
Aggregated coordination polymers of Ag+ with a cysteine derivative ligand containing an AIEgen†
Jing-Jing
Quan
 ,
Qian
Wang
,
Zhao
Li
and
Yun-Bao
Jiang
*
,
Qian
Wang
,
Zhao
Li
and
Yun-Bao
Jiang
*
Department of Chemistry, College of Chemistry and Chemical Engineering, The MOE Key Laboratory of Spectrochemical Analysis and Instrumentation, and iChEM, Xiamen University, Xiamen 361005, China. E-mail: ybjiang@xmu.edu.cn
First published on 8th March 2023
Abstract
We investigated coordination polymers of Ag+ with a cysteine-based thiol ligand designed to contain a tetraphenylethylene AIEgen (L- and D-1). The coordination polymers, forming in a variety of protic and aprotic organic solvents, such as THF, CH3CN and CH3OH, were shown to undergo aggregation in H2O/THF binary solvents at water volume fractions above 50%, where emission was substantially enhanced while the CD profile was reversed, yet the dependence of the CD signal on ee remained S-shaped for the polymers in the aprotic organic solvents THF and CH3CN, in contrast to that in protic solvents CH3OH and C2H5OH.
We have recently shown that the coordination polymers of Ag+ with chiral thiol ligands exhibit interesting properties,1–10 including overall supramolecular chirality with respect to the molecular and supramolecular chirality of the ligands,6 properties consistent with their serving as functional materials for sensing,1–4,8–10 and their being amenable to having their morphologies controlled.7 We therefore considered extending these efforts to the study of aggregates of these coordination polymers, a subject not yet pursued despite the wealth of investigations on the aggregates of the covalent polymers. In this regard, it was critical to design a thiol ligand whose properties would be sensitive to the existing forms of the polymers. We thus considered a cysteine-based chiral ligand containing an AIEgen whose emission has been shown to be sensitive to the extent of aggregation.5,11,12 In the current work, we investigated coordination polymers of Ag+ with L- and D-1 forms of a chiral ligand (Scheme 1) in organic solvents and the aggregates of these polymers in water-rich tetrahydrofuran (THF) solutions, and investigated their substantially differing properties, and intriguing chirality.
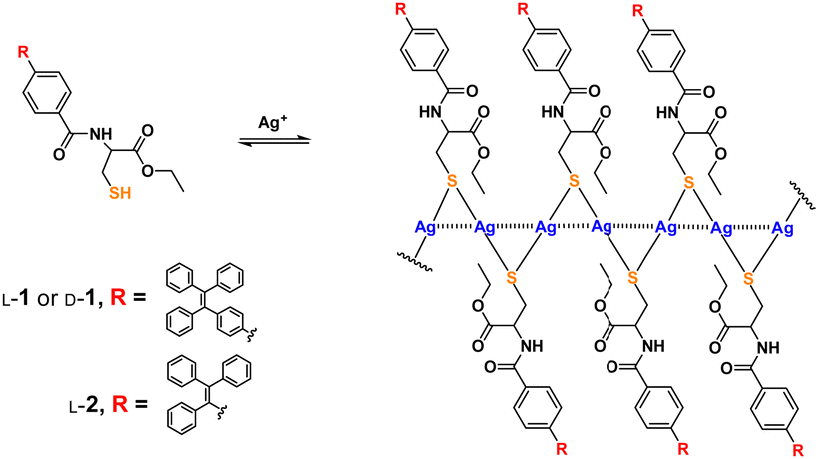 | ||
| Scheme 1 Formation of coordination polymers of Ag+ with thiol ligand 1 and its control compound 2. Dashed lines through the backbone represent Ag+⋯Ag+ interactions.16 | ||
We previously showed the coordination polymers of Ag+ with cysteine in aqueous solutions to be stabilized by both the Ag+⋯Ag+ interactions (the argentophilic interactions)13–15 along the polymeric backbone and by electrostatic interactions between the ligands attached to the backbone.2,10 The interaction network has been assumed to be responsible for allowing the molecular chirality of the ligand to be transferred into the polymeric backbone as a supramolecular chirality—with this assumption based on, for example, 350 nm-wavelength CD signals of the chromophores related to the argentophilic interactions in the Ag+ cysteine coordination polymers in aqueous solutions.2 This feature has allowed for a variety of photofunctional applications, such as spectral sensing, via tuning the interactions between ligands on the polymeric backbone.1–4,8–10 In those investigations, the coordination polymers were made to behave as individual polymeric species. We therefore proposed to investigate the behavior of the coordination polymers in their aggregated forms, a subject not much pursued. For this purpose, we designed a cysteine-based ligand (L-/D-1, Scheme 1) by introducing an AIEgen, namely tetraphenylethylene (TPE), whose emission was expected to be sensitive to the extent of aggregation. A control compound, namely L-2 bearing an AIEgen directly linked to the cysteine residue5 and having only three instead of four phenyl spacers, was employed (Scheme 1). Syntheses of these ligands are described in the ESI† (Schemes S1 and S2). The new compounds were fully characterized by performing 1H and 13C NMR and mass spectrometry (MS) experiments (Fig. S36–S44, ESI†) and the Ag+–L-1 coordination polymers were characterized using MALDI-TOF MS (Fig. S45, ESI†).
Absorption and CD spectra of D-1 in the presence of increasing concentrations of Ag+ were first examined for the compound in THF (Fig. 1a). D-1 by itself exhibited an absorption at 325 nm; this absorption red-shifted, specifically to 340 nm, when Ag+ cation was introduced, suggesting the occurrence of the Ag+⋯Ag+ interactions.16–18 Meanwhile, negative and positive CD signals appeared at 350 nm and 295 nm, respectively. The CD profile of L-1 in the presence of Ag+ in THF was found to be the mirror image of that of D-1 with Ag+, confirming that the chirality of the formed species originated from the molecular chirality of ligand L-1 or D-1.19–21 Plots of the absorbance or CD signals against concentration of Ag+ (Fig. S1, ESI†) suggested a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry, agreeing with that deduced from the Job plots (Fig. S2, ESI†). DLS results (Fig. S3, ESI†) and TEM (Fig. S4, ESI†), SEM and AFM images (Fig. 2) indicated the formation of helical fiber structures with diameters of ca. 28 nm and lengths of up to tens of micrometers, i.e., the coordination polymers of Ag+ with L-1 or D-1 (Scheme 1). The helical senses of the Ag+–L-1 and Ag+–D-1 coordination polymers formed in THF were left- (M) and right-handed (P), respectively (Fig. 2a–d). The acquired 1H NMR spectrum of the ligand in THF-d8 showed well-resolved signals (Fig. S5, ESI†), which increasingly broadened and became less well-resolved when increasing concentrations of Ag+ were added, evident for the formation of the coordination polymers.22 The efficient communication of the molecular chirality of the ligand into the chromophore relating to the Ag+⋯Ag+ interactions along the polymeric backbone thus suggested the occurrence of interactions between ligands and their networking with the Ag+⋯Ag+ interactions.2,4–6,9 Fluorescence of D-1 in THF was observed at 490 nm upon excitation at 350 nm, and was increasingly enhanced when increasing amounts of Ag+ were added up to 1 equivalent of Ag+, at which point the fluorescence enhancement finally levelled off (Imax/I0) at 2.3 (Fig. S6, ESI†). Absorption and CD spectra recorded next of the coordination polymers in a variety of solvents (Fig. 1b) showed profiles similar to those in THF, except for an extremely weak CD signal in water. We thus tentatively concluded that the coordination polymers formed in all the tested solvents, despite no crystal structures obtained of the coordination polymers even after many trials. The Ag+-thiolate coordination polymers may have formed intertwined polymeric structures (see ref. 19 and 23).
:1 stoichiometry, agreeing with that deduced from the Job plots (Fig. S2, ESI†). DLS results (Fig. S3, ESI†) and TEM (Fig. S4, ESI†), SEM and AFM images (Fig. 2) indicated the formation of helical fiber structures with diameters of ca. 28 nm and lengths of up to tens of micrometers, i.e., the coordination polymers of Ag+ with L-1 or D-1 (Scheme 1). The helical senses of the Ag+–L-1 and Ag+–D-1 coordination polymers formed in THF were left- (M) and right-handed (P), respectively (Fig. 2a–d). The acquired 1H NMR spectrum of the ligand in THF-d8 showed well-resolved signals (Fig. S5, ESI†), which increasingly broadened and became less well-resolved when increasing concentrations of Ag+ were added, evident for the formation of the coordination polymers.22 The efficient communication of the molecular chirality of the ligand into the chromophore relating to the Ag+⋯Ag+ interactions along the polymeric backbone thus suggested the occurrence of interactions between ligands and their networking with the Ag+⋯Ag+ interactions.2,4–6,9 Fluorescence of D-1 in THF was observed at 490 nm upon excitation at 350 nm, and was increasingly enhanced when increasing amounts of Ag+ were added up to 1 equivalent of Ag+, at which point the fluorescence enhancement finally levelled off (Imax/I0) at 2.3 (Fig. S6, ESI†). Absorption and CD spectra recorded next of the coordination polymers in a variety of solvents (Fig. 1b) showed profiles similar to those in THF, except for an extremely weak CD signal in water. We thus tentatively concluded that the coordination polymers formed in all the tested solvents, despite no crystal structures obtained of the coordination polymers even after many trials. The Ag+-thiolate coordination polymers may have formed intertwined polymeric structures (see ref. 19 and 23).
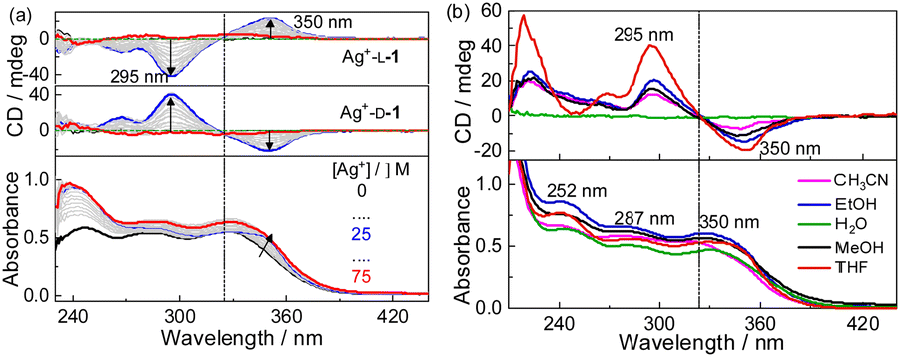 | ||
| Fig. 1 (a) Absorption (bottom panel) and CD (top panel) spectra of D-1 in the presence of increasing concentrations Ag+ in THF and (b) absorption and CD spectra of Ag+–D-1 coordination polymers in indicated solvents. Dissymmetry factor |gabs| values of the coordination polymers in THF were calculated from (a), and found to be 3.39 × 10−3 at 295 nm and 1.22 × 10−3 at 350 nm. [L-1] = [D-1] = 25 μM, [Ag+] = 0–75 μM (in panel a) or 25 μM (in panel b). | ||
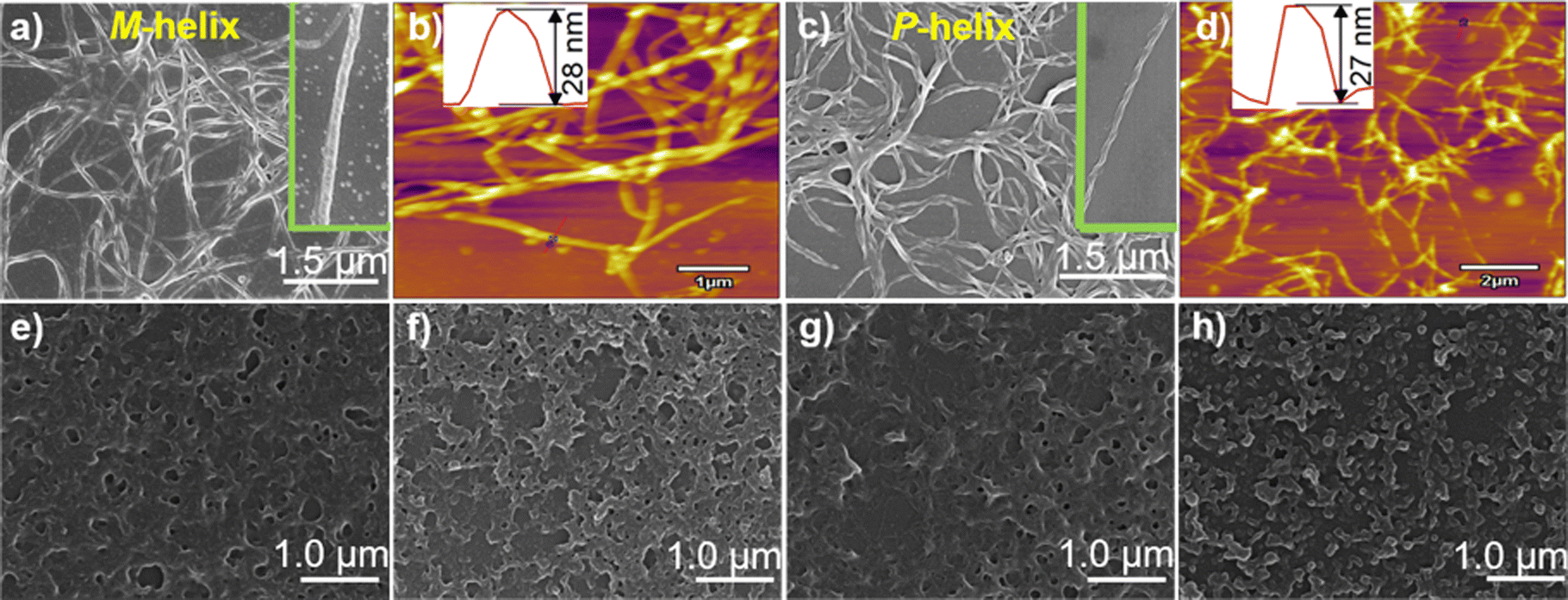 | ||
| Fig. 2 SEM and AFM images of supramolecular structures of (a and b) Ag+–L-1 formed in THF, and (c–h) Ag+–D-1 in (c and d) THF, (e) EtOH, (f) MeOH, (g) CH3CN and (h) H2O. The insets in (a) and (c) show, respectively, two types of polymeric structures, namely the M-helix of Ag+–L-1 and P-helix of Ag+–D-1. [Ag+–L-1] = [Ag+–D-1] = 25 μM. | ||
To exclude the effect of self-aggregation of the coordination polymers due to stacking of the large aromatic TPE moieties, various concentrations were tested. As illustrated in Fig. S7–S10 (ESI†), addition of Ag+ to THF solutions of 1 at low concentrations (5 and 15 μM) led to spectral changes similar to those observed at the higher concentration of 25 μM. Furthermore, the experiments with coordination polymers at the different concentrations all showed nearly the same maximum fluorescence enhancement, Imax/I0, of 2.3.
These measurements thus confirmed that it was the Ag+⋯Ag+ interactions that induced the assembly of L-1 or D-1 in the presence of Ag+.
Hydrophobic TPE derivatives have been shown to undergo aggregation in water-rich solutions.24–26 However, D-1 at 25 μM was found to be CD silent in H2O/THF mixtures of various compositions (Fig. S11, ESI†), indicating that D-1 at 25 μM existed in a monomeric form. In contrast, Ag+–D-1 and Ag+–L-1 coordination polymers at the same ligand concentration of 25 μM were observed to be CD active—and, more interestingly, signs of the Cotton effects related to the ligand-to-metal–metal charge transfer (LMMCT) at 350 nm and N-benzamide moiety at 272 nm were found inverted when the volume content of H2O reached 50% (Fig. 3a and b). Meanwhile, hypochromatic shifts by ca. 10 nm, specifically from 272 nm to 260 nm and 350 nm to 340 nm, were observed together with a remarkable reduction in the CD intensity, compared to those in pure THF (Fig. 3 and Fig. S12a, ESI†). Compared to the CD curves of Ag+–L-1, those of Ag+–D-1 showed a reversed, mirror image relationship (Fig. 3b and Fig. S12b, ESI†). In both cases, the CD profiles remained unchanged for solvent composition VTHF/VH2O values varying from 10:0 to 6:4, but then the signal was weakened and then reversed sign as the volume ratio was further changed to 5:5 and then 4:6, respectively. Similar observations were made at the same volume composition of 5:5 in solutions with a lower concentration of the coordination polymers (15 μM, Fig. S13 and S14, ESI†).
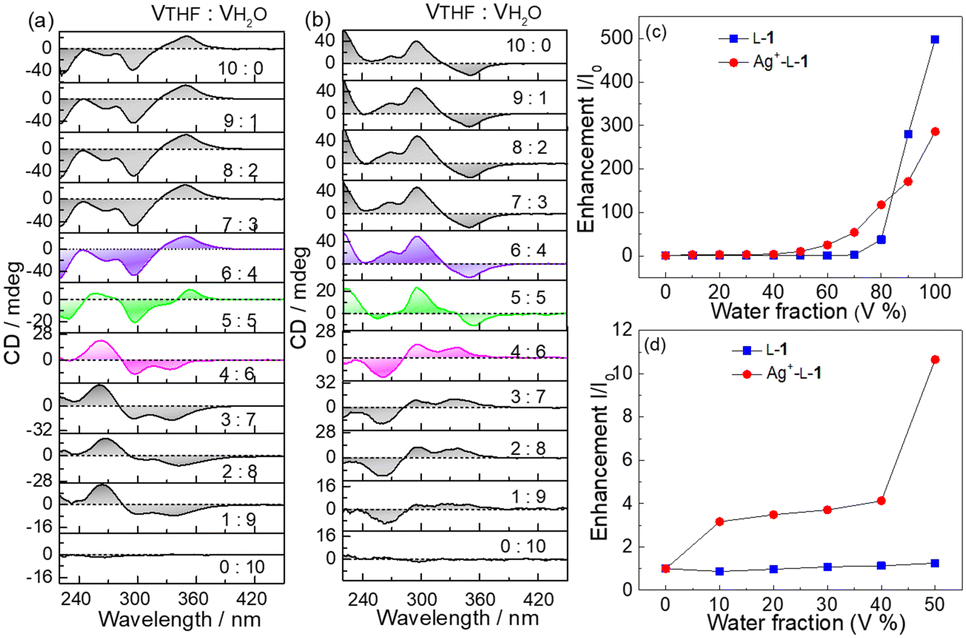 | ||
| Fig. 3 (a and b) CD spectra of (a) Ag+–L-1 and (b) Ag+–D-1 in THF containing increasing volume fractions of water. (c and d) Plots of the enhancements of fluorescence of L-1 and Ag+–L-1 at 484 nm versus water volume fraction. λex = 350 nm, [Ag+] = [L-1] = [D-1] = 25 μM. | ||
SEM images indicated that with increasing water volume fraction, the helical polymeric structure of the Ag+–D-1 coordination polymers formed in pure THF gradually transformed into nanoparticles in the water-rich solutions (Fig. S15, ESI†). A plot of DLS-derived hydrodynamic diameter versus water volume fraction showed a “bell” shape, with a plateau between water volume fractions of 20% and 70% (Fig. S16, ESI†). Fluorescence spectra of L-1 in H2O/THF binary mixtures in the absence (Fig. 4a) and presence of Ag+ (Fig. 4b) suggested that the Ag+–L-1 coordination polymers aggregated in H2O/THF volume compositions higher than 5:5, while L-1 itself aggregated at a much higher water volume fraction of 70% (Fig. 3c and d), following a model of the aggregation-induced emission (AIE).11,12,27–29 This result indicated that the formation of coordination polymers of the thiol ligand promoted the aggregation. The observation of the inversion of the CD profile of the coordination polymers at the solvent composition at which aggregation occurred suggested that it was the aggregation of the coordination polymers that led to the inversion of the supramolecular chirality. Also note that the aggregation of the coordination polymers in water-rich H2O/THF solutions resulted in relatively high enhancements of the emission, specifically I/I0 > 3 (Fig. 3c, d and 4 versus Fig. S6, ESI†). A similar CD profile inversion was not observed in the same series of H2O/THF solutions of the coordination polymers of Ag+ with the control ligand L-2, i.e., that with three instead of four phenyl groups (Ag+–L-2, Fig. S17, ESI†). The signals of the fluorescence spectra of the Ag+–L-2 coordination polymers did not undergo a dramatic enhancement when in the water-rich solvents, i.e., up to 80% by volume of H2O (Fig. S18–S20, ESI†). The difference in aggregation between the coordination polymers of Ag+ with 1 and 2 indicated a contribution of the additional benzene spacer in 1 to increasing the flexibility of the TPE AIEgen so that its emission could be more sensitive to the occurrence of aggregation, in addition to its contribution to increasing the hydrophobicity of ligand 1. This feature is expected to be important for future applications in developing photofunctional materials in terms of ligand structure design. For this purpose, pH and temperature effects on the Ag+–D-1 or Ag+–L-1 coordination polymers in THF and/or 3:7 (v/v) THF/H2O by using their CD and emission spectral variations were examined. As shown in Fig. S21–S23 (ESI†), the chiroptical activity of Ag+–D-1 coordination polymers did not change much as the pH was changed in the range 3.6–9.0 while the emission intensity was slightly quenched; at higher pH, precipitations started to occur. We concluded there to be a negligible influence of pH in the 3.6–9.0 range on the stability of coordination polymers in 3:7 (v/v) THF/H2O. Moreover, effect of temperature on CD and fluorescence of coordination polymers in THF and 3:7 (v/v) THF/H2O binary mixtures were investigated. Upon increasing the solution temperature from 20 °C to 60 °C, the intensities of the CD signals of Ag+–L-1 coordination polymers in THF slightly decreased (Fig. S24 and S25, ESI†), suggesting a gradual disassembly of the coordination polymers. When the temperature was then lowered back to 20 °C, the original CD signals were nearly fully restored. The fluorescence also displayed a reversible change with change in temperature (Fig. S26 and S27, ESI†), both suggesting an excellent thermostability of the Ag+–L-1 coordination polymers within the tested temperature range. In 3:7 (v/v) THF/H2O, however, an irreversible CD change was observed (Fig. S28 and S29, ESI†), and the emission intensity of the coordination polymers failed to recover to the original value after a cycle of heating and cooling (Fig. S30 and S31, ESI†), indicating a poorer stability of the aggregates of the coordination polymers when in water-rich THF solutions.
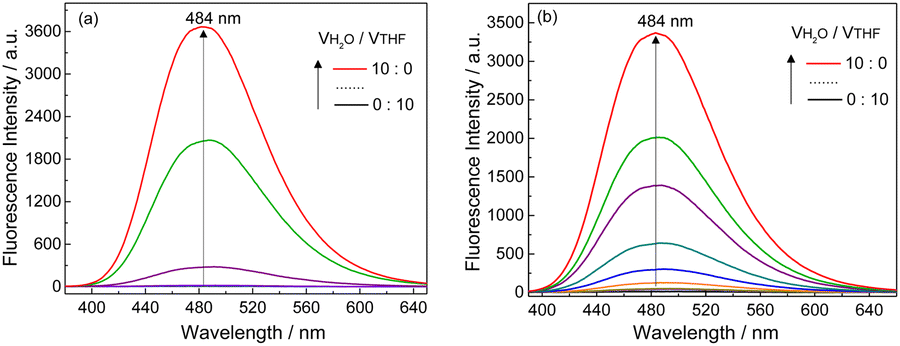 | ||
| Fig. 4 Fluorescence spectra of (a) L-1 and (b) Ag+–L-1 in H2O/THF mixtures with increasing volume fractions of H2O. λex = 350 nm, [L-1] = [Ag+–L-1] = 25 μM. | ||
The supramolecular chirality of the coordination polymers was further probed by examining their CD profiles as a function of the enantiomeric excess (ee) of the ligand.30 In aprotic organic solvents, namely THF (Fig. 5a and b and Fig. S32, ESI†) and CH3CN (Fig. 5c and d and Fig. S33, ESI†), the CD spectrum of the coordination polymers of Ag+ formed with mixtures of the enantiomer pair L-1 and D-1 exhibited an “S”- or “Z”-shaped dependence on the ee of ligand, whereas an anti-“S”- or anti-“Z”-shaped dependence was observed in protic solvents, namely CH3OH (Fig. 5c and d and Fig. S34, ESI†) and C2H5OH (Fig. 5c and d and Fig. S35, ESI†). Interestingly, such dependence in water-rich 7:3 (v/v) H2O/THF, in which the coordination polymers aggregated, was found to be “S”-shaped (Fig. 5e and f), despite the protic nature of the bulk solvent that would have led to an anti-“S”-shaped dependence as that observed for the individual coordination polymers in CH3OH or C2H5OH (Fig. 5c and d and Fig. S34 and S35, ESI†). This result meant that the chromophores in the aggregated coordination polymers in water-rich protic solutions experienced a microenvironment similar to that in aprotic solvent.
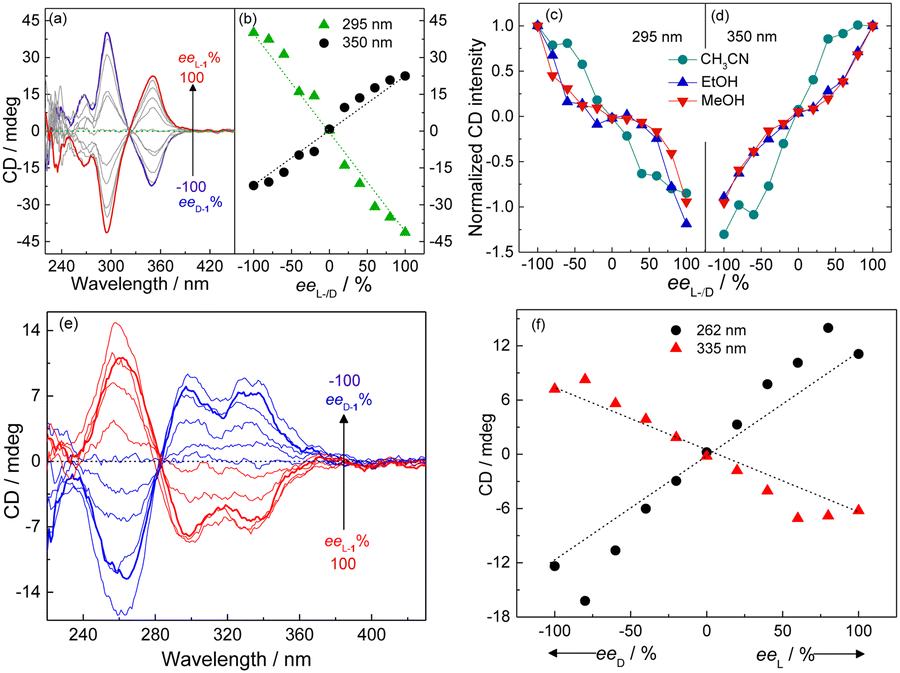 | ||
| Fig. 5 (a and e) CD spectra and (b–d and f) plots of CD signals of Ag+-1 coordination polymers of varying ee of 1 in (a and b) THF, (e and f) 7:3 (v/v) H2O/THF and (c and d) other protic (CH3OH, C2H5OH) and aprotic (CH3CN) organic solvents. [Ag+] = 25 μM, [D-1] + [L-1] = 25 μM. | ||
In summary, we have reported coordination polymers of Ag+ with a chiral cysteine-based ligand containing a TPE AIEgen. The formation of the coordination polymers in a variety of aprotic and protic organic solvents were indicated by spectral signals, e.g., absorption, CD and emission signals, to exist in the form of individual polymeric species. In binary H2O/THF solutions with water volume fractions above 50%, the coordination polymers were indicated to aggregate by morphology observations, a dramatic enhancement of the emission, characteristic of the AIE character of the TPE-fluorophore attached to the thiol ligand, and the inversion of the CD profile. Such behavior did not happen with the coordination polymers of the ligand with one phenyl group absent between the TPE-fluorophore and the cysteine residue, suggesting the importance of the structural design in which the ligand would more sensitively experience the extent of aggregation by extruding more into the bulk solution. An interesting observation made with the supramolecular chirality of the aggregated coordination polymers in the water-rich THF solution was that the CD-ee dependence remained S-shaped, opposite to that observed in protic solvents such as CH3OH and C2H5OH in which the coordination polymers existed as individual polymeric species. This observation suggested a shielding of the chromophore of the coordination polymers in the aggregate forms from the protic solvent components. Our data thus showed that the aggregation of the dynamic coordination polymers resulted in substantial spectral changes, providing a new avenue to develop smart photofunctional materials by controlling the extent of aggregation of the coordination polymers. Creating a large variety of structural modifications on the thiol ligand would be expected to lead to many such opportunities, and with eventually tunable spectral properties.
This work has been supported by the National Natural Science Foundation of China (grant no. 21820102006, 91856118, 21904111 and 22241503) and the fundamental research funds for the central universities (grant no. 20720220005 and 20720220121).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J.-S. Shen, D.-H. Li, Q.-G. Cai and Y.-B. Jiang, J. Mater. Chem., 2009, 19, 6219 RSC.
- J.-S. Shen, D.-H. Li, M.-B. Zhang, J. Zhou, H. Zhang and Y.-B. Jiang, Langmuir, 2011, 27, 481 CrossRef CAS PubMed.
- D.-H. Li, J.-S. Shen, N. Chen, Y.-B. Ruan and Y.-B. Jiang, Chem. Commun., 2011, 47, 5900 RSC.
- Q. Zhang, Y. Hong, N. Chen, D.-D. Tao, Z. Li and Y.-B. Jiang, Chem. Commun., 2015, 51, 8017 RSC.
- D.-D. Tao, Q. Wang, X.-S. Yan, N. Chen, Z. Li and Y.-B. Jiang, Chem. Commun., 2017, 53, 255 RSC.
- Y. Yuan, Y.-W. Xiao, X.-S. Yan, S.-X. Wu, H. Luo, J.-B. Lin, Z. Li and Y.-B. Jiang, Chem. Commun., 2019, 55, 12849 RSC.
- D.-D. Tao, J.-H. Wei, X.-S. Yan, Q. Wang, B.-H. Kou, N. Chen and Y.-B. Jiang, Chem. Commun., 2020, 56, 15133 RSC.
- Q. Wang, S.-L. Dong, D.-D. Tao, Z. Li and Y.-B. Jiang, Coord. Chem. Rev., 2021, 432, 213717 CrossRef CAS.
- S.-L. Dong, Y. Xu, Y.-Z. Chen, X.-S. Yan, Z. Li, J.-W. Xie and Y.-B. Jiang, Inorg. Chem., 2021, 60, 5413 CrossRef CAS PubMed.
- Y. Xu, S.-L. Dong, X.-S. Yan, Q. Wang, Z. Li and Y.-B. Jiang, Chem. Commun., 2021, 57, 4311 RSC.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC.
- Z. Zhao, H.-K. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746 CrossRef CAS PubMed.
- A. K. Jassal, Inorg. Chem. Front., 2020, 7, 3735 RSC.
- O. Veselska, C. Dessal, S. Melizi, N. Guillou, D. Podbevsek, G. Ledoux, E. Elkaim, A. Fateeva and A. Demessence, Inorg. Chem., 2019, 58, 99 CrossRef CAS PubMed.
- C. Streb, C. Ritchie, D.-L. Long, P. Kögerler and L. Cronin, Angew. Chem., Int. Ed., 2007, 46, 7579 CrossRef CAS PubMed.
- C.-M. Che, M.-C. Tse, M. C. W. Chan, K.-K. Cheung, D. L. Phillips and K.-H. Leung, J. Am. Chem. Soc., 2000, 122, 2464 CrossRef CAS.
- Y. Akanuma, T. Imaoka, H. Sato and K. Yamamoto, Angew. Chem., Int. Ed., 2021, 60, 4551 CrossRef CAS PubMed.
- C. Li, K. Deng, Z. Tang and L. Jiang, J. Am. Chem. Soc., 2010, 132, 8202 CrossRef CAS PubMed.
- W. Jiang, Z.-b Qu, P. Kumar, D. Vecchio, Y. Wang, Y. Ma, J. H. Bahng, K. Bernardino, W. R. Gomes, F. M. Colombari, A. Lozada-Blanco, M. Veksler, E. Marino, A. Simon, C. Murray, S. R. Muniz, A. F. de Moura and N. A. Kotov, Science, 2020, 368, 642 CrossRef CAS PubMed.
- M. Jakob, A. von Weber, A. Kartouzian and U. Heiz, Phys. Chem. Chem. Phys., 2018, 20, 20347 RSC.
- H. Ju, Y. Tsuruoka, M. Hayano, E. Lee, K. M. Park, M. Ikeda, I. J. Ishi, S. Kuwahara and Y. Habata, Angew. Chem., Int. Ed., 2021, 60, 650 CrossRef CAS PubMed.
- B. O. Leung, F. Jalilehvand, V. Mah, M. Parvez and Q. Wu, Inorg. Chem., 2013, 52, 4593 CrossRef CAS PubMed.
- J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144 RSC.
- N. Lu, X. Gao, M. Pan, B. Zhao and J. Deng, Macromolecules, 2020, 53, 8041 CrossRef CAS.
- Y. Wang, Y. Cai, L. Cao, M. Cen, Y. Chen, R. Zhang, T. Chen, H. Dai, L. Hu and Y. Yao, Chem. Commun., 2019, 55, 10132 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- Q. C. Peng, X. M. Luo, Y. J. Qin, T. Wang, B. Bai, X. L. Wei, K. Li and S. Q. Zang, CCS Chem., 2022, 4, 3686 CrossRef CAS.
- Y. J. Kong, Z. P. Yan, S. Li, H. F. Su, K. Li, Y. X. Zheng and S. Q. Zang, Angew. Chem., Int. Ed., 2020, 59, 5336 CrossRef CAS PubMed.
- X. Yan, Q. Wang, X.-X. Chen and Y.-B. Jiang, Adv. Mater., 2020, 32, 1904667 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc00474k |
| This journal is © The Royal Society of Chemistry 2023 |