
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic CO2 reduction sensitized by a special-pair mimic porphyrin connected with a rhenium(I) tricarbonyl complex†
Yusuke
Kuramochi
 *ab,
Ren
Sato
a,
Hiroki
Sakuma
b and
Akiharu
Satake
*ab
*ab,
Ren
Sato
a,
Hiroki
Sakuma
b and
Akiharu
Satake
*ab
aGraduate School of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo, 162-8621, Japan
bDepartment of Chemistry, Faculty of Science Division II, Tokyo University of Science, Japan. E-mail: kuramochiy@rs.tus.ac.jp; asatake@rs.tus.ac.jp
First published on 3rd August 2022
Abstract
Zn porphyrins with an imidazolyl group at the meso position generate a highly stable porphyrin dimer by complementary coordination from the imidazolyl to the Zn ion in noncoordinating solvents such as chloroform, which mimics the natural special pair in photosynthesis. In this work, we have synthesized an imidazolyl-substituted Zn porphyrin connected with a Re 2,2-bipyridine tricarbonyl complex as a CO2 reduction catalyst via a p-phenylene linker, affording a homodimer with two Re complexes on both sides (ReDRe). The dimeric structure is easily dissociated into the corresponding monomers in coordinating solvents. Therefore, we prepared a mixture containing a heterodimer with the Re carbonyl complex on one side (ReD) by simple mixing with an imidazolyl Zn porphyrin and evaporating the solvent. Using the Grubbs catalyst, the subsequent olefin metathesis reaction of the mixture gave covalently linked porphyrin dimers through the allyloxy side chains, enabling the isolation of the stable hetero- (ReD′) and homo-dimers (ReD′Re) with gel permeation chromatography. The Zn porphyrin dimers have intense absorption bands in the visible light region and acted as good photosensitizers in photocatalytic CO2 reduction in a mixture of N,N-dimethylacetamide and triethanolamine (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) containing 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole as the electron donor, giving CO with high selectivity and durability. Under irradiation with strong light intensity, the reaction rate in ReD′ exceeded that of the previous porphyrin
:1 v/v) containing 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole as the electron donor, giving CO with high selectivity and durability. Under irradiation with strong light intensity, the reaction rate in ReD′ exceeded that of the previous porphyrin![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Re complex dyad, ZnP-phen=Re. For instance, after irradiation at 560 nm for 18 h, the turnover number (TONCO) of ReD′ reached 2800, whereas the TONCO of ZnP-phen=Re was 170. The high activity in the system using the porphyrin dimer originates from no accumulation of the one-electron reduced species of the porphyrin that inhibit light absorption due to the inner-filter effect.
Re complex dyad, ZnP-phen=Re. For instance, after irradiation at 560 nm for 18 h, the turnover number (TONCO) of ReD′ reached 2800, whereas the TONCO of ZnP-phen=Re was 170. The high activity in the system using the porphyrin dimer originates from no accumulation of the one-electron reduced species of the porphyrin that inhibit light absorption due to the inner-filter effect.
Introduction
Undoubtedly, CO2 emissions caused by human activity have drastically changed the global climate. Various systems have been developed to reduce CO2 in the atmosphere, and systems converting CO2 into energy-rich compounds with the aid of light (solar energy) have much potential to solve not only the problem of global warming, but simultaneously also that of a shortage of fossil-fuel resources.1 Sunlight consists mainly of visible light with low photon density. The low photon density can be disadvantageous, in particular in multielectron reactions, and therefore photosensitizers capturing visible light with high efficiency are essential.2 In natural photosynthesis, dilute sunlight is collected by networks of pigments—the so-called light-harvesting complexes.3 The collected energy is transferred to the chlorophyll dimer in the reaction center, the so-called “special pair”, that initiates photoinduced electron transfer to give a charge-separated (CS) state in the reaction center.4In 1994, Kobuke et al. reported that Zn porphyrins having an imidazolyl group at the meso position form a slipped-cofacial dimeric structure by complementary coordination from the imidazolyl to the Zn ion of the porphyrin center in noncoordinating solvents, and that the dimeric structure mimics the special pair in photosynthetic systems.5 When the dimer is one-electron oxidized, the produced radical cation delocalizes over two porphyrin units. The CS rate is accelerated in the photoinduced electron transfer, while the charge-recombination rate decelerates to give the ground state.6 The dimeric structure is sufficiently stable to be treated as a photosensitizer in noncoordinating solvents such as chloroform and toluene, but it easily dissociates into monomeric units in coordinating solvents such as pyridine and dimethyl sulfoxide (DMSO). These properties are somewhat advantageous for preparing various combinations of dimers.7 The porphyrin units can be freely exchanged by simply mixing two porphyrin components in a coordinating solvent followed by evaporating the solvent, giving a hetero-porphyrin dimer. For instance, a triad system consisting of a porphyrin dimer with ferrocene as an electron donor on the one side and fullerene as an electron acceptor on the other side was easily formed by the simple mixing of two kinds of Zn porphyrin having either ferrocene or fullerene. The supramolecular triad gave long-lived CS species after efficient photoinduced electron transfer and charge shift.8 In addition, this dimer-based supramolecular methodology is applicable in the construction of various self-assembled porphyrin arrays having cyclic, chain, and sheet structures for photosynthetic antenna models,9–11 two-photon absorption materials,12 solar cell materials,13 solvation/desolvation indicators,14 and so on.
Zn porphyrin has been used as the photosensitizer in homogeneous photoredox reactions.15,16 For example, dyads combining Zn porphyrin and the Re diimine tricarbonyl complex catalyst have been developed for photocatalytic CO2 reduction.16–18 Among them, we have recently reported on a Zn porphyrinRe complex dyad, ZnP-phen=Re (Fig. 1), in which fac-Re(phen)(CO)3Br (where phen = 1,10-phenanthroline) is connected with a Zn porphyrin, and affords CO with high selectivity (>99.9%) and efficiency (ΦCO = 8% and turnover number (TONCO) > 1300) in photocatalytic CO2 reduction using 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) as an electron donor, phenol as a proton source, and N,N-dimethylacetamide (DMA) as a solvent.18
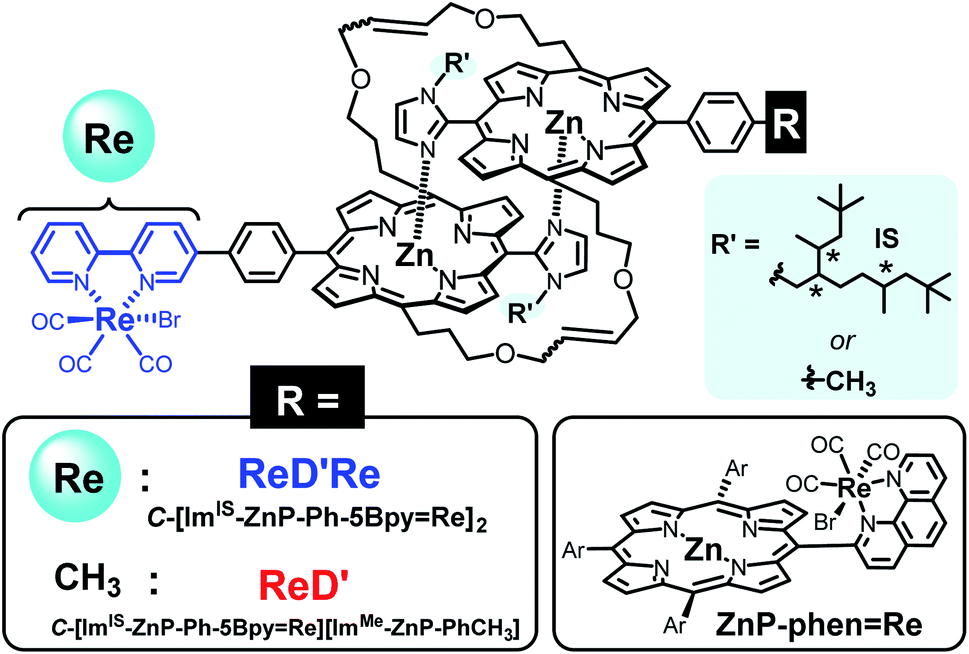 | ||
| Fig. 1 Structures of the porphyrin dimers having the Re complex(es) (ReD′Re and ReD′) and the porphyrinRe complex dyad (ZnP-phen=Re). The asterisks in the IS group indicate asymmetric carbons. | ||
In this work, Kobuke's special-pair mimic porphyrin was first used as the photosensitizer for photocatalytic CO2 reduction. It is expected that the slipped-cofacial dimer has the advantage that the dimer can utilize light of a wider wavelength because the intense Soret band appearing at ∼420 nm of monomeric porphyrin is split by strong excitonic coupling between two Zn porphyrins in close proximity.19 In addition, because the dimeric structure can be easily dissociated in coordinating solvents, unsymmetric heterodimers can be constructed by simply mixing two kinds of porphyrin in a coordinating solvent and then evaporating the solvent. Furthermore, a multisite ring-closing metathesis reaction using the Grubbs catalyst on olefin moieties at the meso positions of the porphyrin fixes the dimeric structure through covalent bonds, giving a stable dimer that does not dissociate even in coordinating solvents such as pyridine or DMSO.20 We have synthesized a Re bipyridine tricarbonyl complex connected to the opposite meso position of the imidazolyl Zn porphyrin through a p-phenylene linker. The porphyrin forms a stable dimer having the Re complex on both sides (ReDRe). The reorganizing procedure by mixing ReDRe and a dimer without the Re complex (D) in pyridine and evaporating the solvent gave a mixture of ReDRe, D, and a hetero-dimer having the Re complex on one side (ReD). The ring-closing metathesis reaction followed by separation of the mixture with gel permeation chromatography (GPC) produced covalently linked dimers, ReD′ and ReD′Re (Fig. 1). Here, a bulky isostearyl group (IS group) was introduced on the imidazolyl group in ReDRe to be soluble in practical organic solvents. The photocatalytic reaction in CO2-saturated DMA containing BIH showed that the triethanolamine (TEOA) presence dramatically improved the catalytic activity. In addition, we performed photocatalytic CO2 reduction using ReDRe and ReD′Re in DMSO, and ReD′Re had higher durability than ReDRe, which was dissociated to the monomers in DMSO, demonstrating that the dimeric structure plays an important role for the high activity in photocatalytic reactions.
Results and discussion
Synthesis of ReDRe
First, we attempted to synthesize the porphyrin dimer having 1-methyl-imidazolyl groups at the meso positions of the porphyrins (see the ESI†). However, when the Re tricarbonyl complexes were introduced into the bpy groups of [ImMe-ZnP-Ph-5Bpy]2, the product became insoluble in preferred solvents (chloroform, dichloromethane, toluene, and acetonitrile) for the ring-closing metathesis reaction using the Grubbs catalyst as the dimer. [ImMe-ZnP-Ph-5Bpy=Re]2 was soluble in N,N-dimethylformamide (DMF) and the 1H NMR spectrum recorded under high-concentration conditions showed an almost dimeric structure in DMF-d7 (Fig. S14†). However, [ImMe-ZnP-Ph-5Bpy=Re]2 was partially dissociated intothe monomer in the concentration range for the intramolecular metathesis reaction of the dimer. We also tried the ring-closing metathesis reaction with the dimer, [ImMe-ZnP-Ph-5Bpy]2, before the introduction of the Re complex, but the reaction did not proceed, probably due to the deactivation of the Grubbs catalyst (1st generation) by coordination of the bpy groups.21 Therefore, we changed the substituent group on the imidazolyl moiety from the methyl group to a bulky IS group (Fig. 1) to improve organic solvent solubility. Scheme 1 shows the synthetic route for the porphyrin dimer having the IS groups at the imidazolyl groups and Re tricarbonyl complexes at the bpy groups ([ImIS-ZnP-Ph-5Bpy=Re]2, ReDRe). The free-base porphyrin ImIS-H2P-PhI was synthesized with 19% yield by condensation of 4-iodobenzaldehyde and ImIS-CHO with meso-(3-allyloxypropyl)dipyrromethane at room temperature (rt). The three asymmetric C atoms of the IS groups provide 23 = 8 stereoisomers and their conformational isomers, resulting in broadened and complicated signals in their 1H and 13C NMR spectra. Successful syntheses of the porphyrins were confirmed by not only using 2-dimensional (2D) NMR but also by comparing with the spectra of porphyrins substituted by 1-methyl-imidazolyl. The complicated signals appearing from 1.38 to −0.80 ppm in the 1H NMR spectrum of ImIS-H2P-PhI were assigned to the methine, methylene, and methyl signals of the IS groups (Fig. S36†). The Zn(II) ion was introduced into ImIS-H2P-PhI by treatment with a methanol solution of Zn(OAc)2 in CHCl3. The 1H NMR spectrum showed that the Im-4 and Im-5 protons that appeared at 7.74–7.72 ppm and 7.52–7.50 ppm in ImIS-H2P-PhI were shifted to a higher field by the Zn insertion to show the peaks at 2.06 ppm for the Im-4 proton and at 5.56 ppm for the Im-5 proton. The spectral change was similar to that of [ImMe-ZnP-PhI]2, indicating that all the stereoisomers quantitatively construct the slipped-cofacial dimer, [ImIS-ZnP-PhI]2 (Fig. S42†). The dimeric structure was also clarified by the characteristic split Soret band in the ultraviolet visible (UV-vis) absorption spectrum (Fig. S46†).19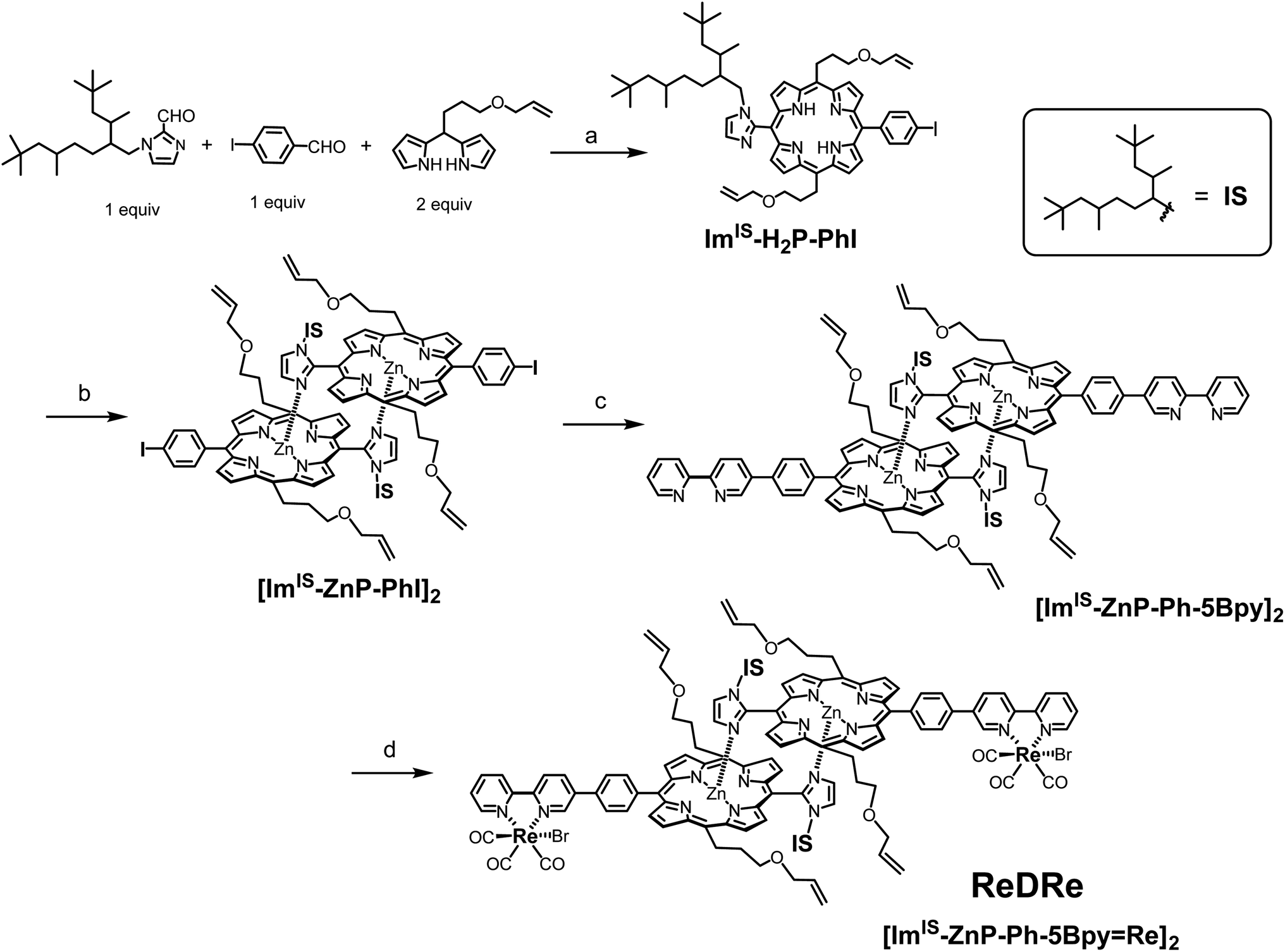 | ||
| Scheme 1 Synthetic route of the noncovalently linked porphyrin dimer, [ImIS-ZnP-Ph-5Bpy=Re]2 (ReDRe). Reagents and conditions: (a) (i) trifluoroacetic acid/CHCl3, rt, and 3.5 h, (ii) Et3N, p-chloranil, rt, overnight, and 19%; (b) Zn(OAc)2 (8 equivalent (equiv.)), CHCl3/CH3OH, rt, overnight, and 96%; (c) 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,2′-bipyridine (5Bpy-B(OR)2, 1.5 equiv.), Pd(PPh3)4 (0.2 equiv.), CsCO3 (2.9 equiv.), toluene/DMF/water, 90 °C, 4 h, and 89%; (d) Re(CO)5Br (0.97 equiv.), toluene, 90 °C, 2 d, and 72%. | ||
The porphyrin dimer with the bpy groups, [ImIS-ZnP-Ph-5Bpy]2, was synthesized by the Suzuki–Miyaura coupling reaction of [ImIS-ZnP-PhI]2 and 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2,2′-bipyridine (5Bpy-B(OR)2) in toluene and DMF. The reaction was first performed under anhydrous conditions, but the coupling reaction did not proceed. The coupling reaction proceeded with water addition, giving [ImIS-ZnP-Ph-5Bpy]2 at 89% yield. The product, [ImIS-ZnP-Ph-5Bpy]2, also showed the split Soret bands in the UV-vis absorption spectrum and the upper-field shifts of the β-pyrrole and imidazolyl protons compared with the free-base porphyrin in the 1H NMR spectrum (Fig. S48†), indicating that the bpy group does not interrupt the formation of the dimeric structure. Here, 5Bpy-B(OR)2 was difficult to purify with column-chromatographic techniques due to the decomposition of boron derivatives on the silica gel column.22 Thus, 5Bpy-B(OR)2 was purified by partitioning between the aqueous and organic layers. The lithiation of 5-bromo-2,2′-bipyridine followed by addition of triisopropyl borate gave a boronic acid derivative, which was extracted at pH = 11 to the aqueous layer to remove the starting material and by-products existing the organic layer. Next, the aqueous layer was evaporated to dryness, and the residue was reacted with pinacol. Pure 5Bpy-B(OR)2 was extracted with toluene and other impurities remained as insoluble materials (see the ESI†).
The introduction of the Re(I) tricarbonyl complex into the bpy groups of [ImIS-ZnP-Ph-5Bpy]2 was performed by refluxing with Re(CO)5Br in toluene. The use of excess amounts (1.5 equiv.) of Re(CO)5Br against the bpy group made the characteristic split Soret band of the dimeric structure change into a single Soret band (Fig. S55†). This observation indicates that the coordination of the excess Re(I) ion to the imidazolyl group of the porphyrin dissociates the dimer to give a monomeric species. The reflux of the mixture in pyridine recovered the split Soret band and gave the target compound, [ImIS-ZnP-Ph-5Bpy=Re]2 (ReDRe, Fig. S55 and 56†), indicating that the monodentate imidazole Re complex dissociated in pyridine, whereas the bidentate bpy complex was stable in pyridine even under reflux conditions. The use of 0.97 equiv. of Re(CO)5Br selectively gave the target compound without forming the monomeric species, (Fig. S55†), and the purification on a silica gel column afforded ReDRe at 72% yield. Three CO peaks at 197.17, 196.88, and 189.20 ppm in the 13C NMR spectrum (Fig. S59†) and three CO stretching peaks at 1901, 1923, and 2020 cm−1 in the infrared (IR) spectrum (Fig. S63†) indicate that the Re tricarbonyl complex has an fac-Re(bpy)(CO)3Br-type structure.23 The bulky IS substituent on the imidazolyl group significantly improved the solubility for organic solvents, and ReDRe was easily soluble in chloroform, dichloromethane, toluene, and acetonitrile over a 0.1 mM concentration.
Fixation of the dimers via covalent bonds in the side chains
To prevent the dissociation of the coordination bond during the catalytic reaction in polar solvents, the dimeric structure was fixed by the ring-closing olefin metathesis reaction on the allyloxy groups at the meso positions of the porphyrin.20,21 First, the metathesis reaction for ReDRe was performed using the Grubbs catalyst (1st generation) in dichloromethane (Scheme 2). The reaction progress was monitored with the UV-vis absorption spectrum in pyridine. The split Soret band was observed even in pyridine after the overnight reaction, indicating that all four allyloxy side chains were covalently linked (Fig. S64†). GPC using a pyridine eluent showed a single peak at 21.2 min, suggesting that the reaction proceeded almost perfectly (Fig. S65†). The matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry using trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]-malononitrile (DCTB) as a matrix gave signals corresponding to the covalently linked dimer, C-[ImIS-ZnP-Ph-5Bpy=Re]2 (ReD′Re) in Scheme 2 (Fig. S66†). The 1H NMR spectrum of the purified ReD′Re was compared with that of the nonmetathesized compound ReDRe (Fig. S68†). The elimination of the exomethylene signals and a change in the methine signals of the allyl ether groups were observed. The broad signals coupled with the neighboring sp3-methylene and terminal exomethylene protons were split into two signals at 6.46 and 6.11 ppm, which were assigned to the E- and Z-olefin isomers, respectively. The molar ratio of the E- and Z-isomers was 1.9:1, suggesting that three isomers exist in a 0.96:1:0.27 ratio (Fig. S69†). Therefore, ReD′Re is a diastereo-mixture formed from the two IS groups and the two olefin parts. The diastereomers are difficult to separate in chromatography and are used as a mixture for a catalytic reaction. In addition, three CO peaks for ReDRe in the 13C NMR spectrum were split into six peaks for ReD′Re, which would result from the E- and Z-olefin isomers (Fig. S70†).
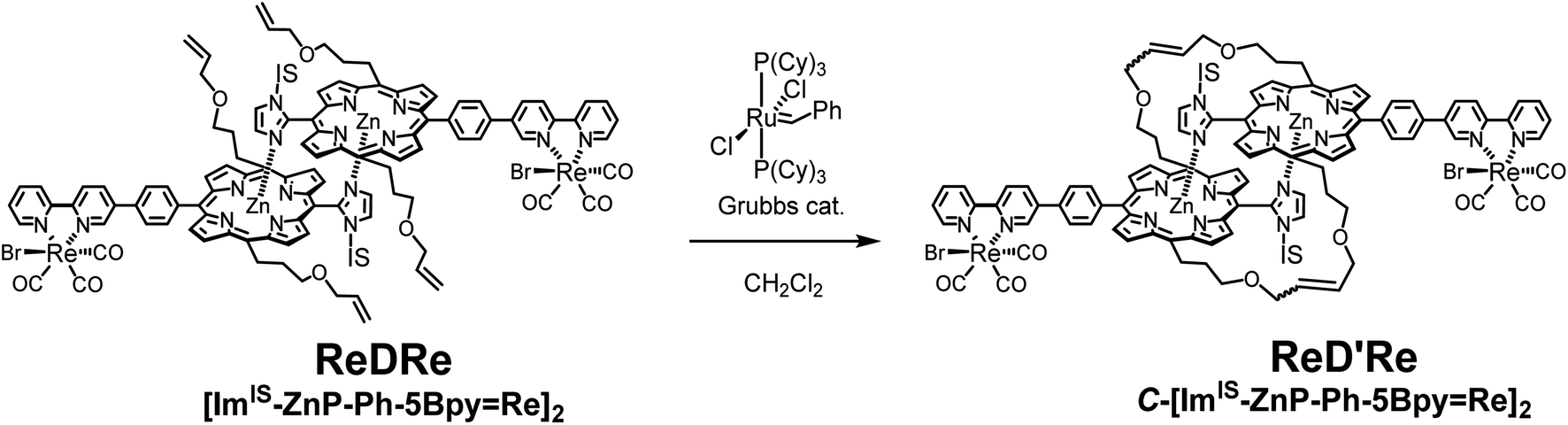 | ||
| Scheme 2 Ring-closing metathesis reaction of ReDRe. | ||
We confirmed that the ring-closing metathesis reaction of the homodimer ReDRe quantitatively proceeds to give the covalently linked dimer, ReD′Re. Next, a covalently linked heterodimer in which two types of imidazolyl Zn porphyrins are paired was prepared. Here, to investigate whether a heterodimer can be formed using the porphyrin with the Re complex, a simple p-tolyl-substituted Zn porphyrin dimer was used (D in Scheme 3). Two types of dimer, ReDRe and D (1:1 molar ratio), were dissolved in pyridine and then the solvent was evaporated. In pyridine, the dimer is dissociated into a pyridine-coordinated monomer. The evaporation of pyridine gives the reorganized dimers. In addition, the ring-closing metathesis reaction was applied to the resulting product in chloroform, giving the heterodimer ReD′Re, accompanied by the homodimers ReD′Re and D′. In the analytical GPC traces of the crude product (Fig. 2), the peaks at 21.2 and 23.8 min were respectively assigned to be ReD′Re and D′ by comparing with the corresponding homodimers. The central peak at 22.1 min was expected as the target ReD′. Larger components (oligomers), which were probably formed by the intermolecular metathesis reaction of the dimers, were also observed before 21 min. The MALDI-TOF mass spectrum of each isolated component proved that the species at 22.1 min is ReD′. The peak area ratio of ReD′Re, ReD′, and D′ in Fig. 2 is a statistical ratio of 0.5:1:0.5. When ReDRe and D were used in a 1:4 molar ratio as the starting dimers, the ratio of ReD′Re, ReD′, and D′ was 0.04:0.26:0.70, which was consistent with the ratio (0.04:0.32:0.64) assumed for the covalently linked dimers to be statistically obtained (Fig. S75†). Thus, if the proportion of D, which can be easily synthesized, is increased, ReD′ can be efficiently obtained from ReDRe. The isolated peaks of ReD′Re and ReD′ have a single Gaussian shape, indicating that the diastereomers originating from the IS and alkene groups hardly affect the molecular size or the hydrodynamic volume.
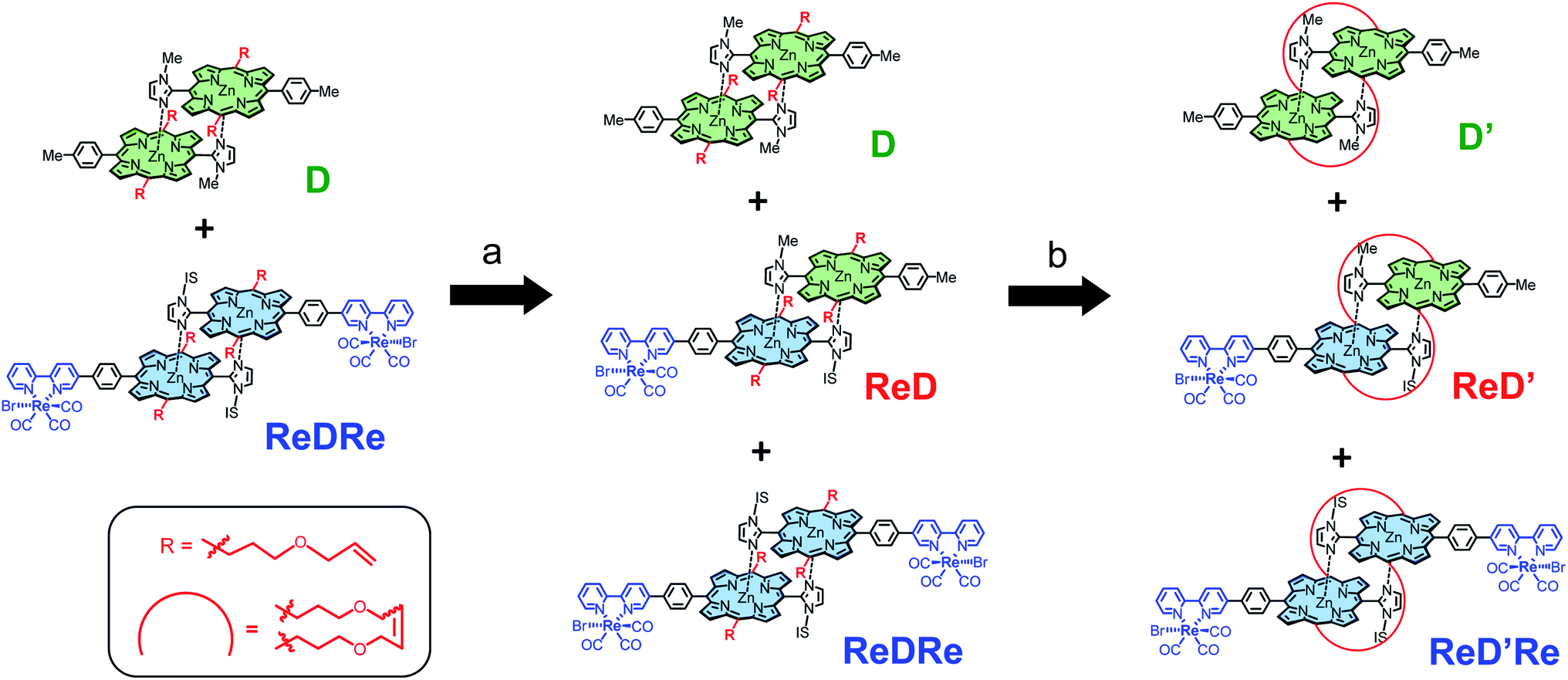 | ||
| Scheme 3 Synthesis of the heterodimer, ReD′. Reagents and conditions: (a) (i) mixing in pyridine at rt, (ii) evaporating the solvent in vacuo; (b) Grubbs catalyst (0.5 equiv.), CHCl3, and rt. | ||
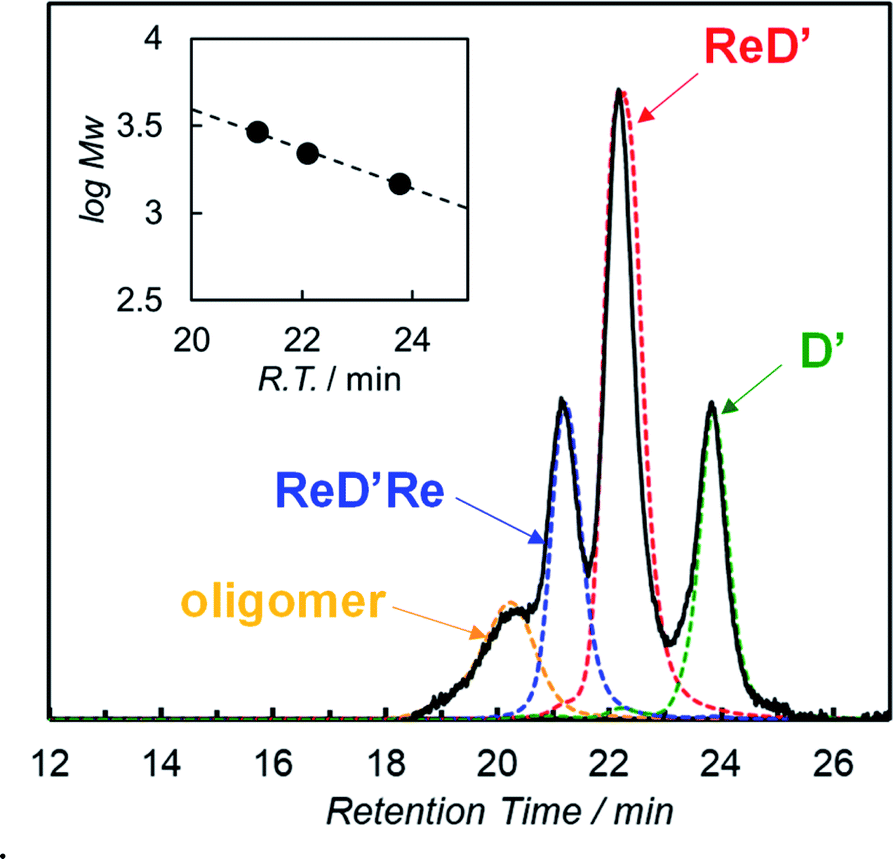 | ||
| Fig. 2 GPC-high-performance liquid chromatography (HPLC) charts (column: TSK gel G2500HHR × 2 + G2000HHR × 1, eluent: pyridine, flow rate: 1.0 mL min−1, and detection: 440 nm) of the crude product obtained after the metathesis reaction using ReDRe and D (molar ratio 1:1), and the isolated samples by preparative GPC-HPLC. The inset shows the logarithm of molecular weight against the retention time.9e,24 | ||
Photochemical properties
To investigate the substituent effect on the imidazolyl group on the photochemical properties, the UV-vis absorption spectrum of [ImIS-ZnP-Ph-5Bpy]2 was compared with that of [ImMe-ZnP-Ph-5Bpy]2 in chloroform (Fig. S83†). Here, [ImMe-ZnP-Ph-5Bpy=Re]2 is insoluble in chloroform. The two spectra overlap almost completely, suggesting that there is no strong electronic interaction between the IS group and the porphyrin unit and that differences in the photochemical properties due to the presence of isomers of the IS groups would be negligible. Previous studies have reported that the E- and Z-olefin isomers at the meso positions of the porphyrins do not affect the electrochemical and photochemical properties of porphyrins.20 In fact, the absorption and fluorescence spectra between ReDRe and ReD′Re in chloroform are almost the same, indicating that the olefin side chains do not affect the electronic state of the porphyrin ring (Fig. S84†).Fig. 3a shows the UV-vis absorption spectra of D′, ReD′Re, ReD′, and fac-Re(bpy)(CO)3Br in DMA, a solvent used for photocatalytic CO2 reduction.25 Because DMA weakly coordinates to Zn porphyrin,18a the noncovalently linked dimer, D, was mostly dissociated into the monomer in the micromolar concentration range. The inset shows a spectral comparison between the Zn porphyrin dimer and fac-Re(bpy)(CO)3Br, indicating that the Zn porphyrin unit has a much larger absorption coefficient than the Re(bpy)(CO)3Br unit. The Soret bands split into two peaks (at 414 and 437 nm; ΔE = 1270 cm−1 for ReD′) due to strong exciton coupling between the two porphyrins in the dimer.19 The excitonic coupling in the Q bands was negligibly small due to the smaller oscillator strengths, and the spectral shape of the Q bands in the dimer was similar to that of the monomeric porphyrin.9e,19 The introduction of the Re(bpy)(CO)3Br unit caused a slight broadening of the Soret bands and a slight red-shift of the peaks throughout the spectrum (Table 1). The dimer shows a much broader Soret band (caused by the exciton coupling) than that of monomeric Zn porphyrin, enabling the use of a very wide wavelength range of visible light for the photocatalytic reactions.
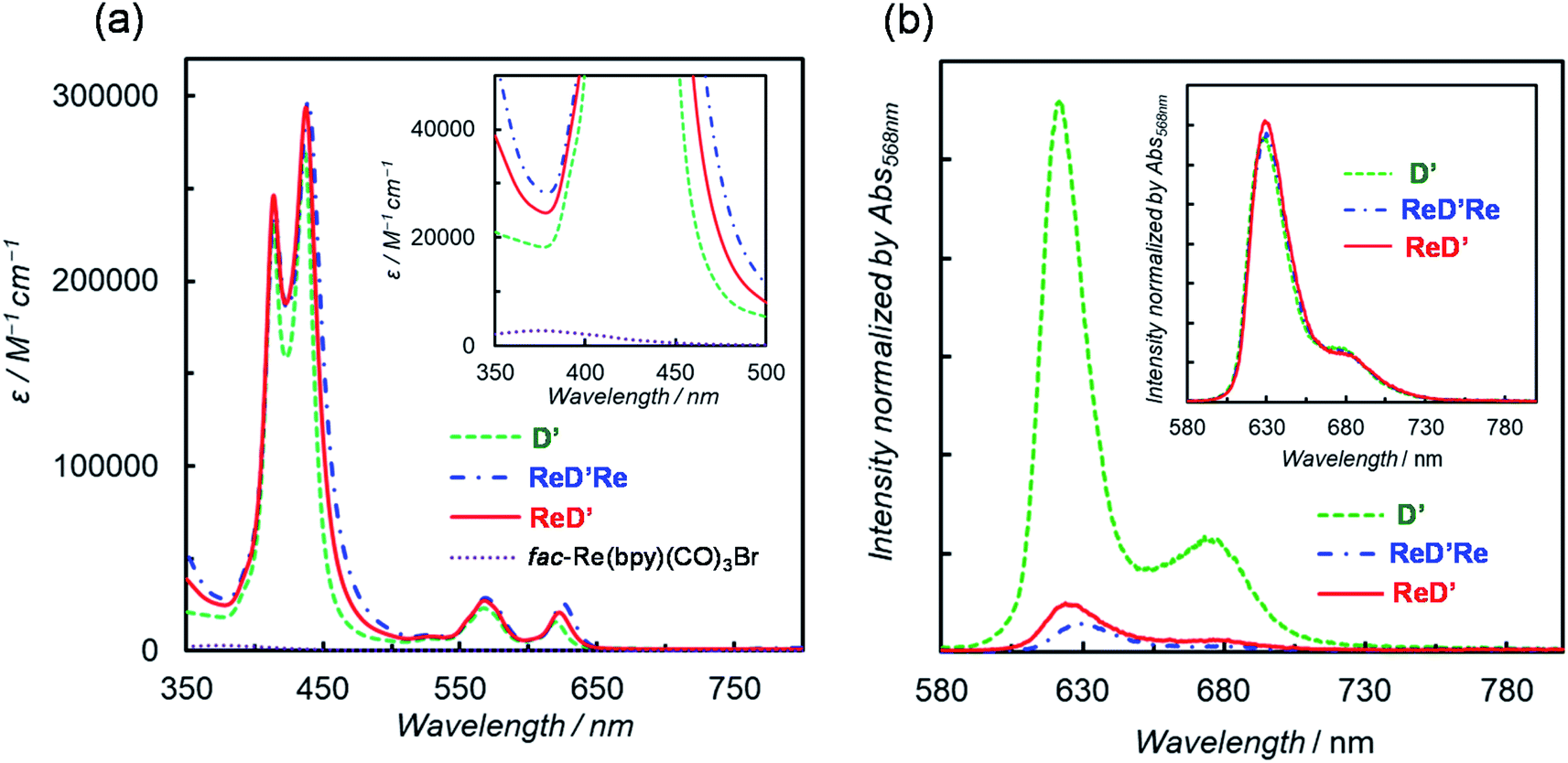 | ||
| Fig. 3 (a) UV-vis absorption spectra of D′, ReD′, ReD′, and fac-Re(bpy)(CO)3Br in DMA. The inset presents the magnification of the near-UV region. (b) Fluorescence spectra of D′, ReD′Re, and ReD′ in DMA at 298 K. The inset shows the spectra in toluene. The fluorescence spectra were normalized with the absorbance at the excitation wavelength (λex = 568 nm). | ||
| λ abs/nm (ε/104 M−1 cm−1) | λ em/nmb | Φ f | ||
|---|---|---|---|---|
| Toluene | DMAc | |||
| a Excited at 568 nm. b In DMA. c The values in parentheses are the ratio to the fluorescence quantum yield of D. | ||||
| D | 413 (23), 436 (27) | 622 | 7.9% | 4.2% (1) |
| 567 (2.3), 620 (1.6) | ||||
| ReD′ | 414 (25), 437 (29) | 625 | 8.1% | 0.50% (0.12) |
| 568 (2.7), 622 (2.0) | ||||
| ReD′Re | 414 (24), 439 (30) | 629 | 8.0% | 0.31% (0.073) |
| 568 (2.9), 625 (2.6) | ||||
The fluorescence spectra of D′, ReD′Re, and ReD′ are shown in Fig. 3b and their fluorescence quantum yields (Φf) in DMA and toluene are summarized in Table 1. The fluorescence intensity decreases in polar DMA (dielectric constant: εs = 38.3) as the Re complex is introduced, whereas the fluorescence quenching by the introduction of the Re complex is not observed in nonpolar toluene (εs = 2.43) (inset of Fig. 3b). The behavior suggests that the intramolecular electron transfer occurs from the lowest excited singlet state (S1) of the porphyrin dimer unit to the Re complex unit in ReD′Re and ReD′.9a,26 According to the energy levels of the CS state (1.9 eV) estimated from the electrochemical data of D′ and fac-Re(bpy)(CO)3Br and of the S1 (2.0 eV) of D′,26 the intramolecular electron transfer, the so-called “oxidative quenching”,28 is thermodynamically possible (Fig. S85†). In DMA, the quenching efficiencies of ReD′ and ReD′Re compared to D′ were 88% and 93%, respectively (Table 1). Assuming that the value of the CS rate constant (kCS) in ReD′Re is twice the value in ReD′, the ratio of the fluorescence quantum yields of ReD′Re to D was calculated to be 0.064 (see p. S68 in the ESI†). The value is consistent with the experimental value (0.073), indicating that each Re complex acts independently to quench the S1 of the porphyrin unit in ReD′Re. The quenching efficiencies show that most of the excited energy on the porphyrin mainly produces the CS state between the porphyrin and the Re complex both in ReD′Re and ReD′. The fluorescence quenching experiments of D′, ReD′, and ReD′Re by BIH gave the Stern–Volmer constants KSV = 3.5 M−1, 0.65 M−1, and 0.32 M−1, respectively (Fig. S86†). The KSV values of ReD′ and ReD′Re were smaller compared with that of D′ and reflect that the lifetime of the S1 of the porphyrin is shortened by the formation of the CS state. The lifetimes of ReD′ and ReD′Re were estimated to be 0.37 and 0.18 ns from the Stern–Volmer plots by assuming that the fluorescence quenching process is diffusion-controlled,18b which was well consistent with the lifetimes (0.24 and 0.13 ns for ReD′ and ReD′Re, respectively) estimated from the fluorescence quantum yields (see p. S68 in the ESI†).
In a previous report, ZnP-phen=Re showed phosphorescence from the Zn porphyrin in Ar-saturated DMA even at rt by strong spin–orbit coupling imposed by the large Re atom in the vicinity.18b The phosphorescence was efficiently quenched by an electron donor used in photocatalytic CO2 reduction, i.e., BIH, indicating that quantitative photoinduced electron transfer occurred from BIH to the long-lived excited triplet state (T1) of ZnP-phen=Re. In the present systems, no phosphorescence was observed in ReD′Re and ReD′. It is considered that the distance between the Re atom and the porphyrin unit is too long to induce rt phosphorescence from the Zn porphyrin. In fact, the fluorescence quantum yields of ReD′Re and ReD′ are almost the same as that of D′ in toluene (Table 1), indicating that the heavy atom effect of the Re atom on porphyrin was negligible. This also supports that the S1 state of Zn porphyrin was quenched to predominantly produce the CS states in ReD′Re and ReD′. According to the energy diagram using the phosphorescence spectrum of D′ at 77 K,27 the charge-recombination process from the CS states to generate the corresponding T1 state is thermodynamically possible, as is the generation of ground states (Fig. S85†).
Photocatalytic CO2 reduction in DMA
Photocatalytic CO2 reduction was conducted in a DMA solution containing ReD′ (0.025 mM) and BIH (0.1 M) as the electron donor18,28 under photoirradiation at 560 nm using light-emitting diode (LED) lamps. A trace amount of CO was produced and its turnover number against the Re atom was only TONCO = 15 after 18 h. By contrast, the addition of either phenol (PhOH, 0.1 M) or TEOA (17 vol%) dramatically increased CO production, and its TONCO reached 780 and 2800 after 18 h, respectively (Fig. 4). PhOH has been reported to act as a proton source to promote the reaction with CO2 on the Re complex,29 and TEOA assists trapping CO2 by forming the Re(bpy)(CO)3(CO2-TEOA) species.30 Herein, CO was selectively produced without forming detectable amounts of H2, CH4, and HCOOH. Because TEOA can work as an electron donor,16c,28 irradiation of a CO2-saturated DMA-TEOA solution containing ReD′ was carried out in the absence of BIH as a control experiment. The reaction produced a trace amount of CO (TONCO = 14 after 48 h (Table S1 entry 5†)), indicating that BIH acts as the major electron donor.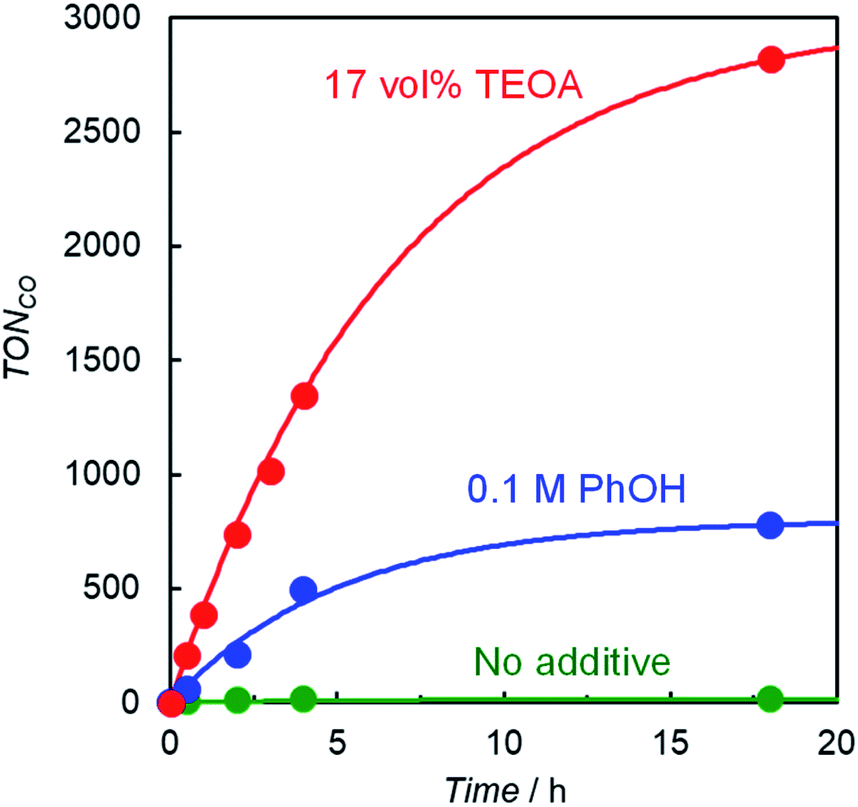 | ||
| Fig. 4 CO formation with time during irradiation at 560 nm from LED lamps with a merry-go-round apparatus for CO2-saturated DMA solutions (total 1.0 mL) containing ReD′ (0.025 mM) and BIH (0.1 M) in the absence (green) and presence of additives ([PhOH] = 0.1 M (blue) and DMA:TEOA = 5:1 v/v (red)). | ||
The formation of CO using ReD′, ReD′Re, mixed systems of D′ and fac-Re(bpy)(CO)3Br (1 and 2 equiv. against D′), and ZnP-phen=Re in CO2-saturated DMA-TEOA (5:1 v/v) solutions containing BIH (0.1 M) is illustrated in Fig. 5. All systems photocatalyzed CO2 reduction, selectively giving CO. The time profiles of the CO production between ReD′ and ReD′Re were similar, indicating that the absorbed light energy can be used by one Re complex site for the CO2 reduction reaction and that the catalytic activity of the Re complex is unaffected by another Re complex. From the fluorescence quenching experiment (Fig. S86†), the quenching efficiencies (ηq) of the S1 states in the presence of [BIH] = 0.1 M can be estimated to be 6% and 3% for ReD′ and ReD′Re, respectively.28 The quenching efficiency of ReD′ having one Re complex was higher than that of ReD′Re by a factor of 2. If the initial photoinduced electron transfer from BIH occurred mainly through the S1 state of the porphyrin dimer (reductive quenching),28 the difference in photocatalytic activities between ReD′ and ReD′Re would be larger. The results indicate that the reductive quenching mechanism can be excluded. Thus, the electron transfer from BIH is expected to occur mainly through the subsequent state after the intramolecular electron transfer (Fig. S85†) (oxidative quenching). The mixed systems of D′ and fac-Re(bpy)(CO)3Br (Re) show a mainly relatively lower activity than the connected systems, ReD′ and ReD′Re, but the mixed system using only two equivalents of Re shows a moderate activity. In the mixed system, the intermolecular electron transfer from the S1 state of D′ to Re (oxidative quenching) is unlikely to occur due to the low concentration of Re. Considering that the reaction through the T1 state of D′ is thermodynamically difficult (vide infra), the reductive quenching from BIH to the S1 state of D′ would start under the condition of high BIH concentration (0.1 M). We believe that the S1 state of D′ has a sufficient lifetime to be quenched by the concentrated BIH (ηq = 26% at 0.1 M from Fig. S86†).
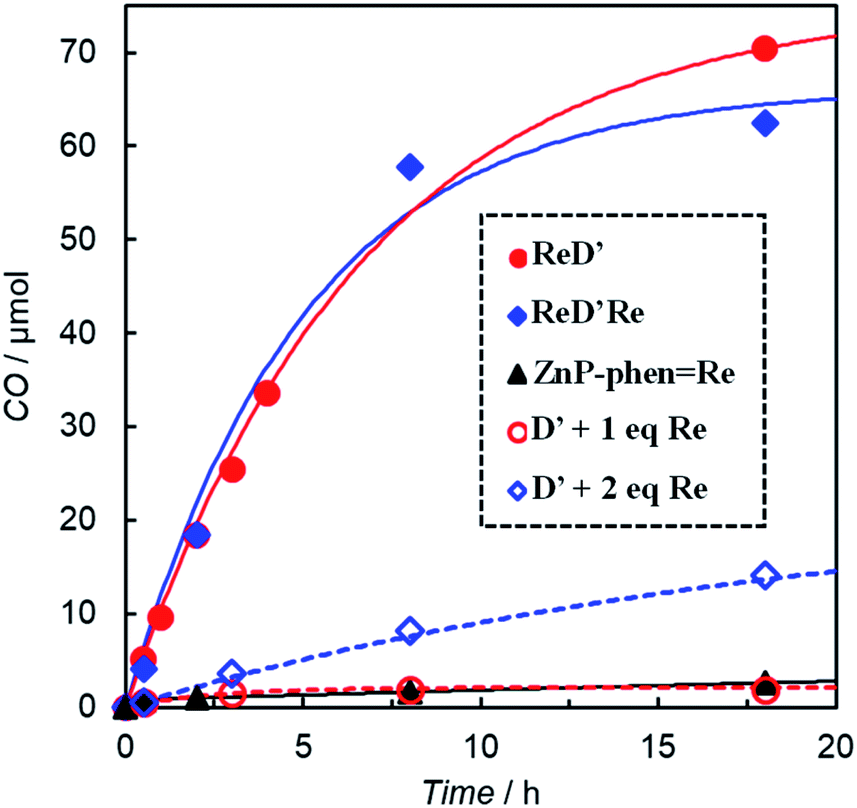 | ||
| Fig. 5 CO formation with time under irradiation from LED lamps at 560 nm with a merry-go-round apparatus for CO2-saturated DMA-TEOA (5:1 v/v, total 1.0 mL) solutions containing BIH (0.1 M) in the presence of ReD′ (0.025 mM), ReD′Re (0.025 mM), ZnP-phen=Re (0.050 mM), and a mixed system of D′ (0.025 mM) and fac-Re(bpy)(CO)3Br (Re: 0.025 mM or 0.05 mM). | ||
The CO production by ReD′ is much larger than that of ZnP-phen=Re (Fig. 5). The photocatalytic CO2 reduction upon irradiation at 560 nm of DMA solutions (1.0 mL) containing ZnP-phen=Re (0.05 mM) and BIH (0.1 M) gave a linear increase in CO production in both the absence and presence of PhOH, whereas the addition of TEOA enhanced the CO production at the initial stage but decreased the durability (Fig. S87†). The TONCO of ZnP-phen=Re reached 172 after 18 h when PhOH (0.1 M) was added but was 1/16 compared with that of ReD′ in DMA-TEOA (Table S1†).
The UV-vis absorption spectral changes of the reaction solutions containing either ReD′ or ZnP-phen=Re during the photocatalytic reactions under CO2 and Ar atmospheres are shown in Fig. 6. Under the CO2 atmosphere, the CO formation catalytically proceeded even under micromolar-order concentration conditions of the photocatalysts. The spectrum of ReD′ was almost unchanged (Fig. 6a), whereas the original bands of ZnP-phen=Re significantly decreased, accompanied by the appearance of a band at 620 nm and a featureless broad band at about 700 nm (Fig. 6c), which are assignable to Zn chlorins16c,d,31 and the one-electron reduced species (OERS) of Zn porphyrin,32 respectively. Even under an Ar atmosphere, ReD′ showed no spectral change (Fig. 6b), suggesting that there is a rapid back-electron transfer process returning to the ground state in ReD′. In addition, in ZnP-phen=Re, TEOA promotes the formation of chlorins and tends to decrease the OERS of porphyrin (Fig. 6c and d). Visible-light irradiation of tetraphenylporphyrin in the presence of aliphatic amine produces chlorins via an adduct of porphyrin and amine.33 Thus, the low catalytic durability of ZnP-phen=Re in the presence of TEOA (Fig. S87a†) would come from chlorin formation. In the absence of TEOA, ZnP-phen=Re shows high durability (Fig. S87a†), indicating that the OERS of porphyrin is not a species that directly decomposes the photocatalyst. In contrast to ZnP-phen=Re, TEOA is essential for ReD′ and ReD′Re to obtain high catalytic activity and would not react with the porphyrin dimer to form chlorin-producing adducts.
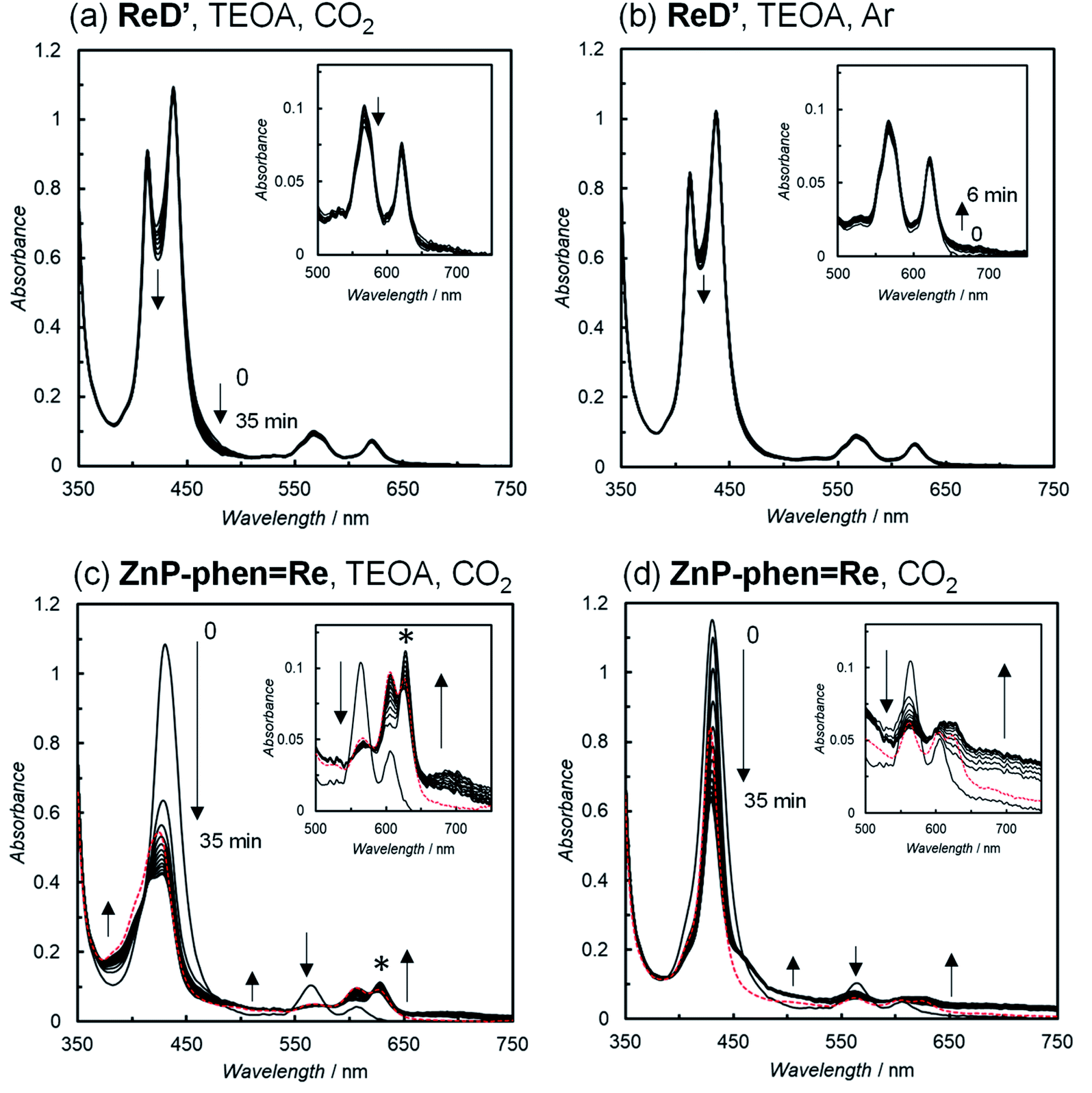 | ||
| Fig. 6 UV-vis absorption spectral changes of (a, c and d) CO2– and (b) Ar-saturated DMA solutions in the (a, b and c) presence and (d) absence of 17 vol% TEOA containing the photocatalysts; ReD′(a and b; 2.5 μM), ZnP-phen=Re (c and d; 5.0 μM), and 0.01 M BIH during irradiation at 560 nm (3.5 × 10−8 einstein s−1) from a Xe lamp, 25 °C. The peak marked with an asterisk (*) in Fig. 6c is assigned to chlorins. Red dotted lines show the spectra of resulting solutions after standing for 2 d in the dark. | ||
Table 2 summarizes the reaction quantum yields (ΦCO) of CO and the TONCO using ReD′ and ReD′Re in DMA-TEOA (5:1 v/v). The ΦCO upon excitations to the upper excited singlet states (S2) by λex = 420 and 450 nm and to S1 by λex = 560 nm has similar values of 2%, indicating that there is no deactivation process from S2 to S1. In addition, the ΦCO values are similar between ReD′Re and ReD′, indicating that one Re complex site is sufficient for the CO2 reduction reaction and that the electron transfer from BIH to the S1 of the porphyrin dimer is a minor process in the photocatalytic CO2 reduction described above. Considering that the ΦCO of ZnP-phen=Re is 8% in DMA containing PhOH,18b the ΦCO of ReD′ does not seem to reflect a TONCO of ReD′ much larger than that of ZnP-phen=Re using the merry-go-round apparatus (Fig. 5 and Table S1†). Focusing on the fact that the most different factor between the ΦCO and TONCO measurements is light irradiation intensity,34 we investigated the dependence of ΦCO on the light intensity and found that the dependence between ReD′ and ZnP-phen=Re was significantly different.
| Quantum yield (ΦCO)a | Product/μmol (TON(dimer)b) | |||||
|---|---|---|---|---|---|---|
| 420 nm | 450 nm | 560 nm | CO | H2 | HCOOH | |
|
a Dependence of quantum yield on the excitation wavelength. Quantum yield as a ratio of number of CO and absorbed photons (light intensity: 2.6 × 10−8, 2.6 × 10−8, and 3.6 × 10−8 einstein s−1 for 420, 450, and 560 nm, respectively (Fig. S89)). A Xe lamp with band path filters was used.
b A merry-go-round apparatus equipped with LED lamps was used (λex = 560 nm). [ReD′] = [ReD′Re] = 0.025 mM and [BIH] = 0.1 M in CO2-saturated DMA-TEOA (5:1 v/v). The turnover number is based on the porphyrin dimer unit.
c n.d., not detected.
|
||||||
| ReD′ | 0.023 | 0.032 | 0.024 | 70.5 (2820) | n.d.c | n.d.c |
| ReD′Re | 0.020 | 0.019 | 0.016 | 62.5 (2500) | n.d.c | n.d.c |
The plots of ΦCO under favorable conditions for ReD′ and ZnP-phen=Re with respect to the light irradiation intensity are shown in Fig. 7. We controlled the light intensity using neutral density filters of 560 nm monochromic light from a Xe lamp with a band path filter. The ΦCO of ReD′ was independent of the light intensity (red circles in Fig. 7 and S90†), whereas the ΦCO of ZnP-phen=Re significantly decreases as the light intensity increases. The decrease in the reaction quantum yield with increasing light intensity is rather commonly observed in photocatalytic systems, because OERS accumulation reduces the catalytic activity due to the inner-filter effect, which is caused by OERS absorption.35,36 By contrast, the OERS of ReD′, which absorbs 560 nm light, did not form during the irradiation (Fig. 6a), resulting in the independence of ΦCO on the light intensity. The strong light from LED lamps with the merry-go-round apparatus lowered the photocatalytic activity of ZnP-phen=Re by the inner-filter effect of the OERS, whereas the photocatalytic activity of ReD′ was not lowered due to no OERS accumulation (Fig. 6a). The independence of ReD′ on light intensity was also observed on the TONCO using the merry-go-round apparatus (Fig S91†).
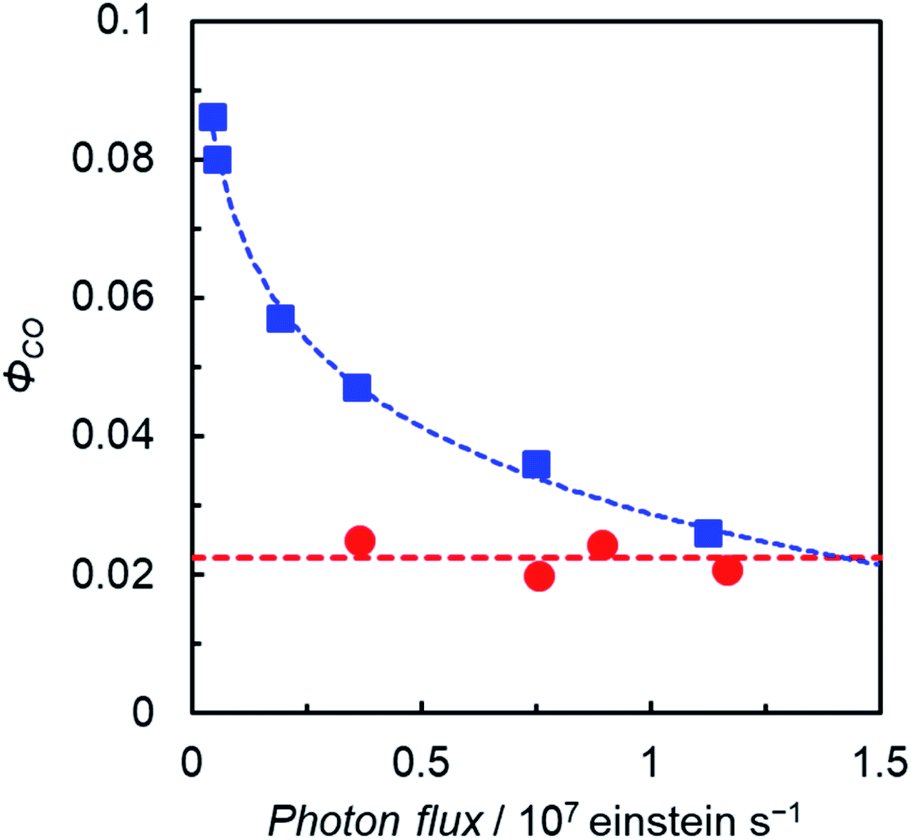 | ||
| Fig. 7
Φ
CO as a function of light irradiation intensity (Xe lamp, λex = 560 nm) for ReD′ (0.1 mM) in DMA-TEOA (5:1 v/v) solutions containing BIH (0.1 M) (red circles) and ZnP-phen=Re (0.3 mM) in DMA solutions containing BIH (0.05 M) and PhOH (0.1 M) (blue squares). | ||
Another interesting difference between ReD′ and ZnP-phen=Re is the dependence of the catalytic activity on the BIH concentration. The catalytic activity of ZnP-phen=Re does not depend on the concentration of BIH because the photoinduced electron transfer from BIH occurs via the long-lived T1 (KSV = 180000 M−1) with reductive quenching from the T1 state.18b Upon excitation at 560 nm, the overlaid plots of CO production against the absorbed photon using 0.01 M and 0.05 M BIH show the independence of the BIH concentration in ZnP-phen=Re (Fig. S92†). By contrast, the ΦCO and TONCO strongly depend on the BIH concentration in ReD′ (Fig. 8 and S93†). The simulation of the quenching efficiencies on the state that mediates the electron transfer from BIH gave the same order values of KSV = 8 M−1 and 15 M−1 from the plots of ΦCO and TONCO, respectively. The obtained value was much smaller than that of ZnP-phen=Re, suggesting that a relatively short-lived state such as the CS state (ZnP˙+−Re˙− in Fig. S85†) contributes to the electron transfer from BIH. In fact, the coordination of the imidazolyl group to the Zn porphyrin lowers the energy level of T1 (ref. 27) and shifts the reduction potential of the porphyrin to the negative side,26,37 so that the electron transfer from BIH to the T1 of the porphyrin dimer forming the OERS of the porphyrin is thermodynamically unfavorable (Fig. S94†).
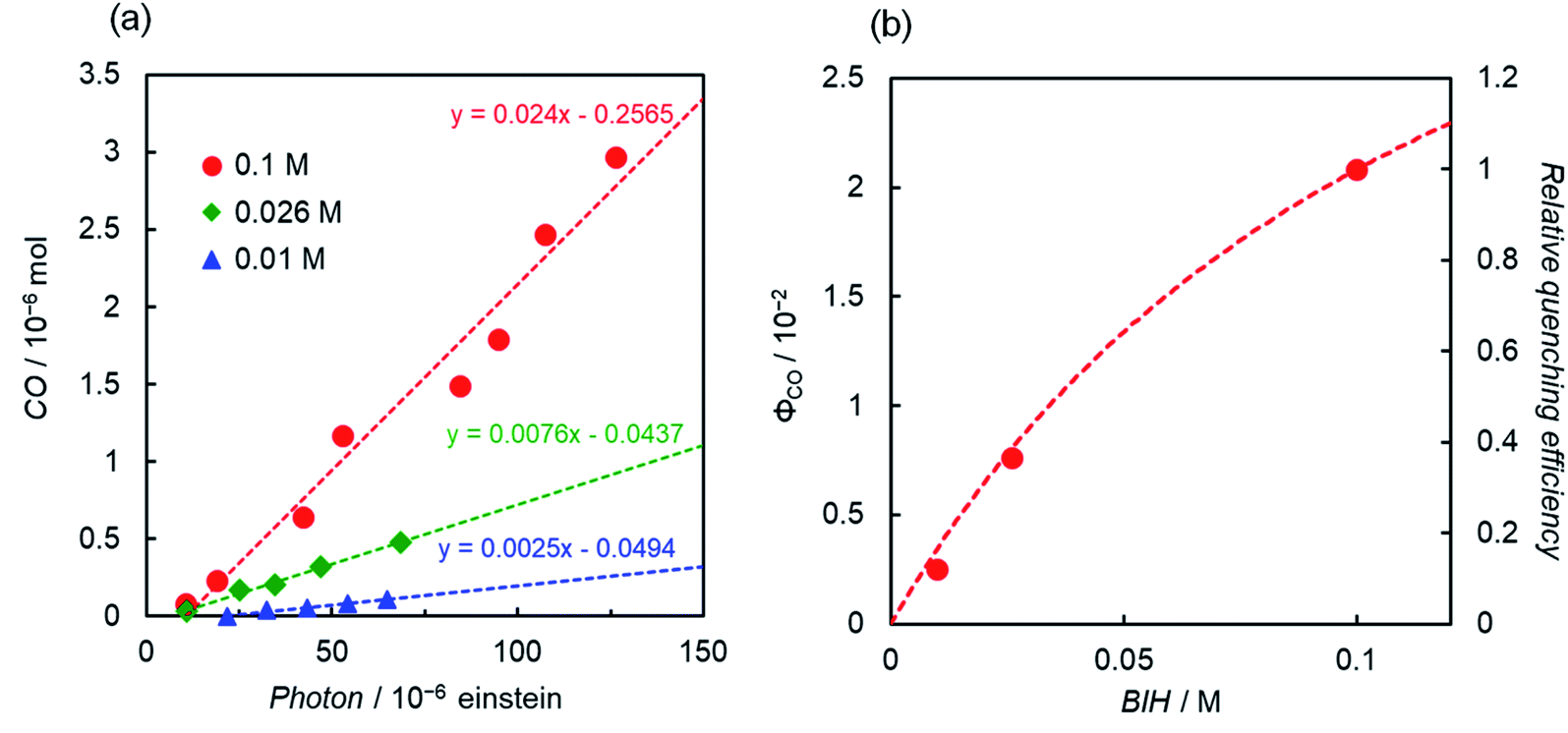 | ||
| Fig. 8 (a) CO formation during irradiation at 560 nm (Xe lamp, 3.6 × 10−8 einstein s−1) using BIH (red circle, 0.1 M; green diamond. 0.026 M; blue triangle, 0.01 M) and ReD′ (0.1 mM) in CO2-saturated DMA-TEOA (5:1 v/v). (b) The reaction quantum yield as a function of BIH concentration. The quenching efficiency relative to ηq at [BIH] = 0.1 M was calculated using ηq = [BIH] KSV/(1 + [BIH] KSV) assuming KSV = 8 M−1. | ||
Photocatalytic CO2 reduction in DMSO
The above results indicate that ReD′ (ReD′Re), which has the porphyrin dimer as the photosensitizer, proceeds by a different mechanism from that of ZnP-phen=Re, and shows an extremely high activity under irradiation with strong light. However, the bond distance and direction between the porphyrin and the Re complex in ReD′ (ReD′Re) were different from those of ZnP-phen=Re. The differences could affect the catalytic activity. Therefore, to clarify whether the dimeric structure of the porphyrin contributes to the high activity, photocatalytic reactions were performed under the same conditions using ReDRe and ReD′Re (Scheme 2) in a coordination solvent of DMSO. Here, DMSO is a suitable solvent for photocatalytic CO2 reduction using a Re diimine tricarbonyl complex in the presence of BIH and TEOA.38 Under micromolar-order concentration conditions, ReDRe dissociated into the corresponding monomer (Fig. 9). The first oxidation and reduction potentials of the porphyrin dimer D′ and monomer D (Scheme 3) were almost the same in DMSO in differential pulse voltammetry (DPV) (Fig. S95†) and the potentials of ReD′Re were similar to those in DMA. In addition, the fluorescence of the monomer prepared from ReDRe in DMSO was quenched by the Re complex unit, indicating that the intramolecular electron transfer from the S1 of the porphyrin unit to the Re complex unit would occur in the monomer as well as in the dimer (Fig. S96†).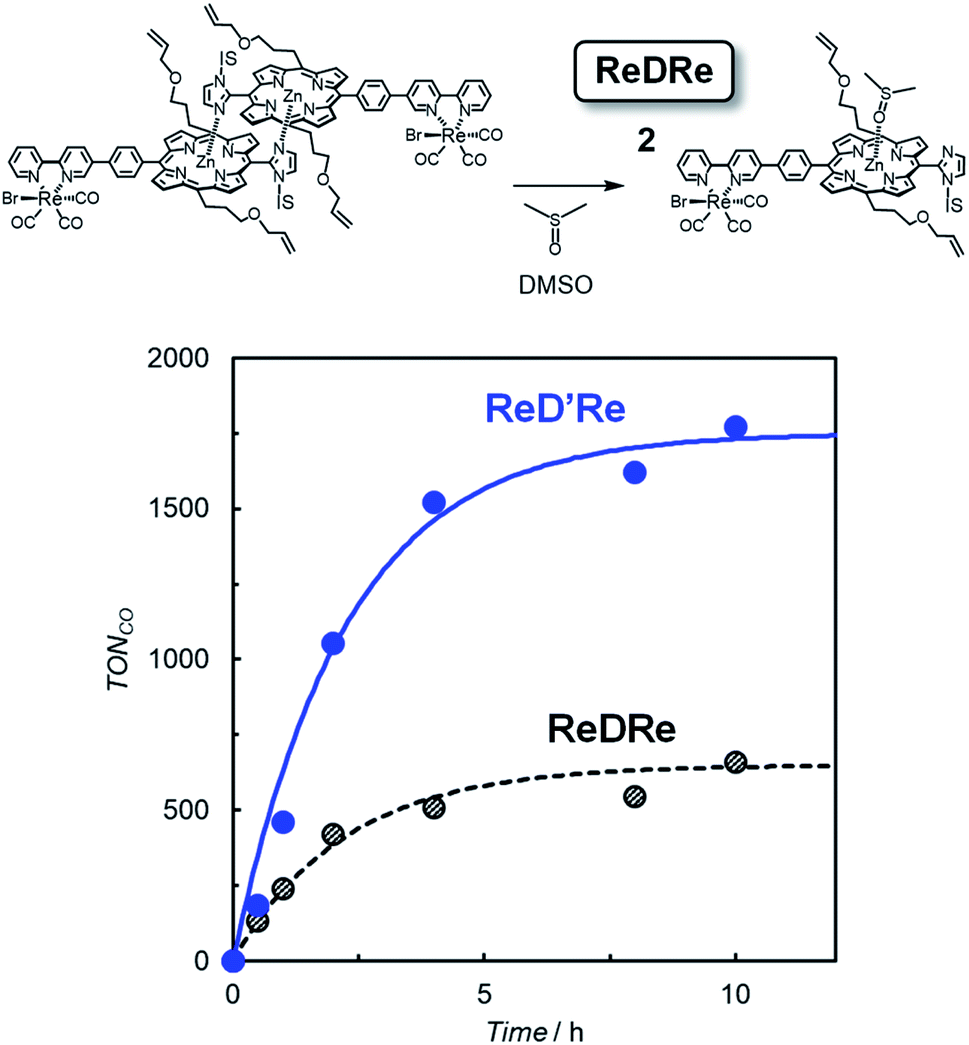 | ||
| Fig. 9 (Top) dissociation of the porphyrin dimer (ReDRe) into porphyrin monomers in DMSO. (Bottom) CO formation with time during irradiation at 560 nm from LED lamps with a merry-go-round apparatus for CO2-saturated DMSO-TEOA (5:1 v/v) solutions containing ReD′Re (2.5 μM) and ReDRe (2.5 μM) in the presence of BIH (0.01 M). The TONCO is calculated against the Re atom. | ||
The CO production with time using dilute ReDRe and ReD′Re of the micromolar range (2.5 μM) is shown in Fig. 9. From the UV-vis absorption spectrum (Fig. 10a inset), ReDRe completely dissociates into the monomeric structure under these reaction conditions (2.5 μM ReDRe). The Q band is inert to whether the porphyrin is the monomer or dimer, and the initial absorbance of ReDRe and ReD′Re at 560 nm is almost the same even in DMSO. Thus, 560 nm was selected as the excitation wavelength of the photocatalytic reaction. In the system using the porphyrin dimer (ReD′Re), the TONCO was much larger than in the system using the porphyrin monomer (ReDRe). The UV-vis spectral changes during irradiation show that the spectrum of ReD′Re hardly changed (Fig. 10), but the monomer spectrum shows significant reductions in both the Soret and Q bands (Fig. 10a inset), indicating that the porphyrin monomer decomposed into colorless species. To investigate the degradation process, we carried out the irradiation of a CO2-saturated DMSO-TEOA (5:1 v/v) containing a higher concentration of ReDRe (10 μM) in the presence of BIH (0.05 M). The UV-vis absorption spectrum after the irradiation for 7 h (Fig. S97†), when the rate of CO formation became slow, showed the appearance of distinct absorption peaks at 500 and 760 nm for bacteriochlorins39 and 610 nm for chlorins.16c,40 Thus, as shown in the literature,16c it is though that the monomeric porphyrin is degraded to colorless species via chlorins and bacteriochlorins, whose process is initiated by two electron reduction of Zn porphyrin (Fig. S98a†). The system using ReD′Re reaches TONCO ∼ 1800 (based on the Re atom). Considering that the equivalent of added BIH against the Re atom was 2000, BIH donated two electrons and an almost quantitative amount of added BIH was consumed.28,41
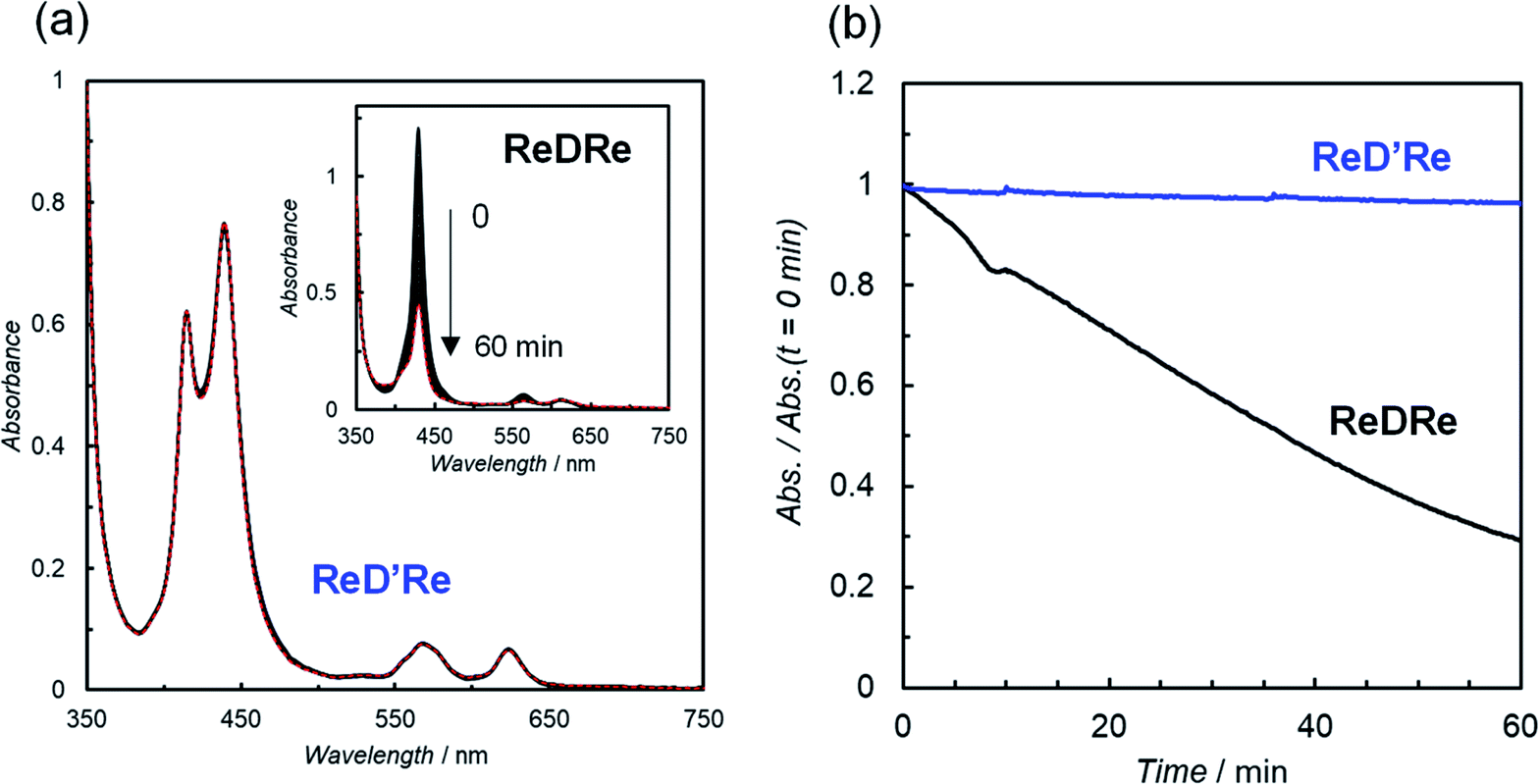 | ||
| Fig. 10 (a) UV-vis absorption spectral change of a CO2-saturated DMSO-TEOA (5:1 v/v) solution containing BIH (0.01 M) and ReD′Re (2.5 μM) during irradiation at 560 nm (Xe lamp, 3.5 × 10−8 einstein s−1) and 25 °C. Inset shows change using ReDRe instead of ReD′Re. Red dotted lines show the spectra after the irradiation for 60 min. (b) Relative absorbance with time at 430 nm of the reaction solutions during the irradiation. | ||
Reaction mechanisms in systems using the porphyrin dimer
Based on the above results, a plausible reaction mechanism is shown in Fig. 11. The porphyrin dimer in ReD′ and ReD′Re can absorb a wide wavelength range of visible light to give the S1 state, which would efficiently produce the CS state by intramolecular electron transfer. Although the CS state can be stabilized by the dimeric structure in ReD′ and ReD′Re,7 a rapid back-electron transfer process generally competes with the catalytic reaction. The nonspectral change in ReD′ under an Ar atmosphere (Fig. 6b) is explained by the rapid back-electron transfer process. The proton promotes the formation of an adduct between the reduced Re complex and CO2,42 and TEOA coordinates to the Re complex in the ground state to form an adduct with CO2.30,38 Thus, with the assistance of PhOH or TEOA, the transient reduced Re complex would rapidly react with CO2 to afford a more stable reaction intermediate that suppresses the back-electron transfer. Considering the fact that the formation of Re–COOH should be rather slower as reported in the literature,43 in the presence of PhOH the hole scavenging process by BIH more probably occurs before the Re–C bond formation. In this mechanism, BIH is used to neutralize the cation radical of the porphyrin to give the ground state porphyrin, and no long-lived OERS of the porphyrin was formed.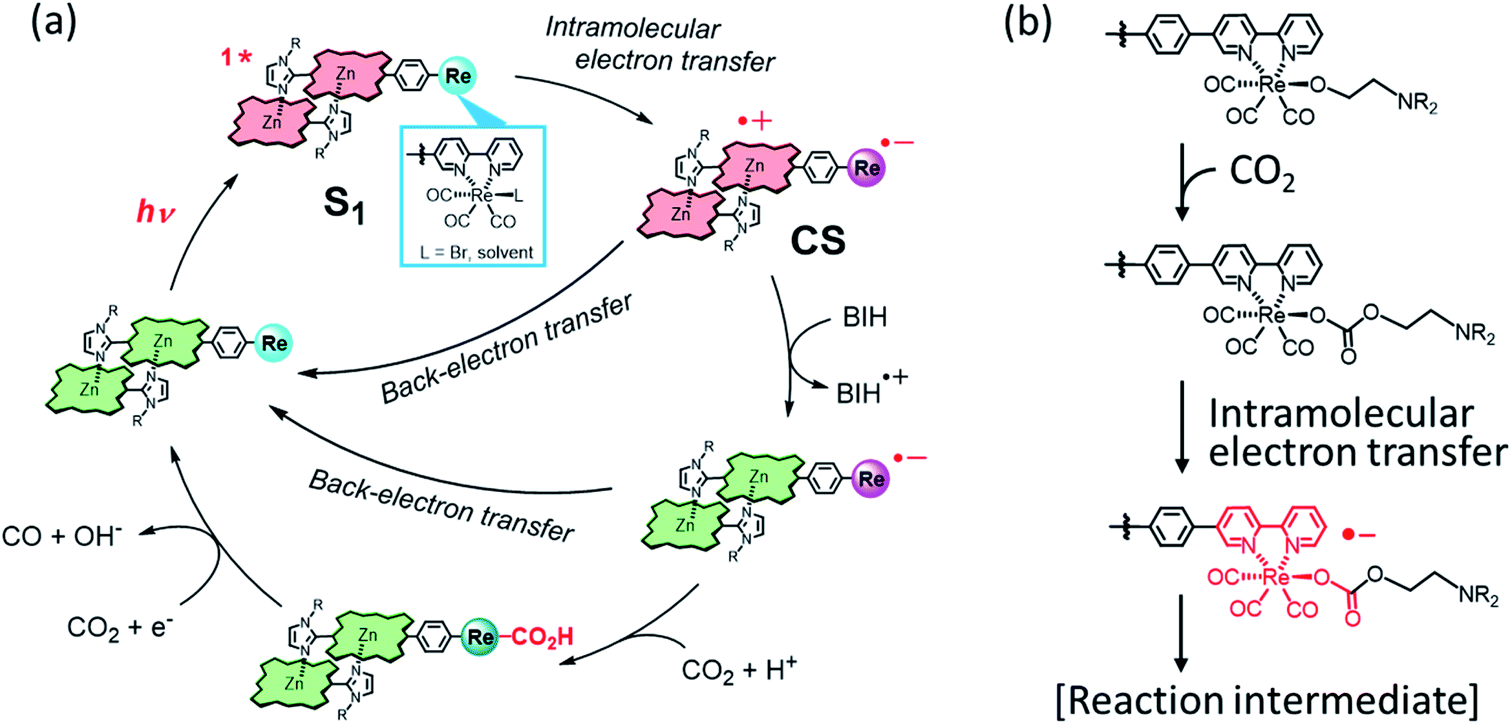 | ||
| Fig. 11 (a) A plausible reaction mechanism of ReD′ (ReD′Re) in the presence of a proton source such as PhOH.42,43 (b) The CO2-capturing reaction with the Re(I) complex with the assistance of TEOA.30,38 | ||
In ZnP-phen=Re, it is thought that the adjacent Re atom enhances the intersystem crossing from the S1 to the T1 of porphyrin and the initial electron transfer mainly occurs via the long-lived T1.18b Thus, the efficient electron transfer from BIH to T1 allows the catalytic reaction to proceed with the same activity even at a low concentration of the electron donor. However, although the accumulation of the porphyrin's OERS would not directly lead to a decrease in the catalytic durability, the inner-filter effect by the OERS under strong light irradiation lowers the reaction efficiency. By contrast, the formation of the CS state in ReD′ and ReD′Re causes independence of the CO2 reduction reaction rate on light intensity because no species that inhibits light absorption is accumulated.
The porphyrin dimer delocalizes the radical cation over the two porphyrin units, giving the stable CS state that can promote the subsequent reaction before the back-electron transfer. The dimeric structure has been reported to accelerate the CS rate and decelerate the charge-recombination rate.7ReD′ had a higher quenching efficiency in DMA against D′ (88% in Fig. 3b and Table 1) than ReDRe against D in DMSO (73% in Fig. S96†), which might support the acceleration of the CS rate. The dimer is superior to the monomer in durability (Fig. 9). Judging from the UV-vis spectrum (Fig. S97†), the irradiation of the DMSO-TEOA solution containing ReDRe (monomer) in the presence of BIH gave chlorins and bacteriochlorins. Because BIH produces a very strong reductant BI˙,28,41 the two-electron reduced species on the porphyrin unit can be generated. Thus, it is thought that the monomeric structure is easily decomposed by two electron reduction, whereas the dimeric structure can suppress degradation by sharing electrons between the two porphyrins of the dimer (Fig. S98b†), even when a two-electron reduced species is produced on the porphyrin unit.44 In the case of ZnP-phen=Re, the high durability might be due to the proximity between Re and the porphyrin, causing rapid electron transfer to the Re complex unit and preventing the accumulation of two electrons on the porphyrin. The detailed reaction mechanisms of ReD′ and ReD′Re in addition to ZnP-phen=Re are currently under investigation using time-resolved measurements.
Conclusion
The special-pair mimic porphyrin dimer was used here as a photosensitizer in a photocatalytic reaction for the first time. Using the fact that the coordination bond of the dimer can be easily rearranged and that the coordination structure can be fixed by a covalent bond by the ring-closing metathesis reaction, dimers having Re complex(es) on either one side (ReD′) or two sides (ReD′Re) have been synthesized. The photocatalytic CO2 reductions using ReD′ and ReD′Re selectively gave CO, the reaction quantum yield of which was estimated to be 2%, independent of the excitation wavelength, light intensity, and number of Re complex units. A high TONCO reaching >2800 after 18 h was observed in ReD′ under irradiation of relatively strong light, which is an order of magnitude higher than the value (172 after 18 h) of ZnP-phen=Re under the optimal conditions. The high catalytic activity stems from the fact that the absorption spectrum of the dimer remains almost unchanged during the irradiation and there is no deactivation caused by the internal filter effect that the OERS absorbs light as observed in general photosensitizers. It is thought that the catalytic reaction proceeds via the CS state formed by the intramolecular electron transfer from the S1 of the porphyrin unit to the Re complex unit, and that BIH neutralizes the cation radical of the porphyrin. Thus, the excessive OERS does not form during the irradiation. In addition, it was shown that the dimeric structure contributes to the improvement of the catalytic durability of CO2 reduction by sharing electrons between the two porphyrins of the dimer. In this work, we prepared a heterodimer of ReD′ using a simple porphyrin D having a tolyl group. The simple porphyrin can be substituted with an imidazolyl Zn porphyrin having more functional units. As a result, it may be possible to create functional heterodimers that can bind to semiconductors and supramolecular systems such as chain structures and cyclic structures with a higher absorption ability.Experimental section
General procedure
All chemicals and solvents were of commercial reagent quality and were used without further purification unless otherwise stated. Dry diethyl ether was prepared by distillation over benzophenone/Na. DMA was dried over molecular sieves of size 4 Å and distilled under reduced pressure. The Grubbs catalyst (1st generation), benzylidene-bis(tricyclohexylphosphine)dichlororuthenium, was purchased from Aldrich. 1-Methylimidazole-2-carbaldehyde, meso-(3-allyloxypropyl)dipyrromethane, and BIH were synthesized according to the literature.9,45 FINEOXOCOL180 was provided by Nissan Chemical Corporation. Super dehydrated N,N-dimethylformamide (DMF) and tetrahydrofuran (THF) were purchased from Fujifilm Wako Pure Chemical Corporation. TEOA was distilled under reduced pressure. The reaction was monitored on silica-gel 60F254 TLC plates (Merck). Silica-gels utilized for column chromatography were purchased from Kanto Chemical Co. Inc.: Silica-Gel 60N (Spherical, Neutral) 63–210 μm and 40–50 μm (Flash). 1H and 13C NMR, distortionless enhancement by polarization transfer 135 (DEPT 135), 1H–1H correlation spectroscopy (COSY), 1H–13C heteronuclear single quantum correlation (HSQC), and 1H–13C heteronuclear multiple bond correlation (HMBC) spectra were recorded by using a JEOL JNMECS-300, JNMECZ-400, and JNMECS-500. Chemical shifts were recorded in parts per million (ppm) relative to tetramethylsilane. UV-vis absorption spectra were collected using square cells (path length = 1.0 cm) on JASCO V-650 and V-660 spectrometers, and a Asahi Spectra PRA-201. Steady-state emission spectra were collected on a Hitachi F-4500 spectrometer and corrected for the response of the detector system. The fluorescence intensities were normalized at the absorption of the excitation wavelength. Fluorescence quantum yields were determined from the integrated ratios of the fluorescence spectra relative to that of ZnTPP (Φf = 3.3% in toluene).46 Their values were corrected with the refractive indices of the used solvents. Hi-resolution MALDI–TOF mass spectra were collected on a JEOL JMS S-3000 with dithranol or trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]-malononitrile (DCTB) as a matrix. FTIR spectra were recorded in KBr using a JASCO FT/IR-4600. Analytical GPC-HPLCs using pyridine as the eluent were performed on a PU-2080plus and MD-2018plus (JASCO) system equipped with two TSK G2500HHR (Tosoh, exclusion limit: 20000 Da) and one TSK G2000HHR (Tosoh, exclusion limit: 10000 Da) columns. Preparative HPLCs were carried out on an LC-908 (JAI) attached to one TSK G2500HHR and one G2000HHR columns eluted with pyridine. The differential pulse voltammogram (DPV) was recorded using an ALS-H/CHI Model 612E electrochemical analyzer in a micro-cell equipped with a glassy carbon working electrode (ϕ 1.6 mm) and a Pt counter electrode. The micro-cell was connected via a Luggin capillary with a reference electrode of Ag/AgNO3 (10 mM in DMSO). Tetrabutylammonium hexafluorophosphate (nBu4NPF6) recrystallized from ethyl acetate was used as a supporting electrolyte. Ferrocene was used as an external standard, and all potentials were referenced to the ferrocene/ferrocenium couple (E1/2 = +0.191 V vs. Ag/AgNO3).
), 5.44 (d, J = 17 Hz, 2H, CH2), 5.28 (d, J = 10 Hz, 2H, CH2), 5.10 (m, 4H, –CH2CH2CH2–), 4.08 (d, J = 5.5 Hz, 4H, –OCH2–), 3.85–8.40 (m, 2H, CH2), 3.66 (m, 4H, –CH2CH2CH2–), 2.78 (m, 4H, –CH2CH2CH2–), 1.38–8.40 (m, 2H, CH2), 1.38–(–0.80) (m, 35H), −2.64 (s, 0.6H, NH), −2.66 (s, 0.4H, NH);13C NMR (100 MHz, CDCl3) δ/ppm = 150–145 (C), 148.95 (C), 148.66 (C), 142.30 (C), 142.27 (C), 136.10 (CH), 135.85 (CH), 135.17 (CH), 131.99–131.50 (CH), 131.22–130.74 (CH), 129.66–128.90 (CH), 128.57 (CH), 128.51 (CH), 128.12 (CH), 128.10 (CH), 128.02–127.55 (CH), 121.41 (CH), 121.29 (CH), 120.33 (CH), 120.27 (CH), 119.61 (C), 119.56 (C), 119.48 (C), 119.09 (C), 119.02 (C), 117.00 (CH2), 105.31 (C), 105.26 (C), 105.11 (C), 94.21 (C), 72.14 (CH2), 69.29 (CH2), 51.20 (CH2), 50.75 (CH2), 50.68 (CH2), 50.46 (CH2), 49.39 (CH2), 49.29 (CH2), 49.20 (CH2), 49.12 (CH2), 48.93 (CH2), 47.70 (CH or CH3), 47.42 (CH2), 47.21 (CH2), 46.72 (CH2), 46.32 (CH2), 37.96 (CH2), 37.47 (CH2), 37.23 (CH2), 36.90 (CH2), 36.59 (CH2), 31.44 (CH2), 31.18 (C), 30.93 (C), 30.75 (C), 30.61 (C), 30.55 (C), 30.23 (C), 30.03 (CH or CH3), 29.89 (CH or CH3), 29.86 (CH or CH3), 29.64 (CH or CH3), 29.41 (CH or CH3), 29.18 (CH or CH3), 29.10 (CH or CH3), 29.03 (CH or CH3), 28.61 (CH or CH3), 28.38 (CH or CH3), 28.16 (CH or CH3), 28.00 (CH or CH3), 27.77 (CH or CH3), 26.85 (CH2), 26.72 (CH2), 24.61 (CH2), 24.55 (CH2), 22.51 (CH or CH3), 22.15 (CH or CH3), 22.08 (CH or CH3), 22.01 (CH or CH3), 16.61 (CH or CH3), 16.51 (CH or CH3), 16.40 (CH or CH3), 16.22 (CH or CH3).
), 5.57 (m, 2H, CH2), 5.56 (s, 1H, Im), 5.42 (m, 2H, β-pyrrole), 5.36 (d, J = 10 Hz, 2H, CH2), 5.22 (m, 4H, –CH2CH2CH2–), 4.24 (m, 4H, –OCH2–), 3.92 (m, 4H, –CH2CH2CH2–), 3.02 (m, 4H, –CH2CH2CH2–), 2.06 (m, 1H, Im), 1.84–1.55 (m, 2H, CH2), 0.30–(–1.77) (m, 35H); 13C NMR (100 MHz, CDCl3) δ/ppm = 151.14 (C), 150.05 (C), 148.65 (C), 148.38 (C), 146.02 (C), 144.10 (C), 136.47 (CH), 136.41 (CH), 135.59 (CH), 135.53 (CH), 135.46 (CH), 135.32 (CH), 131.63 (CH), 131.50 (CH), 129.45 (CH), 129.31 (CH), 128.48 (CH), 127.84 (CH), 121.20 (CH), 119.60 (C), 119.33 (C), 117.66 (CH), 116.88 (CH2), 96.93 (C), 93.41 (C), 72.18 (CH2), 70.20 (CH2), 50.53 (CH2), 50.22 (CH2), 50.01 (CH2), 49.91 (CH2), 47.75 (CH2), 47.58 (CH2), 47.38 (CH2), 47.31 (CH2), 46.50 (CH2), 45.81 (CH or CH3), 45.40 (CH or CH3), 45.21 (CH or CH3), 44.98 (CH or CH3), 44.800 (CH or CH3), 38.74 (CH2), 35.60 (CH2), 35.44 (CH2), 32.16 (CH2), 30.26 (C), 30.21 (C), 30.16 (C), 30.05 (C), 29.80 (CH2), 29.60 (CH or CH3), 29.44 (CH or CH3), 29.39 (CH or CH3), 29.31 (CH or CH3), 28.65 (CH or CH3), 28.48 (CH or CH3), 29.21 (CH or CH3), 27.01 (CH or CH3), 26.86 (CH or CH3), 26.63 (CH or CH3), 21.92 (CH or CH3), 21.56 (CH or CH3), 21.27 (CH or CH3), 15.74 (CH or CH3).
), 5.56 (s, 1H, Im), 5.54 (m, 2H, CH2), 5.44 (m, 2H, β-pyrrole), 5.37 (m, 2H, CH2), 5.26 (m, 4H, –CH2CH2CH2–), 4.26 (m, 4H, –OCH2–), 3.94 (m, 4H, –CH2CH2CH2–), 3.05 (m, 4H, –CH2CH2CH2–), 2.16 (m, 1H, Im), 2.09–(–1.72) (m, 35H); 13C NMR (125 MHz, CDCl3) δ/ppm = 156.20 (C), 155.23 (C), 151.15 (C), 150.05 (C), 149.50 (CH), 148.92 (C), 148.87 (C), 148.34 (C), 148.13 (CH), 146.28 (C), 146.09 (C), 144.68 (C), 137.20 (CH), 136.79 (C), 136.29 (C), 135.65 (CH), 135.57 (CH), 135.48 (CH), 131.74 (CH), 129.60 (CH), 129.43 (CH), 129.29 (CH), 128.46 (CH), 127.84 (CH), 125.12 (CH), 124.90 (CH), 123.92 (CH), 121.41 (CH), 121.33 (CH), 120.37 (C), 119.32 (C), 119.24 (C), 117.59 (CH), 116.87 (CH2), 116.84 (CH2), 96.86 (C), 72.17 (CH2), 70.22 (CH2), 67.87 (CH2), 50.58 (CH2), 50.25 (CH2), 50.08 (CH2), 49.91 (CH2), 47.82 (CH2), 47.39 (CH2), 46.53 (CH2), 45.40 (CH or CH3), 45.25 (CH or CH3), 45.01 (CH or CH3), 44.83 (CH or CH3), 39.01 (CH or CH3), 38.75 (CH2), 36.46 (CH2), 36.24 (CH2), 35.64 (CH2), 35.50 (CH2), 32.19 (CH or CH3), 30.67 (C), 30.49 (C), 30.42 (C), 30.29 (C), 30.21 (C), 29.81 (C), 29.63 (CH or CH3), 29.53 (CH or CH3), 29.48 (CH or CH3), 29.45 (CH or CH3), 29.41 (CH or CH3), 29.34 (CH or CH3), 29.09 (CH2), 28.63 (CH or CH3), 28.50 (CH or CH3), 28.25 (CH or CH3), 28.06 (CH or CH3), 27.03 (CH or CH3), 26.91 (CH or CH3), 26.67 (CH or CH3), 25.49 (CH2), 24.09 (CH2), 23.95 (CH2), 23.81 (CH2), 23.75 (CH2), 23.07 (CH2), 21.95 (CH or CH3), 21.91 (CH or CH3), 21.60 (CH or CH3), 21.34 (CH or CH3), 21.28 (CH or CH3), 16.11 (CH or CH3), 15.78 (CH or CH3).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) Re complex dyad ([ImIS-ZnP-Ph-5Bpy=Re]2, ReDRe).
In a 200 mL flask, [ImIS-ZnP-Ph-5Bpy]2 (130 mg, 0.12 mmol), Re(CO)5Br (46 mg, 0.11 mmol), and toluene (135 mL) were placed. The mixture was heated to 90 °C and stirred for 2 days. Pyridine (2 mL) was added to the resulting solution and the solvent was evaporated to dryness. The residue was purified with a flush silica gel column (eluent: CHCl3 → CHCl3/acetone = 20/1). The collected fractions were evaporated to dryness, giving the titular compound as a purple solid (120 mg, 72% yield). TLC (silica gel, CHCl3/acetone = 10/1) Rf = 0.5; HPLC (GPC, pyridine, 1.0 mL min−1) RT = 23.4 min; MALDI-TOF-mass (dithranol) m/z [M + H]+ 1465.4329, calcd for [C72H81BrN8O5ReZn]+ 1465.4329; 1H NMR (500 MHz, CDCl3) δ/ppm = 9.69 (m, 3H, β-pyrrole and bpy 6), 9.17 (d, J = 5.6 Hz, 1H, bpy 6′), 9.07 (m, 2H, β-pyrrole), 8.96 (m, 2H, β-pyrrole), 8.90 (m, 1H, phenylene), 8.53 (q, J = 7.7 Hz, 1H, bpy 3), 8.35 (m, 1H, bpy 3′), 8.27 (m, 2H, phenylene and bpy 4), 8.18 (m, 1H, phenylene), 8.06 (m, 2H, phenylene and bpy 4′), 7.57 (t, J = 6.6 Hz, 1H, bpy 5′), 6.21 (m, 2H, –CH), 5.58 (s, 1H, Im), 5.54 (m, 2H, CH2), 5.44 (m, 2H, β-pyrrole), 5.36 (m, 2H, CH2), 5.25 (m, 4H, –CH2CH2CH2–), 4.25 (m, 4H, –OCH2–), 3.94 (m, 4H, –CH2CH2CH2–), 3.05 (m, 4H, –CH2CH2CH2–), 2.13 (m, 1H, Im), 2.09–(–1.77) (m, 35H); 13C NMR (125 MHz, CDCl3) δ/ppm = 197.17 (CO), 196.88 (CO), 189.20 (CO), 155.80 (C), 153.91 (C), 153.52 (CH), 151.86 (CH), 151.20 (C), 150.25 (C), 148.65 (C), 148.34 (C), 146.61 (C), 145.97 (C), 140.79 (C), 139.00 (CH), 137.04 (CH), 135.99 (CH), 135.88 (CH), 135.43 (CH), 133.18 (C), 131.64 (CH), 131.51 (CH), 129.60 (C), 129.59 (CH), 129.40 (CH), 128.75 (CH), 127.91 (CH), 127.00 (CH), 125.40 (CH), 125.15 (CH), 123.42 (CH), 123.19 (CH), 121.26 (CH), 119.47 (C), 116.94 (CH2), 97.08 (C), 72.19 (CH2), 70.17 (CH2), 67.85 (CH2), 50.58 (CH2), 50.24 (CH2), 50.25 (CH2), 49.88 (CH2), 47.84 (CH2), 47.39 (CH2), 46.53 (CH2), 45.24 (CH3), 45.04 (CH3), 44.84 (CH3), 38.78 (CH2), 36.49 (CH2), 36.23 (CH2), 35.62 (CH2), 35.49 (CH2), 32.20 (CH2), 30.65 (C), 30.49 (C), 30.43 (C), 30.23 (C), 29.80 (C), 29.63 (CH2), 29.45 (CH or CH3), 29.40 (CH or CH3), 29.34 (CH or CH3), 29.08 (C), 28.61 (CH or CH3), 28.48 (CH or CH3), 28.22 (CH or CH3), 28.04 (CH or CH3), 26.84 (CH or CH3), 26.61 (CH or CH3), 25.48 (CH2), 24.06 (CH2), 23.94 (CH2), 23.80 (CH2), 23.07 (CH2), 21.94 (CH or CH3), 21.61 (CH or CH3), 21.29 (CH or CH3), 16.09 (CH or CH3), 15.75 (CH or CH3); FT-IR (KBr) νCO = 1900, 1920, 2022 cm−1.
(E form)), 6.11 (s, 1.4H, –CH (Z form)), 5.56 (s, 2H, Im), 5.42 (m, 8H, β-pyrrole and –CH2CH2CH2–), 5.15 (m, 4H, –CH2CH2CH2–), 4.69 (m, 2.8H, –OCH2-(Z form)), 4.44 (m, 5.2H, –OCH2-(E form)), 4.21 (m, 7H, –CH2CH2CH2–), 3.95 (m, 1H, –CH2CH2CH2–), 3.29 (m, 1H, –CH2CH2CH2–), 3.13 (m, 3H, –CH2CH2CH2–), 3.00 (m, 4H, –CH2CH2CH2–), 2.12 (m, 2H, Im), 2.07–(–1.33) (m, 70H); 13C NMR (125 MHz, CDCl3) δ/ppm = 197.53 (CO), 197.23 (CO), 197.14 (CO), 196.86 (CO), 189.79 (CO), 189.19 (CO), 155.82 (C), 153.93 (C), 153.51 (CH), 153.40 (CH), 151.85 (C), 150.91 (C), 150.13 (C), 148.38 (C), 146.54 (C), 140.84 (C), 140.78 (C), 139.10 (CH), 138.98 (CH), 137.10 (CH), 137.05 (CH), 135.93 (CH), 135.84 (CH), 133.23 (C), 131.58 (CH), 129.60 (CH), 128.32 (CH), 127.88 (CH), 127.05 (CH), 125.38 (CH), 125.16 (CH), 123.43 (CH), 123.20 (CH), 119.70 (C), 116.91 (CH2), 70.80 (CH2), 70.58 (CH2), 67.24 (CH2), 49.93 (CH2), 47.75 (CH2), 40.06 (CH2), 39.01 (CH2), 32.82 (CH2), 30.50 (C), 30.30 (C), 29.70 (CH or CH3), 29.40 (CH or CH3), 28.25 (CH or CH3), 27.04 (CH or CH3), 21.58, (CH or CH3) 15.97 (CH or CH3); FT-IR (KBr) νCO = 1901, 1919, 2021 cm−1. The third fraction of the preparative GPC was collected and the solvent was evaporated to dryness, giving a purple solid (36 mg, 35% yield) as ReD′: HPLC (GPC, pyridine, 1.0 mL min−1) RT = 22.1 min; MALDI-TOF-mass (DCTB) m/z [M]+ 2146.6340, calcd for [C111H114BrN14O7ReZn2]+ 2146.6292; 1H NMR (500 MHz, CDCl3) δ/ppm = 9.70–9.62 (m, 2H, β-pyrrole), 9.56 (m, 2H, β-pyrrole), 9.20 (m, 1H, bpy 6), 9.15 (m, 2H, β-pyrrole), 9.08 (m, 4H, β-pyrrole), 8.90 (m, 3H, β-pyrrole and bpy 6′), 8.64 (m, 1H, bpy 3), 8.50 (brs, 2H, phenylene and bpy 3′), 8.18 (m, 1H, phenylene), 8.03 (m, 1H, phenylene), 7.80 (m, 1H, bpy 4), 7.76–7.60 (m, 5H, phenylene and bpy 4′), 7.36 (m, 2H, bpy 5′), 7.25 (brs, 1H, phenylene), 6.49 (s, 2.75H, –CH (E form)), 6.14 (s, 1.25H, –CH (Z form)), 5.57 (m, 3H, β-pyrrole and Im), 5.51 (m, 2H, β-pyrrole), 5.41 (m, 1H, Im), 5.15 (m, 8H, –CH2CH2CH2–), 4.70 (m, 2.5H, –OCH2–(Z form)), 4.45 (m, 5.5H, –OCH2–(E form)), 4.28–4.21 (m, 8H, –CH2CH2CH2–), 3.33–3.034 (m, 8H, –CH2CH2CH2–), 2.85 (s, 3H, CH3), 2.16 (m, 1H, Im), 1.94 (m, 1H, Im), 1.64 (m, 3H, CH3), 2.09–(–1.77) (m, 35H); 13C NMR (125 MHz, CDCl3) δ/ppm = 197.36 (CO), 197.16 (CO), 197.00 (CO), 196.80 (CO), 189.60 (CO), 188.98 (CO), 155.25 (C), 153.04 (CH), 151.05 (CH), 150.88 (CH), 150.21 (C), 150.10 (C), 149.81 (CH), 149.27 (C), 149.25 (C), 148.42 (C), 148.16 (C), 146.14 (C), 146.06 (C), 141.16 (C), 138.94 (CH), 138.85 (CH), 137.07 (C), 136.08 (CH), 135.90 (CH), 135.46 (CH), 134.82 (CH), 134.61 (CH), 132.84 (C), 132.50 (CH), 132.40 (CH), 131.47 (CH), 129.97 (CH), 129.62 (CH), 128.29 (CH), 127.87 (CH), 127.64 (CH), 127.35 (CH), 127.26 (CH), 126.54 (CH), 125.06 (CH), 123.82 (CH), 122.77 (CH), 121.47 (CH), 119.81 (C), 118.14 (CH), 116.91 (CH2), 96.94 (C), 96.47 (C), 72.21 (CH2), 70.83 (CH2), 70.60 (CH2), 67.30 (CH2), 50.58 (CH2), 49.90 (CH2), 47.85 (CH2), 47.34 (CH2), 45.02 (CH3), 40.06 (CH2), 39.01 (CH2), 35.63 (CH2), 32.91 (CH2), 32.03 (C), 30.50 (C), 30.26 (C), 30.19 (C), 29.81 (CH2), 29.66 (CH or CH3), 29.61 (CH or CH3), 29.50 (CH or CH3), 29.44 (CH or CH3), 29.40 (CH or CH3), 29.35 (CH or CH3), 28.51 (CH or CH3), 28.37 (CH or CH3), 28.28 (CH or CH3), 28.13 (CH or CH3), 26.93 (CH or CH3), 21.94 (CH or CH3), 21.79 (CH or CH3), 21.60 (CH or CH3), 21.29 (CH or CH3), 15.80 (CH or CH3); FT-IR (KBr) νCO = 1900, 1920, 2022 cm−1.
Re complex dyad ([ImIS-ZnP-Ph-5Bpy=Re]2, ReDRe).
In a 200 mL flask, [ImIS-ZnP-Ph-5Bpy]2 (130 mg, 0.12 mmol), Re(CO)5Br (46 mg, 0.11 mmol), and toluene (135 mL) were placed. The mixture was heated to 90 °C and stirred for 2 days. Pyridine (2 mL) was added to the resulting solution and the solvent was evaporated to dryness. The residue was purified with a flush silica gel column (eluent: CHCl3 → CHCl3/acetone = 20/1). The collected fractions were evaporated to dryness, giving the titular compound as a purple solid (120 mg, 72% yield). TLC (silica gel, CHCl3/acetone = 10/1) Rf = 0.5; HPLC (GPC, pyridine, 1.0 mL min−1) RT = 23.4 min; MALDI-TOF-mass (dithranol) m/z [M + H]+ 1465.4329, calcd for [C72H81BrN8O5ReZn]+ 1465.4329; 1H NMR (500 MHz, CDCl3) δ/ppm = 9.69 (m, 3H, β-pyrrole and bpy 6), 9.17 (d, J = 5.6 Hz, 1H, bpy 6′), 9.07 (m, 2H, β-pyrrole), 8.96 (m, 2H, β-pyrrole), 8.90 (m, 1H, phenylene), 8.53 (q, J = 7.7 Hz, 1H, bpy 3), 8.35 (m, 1H, bpy 3′), 8.27 (m, 2H, phenylene and bpy 4), 8.18 (m, 1H, phenylene), 8.06 (m, 2H, phenylene and bpy 4′), 7.57 (t, J = 6.6 Hz, 1H, bpy 5′), 6.21 (m, 2H, –CH), 5.58 (s, 1H, Im), 5.54 (m, 2H, CH2), 5.44 (m, 2H, β-pyrrole), 5.36 (m, 2H, CH2), 5.25 (m, 4H, –CH2CH2CH2–), 4.25 (m, 4H, –OCH2–), 3.94 (m, 4H, –CH2CH2CH2–), 3.05 (m, 4H, –CH2CH2CH2–), 2.13 (m, 1H, Im), 2.09–(–1.77) (m, 35H); 13C NMR (125 MHz, CDCl3) δ/ppm = 197.17 (CO), 196.88 (CO), 189.20 (CO), 155.80 (C), 153.91 (C), 153.52 (CH), 151.86 (CH), 151.20 (C), 150.25 (C), 148.65 (C), 148.34 (C), 146.61 (C), 145.97 (C), 140.79 (C), 139.00 (CH), 137.04 (CH), 135.99 (CH), 135.88 (CH), 135.43 (CH), 133.18 (C), 131.64 (CH), 131.51 (CH), 129.60 (C), 129.59 (CH), 129.40 (CH), 128.75 (CH), 127.91 (CH), 127.00 (CH), 125.40 (CH), 125.15 (CH), 123.42 (CH), 123.19 (CH), 121.26 (CH), 119.47 (C), 116.94 (CH2), 97.08 (C), 72.19 (CH2), 70.17 (CH2), 67.85 (CH2), 50.58 (CH2), 50.24 (CH2), 50.25 (CH2), 49.88 (CH2), 47.84 (CH2), 47.39 (CH2), 46.53 (CH2), 45.24 (CH3), 45.04 (CH3), 44.84 (CH3), 38.78 (CH2), 36.49 (CH2), 36.23 (CH2), 35.62 (CH2), 35.49 (CH2), 32.20 (CH2), 30.65 (C), 30.49 (C), 30.43 (C), 30.23 (C), 29.80 (C), 29.63 (CH2), 29.45 (CH or CH3), 29.40 (CH or CH3), 29.34 (CH or CH3), 29.08 (C), 28.61 (CH or CH3), 28.48 (CH or CH3), 28.22 (CH or CH3), 28.04 (CH or CH3), 26.84 (CH or CH3), 26.61 (CH or CH3), 25.48 (CH2), 24.06 (CH2), 23.94 (CH2), 23.80 (CH2), 23.07 (CH2), 21.94 (CH or CH3), 21.61 (CH or CH3), 21.29 (CH or CH3), 16.09 (CH or CH3), 15.75 (CH or CH3); FT-IR (KBr) νCO = 1900, 1920, 2022 cm−1.
(E form)), 6.11 (s, 1.4H, –CH (Z form)), 5.56 (s, 2H, Im), 5.42 (m, 8H, β-pyrrole and –CH2CH2CH2–), 5.15 (m, 4H, –CH2CH2CH2–), 4.69 (m, 2.8H, –OCH2-(Z form)), 4.44 (m, 5.2H, –OCH2-(E form)), 4.21 (m, 7H, –CH2CH2CH2–), 3.95 (m, 1H, –CH2CH2CH2–), 3.29 (m, 1H, –CH2CH2CH2–), 3.13 (m, 3H, –CH2CH2CH2–), 3.00 (m, 4H, –CH2CH2CH2–), 2.12 (m, 2H, Im), 2.07–(–1.33) (m, 70H); 13C NMR (125 MHz, CDCl3) δ/ppm = 197.53 (CO), 197.23 (CO), 197.14 (CO), 196.86 (CO), 189.79 (CO), 189.19 (CO), 155.82 (C), 153.93 (C), 153.51 (CH), 153.40 (CH), 151.85 (C), 150.91 (C), 150.13 (C), 148.38 (C), 146.54 (C), 140.84 (C), 140.78 (C), 139.10 (CH), 138.98 (CH), 137.10 (CH), 137.05 (CH), 135.93 (CH), 135.84 (CH), 133.23 (C), 131.58 (CH), 129.60 (CH), 128.32 (CH), 127.88 (CH), 127.05 (CH), 125.38 (CH), 125.16 (CH), 123.43 (CH), 123.20 (CH), 119.70 (C), 116.91 (CH2), 70.80 (CH2), 70.58 (CH2), 67.24 (CH2), 49.93 (CH2), 47.75 (CH2), 40.06 (CH2), 39.01 (CH2), 32.82 (CH2), 30.50 (C), 30.30 (C), 29.70 (CH or CH3), 29.40 (CH or CH3), 28.25 (CH or CH3), 27.04 (CH or CH3), 21.58, (CH or CH3) 15.97 (CH or CH3); FT-IR (KBr) νCO = 1901, 1919, 2021 cm−1. The third fraction of the preparative GPC was collected and the solvent was evaporated to dryness, giving a purple solid (36 mg, 35% yield) as ReD′: HPLC (GPC, pyridine, 1.0 mL min−1) RT = 22.1 min; MALDI-TOF-mass (DCTB) m/z [M]+ 2146.6340, calcd for [C111H114BrN14O7ReZn2]+ 2146.6292; 1H NMR (500 MHz, CDCl3) δ/ppm = 9.70–9.62 (m, 2H, β-pyrrole), 9.56 (m, 2H, β-pyrrole), 9.20 (m, 1H, bpy 6), 9.15 (m, 2H, β-pyrrole), 9.08 (m, 4H, β-pyrrole), 8.90 (m, 3H, β-pyrrole and bpy 6′), 8.64 (m, 1H, bpy 3), 8.50 (brs, 2H, phenylene and bpy 3′), 8.18 (m, 1H, phenylene), 8.03 (m, 1H, phenylene), 7.80 (m, 1H, bpy 4), 7.76–7.60 (m, 5H, phenylene and bpy 4′), 7.36 (m, 2H, bpy 5′), 7.25 (brs, 1H, phenylene), 6.49 (s, 2.75H, –CH (E form)), 6.14 (s, 1.25H, –CH (Z form)), 5.57 (m, 3H, β-pyrrole and Im), 5.51 (m, 2H, β-pyrrole), 5.41 (m, 1H, Im), 5.15 (m, 8H, –CH2CH2CH2–), 4.70 (m, 2.5H, –OCH2–(Z form)), 4.45 (m, 5.5H, –OCH2–(E form)), 4.28–4.21 (m, 8H, –CH2CH2CH2–), 3.33–3.034 (m, 8H, –CH2CH2CH2–), 2.85 (s, 3H, CH3), 2.16 (m, 1H, Im), 1.94 (m, 1H, Im), 1.64 (m, 3H, CH3), 2.09–(–1.77) (m, 35H); 13C NMR (125 MHz, CDCl3) δ/ppm = 197.36 (CO), 197.16 (CO), 197.00 (CO), 196.80 (CO), 189.60 (CO), 188.98 (CO), 155.25 (C), 153.04 (CH), 151.05 (CH), 150.88 (CH), 150.21 (C), 150.10 (C), 149.81 (CH), 149.27 (C), 149.25 (C), 148.42 (C), 148.16 (C), 146.14 (C), 146.06 (C), 141.16 (C), 138.94 (CH), 138.85 (CH), 137.07 (C), 136.08 (CH), 135.90 (CH), 135.46 (CH), 134.82 (CH), 134.61 (CH), 132.84 (C), 132.50 (CH), 132.40 (CH), 131.47 (CH), 129.97 (CH), 129.62 (CH), 128.29 (CH), 127.87 (CH), 127.64 (CH), 127.35 (CH), 127.26 (CH), 126.54 (CH), 125.06 (CH), 123.82 (CH), 122.77 (CH), 121.47 (CH), 119.81 (C), 118.14 (CH), 116.91 (CH2), 96.94 (C), 96.47 (C), 72.21 (CH2), 70.83 (CH2), 70.60 (CH2), 67.30 (CH2), 50.58 (CH2), 49.90 (CH2), 47.85 (CH2), 47.34 (CH2), 45.02 (CH3), 40.06 (CH2), 39.01 (CH2), 35.63 (CH2), 32.91 (CH2), 32.03 (C), 30.50 (C), 30.26 (C), 30.19 (C), 29.81 (CH2), 29.66 (CH or CH3), 29.61 (CH or CH3), 29.50 (CH or CH3), 29.44 (CH or CH3), 29.40 (CH or CH3), 29.35 (CH or CH3), 28.51 (CH or CH3), 28.37 (CH or CH3), 28.28 (CH or CH3), 28.13 (CH or CH3), 26.93 (CH or CH3), 21.94 (CH or CH3), 21.79 (CH or CH3), 21.60 (CH or CH3), 21.29 (CH or CH3), 15.80 (CH or CH3); FT-IR (KBr) νCO = 1900, 1920, 2022 cm−1.
| Φo/Φ = 1 + Ksv[BIH] = 1 + kqτ[BIH] | (1) |
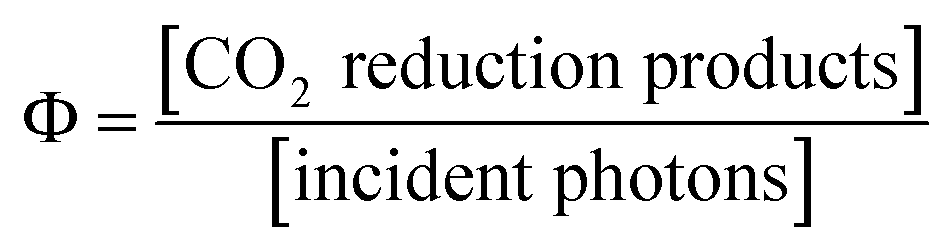 | (2) |
The solutions in 7 mL quartz cubic cells (optical path length: 1.0 cm) were irradiated with light at 420, 450 or 560 nm using bandpass filters. The concentrations of the catalysts were 0.025 mM (420 and 450 nm) and 0.1 mM (560 nm). The gaseous reaction products (CO and H2) were quantified with a gas chromatography system (GC-2014, Shimadzu Science) equipped with a Shincarbon column (i.d. 3.0 mm × 3.0 m) and a thermal conductivity detector (TCD). The product (formate) in the solutions was analyzed with a capillary electrophoresis system (Otuka Electronics Co. CAPI-3300I).
Author contributions
Y. K. conceived the idea, designed and performed some of the photocatalytic experiments, and wrote the manuscript. R. S. synthesized and characterized the isostearyl-substituted porphyrins, and performed most of the photocatalytic experiments. H. S. synthesized the methyl-substituted porphyrins. A. S. supervised the project, and provided suggestions on the experiments and writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP19K05677 and JP22H02186. We thank Prof. Akihiko Kudo (Tokyo University of Science) for help with capillary electrophoresis analyses.References
- (a) T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Solar Energy Supply and Storage for the Legacy and Nonlegacy Worlds, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed; (b) G. A. Olah, G. K. S. Prakash and A. Goeppert, Anthropogenic Chemical Carbon Cycle for a Sustainable Future, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- (a) H. Inoue, T. Shimada, Y. Kou, Y. Nabetani, D. Masui, S. Takagi and H. Tachibana, The Water Oxidation Bottleneck in Artificial Photosynthesis: How Can We Get Through It? An Alternative Route Involving a Two-Electron Process, ChemSusChem, 2011, 4, 173–179 CAS; (b) J.-H. Alstrum-Acevedo, M. K. Brennaman and T. J. Meyer, Chemical Approaches to Artificial Photosynthesis. 2, Inorg. Chem., 2005, 44, 6802–6827 CrossRef CAS PubMed.
- (a) A. W. Roszak, T. D. Howard, J. Southall, A. T. Gardiner, C. J. Law, N. W. Isaacs and R. J. Cogdell, Crystal Structure of the RC-LH1 Core Complex from Rhodopseudomonas palustris, Science, 2003, 302, 1969–1972 CrossRef CAS PubMed; (b) G. McDermott, S. M. Prince, A. A. Freer, A. M. Hawthornthwaite-Lawless, M. Z. Papiz, R. J. Cogdell and N. W. Isaacs, Crystal Structure of an Integral Membrane Light-Harvesting Complex from Photosynthetic Bacteria, Nature, 1995, 374, 517–521 CrossRef CAS.
- (a) R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Blackwell, Oxford, 2001 Search PubMed; (b) J. Deisenhofer, O. Epp, K. Miki, R. Huber and H. Michel, Structure of the Protein Subunits in the Photosynthetic Reaction Centre of Rhodopseudomonas viridis at 3Å Resolution, Nature, 1985, 318, 618–624 CrossRef CAS PubMed.
- Y. Kobuke and H. Miyaji, Supramolecular Organization of Imidazolyl-Porphyrin to a Slipped Cofacial Dimer, J. Am. Chem. Soc., 1994, 116, 4111–4112 CrossRef CAS.
- (a) F. Ito, Y. Ishibashi, S. R. Khan, H. Miyasaka, K. Kameyama, M. Morisue, A. Satake, K. Ogawa and Y. Kobuke, Photoinduced Electron Transfer and Excitation Energy Transfer in Directly Linked Zinc Porphyrin/Zinc Phthalocyanine Composite, J. Phys. Chem. A, 2006, 110, 12734–12742 CrossRef CAS PubMed; (b) H. Ozeki, A. Nomoto, K. Ogawa, Y. Kobuke, M. Murakami, K. Hosoda, M. Ohtani, S. Nakashima, H. Miyasaka and T. Okada, Role of the Special Pair in the Charge-Separating Event in Photosynthesis, Chem. – Eur. J., 2004, 10, 6393–6401 CrossRef CAS PubMed.
- K. Yoneyama, R. Suzuki, Y. Kuramochi and A. Satake, A Candidate for Multitopic Probes for Ligand Discovery in Dynamic Combinatorial Chemistry, Molecules, 2019, 24, 2166 CrossRef CAS PubMed.
- H. Nakagawa, K. Ogawa, A. Satake and Y. Kobuke, A Supramolecular Photosynthetic Triad of Slipped Cofacial Porphyrin Dimer, Ferrocene, and Fullerene, Chem. Commun., 2006, 1560–1562 RSC.
- (a) Y. Kuramochi, A. Satake, A. S. D. Sandanayaka, Y. Araki, O. Ito and Y. Kobuke, Fullerene- and Pyromellitdiimide-Appended Tripodal Ligands Embedded in Light-Harvesting Porphyrin Macrorings, Inorg. Chem., 2011, 50, 10249–10258 CrossRef CAS PubMed; (b) A. Satake, S. Azuma, Y. Kuramochi, S. Hirota and Y. Kobuke, Supramolecular Organization of Light-Harvesting Porphyrin Macrorings, Chem. – Eur. J., 2011, 17, 855–865 CrossRef CAS PubMed; (c) Y. Kuramochi, A. S. D. Sandanayaka, A. Satake, Y. Araki, K. Ogawa, O. Ito and Y. Kobuke, Energy Transfer Followed by Electron Transfer in a Porphyrin Macrocycle and Central Acceptor Ligand: A Model for a Photosynthetic Composite of the Light-Harvesting Complex and Reaction Center, Chem. – Eur. J., 2009, 15, 2317–2327 CrossRef CAS PubMed; (d) N. Nagata, Y. Kuramochi and Y. Kobuke, Energy Transfer among Light-Harvesting Macrorings Incorporated into a Bilayer Membrane, J. Am. Chem. Soc., 2009, 131, 10–11 CrossRef CAS PubMed; (e) Y. Kuramochi, A. Satake, M. Itou, K. Ogawa, Y. Araki, O. Ito and Y. Kobuke, Light-Harvesting Supramolecular Porphyrin Macrocycle Accommodating a Fullerene−Tripodal Ligand, Chem. – Eur. J., 2008, 14, 2827–2841 CrossRef CAS PubMed; (f) Y. Kuramochi, A. Satake and Y. Kobuke, Light-Harvesting Macroring Accommodating a Tetrapodal Ligand Based on Complementary and Cooperative Coordinations, J. Am. Chem. Soc., 2004, 126, 8668–8669 CrossRef CAS PubMed.
- (a) F. Hajjaj, Z. S. Yoon, M.-C. Yoon, J. Park, A. Satake, D. Kim and Y. Kobuke, Assemblies of Supramolecular Porphyrin Dimers in Pentagonal and Hexagonal Arrays Exhibiting Light-Harvesting Antenna Function, J. Am. Chem. Soc., 2006, 128, 4612–4623 CrossRef CAS PubMed; (b) R. Takahashi and Y. Kobuke, Hexameric Macroring of Gable-Porphyrins as a Light-Harvesting Antenna Mimic, J. Am. Chem. Soc., 2003, 125, 2372–2373 CrossRef CAS PubMed.
- S. Morikawa, C. Ikeda, K. Ogawa and Y. Kobuke, Two-Dimensional Porphyrin Array Assembled by Self-Coordination, Lett. Org. Chem., 2004, 1, 6–11 CrossRef CAS.
- (a) J. E. Raymond, A. Bhaskar, T. Goodson III, N. Makiuchi, K. Ogawa and Y. Kobuke, Synthesis and Two-Photon Absorption Enhancement of Porphyrin Macrocycles, J. Am. Chem. Soc., 2008, 130, 17212–17213 CrossRef CAS PubMed; (b) K. Ogawa, A. Ohashi, Y. Kobuke, K. Kamada and K. Ohta, Strong Two-Photon Absorption of Self-Assembled Butadiyne-Linked Bisporphyrin, J. Am. Chem. Soc., 2003, 125, 13356–13357 CrossRef CAS PubMed; (c) K. Ogawa, T. Zhang, K. Yoshihara and Y. Kobuke, Large Third-Order Optical Nonlinearity of Self-Assembled Porphyrin Oligomers, J. Am. Chem. Soc., 2002, 124, 22–23 CrossRef CAS PubMed.
- (a) M. Morisue, D. Kalita, N. Haruta and Y. Kobuke, Fine-Tuning of a Ferrocene|porphyrin|ITO Redox Cascade for Efficient Sequential Electron Transfer Commenced by an S2 Photoexcited Special Pair Mimic, Chem. Commun., 2007, 2348–2350 RSC; (b) M. Morisue, N. Haruta, D. Kalita and Y. Kobuke, Efficient Charge Injection from the S2 Photoexcited State of Special-Pair Mimic Porphyrin Assemblies Anchored on a Titanium-Modified ITO Anode, Chem. – Eur. J., 2006, 12, 8123–8135 CrossRef CAS PubMed.
- (a) M. Sugimoto, Y. Kuramochi and A. Satake, Measurement of Solvation Ability of Solvents by Porphyrin-Based Solvation/Desolvation Indicators, ACS Omega, 2020, 5, 6045–6050 CrossRef CAS PubMed; (b) A. Satake, Y. Suzuki, M. Sugimoto and Y. Kuramochi, Mechanistic Study of the Solvent-Dependent Formation of Extended and Stacked Supramolecular Polymers Composed of Bis(imidazolylporphyrinatozinc) Molecules, Chem. – Eur. J., 2020, 26, 669–684 CrossRef CAS PubMed; (c) A. Satake, Y. Suzuki, M. Sugimoto, T. Shimazaki, H. Ishii and Y. Kuramochi, A Solvation/Desolvation Indicator Based on van der Waals Interactions between Solvents and Porphyrins, Chem. – Eur. J., 2018, 24, 14733–14741 CrossRef CAS PubMed.
- (a) K. Rybicka-Jasinska, W. Shan, K. Zawada, K. M. Kadish and D. Gryko, Porphyrins as Photoredox Catalysts: Experimental and Theoretical Studies, J. Am. Chem. Soc., 2016, 138, 15451–15458 CrossRef CAS PubMed; (b) S. Shanmugam, J. Xu and C. Boyer, Exploiting Metalloporphyrins for Selective Living Radical Polymerization Tunable over Visible Wavelengths, J. Am. Chem. Soc., 2015, 137, 9174–9185 CrossRef CAS PubMed; (c) T. Lazarides, I. V. Sazanovich, A. J. Simaan, M. C. Kafentzi, M. Delor, Y. Mekmouche, B. Faure, M. Reglier, J. A. Weinstein, A. G. Coutsolelos and T. Tron, Visible Light-Driven O2 Reduction by a Porphyrin−Laccase System, J. Am. Chem. Soc., 2013, 135, 3095–3103 CrossRef CAS PubMed; (d) R. Miyatani and Y. Amao, Photochemical Synthesis of Formic Acid from CO2 with Formate Dehydrogenase and Water-Soluble Zinc Porphyrin, J. Mol. Catal. B: Enzym., 2004, 27, 121–125 CrossRef CAS.
- (a) P. Lang, M. Pfrunder, G. Quach, B. Braun-Cula, E. G. Moore and M. Schwalbe, Sensitized Photochemical CO2 Reduction by Hetero−Pacman Compounds Linking a ReI Tricarbonyl with a Porphyrin Unit, Chem. – Eur. J., 2019, 25, 4509–4519 CrossRef CAS PubMed; (b) C. Matlachowski, B. Braun, S. Tschierlei and M. Schwalbe, Photochemical CO2 Reduction Catalyzed by Phenanthroline Extended Tetramesityl Porphyrin Complexes Linked with a Rhenium(I) Tricarbonyl Unit, Inorg. Chem., 2015, 54, 10351–10360 CrossRef CAS PubMed; (c) C. D. Windle, M. W. George, R. N. Perutz, P. A. Summers, X. Z. Sun and A. C. Whitwood, Comparison of Rhenium−Porphyrin Dyads for CO2 Photoreduction: Photocatalytic Studies and Charge Separation Dynamics Studied by Time-Resolved IR Spectroscopy, Chem. Sci., 2015, 6, 6847–6864 RSC; (d) C. D. Windle, M. V. Campian, A.-K. Duhme-Klair, E. A. Gibson, R. N. Perutz and J. Schneider, CO2 Photoreduction with Long-Wavelength Light: Dyads and Monomers of Zinc Porphyrin and Rhenium Bipyridine, Chem. Commun., 2012, 48, 8189–8191 RSC; (e) K. Kiyosawa, N. Shiraishi, T. Shimada, D. Masui, H. Tachibana, S. Takagi, O. Ishitani, D. A. Tryk and H. Inoue, Electron Transfer from the Porphyrin S2 State in a Zinc Porphyrin-Rhenium Bipyridyl Dyad having Carbon Dioxide Reduction Activity, J. Phys. Chem. C, 2009, 113, 11667–11673 CrossRef CAS.
- (a) S. H. Choi, C. H. Kim, J.-O. Baeg, H. J. Son, C. Pac and S. O. Kang, Collisional Electron Transfer Route between Homogeneous Porphyrin Dye and Catalytic TiO2/Re(I) Particles for CO2 Reduction, ACS Appl. Energy Mater., 2020, 3, 11581–11596 CrossRef CAS; (b) D.-I. Won, J.-S. Lee, Q. Ba, Y.-J. Cho, H.-Y. Cheong, S. Choi, C. H. Kim, H.-J. Son, C. Pac and S. O. Kang, Development of a Lower Energy Photosensitizer for Photocatalytic CO2 Reduction: Modification of Porphyrin Dye in Hybrid Catalyst System, ACS Catal., 2018, 8, 1018–1030 CrossRef CAS.
- (a) Y. Kuramochi and A. Satake, Photocatalytic CO2 Reductions Catalyzed by meso-(1,10-Phenanthrolin-2-yl)-Porphyrins Having a Rhenium(I) Tricarbonyl Complex, Chem. – Eur. J., 2020, 26, 16365–16373 CrossRef CAS PubMed; (b) Y. Kuramochi and Y. Fujisawa, A. Satake, Photocatalytic CO2 Reduction Mediated by Electron Transfer via the Excited Triplet State of Zn(II) Porphyrin, J. Am. Chem. Soc., 2020, 142, 705–709 CrossRef CAS PubMed.
- A. Satake and Y. Kobuke, Artificial Photosynthetic Systems: Assemblies of Slipped Cofacial Porphyrins and Phthalocyanines Showing Strong Electronic Coupling, Org. Biomol. Chem., 2007, 5, 1679–1691 RSC.
- A. Ohashi, A. Satake and Y. Kobuke, Covalent Linking of Coordination-Organized Slipped Cofacial Porphyrin Dimers, Bull. Chem. Soc. Jpn., 2004, 77, 365–374 CrossRef CAS.
- We used 2nd generation Grubbs catalyst, benzylidene[1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(tricyclohexylphosphine)ruthenium, instead of 1st generation Grubbs catalyst but it did not improve the reaction progress. It was reported that 2nd generation Grubbs catalyst gave benzylidene-substituted byproducts in the metathesis reaction for the porphyrin dimer. See: C. Ikeda, A. Satake and Y. Kobuke, Proofs of Macrocyclization of Gable Porphyrins as Mimics of Photosynthetic Light-Harvesting Complexes, Org. Lett., 2003, 5, 4935–4938 CrossRef CAS PubMed.
- (a) S. Hitosugi, D. Tanimoto, W. Nakanishi and H. Isobe, A Facile Chromatographic Method for Purification of Pinacol Boronic Esters, Chem. Lett., 2012, 41, 972–973 CrossRef CAS; (b) A. Lützen, M. Hapke, H. Staats and J. Bunzen, Synthesis of Differently Disubstituted 2,2’-Bipyridines by a Modified Negishi Cross-Coupling Reaction, Eur. J. Org. Chem., 2003, 3948–3957 CrossRef.
- S. Sato, T. Morimoto and O. Ishitani, Photochemical Synthesis of mer-[Re(bpy)(CO)3Cl], Inorg. Chem., 2007, 46, 9051–9053 CrossRef CAS PubMed.
- O. Shoji, S. Okada, A. Satake and Y. Kobuke, Coordination Assembled Rings of Ferrocene-Bridged Trisporphyrin with Flexible Hinge-like Motion: Selective Dimer Ring Formation, Its Transformation to Larger Rings, and Vice Versa, J. Am. Chem. Soc., 2005, 127, 2201–2210 CrossRef CAS PubMed.
- Y. Kuramochi, M. Kamiya and H. Ishida, Photocatalytic CO2 Reduction in N,N-Dimethylacetamide/Water as an Alternative Solvent System, Inorg. Chem., 2014, 53, 3326–3332 CrossRef CAS PubMed.
- Y. Kuramochi, Y. Kawakami and A. Satake, Synthesis and Photophysical Properties of Porphyrin Macrorings Composed of Freebase Porphyrins and Slipped-Cofacial Zinc Porphyrin Dimers, Inorg. Chem., 2017, 56, 11008–11018 CrossRef CAS PubMed.
- Y. Kuramochi, S. Hashimoto, Y. Kawakami, M. S. Asano and A. Satake, Visualization of Nonemissive Triplet Species of Zn(II) Porphyrins through Cu(II) Porphyrin Emission via the Reservoir Mechanism in a Porphyrin Macroring, Photochem. Photobiol. Sci., 2018, 17, 883–888 CrossRef CAS PubMed.
- Y. Kuramochi, O. Ishitani and H. Ishida, Reaction Mechanisms of Catalytic Photochemical CO2 Reduction using Re(I) and Ru(II) Complexes, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS.
- J. M. Smieja, E. E. Benson, B. Kumar, K. A. Grice, C. S. Seu, A. J. M. Miller, J. M. Mayer and C. P. Kubiak, Kinetic and Structural Studies, Origins of Selectivity, and Interfacial Charge Transfer in the Artificial Photosynthesis of CO, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15646–15650 CrossRef CAS PubMed.
- (a) T. Nakajima, Y. Tamaki, K. Ueno, E. Kato, T. Nishikawa, K. Ohkubo, Y. Yamazaki, T. Morimoto and O. Ishitani, Photocatalytic Reduction of Low Concentration of CO2, J. Am. Chem. Soc., 2016, 138, 13818–13821 CrossRef CAS PubMed; (b) T. Morimoto, T. Nakajima, S. Sawa, R. Nakanishi, D. Imori and O. Ishitani, CO2 Capture by a Rhenium(I) Complex with the Aid of Triethanolamine, J. Am. Chem. Soc., 2013, 135, 16825–16828 CrossRef CAS PubMed.
- H. W. Whitlock and M. Y. Oester, Chemistry of Porphyrins. IV. Behavior of Di- and Tetrahydroporphyrins under Alkaline Conditions. Direct Observation of the Chlorin-Phlorin Equilibrium, J. Am. Chem. Soc., 1973, 95, 5738–5741 CrossRef CAS PubMed.
- (a) H. Yamaguchi, A. Soeta, H. Toeda and K. Itoh, Raman Scattering Study on Electrochemical Reduction Products of Magnesium, Zinc and Copper Tetraphenylporphines, J. Electroanal. Chem. Interfacial Electrochem., 1983, 159, 347–359 CrossRef CAS; (b) J. G. Lanese and G. S. Wilson, Electrochemical Studies of Zinc Tetraphenylporphin, J. Electrochem. Soc., 1972, 119, 1039–1043 CrossRef CAS; (c) G. L. Closs and L. E. Closs, Negative Ions of Porphin Metal Complexes, J. Am. Chem. Soc., 1963, 85, 818–819 CrossRef CAS.
- Y. Harel and J. Manassen, Photoreduction of Tetraphenylporphyrins by Amines in the Visible. Photochemical Syntheses of Reduced Tetraphenylporphyrins and the Mechanism of Photoreduction, J. Am. Chem. Soc., 1978, 100, 6228–6234 CrossRef CAS.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Photocatalytic CO2 Reduction with High Turnover Frequency and Selectivity of Formic Acid Formation using Ru(II) Multinuclear Complexes, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- (a) J. Rohacova and O. Ishitani, Rhenium(I) Trinuclear Rings as Highly Efficient Redox Photosensitizers for Photocatalytic CO2 Reduction, Chem. Sci., 2016, 7, 6728–6739 RSC; (b) V. S. Thoi, N. Kornienko, C. G. Margarit, P. Yang and C. J. Chang, Visible-Light Photoredox Catalysis: Selective Reduction of Carbon Dioxide to Carbon Monoxide by a Nickel N-Heterocyclic Carbene−Isoquinoline Complex, J. Am. Chem. Soc., 2013, 135, 14413–14424 CrossRef CAS PubMed; (c) Y. Tamaki, K. Watanabe, K. Koike, H. Inoue, T. Morimoto and O. Ishitani, Development of Highly Efficient Supramolecular CO2 Reduction Photocatalysts with High Turnover Frequency and Durability, Faraday Discuss., 2012, 155, 115–127 RSC; (d) H. Hori, F. P. A. Johnson, K. Koike, O. Ishitani and T. Ibusuki, Efficient Photocatalytic CO2 Reduction using [Re(bpy)(CO)3{P(OEt)3}]+, J. Photochem. Photobiol., A, 1996, 96, 171–174 CrossRef CAS.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, Development of an Efficient Photocatalytic System for CO2 Reduction Using Rhenium(I) Complexes Based on Mechanistic Studies, J. Am. Chem. Soc., 2008, 130, 2023–2031 CrossRef CAS PubMed.
- K. M. Kadish, L. R. Shiue, R. K. Rhodes and L. A. Bottomley, Reactions of Metalloporphyrin .pi. Radicals. 1. Complexation of Zinc Tetraphenylporphyrin Cation and Anion Radicals with Nitrogenous Bases, Inorg. Chem., 1981, 20, 1274–1277 CrossRef CAS.
- K. Kamogawa, Y. Shimoda, K. Miyata, K. Onda, Y. Yamazaki, Y. Tamaki and O. Ishitani, Mechanistic Study of Photocatalytic CO2 Reduction using a Ru(II)-Re(I) Supramolecular Photocatalyst, Chem. Sci., 2021, 12, 9682–9693 RSC.
- (a) H. Jing, S. Liu, J. Jiang, V.-P. Tran, J. Rong, P. Wang and J. S. Lindsey, Meso Bromination and Derivatization of Synthetic Bacteriochlorins, New J. Chem., 2022, 46, 5556–5572 RSC; (b) J. M. Dąbrowski, L. G. Arnaut, M. M. Pereira, C. J. P. Monteiro, K. Urbańska, S. Simões and G. Stochel, New Halogenated Water-Soluble Chlorin and Bacteriochlorin as Photostable PDT Sensitizers: Synthesis, Spectroscopy, Photophysics, and in vitro Photosensitizing Efficacy, ChemMedChem, 2010, 5, 1770–1780 CrossRef PubMed; (c) J. R. McCarthy, J. Bhaumik, N. Merbouh and R. Weissleder, High-yielding Syntheses of Hydrophilic Conjugatable Chlorins and Bacteriochlorins, Org. Biomol. Chem., 2009, 7, 3430–3436 RSC.
- M. Taniguchi and J. S. Lindsey, Synthetic Chlorins, Possible Surrogates for Chlorophylls, Prepared by Derivatization of Porphyrins, Chem. Rev., 2017, 117, 344–535 CrossRef CAS PubMed.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, Substantial Improvement in the Efficiency and Durability of a Photocatalyst for Carbon Dioxide Reduction Using a Benzoimidazole Derivative as an Electron Donor, J. Catal., 2013, 304, 22–28 CrossRef CAS.
- Y. Kou, Y. Nabetani, D. Masui, T. Shimada, S. Takagi, H. Tachibana and H. Inoue, Direct Detection of Key Reaction Intermediates in Photochemical CO2 Reduction Sensitized by a Rhenium Bipyridine Complex, J. Am. Chem. Soc., 2014, 136, 6021–6030 CrossRef CAS PubMed.
- (a) Y. Kou, Y. Nabetani, R. Nakazato, N. V. Pratheesh, T. Sato, S. Nozawa, S. Adachi, H. Tachibana and H. Inoue, Mechanism of the Photoreduction of Carbon Dioxide Catalyzed by the Benchmarking Rhenium Dimethylbipyridine Complexes; Operando Measurements by XAFS and FT-IR, J. Catal., 2022, 405, 508–519 CrossRef CAS; (b) C. D. Windle, E. Pastor, A. Reynal, A. C. Whitwood, Y. Vaynzof, J. R. Durrant, R. N. Perutz and E. Reisner, Improving the Photocatalytic Reduction of CO2 to CO through Immobilisation of a Molecular Re Catalyst on TiO2, Chem.–Eur. J., 2015, 21, 3746–3754 CrossRef CAS PubMed.
- D. Kalita, M. Morisue and Y. Kobuke, Synthesis and Electrochemical Properties of Slipped-Cofacial Porphyrin Dimers of Ferrocene-Functionalized Zn-Imidazolyl-Porphyrins as Potential Terminal Electron Donors in Photosynthetic Models, New J. Chem., 2006, 30, 77–92 RSC.
- (a) X.-Q. Zhu, M.-T. Zhang, A. Yu, C.-H. Wang and J.-P. Cheng, Hydride, Hydrogen Atom, Proton, and Electron Transfer Driving Forces of Various Five-Membered Heterocyclic Organic Hydrides and Their Reaction Intermediates in Acetonitrile, J. Am. Chem. Soc., 2008, 130, 2501–2516 CrossRef CAS PubMed; (b) E. Hasegawa, T. Seida, N. Chiba, T. Takahashi and H. Ikeda, Contrastive Photoreduction Pathways of Benzophenones Governed by Regiospecific Deprotonation of Imidazoline Radical Cations and Additive Effects, J. Org. Chem., 2005, 70, 9632–9635 CrossRef CAS PubMed.
- D. J. Quimby and F. R. Longo, Luminescence Studies on Several Tetraarylporphins and Their Zinc Derivatives, J. Am. Chem. Soc., 1975, 97, 5111–5117 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR, MALDI-TOF mass, IR, UV-vis and fluorescence spectra, GPC-HPLC charts, calculations for fluorescence quenching experiments, CV, DPV, energy diagrams, and photocatalytic CO2 reduction. See https://doi.org/10.1039/d2sc03251a |
| This journal is © The Royal Society of Chemistry 2022 |