
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring a bio-based adsorbent for sequestration of late transition and rare earth elements†
Troy C.
Breijaert
a,
Tetyana M.
Budnyak
b,
Vadim K.
Kessler
 a and
Gulaim A.
Seisenbaeva
*a
a and
Gulaim A.
Seisenbaeva
*a
aDepartment of Molecular Sciences, Biocentrum, Swedish University of Agricultural Sciences, Almas Allé 5, Box 7015, SE-750 07 Uppsala, Sweden. E-mail: gulaim.seisenbaeva@slu.se
bDivision of Nanotechnology and Functional Materials, Department of Materials Science and Engineering, Uppsala University, Box 35, 751 03 Uppsala, Sweden
First published on 15th November 2022
Abstract
The demand for new renewable energy sources, improved energy storage and exhaust-free transportation requires the use of large quantities of rare earth (REE) and late transition (LTM, group 8–12) elements. In order to achieve sustainability in their use, an efficient green recycling technology is required. Here, an approach, a synthetic route and an evaluation of the designed bio-based material are reported. Cotton-derived nano cellulose particles were functionalized with a polyamino ligand, tris(2-aminoethyl) amine (TAEA), achieving ligand content of up to ca. 0.8 mmol g−1. The morphology and structure of the produced adsorbent were revealed by PXRD, SEM-EDS, AFM and FTIR techniques. The adsorption capacity and kinetics of REE and LTM were investigated by conductometric photometric titrations, revealing quick uptake, high adsorption capacity and pronounced selectivity for LTM compared to REE. Molecular insights into the mode of action of the adsorbent were obtained via the investigation of the molecular structure of the Ni(II)–TAEA complex by an X-ray single crystal study. The bio-based adsorbent nanomaterial demonstrated in this work opens up a perspective for tailoring specific adsorbents in the sequestration of REE and LTM for their sustainable recycling.
Introduction
Rare earth elements (REE) are critical elements for the development of modern technology and industries.1,2 Their application in energy production and storage3 in the form of magnetic materials, permanent magnets4,5 and as components in NiMH batteries6,7 has resulted in an increased demand for these materials.2 This has resulted in an increase in an acquisition by mining, which is inherently unsustainable. In that regard, recycling the existing REE-containing materials such as common REE-based magnets needs to be promoted.8–10 However, both mining and recycling require the use of extraction and separation techniques involving large amounts of hazardous reagents and solvents.5,7,11 The most common technology for REE separation is acidic leaching with various acidic leaching agents, using several precipitation steps to remove unwanted components, after which the product can be calcined and re-dissolved.5,12–14 However, most established methods require repeated steps to obtain the desired metal purity as it is difficult to separate REE from late transition metals (LTM, groups 8–12), which are commonly found with REEs in REE-containing recyclable materials.15–17An alternative to the use of liquid-phase extraction and separation of REEs from late transition metals is the use of solid adsorbent materials for the extraction of metals from solution.18 Today a wide range of adsorbent materials are available that can remove metals from solution, ranging from inorganic materials such as fly ash,19 steel slag materials,20 olivine and zeolites21 to organic materials such as tree bark,22 chitin/chitosan,23–25 ion exchange resins, polymers,26,27 protein-based adsorbents,28,29 lignocellulosic sources30 and peat.31 With purely inorganic materials, it can be difficult to tune metal adsorption capacity and selectivity beyond what is inherent to the material but in some cases, at least with silicate materials32–36 and metal oxides,37,38 it is possible to graft the surface with organic ligands to improve metal uptake and selectivity. Organic adsorbents naturally contain reactive groups on the surface onto which ligands may be grafted. By choosing an appropriate type of coordinating ligand, it is possible to increase the adsorption capacity of the adsorbent materials and selectivity and therefore separate LTM from REE.34,37 Creating a tailored ligand monolayer on the surface of the adsorbent opens up the possibility to adjust it specifically for selective adsorption of target metal species, realizing the so-called molecular recognition approach.39–41
Natural bio-based polymer materials are especially suitable from an environmental point of view as they are considered renewable resources.42–44 Cellulose, for example, is the most abundant, renewable polymer on the planet, accounting for several terra tons of annual biomass production.45 It is found in plants as a major constituent, serving as a structural polymer, but it can also be produced by bacteria with the morphology depending on the source.45,46 In addition, varying the original morphology depending on the source, cellulose can be processed in order to form more advanced structures such as networks, gels and fibres. In terms of the chemical structure, it consists of polymeric β-1,4-linked D-glucose which self-assembles into higher-order structures which can be chemically modified for specific applications.47,48 The wide availability of cellulosic materials in nature combined with its renewable nature and low cost makes cellulose a very promising raw material for the preparation of advanced adsorbent materials.49–51 By grafting the surface of cellulose with ligands that have high affinity to specific metals and preferentially binding them, the recycling and separation of magnetic and battery materials can be made more environmentally sustainable.21,51 Amines, for example, are well known to strongly bind to late transition metals via the formation of coordinate covalent bonds while specific coordination geometries are known to stabilize certain metal cations. An example of such a ligand is tris(2-aminoethyl)amine, which contains four amino functions, three of which are located in freely rotating amine-containing arms and the ligand is known to form stable complexes with transition metals.52–55
In this study, we present the development and characterization of a cellulose-based nanoadsorbent material containing tris(2-aminoethyl)amine as a ligand that can rapidly form stable complexes with transition metals. Additionally, the mode of surface binding and possibility of ion exchange processes have been demonstrated by the single-crystal X-ray study of the model compound. The adsorption capacity, kinetics and metal uptake selectivity of REE and LTM and the pH-dependent desorption of metals from the produced cellulosic material have been studied.
Materials and methods
Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 min−1, 10 minutes). The particles were collected via centrifugation (10 min, 10k rpm) and washed several times with miliQ, re-suspending between each washing cycle. The sample was lyophilized for further analysis.
500 min−1, 10 minutes). The particles were collected via centrifugation (10 min, 10k rpm) and washed several times with miliQ, re-suspending between each washing cycle. The sample was lyophilized for further analysis.
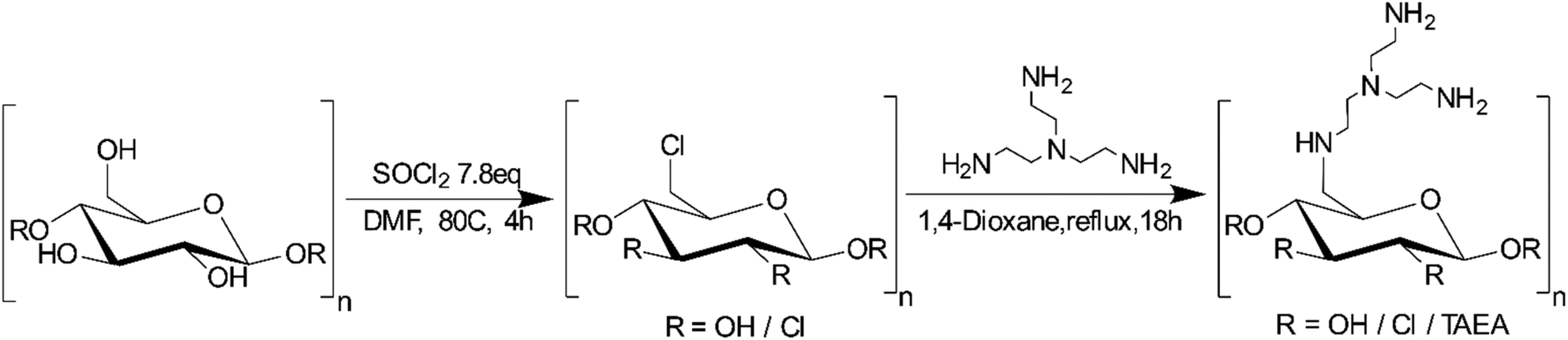 | ||
| Scheme 1 Preparation route to the synthesis of tris(2-aminoethyl)amine modified cellulose. | ||
[Ni(II)(TAEA)(H2O)(NO3)]NO3 (1). Ni(II)(NO3)2·6H2O (279.8 mg, 1 mmol) was dissolved in 4 mL of miliQ, followed by the addition of 1 molar equivalent (143 μl) of the ligand and the solutions were allowed to slowly evaporate at room temperature, yielding violet block-shaped (prismatic) crystals.
[N(C2H4NH3)3](NO3)3 (2). 1 mmol of REE(III)(NO3)2·6H2O (REE = Sm, Dy) was dissolved in 4 mL of miliQ, followed by the addition of 1 molar equivalent (143 μl) of the ligand, and the solutions were allowed to slowly evaporate at room temperature, yielding colorless plate-shaped crystals in a gel-like matrix.
Characterization
Results and discussion
Physical characterization of the adsorbent
One potential issue, depending on the application, with this approach, however, is that the chlorination of cellulose leads to a reduction of hydrogen-bonding along the solvent-accessible surface, which in turn allows the outer regions to dissolve in DMF. This will then precipitate out when exchanging solvent during washing or workup, leading to a change in morphology and crystallinity of the cellulose samples. The change in the cellulose structure as a result of reactive dissolution was clearly observed by pXRD (Fig. S1†). Here, we see a change in crystallinity and phase between pristine cellulose nanocrystals, composed of the Cellulose I phase, and the chlorinated product as a result of the reactive dissolution and subsequent regeneration of cellulose by precipitation in water. Further reaction of the chlorinated material with tris(2-aminoethyl)amine leads to a certain increase in crystallinity. X-ray patterns of modified products, as expected for a modified less crystalline polymer, showed essentially one broad peak with a slight shift to larger 2Theta due to the compression of hydrogen bonding as a result of the substitution of hydroxyl groups.
Textural characteristics of the cellulose–TAEA sample were investigated (Fig. S3†). Thus, the Brunauer–Emmett–Teller specific surface area (SBET) was determined from the low-temperature nitrogen adsorption/desorption isotherms and found to be 11 m2 g−1. The shape of the isotherm confirms the non-porous or macroporous surface of the material.57 The pore-size distribution by volume and area was obtained using the Barret–Joyner–Halenda (BJH) method58 by the desorption branch of the isotherms for the cellulose–TAEA sample. The BJH cumulative surface area and volume of pores between 1.7 nm and 300 nm were found to be 10.3 m2 g−1 and 0.025 cm3 g−1, whereas the BJH average pore width was 9.63 nm. Determination of the nano cellulose surface by nitrogen sorption is challenging because this material collapses, losing porosity even on freeze-drying that was applied. The data indicate that the surface of nano cellulose is not an active player in the adsorption process as the uptake seems to be related firsthand to the content of the functional ligand.
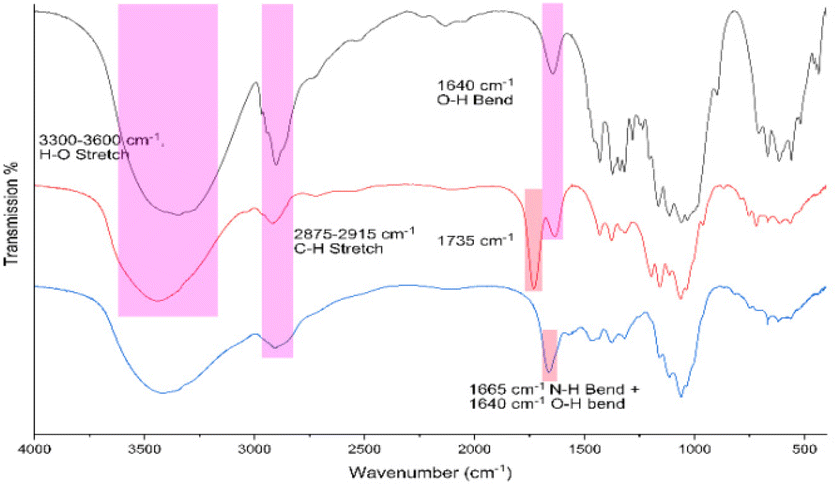 | ||
| Fig. 1 FTIR spectrum of pristine cellulose nanocrystals derived from cotton by sulfuric acid hydrolysis (black), chlorinated CNCs from SOCl2 in DMF (red) and TAEA-modified cellulose (blue). | ||
The relative position of most observed vibrations in the 3000–2800 cm−1 window remains essentially unchanged except for their transmission intensity, which may be attributed to the regeneration of the cellulose matrix observed via PXRD (Fig. S1†). After chlorination, a distinctly new peak appeared around 1725 cm−1, which disappeared after amination. In addition, a small shift from 1650 to 1665 cm−1 was observed which may be attributed to N–H bend vibrations, indicating the successful grafting of tris(2-aminoethyl)amine onto the cellulose matrix.
 | ||
| Fig. 2 AFM images of pristine CNCs derived from cotton by acid hydrolysis (A and B), and scanning electron microscopy images of chlorinated CNC derived from cotton CNCs in DMF (C and D) and TAEA-aminated cellulose (E and F). | ||
The resulting chlorinated product was aminated and after lyophilisation of the sample; the final product appeared to consist of globular nanoparticles in the 150–250 nm range. EDS analysis of the resulting aminated product showed an appreciable amine content of approximately 4.67 ± 1.55 mol g−1 nitrogen, corresponding to 1.17 ± 0.39 mmol g−1 amine. The resulting material still contains 1.76 ± 0.06 mmol g−1 chlorine and 0.12 ± 0.02 mmol g−1 sulphur in addition to the newly installed amine. The remaining chlorine in the sample may be attributed in part to the reaction being incomplete and in addition, partly attributed to the internalization of chlorine-containing cellulose chains, making it inaccessible for the subsequent amination step. For metal selectivity experiments, it is important to note the presence of elements not associated with the ligand as these may in turn interact with metal cations by coordinative covalent bonds or electrostatic interactions. Conductometric titration of the final aminated product leads to an amine content of 0.84 mmol g−1 (Fig. S2†). The discrepancy between EDS and conductometric titrations may be explained by the surface availability of the amine in aqueous media after lyophilisation, requiring a longer equilibration time.
Metal sequestration characterization
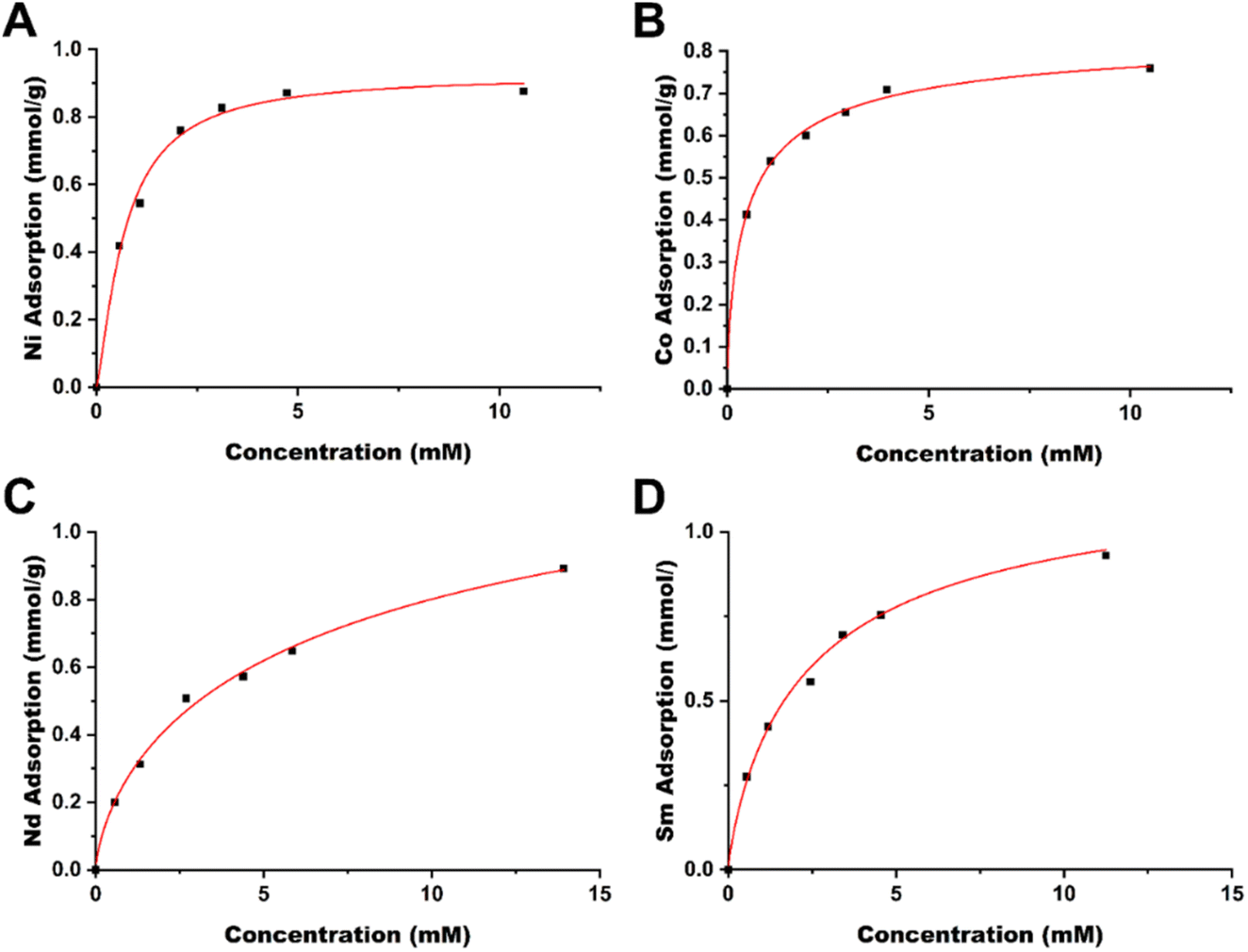 | ||
| Fig. 3 Langmuir isotherms of late transition metals (A) Ni and (B) Co and rare earth elements (C) Nd and (D) Sm. | ||
| Metal | Adsorption capacity [mmol g−1] | Metal/ligand stoichiometry |
|---|---|---|
| Co | 0.76 | 0.90 |
| Ni | 0.88 | 1.04 |
| Nd | 0.79 | 0.94 |
| Sm | 0.82 | 0.97 |
Based on these results, the TAEA-modified cellulose material is promising for metal sequestering, obtaining near 1:1 ligand to metal stoichiometry, indicating that the adsorption capacity of the material is inherently tied to the ligand grafting, i.e. higher ligand grafting will increase the metal adsorption capacity.
To evaluate the rate at which metals are removed from solution, the adsorption kinetics were studied (see Fig. 4), which showed that for cobalt(II), the adsorption occurs rapidly, reaching 53% within an hour and 83% within 6 hours. The next metal with the highest rate of adsorption is neodymium, showing 40% adsorption in 6 hours, followed by nickel(II) at 34% in 6 hours. Samarium in comparison is rather slow to adsorb, reaching 61% adsorption in a 24-hour window. As cobalt(II) is rapidly adsorbed by TAEA-functionalized cellulose, it may provide some kinetic selectivity between the separation of cobalt from samarium in CoSm magnets and materials with similar compositions. The separation between nickel and neodymium in comparison, however, does not yield any significant kinetic selectivity.
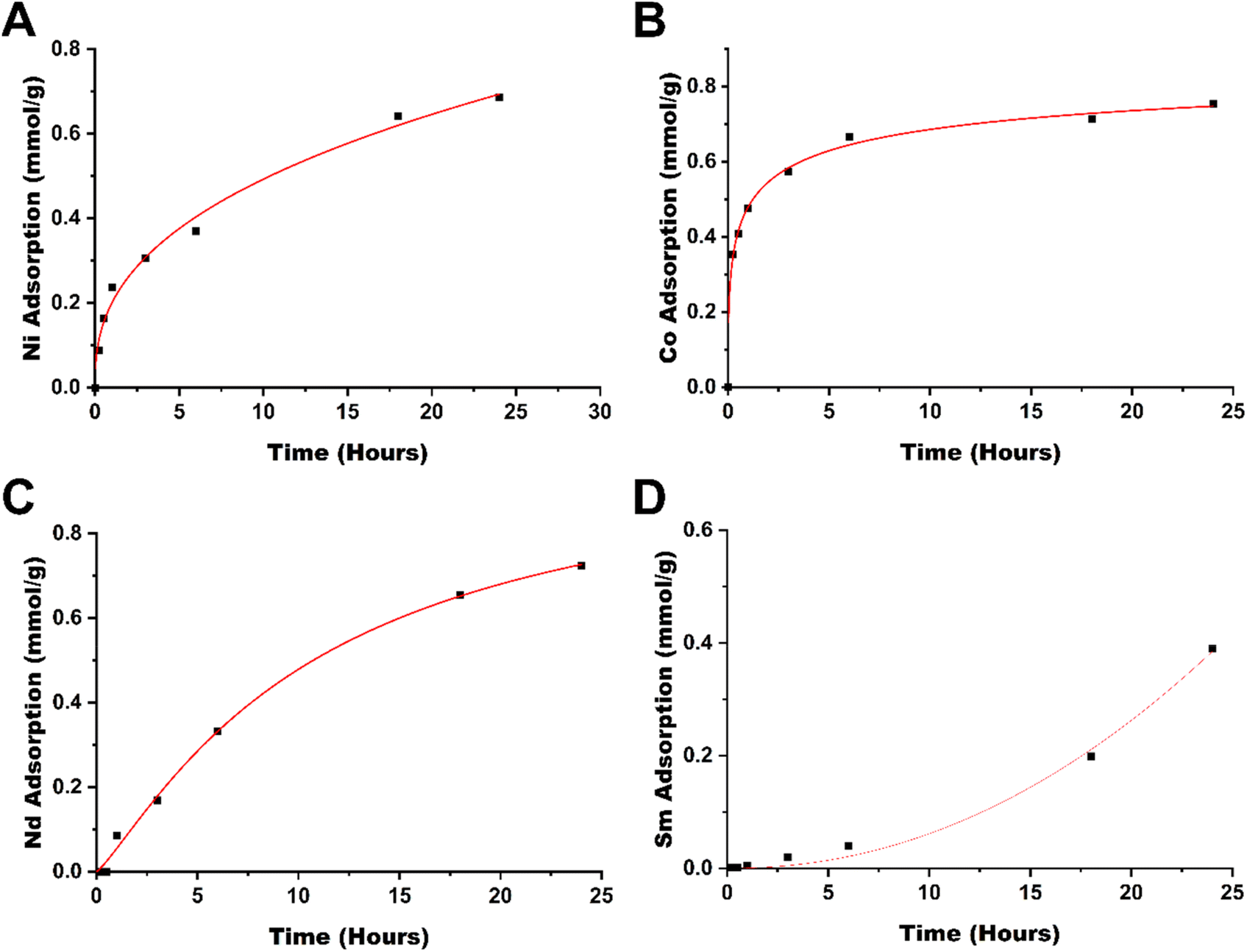 | ||
| Fig. 4 Adsorption kinetics of (A) Ni, (B) Co, (C) Nd, and (D) Sm by cell-TAEA in a 24 h period. | ||
As the intended purpose of the material is to selectively remove one class of metal (LTM/REE) from solution, the adsorption selectivity between equimolar mixtures of different metal ions by the material at a solution equilibrium was studied by EDS spectroscopy. By looking at the elemental ratios between metals on different spots we can estimate the average selectivity of the material. The result of EDS analysis can be viewed in Table 2. Briefly, when presented with an equimolar mixture between a lanthanide and a late transition metal, the TAEA functionalized material has a 5:1 preference towards the adsorption of late transition metals (Fig. S4 and S5†). When presented with an equimolar mixture of cobalt(II) and nickel(II), the product shows negligible selectivity towards cobalt (Fig. S6†). Similarly for a samarium(III)/neodymium(III) mixture, there is a negligible preference towards neodymium (Fig. S7†), indicating that the material is suitable for selectively adsorbing late transition metals over lanthanides from solution.
| Metal mixture | Ratio |
|---|---|
| Ni:Co |
1:1.1 |
| Nd:Ni |
1:5.1 |
| Sm:Co |
1:5.7 |
| Sm:Nd |
1:1.2 |
The difference between the adsorption kinetics of Sm(III) and Nd(III) is difficult to explain. It might originate from different features in the nucleation and growth of the corresponding hydroxides formed on the interaction of the ions with a local basic medium in the proximity of poly-amino ligands (please see below).
In addition, desorption tests were performed at pH values 0, 1, 2, 3, 4 and 5 using nitric acid in 50 mL Falcon tubes. After the samples were equilibrated for 48 hours, the samples were centrifuged and the pH was adjusted to predetermined values.
The samples were placed on an orbital shaker for 24 hours, after which the nanoparticles were separated by centrifugation (5500 rpm, 10 min), the supernatant was collected and the pH was adjusted with a base and titrated against EDTA to determine the desorbed metal amount. The results of the desorption experiments can be seen in Fig. 5 and are summarized in Table TS1.† For most metals at least 50% recovery is possible, expect for cobalt, which appears to be more resilient to acid treatment for recycling of the material.
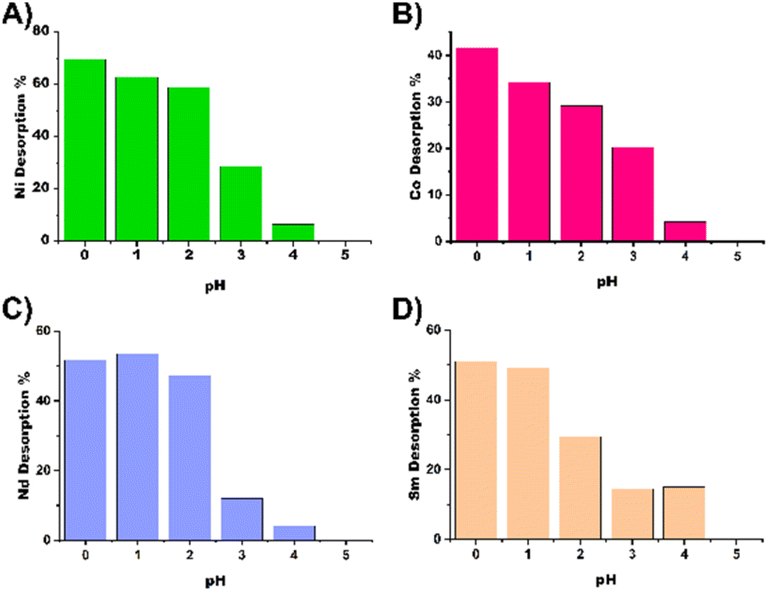 | ||
| Fig. 5 pH-Dependent desorption of (A) Ni, (B) Co, (C) Nd and (D) Sm from cell TAEA. | ||
The single crystal X-ray study revealed a molecular structure (see Fig. 6), where the Ni(II) cation is octahedrally coordinated with the coordination sphere composed of the four nitrogen atoms of the TAEA ligand, one oxygen atom of the coordinated water molecule and one more oxygen atom from the inner-sphere coordinated nitrate ion. The bond lengths are essentially equivalent for all the Ni–N contacts (Ni(1)–N(2) 2.085(6), Ni(1)–N(3) 2.109(6), Ni(1)–N(4) 2.072(6), and Ni(1)–N(5) 2.095(6) Å) and the coordinated nitrate oxygen atom (Ni(1)–O(1) 2.101(5) Å). The bond to hydrating the water molecule is considerably longer, Ni(1)–O(1B) 2.201(5) Å, which is a commonly observed feature. Bonding within the Ni(TAEA) fragment is comparable with that observed earlier in the structures of Ni(II) with this ligand such as Ni(TAEA)(NO3)254 and [Ni3(TAEA)4(H2O)2(TMEDA)2]Cl6.59 In these latter structures, the nature of bonding was very analogous with essentially equal bonds to the five neighbours, four N-atoms of the TAEA ligands in both cases (Ni–N 2.055–2.095 and 2.080–2.117 Å respectively) and one more atom – either an O-atom in one of the nitrate ligands or a N-atom of the additional TMEDA ligand (2.075 Å in both cases). The sixth bond is much longer and stays for bonding either to an oxygen atom in an additional nitrate ion or to that in a water molecule. Observation of this manner of bonding actually is rather peculiar, indicating the most probable Jahn–Teller effect in the 3d8 configuration of the Ni(II) cation, resulting in the weakening of one bond in the octahedral coordination sphere permitting facile ligand exchange between the hydrating water and the nitrate ligand (Table 3).
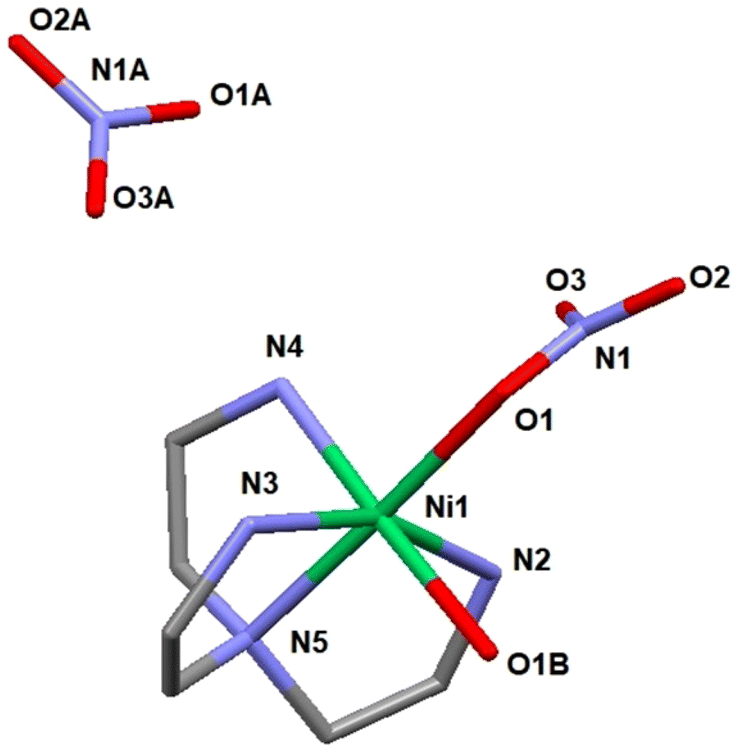 | ||
| Fig. 6 Molecular structure of [Ni(TAEA)(H2O)(NO3)](NO3) (1). | ||
| Compound | 1 |
| Chemical composition | C6H20N6NiO7 |
| Formula weight | 347.09 |
| Crystal system | Monoclinic |
| Space group | P2(1)/c (14) |
| R 1 | 0.0836 |
| wR2 | 0.1990 |
| a [Å] | 8.3129(6) |
| b [Å] | 14.3595(11) |
| c [Å] | 11.7358(9) |
| α [°] | 90 |
| β [°] | 91.8660(10) |
| γ [°] | 90 |
| V [Å3] | 1400.15(18) |
| T [K] | 296(2) |
| Z | 4 |
| No. of obs. independent refl., I ≥ 2σ(I) | 2364 |
| Residual electron density max | 1.635 |
Crystallization in a more humid environment in this case led to a structure with the water molecule included in the coordination sphere and “free” nitrate ion. The late transition metal cations (an analogous Co(II) structure has been reported with two nitrite ligands bound in the coordination sphere via N-atoms, Co(TAEA)(NO2)255 – all bonds essentially equal in length) are apparently forming stable complexes with the chelating TAEA ligand, which explains the stronger selectivity of bonding towards Ni(II) and Co(II) compared to REE of the reported adsorbent.
In the case of REE, the product of mass crystallization from an equimolar mixture of metal nitrate and TAEA ligand turned out to be solely [N(C2H4NH3)3](NO3)3 (2). The nature of the obtained product was the same for both Dy(III) and Sm(III) as indicated by unit cell parameter determination for multiple single crystals from the reaction mixture (see Table TS2 and Fig. S8†). The obtained structure is identical to that described by Bianchi et al.60 This shows that the sorption mechanism of REE is supposedly principally different from that for LTM and is not involving inner sphere complexation with the grafted poly-amino ligand. The REE sorption may involve retention of either the hydrated cations or hydroxide forms. An additional argument for this option is provided by the work of Roesky et al.,61 where the structures of several mixed-ligand complexes of Ni(II) and REE(III) with TAEA and Schiff base carboxylate were reported. In the competition between ligands, the Ni(II) cations were binding exclusively to nitrogen atoms in TAEA and REE – to the carboxylate oxygen atoms (see Fig. S9†). Such behavior is well in line with Pearson's hard and soft acid–base theory62 as Ni(II) is a soft acid binding to a soft amino ligand, while REE cations are typically hard acids binding to hard oxygen donor bases.
Conclusions
The possibility of grafting a specific ligand permitting sequestration and separation of REE and LTM on a bio-based adsorbent matrix of nano cellulose has been demonstrated. Tris(2-aminoethyl)amine was attached to cellulose nanocrystals via a two-step route involving chlorination and amination steps. The morphology and crystallinity of the matrix were preserved, as demonstrated by PXRD and microscopy techniques. The produced adsorbent showed relatively quick adsorption kinetics at room temperature and pH = 6 for LTM, especially for Co(II), and considerably slower for REE. The adsorption capacity achieved values of ca. 0.8 mmol g−1, corresponding to the functional ligand content and thus demonstrating the formation of surface complexes with 1:1 composition for all studied cations. The adsorbent revealed pronounced selectivity towards LTM compared to REE. The desorption of target cations was investigated as a function of pH, demonstrating comparably stronger retention of LTM compared to REE. The X-ray single crystal studies of molecular model compounds, [Ni(TAEA)(H2O)(NO3)](NO3) (1) and [N(C2H4NH3)3](NO3)3 (2), gave insights into the possible mode of action of the adsorbent and its cation and anion exchange processes.
Author contributions
Investigation, analysis, writing, and draft preparation – T. C. B.; Crystallography and writing – review and editing – V. G. K.; nitrogen adsorption/desorption and writing – T. M. B.; FTIR and AFM investigations, funding acquisition, and writing – review and editing – G. A. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express their gratitude to Andreas Orthaber at Uppsala University for access to the single crystal X-ray diffractometer. In addition, the authors would like to express their gratitude to the Swedish Research Council STINT for the support of the grant Nanocellulose Based Materials for Environmental and Theranostic Applications and the Faculty of Natural Resources and Agricultural Sciences, SLU for the support of T. C. B.'s PhD position. The ÅForsk Research Foundation is gratefully acknowledged for the support of the grant Separation and Recycling of Rare Earth Elements and Nickel and Cobalt from magnets, ÅF 21-75.References
- J. B. H. Gordon, B. Haxel and G. J. Orris, Rare Earth Elements – Critical Resources for High Technology, https://pubs.usgs.gov/fs/2002/fs087-02/, (accessed 26/09/2022, 2022).
- V. Balaram, Geosci. Front., 2019, 10, 1285–1303 CrossRef CAS.
- O. Serpell, W. Y. Chu and B. Paren, Rare Earth Elements: A Resource Constraint of the Energy Transition, https://kleinmanenergy.upenn.edu/research/publications/rare-earth-elements-a-resource-constraint-of-the-energy-transition/, (accessed 26/09/2022, 2022).
- J. Cui, M. Kramer, L. Zhou, F. Liu, A. Gabay, G. Hadjipanayis, B. Balasubramanian and D. Sellmyer, Acta Mater., 2018, 158, 118–137 CrossRef CAS.
- Y. Yang, A. Walton, R. Sheridan, K. Güth, R. Gauß, O. Gutfleisch, M. Buchert, B.-M. Steenari, T. Van Gerven, P. T. Jones and K. Binnemans, J. Sustainable Metall., 2016, 3, 122–149 CrossRef.
- A. Porvali, S. Ojanen, B. P. Wilson, R. Serna-Guerrero and M. Lundström, J. Sustainable Metall., 2020, 6, 78–90 CrossRef.
- F. Holmberg, Licentiate, Chalmers University of Technology, 2017.
- A. B. Patil, V. Paetzel, R. P. W. J. Struis and C. Ludwig, Separations, 2022, 9, 56 CrossRef CAS.
- B. Deng, X. Wang, D. X. Luong, R. A. Carter, Z. Wang, M. B. Tomson and J. M. Tour, Sci. Adv., 2022, 8, eabm3132 CrossRef CAS PubMed.
- A. Porvali, V. Agarwal and M. Lundstrom, Waste Manage., 2020, 107, 66–73 CrossRef CAS PubMed.
- K. Korkmaz, M. Alemrajabi, Å. C. Rasmuson and K. M. Forsberg, Sep. Purif. Technol., 2020, 234, 115812 CrossRef CAS.
- S. Peelman, Z. H. I. Sun, J. Sietsma and Y. Yang, Presented in part at the 1st European Rare Earth Resources Conference, Milos, 07/09/2014, 2014.
- R. Q. Honaker, W. Zhang and J. Werner, Energy Fuels, 2019, 33, 5971–5980 CrossRef CAS.
- A. Yuksekdag, B. Kose-Mutlu, B. Zeytuncu-Gokoglu, M. Kumral, M. R. Wiesner and I. Koyuncu, Environ. Sci. Pollut. Res. Int., 2022, 29, 7772–7781 CrossRef CAS PubMed.
- M. Gergoric, C. Ekberg, M. R. S. J. Foreman, B.-M. Steenari and T. Retegan, J. Sustainable Metall., 2017, 3, 638–645 CrossRef.
- N. Swain and S. Mishra, J. Cleaner Prod., 2019, 220, 884–898 CrossRef CAS.
- A. Rout and K. Binnemans, Dalton Trans., 2014, 43, 3186–3195 RSC.
- Y. Hu, J. Florek, D. Lariviere, F. G. Fontaine and F. Kleitz, Chem. Rec., 2018 DOI:10.1002/tcr.201800012.
- U. O. Aigbe, K. E. Ukhurebor, R. B. Onyancha, O. A. Osibote, H. Darmokoesoemo and H. S. Kusuma, J. Mater. Res. Technol., 2021, 14, 2751–2774 CrossRef CAS.
- H.-J. Kang, K.-G. An and D.-S. Kim, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2004, 39, 3015–3028 CrossRef.
- L. J. Westholm, E. Repo and M. Sillanpaa, Environ. Sci. Pollut. Res. Int., 2014, 21, 9109–9128 CrossRef CAS.
- S. Martini, S. Afroze and K. Ahmad Roni, Alexandria Eng. J., 2020, 59, 1637–1648 CrossRef.
- S. Feng, X. Du, M. Bat-Amgalan, H. Zhang, N. Miyamoto and N. Kano, Int. J. Mol. Sci., 2021, 22, 3447 CrossRef CAS PubMed.
- D. L. Ramasamy, A. Wojtuś, E. Repo, S. Kalliola, V. Srivastava and M. Sillanpää, Chem. Eng. J., 2017, 330, 1370–1379 CrossRef CAS.
- D. C. da Silva Alves, B. Healy, L. A. A. Pinto, T. R. S. Cadaval Jr. and C. B. Breslin, Molecules, 2021, 26, 594 CrossRef CAS PubMed.
- A. Chowdhury, S. K. Das, S. Mondal, S. Ruidas, D. Chakraborty, S. Chatterjee, M. K. Bhunia, D. Chandra, M. Hara and A. Bhaumik, Environ. Sci.: Nano, 2021, 8, 2641–2649 RSC.
- S. Mondal, S. Chatterjee, S. Mondal and A. Bhaumik, ACS Sustainable Chem. Eng., 2019, 7, 7353–7361 CrossRef CAS.
- Z. Dong, J. A. Mattocks, G. J. P. Deblonde, D. Hu, Y. Jiao, J. A. Cotruvo and D. M. Park, ACS Cent. Sci., 2021, 7, 1798–1808 CrossRef CAS PubMed.
- X. Xie, K. Yang, Y. Lu, Y. Li, J. Yan, J. Huang, L. Xu, M. Yang and Y. Yan, J. Hazard. Mater., 2022, 438, 129561 CrossRef CAS PubMed.
- M. L. Pitcher, B. Huntington, J. Dominick and A. Sheikhi, Chem. Eng. J., 2022, 447, 137418 CrossRef CAS.
- L. Ringqvist, A. Holmgren and I. Öborn, Water Res., 2002, 36, 2394–2404 CrossRef CAS.
- R. M. Ashour, M. Samouhos, E. Polido Legaria, M. Svärd, J. Högblom, K. Forsberg, M. Palmlöf, V. G. Kessler, G. A. Seisenbaeva and Å. C. Rasmuson, ACS Sustainable Chem. Eng., 2018, 6, 6889–6900 CrossRef CAS.
- E. Polido Legaria, I. Saldan, P. Svedlindh, E. Wetterskog, K. Gunnarsson, V. G. Kessler and G. A. Seisenbaeva, Dalton Trans., 2018, 47, 1312–1320 RSC.
- A. Vardanyan, A. Guillon, T. Budnyak and G. A. Seisenbaeva, Nanomaterials, 2022, 12, 974 CrossRef CAS.
- I. Anastopoulos, A. Bhatnagar and E. C. Lima, J. Mol. Liq., 2016, 221, 954–962 CrossRef CAS.
- D. Dupont, W. Brullot, M. Bloemen, T. Verbiest and K. Binnemans, ACS Appl. Mater. Interfaces, 2014, 6, 4980–4988 CrossRef CAS PubMed.
- A. Mezy, A. Vardanyan, A. Garcia, C. Schmitt, M. Lakić, S. Krajnc, G. Daniel, A. Košak, A. Lobnik and G. A. Seisenbaeva, Sep. Purif. Technol., 2021, 276, 119340 CrossRef CAS.
- G. A. Seisenbaeva, I. V. Melnyk, N. Hedin, Y. Chen, P. Eriksson, E. Trzop, Y. L. Zub and V. G. Kessler, RSC Adv., 2015, 5, 24575–24585 RSC.
- E. Polido Legaria, M. Samouhos, V. G. Kessler and G. A. Seisenbaeva, Inorg. Chem., 2017, 56, 13938–13948 CrossRef CAS.
- S. H. Gellman, Chem. Rev., 1997, 97, 1231–1232 CrossRef CAS.
- Y. M. Chen, C. Z. Wang, Q. Y. Wu, J. H. Lan, Z. F. Chai and W. Q. Shi, Dalton Trans., 2021, 50, 15576–15584 RSC.
- N. Morin-Crini, S. Loiacono, V. Placet, G. Torri, C. Bradu, M. Kostić, C. Cosentino, G. Chanet, B. Martel, E. Lichtfouse and G. Crini, Environ. Chem. Lett., 2018, 17, 393–408 CrossRef.
- D. Stewart, Ind. Crops Prod., 2008, 27, 202–207 CrossRef CAS.
- A. Boujemaoui, S. Mongkhontreerat, E. Malmstrom and A. Carlmark, Carbohydr. Polym., 2015, 115, 457–464 CrossRef CAS.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- A. F. Jozala, L. C. de Lencastre-Novaes, A. M. Lopes, V. de Carvalho Santos-Ebinuma, P. G. Mazzola, A. Pessoa Jr., D. Grotto, M. Gerenutti and M. V. Chaud, Appl. Microbiol. Biotechnol., 2016, 100, 2063–2072 CrossRef CAS.
- A. C. O'Sullivan, Cellulose, 1997, 4, 173–207 CrossRef.
- K. H. Gardner and J. Blackwell, Biopolymers, 1974, 13, 1975–2001 CrossRef CAS.
- R. D. S. Bezerra, R. C. Leal, M. S. da Silva, A. I. S. Morais, T. H. C. Marques, J. A. Osajima, A. B. Meneguin, H. da Silva Barud and E. C. da Silva Filho, Molecules, 2017, 22, 2039 CrossRef PubMed.
- O. Hamed, B. A. Lail, A. Deghles, B. Qasem, K. Azzaoui, A. A. Obied, M. Algarra and S. Jodeh, Environ. Sci. Pollut. Res. Int., 2019, 26, 28080–28091 CrossRef CAS.
- T. Oshima, K. Kondo, K. Ohto, K. Inoue and Y. Baba, React. Funct. Polym., 2008, 68, 376–383 CrossRef CAS.
- X. Huang, X. Chang, Q. He, Y. Cui, Y. Zhai and N. Jiang, J. Hazard. Mater., 2008, 157, 154–160 CrossRef CAS PubMed.
- A. Badiei, A. Mirahsani, A. Shahbazi, H. Younesi and M. Alizadeh, Environ. Prog. Sustainable Energy, 2014, 33, 1242–1250 CAS.
- D. Sivanesan, K. H. Song, S. K. Jeong and H. J. Kim, Catal. Commun., 2019, 120, 66–71 CrossRef CAS.
- M. K. Saha and I. Bernal, C. R. Chim., 2007, 10, 1170–1179 CrossRef CAS.
- M. S. Reid, M. Villalobos and E. D. Cranston, Langmuir, 2017, 33, 1583–1598 CrossRef CAS PubMed.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- K. Matelková, J. Moncol, R. Herchel, Ľ. Dlháň, R. Ivaniková, I. Svoboda, Z. Padělková and A. Mašlejová, Polyhedron, 2013, 56, 1–8 CrossRef.
- C. Bazzicalupi, A. Bencini, A. Bianchi, A. Danesi, C. Giorgi and B. Valtancoli, Inorg. Chem., 2009, 48, 2391–2398 CrossRef CAS PubMed.
- A. Bhunia, M. Yadav, Y. Lan, A. K. Powell, F. Menges, C. Riehn, G. Niedner-Schatteburg, P. P. Jana, R. Riedel, K. Harms, S. Dehnen and P. W. Roesky, Dalton Trans., 2013, 42, 2445–2450 RSC.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2207506. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03150g |
| This journal is © The Royal Society of Chemistry 2022 |