
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rearrangement and decomposition pathways of bare and hydrogenated molybdenum oxysulfides in the gas phase†
Marco
Pritzi
 a,
Tobias F.
Pascher
a,
Marie-Luise
Grutza
b,
Philipp
Kurz
b,
Milan
Ončák
*a and
Martin K.
Beyer
*a
a,
Tobias F.
Pascher
a,
Marie-Luise
Grutza
b,
Philipp
Kurz
b,
Milan
Ončák
*a and
Martin K.
Beyer
*a
aInstitut für Ionenphysik und Angewandte Physik, Universität Innsbruck, Technikerstrasse 25, 6020 Innsbruck, Austria. E-mail: milan.oncak@uibk.ac.at; martin.beyer@uibk.ac.at
bInstitut für Anorganische und Analytische Chemie, Albert-Ludwigs-Universität Freiburg, Albertstrasse 21, 79104 Freiburg, Germany
First published on 24th June 2022
Abstract
Molybdenum sulfides and molybdenum oxysulfides are considered a promising and cheap alternative to platinum as a catalyst for the hydrogen evolution reaction (HER). To better understand possible rearrangements during catalyst activation, we perform collision induced dissociation experiments in the gas phase with eight different molybdenum oxysulfides, namely [Mo2O2S6]2−, [Mo2O2S6]−, [Mo2O2S5]2−, [Mo2O2S5]−, [Mo2O2S4]−, [HMo2O2S6]−, [HMo2O2S5]− and [HMo2O2S4]−, on a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer. We identify fragmentation channels of the molybdenum oxysulfides and their interconnections. Together with quantum chemical calculations, the results show that [Mo2O2S4]− is a particularly stable species against further dissociation, which is reached from all starting species with relatively low collision energies. Most interestingly, H atom loss is the only fragmentation channel observed for [HMo2O2S4]− at low collision energies, which relates to potential HER activity, since two such H atom binding sites on a surface may act together to release H2. The calculations reveal that multiple isomers are often very close in energy, especially for the hydrogenated species, i.e., atomic hydrogen can bind at various sites of the clusters. S2 groups play a decisive role in hydrogen adsorption. These are further features with potential relevance for HER catalysis.
Introduction
Hydrogen plays a major role in sustainable energy economy schemes,1e.g. for the storage of renewable electricity from solar or wind farms or for transportation, either directly or via power-to-gas processes.2 Water electrolysis is crucial for these plans, and both half-cell reactions, the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER), must be optimized to increase efficiency and reduce costs.3,4 The standard HER catalyst under acidic conditions, where the highest efficiencies are obtained, is platinum.5 Given the limited availability of this element in the Earth's crust, large-scale electrolyzers often rely on more abundant catalyst materials which only work under alkaline conditions,6 which reduces the energy efficiency of the process.Molybdenum sulfides7 are extensively investigated as a promising alternative for platinum.8 After electrochemical reduction, they catalyze the HER in acidic solution.7 [Mo3S13]2− clusters have been synthesized to optimize the number of edge sites in an effort to increase the number of active sites for HER catalysis,9 which are also suitable for visible light driven HER.10 Also amorphous molybdenum sulfides MoS2+x, x < 1, show high catalytic activity.11–14 Replacing terminal disulfides by halogenides results in [Mo3S7Cl6]2− and [Mo3S7Br6]2−, which have significantly reduced catalytic activity, illustrating the importance of terminal S2 units in HER catalysis.10
Gas phase ion chemistry offers unique ways to study elementary steps of homogeneous as well as heterogeneous catalysis at a molecular level.15–19 Reactions of small cationic iron sulfide clusters with small alkanes elucidated the oxidative power of these compounds.20 Also molybdenum chalcogenide cations were investigated with gas-phase techniques.21 Jarrold, Raghavachari and co-workers have focused on molybdenum oxide and sulphide cluster reactivity,22–26 with studies involving H2 and H2O bearing relevance for HER catalysis.24,27–31 We have shown that cationic molybdenum clusters readily react with dimethyl disulfide, which reflects the high sulfur affinity of molybdenum.32
[Mo3S13]2− species are particularly attractive for mass spectrometric studies since the ions and their protonated counterparts are readily transferred to the gas phase by electrospray ionization.33 Their stability, decomposition pathways and chemical rearrangements were studied extensively by collision induced dissociation in combination with quantum chemistry.33 Terminal disulfide was identified as the protonation site by infrared multiple photon dissociation spectroscopy of [HMo3S13]−, again guided by quantum chemistry.34
Since the synthesis of molybdenum sulfide cluster compounds starts from the heptamolybdate, it is not surprising that a variety of oxysulfides can be obtained by slightly modified synthesis routes. These also exhibit HER catalytic activity35,36 and have been shown to be capable of catalyst self-healing.37
In the gas phase, we recently probed protonation of [Mo2O2S6]2− and [Mo2O2S5]2− in reactions with organic acids, which proceeds via Coulomb explosion into the protonated species and the deprotonated acid anion.38 The fragmentation pathways of molybdenum oxide clusters in the gas phase provide important additional information.39 To obtain insight into catalytic properties of molybdenum oxysulfides, we investigate here their structure, flexibility and chemical reactivity as well as hydrogen adsorption/desorption through collision-induced dissociation of [Mo2O2S6]2−, [HMo2O2S6]− and their fragments, as well as quantum chemical calculations. The results suggest that these species represent potential reactive intermediates in HER catalysis.
Experimental and computational details
The experiments were performed on a Bruker APEX Qe FT-ICR mass spectrometer, equipped with a Nanobay Console, a 9.4 T superconducting magnet and an Apollo Dual Source II.40 The ions are brought to the gas phase by electrospray ionization (ESI†) of a ∼0.3 mM solution of the tetramethylammonium salt of [92Mo2O2S6]2− in acetonitrile (ACN) and transferred into the ICR cell. Isotopically enriched [(CH3)4N]2[92Mo2O2S6] was prepared from 92Mo2O3 (STB Isotope Germany GmbH), following the procedure established by Müller et al.,41–43 with the modifications described in detail before.38The ultra-high vacuum region of the ICR cell has a base pressure in the low 10−9 mbar range. For sustained off-resonance irradiation (SORI)44 collision induced dissociation (CID), the collision gas argon is introduced via a leak valve at a constant pressure in the 10−8 mbar range. SORI-CID was performed with a fixed frequency offset of 500 Hz and an irradiation pulse length of 1 s. The amplitude of the SORI excitation is controlled via the parameter “SORI power” in % values in the control software Compass 3.0.0 of the instrument. The acquisition of multiple mass spectra yields fragmentation curves as a function of gradually increased SORI power, which provide qualitative information on the relative stability of the different ions.
The high mass resolving power afforded by the 9.4 T magnet proved advantageous for the study of oxysulfides, as the mass of O2 and S differs by only 0.01776 amu. We were able to distinguish between ions where one S atom is replaced by O2, for example [92Mo2O2S5]− and [92Mo2S6]−, even with the collision gas background which reduces the coherence time of the transient ion signal during detection and thus the mass resolution.
The mass spectrum obtained from ESI† of the sample solution contains numerous peaks, as shown in Fig. S1 (ESI†). Based on their exact mass, we identified different molybdenum oxysulfides of interest in the mass spectrum, namely [92Mo2O2S6]2−, [92Mo2O2S5]2−, [92Mo2O2S6]−, [92Mo2O2S5]− and [92Mo2O2S4]−. To obtain protonated species, we added 5% formic acid to the sample solution, which yielded [H92Mo2O2S6]−, [H92Mo2O2S5]− and [H92Mo2O2S4]−. SORI-CID experiments were performed on these eight molybdenum oxysulfide species. Prior to SORI excitation, the ions are mass-selected by ejecting all unwanted ions from the cell via broad-band frequency sweep and single-frequency shot excitation.
The geometries of all precursor and fragment ions were optimized at the B3LYP/def2TZVP level of theory using the D3 dispersion as suggested by Grimme et al.,45 which performed well for molybdenum sulfides.33 Candidate structures were derived from known crystal structure data, modified by educated guess. From our previous studies, we know that molybdenum oxysulfides tend to be low-spin compounds, therefore only the two lowest spin multiplicities, namely singlet and triplet or doublet and quartet, were considered; other that singlet and doublet spin multiplicities are given explicitly in the respective tables and figures; all spin multiplicities are given in the ESI.† Spin contamination in open-shell species was mostly below 4% before spin projection. Among ten structures with higher spin contamination, there are [MoOS3]− and the least stable [Mo2O2S5]2− isomer found, two structures of [Mo2O2S4]− with a loosely bound S2 and six structures of [H2Mo2O2S6] or [Mo2O2S6]. To estimate the induced error, we performed benchmark calculations for S2 dissociation from [Mo2O2S6]− using the BLYP functional without Hartree–Fock exchange (Table S2, ESI†). Reaction energies are calculated from electronic energies corrected for zero-point energy, without thermal corrections.
For evaluation of electron affinities of doubly charged species, a larger basis set is required. Therefore, we performed further calculations using the aug-cc-pVDZ basis set for sulfur and oxygen, and aug-cc-pVDZ-PP for Mo, as found on the Basis Set Exchange Website,46 for these species. The stability of the wave function was verified in all calculations. The connection of the transition state (with a single imaginary frequency) with the local minima was verified, small structural changes (e.g., rotation of an OH group) were ignored. However, intrinsic reaction coordinate (IRC) calculations failed to start in many cases because of the shallow shape of the potential energy surface. In those cases, a small offset of the transition state geometry was generated in both directions along the displacement vector of the vibrational mode carrying the imaginary frequency, followed by optimization. The Gaussian 16 program package was used for all calculations.47 Cartesian coordinates and electronic energies of the optimized structures are available in the ESI.†
Results and discussion
[Mo2O2S6]2−
The mass-selected spectrum of [Mo2O2S6]2− that serves as starting point of the SORI-CID experiment is given as an example in Fig. S2 (ESI†) while Fig. S3 (ESI†) shows a typical decomposition mass spectrum at 0.5% SORI power. The full fragmentation curve is illustrated in Fig. 1(a). The fragmentation curve shows significant intensity of [Mo2O2S6]− at 0% SORI power, which indicates that thermal collisions and residual kinetic excitation of some ions from the mass selection procedure is sufficient to induce electron detachment. Subsequent S2 loss leads to [Mo2O2S4]−. Direct S2− loss is not likely, as S2− was not observed in the mass spectrum. In a potential Coulomb explosion of [Mo2O2S6]2− into [Mo2O2S4]− and S2−, the latter would carry most of the kinetic energy release due to momentum conservation, and we cannot rule out that all S2− escapes from the cell. However, since [Mo2O2S4]− only becomes the dominant fragment above a SORI power of about 0.3%, where the [Mo2O2S6]− curve drops in intensity, the sequence of electron detachment followed by S2 loss at increasing energies is more plausible.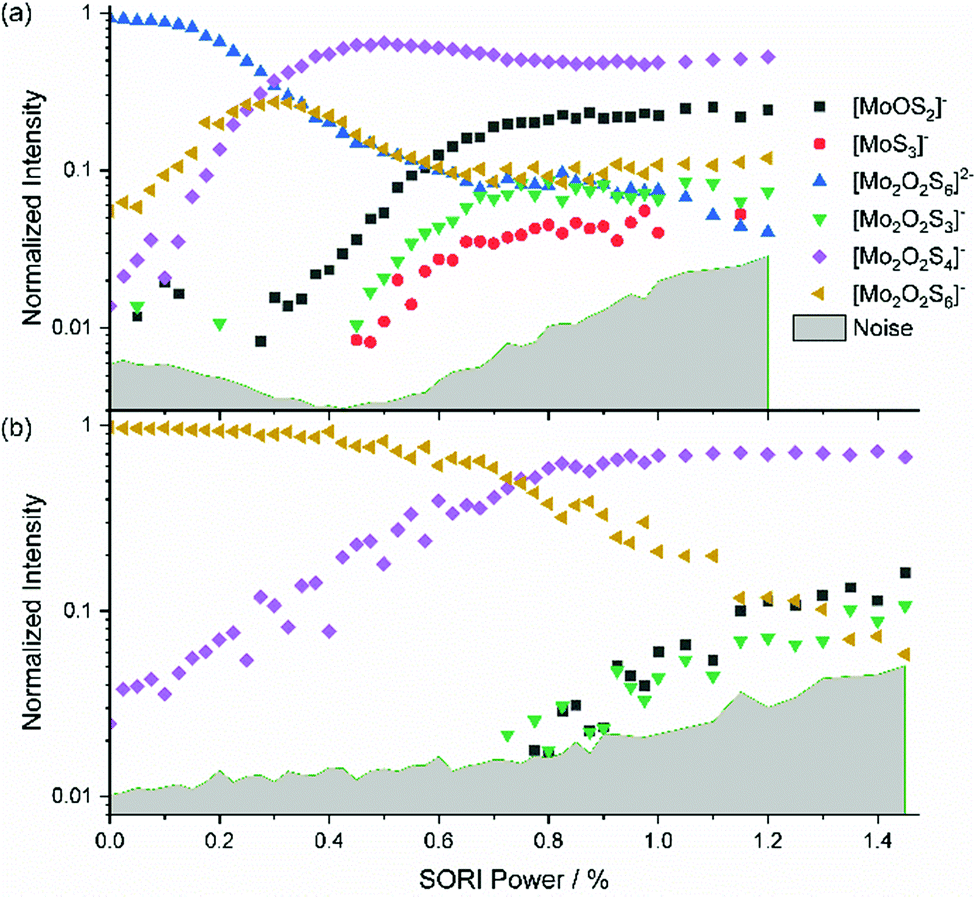 | ||
| Fig. 1 Fragmentation curve obtained from the SORI-CID experiment of (a) [Mo2O2S6]2− and (b) [Mo2O2S6]−. | ||
[Mo2O2S4]− undergoes splitting into [MoOS2]− and, most likely, neutral [MoOS2]. The isobaric [Mo2O2S4]2− structure can be ruled out, based on the late appearance of the peak in the SORI-CID fragmentation curve. It should be noted that secondary fragments are mostly formed in the same collision event as the primary fragment. At high enough collision energies, the first neutral fragment is lost, taking away a small amount of kinetic and internal energy in addition to its binding energy. The major part of the excess energy, however, remains as internal energy of the fragment with the higher density of states, in this case the charged molybdenum oxysulfide fragment. Once dissociation has taken place, however, the SORI irradiation is too far off resonance to yield appreciable collision energies for the fragment ions. Of course, the kinetic energy of the larger fragment remaining after the first collision and dissociation event can be converted into internal energy in subsequent collisions with argon, but this process is likely playing a minor role.
In Fig. 1(a), at around 0.5% SORI power a decrease of the [Mo2O2S4]− intensity is observed, while three other fragments rise. This suggests that the three fragments are formed by fragmentation of [Mo2O2S4]−. To verify this hypothesis, SORI-CID of [Mo2O2S6]− and [Mo2O2S4]− was performed.
[Mo2O2S6]−
The fragmentation curve of the singly charged ion [Mo2O2S6]−, shown in Fig. 1(b), documents a sharp increase in the [Mo2O2S4]− signal starting at 0% SORI power, consistent with the sequential electron detachment and S2 loss mechanism discussed above. At 0.8% SORI power, two other fragments arise, most likely secondary fragments of [Mo2O2S4]−. The [MoS3]− ion observed before is missing here, probably due to the higher noise level. Comparison of threshold energies in Fig. 1(a) and (b) is difficult since the kinetic energy of ions excited in an FT-ICR instrument depends on the square of the charge, i.e. significantly higher collision energies are reached for doubly charged ions compared to singly charged at nominally identical SORI power.[Mo2O2S4]−
[Mo2O2S4]− is obtained in high yields from ESI,† yet no fragment is present at 0% as shown in Fig. 2(a). This underlines the considerable stability of [Mo2O2S4]−. The fragments [MoOS2]−, [Mo2O2S3]− and [MoS3]− emerge at similar thresholds, confirming that [MoS3]− is a direct fragment of [Mo2O2S4]−. At higher SORI power more fragments arise, namely [MoO2S]−, [MoOS]− and [MoS2]− in the order of their appearance. It is interesting to note that all but one fragment contain only one molybdenum atom.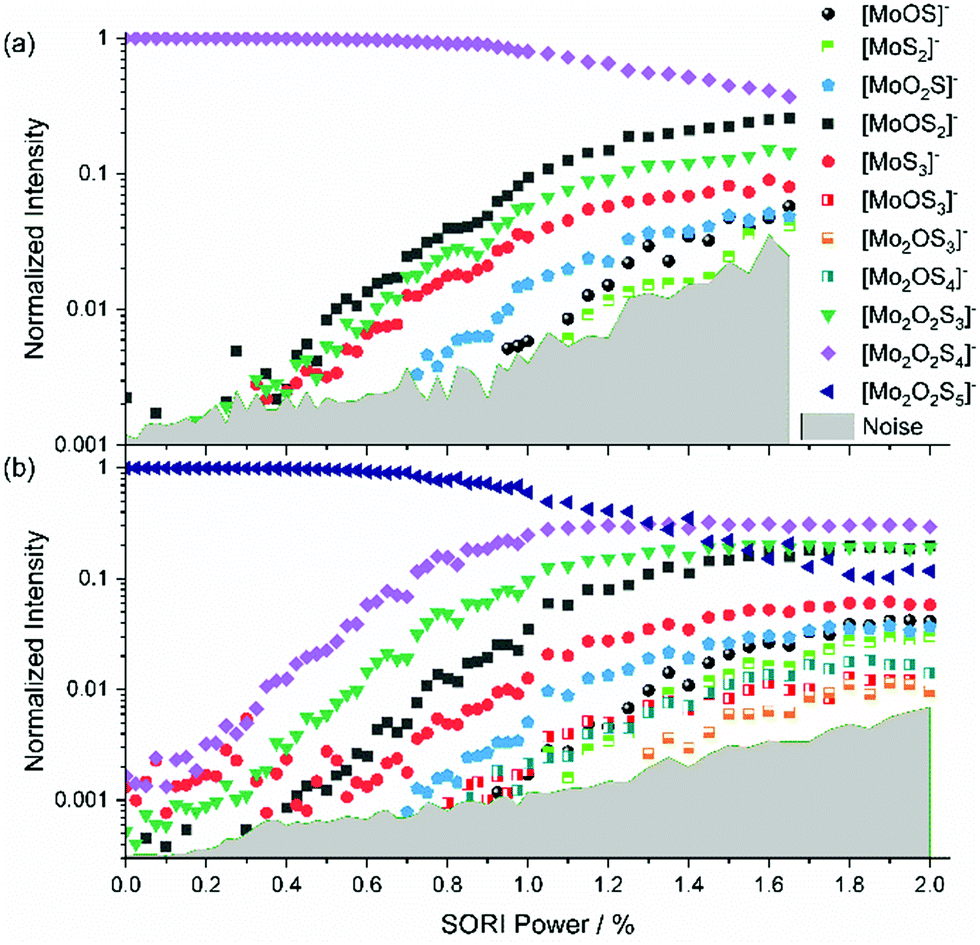 | ||
| Fig. 2 SORI-CID fragmentation curve of (a) [Mo2O2S4]− and (b) [Mo2O2S5]−. | ||
[Mo2O2S5]2− and [Mo2O2S5]−
The intensity of [Mo2O2S5]2− was very weak and accompanied by two other peaks at the same nominal mass. Therefore, proper isolation and a complete SORI-CID experiment was not possible with [Mo2O2S5]2−. Nevertheless, we were able to identify electron detachment as the first fragmentation step. Fig. S4 (ESI†) shows the mass spectrum of mass-selected [Mo2O2S5]2− exposed to collisions with argon at 0% SORI power. Electron detachment is proceeding efficiently, leading to significant intensity of [Mo2O2S5]−. This suggests that electron detachment in this case is even easier than for [Mo2O2S6]2−. A 1000-fold zoom of the m/z axis reveals that the peak at m/z 187.83 actually consists of three separated peaks. The ion of interest [Mo2O2S5]2− is assigned to the center peak, found at m/z 187.831. The peak at m/z 187.823 corresponds to either [Mo2S6]2− or [MoS3]−, and the peak at m/z 187.840 is due to [Mo2O4S4]2− or [MoO2S2]−. At a SORI power of 0.2% the [Mo2O2S5]− peak is already significantly higher than the precursor, Fig. S5 (ESI†). We did not observe the corresponding side peaks for [Mo2O2S5]−, suggesting that the side peaks of [Mo2O2S5]2− are singly charged ions, i.e., [MoS3]− and [MoO2S2]−. Also [Mo2O2S4]− is observed in small amounts.We were able to isolate the singly charged species with considerably higher intensity than the doubly charged one, with no side peak issues. This made SORI-CID feasible, see Fig. 2(b). The first fragment observed in the fragmentation curve of [Mo2O2S5]− is again [Mo2O2S4]−. The fact that this structure has been seen in every fragmentation curve so far further hints at its high stability against further dissociation. Next in the fragmentation curve, the six fragments of [Mo2O2S4]− can be seen again, namely [Mo2O2S3]−, [MoOS2]−, [MoS3]−, [MoO2S]−, [MoOS]− and [MoS2]−. However, the order of appearance of [Mo2O2S3]− and [MoOS2]− is reversed, with the former arising first. This suggests that the precursor [Mo2O2S5]− also undergoes direct fragmentation into [Mo2O2S3]− by loss of S2. Around 1.0% SORI power, three additional fragments appear which were not seen in any other experiment, namely [MoOS3]−, [Mo2OS4]− and [Mo2OS3]−. These ions are most likely direct fragments of the precursor [Mo2O2S5]−, implying loss of MoOS2, SO and S2O, respectively.
Protonated species [HMo2O2S6]−, [HMo2O2S5]− and [HMo2O2S4]−
Addition of 5% formic acid to the sample solution led to the appearance of protonated species in the mass spectrum. The first experiment discussed here is the SORI-CID of [HMo2O2S6]−, with the fragmentation curve shown in Fig. 3(a). Similar to [Mo2O2S6]−, S2 loss has the lowest threshold, leading to [HMo2O2S4]−. The next fragment is [Mo2O2S4]−, which appears at around 0.3% SORI power in the mass spectrum. It is presumably not a direct fragment of the precursor but a fragment of [HMo2O2S4]− as the intensity of the latter decreases above 0.6% SORI power. This suggests that a hydrogen atom can be removed from [HMo2O2S4]− without destroying the [Mo2O2S4]− structure. SORI-CID of [HMo2O2S4]− is discussed below to validate this interpretation. Interestingly, beginning at a SORI power of about 0.6%, [Mo2O2S5]− is observed. This suggests that here the loss of HS is more favorable than the loss of S or H. After that, four fragments of [Mo2O2S4]− appear, where [Mo2O2S3]− is formed as a secondary fragment of both [Mo2O2S5]− and [Mo2O2S4]−, as discussed previously.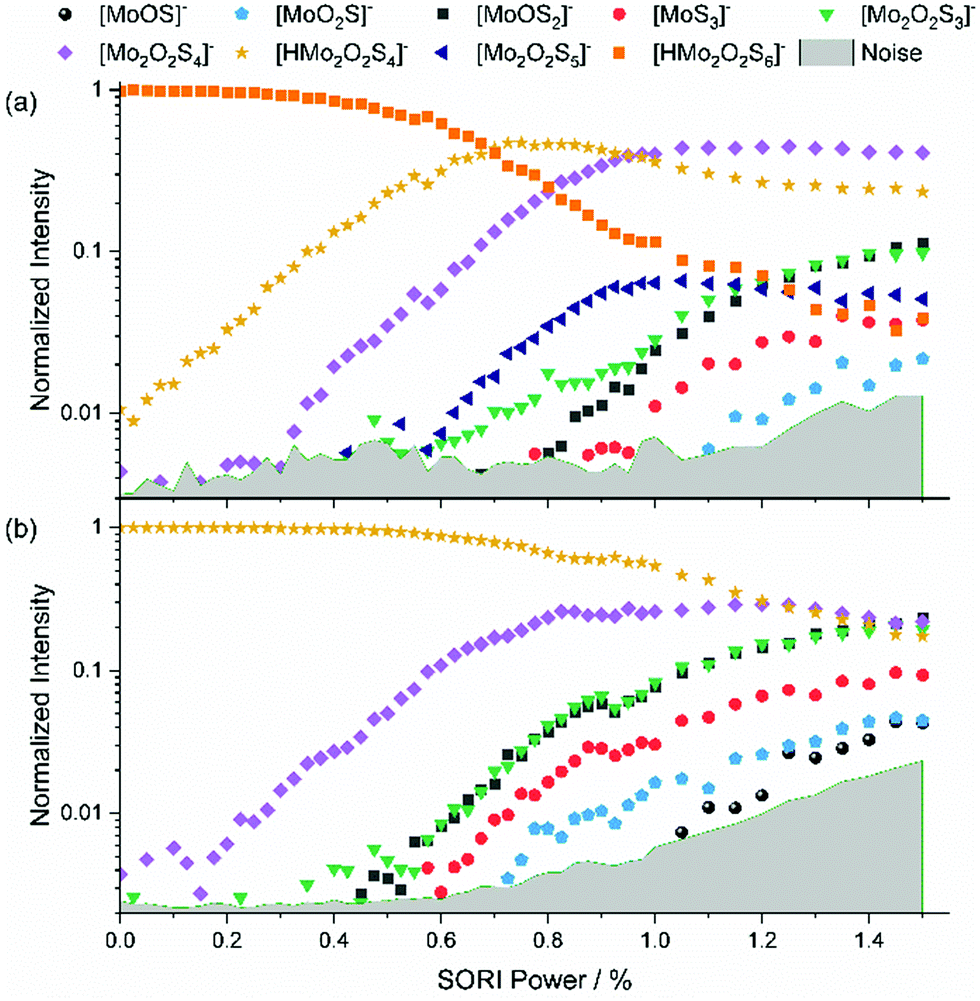 | ||
| Fig. 3 (a) SORI-CID fragmentation curve of (a) [HMo2O2S6]− and (b) [HMo2O2S4]−. | ||
We were also able to perform a SORI-CID experiment with [HMo2O2S5]−, shown in Fig. S6 (ESI†). Here, [Mo2O2S4]− is formed via loss of HS. Fragments of [Mo2O2S4]− again follow at higher SORI power. SORI-CID of [HMo2O2S4]−, Fig. 3(b), confirms the interpretation of the [HMo2O2S6]− experiment. As expected, hydrogen atom loss is the first, and over a wide range dominant, fragmentation channel. Again, the well-known fragments of [Mo2O2S4]− arise above 0.4% SORI power, namely [Mo2O2S3]−, [MoOS2]−, [MoS3]−, [MoO2S]−, and [MoOS]−.
Quantum chemical calculations
Optimized structures and relative energies of low-lying isomers for the most important species investigated are summarized in Fig. 4. In most cases, several isomers are found within 0.2 eV of the lowest energy structure, especially for protonated species. For [Mo2O2S6]2−, the lowest-lying structure found in our calculations corresponds closely to the crystal structure.36,48 The two molybdenum atoms are connected by two bridging sulfur atoms. Each molybdenum center is additionally bound to an oxygen atom and a terminal disulfide unit, overall in C2v symmetry. However, turning one half of the molecule by 180 degrees results in a C2h structure which lies only 0.17 eV higher. Isomers with a bridging disulfide unit or bridging oxygen atoms lie significantly higher in energy.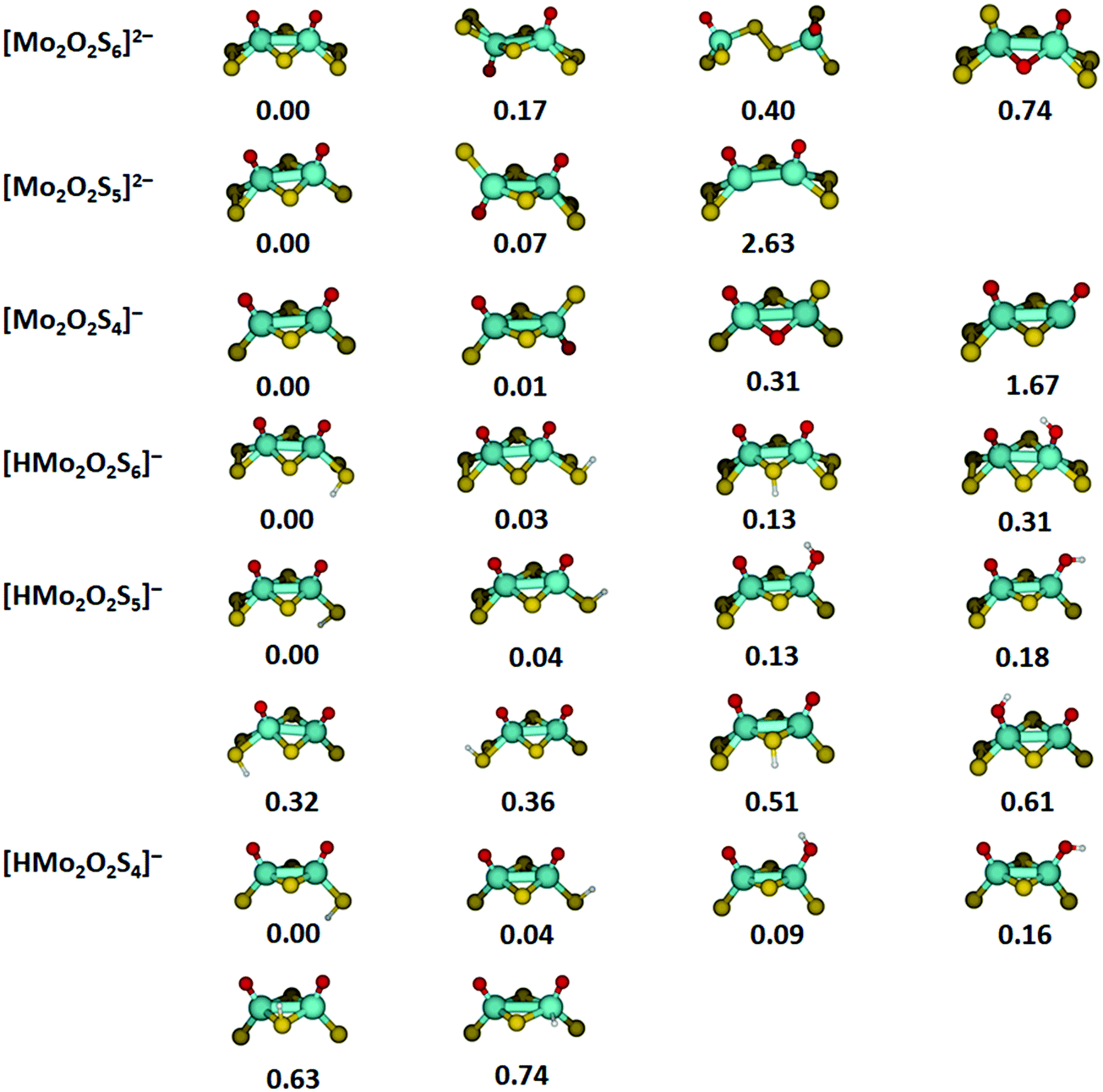 | ||
| Fig. 4 Low-lying isomers for six of the eight studied molybdenum oxysulfides optimized at the B3LYP+D3/def2TZVP level. Zero-point corrected energies are given in eV. Color code: Mo – cyan, S – yellow, O – red, H – white. | ||
The energetically most favorable structure of [Mo2O2S5]2− is reached by removing a terminal sulfur atom from [Mo2O2S6]2−. The loss of a bridging sulfur atom is significantly more demanding and can be ruled out. Removing a terminal sulfur atom from the second molybdenum center together with an electron leads to the lowest-energy structures of [Mo2O2S4]−. For this ion, the energy difference between the C2v and C2h structure is reduced to 0.01 eV. Structures with two non-equivalent molybdenum centers are energetically not competitive.
Fig. 5(a) and (b) shows the isomerization pathway between the C2v and C2h structures of [Mo2O2S6]2− and [Mo2O2S4]−, respectively. The predicted pathway for [Mo2O2S6]2− includes an exchange of a bridging sulfur atom with an oxygen atom followed by a transfer of a terminal sulfur atom and returning of the bridging oxygen atom to the terminal position. For [Mo2O2S4]−, a similar pathway is obtained, without the need of sulfur exchange on the terminal groups as no S2 unit is present. Still, the oxygen transfer reactions in both clusters are energetically demanding, requiring more than 1.6 eV.
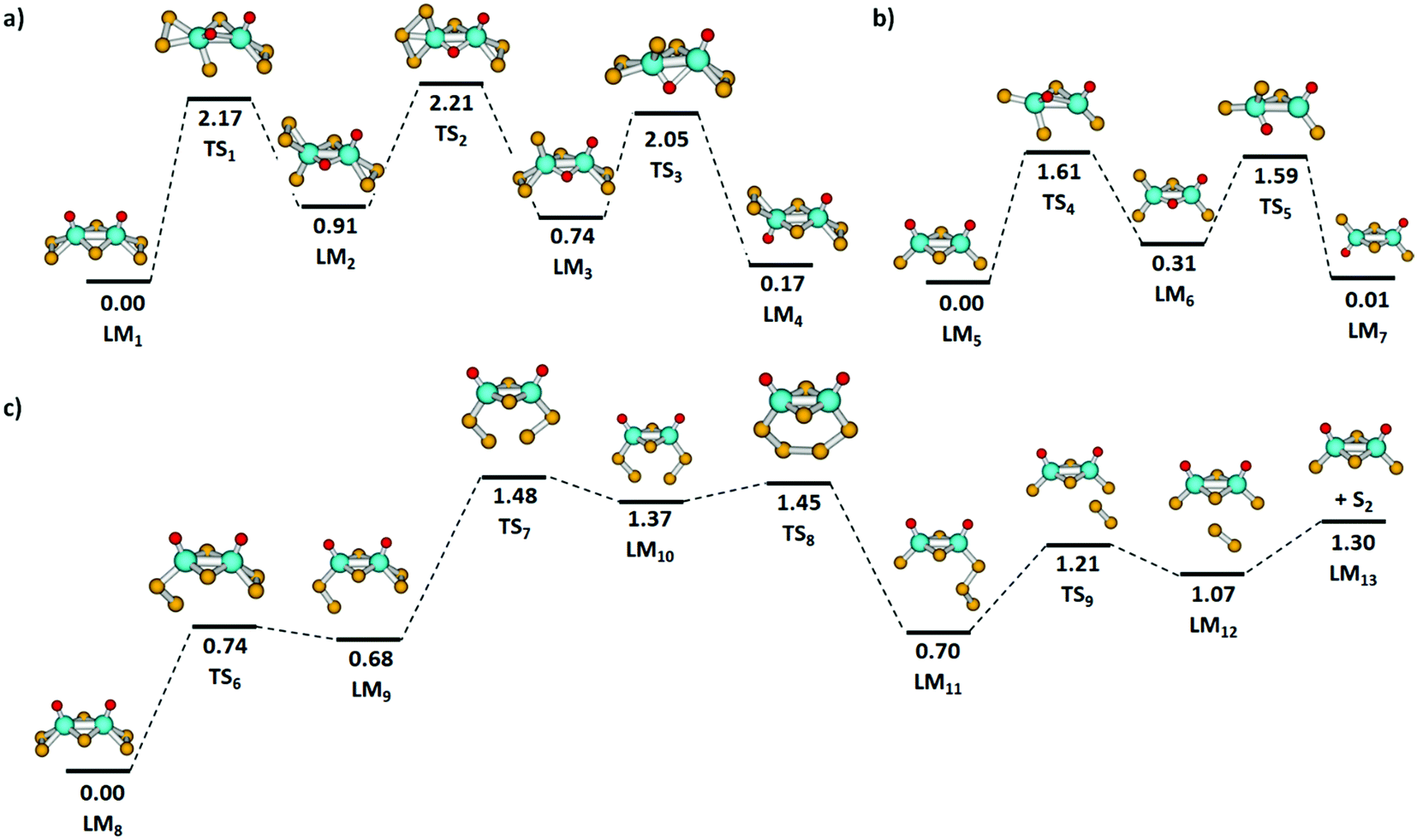 | ||
| Fig. 5 Reaction pathways for isomerization reactions in (a) [Mo2O2S6]2− and (b) [Mo2O2S4]− clusters. (c) Simplified reaction profile from [Mo2O2S6]− to [Mo2O2S4]− + S2. Calculated at the B3LYP+D3/def2TZVP level; relative energies are given in eV, local minima (LM) and transition states (TS) are denoted. Color code: Mo – cyan, S – yellow, O – red, H – white. | ||
For the protonated species, the protonation site and the orientation of the S–H or O–H bond have a small effect on the energetics, corresponding to the dispersed distribution of the negative charge across all sulfur and oxygen atoms. In all cases, the energetically favorable protonation site is a terminal sulfur atom with the hydrogen atom pointing away from the nearest oxygen atom. However, changing the orientation of the S–H bond towards the oxygen atom is only 0.03–0.04 eV higher in energy for the three ions.
Protonation of a bridging sulfur atom lies by 0.13 eV higher than terminal disulfide for [HMo2O2S6]−, but the energy difference increases significantly if single terminal sulfur atoms are available. In this case, also the neighboring oxygen atoms are good proton acceptors, only about 0.1 eV above the preferred structures for [HMo2O2S5]− and [HMo2O2S4]−. The affinity of the oxygen atoms towards a proton depends critically on the coordination of the molybdenum center with single sulfur atoms vs. terminal disulfide units. Protonation of a molybdenum atom seems generally unfeasible, lying 0.74 eV above the minimum for [HMo2O2S4]−.
As mentioned above, the fragmentation of [Mo2O2S6]2− to [Mo2O2S4]− presumably happens in two steps, with electron detachment followed by loss of S2. To reach a favorable [Mo2O2S4]− structure, however, the two sulfur atoms must come from the two terminal disulfide units. The lowest energy pathway for S2 elimination from [Mo2O2S6]− that was found is depicted in Fig. 5(c). First, a terminal disulfide group rotates, reaching LM9 at 0.68 eV. Then, the other terminal disulfide also rotates so that two sulfur atoms of the disulfide units can form a bond, LM10. The strain involved in this rearrangement leads to the highest barrier TS7 along the reaction path, 1.48 eV. Passing through TS8, the newly formed trisulfur unit rotates away from the molybdenum atoms to reach LM11 and dissociates S2via TS9. For comparison, the direct dissociation of a terminal disulfide unit would require at least 2.97 eV in energy without accounting for any transition state or rearrangement. The fragmentation pathway shown in Fig. 5(c) requires considerably less energy.
Additional quantum chemical calculations were performed for all charged fragments observed and the corresponding neutrals. This results in a concise picture of the decomposition of the studied species, summarized in Fig. 6 for major pathways along with Table S1 and Fig. S7 (ESI†) that include also minor fragmentation channels. The energies reported in the figure compare reactant and product energies without considering any higher-lying transition states involved in the fragmentation. However, the reactions represent mostly direct dissociation reactions or require rearrangement of S2 or O groups (similar to Fig. 5) or hydrogen atom transfer (see below), which involve transition states that most likely lie below or close to the respective dissociation energies. The doubly charged species [Mo2O2S6]2− and [Mo2O2S5]2− undergo electron detachment with low energetic thresholds in the experiment. With the basis set used for most calculations, electron detachment is slightly exothermic in both cases, −0.02 eV for [Mo2O2S6]2− and −0.12 eV for [Mo2O2S5]2−. Using a larger basis set, aug-cc-pVDZ on O, S and aug-cc-pVDZ-PP on Mo, the electron detachment energies increase to 0.19 eV and 0.03 eV, respectively. In addition to the binding energy, however, the doubly charged structures are stabilized by a Coulomb barrier against electron detachment.49 The calculations also explain why we were not able to obtain any signal for the doubly charged [Mo2O2S4]2−. Electron detachment for this ion is with −0.39 eV even more exothermic at the B3LYP/def2TZVP theory level, and still −0.26 eV with the bigger basis set defined above. This also shifts the Coulomb barrier down so that it can be overcome in low energy collisions, assisted by tunneling.
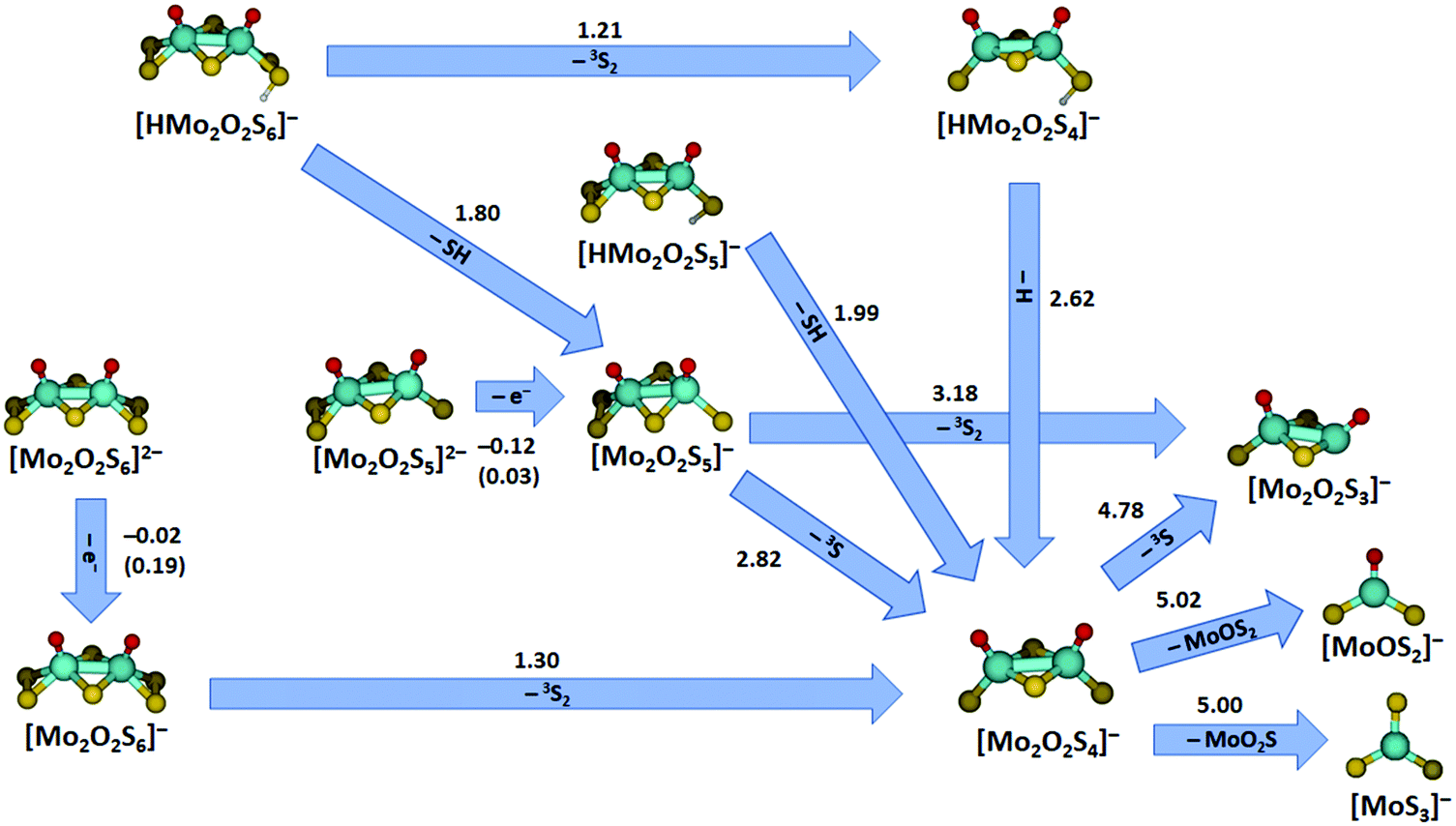 | ||
| Fig. 6 Major fragmentation pathways of the molybdenum oxysulfides studied by SORI-CID along with reaction energies at the B3LYP+D3/def2TZVP level; for electron detachment, values calculated with aug-cc-pVDZ(O,S), aug-cc-pVDZ-PP(Mo) basis sets are given in parentheses. Reaction energies are given in eV. For all minor fragmentation paths, see Fig. S7 and Table S1 (ESI†). Color code: Mo – cyan, S – yellow, O – red, H – white. Other than singlet or doublet spin multiplicities are given explicitly. | ||
Turning to the singly charged species, many different fragments were seen in SORI-CID of [Mo2O2S5]−, five of which have been assigned as direct fragments. The energies needed for the fragmentation correspond to the order of appearance in the fragmentation curve, consistent with this interpretation. The first and energetically least demanding fragment is [Mo2O2S4]− with 2.82 eV, followed by [Mo2O2S3]− with 3.18 eV. Other fragments observed in minor intensity are [Mo2OS4]− with 3.94 eV, [MoOS3]− with 4.23 eV, and [Mo2OS3]− with 5.79 eV.
Loss of S2 from [Mo2O2S6]− to form [Mo2O2S4]− is endothermic by 1.30 eV. In agreement with experiment, fragmentation of [Mo2O2S4]− requires higher energies, see Table S1 (ESI†). The first three fragments appear at very similar SORI power, perfectly reflected in the calculations: formation of [Mo2O2S3]− is endothermic with 4.78 eV, [MoOS2]− with 5.02 eV and [MoS3]− with 5.00 eV. The next fragments appear at significantly higher collision energies, corresponding to the increased energy demand forming [MoO2S]− with 5.13 eV, [MoOS]− with 7.51 eV and [MoS2]− with 7.53 eV, see Table S1 (ESI†).
The protonated species complete the picture. [HMo2O2S4]− decays by H atom loss to [Mo2O2S4]− with 2.62 eV, quite demanding compared to SH loss from [HMo2O2S5]− with 1.99 eV. The SH loss from [HMo2O2S4]−, however, would be quite expensive with 3.77 eV. For comparison, the dissociation of a hydrogen atom from [HMo2O2S5]− requires 2.81 eV while the dissociation of a terminal sulfur atom with 3.01 eV is even more expensive, in agreement with the experimental observation.
The first fragmentation channel of [HMo2O2S6]− in the experiment corresponding to S2 elimination is calculated to be 1.21 eV endothermic. However, a barrier similar to the one shown in Fig. 5(c) may be expected. At higher SORI power, [Mo2O2S5]− appeared in the mass spectrum, most likely resulting from HS loss, which is 1.80 eV endothermic according to calculations and becomes entropically favored compared to a tight transition state involved in S2 elimination, like TS7 in Fig. 5(b). Interestingly, hydrogen atom loss from [HMo2O2S6]− requires 2.52 eV, 0.10 eV less than from [HMo2O2S4]−. Since two energetically lower lying channels are available for [HMo2O2S6]−, H atom loss is not observed in this case. It is intriguing to note that the S–H bond dissociation energy is 55% of the bond dissociation energy of H2, 4.52 eV. This means that the elimination of two H atoms from such neighboring S–H groups with concomitant formation of H2 in a catalytic environment would be 0.52 eV endothermic, which lies in the thermally accessible regime.
The fragmentation of molybdenum oxysulfides bears some similarities with [Mo3S13]2−, [HMo3S13]− and [H3Mo3S13]+ studied previously.33 In both cases the energetically favorable protonation/hydrogenation site was found to be a terminal sulfur atom or a terminal disulfide unit. Also, in both cases, HxSy elimination is primarily seen, and all molybdenum atoms are retained in the ionic fragment at low collision energies. A completely new fragmentation channel in the present study is the loss of a single hydrogen atom for [HMo2O2S4]−. While [Mo3S13]2− do not undergo electron detachment during CID, this pathway was readily observed for the doubly charged molybdenum oxysulfides.
Parallels can also be drawn to our proton transfer reactivity study of [Mo2O2S6]2− and [Mo2O2S5]2−.38 A terminal sulfur atom was identified as the protonation site, and a Coulomb barrier hindered charge separation reactions.38,50–54
The thermochemistry of H atom loss prompted us to calculate H2 elimination pathways from clusters containing two H atoms. Although we do not have any experimental data for these species, we found it worthwhile to gain more insight into potential final steps of HER. We calculated a series of [H2Mo2O2S6]−, [H2Mo2O2S4]−, [H2Mo2O2S6], and [H2Mo2O2S4] clusters, see Fig. 7(a) for low-lying isomers. For all investigated species, formation of two S–H groups leads to a very stable cluster, while clusters with one S–H and one O–H groups are close in energy (see Fig. S8, ESI† for further structures), with S2 groups playing a prominent role in H2 adsorption. For the anionic clusters, H2 adsorption is only mildly exothermic, with a very low adsorption energy. In neutral clusters, H2 is bound much stronger. This indicates that in an electrochemical environment, addition of an electron will promote H2 elimination from these clusters.
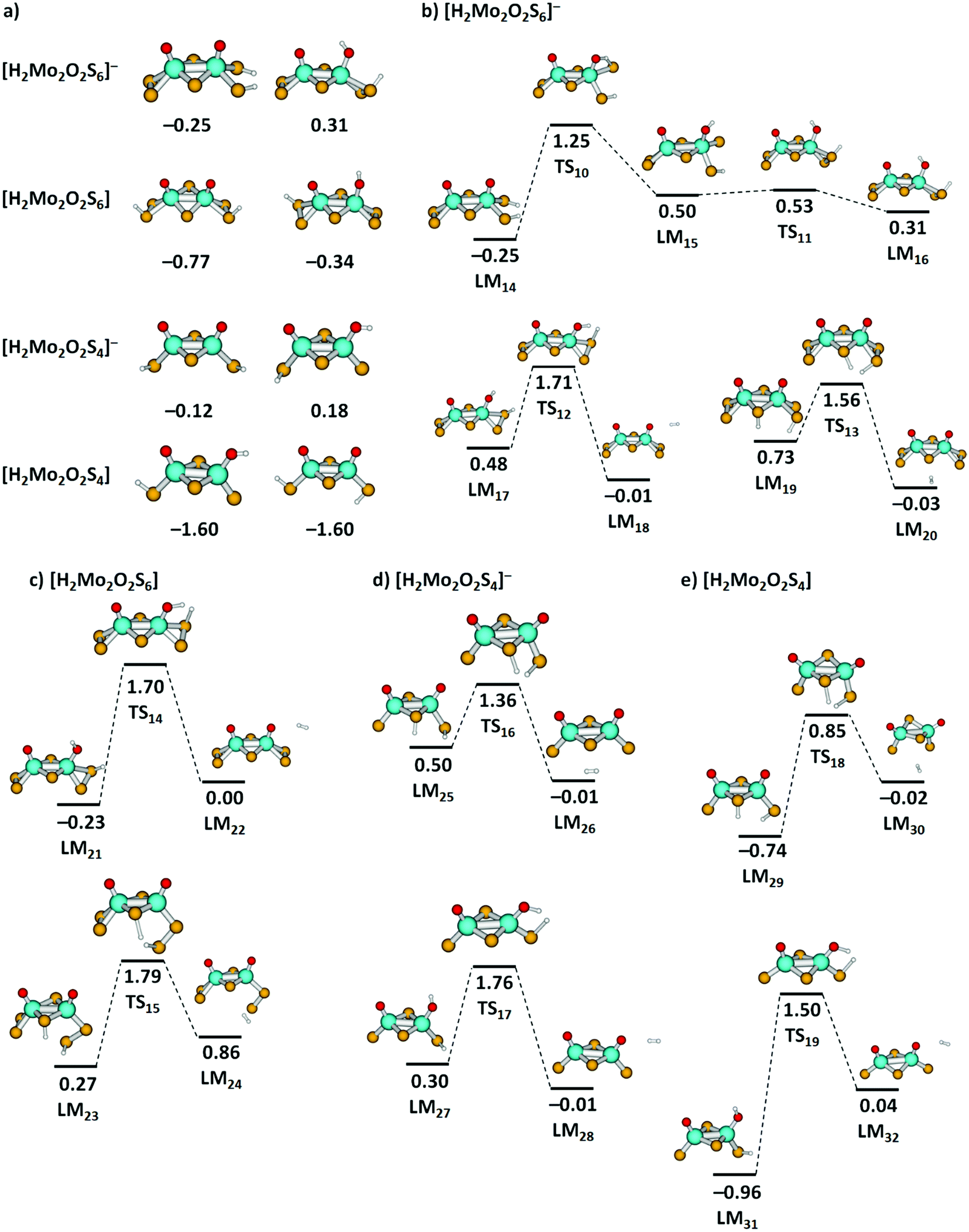 | ||
| Fig. 7 (a) Two most stable located minima of [H2Mo2O2S6]−, [H2Mo2O2S6], [H2Mo2O2S4]−, and [H2Mo2O2S4] clusters, further structures are shown in Fig. S8 (ESI†). (b)–(e) Reaction pathways for H2 dissociation from [H2Mo2O2S6]−, [H2Mo2O2S6], [H2Mo2O2S4]−, and [H2Mo2O2S4] clusters. For [H2Mo2O2S6]−, a simplified reaction pathway for hydrogen transfer is also shown. Calculated at the B3LYP+D3/def2TZVP level; relative energies are given in eV with respect to the limit of [cluster + H2]; local minima (LM) and transition states (TS) are denoted. Color code: Mo – cyan, S – yellow, O – red, H – white. | ||
For selected H2 dissociation pathways from the clusters starting either from two S–H groups or one S–H and one O–H group, we see consistently lower barriers for H2 dissociation for the anionic species. Particularly low barriers are obtained for anionic species with H atoms attached to a bridging and a terminal sulfur unit, with 0.83 eV and 0.86 eV from LM19via TS13 and LM25via TS16, respectively.
Conclusion
SORI-CID experiments on eight different molybdenum oxysulfide species showed that [Mo2O2S4]− is a common fragment of all larger molybdenum oxysulfides. While [Mo2O2S6]− and [HMo2O2S6]− readily lose an S2 unit after some rearrangement at low SORI power, [Mo2O2S4]− shows fragmentation only at relatively high collision energies. Overall, the OMoS2MoO backbone is retained up to high collision energies, while quantum chemical calculations confirm that all decompositions of the [Mo2O2S4]− structure require at least 5 eV, indicating a particular stability of this species with respect to further dissociation reactions. [Mo2O2S4]− is the smallest unit with two fully saturated Mo centers, and may be representative of the structures formed during catalyst activation in electrocatalytic environments. Electron detachment is almost thermoneutral across the doubly charged species, consistent with the catalytically relevant redox properties of these species. Electrons can readily be accepted or donated. An interesting fragmentation channel of [HMo2O2S4]− is the dissociation of an H atom, leaving the [Mo2O2S4]− structure intact. Calculations reveal that the hydrogen atom can favorably bind to a variety of almost isoenergetic sites. We also show that S2 groups play a prominent role as a protonation site. In the calculations, it was possible to add a second H atom to the protonated species, and we investigated H2 elimination from neutral as well as negatively charged clusters. Activation energies for H2 formation below 1 eV were obtained in anionic species, with H atoms attached to a bridging and a terminal sulfur unit. Facile electron attachment and removal, a variety of H atom adsorption sites and low barriers for H2 elimination are all promising properties of a HER catalyst.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project is supported by the Klima- und Energiefonds, administered by the FFG under Project No. 853639, and put into effect in the framework of the programme “ENERGIE DER ZUKUNFT”. We thank Dr Aristeidis Baloglou for preliminary work and Reviewer #2 for providing us with several calculated structures of [Mo2O2S4]−. The computational results presented have been achieved using the HPC infrastructure LEO of the University of Innsbruck.References
- J. A. Turner, Science, 2004, 305, 972 CrossRef CAS PubMed.
- C. Wulf, J. Linssen and P. Zapp, Hydrogen Supply Chains, Elsevier, 2018, pp. 309–345 Search PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- Z. Shi, X. Wang, J. Ge, C. Liu and W. Xing, Nanoscale, 2020, 12, 13249 RSC.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- M. Bodner, A. Hofer and V. Hacker, Wiley Interdiscip. Rev.: Energy Environ., 2015, 4, 365 CAS.
- H. I. Karunadasa, E. Montalvo, Y. Sun, M. Majda, J. R. Long and C. J. Chang, Science, 2012, 335, 698 CrossRef CAS PubMed.
- P. C. K. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951 CrossRef CAS PubMed.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Nat. Chem., 2014, 6, 248 CrossRef CAS PubMed.
- M. Dave, A. Rajagopal, M. Damm-Ruttensperger, B. Schwarz, F. Nägele, L. Daccache, D. Fantauzzi, T. Jacob and C. Streb, Sustainable Energy Fuels, 2018, 2, 1020 RSC.
- J. D. Benck, Z. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, ACS Catal., 2012, 2, 1916 CrossRef CAS.
- C. G. Morales-Guio and X. Hu, Acc. Chem. Res., 2014, 47, 2671 CrossRef CAS PubMed.
- D. Merki, S. Fierro, H. Vrubel and X. Hu, Chem. Sci., 2011, 2, 1262 RSC.
- P. D. Tran, T. V. Tran, M. Orio, S. Torelli, Q. D. Truong, K. Nayuki, Y. Sasaki, S. Y. Chiam, R. Yi, I. Honma, J. Barber and V. Artero, Nat. Mater., 2016, 15, 640–646 CrossRef CAS PubMed.
- D. K. Bohme and H. Schwarz, Angew. Chem., Int. Ed., 2005, 44, 2336 CrossRef PubMed.
- S. M. Lang and T. M. Bernhardt, Phys. Chem. Chem. Phys., 2012, 14, 9255 RSC.
- T. F. Pascher, M. Ončák, C. van der Linde and M. K. Beyer, ChemPhysChem, 2019, 20, 1420 CAS.
- T. F. Pascher, M. Ončák, C. van der Linde and M. K. Beyer, ChemistryOpen, 2019, 8, 1453–1459 CrossRef CAS PubMed.
- E. Barwa, T. F. Pascher, M. Ončák, C. van der Linde and M. K. Beyer, Angew. Chem., Int. Ed., 2020, 59, 7467–7471 CrossRef CAS PubMed.
- S. M. Lang, S. Zhou and H. Schwarz, Phys. Chem. Chem. Phys., 2017, 19, 8055 RSC.
- I. Kretzschmar, A. Fiedler, J. N. Harvey, D. Schröder and H. Schwarz, J. Phys. Chem. A, 1997, 101, 6252 CrossRef CAS.
- N. J. Mayhall, E. L. Becher, A. Chowdhury and K. Raghavachari, J. Phys. Chem. A, 2011, 115, 2291 CrossRef CAS PubMed.
- N. J. Mayhall, E. L. Becher III., A. Chowdhury and K. Raghavachari, J. Phys. Chem. A, 2011, 115, 2291 CrossRef CAS PubMed.
- A. K. Gupta, J. E. Topolski, K. A. Nickson, C. C. Jarrold and K. Raghavachari, J. Phys. Chem. A, 2019, 123, 7261 CrossRef CAS PubMed.
- J. L. Mason, A. K. Gupta, A. J. McMahon, C. N. Folluo, K. Raghavachari and C. C. Jarrold, J. Chem. Phys., 2020, 152, 54301 CrossRef CAS PubMed.
- S. E. Waller and C. C. Jarrold, J. Phys. Chem. A, 2014, 118, 8493 CrossRef CAS PubMed.
- J. E. Topolski, A. K. Gupta, K. A. Nickson, K. Raghavachari and C. C. Jarrold, Int, J. Mass Spectrom., 2018, 434, 193 CAS.
- A. Saha and K. Raghavachari, J. Chem. Phys., 2013, 139, 204301 CrossRef PubMed.
- M. Ray, S. E. Waller, A. Saha, K. Raghavachari and C. C. Jarrold, J. Chem. Phys., 2014, 141, 104310 CrossRef PubMed.
- M. Ray, A. Saha and K. Raghavachari, Phys. Chem. Chem. Phys., 2016, 18, 25687 RSC.
- M. Ray, R. N. Schaugaard, J. E. Topolski, J. O. Kafader, K. Raghavachari and C. C. Jarrold, J. Phys. Chem. A, 2018, 122, 41 CrossRef CAS PubMed.
- A. Baloglou, M. Ončák, C. van der Linde and M. K. Beyer, Top. Catal., 2018, 61, 20 CrossRef CAS PubMed.
- A. Baloglou, M. Ončák, M.-L. Grutza, C. van der Linde, P. Kurz and M. K. Beyer, J. Phys. Chem. C, 2019, 8177 CrossRef CAS PubMed.
- A. Baloglou, M. Plattner, M. Ončák, M.-L. Grutza, P. Kurz and M. K. Beyer, Angew. Chem., Int. Ed., 2021, 60, 5074 CrossRef CAS PubMed.
- D. N. Chirdon, R. F. Lalisse, J. Sun, S. Zhang, B. R. Garrett, C. M. Hadad and Y. Wu, SN Appl. Sci., 2020, 2, 889 CrossRef CAS.
- J. McAllister, N. A. G. Bandeira, J. C. McGlynn, A. Y. Ganin, Y.-F. Song, C. Bo and H. N. Miras, Nat. Commun., 2019, 10, 370 CrossRef CAS PubMed.
- J. Barros Barbosa, P. L. Taberna, V. Bourdon, I. C. Gerber, R. Poteau, A. Balocchi, X. Marie, J. Esvan, P. Puech, A. Barnabé, L. Da Gama Fernandes Vieira, I.-T. Moraru and J. Y. Chane-Ching, Appl. Catal., B, 2020, 278, 119288 CrossRef CAS.
- A. Baloglou, M. Pritzi, T. F. Pascher, J. C. Hartmann, M.-L. Grutza, M. Ončák, P. Kurz and M. K. Beyer, Int. J. Mass Spectrom., 2021, 464, 116558 CrossRef CAS.
- M. Plattner, A. Baloglou, M. Ončák, C. van der Linde and M. K. Beyer, J. Am. Soc. Mass Spectrom., 2019, 30, 1946 CrossRef CAS PubMed.
- A. Herburger, C. van der Linde and M. K. Beyer, Phys. Chem. Chem. Phys., 2017, 19, 10786 RSC.
- A. Müller, R. G. Bhattacharyya and B. Pfefferkorn, Chem. Ber., 1979, 112, 778 CrossRef.
- A. Müller, E. Krickemeyer and U. Reinsch, Z. Anorg. Allg. Chem., 1980, 470, 35 CrossRef.
- A. Müller, S. Sarkar, R. G. Bhattacharyya, S. Pohl and M. Dartmann, Angew. Chem., Int. Ed. Engl., 1978, 17, 535 CrossRef.
- J. W. Gauthier, T. R. Trautman and D. B. Jacobson, Anal. Chim. Acta, 1991, 246, 211 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision A.03, 2016 Search PubMed.
- W. Rittner, A. Müller, A. Neumann, W. Bäther and R. C. Sharma, Angew. Chem., Int. Ed. Engl., 1979, 18, 530 CrossRef.
- P. Weis, O. Hampe, S. Gilb and M. M. Kappes, Chem. Phys. Lett., 2000, 321, 426 CrossRef CAS.
- A. Révész, B. Sztáray, D. Schröder, K. Franzreb, J. Fiser, S. D. Price and J. Roithová, Phys. Chem. Chem. Phys., 2009, 11, 6192 RSC.
- M. K. Beyer and R. B. Metz, J. Phys. Chem. A, 2003, 107, 1760 CrossRef CAS.
- M. Beyer, E. R. Williams and V. E. Bondybey, J. Am. Chem. Soc., 1999, 121, 1565 CrossRef CAS PubMed.
- D. Schröder and H. Schwarz, J. Phys. Chem. A, 1999, 103, 7385 CrossRef.
- B. L. J. Poad, N. D. Reed, C. S. Hansen, A. J. Trevitt, S. J. Blanksby, E. G. Mackay, M. S. Sherburn, B. Chan and L. Radom, Chem. Sci., 2016, 7, 6245 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01189a |
| This journal is © the Owner Societies 2022 |