
Amorphization engineered VSe2−x nanosheets with abundant Se-vacancies for enhanced N2 electroreduction†
Yaojing
Luo‡
a,
Qingqing
Li‡
a,
Ye
Tian‡
 b,
Yaping
Liu‡
a and
Ke
Chu‡
*a
b,
Yaping
Liu‡
a and
Ke
Chu‡
*a
aSchool of Materials Science and Engineering, Lanzhou Jiaotong University, Lanzhou 730070, China. E-mail: chuk630@mail.lzjtu.cn
bDepartment of Physics, College of Science, Hebei North University, Zhangjiakou 075000, Hebei, China
First published on 24th December 2021
Abstract
Electrochemical N2 fixation through the nitrogen reduction reaction (NRR) is a promising route for sustainable NH3 synthesis, while exploring high-performance NRR catalysts lies at the heart of achieving high-efficiency NRR electrocatalysis. Herein, we reported the structural regulation of VSe2 by amorphization engineering, which simultaneously triggered the enriched Se-vacancies. The developed amorphous VSe2−x nanosheets with abundant Se-vacancies (a-VSe2−x) delivered a much enhanced NRR activity with an NH3 yield of 65.7 μg h−1 mg−1 and a faradaic efficiency of 16.3% at −0.4 V, being 8.8- and 3.5-fold higher than those of their crystalline counterparts, respectively. Density functional theory computations combined with molecular dynamics simulations revealed that the amorphization-triggered Se-vacancies could induce the upraised d-band center of unsaturated V atoms, capable of promoting the binding of key *N2/*NNH species to result in an energetically favorable NRR process.
Nitrogen fixation to ammonia is a vital chemical process since NH3 is a crucial nitrogen source for producing fertilizers and is also a highly viable chemical energy carrier.1–3 However, the Haber–Bosch (H–B) process for industrial N2 fixation is performed under high temperature and pressure conditions, which entail a large burden for global energy consumption.4–6 Meanwhile, the hydrogen feedstock of the H–B process primarily originates from coal gasification and methane reforming which causes severe CO2 emissions.7 Alternatively, electrochemical N2 fixation through the nitrogen reduction reaction (NRR) has gained noticeable popularity as a promising route for sustainable NH3 synthesis.8–15 Despite recent progress, the electrocatalytic NRR properties in terms of the NH3 production rate and N2-to-NH3 faradaic efficiency still fall short of the practical implementation, arising from the low N2 solubility in aqueous electrolyte, sluggish N
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond dissociation kinetics and competitive hydrogen evolution reaction (HER).16–29 Thus, it is of great significance to explore advanced electrocatalysts that can reduce the energy barriers for NN bond activation and prohibit the occurrence of the HER simultaneously.30–42
N bond dissociation kinetics and competitive hydrogen evolution reaction (HER).16–29 Thus, it is of great significance to explore advanced electrocatalysts that can reduce the energy barriers for NN bond activation and prohibit the occurrence of the HER simultaneously.30–42
Transition metal selenides have emerged as promising electrocatalysts by virtue of their intriguing physicochemical properties, tunable electronic structure, facile synthesis and good durability.43 Although selenides have been extensively investigated for a series of electrocatalytic reactions,44–46 studies on selenides are rarely related to the electrocatalytic NRR to date. In addition, the NRR activity of the earlier reported selenides is still far from satisfactory,47 leaving much room for further improvement. Amorphization engineering by brokering the atomic long-range order has been regarded as one of the effective strategies for fine-tuning the catalyst electronic structure towards improved catalytic performance.48–51 The amorphous electrocatalysts exhibit the enhanced NRR activity with respect to their crystalline counterparts, attributed primarily to the amorphization-induced unsaturated electronic configuration which promotes the N2 adsorption/activation and optimizes the binding energies of reaction intermediates for the efficient NRR process. Inspired by this, the amorphization engineering may provide the feasibility for regulating the structure and electronic configuration of selenides to achieve the potentially high NRR performance.
Here, we report an amorphization engineering strategy to improve the NRR activity and selectivity of VSe2. Supercritical CO2 (SC-CO2) treatment was utilized to transform crystalline VSe2 nanosheets into amorphous ones composed of abundant Se-vacancies (a-VSe2−x). Such a-VSe2−x exhibited an attractive NRR activity with an NH3 yield of 65.7 μg h−1 mg−1 and an FE of 16.3% at −0.4 V, far better than those of their crystalline counterparts. The electronic structure and NRR mechanism of a-VSe2−x were further unraveled by the density functional theory (DFT) calculations and molecular dynamics (MD) simulations.
The crystalline VSe2 nanosheets (c-VSe2) were first prepared by a facile solvothermal method. As a low-cost and non-flammable reaction solvent, SC-CO2 has unique properties of low viscosity, high diffusion coefficients and good wetting of surfaces, which can be used for liquid exfoliation, phase engineering, and the preparation of amorphous materials.52 During the SC-CO2 treatment process, the SC-CO2 molecules can easily penetrate into the crystal layers of c-VSe2 due to the high diffusion coefficients of SC-CO2 and the assistance of external pressure. Meanwhile, water molecules can also be carried by SC-CO2 and transferred into the layers of c-VSe2.52,53 With the increase in the number of impregnating SC-CO2 and water molecules at the interlayer of c-VSe2, the periodic atom arrangement of c-VSe2 can be destroyed, leading to the crystal-to-amorphous phase conversion with simultaneous removal of plentiful Se atoms,54 obtaining a-VSe2−x. The transmission electron microscopy (TEM) images show that both c-VSe2 (Fig. 1a) and a-VSe2−x (Fig. 1b) present a typical two-dimensional (2D) sheet-like morphology, indicating that amorphization does not change the original 2D morphology of c-VSe2, whereas the structural differences between c-VSe2 and a-VSe2−x are unveiled by selected area electron diffraction (SAED), X-ray diffraction (XRD) and high-resolution TEM (HRTEM) characterization studies.
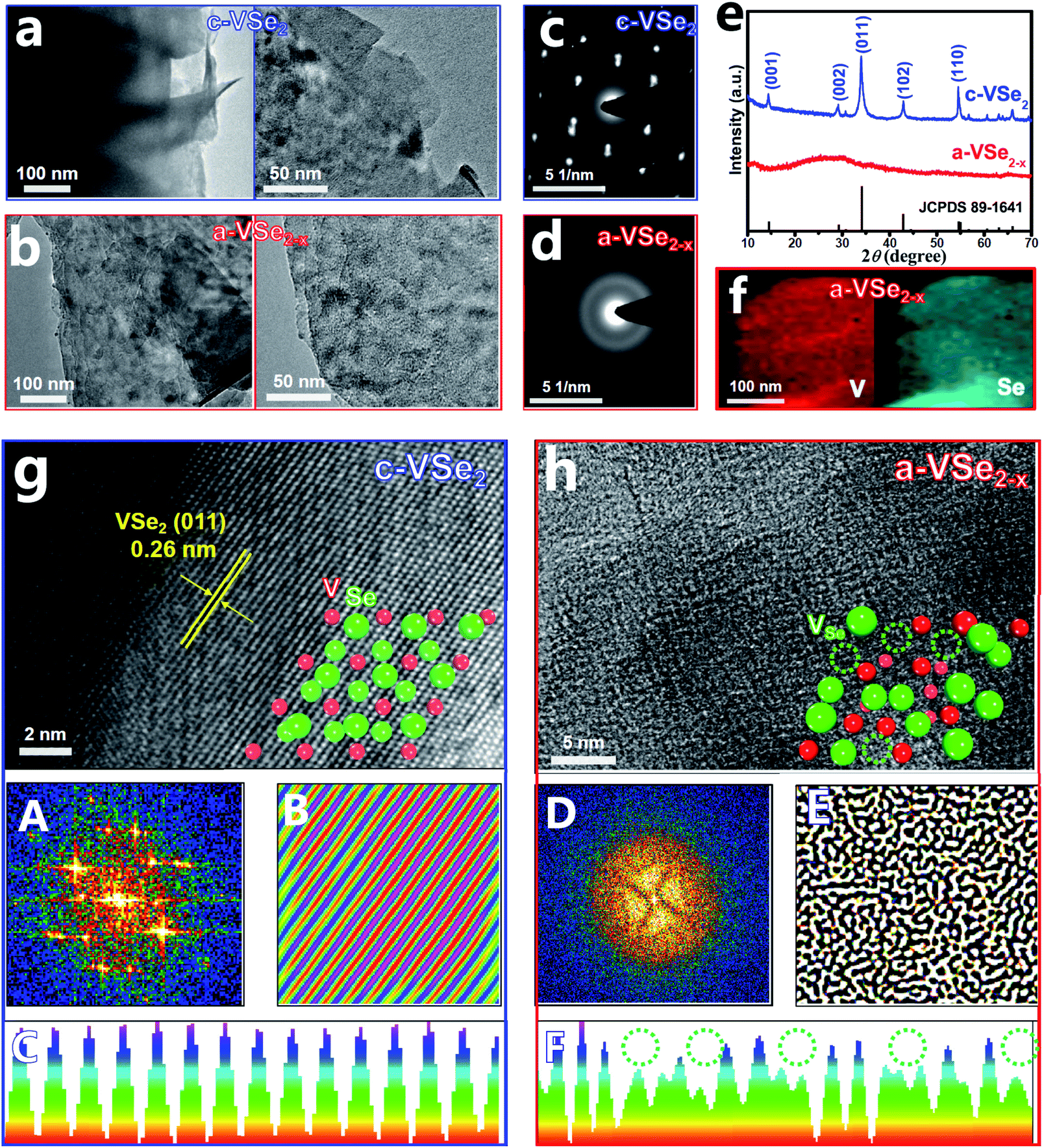 | ||
| Fig. 1 (a and b) TEM images of c-VSe2 and a-VSe2−x at various magnifications. (c and d) SAED patterns of c-VSe2 and a-VSe2−x. (e) XRD patterns of c-VSe2 and a-VSe2−x. (f) Elemental mapping images of a-VSe2−x. (g and h) HRTEM images of c-VSe2 and a-VSe2−x (inset: atomic structure models) and corresponding FFT, IFFT and lattice line scanning analyses (panel A–F). | ||
The SAED pattern of c-VSe2 (Fig. 1c) shows well-defined spots, while a halo feature is presented by a-VSe2−x (Fig. 1d), indicating its amorphous structure. As revealed by the XRD patterns (Fig. 1e), in comparison with the distinct diffraction peaks of c-VSe2, no obvious peaks are found for a-VSe2−x, indicative of its amorphous phase. The elemental mapping images of a-VSe2−x (Fig. 1f) show the uniform distribution of V and Se elements over the entire nanosheets. Further, the HRTEM image of c-VSe2 (Fig. 1g) shows the well-resolved lattice fringes with a d spacing of 0.26 nm, corresponding to the (011) facet of a-VSe2−x. A randomly selected region in the HRTEM image of c-VSe2 is further analyzed by Fast Fourier Transform (FFT) and Inverse Fast Fourier Transform (IFFT),55–57 showing the diffraction dots (FFT, panel A) and crisscross lattice stripes (IFFT, panel B). Further lattice line scanning analysis (panel C) reveals the periodically arranged lattice atoms. All these HRTEM-related analyses verify the highly crystalline structure of c-VSe2. In stark contrast, the HRTEM image of a-VSe2−x (Fig. 1h) shows a rather disordered lattice structure. The corresponding FFT/IFFT analyses show the sun-like pattern (FFT, panel D) and turbostratic structure (IFFT, panel E),51 suggesting the amorphous structure of a-VSe2−x. The lattice line scanning analysis (panel F) reveals the loss of many lattice atoms (dotted circles), implying the generation of enriched vacancies on a-VSe2−x. The elemental analysis of a-VSe2−x unravels a reduced Se/V molar ratio of 1.82 (1.75 for a-VSe2−x and 1.98 for c-VSe2 based on the XPS analyses) relative to its nominal ratio (2), suggesting the Se-deficient nature of a-VSe2−x which comprise abundant Se-vacancies.
X-Ray photoelectron spectroscopy (XPS) is used to investigate the electronic bonding states. For both c-VSe2 and a-VSe2−x, the V 2p spectra (Fig. 2a) show two V 2p3/2 and V 2p1/2 peaks centered at ∼515.5 and ∼523.1 eV, respectively. The deconvoluted peaks at 54.5 and 55.3 eV are associated with the Se 3d state (Fig. 2b). Obviously, both V 2p and Se 3d spectra of a-VSe2−x are found to present an obvious peak-shift toward the low binding energies compared to those of c-VSe2. The increased electronic states of a-VSe2−x are mainly caused by the amorphization-triggered Se-vacancies. The electron paramagnetic resonance (EPR, Fig. 2c) spectroscopy shows that a-VSe2−x exhibits a much stronger EPR signal than c-VSe2, further confirming the amounts of Se-vacancies generated on a-VSe2−x.
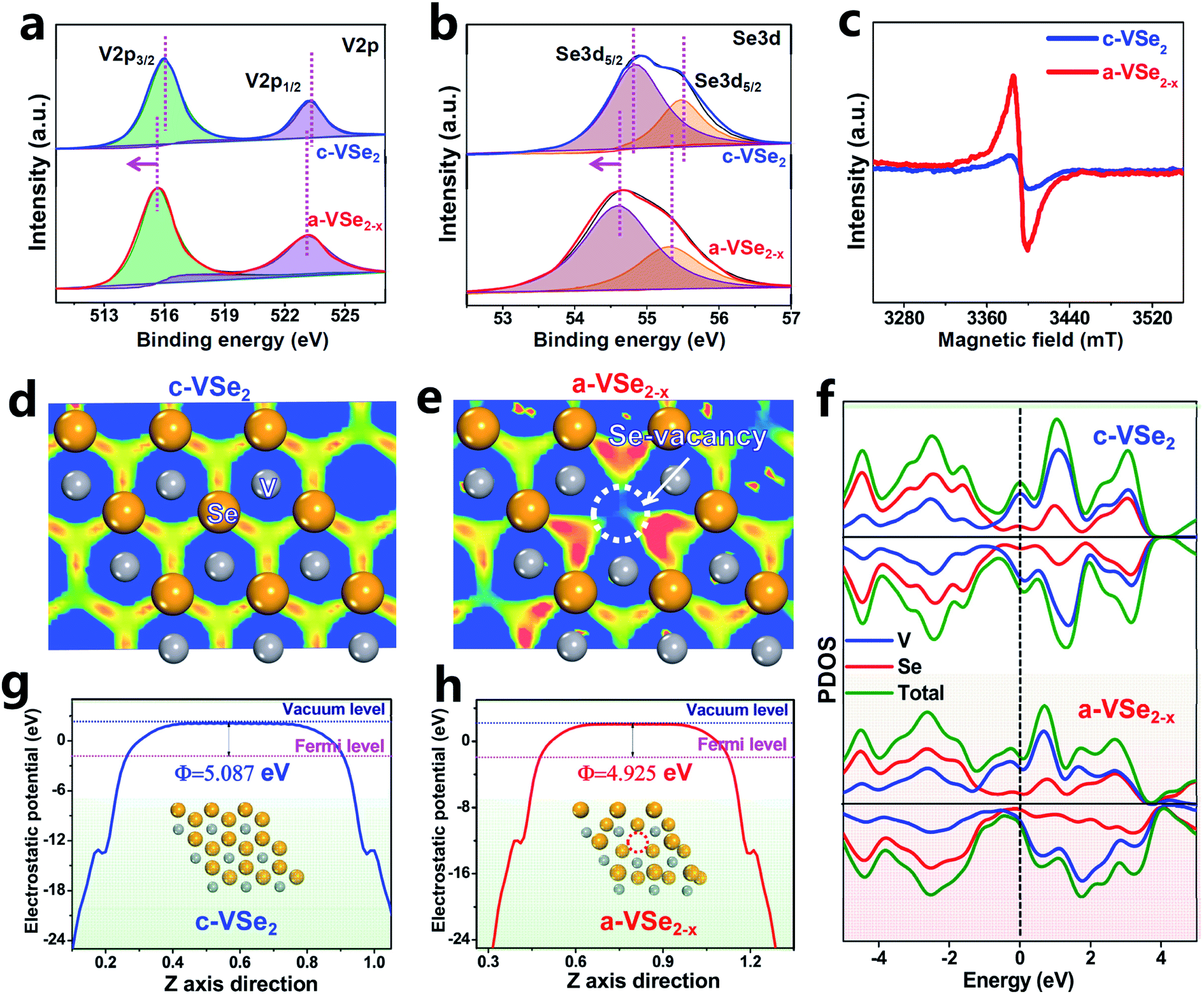 | ||
| Fig. 2 (a) XPS V 2p spectra of c-VSe2 and a-VSe2−x. (b) XPS Se 3d spectra of c-VSe2 and a-VSe2−x. (c) EPR spectra of c-VSe2 and a-VSe2−x. (d and e) Electron localization function images of c-VSe2 and a-VSe2−x. Blue and red regions represent the electron depletion and accumulation, respectively. (f) PDOS of c-VSe2 and a-VSe2−x. (g and h) Average potential profiles of c-VSe2 and a-VSe2−x. | ||
DFT calculations are performed to investigate the electronic structures of c-VSe2 and a-VSe2−x. According to the electron localization function (ELF) images sliced along the (001) plane, c-VSe2 (Fig. 2d) displays an electron localization feature with a uniform distribution of electrons around V atoms. As for a-VSe2−x (Fig. 2e), the electrons tend to flow toward Se-vacancies, leading to the delocalized electron gathering at the Se-vacancy region. Such Se-vacancy-induced delocalized electrons are most likely transferred into the antibonding orbitals of N2 molecules for polarization of the NN covalent bond.58 As illustrated by the projected density of states (PDOS, Fig. 2f), both c-VSe2 and a-VSe2−x deliver higher occupied states across the Fermi level, suggesting their high conductivity which is favorable for accelerated proton-coupled electron transfer. This can also be experimentally confirmed by electrochemical impedance spectroscopy (EIS, Fig. S1†). The good conductivity of c-VSe2 and a-VSe2−x is mainly attributed to the strong electron coupling interactions involved in V4+–V4+ pairs of VSe2 inducing the metallic properties.43 Additionally, in contrast to the symmetry spin-up/spin-down states of c-VSe2, the spin-up and spin-down states are asymmetrical for a-VSe2−x, manifesting that a-VSe2−x is spin-polarized with the existence of magnetic spin moments.59 The spin moments of a-VSe2−x can loosen the orbital electrons in the NN covalent bond, favorable for activating and polarizing the N2 molecules.59 Moreover, the average potential profiles reveal that a-VSe2−x has a lower work function (4.925 eV, Fig. 2g) than c-VSe2 (5.087 eV, Fig. 2h), indicating that a-VSe2−x has higher electronic energy levels and a stronger electron-donating ability to enhance the π back-donation and promote the NRR activity.60–62
Electrochemical NRR measurements are performed in N2-saturated 0.5 M LiClO4 solution in a gas-tight two-compartment electrolytic cell.39 All potentials reported in this work were converted to the reversible hydrogen electrode (RHE) scale. Prior to NRR tests, all feed gases are purified through acid/alkaline traps to remove any possible NOx contaminants (Fig. S2†).63 As shown in the linear sweep voltammetry (LSV, Fig. S3†) curves, a cathodic current onset is present at around −0.2 V due to the occurrence of the HER in both Ar and N2 saturated electrolytes, while N2 electrolyte presents a noticeably higher current density than Ar solution, confirming the feasibility of a-VSe2−x for N2 electroreduction. When the potential becomes more negative, the LSV curves in Ar and N2 saturated electrolytes gradually coincide due to the enhanced HER that dominates the electrode process.29 Electrolysis for quantitatively evaluating the NRR performance of a-VSe2−x is conducted by chronoamperometry at various potentials for 2 h (Fig. 3a). The produced NH3 in each resulting electrolyte is analyzed by a colorimetric method (Fig. S4†). Fig. 3b shows the corresponding UV-vis absorption spectra of the electrolytes stained with an indophenol indicator, and the calculated data of NH3 yields and FEs are plotted in Fig. 3c. Notably, a-VSe2−x attains the highest NH3 yield (65.7 μg h−1 mg−1) and FE (16.3%) at −0.4 V, which are compared favorably with those of most reported electrocatalysts. Moreover, no N2H4 by-product can be detected after NRR electrolysis (Fig. S5 and S6†), confirming a high selectivity of a-VSe2−x for N2-to-NH3 electroreduction.
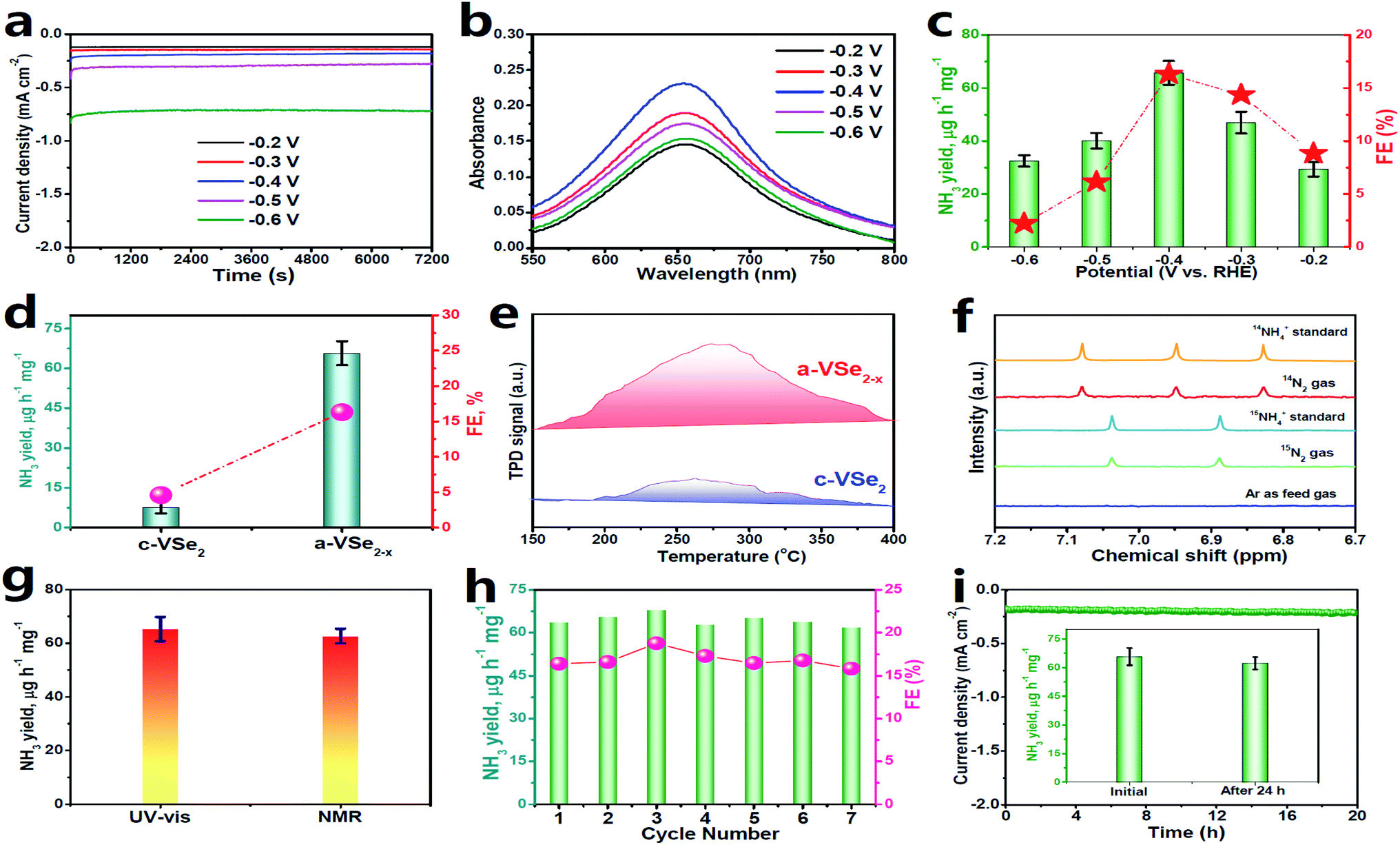 | ||
| Fig. 3 (a) Chronoamperometry curves of a-VSe2−x at various potentials after 2 h of NRR electrocatalysis, corresponding (b) UV-vis absorption spectra, and (c) obtained NH3 yields and FEs. (d) Comparison of the NH3 yields/FEs of c-VSe2 and a-VSe2−x. (e) N2-TPD spectra of c-VSe2 and a-VSe2−x. (f) Isotope-labeling NMR spectra of the electrolytes fed by 15N2/14N2 and Ar gases. (g) Comparison of NH3 yields by UV-vis and NMR tests (Fig. S9†). (h) Cycling test. (i) Chronoamperometry test for 24 h (inset: NH3 yields of initial and post-NRR electrolysis). (d and f–i) are performed at −0.4 V. | ||
In comparison with a-VSe2−x (Fig. 3d), c-VSe2 presents a much inferior NRR activity with the optimum NH3 yield (4.5 μg h−1 mg−1) and FE (2.3%) being 8.8- and 3.5-fold lower than those of a-VSe2−x, respectively, suggesting the enhanced NRR activity of a-VSe2−x. As seen in Fig. S7,† a-VSe2−x shows a double layer capacitance (Cdl) of 7.03 mF cm−2, 2.2 times higher than that of c-VSe2 (3.14 mF cm−2), suggesting the increased electrochemical surface area (ECSA) of a-VSe2−x for electrocatalytic NRR. The increased ECSA of a-VSe2−x reflects its large number of exposed active sites derived primarily from the abundant unsaturated V atoms and Se-vacancies, which would facilitate the N2 adsorption that is the prerequisite for the electrocatalytic NRR process. As shown in Fig. 3e, a-VSe2−x delivers a stronger chemical desorption peak relative to c-VSe2, indicating that the amorphous structure enables the substantially improved adsorption of N2, rendering a-VSe2−x with the boosted NRR performance.
A series of control experiments are conducted to verify that NH3 production is produced through the NRR process. First, no NH3 can be detected in three control colorimetric tests (Fig. S8†). Isotope-labelling electrocatalysis based on 1H nuclear magnetic resonance (1H-NMR) spectroscopy is performed by employing 15N2 as the feed gas. As displayed in Fig. 3f, the resultant electrolytes show doublet coupling peaks assignable to 15NH4+, whereas no doublet 15NH4+ peaks can be detected in the electrolyte saturated with Ar under the same electrochemical conditions, indicating that the 15NH4+ products are derived from the NRR. The same results can also be obtained by employing 14N2 as the feed gas. We also use the 1H-NMR technique to quantify the 15NH4+ concentration, and the 15NH4+ production rate catalyzed by a-VSe2−x is determined to correlate well with that obtained by the UV-vis results (Fig. 3g and S9†). Moreover, an N2–Ar gas switching experiment (Fig. S10†) shows that the NH3 products are only obtained in the periods of N2-saturated electrolyte, evidencing the electrocatalytic NRR process. All these control experiments unequivocally validate the N2-to-NH3 electroreduction.
The stability of a catalyst in the electrolysis process also represents a critical factor for practical applications. For the consecutive seven cycles of electrolysis (Fig. 3h), no considerable fluctuations in NH3 yield/FEs can be found, implying the good cycling stability of a-VSe2−x. In addition, the chronoamperometry test for 24 h of continuous NRR electrolysis (Fig. 3i) shows a steady current density with no significant decay, and the corresponding NH3 yield (Fig. 3i, inset) exhibits a very small change with respect to the initial one, proving the excellent long-term durability of a-VSe2−x. Moreover, no obvious change in the structure of a-VSe2−x for the post-NRR is found in terms of XRD (Fig. S11†), TEM (Fig. S12†) and XPS (Fig. S13†) characterization studies, signifying the good structural durability.
Theoretical investigations are carried out to provide more insights into the NRR mechanism of a-VSe2−x. Fig. 4a shows that c-VSe2 exhibits a weak N2 adsorption with an ignorable N–N elongation (1.11 Å). The poor N2 adsorption is also found for amorphous VSe2 without Se-vacancies (Fig. S14†). With Se-vacancies on a-VSe2−x (Fig. 4b), however, N2 can be favorably trapped into the Se-vacancy site where *N2 is bonded with three unsaturated V atoms to generate a trinuclear side-on coordinated configuration (Fig. S15†). Electron deformation density (Fig. 4c) displays the distinct charge accumulation and depletion around *N2 and its adjacent unsaturated V atoms, suggesting that Se-vacancies on a-VSe2−x play a crucial role in creating unsaturated V atoms as electron back-donation centers for effective N2 activation. The PDOS analysis (Fig. 4d) shows that a-VSe2−x presents an upshift of the d-band center (unsaturated V atoms), suggesting that the antibonding states between unsaturated V atoms of a-VSe2−x and adsorbed intermediates are less occupied, resulting in their enhanced binding strength. In this scenario, the V-3d orbital of a-VSe2−x can favorably interact with the N2-2p orbital to generate hybrid states that facilitate the sufficient adsorption and activation of N2 (Fig. S16†). Thermodynamically speaking, *NNH is generally deemed as a key intermediate for the NRR protonation process.21,39,64 With the strong N2 activation over a-VSe2−x, a prominent charge interaction can also be found between *NNH and unsaturated V atoms (Fig. S17†), leading to the effective *NNH stabilization. The favorable stabilization of *N2/*NNH species over a-VSe2−x can also be confirmed by the MD simulations (Fig. 4e and S18†), showing that a-VSe2−x simulation systems display stronger N2-catalyst and NNH-catalyst interactions (Fig. 4f, corresponding to a higher radial distribution function (RDF)) with respect to their c-VSe2 counterparts. Therefore, the amorphization-triggered Se-vacancies can induce the upraised d-band center of unsaturated V atoms to promote N2 activation and *NNH stabilization, favorable for the expedited NRR activity of a-VSe2−x.
 | ||
| Fig. 4 (a and b) Optimized structures of *N2 on c-VSe2 and a-VSe2−x. (c) Electron deformation density of *N2 on a-VSe2−x. Yellow and cyan iso-surfaces represent electron accumulation and electron depletion, respectively. (d) PDOS of V-3d in c-VSe2 and a-VSe2−x for determining the d-band center (εd). (e) Snapshots of the dynamic process of N2/NNH adsorption on c-VSe2 and a-VSe2−x in LiClO4 electrolyte after 5 ns MD simulations, and corresponding (f) RDF curves of N2-catalyst and NNH-catalyst interactions. (g) Free energy diagrams of energetically favorable NRR pathways on c-VSe2 and a-VSe2-x at U = −0.4 V and pH = 7. (h) UL(NRR) − UL(HER) of c-VSe2 and a-VSe2−x. | ||
The Gibbs free energies for all the NRR reaction steps are explored on c-VSe2 and a-VSe2−x (Fig. 4g). Based on their respective energetically favorable NRR pathways (alternating pathway for c-VSe2 (Fig. S19†) and consecutive for a-VSe2−x (Fig. S20†)),65 the transition from *N2 to *NNH is determined to be the rate-determining step (RDS) for both c-VSe2 and a-VSe2−x. The large RDS energy barrier (2.21 eV) of c-VSe2 implies its sluggish hydrogenation process with low NRR activity. Remarkably, the RDS energy barrier of a-VSe2−x is largely decreased to 0.96 eV, indicating more favorable NRR energetics and enhanced NRR activity on a-VSe2−x. The much reduced RDS barrier of a-VSe2−x is attributed to the enhanced binding of key *N2/*NNH species derived from the Se-vacancy-induced upshift of the d-band center for active unsaturated V atoms. Considering the competitive reaction of H2 evolution, we finally examine the HER activity by estimating the limiting potential difference between the NRR and HER (UL(NRR) − UL(HER)) which is a good indicator for assessing the NRR selectivity.66 The more positive UL(NRR) − UL(HER) implies a higher NRR selectivity. As displayed in Fig. 4h, a-VSe2−x presents a more positive UL(NRR) − UL(HER) than c-VSe2, suggesting a-VSe2−x can effectively alleviate the competition from the HER. Therefore, the amorphization-triggered Se-vacancies can boost the NRR and impede the HER synchronously, rendering a-VSe2−x with high NRR activity and selectivity.
In summary, a-VSe2−x has been verified to be a highly active and selective NRR catalyst, significantly outperforming the c-VSe2 counterpart, together with the favorable cycling and long-term stability. Theoretical investigations revealed that the largely enhanced NRR activity of a-VSe2−x originated from the amorphization-triggered Se-vacancies that could induce the upraised d-band center of unsaturated V atoms, promoting the NRR and impeding the HER synchronously. This work underscores the combined amorphization/vacancy engineering for exploring highly active and selective catalysts for N2-to-NH3 electrocatalysis.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51761024 and 52161025), Natural Science Foundation of Gansu Province (20JR10RA241), Hebei North University General Project (YB2020004) and the “Longyuan Young Talents” Program of Gansu Province.References
- J. A. Brandes, N. Z. Boctor, G. D. Cody, B. A. Cooper, R. M. Hazen and H. S. Yoder Jr, Nature, 1998, 395, 365 CrossRef CAS PubMed.
- J. B. Howard and D. C. Rees, Chem. Rev., 1996, 96, 2965–2982 CrossRef CAS PubMed.
- R. D. Milton, R. Cai, S. Abdellaoui, D. Leech, A. L. De Lacey, M. Pita and S. D. Minteer, Angew. Chem., Int. Ed., 2017, 56, 2680–2683 CrossRef CAS PubMed.
- B. N. Van, Nature, 2002, 415, 381–382 CrossRef PubMed.
- C. J. Van der Ham, M. T. Koper and D. G. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- T. Kandemir, M. E. Schuster, A. Senyshyn, M. Behrens and R. Schlögl, Angew. Chem., Int. Ed., 2013, 52, 12723–12726 CrossRef CAS PubMed.
- P. J. Chirik, Nat. Chem., 2009, 1, 520 CrossRef CAS PubMed.
- T. Xu, J. Liang, S. Li, Z. Xu, L. Yue, T. Li, Y. Luo, Q. Liu, X. Shi, A. M. Asiri, C. Yang and X. Sun, Small Science, 2021, 1, 2000069 CrossRef.
- Q. Liu, T. Xu, Y. Luo, Q. Kong, T. Li, S. Lu, A. A. Alshehri, K. A. Alzahrani and X. Sun, Curr. Opin. Electrochem., 2021, 29, 100766 CrossRef CAS.
- T. Xu, B. Ma, J. Liang, L. Yue, Q. Liu, T. Li, H. Zhao, Y. Luo, S. Lu and X. Sun, Acta Phys.-Chim. Sin., 2020, 37, 2009043 Search PubMed.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C.-P. Chen, M. R. Smith and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed.
- C. Yang, Y. Zhu, J. Liu, Y. Qin, H. Wang, H. Liu, Y. Chen, Z. Zhang and W. Hu, Nano Energy, 2020, 77, 105126 CrossRef CAS.
- Y. Ren, C. Yu, X. Tan, H. Huang, Q. Wei and J. Qiu, Energy Environ. Sci., 2021, 14, 1176–1193 RSC.
- K. Tanifuji and Y. Ohki, Chem. Rev., 2020, 120, 5194–5251 CrossRef CAS PubMed.
- Y. Li, H. Wang, C. Priest, S. Li, P. Xu and G. Wu, Adv. Mater., 2021, 33, 2000381 CrossRef CAS PubMed.
- T. Wang, S. Li, B. He, X. Zhu, Y. Luo, Q. Liu, T. Li, S. Lu, C. Ye, A. M. Asiri and X. Sun, Chin. J. Catal., 2021, 42, 1024–1029 CrossRef CAS.
- T. Wang, Q. Liu, T. Li, S. Lu, G. Chen, X. Shi, A. M. Asiri, Y. Luo, D. Ma and X. Sun, J. Mater. Chem. A, 2021, 9, 884–888 RSC.
- Z. Xiao, Y. Jia, M. Lin, Y. Xia and C. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 8129–8137 CrossRef CAS PubMed.
- L. Xiao, S. Zhu, Y. Liang, Z. Li, S. Wu, S. Luo, C. Chang and Z. Cui, ACS Appl. Mater. Interfaces, 2021, 13, 30722–30730 CrossRef CAS PubMed.
- K. Chu, Y. Liu, J. Wang and H. Zhang, ACS Appl. Energy Mater., 2019, 2, 2288–2295 CrossRef CAS.
- K. Chu, Y. Liu, Y. Chen and Q. Li, J. Mater. Chem. A, 2020, 8, 5200–5208 RSC.
- S. Li, Y. Wang, J. Liang, T. Xu, D. Ma, Q. Liu, T. Li, S. Xu, G. Chen, A. M. Asiri, Y. Luo, Q. Wu and X. Sun, Mater. Today Phys., 2021, 18, 100396 CrossRef CAS.
- X. Zhu, S. Mou, Q. Peng, Q. Liu, Y. Luo, G. Chen, S. Gao and X. Sun, J. Mater. Chem. A, 2020, 8, 1545–1556 RSC.
- W. Guo, K. Zhang, Z. Liang, R. Zou and Q. Xu, Chem. Soc. Rev., 2019, 48, 5658–5716 RSC.
- G. F. Chen, S. Y. Ren, L. L. Zhang, H. Cheng, Y. R. Luo, K. H. Zhu, L. X. Ding and H. H. Wang, Small Methods, 2019, 3, 1800337 CrossRef.
- D. Yan, H. Li, C. Chen, Y. Zou and S. Wang, Small Methods, 2018, 1800331 Search PubMed.
- C. Guo, J. Ran, A. Vasileff and S.-Z. Qiao, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- J. Deng, J. A. Iñiguez and C. Liu, Joule, 2018, 2, 846–856 CrossRef CAS.
- L. Li, C. Tang, B. Xia, H. Jin, Y. Zheng and S.-Z. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS.
- Q. Li, Y. Guo, Y. Tian, W. Liu and K. Chu, J. Mater. Chem. A, 2020, 8, 16195–16202 RSC.
- Z. Du, J. Liang, S. Li, Z. Xu, T. Li, Q. Liu, Y. Luo, F. Zhang, Y. Liu, Q. Kong, X. Shi, B. Tang, A. M. Asiri, B. Li and X. Sun, J. Mater. Chem. A, 2021, 9, 13861–13866 RSC.
- K. Chu, Q. Q. Li, Y. H. Cheng and Y. P. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 11789–11796 CrossRef CAS PubMed.
- K. Chu, Y. Cheng, Q. Li, Y. Liu and Y. Tian, J. Mater. Chem. A, 2020, 8, 5865–5873 RSC.
- X. Cheng, J. Wang, W. Xiong, T. Wang, T. Wu, Y. Luo, S. Lu, G. Chen, S. Gao, X. Shi, Z. Jiang, X. Niu and X. Sun, ChemNanoMat, 2020, 6, 1315–1319 CrossRef CAS.
- P. Wang, Q. Q. Li, Y. H. Cheng and K. Chu, J. Mater. Sci., 2020, 55, 4624–4632 CrossRef CAS.
- W. Gu, Y. Guo, Q. Li, Y. Tian and K. Chu, ACS Appl. Mater. Interfaces, 2020, 12, 37258–37264 CrossRef CAS PubMed.
- X. Li, Y. Tian, X. Wang, Y. Guo and K. Chu, Sustainable Energy Fuels, 2021, 5, 4277–4283 RSC.
- K. Chu, X. Li, Q. Li, Y. Guo and H. Zhang, Small, 2021, 17, 2102363 CrossRef CAS PubMed.
- X. Li, Y. Luo, Q. Li, Y. Guo and K. Chu, J. Mater. Chem. A, 2021, 9, 15955–15962 RSC.
- Q. Li, P. Shen, Y. Tian, X. Li and K. Chu, J. Colloid Interface Sci., 2022, 606, 204–212 CrossRef CAS PubMed.
- K. Chu, Q. Li, Y. Liu, J. Wang and Y. Cheng, Appl. Catal., B, 2020, 267, 118693 CrossRef CAS.
- Y. Luo, P. Shen, X. Li, Y.-l. Guo and K. Chu, Chem. Commun., 2021, 57, 9930–9933 RSC.
- X. Peng, Y. Yan, X. Jin, C. Huang, W. Jin, B. Gao and P. K. Chu, Nano Energy, 2020, 105234 CrossRef CAS.
- B. Wang, K. Srinivas, X. Wang, Z. Su, B. Yu, Y. Liu, F. Ma, D. Yang and Y. Chen, Nanoscale, 2021, 13, 9651–9658 RSC.
- K. R. Park, D. T. Tran, T. T. Nguyen, N. H. Kim and J. H. Lee, Chem. Eng. J., 2021, 422, 130048 CrossRef CAS.
- Y. Li, Y. Zhao, F.-M. Li, Z. Dang and P. Gao, ACS Appl. Mater. Interfaces, 2021, 13, 34457–34467 CrossRef CAS PubMed.
- S. Yang, W. Ye, D. Zhang, X. Fang and D. Yan, Inorg. Chem. Front., 2021, 8, 1762–1770 RSC.
- A. Bergmann, E. Martinez-Moreno, D. Teschner, P. Chernev, M. Gliech, J. F. De Araújo, T. Reier, H. Dau and P. Strasser, Nat. Commun., 2015, 6, 8625 CrossRef CAS PubMed.
- J. Zhang, R. Yin, Q. Shao, T. Zhu and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 5609–5613 CrossRef CAS PubMed.
- J. Wang, L. Han, B. Huang, Q. Shao, H. L. Xin and X. Huang, Nat. Commun., 2019, 10, 1–11 CrossRef PubMed.
- K. Chu, W. Gu, Q. Li, Y. Liu, Y. Tian and W. Liu, J. Energy Chem., 2021, 53, 82–89 CrossRef.
- Y. Ren and Q. Xu, Energy Environ. Mater., 2018, 1, 46–60 CrossRef.
- W. Liu and Q. Xu, Chem.–Eur. J., 2018, 24, 13693–13700 CrossRef CAS PubMed.
- W. Liu, Q. Xu, W. Cui, C. Zhu and Y. Qi, Angew. Chem., Int. Ed., 2017, 56, 1600–1604 CrossRef CAS PubMed.
- K. Chu, H. Nan, Q. Li, Y. Guo, Y. Tian and W. Liu, J. Energy Chem., 2021, 53, 132–138 CrossRef.
- K. Chu, X. H. Wang, Y. B. Li, D. J. Huang, Z. R. Geng, X. L. Zhao, H. Liu and H. Zhang, Mater. Des., 2018, 140, 85–94 CrossRef CAS.
- K. Chu, F. Wang, Y. B. Li, X. H. Wang, D. J. Huang and Z. R. Geng, Composites, Part A, 2018, 109, 267–279 CrossRef CAS.
- Z. Sun, R. Huo, C. Choi, S. Hong, T.-S. Wu, J. Qiu, C. Yan, Z. Han, Y. Liu, Y.-L. Soo and Y. Jung, Nano Energy, 2019, 62, 869–875 CrossRef CAS.
- Z. Chen, J. Zhao, C. R. Cabrera and Z. Chen, Small Methods, 2019, 3, 1800368 CrossRef.
- K. Chu, Y. Liu, Y. Li, J. Wang and H. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 31806–31815 CrossRef CAS PubMed.
- K. Chu, J. Wang, Y. P. Liu, Q. Q. Li and Y. L. Guo, J. Mater. Chem. A, 2020, 8, 7117–7124 RSC.
- K. Chu, Y. P. Liu, Y. B. Li, Y. L. Guo and Y. Tian, ACS Appl. Mater. Interfaces, 2020, 12, 7081–7090 CrossRef CAS PubMed.
- M. Wang, S. Liu, H. Ji, T. Yang, T. Qian and C. Yan, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed.
- K. Chu, Y. Liu, Y. Li, H. Zhang and Y. Tian, J. Mater. Chem. A, 2019, 7, 4389–4394 RSC.
- J. Wang, H. Nan, Y. Tian and K. Chu, ACS Sustainable Chem. Eng., 2020, 8, 12733–12740 CrossRef CAS.
- Y. Liu, X. Kong, X. Guo, Q. Li, J. Ke, R. Wang, Q. Li, Z. Geng and J. Zeng, ACS Catal., 2019, 10, 1077–1085 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta06746j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |