DOI:
10.1039/D1DT01671G
(Paper)
Dalton Trans., 2021,
50, 9236-9249
Pseudotetrahedral copper(II)-complexes with enantiopure (R or S)-2-(((aryl)ethylimino)ethyl)phenolate Schiff base ligands†
Received
23rd May 2021
, Accepted 8th June 2021
First published on 8th June 2021
Abstract
Condensation of 2-hydroxy-benzophenone (HL′) with (R or S)-(Ar)ethylamine yields the enantiopure Schiff bases (S or R)-2-((E)-1-(1-(Ar)ethylimino)ethyl)phenol {Ar = C6H5 (S- or R-HL1), p-CH3OC6H4 (S- or R-HL2)}. These Schiff bases react with copper(II) acetate under reflux to give green microcrystals of bis[(R or S)-2-((E)-1-(1-(Ar)ethylimino)ethyl)phenolato-κ2N,O]-Λ/Δ-copper(II), {Ar = C6H5 (Λ/Δ-Cu-R- or S-L1), p-CH3OC6H4 (Λ/Δ-Cu-R- or S-L2)} with induction of Λ/Δ-chirality at-metal. The presence of Schiff base ligands in the paramagnetic green microcrystals is confirmed by decomplexation reaction with NaCN via reduction of Cu(II) to Cu(I) in DMSO-d6 solution. Crystallization attempts of the green microcrystalline Schiff-base Cu complexes provide deep-green block-shaped crystals of an about equal admixture of bis[2-oxo-benzophenonato-κ2O,O′]-copper(II), (CuL′2) and bis[2-(imino(phenyl)methyl)phenolato-κ2N,O]copper(II), (CuL′′2) via in situ hydrolysis of the coordinated Schiff base ligands back to 2-hydroxy-benzophenone (HL′) and to 2-(imino(phenyl)methyl)phenol (HL′′), which in-turn bind with the copper(II) ion. Powder X-ray diffraction (PXRD) patterns of R-HL1 and Cu-R-L1 allowed their structure determinations using the program Expo-2014 followed by Rietveld refinement. The Cu structures refined to four-coordinated Λ/Δ-copper(II)-complexes by the two phenolate-oxygen and two imine-nitrogen atoms from two Schiff base ligands in a pseudotetrahedral geometry. DFT optimized structures (at gas-phase) reveal the Δ-Cu-S-L1 or Λ-Cu-R-L1 diastereomer as slightly more stable than the corresponding Λ-Cu-S-L1 or Δ-Cu-R-L1 by ca. 7.60 kcal mol−1, resulting from diastereoselectively induced Λ vs. Δ-chirality at-metal. Electronic circular dichroism (ECD) spectra display mirror-image relationships and comparisons of experimental and simulated ECD spectra by TDDFT suggest an excess of the Δ-Cu-S-L1 or Λ-Cu-R-L1 diastereomer in solution. The cyclic voltammograms demonstrate two one electron charge transfer processes for Cu2+/Cu+ and Cu+/Cu0 couples in acetonitrile, respectively.
Introduction
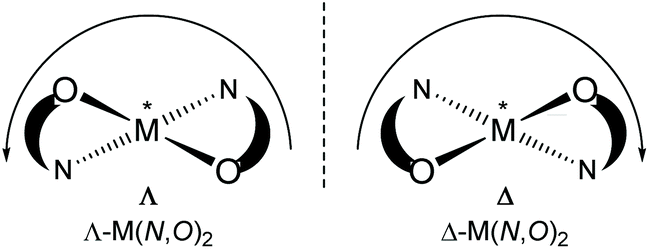
Chiral/achiral bidentate N,O-chelate Schiff base ligands can provide four coordinated non-planar C2-symmetrical transition metal(II)-complexes with tetrahedral or pseudotetrahedral geometry with Fe(II), Co(II), Ni(II), Cu(II) and Zn(II). The molecular structures of these complexes are of continued interest in the context of the chiroptical properties, metal-centred induced Λ vs. Δ-chirality and concomitant (dia)stereoselectivity.1,2 With enantiopure R- or S-N,O chelate ligands, an induced chirality at-metal adds right (Δ)- and left (Λ)-handed helical isomers and can give two opposite configured diastereomers Δ-M(R-N,O)2 and Λ-M(R-N,O)2 {or Λ-M(S-N,O)2 and Δ-M(S-N,O)2} whose relative amount can be different in solution and in the solid state (Scheme 1).3–14
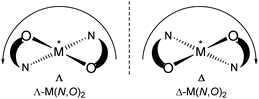 |
| | Scheme 1 Enantiomeric configurations at the metal of a non-planar bis-N,O-chelate complex viewed along the C2 axis (perpendicular to the paper plane): Λ left-handed helicity, Δ right-handed helicity along the C2 axis. | |
The non-covalent inter- and intra-complex interactions and the interactions to the solvent molecules in solution result in free energy differences between the two diastereomers, leading to one of the diastereomers being thermodynamically favoured. In this connection, ligand chirality and design, substituents, steric constraints, metal ion and counter anion selection, solute-solvent interactions, reaction environment, crystallization protocol, pH of solution and redox reactions can significantly control the phenomenon.3–14
Concerning the chirality induction at-metal and thereby diastereoselection in M(N,O)2 complexes with N,O = chiral bidentate salicylaldiminate (Schiff base) ligand the crucial question is the diastereomeric purity. Typically, solid-state single-crystal structures (from one crystal) are taken as the basis to assign the diastereomer as Δ-M(R-N,O)2 or Λ-M(R-N,O)2, etc. This does not rule out that already the single crystal batch may form a mixture of diastereomers and that the preferred diastereomer may be different in solution, including diastereomer equilibria with Λ/Δ-helicity inversion.3,4,9a Enantiopure salicylaldimine ligands can give single diastereomers in one crystal, but the solution diastereoselection can be very poor. This was addressed by Scott et al. for the solid state by matching simulated and experimental powder X-ray data under the assumption that the other diastereomer will have an independent crystal structure and display a distinctly different powder X-ray diffraction pattern.9b
Solid-state versus solution studies with X-ray, vibrational circular dichroism and NMR along with theoretical calculations revealed that diastereoselection is phase dependent for the explored solvation-induced helicity inversion at-metal from Λ-Cu-R or Δ-Cu-S at solid-state to Δ-Cu-R or Λ-Cu-S in solution for Schiff base complexes.3,15 Solvent dependent helicity inversion from Δ-Ni (in acetonitrile) to Λ-Ni (in DMSO) was reported in tris(diamine)nickel(II), whereas both stereoisomers coexist with almost equal ratio at the solid-state.13a Similarly, variation of pH of the solution or solvent polarity led to an epimerization of metal centred chirality in Ti(IV)/Re(I)-complexes.13b,c Indeed, certain redox-reactions led to changes in the chirality at-metal in Co/Cu(II)-chiral-ligand complexes.14
Our recent works along induced chirality at-metal and diastereoselection showed a phase-dependent preferred formation of the Δ-M or Λ-M diastereomer in non-planar or pseudotetrahedral mononuclear [M(R or S-N,O)2] (M = Fe, Co, Ni, Cu, Zn and N,O− = salicylaldiminato/oxo-naphthaldiminato ligands16), examined by X-ray, 1H NMR-/ECD-/VCD-spectroscopy, DSC and DFT/TDDFT.3–8,15 X-ray structure determinations, the most reliable method to assign the absolute configuration in the solid state, reveal that coordination of R- or S-N,O ligands to the metal ions typically gives Λ-M-R or Δ-M-S as major diastereomer in an investigated enantiopure single crystal (not ruling out the co-existence of crystals of the other diastereomer in the batch).3–8,15 Only in a few cases, both diastereomers co-exist in a single crystal.1i,5 In solution, a dynamic equilibrium between two diastereomers can lead to Λ/Δ-helicity inversion, as shown by variable time and/or temperature 1H NMR and ECD spectra.3b,4,7a,8a
Synthesis of Schiff base ligands is a reversible process, and hydrolysis shifts the equilibrium to the reverse direction with reformation of the aldehyde or ketone and the amine.17 The in situ hydrolysis of a Schiff base during complexation and/or a slow crystallization process, due to presence of small amounts of water, has been described before.17,18 The hydrolytic cleavage of the imine-bond in the presence of a metal ion or in the (coordinated) Schiff base can then lead to the formation of the metal(II)-aldehydate or -ketonate complexes.17b,18d We have recently reported two cases of in situ hydrolysis of Schiff base ligands during the complexation and/or crystallization process with a copper(II)-salt, which provided mononuclear bis[salicylaldehydato-κ2O,O′]copper(II)19 and bis[2-oxo-1-naphthaldehydato-κ2O,O′]copper(II)19b and dinuclear (μ-salicylaldehydato)(2,2′-bipyridine)(nitrato)-copper(II).19a
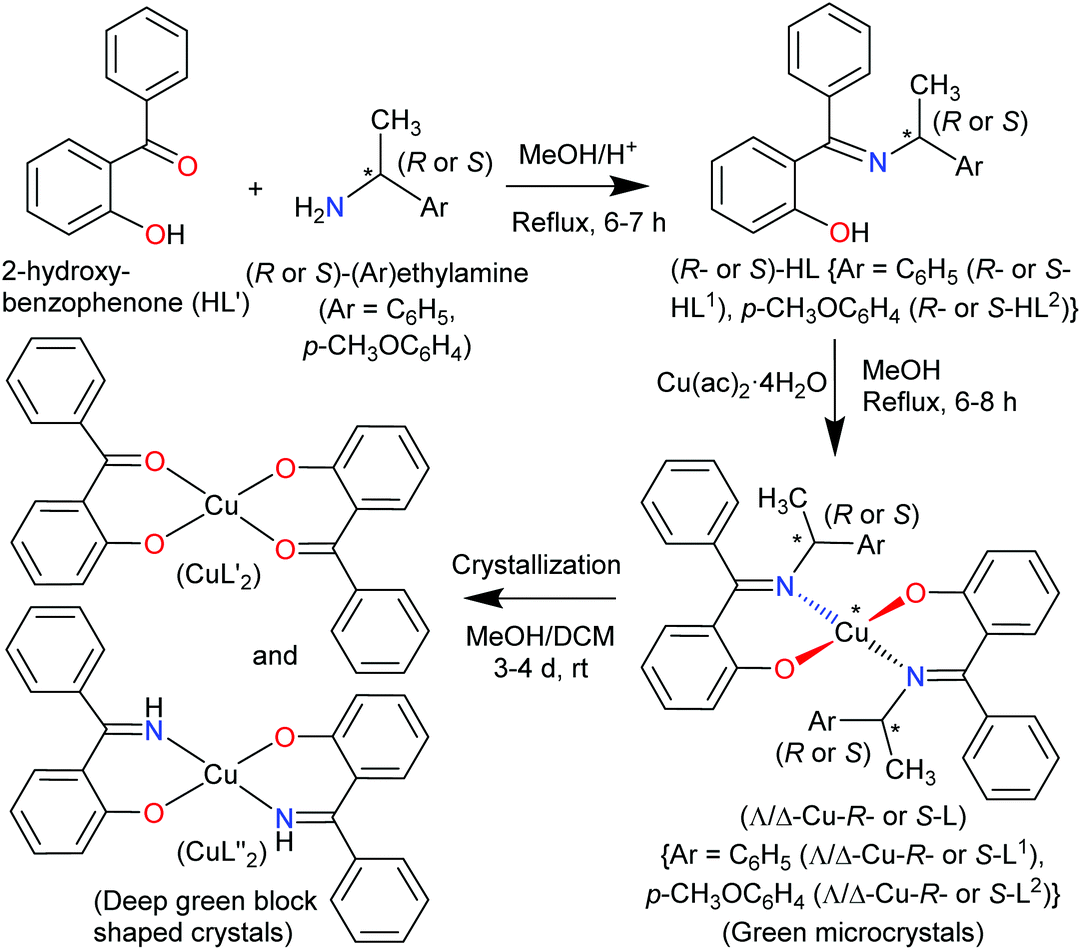
The present paper reports the results of the synthesis and spectroscopic characterization of the enantiopure Schiff base ligands (S or R)-2-((E)-1-(1-(Ar)ethylimino)ethyl)phenol (S- or R-HL) and their complexes bis[(R or S)-2-((E)-1-(1-(Ar)ethylimino)ethyl)phenolato-κ2N,O]-Λ/Δ-copper(II), [Λ/Δ-Cu-S- or R-L]. Crystallization of these complexes failed and only the in situ hydrolysis products could be obtained as an almost equal admixture of bis[2-oxo-benzophenonato-κ2O,O′]-copper(II), (CuL′2) and bis[2-(imino(phenyl)methyl)phenolato-κ2O,N], (CuL′′2) (Scheme 2). For the diastereoselection of Λ/Δ-Cu-S- or R-L a combination of experimental (UV-Vis and ECD) and computational procedures including structure optimizations and excited state properties by DFT/TDDFT is employed to assess the chirality induction at-metal. In addition, solid state X-ray and PXRD measurements are carried out to elucidate the molecular structures of the ligands and the complexes.
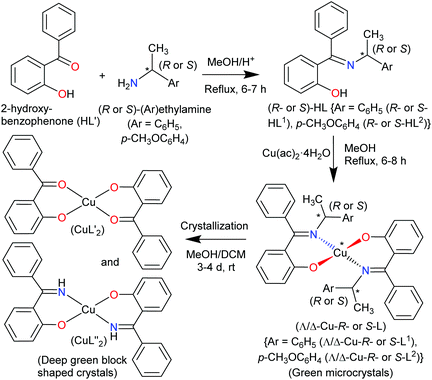 |
| | Scheme 2 Synthetic route to the formation of the copper(II)-complexes. | |
Results and discussion
The enantiopure Schiff base ligands (S or R)-2-((E)-1-(1-(Ar)ethylimino)ethyl)phenol {S- or R-HL: Ar = C6H5 (S- or R-HL1) and p-CH3OC6H4 (S- or R-HL2)} were synthesized from the reaction between 2-hydroxy-benzophenone (HL′) and an enantiopure amine (R or S)-(Ar)ethylamine (Scheme 2). These Schiff base ligands react with copper(II) acetate under reflux to provide deep-green microcrystals of bis[(R or S)-2-((E)-1-(1-(Ar)ethylimino)ethyl)phenolato-κ2N,O]-Λ/Δ-copper(II), (Λ/Δ-Cu-R- or S-L) {Ar = C6H5 (Λ/Δ-Cu-R- or S-L1) and p-CH3OC6H4 (Λ/Δ-Cu-R- or -S-L2)}.
Surprisingly, from single-crystal X-ray diffraction the deep-green block-shaped crystal product of the complex Cu-R-L1 is analyzed as bis[2-oxo-benzophenonato-κ2O,O′]-copper(II), (CuL′2) in an about equimolar mixture with bis[2-(imino(phenyl)methyl)phenolato-κ2N,O]copper(II), (CuL′′2) (Scheme 2). As the formation of the Schiff base ligands and their purity had been established by various methods, obviously an in situ hydrolysis of the (coordinated) Schiff base ligands had occurred during the slow crystallization process due to the presence of a small amount of water in the sample.17–19
The reaction for the normal hydrolysis of a Schiff base ligand, opposite to its formation, is as given in eqn (1) and is initiated by a nucleophilic attack of water on the imine carbon atom.
| | 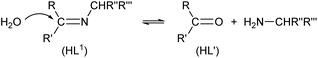 | (1) |
Remarkably, here the in situ hydrolysis had not only occurred along the Ar(Ph)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(alkyl) bond with re-formation of the starting 2-hydroxy-benzophenone (HL′) but also along the N–C(alkyl) bond with formation of 2-(imino(phenyl)methyl)phenol (HL′′). Our results show that apparently for a (Cu-coordinated) imine nitrogen atom, the direction of hydrolysis cannot only occur at the CN bond but also along the N–C(alkyl) bond. The latter leads to an imine and alcohol according to eqn (2) and is initiated by the nucleophilic attack of water on the N-bound alkyl carbon atom. The substitution of two aryl groups on the imine-carbon atom with their steric shielding in the case of L1 may facilitate the nucleophilic attack of water on the N-bound alkyl carbon atom. Following the nucleophilic attacks of H2O, subsequent proton shifts to the imine-N atom then cleave the respective CN or N–C bonds.
N(alkyl) bond with re-formation of the starting 2-hydroxy-benzophenone (HL′) but also along the N–C(alkyl) bond with formation of 2-(imino(phenyl)methyl)phenol (HL′′). Our results show that apparently for a (Cu-coordinated) imine nitrogen atom, the direction of hydrolysis cannot only occur at the CN bond but also along the N–C(alkyl) bond. The latter leads to an imine and alcohol according to eqn (2) and is initiated by the nucleophilic attack of water on the N-bound alkyl carbon atom. The substitution of two aryl groups on the imine-carbon atom with their steric shielding in the case of L1 may facilitate the nucleophilic attack of water on the N-bound alkyl carbon atom. Following the nucleophilic attacks of H2O, subsequent proton shifts to the imine-N atom then cleave the respective CN or N–C bonds.
| | 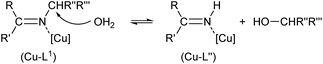 | (2) |
Both deprotonated ligands L′− and L′′− then coordinate to copper and form bis-chelate ligands complexes. Both homoleptic complexes CuL′2 and CuL′′2 crystallized as a mixture sharing the same crystallographic position. Alternatively, a mixed-ligand complex CuL′L′′ could have formed (which would then show a ligand disorder on the centrosymmetrical site). As the Cu atom sits on an inversion symmetry site this renders the two ligands symmetry equivalent and the two possibilities cannot be differentiated by crystallography. Also, a mixed crystal of CuL′L′′ with some Cu(L′)2 is possible, as crystallographically the shared keto O and imine N atoms refine to a ratio of 56/44. Spectroscopy confirms the presence of the imine in the crystal product. IR spectra show the main characteristic bands of νCN at ca. 1605 cm−1 in Schiff base ligands (R- or S-HL) (Fig. S6a, ESI†) and ca. 1607 cm−1 in green microcrystals (Cu-R or S-L) (Fig. S6b, ESI†). Spectra for the deep-green block-shaped crystals (admixture of CuL′2 and CuL′′2) show peaks at ca. 3325 cm−1 (νN–H) and ca. 1601/1579 cm−1 (νCN/O) (Fig. S6c, ESI†). Calculated IR spectra from optimized structures of CuL′2 or CuL′′2 by DFT at the b3lyp/6-31 g(d) level exhibit these bands at a comparable range (Fig. S6d, ESI† and see Experimental section). In electron impact (EI) mass spectra, the deep-green block-shaped crystal sample obtained from the crystallization of Cu-R or S-L1 show the ion peaks of the hydrolysis products at m/z = 198 for [HL′]+, 197 [HL′′]+ and 196 [HL′′–H]+. The complex molecular ion peaks are at m/z = 457 [CuL′2]+, 456 [CuL′2–H or Cu-L′L′′]+ and 455 [CuL′′2]+ (Fig. S7, ESI†). The mass spectra give further ion peaks at m/z = 260 for [CuL′]+, 258 for [CuL′′-H]+, 121 for [{(C6H4OH)CHO}-H]+ and 105 for [{C6H5CHO}-H]+.
1H-NMR spectra
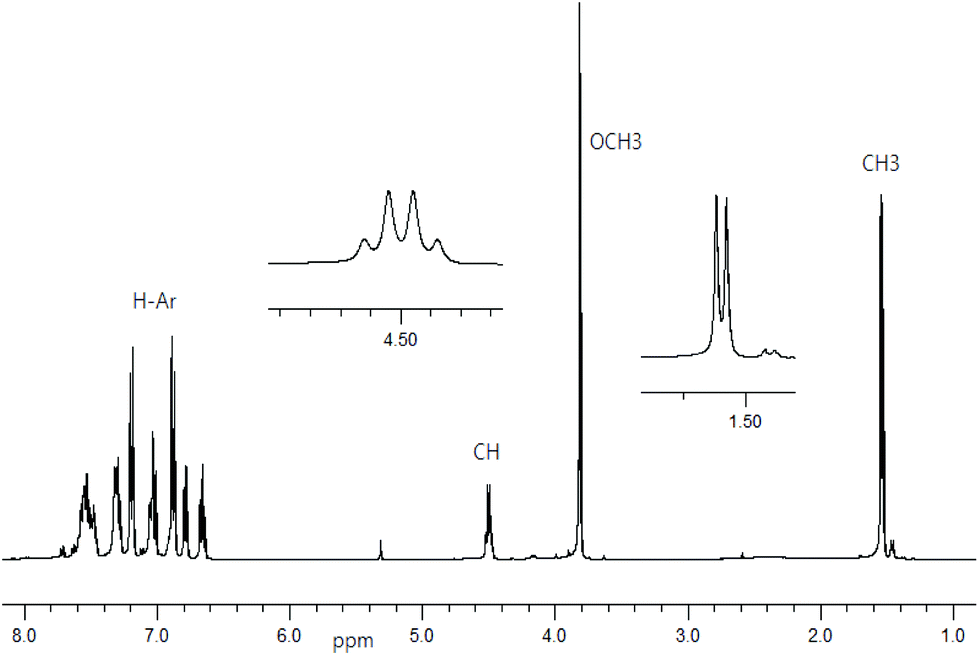
1H NMR spectra for Schiff base ligands show a doublet at δ 1.53–1.57 ppm (JHH = 6.8 Hz) for the methyl protons and a quartet at δ 4.49–4.55 ppm (JHH = 6.8 Hz) for the methine proton (Fig. 1).16 In addition, the spectra show a sharp singlet at δ 3.82 ppm for the p-methoxy protons in R- or S-HL2. The spectra are dominated by several peaks as doublet, triplet, doublet of doublet, doublet of triplet and multiplet at δ 6.66–6.71 ppm for the aromatic protons (see Experimental section). The presence of the p-methoxy group as substituent on the aryl-ring in R- or S-HL2 shifts the methine proton signal upfield by ca. 0.05 ppm in contrast to that in R- or S-HL1 due to an electron inductive effect. However, the signal for the phenolic proton (acidic) could not be detected in CDCl3 solution.
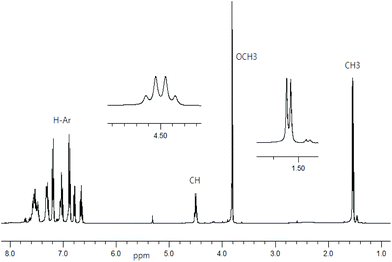 |
| | Fig. 1
1H NMR spectrum for R-HL2 in CDCl3 at 20 °C. | |
Decomplexation reaction of Cu-R-L1 with NaCN
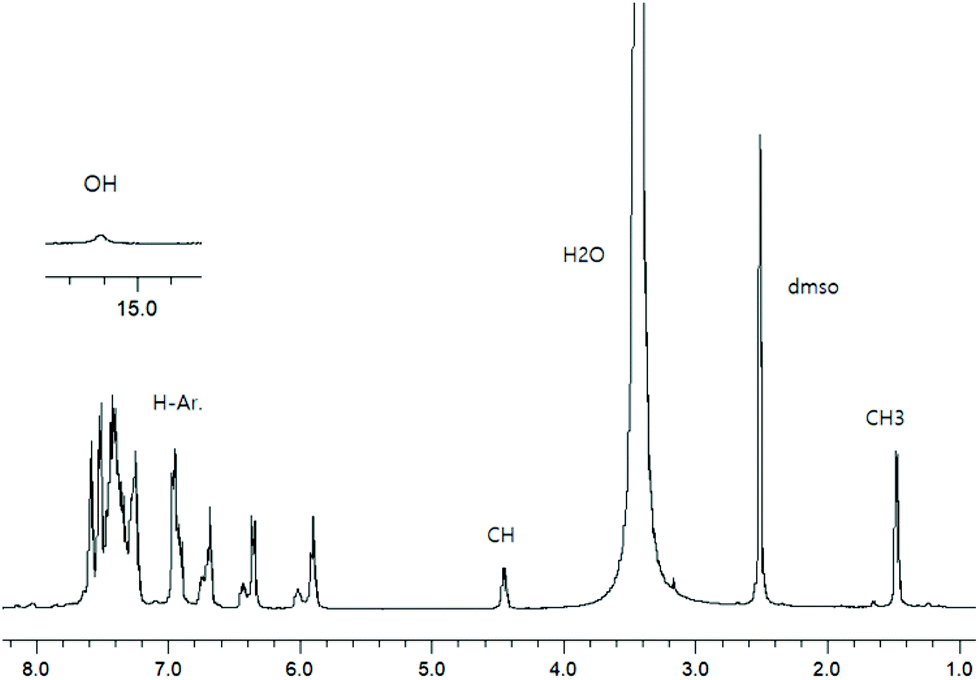
To check the formation of paramagnetic Cu-R-Ļ that is, to verify the presence of the intact Schiff base ligand (HL) in the green microcrystals (Scheme 2) by 1H NMR, we employed a decomplexation reaction of Cu-R-L1 with NaCN with concomitant reduction of Cu(II) to Cu(I) in DMSO-d6 solution.5,6,8b The reaction results in the formation of diamagnetic [CuI(CN)4]3− and free Schiff base (R-HL1) in solution, accompanied by a colour change from deep-green to light-orange. The 1H NMR spectrum of this solution (Fig. 2) is identical to the neat Schiff base (Fig. 1) and shows a doublet at δ 1.47 ppm (JHH = 6.4 Hz) for the methyl protons and a quartet at δ 4.45 ppm (JHH = 6.2 Hz) for the methine proton. The phenolic proton appears as a broad signal at δ 15.55 ppm. Indeed, there is no peak which corresponds to the hydrolyzed ligands 2-hydroxy-benzophenone (HL′) and/or 2-(imino(phenyl)methyl)phenol (HL′′). This result suggests the formation of the Cu-R- or S-L complexes which provide bis[2-oxo-benzophenonato-κ2O,O′]-copper(II), (CuL′2) and bis[2-(imino(phenyl)methyl)phenolato-κ2N,O]copper(II), (CuL′′2) via in situ hydrolysis of the coordinated Schiff base during the subsequent crystallization in MeOH/DCM (Scheme 2).
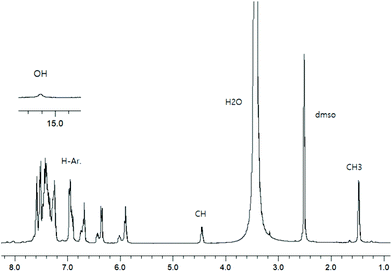 |
| | Fig. 2
1H NMR spectrum from the decomplexation reaction of Cu-R-L1 with NaCN in DMSO-d6 at 20 °C. | |
X-ray structures for R-HL1, CuL′2 and CuL′′2
The molecular structure for R-HL1 is shown in Fig. 3 and selected bond lengths and angles are listed in Table 1. R-HL1 crystallizes in the monoclinic space group P21. The refinement for an absolute structure with a Flack parameter of 0.04(7) (cf.Table 4) indicates that the investigated crystal is enantiopure. A Flack parameter close to zero confirms the absolute structure of the crystal and excludes the presence of the Schiff base molecule with S-chirality at C14. The N1–C7 bond length of 1.288(2) Å indicates the double bond character of the imine, while the C7–N1–C14 bonding angle of 121.73(13)° represents a sp2-hybridization of the imino-nitrogen atom. There is an intramolecular hydrogen bond from the phenol (O-)H atom to the imine N atom.
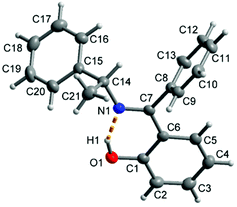 |
| | Fig. 3 Molecular structure of R-HL1 with the dashed orange line showing the intramolecular hydrogen bond with O1–H1 0.91(2) Å, H1⋯N1 1.70(3) Å, O1⋯N1 2.533(2) Å, O1–H1⋯N1 angle 151(2)°. | |
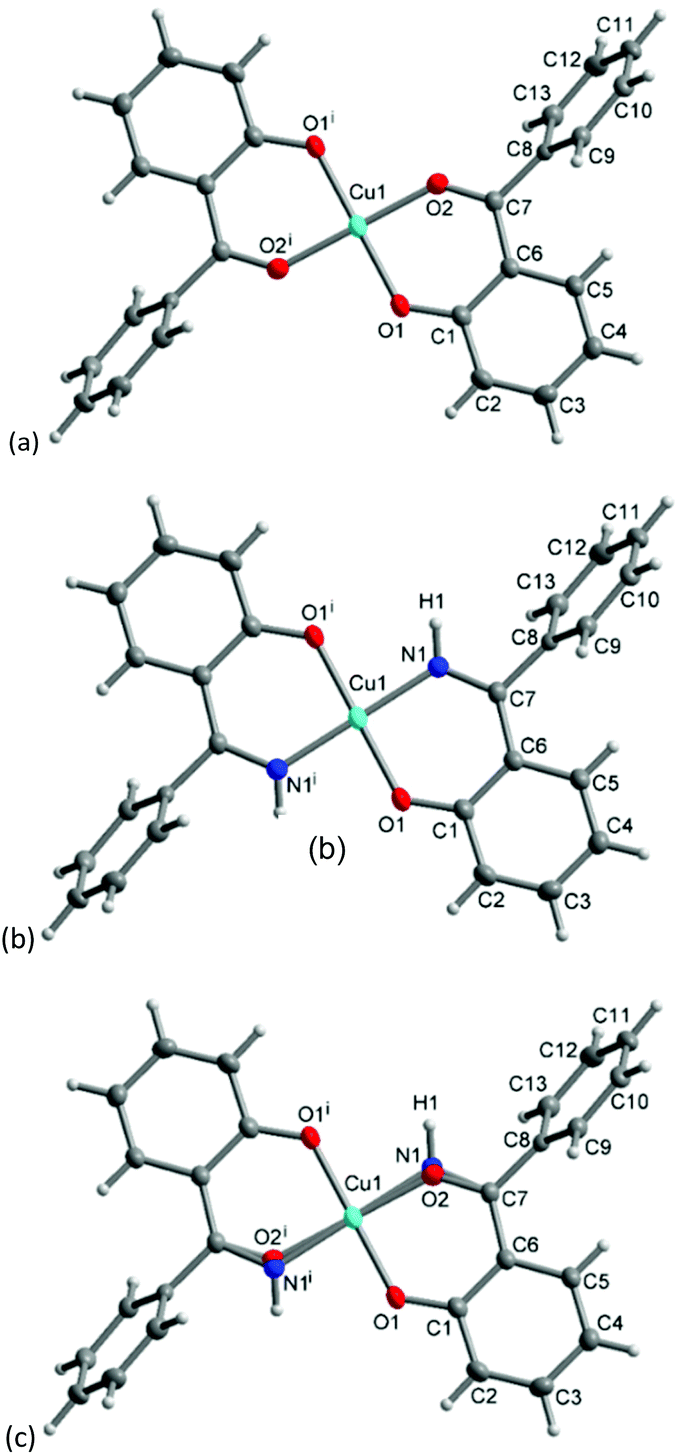
Table 1 Selected bond lengths (Å) and angles (°) in R-HL1, CuL′2 and CuL′′2
| Bond lengths (Å)/angles (°) |
R-HL1 |
CuL′2 |
CuL′′2 |
| Symmetry transformation i = −x + 1, −y + 2, −z + 1. |
| Cu–O1 |
|
1.9070(16) |
1.9070(16) |
| Cu–O2 |
|
1.89(1) |
— |
| Cu–N1 |
|
— |
1.96(2) |
| O1–C1 |
1.347(2) |
1.308(3) |
| N1/O2–C7 |
1.288(2) |
1.25(11) |
1.34(2) |
| N1–C14 |
1.470(2) |
— |
| C6–C7 |
1.473(2) |
1.446(3) |
| C7–N1–C14 |
121.73(13) |
— |
| C1–O1–H1 |
106.2(14) |
— |
| C6–C7–N1 |
118.18(1) |
|
| O1–Cu–O2 |
— |
90.1(3) |
— |
| O1–Cu–N1 |
— |
|
95.1(5) |
| O1–Cu–O2i/N1i |
— |
89.9(3) |
84.9(5) |
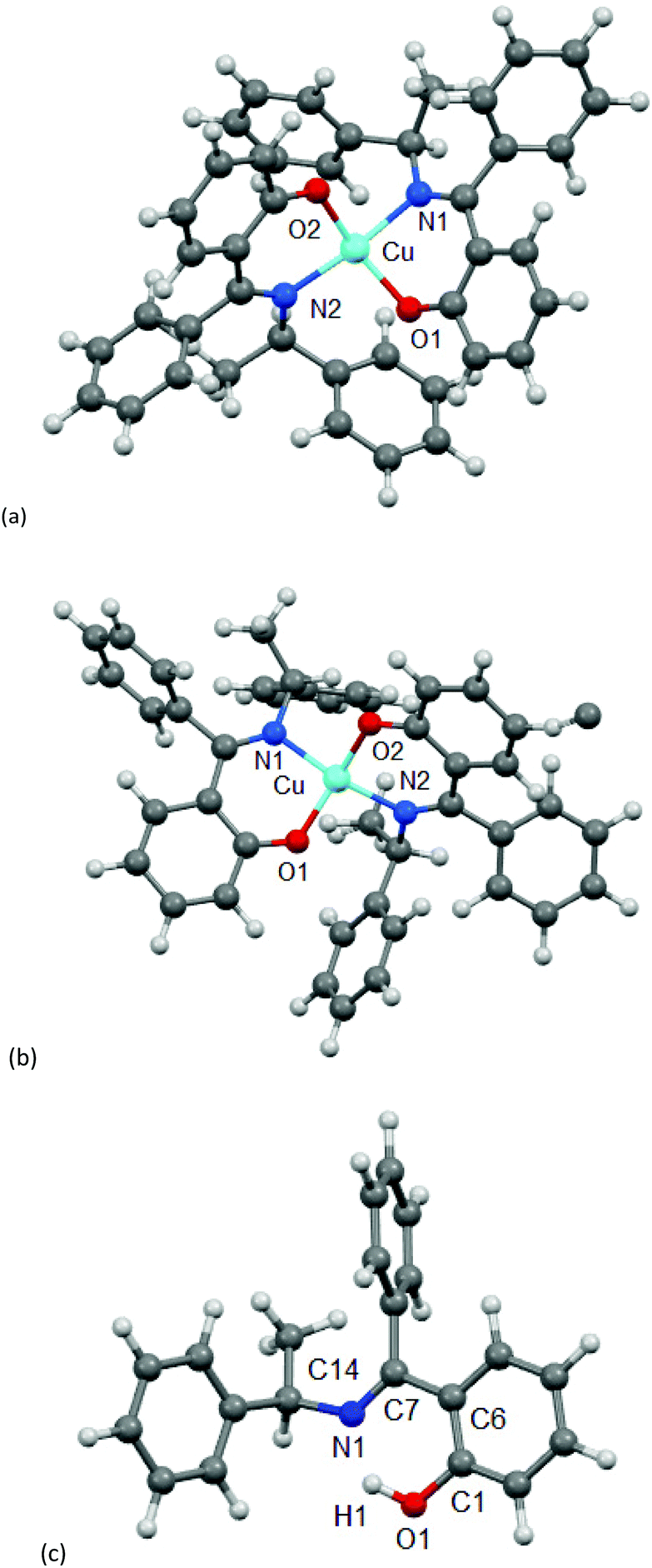
The molecular structure for CuL′2 and CuL′′2 on their shared centrosymmetry position is depicted in Fig. 4. For clarity also the individual CuL′2 and CuL′′2 complexes are shown. Both complexes are square-planar. The Cu atom occupies the crystallographic inversion center so that both ligands are symmetry equivalent. The bond lengths are largely as expected. We note that the imino-N atom could only be refined isotropically. Crystallographically the shared position of the two complexes manifests itself as disorder of the keto-O and imino-N atom which refined to 56/44 occupancy. The disorder then led to a lower accuracy in the involved C–O, C–N and Cu–O, Cu–N bond distances (Table 1).
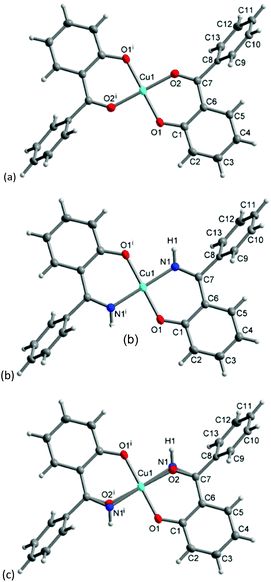 |
| | Fig. 4 Molecular structures of (a) CuL′2 and (b) CuL′′2 shown individually and (c) on their shared crystallographic site. Selected bond lengths and angles are given in Table 1. Symmetry transformation i = −x + 1, −y + 2, −z + 1. | |
PXRD-derived structures for R-HL1 and Cu-R-L1
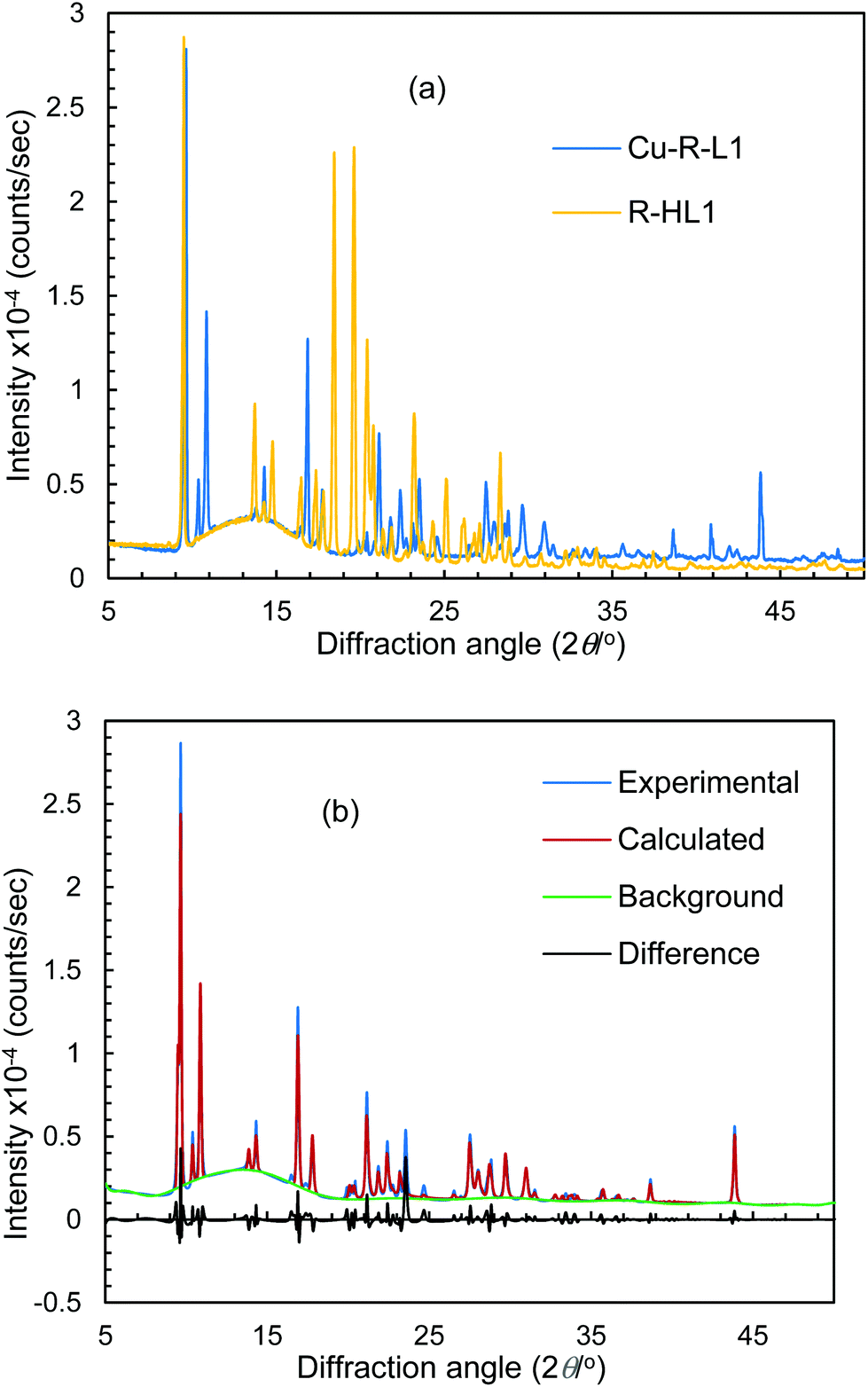
Because of the failure to grow single crystals of Cu-S- or R-L1 in X-ray quality due to the in situ hydrolysis, it was inevitable to determine their structures through powder X-ray diffraction (PXRD) on the green microcrystals of Cu-S- or R-L1. PXRD patterns for the Schiff base ligand (R-HL1) and complex (Cu-R-L1) are shown in Fig. 5a, which reveal two distinct phases and hence confirm the formation of the complex. PXRD patterns reflect a well-defined crystalline nature and allowed for structure determination using the program Expo-201420 followed by Rietveld refinement.21–23 For structure solution calculations, we used the gas-phase optimized structures for R-HL1 and Cu-R-L1 (both Λ-Cu and Δ-Cu diastereomers, see below) as input files, respectively. The experimental and calculated Rietveld refinement plots are in good agreement with very little difference (Fig. 5b, Fig. S8, and S9, ESI†). The structure for Cu-R-L1 reveals that two phenolate-oxygen and two imine-nitrogen atoms from two molecules of Schiff base ligands form a non-planar N2O2-coordination sphere around copper(II) in a pseudotetrahedral geometry (Fig. 6a and b). The structure solved for R-HL1 (Fig. 6c) is very similar to its single crystal X-ray structure (Fig. 2), discussed above. The selected bond lengths and angles in R-HL1 and Cu-R-L1 are listed in Table 2, very close to the X-ray results for R-HL1 (Table 1) and for an analogous Δ-Cu-R-L complex3a (Table 2). In fact, PXRD structures satisfy well to both diastereomers of Λ-Cu-R-L1 and Δ-Cu-R-L1 and obviously cannot determine the absolute configuration of the metal ion (e.g., Λ-Cu or Δ-Cu) as in case of a single-crystal X-ray structure.
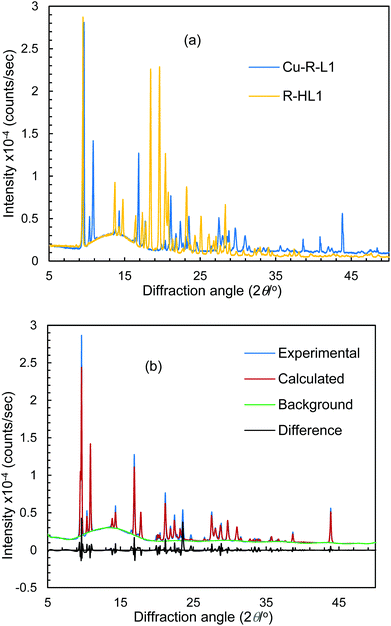 |
| | Fig. 5 (a) PXRD patterns for R-HL1 and Cu-R-L1. (b) Experimental and calculated Rietveld refinement plots for Cu-R-L1 (calculated for Λ-Cu-R-L1; see Fig. S8, S9, ESI† for R-HL1 and Δ-Cu-R-L1). | |
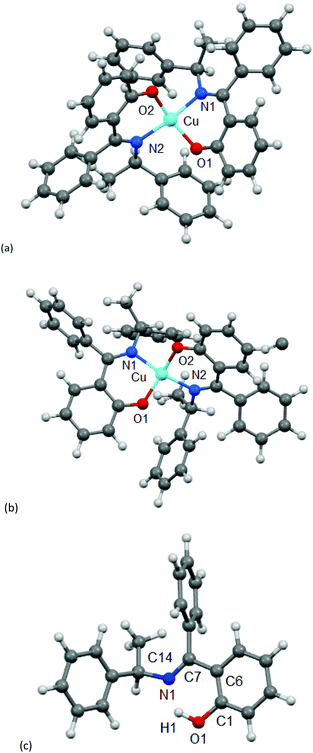 |
| | Fig. 6 PXRD-derived structures for (a) Λ-Cu-R-L1, (b) Δ-Cu-R-L1 (b) and (c) R-HL1. | |
Table 2 Selected bond lengths (Å) and angles (°) in R-HL1 and in the diastereomeric pair Δ-Cu-R-L1/Λ-Cu-R-L1
| Bond length (Å) & angles (°) |
PXRD structures |
X-ray structurea |
R-HL1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
Δ-Cu-R-L1 |
Λ-Cu-R-L1 |
Δ-Cu-R-L |
|
From analogous bis[(R)-N-1-(phenyl)ethyl-salicylaldiminato]-Δ-copper(II), (Δ-Cu-R-L).3a.
See values for X-ray structure in Table 1.
|
| Cu—O1/Cu—O2 |
|
1.873/1.870 |
1.887/1.913 |
1.870(2), /1.880(2) |
| Cu—N1/Cu—N2 |
|
1.993/1.984 |
1.995/1.991 |
2.021(3), /1.976(3) |
| O1—Cu—O2 |
|
151.7 |
149.6 |
151.56(12) |
| N1—Cu—N2 |
|
150.5 |
164.2 |
151.94(13) |
| N1—Cu—O1 |
|
92.7 |
88.2 |
86.13(12) |
| N2—Cu—O2 |
|
93.3 |
92.8 |
84.92(12) |
| N1—Cu—O2 |
|
93.4 |
89.5 |
101.63(12) |
| N2—Cu—O1 |
|
94.8 |
97.6 |
101.10(12) |
| O1—C1/C6—C7 |
1.392/1.503 |
|
|
|
| N1—C7/N1—C14 |
1.289/1.493 |
|
|
|
| C7—N1—C14 |
126.29 |
|
|
|
| C6—C7—N1 |
116.57 |
|
|
|
| C1—O1—H1 |
110.95 |
|
|
|
|
θ/° |
|
40.2 |
33.6 |
42.1 |
|
τ
tet-sq (θ/90°) |
|
0.45 |
0.37 |
0.47 |
For analyses of coordination geometry of Λ/Δ-Cu-R-L1, we measured the degree of distortion from tetrahedral to square-planar based on the dihedral angle θ/° (i.e., angle between two coordinating O1–Cu–N1 and O2–Cu–N2 planes) and its normalized function τtet-sq(θ/90°).3–8,15 The values of θ are 0° (or τtet-sq = 0) for square-planar and 90° (or 1.0) for tetrahedral geometry (not considering the inherent distortion induced by the chelate ring formation). The values are found to be 33.6° (θ)/0.37 (τtet-sq) for Λ-Cu-R-L1 and 40.2°/0.45 for Δ-Cu-R-L1, which are comparable to literature values for the analogous bis[(R)-N-1-(phenyl)ethyl-salicylaldiminato]-Δ-copper(II) (42.1°/0.473a) and suggest a pseudotetrahedral geometry around the copper(II) ion.
Electronic spectra (UV-Vis and ECD)
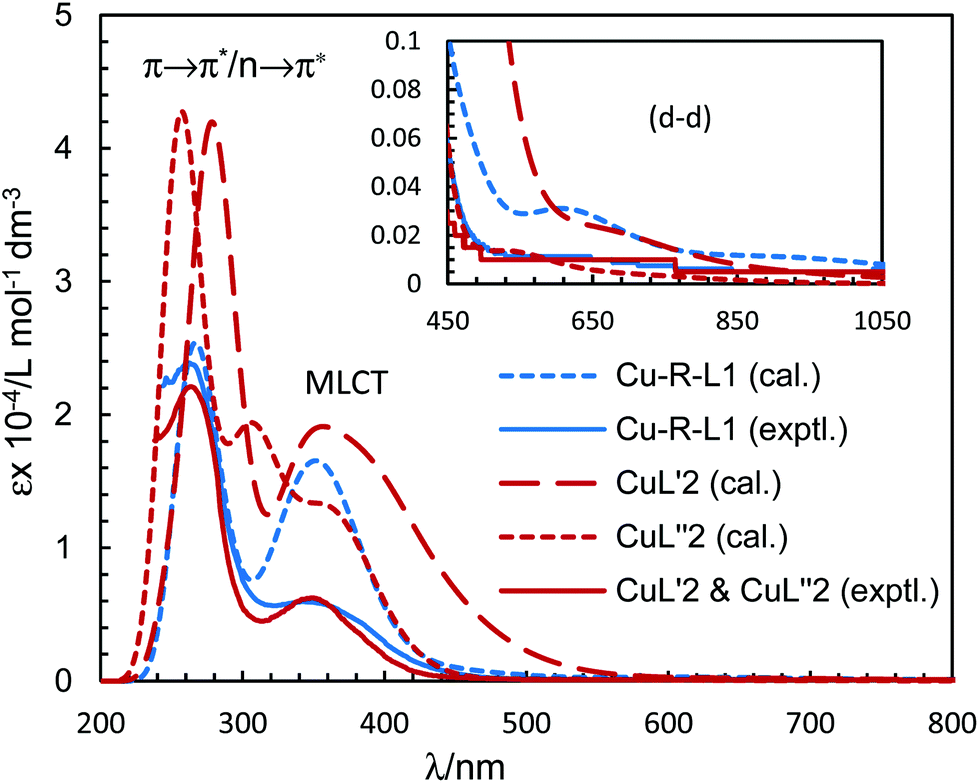
The electronic spectra (UV-Vis) for the green microcrystals Cu-R-L1 or Cu-S-L1 are identical, and comparable to that for the deep-green block-shaped crystals (CuL′2/CuL′′2) in chloroform. The spectra feature a very strong band below ca. 320 nm (λmax/εmax = 260 nm/23925 L mol−1 dm−3 for Cu-R-L1 and 265 nm/22100 L mol−1 dm−3 for CuL′2/CuL′′2), assigned to the intra-ligand π → π*/n → π* transitions (LL) (Fig. 7). A medium broad band at 320–450 nm (λmax/εmax = 342 nm/5963 L mol−1 dm−3 for Cu-R-L1 and 349 nm/6250 L mol−1 dm−3 for CuL′2/CuL′′2), attributes to the metal–ligand (ML) charge transfer transitions. The spectra further show a very weak broad band at visible region 500–1100 nm (λmax/εmaxca. 650 nm/100 L mol−1 dm−3), due to the metal-centred d–d (MM) transitions for the copper(II)-core electrons (Fig. 7, inset).5,7b,24,25,26 The computed UV-Vis spectra by TDDFT, calculated at the m06/sdd//b3lyp/6-31 g(d) level with polarization continuum model (PCM) in chloroform, are shown in Fig. 7, which are very similar to the experimental spectra in solution.
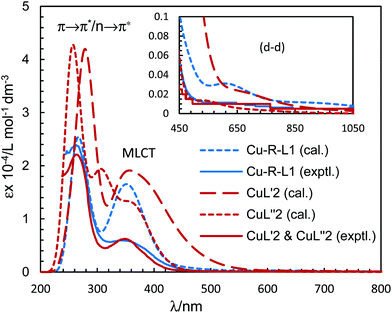 |
| | Fig. 7 Experimental UV-Vis spectrum for Cu-R-L1 (identical to Cu-S-L1) (0.04 mM) and CuL′2/CuL′′2 (0.05 mM) in chloroform at 25 °C. Calculated spectra for Λ- or Δ-Cu-R-L1, CuL′2 and CuL′′2 at the m06/sdd//b3lyp/6-31 g(d) level with PCM in chloroform (Gaussian band shape with exponential half-width, σ = 0.33 eV). | |
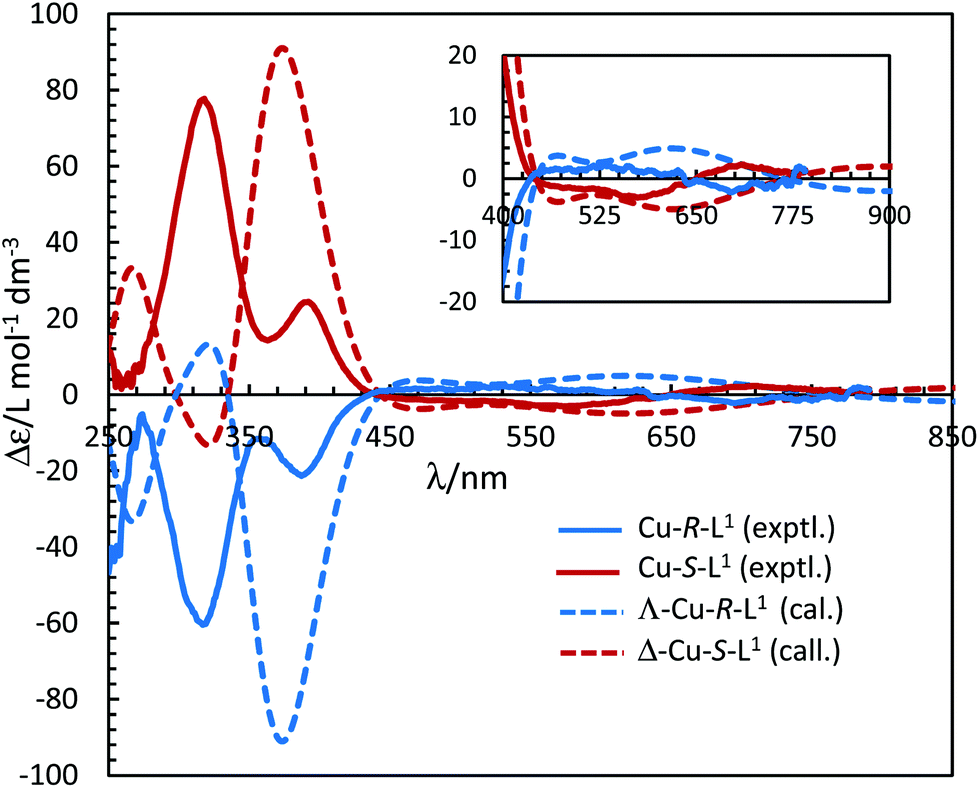
The electronic circular dichroism (ECD) spectra in chloroform exhibit mirror-image relationships for Cu-R-L1 and Cu-S-L1 (Fig. 8). The spectra feature several bands with opposite Cotton effect, associated to the different electronic transitions as observed in the UV-Vis spectra. Indeed, the mirror-image relationships reflect diastereomeric excess of the S- or R-ligated complexes in solution. The ECD spectral data for Cu-R-L1/Cu-S-L1 are characterized with the band maxima and patterns (sign and strength) at ca. 710 nm (±, weak), 570 nm (±, weak), 390 nm (±, medium) and 320 nm (±, strong), respectively. Similar ECD spectral patterns were reported for the analogous transition metal(II)-complexes with chiral-N,O-chelate Schiff base ligands in solution (M = Fe, Co, Ni, Cu, Zn).3,4,5,8b,15
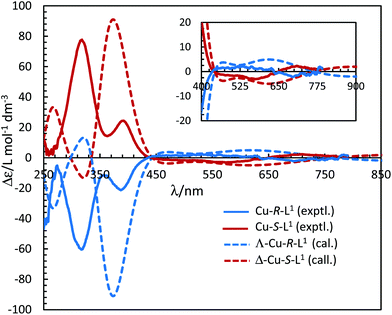 |
| | Fig. 8 Experimental ECD spectra for Cu-R-L1/Cu-S-L1 (1.21/1.33 mM) in chloroform at 25 °C (values of experimental Δε are increased by 5 times). Calculated spectra for Λ-Cu-R-L1/Δ-Cu-S-L1 at the m06/sdd//b3lyp/6-31 g(d) level with PCM in chloroform (Gaussian band shape with exponential half-width σ = 0.33 eV). See Fig. S5b, ESI† for the calculated spectra of the opposite Δ-Cu-R-L1/Λ-Cu-S-L1 configuration. | |
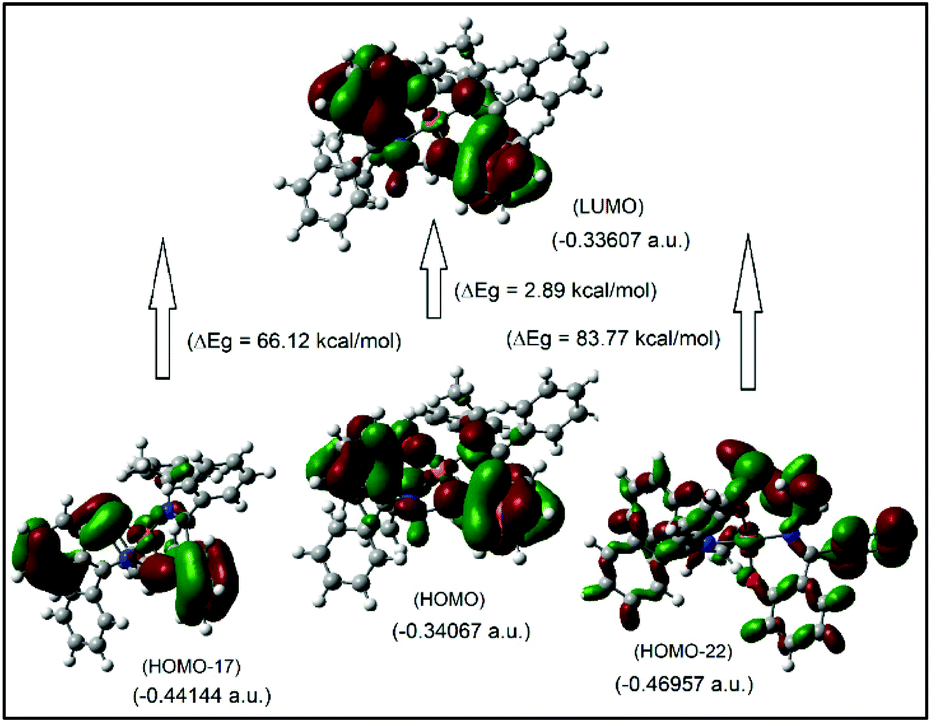
It was attempted to assign the electronic spectra (i.e., different electronic transitions) based on molecular orbital and population analyses for the Λ-Cu-R-L1 diastereomer.5–8,24–26 For open shell transition-metal complexes (as in the present case), a large number of excitations occur at a single excited state (wavelength) (Table S1, ESI†), which make the assignment protocol complicated. However, some selected and simplified assignments on electronic spectra are listed in Table 3, which are comparable to the experimental data. Thus, the band at 681 nm comprises a combination from MM and ML/LM transitions with significant oscillator strength (f) of 0.0011, mostly due to HOMO−17 to LUMO excitation with the highest molecular orbital (MO) contribution of 63%. The HOMO−17 orbital is mainly localized on the metal dxy-/ligand (sal-ring) π-moieties, while the LUMO is localized on the metal dz2-/ligand (sal-ring) π-moieties. This band is very close to the experimental d–d (MM) transition band at ca. 650 nm (Fig. 7, inset, Table 3). Similarly, the band at 607 nm consists of a combination from MM, ML/LM and LL transitions (f = 0.0053), and mainly assigns for metal dz2-/ligand σ-moieties (HOMO−22) to metal dz2-/ligand (sal-ring) π-moieties (LUMO) excitation with MO contributions of 40%. However, the HOMO to LUMO excitation, which occurs at 430 nm (f = 0.0085, MOs = 42%), comprises a combination from MM and ML/LM transitions. This later band is assigned mainly for metal dx2–y2-/ligand (sal-ring) π-moieties (HOMO) to metal dz2-/ligand (sal-ring) π-moieties (LUMO) excitation. Indeed, there are some other intense bands on the computed spectra, very close to the experimental bands (Table 3). The frontier molecular orbitals (MOs) for HOMO, HOMO−17, HOMO−22 and LUMO are illustrated in Fig. 9.
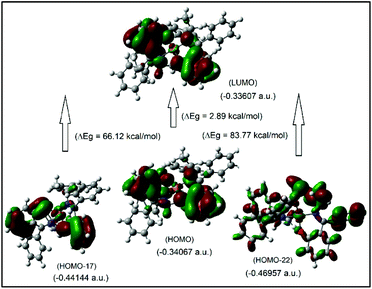 |
| | Fig. 9 The frontier MOs of HOMO, HOMO−17, HOMO−22 and LUMO for Λ-Cu-R-L1 (calculated at m06/sdd//b3lyp/631 g(d)), for β-spin considerations. | |
Table 3 Selected and simplified assignments on electronic spectra calculated for Λ-Cu-R-L1 at the m06/sdd//b3lyp/631 g(d) level
| Bandsa (λ/nm) |
Oscil. strength. (f) |
Excitationsb (MOs contribution, %) |
Assignmentsc,d |
|
Experimental values are in parentheses.
H/L = HOMO/LUMO.
MM = metal centered d–d, ML = metal–ligand and LL = intra-ligand transitions, respectively.
Assigned for β-spin MOs consideration.
|
| 681 (∼650) |
0.0011 |
H-17 → L (63), H-16 → L (35) |
MM, ML/LM |
| 607 |
0.0053 |
H-22 → L (40), H-2 → L (23) |
MM, ML/LM, LL |
| 430 |
0.0085 |
H → L (42), H → L + 2 (16) |
MM, ML/LM |
| 363 (342) |
0.1245 |
H → L + 1 (14), H → L + 2 (26) |
ML, LL |
| 333 |
0.0765 |
H-5 → L (63), H-3 → L (14) |
LM, LL |
| 263 (259) |
0.0852 |
H → L + 7 (20), H → L + 12 (13) |
ML, LL |
Optimized structures and excited state properties (diastereoselection and chirality induction at-metal)
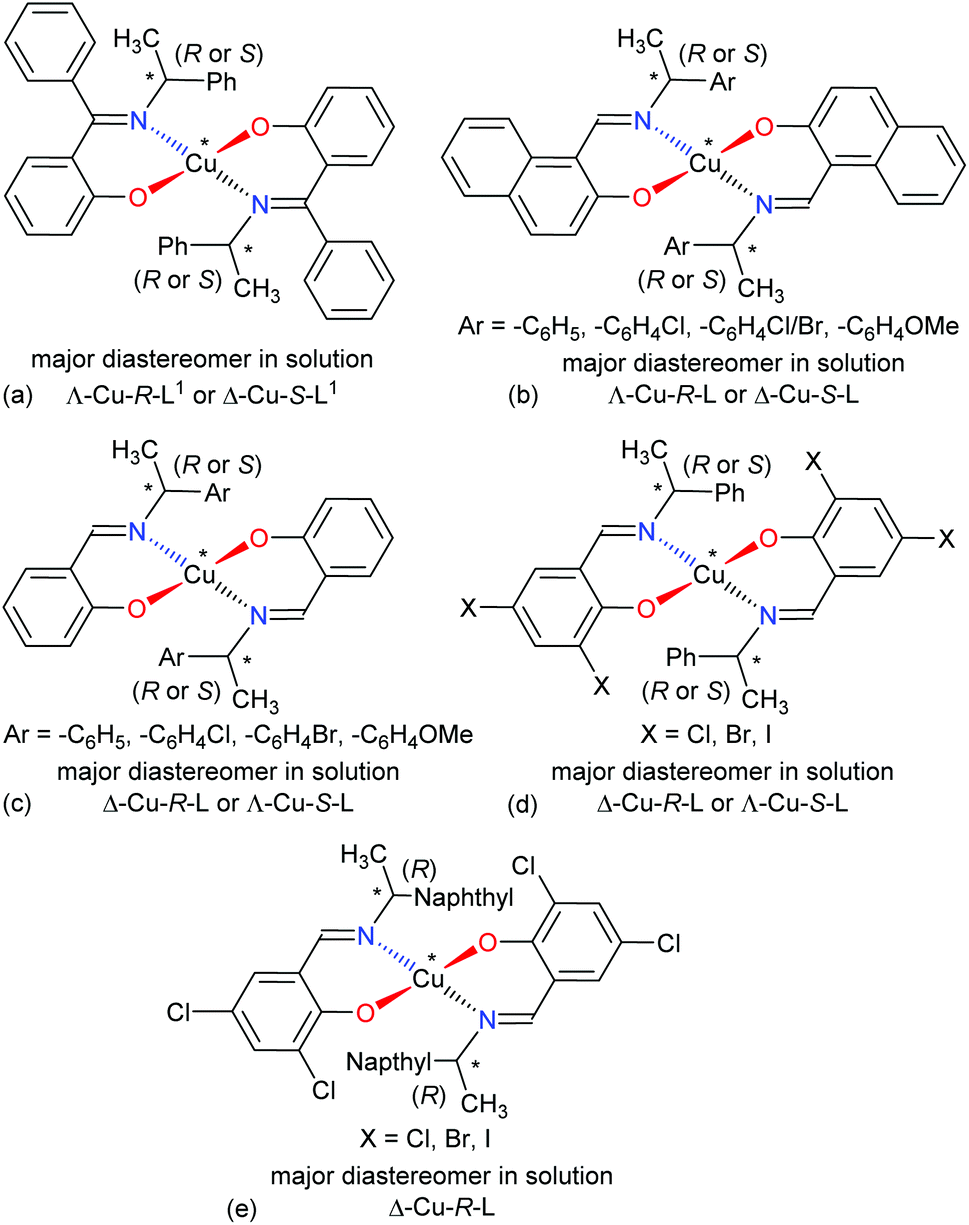
The four-coordinated non-planar metal(II)-complexes with chiral N,O-chelate Schiff base ligands exhibit Λ vs. Δ-chirality induction at-metal. The enantiopure R- or S-N,O ligands provide two opposite configured diastereomers of C2-symmetry with Δ-Cu and Λ-Cu.3–9,15 In order to determine the Λ vs. Δ-chirality, we optimized the structures for diastereomeric pairs Δ-Cu-R-L1/Λ-Cu-R-L1 and Δ-Cu-S-L1/Λ-Cu-S-L1 at the b3lyp/6-31 g(d) level (Fig. S1, ESI†). The results suggest that the Λ-Cu-R-L1 and Δ-Cu-S-L1 diastereomers are more stable than the corresponding Δ-Cu-R-L1 and Λ-Cu-S-L1 diastereomers by ca. 7.60 kcal mol−1. The excited state properties by TDDFT, calculated for the diastereomeric pairs Δ-Cu-R-L1/Λ-Cu-R-L1 and Δ-Cu-S-L1/Λ-Cu-S-L1 at m06/sdd with PCM in chloroform, provide UV-Vis and ECD spectra, which can be compared to the experimental spectra in solution (Fig. 7 and 8). The experimental and calculated UV-Vis spectra for both diastereomeric pairs are, as expected, essentially identical (Fig. S3†), while the ECD spectra show mirror-image relationships (Fig. 8 and Fig. S3, ESI†). ECD spectra indicate diastereomeric excess and allow to determine the absolute configuration by comparison with spectra of known structures or with calculated spectra. Here we compare the experimental and computed ECD spectra to assign the absolute configuration of the metal-centre (i.e., Λ-M vs. Δ-M).3–8,15 The experimental spectra for Cu-R-L1 or Cu-S-L1 fit well to the computed spectra for the Λ-Cu-R-L1 or Δ-Cu-S-L1 diastereomer with slight shifting of the band positions (Fig. 8; the opposite comparison is given in Fig. S5b, ESI†). This and the optimized structures (in the gas phase) strongly indicate Λ-Cu-R-L1 or Δ-Cu-S-L1 as major diastereomers in solution for the R- or S-N,O ligated complexes, respectively. For the calculated ECD spectra for the opposite Δ-Cu-R-L1/Λ-Cu-S-L1 configuration we note that the region of the d–d transition above 450 nm is similarly well reproduced for the Δ-Cu or Λ-Cu configuration regardless of the ligand enantiomer (Fig. S5b, ESI†). In the region of the ligand or charge-transfer transitions below 450 nm it is evident that the sign of Δε reverses with the wrong metal chirality for the given ligand enantiomer (Fig. 8 and Fig. S5b, ESI†). These results are in parallel to analogous bis[{(R or S)-N-(Ar)ethyl-2-oxo-1-naphthaldiminato-κ2N,O}]Cu(II) (Scheme 3b)5 both at solid-state and in solution. In the contrary, the preferred formation of Δ-Cu-R-L or Λ-Cu-S-L was reported (both at solid-state and in solution) in the analogous bis[(R or S)-N-1-(Ar)ethyl-salicylaldiminato-κ2N,O]Cu(II) (Scheme 3c)3a and bis [N-(R)-1-(naphthyl)ethyl-3,5-dichlorosalicydenaminato]Cu(II) (Scheme 3e).2g Di-halogen substituents on the salicylal-ring favor Λ-Cu-R-L or Δ-Cu-S-L in bis[(S or R)-N-1-(phenyl)ethyl-(2,4-di-halogen-salicylaldiminato-κ2N,O)]Cu(II) (Scheme 3d)15 at solid-state, which show helicity inversion to Δ-Cu-R-L or Λ-Cu-S-L in solution. The structural differences to the present complexes in Scheme 3a are (i) imino-hydrogen atom (Scheme 3b–e) instead of phenyl group, (ii) naphthyl ring (Scheme 3b) instead of salicylal ring, (iii) di-halogen substituents (Scheme 3d and 3e) on the salicylal ring, (iv) di-halogen substituents on the salicylal ring plus naphthyl group attached to the chiral carbon center (Scheme 3e). These results strongly suggest that an understanding and design of the ligand substituents can efficiently control the diastereoselection and chirality induction at-metal (i.e., Δ vs. Λ).
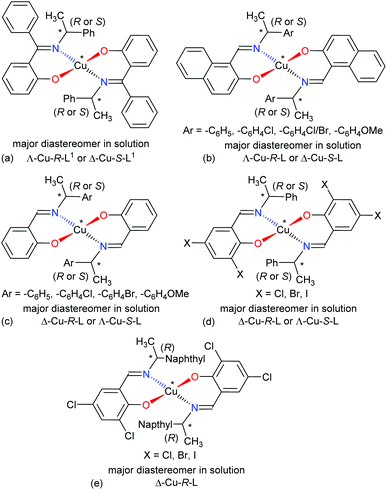 |
| | Scheme 3 Structural comparisons of Cu(II)-chiral Schiff bases complexes and their preferred diastereomer in solution: (a) this work, (b) ref. 5, (c) ref. 3a, (d) ref. 15, (e) ref. 2g. | |
Cyclic voltammetry (CV)
The cyclic voltammograms for Cu-R-L1 were run at −1.00 to +0.80 V (vs. Ag/AgCl) at varying scan rates (i.e., 0.10–0.30 V s−1) in acetonitrile at 25 °C (Fig. 10). The reductive scans exhibit a strong broad cathodic peak centered at ca. −0.60 V at a scan rate of 0.10 V s−1, which becomes more populated and is split into two strong peaks at ca. −0.43 V (Ec1) and −0.67 V (Ec2) with faster scan rates. These peaks are referred to two-electron charge-transfer processes for the [Cu-R-L1]0/[Cu-R-L1]− (Cu2+/Cu+) and [Cu-R-L1]−/[Cu-R-L1]2− (Cu+/Cu0) couples (L1 = deprotonated Schiff base ligand), respectively. The oxidative scans reveal two corresponding weak anodic peaks at ca. −0.25 V (Ea2) and +0.55 V (Ea1) for the [Cu-R-L1]2−/[Cu-R-L1]− and [Cu-R-L1]−/[Cu-R-L1]0 couples, respectively. The relatively weak anodic peaks on the oxidative waves reflect instability of the oxidized species, which undergo chemical transformation and/or decomposition reaction. Indeed, a CV pattern run at the extended range from −1.00 to +1.20 V with a scan rate of 0.10 V s−1 shows a sharp anodic peak at ca. 0.97 V (EaTBAP) for the electrolyte (Fig. S10, ESI†).8b The CV results thus unequivocally demonstrate two quasi-reversible one-electron charge-transfer processes for the Cu2+/Cu+ and Cu+/Cu0 couples in acetonitrile.5,8b,24–27 Analyses of voltammograms at variable scan rates exhibit that the cathodic and anodic peaks become increasingly populated and shift to lower and higher potentials, respectively with faster scan rates (Fig. 10). Indeed, a plot of cathodic peak current versus square root of scan rates (i.e., Ic2 vs. ν1/2) reflects a linear relationship (Fig. S11, ESI†), suggesting a diffusion-controlled electrochemical process in solution.8b,24–26
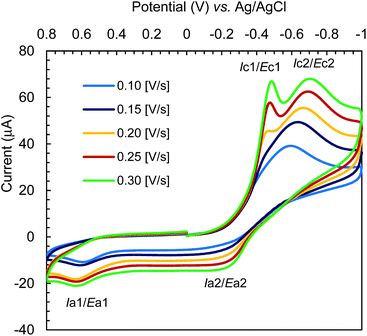 |
| | Fig. 10 Cyclic voltammograms for Cu-R-L1 (0.5 mM); TBAP (0.1 M) at varying scan rates 0.10–0.30 [V s−1] in acetonitrile at 25 °C. | |
Experimental
Materials and methods
IR spectra were recorded on a Nicolet iS10 spectrometer as KBr discs at ambient temperature. UV-Vis spectra were recorded with a Shimadzu UV 1800 spectrophotometer in methanol at 25 °C. ECD spectra were obtained with a JASCO Spectropolarimeter (J715) in chloroform at 20 °C. Elemental analyses were run on a Vario EL instrument from Elementaranalysensysteme. 1H NMR spectra were measured on a Bruker Avance DPX 400 spectrometer at 20 °C with calibration against the residual protonated solvent signal of CDCl3 (δ = 7.26 ppm). Electron impact (EI) mass spectra were recorded with a Thermo-Finnigan TSQ 700. Isotopic distribution patterns for the 63/65Cu containing ions were clearly visible in the spectra. An Epsilon™ instrument (BASi) electrochemical analyser was used for running cyclic voltammograms with tetra-N-butyl-ammonium hexafluorophosphate (TBAP) as supporting electrolyte in acetonitrile at 25 °C. A three-electrode measurement system was used, composed of a platinum disc working electrode, a platinum wire auxiliary electrode and an Ag/AgCl reference electrode, respectively. The solution containing the sample and TBAP was deoxygenated with purging nitrogen gas for about 10 minutes prior to use. The working electrode was cleaned with the supplied solution and dried prior to each scan to avoid any contamination due to deposition of the metal. Reproducibility of the CV patterns was checked at a scan rate of 0.10 V s−1 in acetonitrile.
General procedure to synthesize the Schiff base ligands (R- or S-HL)
Hydroxy-benzophenone (HL′, 12.07 mmol) was dissolved in 20 mL of methanol and 3–4 drops of concentrated H2SO4 were added into the solution which was then stirred for ca. 10 min. An equimolar amount of enantiopure (R or S)-(phenyl)ethylamine (dissolved in 5 mL of methanol) was added into this solution slowly. The reaction mixture was then refluxed for 5–6 h and the colour changed to yellow-orange. Thin-layer chromatography (TLC) was run to monitor the progress of the reaction. After completion of the reaction, the solvent was evaporated to ca. 50% in vacuo and this solution was left standing for crystallization. After 3–4 d, microcrystals were formed, filtered off and washed two times with methanol (1 mL) followed by n-hexane (1 mL). The microcrystals were dried in air for 3–4 days and the obtained solid yellow-orange products were analyzed as 2-{(E)-((S)-1-(phenyl)ethylimino)(phenyl)methyl}phenol (R- or S-HL1). The same procedure was followed for the synthesis of 2-{(E)-((S)-1-(p-methoxyphenyl)ethylimino)(phenyl)methyl}phenol (R- or S-HL2) using (R or S)-(p-methoxyphenyl)ethylamine. Bright-yellow single crystals of R-HL1 in X-ray quality were obtained via recrystallization or slow evaporation from concentrated methanol solution.
2-{(E)-((S)-1-(Phenyl)ethylimino)(phenyl)methyl}phenol (S-HL1).
Yield: 2.10 g (73%, based on HL′). – IR (KBr, cm−1): ν = 3065, 3034, 2979, 2929w (C–H), 1604vs, 1598sh (CN) and 1572vs (CC). – 1H NMR (400 MHz, CDCl3): δ = 1.57 (d, JHH = 6.8 Hz, 3H, CH3), 4.55 (q, JHH = 6.8 Hz, 1H, CH), 6.67 (t, JHH = 7.4 Hz, 1H, HAr), 6.79 (dd, JHH = 8.0, 1.6 Hz, 1H, HAr), 7.11 (d, JHH = 8.0 Hz, 1H, HAr), 7.18 (m, 1H, HAr), 7.25–7.28 (m, 4H, HAr), 7.30–7.36 (m, 3H, HAr) and 7.53–7.56 (m, 3H, HAr).
2-{(E)-((R)-1-(Phenyl)ethylimino)(phenyl)methyl}phenol (R-HL1).
Yield: 2.04 g (71%, based on HL′). – IR (KBr, cm−1): ν = 3061, 3030, 2974, 2922w (C–H), 1605vs, 1595sh (CN) and 1570vs (CC). – 1H NMR (400 MHz, CDCl3): δ = 1.56 (d, JHH = 6.8 Hz, 3H, CH3), 4.54 (q, JHH = 6.8 Hz, 1H, CH), 6.67 (t, JHH = 7.4 Hz, 1H, HAr), 6.79 (dd, JHH = 8.0, 1.6 Hz, 1H, HAr), 7.08 (d, JHH = 8.4 Hz, 1H, HAr), 7.18 (m, 1H, HAr), 7.26–7.28 (m, 4H, HAr), 7.29–7.36 (m, 3H, HAr) and 7.53–7.55 (m, 3H, HAr).
2-{(E)-((S)-1-(p-Methoxyphenyl)ethylimino)(phenyl)methyl}phenol (S-HL2).
Yield: 2.50 g (77%, based on HL′). – IR (KBr, cm−1): ν = 3057, 3030, 2995, 2928w (C–H), 1604 (CN) and 1570vs (CC). – 1H NMR (400 MHz, CDCl3): δ = 1.54 (d, JHH = 6.8 Hz, 3H, CH3), 3.82 (s, 3H, OCH3), 4.51 (q, JHH = 6.6 Hz, 1H, CH), 6.66 (dt, JHH = 8.0, 0.8 Hz, 1H, HAr), 6.80 (dd, JHH = 8.0, 1.6 Hz, 1H, HAr), 6.88 (d, JHH = 8.4 Hz, 2H, HAr), 7.04 (d, JHH = 8.4 Hz, 2H, HAr), 7.20 (d, JHH = 8.4 Hz, 2H, HAr), 7.30 (dt, JHH = 8.4, 1.6 Hz, 2H, HAr) and 7.53–7.55 (m, 3H, HAr).
2-{(E)-((R)-1-(p-Methoxyphenyl)ethylimino)(phenyl)methyl}phenol (R-HL2).
Yield: 2.40 g (74%, based on HL′). – IR (KBr, cm−1): ν = 3057, 3032, 2997, 2930w (C–H), 1605 (CN) and 1568vs (CC). – 1H NMR (400 MHz, CDCl3): δ = 1.53 (d, JHH = 6.8 Hz, 3H, CH3), 3.82 (s, 3H, OCH3), 4.51 (q, JHH = 6.6 Hz, 1H, CH), 6.66 (dt, JHH = 8.0, 0.8 Hz, 1H, HAr), 6.79 (dd, JHH = 8.0, 1.6 Hz, 1H, HAr), 6.88 (d, JHH = 8.8 Hz, 2H, HAr), 7.02 (d, JHH = 8.4 Hz, 1H, HAr), 7.04 (d, JHH = 9.6 Hz, 1H, HAr), 7.19 (d, JHH = 8.8 Hz, 2H, HAr), 7.27–7.32 (m, 2H, HAr) and 7.46–7.57 (m, 3H, HAr).
General procedure to synthesize the complexes (Cu-S- or -R-L)
The enantiopure Schiff base ligand, 2-{(E)-((R or S)-1-(phenyl)ethylimino)(phenyl)methyl}phenol (R- or S-HL1) (239.3 mg, 1 mmol) was dissolved in 10 mL of methanol, which was then poured into 5 mL of a methanolic solution of Cu(II) acetate (99.8 mg, 0.5 mmol). The reaction mixture was refluxed for 6–7 hours and a green product precipitated, which was then cooled at room temperature. The green product was separated by filtration and washed with methanol three times (1 mL each). The product was dried in air for 2–3 days to give deep-green microcrystals of bis[(R or S)-2-((E)-1-(1-(phenyl)ethylimino)ethyl)phenolato-κ2N,O]-Λ/Δ-copper(II) (Λ/Δ-Cu-S- or -R-L1). The same procedure was followed to synthesize bis[(R or S)-2-((E)-1-(1-(p-methoxyphenyl)ethylimino)ethyl)phenolato-κ2N,O]-Λ/Δ-copper(II) (Λ/Δ-Cu-S- or -R-L2) using the enantiopure Schiff base ligands of R- or S-HL2.
Bis[(R or S)-2-((E)-1-(1-(phenyl)ethylimino)ethyl)phenolato-κ2N,O]-Λ/Δ-copper(II), (Λ/Δ-Cu-R- or S-L1).
Yield 225 mg (68%). – IR (KBr, cm−1): ν = 3059, 3028, 2972, 2926w (C–H), 1607vs (CN) and 1570 (CC).
Bis[(R or S)-2-((E)-1-(1-(p-methoxyphenyl)ethylimino)ethyl)phenolato-κ2N,O]-Λ/Δ-copper(II), (Λ/Δ-Cu-R- or S-L2).
Yield 256 mg (71%). – IR (KBr, cm−1): ν = 3152, 2986, 2936w (C–H), 1616sh, 1605 (CN) and 1570 vs (CC).
Crystallization of Cu-S- or -R-L1 to yield CuL′2 and CuL′′2
Deep-green block-shaped single crystals in X-ray quality were obtained via slow diffusion of methanol into a concentrated solution of Cu-S- or -R-L1 in dichloromethane after 3–4 days. Single-crystal X-ray results show the formation of bis[2-oxo-benzophenonato-κ2O,O′]-copper(II), (CuL′2) as product from the crystallization experiment with about equal admixture of bis[2-(imino(phenyl)methyl)phenolato-κ2N,O]copper(II), (CuL′′2) (Scheme 2) via in situ hydrolysis of the coordinated Schiff bases back to 2-hydroxy-benzophenone (HL′) and to 2-(imino(phenyl)methyl)phenol (HL′′), which in-turn bind with the copper(II) ion to form the mixture of complexes during crystallization. However, several attempts were made to grow suitable single crystals of Cu-S- or R-L1 in X-ray quality, but failed due to the in situ hydrolysis of the ligands in the complexes.
Mixed product of bis[2-oxo-benzophenonato-κ2O,O′]-copper(II), (CuL′2) and bis[2-(imino(phenyl)methyl)phenolate-κ2N,O]copper(II), (CuL′′2): – IR (KBr, cm−1): ν = 3325 (N–H), 3056, 3013, 2974w (C–H), 1601/1579vs (CN/O) and 1559s (CC). – IR (calculated for CuL′2, cm−1): ν = 3227, 3208, 3182 (C–H), 1670, 1639 (CC), 1605 (CO) and 1571 (CC). – IR (calculated for CuL′′2, cm−1): ν = 3519, 3517 (N–H), 3222, 3210, 3178 (C–H), 1667, 1640 (CC), 1621 (CN) and 1574 (CC). – EI-MS (at 100 °C): 301 (55) [S- or R-HL1]+, 197 (25) [HL′′]+, 196 (100) [HL′′-H]+ and 105 (60) [{C6H5CHO}-H]+. EI-MS (at 240 °C): 457 (11) [CuL′2 = Cu{(C6H4O)(C6H5)CO}2]+, 456 (10) [CuL′L′′ = Cu{(C6H4O)(C6H5)CO}{(C6H4O)(C6H5)CNH}]+, 455 (7) [CuL′′2 = Cu{(C6H4O)(C6H5)CNH}2]+, 260 (43) [CuL′]+, 258 (33) [CuL′′-H]+, 198 (60) [HL′]+, 197 (100) [HL′′]+, 121 (61) [{(C6H4OH)CHO}-H]+ and 105 (37) [{C6H5CHO}-H]+. Elemental analysis: calc. for CuL′2 (C26H18CuO4, M = 457.96 g mol−1) C 68.19, H 3.96; calc. for CuL′′2 (C26H20CuN2O2, M = 456.01 g mol−1) C 68.49, H 4.42, N 6.14: found C 68.44, H 4.48, N 2.63%.
X-ray crystallography
Suitable single-crystals of R-HL1 and from the crystallization product of Cu-R-L1, that is the mixture of CuL′2 and CuL′′2 were carefully selected under a polarizing microscope and mounted on a loop. Data collection: Kappa APEX2 Duo CCD diffractometer with a microfocus source and multi-layer mirror monochromator for Cu–Kα radiation (λ = 1.54178 Å) for R-HL1 and Mo-Kα radiation (λ = 0.71073 Å) for CuL′2/CuL′′2 at 140(2) K; ω-scans (Table 4). Data collection and cell refinement with APEX2,28 data reduction with SAINT (Bruker).29Structure analysis and refinement: The structures were solved by intrinsic phasing (SHELXT-2015),30 refinement was done by full-matrix least squares on F2 using the SHELXL-2017 program suite,30 empirical (multi-scan) absorption correction with SADABS (Bruker).31 All non-hydrogen atom positions in R-HL1 were refined with anisotropic temperature factors. In the metal complex the Cu atom sits on an inversion center, rendering the two ligands symmetry equivalent. When the metal complex was refined as bis[2-oxo-benzophenonato-κ2O,O′]-copper(II), (CuL′2) a large residual electron density of 0.52 e Å−3 was found in a hydrogen-bond distance to the keto oxygen atom. The IR spectrum showed a sharp band at 3325 cm−1 for ν(N–H) (Fig. S6c, ESI†) and the CHN elemental analysis gave a nitrogen content of 2.63 wt%. Subsequently, the keto O atom has been refined as a split position of O and NH using PART commands, with about equal occupancy (the occupancy was free to refine). This split position of NH then corresponded to the complex bis[2-(imino(phenyl)methyl)phenolato-κ2N,O]copper(II), (CuL′′2). This split N atom could only be refined isotropically, while the rest of the atoms have been refined anisotropically. The hydrogen atom of the NH group was found and refined with Uiso(H) = 1.5Ueq(N). The NH distance had to be restrained with DFIX.
Table 4 X-ray crystal data and structure refinement parameters for R-HL1 and CuL′2/CuL′′2
| Identification code |
R-HL1 |
CuL′2/CuL′′2 |
|
Largest difference peak and hole.
R
1 = [∑(||Fo| − |Fc||)/∑ |Fo|]; wR2 = [∑ [w(Fo2 − Fc2)2]/∑ [w(Fo2)2]]1/2.
Goodness-of-fit = [∑ [w(Fo2 − Fc2)2]/(n − p)]1/2.
Absolute structure parameter.34
|
| Empirical formula |
C21H19NO |
C26H18.88CuN0.88O3.12 |
|
M (g mol−1) |
301.37 |
457.08 |
| Crystal size (mm3) |
0.25 × 0.25 × 0.10 |
0.16 × 0.09 × 0.04 |
| Temperature (K) |
140 |
140 |
|
θ range (°) (completeness) |
4.4–67.6° (0.99) |
2.5–27.6° (0.999) |
|
h, k, l range |
h = −11 → 11 |
h = −24 → 22 |
|
k = −9 → 9 |
k = −7 → 7 |
|
l = −13 → 13 |
l = −25 → 25 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21 |
C2/c |
|
a (Å) |
9.9056(5) Å |
18.4742(15) Å |
|
b (Å) |
8.1390(4) Å |
5.8299(5) Å |
|
c (Å) |
10.8874(5) Å |
19.8803(17) Å |
|
β (°) |
111.182 (2)° |
113.803 (4)° |
|
V (Å3) |
818.46(7) Å3 |
1959.0(3) Å3 |
|
Z
|
2 |
4 |
|
D
calc (g cm−3) |
1.223 |
1.550 |
| Radiation source (Å) |
Cu Kα, λ = 1.54178 |
Mo Kα, λ = 0.71073 |
|
μ (mm−1) |
0.58 |
1.15 |
|
F (000) |
320 |
940 |
| Max./min. transmission |
1.000/0.893 |
1.000/0.908 |
| Reflect. Collected |
22837 |
10309 |
| Independent reflections (Rint) |
2955 (0.034) |
2278 |
| Data/restraints/parameters |
2955/1/212 |
2278/1/150 |
| Max./min. Δρa (e Å−3) |
0.10/−0.14 |
0.46/−0.31 |
|
R
1/wR2 [I > 2σ (I)]b |
0.0258/0.0658 |
0.0376/0.1039 |
|
R
1/wR2 (all data)b |
0.0261/0.0658 |
0.0463/0.1093 |
| Goodness-of-fit (GOF) on F2c |
1.04 |
1.05 |
| Flack parameterd |
0.04 (7) |
— |
Hydrogen atoms for aromatic CH, aliphatic CH and CH3 groups were positioned geometrically (C–H = 0.95 Å for aromatic CH, 1.00 Å for aliphatic CH and 0.98 Å for CH3,) and refined using a riding model (AFIX 43 for aromatic CH, AFIX 13 for aliphatic CH, AFIX 137 for CH3), with Uiso(H) = 1.2Ueq(CH) and Uiso(H) = 1.5Ueq(CH3). The protic hydrogen for the OH group in R-HL1 was positioned and refined freely with Uiso(H) = 1.5Ueq(O). Details of the X-ray structure determinations and refinements are provided in Table 4. Graphics were drawn with DIAMOND (Version 4.6.4).32 Computations on the supramolecular interactions were carried out with PLATON for Windows.33 The structural data for this paper has been deposited with the Cambridge Crystallographic Data Center (CCDC-numbers 2060528 for R-HL1, 2060529 for CuL′2/CuL′′2†).
Powder X-ray diffraction (PXRD)
Powder X-ray diffraction (PXRD) data for R-HL1 and Cu-R-L1 were collected on a GNR Explorer powder X-ray diffractometer, operating in Bragg–Brentano geometry with Cu target at 30 kV and 20 mA. A zero background sample holder was used to filter signals from the sample holder, while a Ni (nickel) filter was used to cut the beta (Kβ) radiation. Data collections were carried out at 298 K with a step size 0.02° and an integration time of 3 s per step over an angular range of 5–50° (2θ). All steps to determine the crystal structure for Cu-R-L1 were performed using the program Expo-2014.20a The background of the diffraction pattern was modelled by a shifted Chebyshev function (number of coefficients 15). The selected 20 peaks within 5–50° were indexed using the N-Treor09 module.20b The results suggested the possibility of three crystal systems with triclinic symmetry, among them the best one was chosen considering the figure of merit (M20), unit cell volume and peak positions. Density consideration indicated only one molecule of Cu-R-L1 in the unit cell. Analysis of the powder pattern by the FINDSPACE module of Expo-201420a suggested the space group either as P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with a non-chiral or P1 with a chiral molecule in the asymmetric unit. Hence, the structure solution calculations were performed with the space group P1. We used DFT optimized structures for both diastereomers of Λ-Cu-R-L1 and Δ-Cu-R-L1 as input files for structure solution calculations, respectively. The solution was carried out by global optimization models using simulated annealing technique.19b,21 A total of 14 structural variables were used for structure solution. The Cu atom was selected as the center of rotation and anti-bump restraints were applied to C, N and O atoms. Among 10 trial structures, the best one was chosen based on crystal packing and minimum difference between calculated and experimental patterns, which was used as the initial structural model for Rietveld refinement.22,23 In Rietveld refinement, standard restraints were applied only to the phenyl rings.21,22d,23b,c Hydrogen atoms bonded to the carbon atoms were placed automatically at calculated positions. The summary of crystal data and structure refinement parameters are listed in Table 5.
with a non-chiral or P1 with a chiral molecule in the asymmetric unit. Hence, the structure solution calculations were performed with the space group P1. We used DFT optimized structures for both diastereomers of Λ-Cu-R-L1 and Δ-Cu-R-L1 as input files for structure solution calculations, respectively. The solution was carried out by global optimization models using simulated annealing technique.19b,21 A total of 14 structural variables were used for structure solution. The Cu atom was selected as the center of rotation and anti-bump restraints were applied to C, N and O atoms. Among 10 trial structures, the best one was chosen based on crystal packing and minimum difference between calculated and experimental patterns, which was used as the initial structural model for Rietveld refinement.22,23 In Rietveld refinement, standard restraints were applied only to the phenyl rings.21,22d,23b,c Hydrogen atoms bonded to the carbon atoms were placed automatically at calculated positions. The summary of crystal data and structure refinement parameters are listed in Table 5.
Table 5 PXRD data and structure refinement parameters for R-HL1, Λ-Cu-R-L1 and Δ-Cu-R-L1
| Compound |
R-HL1 |
Λ-Cu-R-L1 |
Δ-Cu-R-L1 |
| Empirical formula |
C21H19NO |
C42H36CuN2O2 |
|
M (g mol−1) |
301.4 |
664.3 |
| Temperature T (K) |
298 |
| Crystal system |
Monoclinic |
Triclinic |
| Radiation source (Å) |
Cu Kα, 1.54056 |
| Space group |
P21 |
P1 |
|
a (Å) |
10.964 |
10.126 |
10.119 |
|
b (Å) |
8.306 |
9.382 |
9.374 |
|
c (Å) |
9.997 |
9.346 |
9.342 |
|
α (°) |
90.00 |
91.58 |
91.39 |
|
β (°) |
111.45 |
114.45 |
114.42 |
|
γ (°) |
90.00 |
83.31 |
83.30 |
| Volume (Å3) |
847.28 |
802.48 |
801.07 |
|
D
calc (g cm−3) |
1.177 |
1.377 |
|
Z
|
2 |
1 |
| 2θ range (°) |
5.00–50.00 |
| Step size (2θ/°) |
0.02 |
| Counting time (s) |
3 |
| No. of counts |
4502 |
| No. of reflections |
171 |
274 |
| No. of background points |
20 |
|
R
p
|
4.568 |
6.521 |
6.881 |
|
R
wp/
R
exp
|
6.354/2.363 |
10.637/2.312 |
10.992/2.315 |
|
χ
2
|
7.23 |
21.177 |
22.547 |
| Goodness-of-fit |
2.689 |
4.601 |
4.748 |
Computational method
A thorough computational procedure was performed with Gaussian 0935 to rationalize the experimental results and thereby to gain insight into diastereoselection and Λ vs. Δ-chirality induction at-metal. The metal(II)-chiral N,O-chelate Schiff base complexes in tetrahedral or distorted-tetrahedral geometry demonstrate chirality induction at the metal centre (Λ-Cu and Δ-Cu), and hence, provide two diastereomers of opposite configuration along with C2-symmetry of the molecule.3–8,15 Thus, we optimized the diastereomeric pairs Λ-Cu-R-L1/Δ-Cu-R-L1 and Λ-Cu-S-L1/Δ-Cu-S-L1 at the b3lyp/6-31 g(d) level (Fig. S1, ESI†). The gas-phase initial geometries for these diastereomeric pairs were generated from X-ray structures of the analogous bis[(R or S)-N-1-(phenyl)ethyl-salicylaldiminato-κ2N,O]-Λ or Δ-Cu(II)3a by replacing the imino-hydrogen atom with the phenyl group. It was attempted to optimize the hydrolysed products from crystallization of Cu-R-L1 such as bis[2-oxo-benzophenonato-κ2O,O′]-copper(II), (CuL′2) and bis[2-(imino(phenyl)methyl)phenolato-κ2N,O]copper(II) (CuL′′2) at the b3lyp/6-31 g(d) level (Fig. S2, ESI†), respectively. The gas-phase initial geometries for these complexes were generated from their X-ray structures. The excited state properties (e.g., UV-Vis and ECD spectra) for the diastereomeric pairs (Λ-Cu-R-L1/Δ-Cu-R-L1 and Λ-Cu-S-L1/Δ-Cu-S-L1) and for CuL′2/CuL′′2 were calculated by TDDFT at the m06/sdd level (Fig. 7, 8 and Fig. S3, ESI†).5,15 The polarization continuum model (PCM) using chloroform as solvent was employed and 72 excited states (roots) were considered for calculations (Table S1, ESI†). Indeed, excited state properties were calculated at different combinations of the functionals and the basis sets such as b3lyp/sdd, b3lyp/svp and m06/tzvp, respectively, for Λ-Cu-R-L1 (Fig. S4 and S5a, ESI†). The results are almost identical spectra with little shifting of band maxima, which strongly support the reliability and validity of the methods used. However, the best fit to experimental spectra was found at the m06/sdd//b3lyp/6-31 g(d) level (Fig. 7 and 8). In addition, assignments on electronic spectra were performed at the same level of theory for Λ-Cu-R-L1 (Table 5). The spectra were generated using the program SpecDis36 by applying Gaussian band shape with exponential half-width σ = 0.33 eV.
Conclusions
The pseudotetrahedral complex bis[(R or S)-2-((E)-1-(1-(Ar)ethylimino)ethyl)phenolato-κ2N,O]-Λ/Δ-copper(II), (Λ/Δ-Cu-S- or R-L) was investigated as a potential candidate to exhibit induced chirality at-metal, that is formation of the Δ-Cu-S or Λ-Cu-R diastereomer for S- or R-HL ligands. Formation of the intact complexes is confirmed by decomplexation reaction with NaCN, which provides the free Schiff base ligands in DMSO-d6 solution. PXRD structure determinations for Cu-R-L1 showed two phenolate-oxygen and two imine-nitrogen atoms from two Schiff base ligands to coordinate to copper(II) in a pseudotetrahedral geometry with Λ/Δ-chirality induction at-metal. Yet, PXRD structure determination did not allow to determine the absolute configuration unlike single-crystal X-ray diffractometry. Optimized gas-phase structures by DFT indicate the Δ-Cu-S-L or Λ-Cu-R-L diastereomer as slightly more stable. In the absence of single crystals for any of the Cu-S-L or Cu-R-L complexes and also to assess the diastereoselectivity in solution, we had to resort to electronic circular dichroism (ECD) in combination with theoretical calculations. Experimental solution ECD spectra show an expected mirror-image relationship and upon comparison with calculated ECD spectra suggest the preferred formation of the Δ-Cu-S-L or Λ-Cu-R-L diastereomer (i.e., diastereomeric excess) in solution. The present study adds a case to the understanding and design of the ligand substituents for controlling the diastereoselection and chirality induction at-metal (i.e., Δ vs. Λ) in pseudotetrahedral Cu-Schiff-base complexes. Crystallization of Cu-S-L1 or Cu-R-L1 in MeOH/DCM failed due to in situ hydrolysis of the coordinated Schiff base ligands, as evidenced by X-ray structures determinations which yielded only Cu complexes of the hydrolysis products as bis[2-oxo-benzophenonato-κ2O,O′]-copper(II) and bis[2-(imino(phenyl)methyl)phenolato-κ2N,O]copper(II).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the financial support from the Alexander von Humboldt Foundation (AvH), Bonn, Germany under the project “Research Group Linkage Program”. We recognize the “Wazed Miah Science Research Centre” at Jahangirnagar University, Dhaka, Bangladesh for obtaining CV and PXRD data. Our sincere thanks to Professor Walid Houry and Vaibhav Bhandari, Department of Biochemistry, Faculty of Medicine, University of Toronto, for running ECD spectra. We thank Professor A. B. P. Lever, Department of Chemistry, York University, Toronto and the computecanada.ca (https://ccdb.computecanada.ca/) Ontario, for using computational resources.
Notes and references
-
(a)
A. von Zelewsky, Stereochemistry of Coordination Compounds, John Wiley & Sons, Chichester, 1996, p. 69 Search PubMed;
(b)
H. Amouri and M. Gruselle, Chirality in Transition Metal Chemistry: Molecules, supramolecular assemblies and materials, John Wiley & Sons, Chichester, 2008, p. 40 Search PubMed;
(c) E. C. Constable, Chem. Soc. Rev., 2013, 42, 1637 Search PubMed;
(d) H. Song, M. Postings, P. Scott and N. J. Rogers, Chem. Sci., 2021, 12, 1620 Search PubMed;
(e) K. A. Jensen, Inorg. Chem., 1970, 9, 1 Search PubMed;
(f) U. Knof and A. von Zelewsky, Angew. Chem., Int. Ed., 1999, 38, 302 Search PubMed;
(g) H. Brunner, M. Niemetz and M. Zabel, Z. Naturforsch., 2000, 55b, 145 Search PubMed;
(h) C. Evans and D. Luneau, J. Chem. Soc., Dalton Trans., 2002, 83 Search PubMed;
(i) G. Brewer, C. Brewer, R. J. Butcher, G. T. Robichaux and C. Viragh, Inorg. Chim. Acta, 2014, 410, 171 Search PubMed;
(j) R. E. Ernst, M. J. O'Connor and R. H. Holm, J. Am. Chem. Soc., 1967, 89, 6104 Search PubMed;
(k) E. C. Constable, G. Zhang, C. E. Housecroft, M. Neuburger and J. A. Zampese, Chem. Commun., 2010, 46, 3077 Search PubMed;
(l) F. Wang, H. Zhang, L. Li, H.-Q. Hao, X.-Y. Wang and J.-G. Chen, Tetrahedron: Asymmetry, 2006, 17, 2059 Search PubMed.
-
(a) H. Sakiyama, H. Okawa, N. Matsumoto and S. Kida, Bull. Chem. Soc. Jpn., 1991, 64, 2644 Search PubMed;
(b) H. Sakiyama, H. Okawa, N. Matsumoto and S. Kida, J. Chem. Soc., Dalton Trans., 1990, 2935 Search PubMed;
(c) Z. Dezhahang, M. R. Poopari, J. Cheramy and Y. J. Xu, Inorg. Chem., 2015, 54, 4539 Search PubMed;
(d) H. Ōkawa, M. Nakamura and S. Kida, Inorg. Chim. Acta, 1986, 120, 185 Search PubMed;
(e) T. Akitsu and Y. Einaga, Polyhedron, 2005, 24, 2933 Search PubMed;
(f) T. Akitsu and Y. Einaga, Polyhedron, 2005, 24, 1869 Search PubMed;
(g) T. Akitsu, Polyhedron, 2007, 26, 2527 Search PubMed;
(h) T. Ohno, S. Chorazy, K. Imoto and S. Ohkoshi, Cryst. Growth Des., 2016, 16, 4119 Search PubMed.
-
(a) A.-C. Chamayou, G. Makhloufi, L. A. Nafie, C. Janiak and S. Lüdeke, Inorg. Chem., 2015, 54, 2193 Search PubMed;
(b) A.-C. Chamayou, S. Lüdeke, V. Brecht, T. B. Freedman, L. A. Nafie and C. Janiak, Inorg. Chem., 2011, 50, 11363 Search PubMed.
- M. Enamullah, G. Makhloufi, R. Ahmed, B. Alif Joy, M. A. Islam, D. Padula, H. Hunter, G. Pescitelli and C. Janiak, Inorg. Chem., 2016, 55, 6449 Search PubMed.
- M. Enamullah, A. K. M. Royhan Uddin, G. Pescitelli, R. Berardozzi, G. Makhloufi, V. Vasylyeva, A.-C. Chamayou and C. Janiak, Dalton Trans., 2014, 43, 3313 Search PubMed.
- M. Enamullah, M. A. Quddus, M. R. Hasan, G. Pescitelli, R. Berardozzi, G. Makhloufi, V. Vasylyeva and C. Janiak, Dalton Trans., 2016, 45, 667 Search PubMed.
-
(a) G. Pescitelli, S. Lüdeke, A.-C. Chamayou, M. Marolt, V. Justus, M. Górecki, L. Arrico, L. Di Bari, M. A. Islam, I. Gruber, M. Enamullah and C. Janiak, Inorg. Chem., 2018, 57, 13397 Search PubMed;
(b) M. Enamullah, M. A. Islam, B. A. Joy, B. Dittrich, G. J. Reiß and C. Janiak, Inorg. Chim. Acta, 2018, 482, 935 Search PubMed.
-
(a) M. Enamullah, V. Vasylyeva and C. Janiak, Inorg. Chim. Acta, 2013, 408, 109 Search PubMed;
(b) M. Enamullah, M. A. Quddus, M. R. Hasan, G. Pescitelli, R. Berardozzi, G. J. Reiß and C. Janiak, Eur. J. Inorg. Chem., 2015, 2758 Search PubMed.
-
(a) S. E. Howson and P. Scott, Dalton Trans., 2011, 40, 4332 Search PubMed;
(b) J. M. Becker, J. Barker, G. J. Clarkson, R. van Gorkum, G. K. Johal, R. I. Walton and P. Scott, Dalton Trans., 2010, 39, 2309 Search PubMed;
(c) K. S. Min, A. H. Park, J. W. Shin, S. R. Rowthu, S. K. Kim and J. J. Ryoo, Dalton Trans., 2010, 39, 8741 Search PubMed.
-
(a) O. Mamula, A. von Zelewsky, T. Bark, H. Stoeckli-Evans, A. Neels and G. Bernardinelli, Chem. Eur. J., 2000, 6(19), 3575 Search PubMed;
(b) R. Dreos, G. Nardin, L. Randaccio, P. Siega and G. Tauzher, Inorg. Chem., 2004, 43, 3433 Search PubMed;
(c) H. Dong, H. Hu, Y. Liu, J. Zhong, G. Zhang, F. Zhao, X. Sun, Y. Li and Z. Kang, Inorg. Chem., 2014, 53, 3434 Search PubMed;
(d) W. Huang and T. Ogawa, Polyhedron, 2006, 25, 1379 Search PubMed;
(e) T.-Y. Li, Y.-X. Zheng and Y.-H. Zhou, Dalton Trans., 2016, 45, 19234 Search PubMed.
-
(a) I. Katsuki, Y. Motoda, Y. Sunatsuki, N. Matsumoto, T. Nakashima and M. Kojima, J. Am. Chem. Soc., 2002, 124, 629 Search PubMed;
(b) S. Nagasato, I. Katsuki, Y. Motoda, Y. Sunatsuki, N. Matsumoto and M. Kojima, Inorg. Chem., 2001, 40, 2534 Search PubMed;
(c) D.-H. Ren, X.-L. Sun, L. Gu, D. Qiu, Z. Li and Z.-G. Gu, Inorg. Chem. Commun., 2015, 51, 50 Search PubMed.
-
(a) T. Akitsu and Y. Einaga, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, m640 Search PubMed;
(b) C. T. Brewer, G. Brewer, R. J. Butcher, E. E. Carpenter, A. Schmiedekamp and C. Viragh, Dalton Trans., 2007, 295 Search PubMed;
(c) C. T. Brewer, G. Brewer, R. J. Butcher, E. E. Carpenter, A. M. Schmidekamp, C. Schmiedekamp, A. Straka, C. Viragh, Y. Yuzefpolskiy and P. Zavalij, Dalton Trans., 2011, 40, 181 Search PubMed;
(d) B. R. Groves, D. I. Arbuckle, E. Essoun, T. L. Lundrigan, R. Wang and M. A. S. Aquino, Inorg. Chem., 2013, 52, 11563 Search PubMed;
(e) R. Vadavi, E. D. Conrad, D. I. Arbuckle, T. S. Cameron, E. Essoun and M. A. S. Aquino, Inorg. Chem., 2011, 50, 11862 Search PubMed.
-
(a) C. Merten, R. McDonald and Y. Xu, Inorg. Chem., 2014, 53, 3177 Search PubMed;
(b) M. Albrecht, E. Isaak, M. Baumert, V. Gossen, G. Raabe and R. Fröhlich, Angew. Chem., Int. Ed., 2011, 50, 2850 Search PubMed;
(c) C. M. álvarez, R. Carrillo, R. García-Rodríguez and D. Miguel, Chem. Commun., 2011, 47, 12765 Search PubMed.
-
(a) J. Gregoliński, M. Hikita, T. Sakamoto, H. Sugimoto, H. Tsukube and H. Miyake, Inorg. Chem., 2016, 55, 633 Search PubMed;
(b) S. Zahn and J. W. Canary, Science, 2000, 288, 1404 Search PubMed.
- N. Kordestani, H. A. Rudbari, G. Bruno, S. Rosario, J. D. Braun, D. E. Herbert, O. Blacque, I. Correia, M. Al-M. Zaman, M. Mamun Bindu, C. Janiak and M. Enamullah, Dalton Trans., 2020, 49, 8247 Search PubMed.
-
(a) M. Enamullah, A. K. M. Royhan Uddin, A.-C. Chamayou and C. Janiak, Z. Naturforsch., B: Chem. Sci., 2007, 62, 807 Search PubMed;
(b) M. Enamullah, A. K. M. Royhan Uddin, G. Hogarth and C. Janiak, Inorg. Chim. Acta, 2012, 387, 173 Search PubMed.
-
(a) B. M. Drašković, G. A. Bogdanović, M. A. Neelakantan, A.-C. Chamayou, S. Thalamuthu, Y. S. Avadhut, J. Schmedt auf der Günne, S. Banerjee and C. Janiak, Cryst. Growth Des., 2010, 10, 1665 Search PubMed;
(b) K. Sarmah, G. Pandit, A. B. Das, B. Sarma and S. Pratihar, Cryst. Growth Des., 2017, 17, 368 Search PubMed.
-
(a) A. J. McKinnon, T. N. Waters and D. Hall, J. Chem. Soc., 1964, 3290 Search PubMed;
(b) D. Hall, A. J. McKinnon and T. N. Waters, J. Chem. Soc., 1965, 425 Search PubMed;
(c) J. A. Bevan, D. P. Graddon and J. F. McConnell, Nature, 1963, 199, 373 Search PubMed;
(d) G. Q. Zhang, G. Q. Yang and J. S. Ma, J. Chem. Res., 2006, 19 Search PubMed.
-
(a) M. Enamullah, A.-C. Chamayou, K. S. Banu, A.-C. Kautz and C. Janiak, Inorg. Chim. Acta, 2017, 464, 186 Search PubMed;
(b) M. Enamullah, B. Alif Joy and M. K. Islam, J. Mol. Struct., 2019, 1175, 56 Search PubMed.
-
(a) A. Altomare, C. Cuocci, C. Giacovazzo, A. Moliterni, R. Rizzi, N. Corriero and A. Falcicchio, J. Appl. Crystallogr., 2013, 46, 1231 Search PubMed;
(b) A. Altomare, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, R. Rizzi and P.-E. Werner, J. Appl. Crystallogr., 2000, 33, 1180 Search PubMed.
- L. T. Wille, Chem. Phys. Lett., 1987, 133, 405 Search PubMed.
-
(a) H. M. Rietveld, Acta Crystallogr., 1967, 22, 151 Search PubMed;
(b) B. M. Kariuki, D. M. S. Zin, M. Tremayne and K. D. M. Harris, Chem. Mater., 1996, 8, 565 Search PubMed;
(c) K. Užarević, V. Štrukil, C. Mottillo, P. A. Julien, A. Puškarić, T. Friščić and I. Halasz, Cryst. Growth Des., 2016, 16, 2342 Search PubMed;
(d) S. Mondal, M. Mukherjee, K. Dhara, S. Ghosh, J. Ratha, P. Banerjee and A. K. Mukherjee, Cryst. Growth Des., 2007, 7, 1716 Search PubMed.
-
(a) R. Rahmani, A. Djafri, A. Chouaih, A. Djafri, F. Hamzaoui, R. Rizzi and A. Altomare, J. Mol. Struct., 2017, 1143, 259 Search PubMed;
(b) A. E. Watts, K. Maruyoshi, C. E. Hughes, S. P. Brown and K. D. M. Harris, Cryst. Growth Des., 2016, 16, 1798 Search PubMed;
(c) T. Dey, P. Chatterjee, A. Bhattacharya, S. Pal and A. K. Mukherjee, Cryst. Growth Des., 2016, 16, 1442 Search PubMed.
- M. Enamullah, M. Al-M. Zaman, M. M. Bindu, M. K. Islam and M. A. Islam, J. Mol. Struct., 2020, 1201, 127207 Search PubMed.
- M. Enamullah, M. A. Islam, B. A. Joy and G. J. Reiss, Inorg. Chim. Acta, 2016, 453, 202 Search PubMed.
- M. Enamullah, M. A. Islam, A.-C. Kautz and C. Janiak, J. Coord. Chem., 2018, 71, 2557 Search PubMed.
- A. Zianna, G. Psomas, A. Hatzidimitriou and M. Lalia-Kantouri, RSC Adv., 2015, 5, 37495 Search PubMed.
-
APEX2, Data Collection Program for the CCD Area-Detector system, Version 2.1-0, Bruker Analytical X-ray Systems, Madison, Wisconsin, USA, 2006 Search PubMed.
-
SAINT, data reduction and frame integration program for the CCD area-detector system, Bruker Analytical X-ray Systems, Madison (WI), 2006 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 Search PubMed.
-
G. M. Sheldrick, Program SADABS, University of Göttingen: Göttingen, Germany, 1996 Search PubMed.
-
K. Brandenburg, Diamond (Version 4), Crystal and Molecular Structure Visualization, Crystal Impact, K. Brandenburg & H. Putz Gbr, Bonn, Germany, 2009–2020 Search PubMed.
-
(a) A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 Search PubMed;
(b)
A. L. Spek, PLATON – A multipurpose crystallographic tool, Utrecht University, Utrecht, The Netherlands, 2005 Search PubMed.
-
(a) H. D. Flack, M. Sadki, A. L. Thompson and D. J. Watkin, Acta Crystallogr., Sect. A: Found. Crystallogr., 2011, 67, 21 Search PubMed;
(b) H. D. Flack and G. Bernardinelli, Chirality, 2008, 20, 681 Search PubMed;
(c) H. D. Flack and G. Bernardinelli, Acta Crystallogr., Sect. A: Found. Crystallogr., 1999, 55, 908 Search PubMed;
(d) H. D. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 1983, 39, 876 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized structures, IR-spectra, simulated UV-Vis/CD spectra, PXRD data, CV data, cif/checkcif files and excited state properties. CCDC 2060528 for R-HL1 and 2060529 for CuL′2/CuL′′2. For ESI and crystallographic data in CIF or other electronic format, see DOI: 10.1039/d1dt01671g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Mohammad
Anwar Hossain
*a,
Mohammad
Anwar Hossain