
Two-dimensional conductive metal–organic frameworks as efficient electrocatalysts for oxygen evolution and reduction reactions†
Yanan
Zhou
a,
Li
Sheng
b,
Lanlan
Chen
c,
Qiquan
Luo
 d,
Wenhui
Zhao
*e,
Wenhua
Zhang
*c and
Jinlong
Yang
*f
d,
Wenhui
Zhao
*e,
Wenhua
Zhang
*c and
Jinlong
Yang
*f
aSchool of Material Science and Chemical Engineering, Institute of Mass Spectrometry, Ningbo University, Fenghua Road 818, Ningbo 315211, China
bDepartment of Chemical Physics, University of Science and Technology of China, Hefei, Anhui 230026, China
cDepartment of Material Science and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: whhzhang@ustc.edu.cn
dInstitutes of Physical Science and Information Technology, Anhui University, Hefei 230601, China
eDepartment of Physics, Ningbo University, Fenghua Road 818, Ningbo 315211, China. E-mail: zhaowenhui@nbu.edu.cn
fHefei National Laboratory for Physical Sciences at the Microscale, CAS Key Laboratory of Materials for Energy Conversion and Synergetic Innovation Centre of Quantum Information and Quantum Physics, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: jlyang@ustc.edu.cn
First published on 18th July 2023
Abstract
It is vital to search for efficient and stable oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) electrocatalysts for the development of metal–air batteries. Herein, we systematically investigated a series of TMNxO4−x-HTC (TM = Fe, Co, Ni, Ru, Rh, Pd, Ir and Pt; x = 0–4; HTC = hexatribenzocyclyne) analogs of two-dimensional (2D) electrically conductive metal–organic frameworks (MOFs) as potential electrocatalysts for the OER and ORR by using density functional theory calculations. The calculated results exhibit good thermodynamic and electrochemical stabilities of the designed TMNxO4−x-HTC. The OER and ORR catalytic activity of the designed catalyst is governed by the interaction strength between the intermediates and the catalyst, and this interaction can be tuned by adjusting TM atoms and the local coordination number of N/O atoms. CoN3O1-HTC is found to be the best OER catalyst with an overpotential ηOER of 0.29 V, and RhN2O2-HTC exhibits the lowest ORR overpotential ηORR of 0.20 V. Importantly, RhO4-HTC, RhN2O2-HTC and CoN1O3-HTC are predicted as efficient bifunctional catalysts for the OER and ORR. Moreover, the kinetics simulation verifies the four-electron ORR pathway with high activity and selectivity toward H2O production. The results not only contribute to designing and searching for efficient OER and ORR electrocatalysts but shed light on the opportunities to explore electrochemical applications based on 2D MOF materials.
1. Introduction
The development of sustainable and renewable energy technologies is of great significance to solve the increasingly serious global energy crisis and environmental pollution problems.1,2 Among various technologies, rechargeable metal–air batteries have attracted much attention owing to their high theoretical energy density and environmental compatibility.3 Charging and discharging processes are driven by the electrochemical oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) processes.4 The OER and ORR with sluggish kinetics require high overpotential and thus lead to low energy efficiency which hinders the application of metal–air batteries.3 Ir/Ru oxides5,6 and Pt-based materials7,8 are known as the state-of-the-art catalysts for the OER and ORR, respectively. Generally, only the metal atoms on the surface of the bulk material could react with reactive species.9 Therefore, only a small portion of metal atoms in the bulk material could participate in the catalytic reaction. It should be noted that the uniformly exposed metal active sites on two-dimensional (2D) materials compared with that of bulk materials could provide promising avenues for exploring alternative catalysts. Moreover, the current studies on 2D material-based catalysts for the OER and ORR are usually investigated one at a time,10–12 thus, making it difficult to understand the overall catalytic performance of the catalyst. Specifically, to a certain extent, the use of bifunctional electrocatalysts could also reduce costs and simplify procedures since the working conditions of bifunctional catalysts are the same. Hence, exploring efficient 2D material-based bifunctional electrocatalysts for both the OER and ORR is highly desirable for the development of metal–air batteries.2D metal–organic frameworks (MOFs) are a class of layer-stacked materials consisting of well-organized metal centers and organic ligands and have exhibited great potential for application in catalysis and energy storage due to their large surface area, exposed metal active sites and tunable chemical functionality.13–17 Particularly, 2D electrically conductive MOFs are a newly emerging class of electronic materials that not only inherit most merits from conventional 2D MOFs but also show electrical conductivity due to the extended conjugation.18–20 Recently, a new 2D conductive MOF, Co/Ni-based 2,3,8,9,14,15-hexahydroxyltribenzocyclyne (namely Co/NiO4-HTC), was successfully synthesized.21 The active centers for the two materials are transition metal–oxygen (TM–O) linkages. Topologically, 2,3,8,9,14,15-hexahydroxyltribenzocyclyne resembles 2,3,6,7,10,11-hexaiminotriphenylene that we have studied in our previous work,22 an archetype ligand with the same 3-fold symmetry, but possesses large surface area due to its bigger size. Therefore, it is worth investigating the electrochemical catalytic activity of this kind of material. Importantly, a systematic theoretical investigation of using this conductive 2D MOF material as a catalyst for electrochemistry is lacking. Such a theoretical investigation is crucial and necessary given the rapid progress of experimental work in this field. As we demonstrated in our previous work based on 2,3,6,7,10,11-hexaiminotriphenylene,22 due to the structural tunability of 2D MOF materials, the catalytic activity of 2D MOF materials can be tuned by substituting the central active transition metal atoms and the organic ligand to adjust the electronic properties. Hence, different TMO4-HTC materials with TM–O linkage and TMN4-HTC materials with TM–N linkage as well as materials with different local coordination environments between TM and N/O atoms (TMNxO4−x-HTC, x = 0–4) were designed.
In this work, a series of TMNxO4−x-HTC monolayers were constructed via tuning the TM atoms and the local coordination number between TM atoms and N/O linkages, and their reaction mechanisms and catalytic activity for the OER and ORR were systematically investigated by using density functional theory (DFT) calculations. The results show that all the catalysts could exhibit good thermodynamic and electrochemical stabilities. Notably, CoN3O1-HTC and RhN2O2-HTC are predicted to be promising electrocatalysts for the OER and ORR with the calculated overpotential ηOER and ηORR of 0.29 and 0.20 V, respectively. Moreover, RhO4-HTC and RhN2O2-HTC are found to be efficient bifunctional catalysts for the OER and ORR.
2. Computational methods
All calculations were carried out by using the Vienna ab initio Simulation Package (VASP)23,24 based on the spin-polarized density functional theory method. The ion–electron interactions were described by using the projector augmented wave (PAW) method,25 and the electronic exchange–correlation interactions were determined using the Perdew–Burke–Ernzerhof (PBE)26 functional of the generalized gradient approximation (GGA).27 The van der Waals (vdW) interactions were described by using Grimme's DFT-D3 correction method.28 A plane-wave cutoff energy of 500 eV was adopted for all the computations. The convergence criterion for energy and force during geometrical optimization was set to 10−5 eV and 10−2 eV Å−1, respectively. The Brillouin zone was sampled using a 3 × 3 × 1 k-point29 during geometry optimization. A vacuum space of 20 Å was applied to avoid the interaction between the periodic images. Throughout all the calculations, we used an implicit solvent model to consider the solvent effect of the water environment through the polarized continuum model as implemented in VASPsol with a dielectric constant of 78.4.30Ab initio molecular dynamics (AIMD) simulations were performed to demonstrate the thermodynamic stability of the designed catalyst, and the algorithm of the Nose thermostat was carried out to simulate a canonical ensemble31 for 10 ps with a time step of 2 fs. Bader charge analysis was adopted to investigate the charge transfer process.32 The calculation details for the OER and ORR are listed in the ESI† as in our previous study.33,34 The adsorption Gibbs free energy is defined as Gads = Gadsorbent+catalyst − Gcatalyst − Gadsorbent, here Gadsorbent+catalyst, Gcatalyst, and Gadsorbent refer to the Gibbs free energies of the adsorbent on the catalyst, the isolated catalyst, and the isolated adsorbent, respectively.3. Results and discussion
Fig. 1 shows the stable geometric configurations of 2D TMNxO4−x-HTC (x = 0–4). The optimized unit cell of TMNxO4−x-HTC contains three equivalent TM atoms, and each TM atom is surrounded by four nitrogen or oxygen atoms with different ratios. In this work, three 3d transition metals (Fe, Co and Ni), three 4d transition metals (Ru, Rh and Pd), and two 5d transition metals (Ir and Pt) were considered to build catalysts, as these transition metals are commonly used to design OER electrocatalysts.33,34 It should be mentioned that the stability of the designed catalysts is of significant importance for their long-term use. Hence, we calculated the formation energy (Ef) and dissolution potential (Udiss) of all the designed catalysts to evaluate their thermodynamic and electrochemical stabilities.35,36 The Ef and Udiss are defined as Ef = (ETMNxO4−x − ENxO4−x − 3ETM)/3 and Udiss = Udiss(bulk) − Ef/ne, respectively, where ETMNxO4−x and ENxO4−x are the total energies of the TMNxO4−x system and the NxO4−x substrate, respectively. ETM is the total energy of a metal atom in its most stable bulk structure. Udiss(bulk) is the standard dissolution potential of the bulk metal and n is the number of electrons involved during the dissolution process. Since ETM is referenced with respect to that of the bulk metal, systems with negative values of Ef are evaluated to be thermodynamically stable against the clustering of TM atoms. Systems with positive values of Udissvs. the standard hydrogen electrode (SHE) are considered to be electrochemically stable. The calculated results of Ef and Udiss are shown in Fig. 2, which suggest that all the designed catalysts could exhibit good thermodynamic and electrochemical stabilities that meet the stability criteria for electrocatalysts.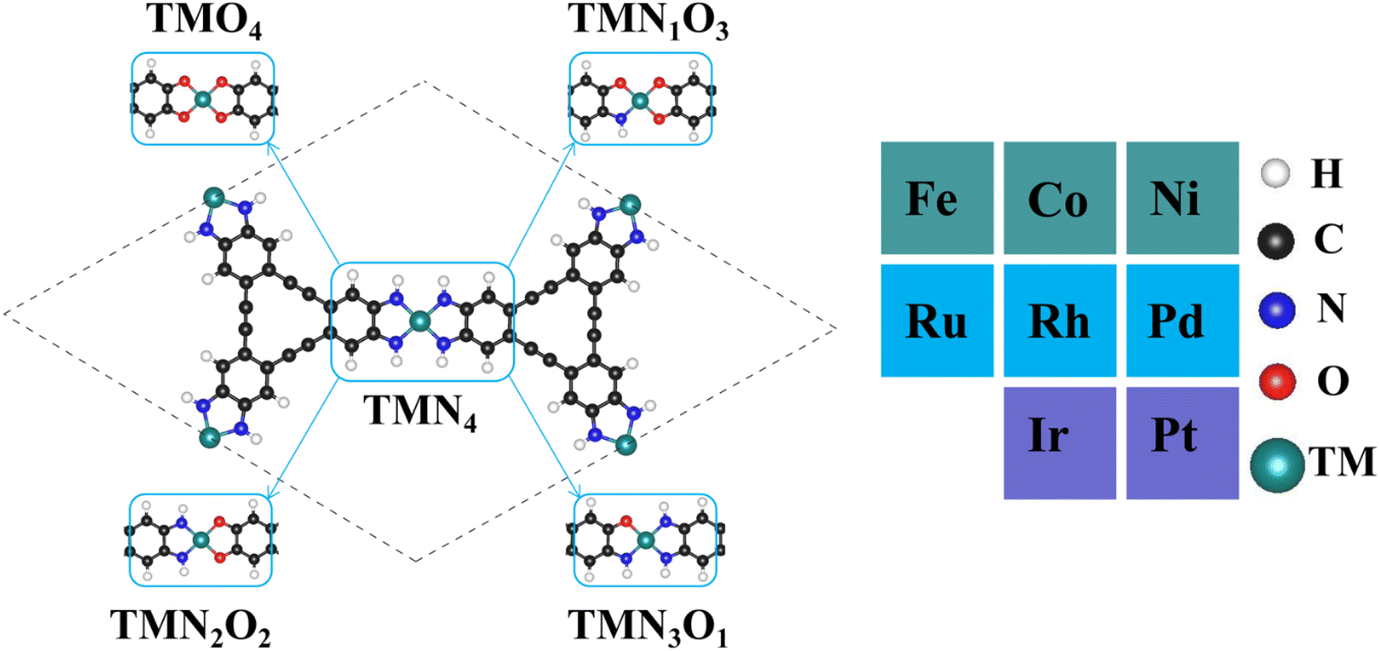 | ||
| Fig. 1 Optimized geometric configurations of 2D TMNxO4−x-HTC (x = 0–4) and the considered dopant transition metal atoms. | ||
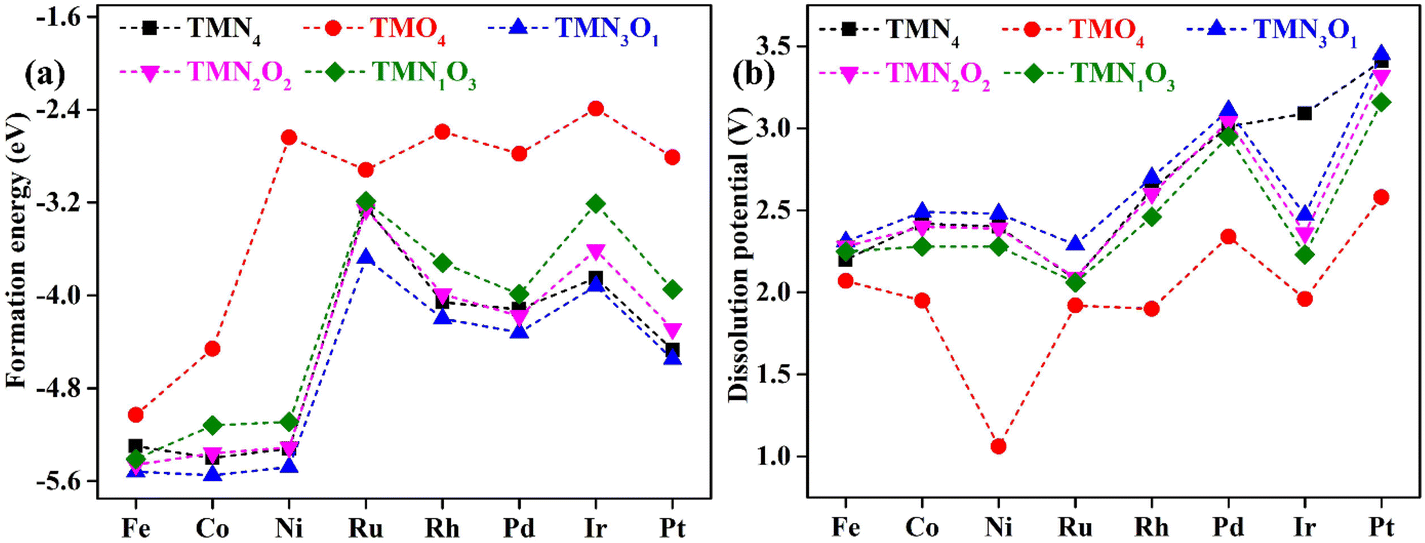 | ||
| Fig. 2 Calculated (a) formation energy and (b) dissolution potential of transition metal atoms for the designed TMNxO4−x-HTC catalysts. | ||
Importantly, the distinct electronic properties of all the designed catalysts were studied to obtain insight into their catalytic performance. As presented in Fig. S1–S5,† all the designed catalysts exhibit metallicity, indicating their good electrical conductivity and ensuring efficient electron transfer during the OER and ORR processes. Additionally, in Fig. S6–S10,† the calculated results of the partial density of states (PDOS) suggest that the different dopant TM atoms show different contributions to the electronic states of the designed catalysts around the Fermi level, and the electronic states of the catalysts across the Fermi level are mainly contributed by the d orbitals of the dopant TM atoms. Moreover, around the Fermi energy, the hybridization between the p orbitals of the O/N atoms and the d orbitals of the TM atoms further demonstrates their strong binding interaction. The charge transfer between the substrates and TM atoms could also reveal their strong interaction. Fig. S11† shows a large amount of charge (0.76–1.33e) transfer from the TM atoms to substrates, making the dopant TM atoms positively charged. These positively charged TM atoms are considered to be active sites in the OER and ORR catalytic processes. In previous literature, the d-band center (εd) was used to analyze the interaction strength between catalysts and adsorbates.37–39 Thus, we calculated the εd values of the designed catalysts and plotted them in Fig. S6–S10.† From Fig. S12a,† it can be concluded that the εd values shift to a lower energy position than the Fermi level with the increase of the number of d-electrons in the TM atoms at least when the TM atoms are in the same row of the periodic table. Generally, a larger d-electron number of the TM atom and lower energy of εd could result in weaker interaction strength between catalysts and adsorbates.40 For the OER and ORR, the calculated adsorption Gibbs free energies of the HO*, O* and HOO* intermediates (ΔGHO*, ΔGO*, and ΔGHOO*) with the corresponding d-electron numbers of TMNxO4−x-HTC catalysts are plotted in Fig. S12b–f.† Moreover, ΔGHO*, ΔGO*, and ΔGHOO* with the corresponding εd values of all TMNxO4−x-HTC systems are plotted in Fig. S13.† It can be concluded that the adsorption Gibbs free energies of the intermediates decrease with the increase of d-electron numbers of the TM atoms when they are in the same row of the periodic table, which also agrees with the position of εd values. Therefore, the adsorption Gibbs free energies of intermediates are negatively correlated with the εd values when the TM atoms are in the same row of the periodic table. This phenomenon was also observed in reported experimental and theoretical literature.41,42 As a consequence, by tuning the doping of TM atoms on the substrate, the catalyst could exhibit the optimal interaction strength with the intermediates for the OER and ORR.
As proposed by Nørskov et al.,43 the adsorption Gibbs free energies of intermediates govern the intrinsic OER and ORR activity of a catalyst. The calculated corresponding adsorption Gibbs free energy values of intermediates on all the designed TMNxO4−x-HTC catalysts are listed in Fig. S14–S18.† According to the Sabatier principle,44 too weak or too strong interaction strength of the intermediates on the catalysts could lead to an adverse effect on the catalytic activity. Therefore, one of our goals is to identify efficient OER/ORR catalysts with moderate intermediate interaction strength. For an ideal OER/ORR catalyst under conditions where the applied potential U equals zero, the adsorption Gibbs free energy value difference between two adjacent intermediates for all the four-electron transfer steps should be 1.23 eV. In other words, for an ideal catalyst, the adsorption Gibbs free energy values of HO*, O* and HOO* intermediates should be 1.23, 2.46 and 3.69 eV, respectively, which makes the four-electron OER/ORR occur at the thermodynamic limit and the overpotential η is zero. However, the reality is that the adsorption Gibbs free energy value difference between two adjacent intermediates is not equal. The OER overpotential (ηOER) is determined using the maximum adsorption Gibbs free energy difference of two adjacent intermediates, while the ORR overpotential (ηORR) is determined using the minimum adsorption Gibbs free energy difference of two adjacent intermediates. The calculated Gibbs free energy diagrams for the OER and ORR on all the designed electrocatalysts are shown in Fig. S14–S18,† and the potential-determining step (PDS) is colored in yellow for the OER and in pink for the ORR. Moreover, the calculated ηOER and ηORR values on all the designed catalysts are summarized in Fig. 3. Notably, among all the designed TMNxO4−x-HTC catalysts, CoN3O1-HTC is predicted to be the best OER catalyst with a calculated ηOER value of 0.29 V, followed by RhN3O1-HTC (ηOER = 0.32 V), CoN2O2-HTC (ηOER = 0.33 V), RhO4-HTC (ηOER = 0.33 V), CoN4-HTC (ηOER = 0.35 V), CoO4-HTC (ηOER = 0.38 V), CoN1O3-HTC (ηOER = 0.39 V), and RhN2O2-HTC (ηOER = 0.43 V). Importantly, the OER overpotential values of all the above-mentioned electrocatalysts are lower than that of the IrO2 (110) catalyst (ηOER = 0.52 V),45 indicating their efficient OER catalytic activity. Additionally, the formation of HOO* from O* is the potential-determining step for all the above-mentioned designed efficient catalysts. As known, the ORR is the reverse reaction of the OER. The calculated Gibbs free energy diagrams of all the designed TMNxO4−x-HTC catalysts toward the ORR are also displayed in Fig. S14–S18.† From Fig. 3b, it can be seen that RhN2O2-HTC is predicted to be the best ORR electrocatalyst with a calculated ηORR value of 0.20 V, followed by RhN1O3-HTC (ηORR = 0.29 V), IrO4-HTC (ηORR = 0.35 V), FeN1O3-HTC (ηORR = 0.37 V), RhO4-HTC (ηORR = 0.39 V), FeN2O2-HTC (ηORR = 0.44 V), CoN1O3-HTC (ηORR = 0.49 V), and IrN2O2-HTC (ηORR = 0.50 V), suggesting their efficient ORR catalytic activity. Remarkably, their ORR overpotentials are lower than or comparable to that of Pt (111) (ηORR = 0.48 V).46 In addition, the formation of HO* is the potential-determining step for RhN2O2-HTC, RhN1O3-HTC, IrO4-HTC, RhO4-HTC, FeN2O2-HTC and IrN2O2-HTC catalysts, and the formation of HOO* is the potential-determining step for FeN1O3-HTC and CoN1O3-HTC catalysts. Hence, based on the above results, it can be noted that RhO4-HTC, CoN3O1-HTC, RhN2O2-HTC and CoN1O3-HTC are predicted as promising and efficient bifunctional electrocatalysts toward both the OER and ORR, and their free energy diagrams for the OER and ORR are shown in Fig. 4. Moreover, for comparison, previous results on the OER and ORR catalytic activity of 2D-MOF materials are listed in Table S2.†
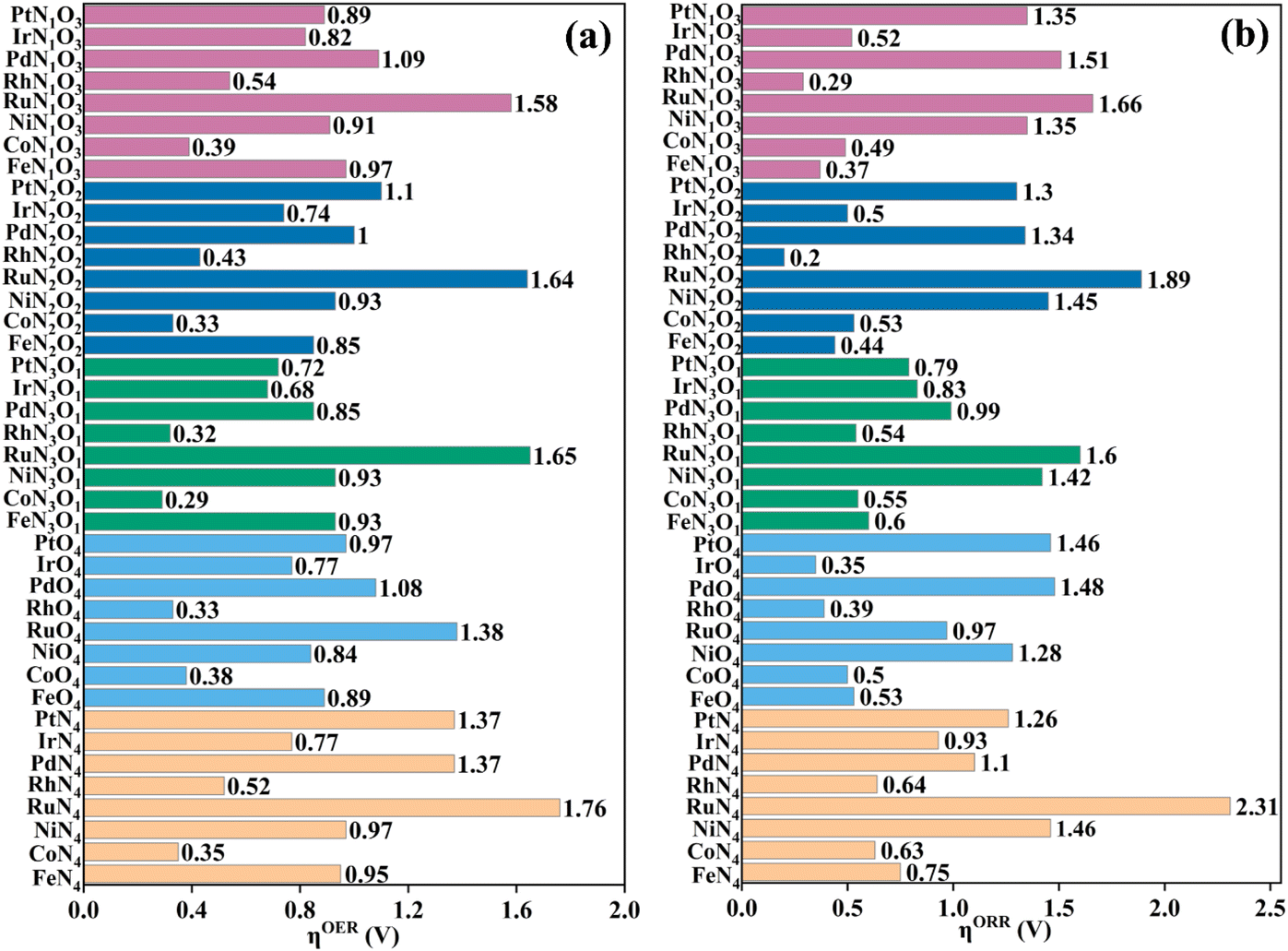 | ||
| Fig. 3 Calculated (a) OER and (b) ORR overpotentials of different designed TMNxO4−x-HTC catalysts. | ||
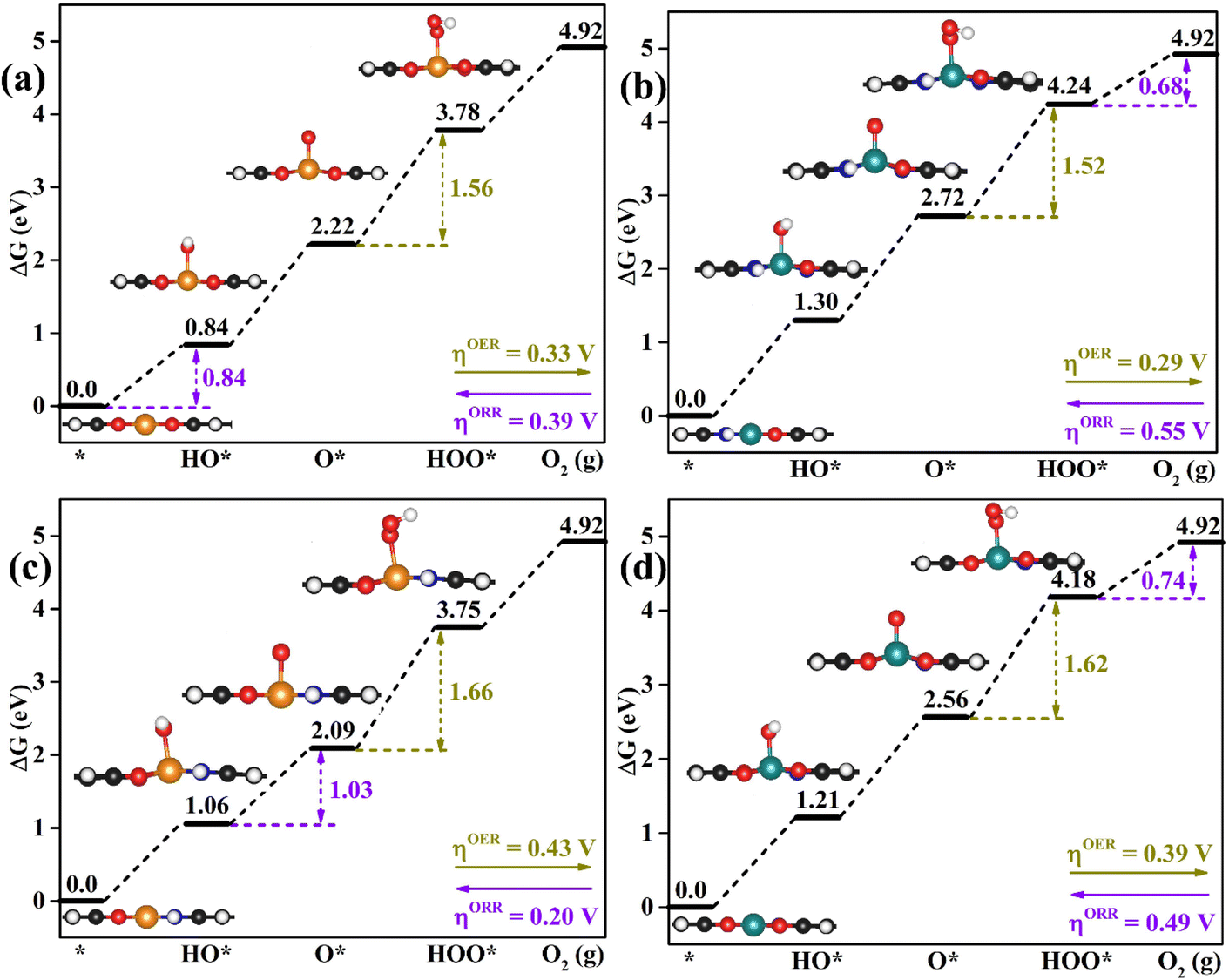 | ||
| Fig. 4 Calculated free energy diagrams of the promising bifunctional catalysts for the OER and ORR: (a) RhO4-HTC, (b) CoN3O1-HTC, (c) RhN2O2-HTC and (d) CoN1O3-HTC. The yellow and pink values are the potential-determining step values for the OER and ORR. The optimized configurations of intermediates on the catalysts are also exhibited. | ||
In-depth understanding of the OER and ORR catalytic performance of different catalysts can guide us to design efficient electrocatalysts. As mentioned above, the catalytic performance for the OER and ORR is determined using the Gibbs free energy of the corresponding intermediates on the catalyst. Hence, identifying the relationship between the Gibbs free energy of the intermediates and catalytic activity is essential for the rational design of efficient catalysts. In this work, by comparing the adsorption Gibbs free energy values of the HO* and HOO* intermediates on all the designed catalysts, we found that ΔGHOO* can be expressed as a function of ΔGHO*via the equation ΔGHOO* = 0.92ΔGHO* + 3.00 eV (Fig. 5a). It is suggested that the calculated adsorption Gibbs free energy values of the HO* and HOO* intermediates show a strong linear relationship mainly due to both intermediates forming single bonds between O and TM atoms (Fig. 4), and the difference between the Gibbs free energy values of the HO* and HOO* intermediates is a constant. The above results are consistent with those of previously reported carbon-based catalysts for the OER and ORR.47,48 Given the fact that most of the OER potential-determining step occurs at the HO* to O* or O* to HOO* step, the OER overpotential could be determined using the difference of ΔGO* − ΔGHO*. This is confirmed by the volcano plot displayed in Fig. 5b, where the overpotential values of the OER fall in a line as a function of ΔGO* − ΔGHO*. Obviously, the designed CoN3O1-HTC, RhN3O1-HTC, CoN2O2-HTC and RhO4-HTC catalysts are located around the peak of the volcano curve with low OER overpotentials and stand out to be promising OER electrocatalysts. For the ORR, the potential-determining step approximately occurs at the * to HO* or HOO* to *+O2 step, and then, the ORR overpotential could be determined using the ΔGHO* value. Indeed, Fig. 5c shows the volcano plot of the ORR overpotential as a function of ΔGHO*. Apparently, RhN2O2-HTC, RhN1O3-HTC, IrO4-HTC and FeN1O3-HTC catalysts with low ORR overpotentials are located around the peak of the volcano plot. Moreover, it can be concluded that the moderate interaction strength of the intermediates on the catalyst could enable good catalytic activity for the OER and ORR. What is more, RhO4-HTC, CoN3O1-HTC, RhN2O2-HTC and CoN1O3-HTC catalysts are located around the top of both volcano plots and screened out to be promising and efficient bifunctional electrocatalysts for both the OER and ORR.
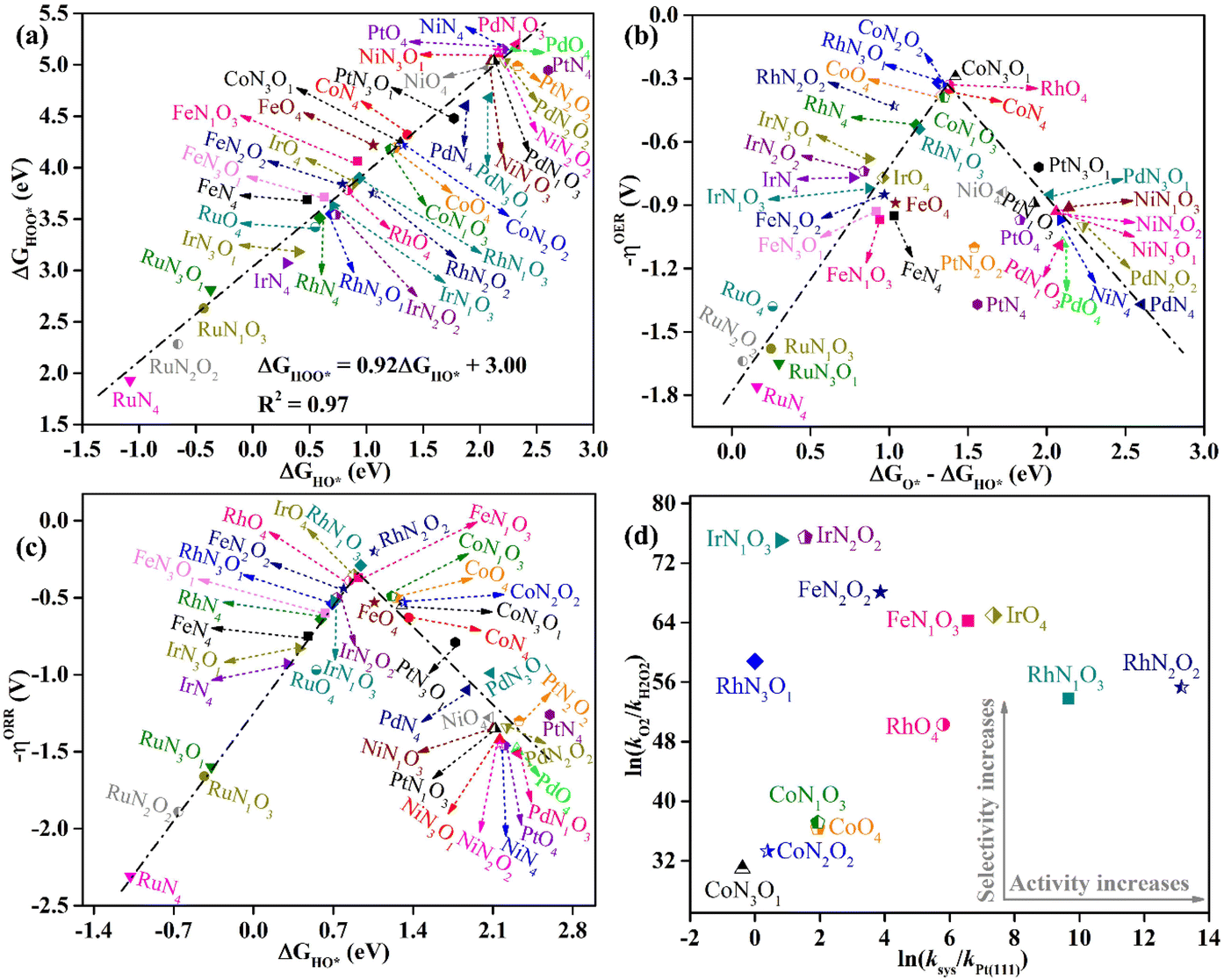 | ||
| Fig. 5 (a) Scaling relationship between ΔGHO* and ΔGHOO* on all the designed TMNxO4−x-HTC catalysts. (b) Calculated OER volcano curve of −ηOER as a function of ΔGO* − ΔGHO* on all the designed catalysts. (c) Calculated ORR volcano curve of −ηORR as a function of ΔGHO* on all the designed catalysts. (d) Variations of the ORR activity vs. selectivity of the potential electrocatalysts. | ||
It is noteworthy that the four-electron ORR pathway from O2 to H2O is particularly important in metal–air batteries, in which hydrogen peroxide (H2O2) is an undesirable product since it could cause the degradation of the catalyst.49 For the above screened potential ORR electrocatalysts with the calculated overpotential lower than 0.55 V (RhN2O2-HTC, RhN1O3-HTC, IrO4-HTC, FeN1O3-HTC, RhO4-HTC, FeN2O2-HTC, IrN2O2-HTC, CoO4-HTC, CoN1O3-HTC, IrN1O3-HTC, RhN3O1-HTC, CoN2O2-HTC and CoN3O1-HTC), the selectivity for the four-electron pathway is verified from the thermodynamic perspective, since the calculated ΔGO* values (2.09, 2.13, 1.84, 1.86, 2.22, 1.76, 1.57, 2.58, 2.56, 1.58, 2.00, 2.66 and 2.72) are all smaller than 3.52 eV (ΔGH2O2 − ΔGH2O).50,51 A kinetics investigation was carried out to understand the catalytic selectivity for the ORR50 and the corresponding calculation details are listed in the ESI.† As shown in Fig. 5d, all the calculated ln(kO2/kH2O2) values are positive, suggesting that the reduction of O2 to H2O is prioritized on these designed catalysts. Especially, the calculated ln(ksys/kPt(111)) values of RhN2O2-HTC, RhN1O3-HTC, IrO4-HTC, FeN1O3-HTC, RhO4-HTC and FeN2O2-HTC are 13.15, 9.67, 7.35, 6.57, 5.80 and 3.87, respectively, indicating that the reaction rate on these designed catalysts is much faster than that on Pt(111).
To explicitly visualize how the OER and ORR catalytic activity of the screened CoN3O1-HTC, RhN3O1-HTC, RhO4-HTC, CoO4-HTC, RhN2O2-HTC, CoN2O2-HTC, RhN1O3-HTC and CoN1O3-HTC catalysts is in practice, the corresponding theoretical OER and ORR polarization curves were simulated based on the reversible hydrogen electrode (RHE) in comparison with those of IrO2(110) for the OER and Pt(111) for the ORR, that is, the change in current density as a function of potential U. The simulation details are listed in the ESI.† As shown in Fig. 6, for the OER, at a current density of 10 mA cm−2, the simulated polarization curves of CoN3O1-HTC, RhN3O1-HTC, RhO4-HTC, CoN2O2-HTC, CoO4-HTC, CoN1O3-HTC, RhN2O2-HTC and RhN1O3-HTC exhibit lower onset potentials of 1.59, 1.65, 1.67, 1.67, 1.78, 1.80, 1.88 and 2.09 V vs. RHE than that of IrO2 (2.27 V) as reported in our previous work, respectively;22 for the ORR, at a current density of 1 mA cm−2, the simulated polarization curves of RhN2O2-HTC, RhN1O3-HTC and RhO4-HTC exhibit higher onset potentials of 1.16, 0.98 and 0.78, vs. RHE than that of Pt(111) (0.67 V) as reported in our previous work, respectively,22 while the simulated polarization curves of CoN1O3-HTC, CoO4-HTC, CoN2O2-HTC, RhN3O1-HTC and CoN3O1-HTC exhibit lower onset potentials of 0.58, 0.55, 0.51, 0.48 and 0.46 V, respectively. The above results indicate that these screened-out catalysts possess efficient OER and ORR catalytic activity, which makes them potential alternatives to IrO2 and Pt electrodes. Additionally, we performed AIMD simulations for the potential catalysts (taking CoN3O1-HTC, RhN3O1-HTC, CoN2O2-HTC, RhN2O2-HTC, CoN1O3-HTC, RhN1O3-HTC, CoO4-HTC and RhO4-HTC as examples) to evaluate their dynamic stabilities. The simulated results (Fig. S19–S22†) show that the energies oscillate near the equilibrium state during the 10 ps simulations and the structures have no obvious structural reconstruction, which suggests their good kinetic stability.
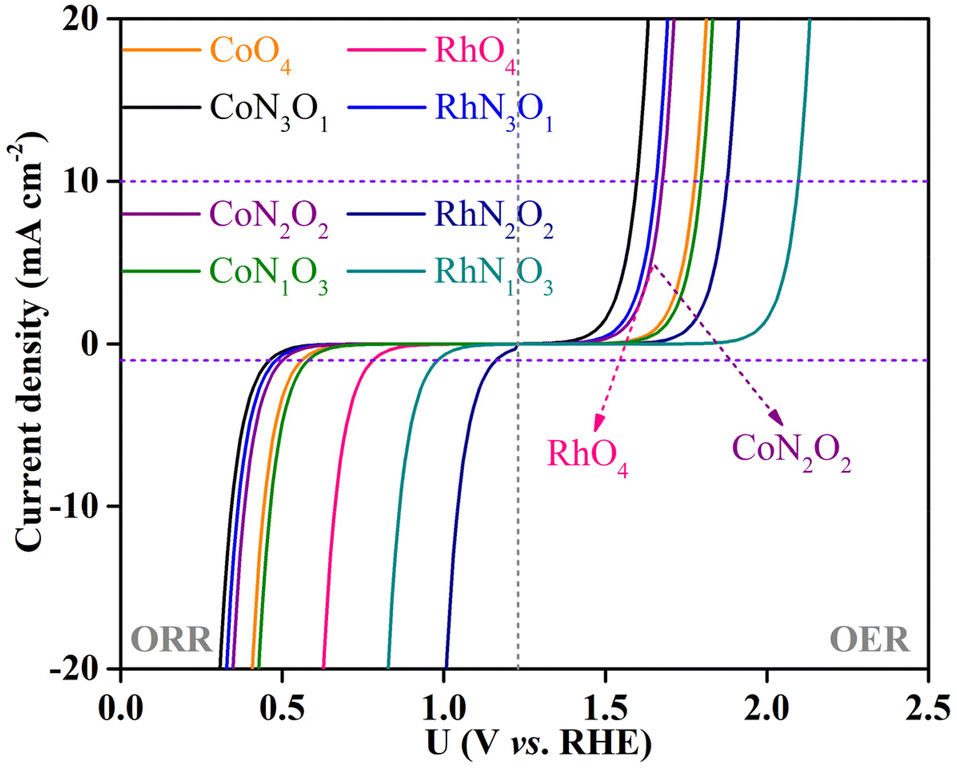 | ||
| Fig. 6 Simulated polarization curves for the screened-out catalysts. | ||
4. Conclusions
In summary, a series of 2D TMNxO4−x-HTC (x = 0–4) electrocatalysts were designed and systematically investigated for their catalytic activity toward the OER and ORR based on DFT calculations. The strong interaction between TM atoms and NxO4−x-HTC could guarantee the stability of TMNxO4−x-HTC. It was found that as the number of d-electrons increases, the d-band value decreases, thereby weakening the interaction between the intermediates and TM atoms. The OER overpotential ηOER follows a volcano plot of ΔGO* − ΔGHO*, and the ORR overpotential ηORR follows a volcano plot of ΔGHO*. Among all the designed TMNxO4−x-HTC electrocatalysts, the best OER catalyst is CoN3O1-HTC with a calculated ηOER value of 0.29 V, followed by RhN3O1-HTC (0.32 V), CoN2O2-HTC (0.33 V) and RhO4-HTC (0.33 V); the best ORR catalyst is RhN2O2-HTC with a calculated ηORR value of 0.20 V, followed by RhN1O3-HTC (0.29 V), IrO4-HTC (0.35 V), FeN1O3-HTC (0.37 V) and RhO4-HTC (0.39 V). Notably, RhO4-HTC, RhN2O2-HTC and CoN1O3-HTC are predicted as promising and efficient bifunctional electrocatalysts for both the OER and ORR. Moreover, by analyzing the relationship between the ORR products H2O and H2O2, the thermodynamically favorable selectivity for H2O is elucidated from the kinetics perspective. Our results shed light on the exploration of 2D-MOF materials as promising OER and ORR electrocatalysts.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22102167 and U21A20317). The calculations were performed at the Supercomputing Center of the University of Science and Technology of China.References
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Design of Electrocatalysts for Oxygen- and Hydrogen-Involving Energy Conversion Reactions, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- C. X. Zhao, J. N. Liu, J. Wang, D. Ren, B. Q. Li and Q. Zhang, Recent Advances of Noble-Metal-Free Bifunctional Oxygen Reduction and Evolution Electrocatalysts, Chem. Soc. Rev., 2021, 50, 7745–7778 RSC.
- F. Y. Cheng and J. Chen, Metal-Air Batteries: from Oxygen Reduction Electrochemistry to Cathode Catalysts, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- J. S. Lee, S. T. Kim, R. G. Cao, N. S. Choi, M. L. Liu, K. T. Lee and H. Cho, Metal-Air Batteries with High Energy Density: Li-Air versus Zn-Air, Adv. Energy Mater., 2011, 1, 34–50 CrossRef CAS.
- E. A. Paoli, F. Masini, R. Frydendal, D. Deiana, C. Schlaup, M. Malizia, T. W. Hansen, S. Horch, I. E. L. Stephens and I. Chorkendorff, Oxygen Evolution on Wellcharacterized Mass-Selected Ru and RuO2 Nanoparticles, Chem. Sci., 2015, 6, 190–196 RSC.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, Synthesis and Activities of Rutile IrO2 and RuO2 Nanoparticles for Oxygen Evolution in Acid and Alkaline Solutions, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- X. Q. Huang, Z. P. Zhao, L. Cao, Y. Chen, E. B. Zhu, Z. Y. Lin, M. F. Li, A. M. Yan, A. Zettl, Y. M. Wang, X. F. Duan, T. Mueller and Y. Huang, High-Performance Transition Metal–Doped Pt3Ni Octahedra for Oxygen Reduction Reaction, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- N. M. Markovic, T. J. Schmidt, V. Stamenkovic and P. N. Ross, Oxygen Reduction Reaction on Pt and Pt Bimetallic Surfaces A Selective Review, Fuel Cells, 2001, 1, 105–116 CrossRef CAS.
- D. Zhao, Z. W. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. D. Li, Atomic Site Electrocatalysts for Water Splitting, Oxygen Reduction and Selective Oxidation, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC.
- Y. N. Zhou, J. Li, X. P. Gao, W. Chu, G. P. Gao and L. W. Wang, Recent Advances in Single-Atom Electrocatalysts Supported on Two-Dimensional Materials for the Oxygen Evolution Reaction, J. Mater. Chem. A, 2021, 9, 9979–9999 RSC.
- L. Tang, X. G. Meng, D. H. Deng and X. H. Bao, Confinement Catalysis with 2D Materials for Energy Conversion, Adv. Mater., 2019, 31, 1901996 CrossRef CAS PubMed.
- H. Y. Jin, C. X. Guo, X. Liu, J. L. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S. Z. Qiao, Emerging Two-Dimensional Nanomaterials for Electrocatalysis, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS PubMed.
- M. T. Zhao, Y. Huang, Y. W. Peng, Z. Q. Huang, Q. L. Ma and H. Zhang, Two-Dimensional Metal-Organic Framework Nanosheets: Synthesis and Applications, Chem. Soc. Rev., 2018, 47, 6267–6295 RSC.
- L. Jiao, Y. Wang, H. L. Jiang and Q. Xu, Metal-Organic Frameworks as Platforms for Catalytic Applications, Adv. Mater., 2018, 30, 1703663 CrossRef PubMed.
- Q. L. Qi, J. Hu, Y. J. Zhang, W. Li, B. L. Huang and C. X. Zhang, Two-Dimensional Metal-Organic Frameworks-Based Electrocatalysts for Oxygen Evolution and Oxygen Reduction Reactions, Adv. Energy Sustainability Res., 2020, 2, 2000067 CrossRef.
- T. C. Li, M. M. Li, X. Y. Zhu, J. Zhang and Y. Jing, Conductive Two-Dimensional M3(C6S3O3)2 Monolayers as Effective Electrocatalysts for the Oxygen Reduction Reaction, J. Mater. Chem. A, 2021, 9, 24887–24894 RSC.
- J. Zhang, X. Y. Zhu, W. X. Geng, T. C. Li, M. M. Li, C. B. Fang, X. C. Shan and Y. Jing, Mo3(C6X6)2 (X = NH,S,O) Monolayers: Two-Dimensional Conductive Metal-Organic Frameworks as Effective Electrocatalysts for the Nitrogen Reduction Reaction, J. Energy Chem., 2021, 61, 71–76 CrossRef CAS.
- H. Huang, Y. Zhao, Y. M. Bai, F. M. Li, Y. Zhang and Y. Chen, Conductive Metal-Organic Frameworks with Extra Metallic Sites as an Efficient Electrocatalyst for the Hydrogen Evolution Reaction, Adv. Sci., 2020, 7, 2000012 CrossRef CAS PubMed.
- M. C. Wang, R. H. Dong and X. L. Feng, Two-Dimensional Conjugated Metal-Organic Frameworks (2D c-MOFs): Chemistry and Function for MOFtronics, Chem. Soc. Rev., 2021, 50, 2764–2793 RSC.
- H. F. Wang, L. Y. Chen, H. Pang, S. Kaskel and Q. Xu, MOF-Derived Electrocatalysts for Oxygen Reduction, Oxygen Evolution and Hydrogen Evolution Reactions, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC.
- H. T. B. Pham, J. Y. Choi, S. F. Huang, X. B. Wang, A. Claman, M. Stodolka, S. Yazdi, S. Sharma, W. Zhang and J. Park, Imparting Functionality and Enhanced Surface Area to a 2D Electrically Conductive MOF via Macrocyclic Linker, J. Am. Chem. Soc., 2022, 144, 10615–10621 CrossRef CAS PubMed.
- Y. N. Zhou, L. Sheng, Q. Q. Luo, W. H. Zhang and J. L. Yang, Improving the Activity of Electrocatalysts toward the Hydrogen Evolution Reaction, the Oxygen Evolution Reaction, and the Oxygen Reduction Reaction via Modification of Metal and Ligand of Conductive Two-Dimensional Metal-Organic Frameworks, J. Phys. Chem. Lett., 2021, 12, 11652–11658 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Efficiency of Ab initio Total Energy Calculations for Metals and Semiconductors Using A Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blochl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, Rationale for Mixing Exact Exchange with Density Functional Approximations, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104–154123 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Special Points for Brillouin-Zone Integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, Implicit Solvation Model for Density-Functional Study of Nanocrystal Surfaces and Reaction Pathways, J. Chem. Phys., 2014, 140, 084106–084114 CrossRef PubMed.
- G. J. Martyna, M. L. Klein and M. Tuckerman, Nosé–Hoover Chains: The Canonical Ensemble via Continuous Dynamics, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, A Grid-Based Bader Analysis Algorithm Without Lattice Bias, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- Y. N. Zhou, G. P. Gao, J. Kang, W. Chu and L. W. Wang, Transition Metal Embedded Two-Dimensional C3N as Highly Active Electrocatalysts for Oxygen Evolution and Reduction Reactions, J. Mater. Chem. A, 2019, 7, 12050–12059 RSC.
- Y. N. Zhou, G. P. Gao, W. Chu and L. W. Wang, Computational Screening of Transition Metal-Doped Phthalocyanine Monolayers for Oxygen Evolution and Reduction, Nanoscale Adv., 2020, 2, 710–716 RSC.
- J. Greeley and J. K. Nørskov, Electrochemical Dissolution of Surface Alloys in Acids: Thermodynamic Trends from First-Principles Calculations, Electrochim. Acta, 2007, 52, 5829–5836 CrossRef CAS.
- X. Y. Guo, J. X. Gu, S. R. Lin, S. L. Zhang, Z. F. Chen and S. P. Huang, Tackling the Activity and Selectivity Challenges of Electrocatalysts toward the Nitrogen Reduction Reaction via Atomically Dispersed Biatom Catalysts, J. Am. Chem. Soc., 2020, 142, 5709–5721 CrossRef CAS PubMed.
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Role of Strain and Ligand Effects in the Modification of the Electronic and Chemical Properties of Bimetallic Surfaces, Phys. Rev. Lett., 2004, 93, 156801 CrossRef CAS PubMed.
- V. Stamenkovic, B. S. Mun, K. L. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Changing the Activity of Electrocatalysts for Oxygen Reduction by Tuning the Surface Electronic Structure, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed.
- Y. Liu, Y. M. Wang, B. I. Yakobson and B. C. Wood, Assessing Carbon-Based Anodes for Lithium-Ion Batteries: A Universal Description of Charge-Transfer Binding, Phys. Rev. Lett., 2014, 113, 028304 CrossRef CAS PubMed.
- F. Calle-Vallejo, A. Krabbe and J. M. Garcia-Lastra, How Covalence Breaks Adsorption-Energy Scaling Relations and Solvation Restores Them, Chem. Sci., 2017, 8, 124–130 RSC.
- M. T. Groot and M. T. M. Koper, Redox Transitions of Chromium, Manganese, Iron, Cobalt and Nickel Protoporphyrins in Aqueous Solution, Phys. Chem. Chem. Phys., 2008, 10, 1023–1031 RSC.
- C. Y. Ling, L. Shi, Y. X. Ouyang, X. C. Zeng and J. L. Wang, Nanosheet Supported Single-Metal Atom Bifunctional Catalyst for Overall Water Splitting, Nano. Lett., 2017, 17, 5133–5139 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir and L. Lindqvist, Origin of the Overpotential for Oxygen Reduction at A Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, Trends in the Exchange Current for Hydrogen Evolution, J. Electrochem. Soc., 2005, 152, 23–26 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Combining Theory and Experiment in Electrocatalysis: Insights into Materials Design, Science, 2017, 355, 4998 CrossRef PubMed.
- J. Greeley, I. E. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Alloys of Platinum and Early Transition Metals as Oxygen Reduction Electrocatalysts, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- H. X. Xu, D. J. Cheng, D. P. Cao and X. C. Zeng, A Universal Principle for A Rational Design of Single-Atom Electrocatalysts, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- T. W. He, S. K. Matta, G. Will and A. J. Du, Transition-Metal Single Atoms Anchored on Graphdiyne as High-Efficiency Electrocatalysts for Water Splitting and Oxygen Reduction, Small Methods, 2019, 3, 1800419 CrossRef.
- R. Zhang and J. J. Warren, Controlling the Oxygen Reduction Selectivity of Asymmetric Cobalt Porphyrins by Using Local Electrostatic Interactions, J. Am. Chem. Soc., 2020, 142, 13426–13434 CrossRef CAS PubMed.
- X. Y. Guo, S. R. Lin, J. X. Gu, S. L. Zhang, Z. F. Chen and S. P. Huang, Simultaneously Achieving High Activity and Selectivity toward Two-Electron O2 Electroreduction: The Power of Single-Atom Catalysts, ACS Catal., 2019, 9, 11042–11054 CrossRef CAS.
- L. H. Zhang, X. Y. Guo, S. L. Zhang and S. P. Huang, Building Up the “Genome” of Bi-Atom Catalysts toward Efficient HER/OER/ORR, J. Mater. Chem. A, 2022, 10, 11600–11612 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qi01112g |
| This journal is © the Partner Organisations 2023 |