnefoy, J. Canivet, E. A. Quadrelli, D. Farrusseng and C. Mellot-Draznieks, Photocatalytic Carbon Dioxide Reduction with Rhodium-based Catalysts in Solution and Heterogenized within Metal–Organic Frameworks, ChemSusChem, 2015, 8, 603–608 CrossRef CAS PubMed.
- B. Yang, X. P. Wu, L. Gagliardi and D. G. Truhlar, Methane functionalization by an Ir(III) catalyst supported on a metal–organic framework: an alternative explanation of steric confinement effects, Theor. Chem. Acc., 2019, 138, 1–11 Search PubMed.
- J. Zhu, Y. Tang, Y. Yang, B. Li, Y. Cui and G. Qian, Post-modified metal-organic framework as a turn-on fluorescent probe for potential diagnosis of neurological diseases, Microporous Mesoporous Mater., 2019, 288, 109610 CrossRef CAS.
- K. M. Choi, D. Kim, B. Rungtaweevoranit, C. A. Trickett, J. T. D. Barmanbek, A. S. Alshammari, P. Yang and O. M. Yaghi, Plasmon-Enhanced Photocatalytic CO 2 Conversion within Metal–Organic Frameworks under Visible Light, J. Am. Chem. Soc., 2017, 139, 356–362 CrossRef CAS PubMed.
- E. S. Gutterød, S. Øien-Ødegaard, K. Bossers, A.-E. Nieuwelink, M. Manzoli, L. Braglia, A. Lazzarini, E. Borfecchia, S. Ahmadigoltapeh, B. Bouchevreau, B. T. Lønstad-Bleken, R. Henry, C. Lamberti, S. Bordiga, B. M. Weckhuysen, K. P. Lillerud and U. Olsbye, CO 2 Hydrogenation over Pt-Containing UiO-67 Zr-MOFs—The Base Case, Ind. Eng. Chem. Res., 2017, 56, 13206–13218 CrossRef.
- R. Mori, CO2 photocatalytic reduction with robust and stable metal–organic framework: a review, Mater. Renewable Sustainable Energy, 2024, 13, 109–132 CrossRef.
- D. Sun, Y. Fu, W. Liu, L. Ye, D. Wang, L. Yang, X. Fu and Z. Li, Studies on photocatalytic CO2 reduction over NH2-Uio-66 (Zr) and its derivatives: towards a better understanding of photocatalysis on metal- organic frameworks, Chem. – Eur. J., 2013, 19, 14279–14285 CrossRef CAS PubMed.
- B. Guo, X. Cheng, Y. Tang, W. Guo, S. Deng, L. Wu and X. Fu, Dehydrated UiO–66(SH) 2: The Zr–O Cluster and Its Photocatalytic Role Mimicking the Biological Nitrogen Fixation, Angew. Chem., Int. Ed., 2022, 61, e202117244 CrossRef CAS PubMed.
- Y. Hidalgo-Rosa, K. Mena-Ulecia, M. A. Treto-Suárez, E. Schott, D. Páez-Hernández and X. Zarate, Insights into the selective sensing mechanism of a luminescent Cd(II)-based MOF chemosensor toward NACs: roles of the host–guest interactions and PET processes, J. Mater. Sci., 2021, 56, 13684–13704 CrossRef CAS.
- S. Chavan, J. G. Vitillo, D. Gianolio, O. Zavorotynska, B. Civalleri, S. Jakobsen, M. H. Nilsen, L. Valenzano, C. Lamberti, K. P. Lillerud and S. Bordiga, H 2 storage in isostructural UiO-67 and UiO-66 MOFs, Phys. Chem. Chem. Phys., 2012, 14, 1614–1626 RSC.
- D. A. Deming, M. J. Hurlock, X. Li, K. W. Kriegsman, G. Ding, X. Guo and Q. Zhang, A facile method to introduce iron secondary metal centers into metal–organic frameworks, J. Organomet. Chem., 2019, 897, 114–119 CrossRef CAS.
- R. Hoffman and D. P. Mathies, Photoexcited structural dynamics of an azobenzene analog 4-nitro-4′-dimethylamino-azobenzene from femtosecond stimulated Raman, Phys. Chem. Chem. Phys., 2012, 14, 6298 RSC.
- M. Kasha, Characterization of Electronic Transitions in Complex Molecules, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- E. Briggs and N. Besley, Density Functional Theory Based Analysis of Photoinduced Electron Transfer in a Triazacryptand Based K\n + \n Sensor, J. Phys. Chem. A, 2015, 119, 2902–2907 CrossRef CAS PubMed.
- M. A. Treto-Suárez, Y. Hidalgo-Rosa, E. Schott, D. Páez-Hernández and X. Zarate, Fluorescence Turn-On and Turn-Off Mechanisms of a Dual-Selective Chemosensor of Bi3 + and pH Changes: Insights from a Theoretical Perspective, Dyes Pigm., 2020, 185, 108934 CrossRef.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- A. D. Ecke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- N. Klinhom, N. Saengsuwan, S. Sriyab, P. Prompinit, S. Hannongbua and S. Suramitr, Photophysical properties for excited-state intramolecular proton transfer (ESIPT) reaction of N-salicylidene-o-aminophenol: Experimental and DFT based approaches, Spectrochim. Acta, Part A, 2019, 206, 359–366 CrossRef CAS PubMed.
- M. A. Treto-Suarez, J. Tapia, Y. Hidalgo-Rosa, D. Paez-Hernandez, E. Molins, X. Zarate and E. Enrique Schott, New Sensitive and Selective Chemical Sensors for Ni2+ and Cu2+ Ions, Sensing Mechanism Insights through DFT Methods, J. Phys. Chem. A, 2020, 124, 6493–6503 CrossRef CAS PubMed.
- R. A. Marcus, Electron Transfer Reactions in Chemistry: Theory and Experiment (Nobel Lecture), Angew. Chem., Int. Ed. Engl., 1993, 32, 1111–1121 CrossRef.
- B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, A complete active space SCF method- (CASSCF) using a density matrix: formulated super-CI approach, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- F. Neese, Software update: The ORCA program system—Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- E. J. Baerends, T. Ziegler, J. Autschbach, D. B. A. Bashford, F. M. Bickelhaupt, C. Bo, P. M. Boerrigter, L. Cavallo and D. P. Chong, ADF 2020, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2020, https://www.Scm.Com.
- M. P. Mitoraj, Bonding in ammonia borane: An analysis based on the natural orbitals for chemical valence and the extended transition state method (ETS-NOCV), J. Phys. Chem. A, 2011, 115, 14708–14716 CrossRef CAS PubMed.
- A. Carreño, E. Solís-Céspedes, D. Páez-Hernández and R. Arratia-Pérez, Exploring the geometrical and optical properties of neutral rhenium(I) tricarbonyl complex of 1,10-phenanthroline-5,6-diol using relativistic methods, Chem. Phys. Lett., 2017, 685, 354–362 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. J. van Setten, M. Giantomassi, E. Bousquet, M. J. Verstraete, D. R. Hamann, X. Gonze and G.-M. Rignanese, The PseudoDojo: Training and grading a 85 element optimized norm-conserving pseudopotential table, Comput. Phys. Commun., 2018, 226, 39–54 CrossRef CAS.
- S. Smidstrup, T. Markussen, P. Vancraeyveld, J. Wellendorff, J. Schneider, T. Gunst, B. Verstichel, D. Stradi, P. A. Khomyakov, U. G. Vej-Hansen, M.-E. Lee, S. T. Chill, F. Rasmussen, G. Penazzi, F. Corsetti, A. Ojanperä, K. Jensen, M. L. N. Palsgaard, U. Martinez, A. Blom, M. Brandbyge and K. Stokbro, QuantumATK: an integrated platform of electronic and atomic-scale modelling tools, J. Phys.: Condens. Matter, 2020, 32, 015901 CrossRef CAS PubMed.
- F. Momma and K. Izumi, VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data, Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef.
- L. Yan, Y. Zhou, W. Du, Z. Kong and Z. Qi, A new turn on coumarin-based fluorescence probe for Ga3 +detection in aqueous solution, Spectrochim. Acta, Part A, 2016, 155, 116–124 CrossRef CAS PubMed.
- J. Hoche, A. Schulz, L. M. Dietrich, A. Humeniuk, M. Stolte, D. Schmidt, T. Brixner, F. Würthner and R. Mitric, The origin of the solvent dependence of fluorescence quantum yields in dipolar merocyanine dyes, Chem. Sci., 2019, 10, 11013–11022 RSC.
- L. Jiao, C. Yu, J. Wang, E. A. Briggs, N. A. Besley, D. Robinson, M. J. Ruedas-Rama, A. Orte, L. Crovetto, E. M. Talavera, J. M. Alvarez-Pez, M. Van der Auweraer and N. Boens, Unusual spectroscopic and photophysical properties of meso-tert-butylBODIPY in comparison to related alkylated BODIPY dyes, RSC Adv., 2015, 5, 89375–89388 RSC.
- R. Ramakrishnan, M. Hartmann, E. Tapavicza and O. A. Von Lilienfeld, Electronic spectra from TDDFT and machine learning in chemical space, J. Chem. Phys., 2015, 143, 084111 CrossRef PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, Rationale for mixing exact exchange with density functional approximations, J. Chem. Phys., 1996, 109, 9982–9985 CrossRef.
- B. O. Roos, P. R. Taylor and P. R. Sigbahn, A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- B. O. Malmqvist and P. Roos, The CASSCF state interaction method., Chem. Phys. Lett., 1989, 155, 189–194 CrossRef.
- J. P. M. C. Angeli, R. Cimiraglia, S. Evangelisti and T. Leininger, Introduction of n-electron valence states for multireference perturbation theory, J. Chem. Phys., 2001, 114, 10252 CrossRef.
- M. J. Beltrán-Leiva, P. Cantero-López, C. Zúñiga, A. Bulhões-Figueira, D. Páez-Hernández and R. Arratia, Theoretical Method for an Accurate Elucidation of Energy Transfer Pathways in Europium(III) Complexes with Dipyridophenazine (dppz) Ligand: One More Step in the Study of the Molecular Antenna Effect, Inorg. Chem., 2017, 56, 9200 CrossRef PubMed.
- Y. Zhang, T. S. Lee, J. L. Petersen and C. Milsmann, A Zirconium Photosensitizer with a Long-Lived Excited State: Mechanistic Insight into Photoinduced Single-Electron Transfer, J. Am. Chem. Soc., 2018, 140, 5934–5947 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt01017e |
| This journal is © The Royal Society of Chemistry 2024 |