
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosynthesis of alkyne-containing natural products
Xinyang
Li
a,
Jian-Ming
Lv
b,
Dan
Hu
 b and
Ikuro
Abe
*ac
b and
Ikuro
Abe
*ac
aGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: abei@mol.f.u-tokyo.ac.jp
bInstitute of Traditional Chinese Medicine and Natural Products, College of Pharmacy, Jinan University, Guangzhou 510632, People's Republic of China
cCollaborative Research Institute for Innovative Microbiology, The University of Tokyo, Yayoi 1-1-1, Bunkyo-ku, Tokyo 113-8657, Japan
First published on 21st December 2020
Abstract
Alkyne-containing natural products are important molecules that are widely distributed in microbes and plants. Inspired by the advantages of acetylenic products used in the fields of medicinal chemistry, organic synthesis and material science, great efforts have focused on discovering the biosynthetic enzymes and pathways for alkyne formation. Here, we summarize the biosyntheses of alkyne-containing natural products and introduce de novo biosynthetic strategies for alkyne-tagged compound production.
 Xinyang Li | Xinyang Li received the BSc degree (2012) from Shaanxi University of Chinese Medicine and MSc degree (2015) from Nanjing University of Chinese Medicine. In 2019, she received her PhD degree in natural product chemistry from Zhejiang University. Then, she joined in Prof. Ikuro Abe's group in the University of Tokyo as a postdoctoral research fellow. Her research interest is the isolation and biosynthetic study of fungi-derived natural products. |
 Jian-Ming Lv | Jian-Ming Lv received his BS degree (2007) from Tianjin University and Master's degree (2010) from Zhejiang University. In 2014, he joined Prof. Xin-Sheng Yao's group at the College of Pharmacy, Jinan University, and earned his PhD degree (2017) in Medicinal Chemistry. Afterwards, he worked as a postdoctoral fellow at the College of Pharmacy, Jinan University, with Prof. Hao Gao and Prof. Dan Hu. His research interest is the isolation and biosynthetic study of fungi-derived natural products. |
 Dan Hu | Dr Dan Hu received his PhD in 2010 in Integrated Biosciences from the University of Tokyo, Japan. He then did his postdoctoral research at the National Institute of Advanced Industrial Science and Technology (AIST). In 2013, he moved back to China and joined the college of pharmacy at Jinan University. In 2018, He was promoted to professor. His research interest has been focused on biosynthesis of fungi-derived biologically important natural products. |
 Ikuro Abe | Ikuro Abe received his BS (1984) and PhD (1989) from The University of Tokyo. After postdoctoral research with Professor Guy Ourisson, and mostly with Professor Michel Rohmer at the Ecole Nationale Supérieure de Chimie de Mulhouse (1989–1991), he worked with Professor Glenn D. Prestwich at the State University of New York at Stony Brook (1991–1996) and then at the University of Utah (1996–1998). In 1998, he moved back to Japan at the University of Shizuoka (1998–2009), and then was appointed as Professor of Natural Products Chemistry at the Graduate School of Pharmaceutical Sciences, The University of Tokyo (2009-current). His research interests mostly focus on exploring and engineering natural product biosynthesis. |
1. Introduction
The alkyne is an important chemical functional moiety found in numerous natural products widely distributed in terrestrial bacteria, fungi, insects, and plants.1 Marine organisms, such as microbes, algae and sponges also represent rich sources for producing acetylenic natural products.1 Many of them display promising biological activities, such as anti-tumor, anti-parasitic, anti-malarial, and anti-HIV activities.1 In addition, the alkyne moiety is a valuable synthetic building block in organic chemistry. Conjugated enynes, including the 1,3-enyne motif and its derivatives found in many bioactive molecules,2,3 are important intermediates in the syntheses of organic compounds and macromolecules.4–7 The terminal alkyne is particularly useful for the alkyne–azide cycloaddition (“click” reaction), which enables the installation of a covalent bond of two chemical fragments to form complex molecular scaffolds for applications in pharmaceutical, cell biology and material sciences research.8–10 The alkyne can be used as an important bio-orthogonal tag and chemically or metabolically installed into biologically and medicinally important molecules, such as proteins, lipids, nucleic acids, and glycans.11 This handle will enable the visualization and quantification of the alkyne-tagged molecules through bio-orthogonal chemistry. Additionally, the modes of action and cellular dynamics of these alkyne-tagged molecules can be investigated through click chemistry or directly by stimulated Raman scattering microscopy.12,13Given the importance of alkynes in both chemistry and biology, chemical synthesis and precursor directed biosynthesis have been used for alkyne-tagged compound production.14–17 Although total and semi-synthesis are powerful tools for chemical production, they are often restricted by the yield of target compounds and the availability of starting materials. For the precursor directed biosynthesis, a set of structural analogues can be obtained by feeding precursor variants to an organism; however, it is often challenging to incorporate the desired precursors into the biosynthetic pathway due to the substrate specificity of the enzymatic machinery. In addition, both the shunt transformation pathway and coexistence of precursors and target compounds pose challenges to selectively probe the target compounds in the system, thus impeding broader applications.18 Alternatively, de novo biosynthesis is another promising strategy for producing alkyne-tagged compounds, without feeding alkynoic precursors. For this purpose, the biosynthesis of alkyne formation should be thoroughly understood. The discovery of the biosynthetic machineries for alkyne moieties has facilitated some illuminating progress in recent years. Hitherto, only a few types of genes have been identified as being involved in alkyne biosynthesis. Especially, the discovery of fatty acid/acyl acetylenases, a unique group of desaturases that catalyze alkyne formation in acetylenic fatty acids, is regarded as the first well studied example of the alkyne-forming enzymatic machinery.19,20 Furthermore, studies on the discovery of enzymes that catalyze alkyne formation in β-ethynylserine and alkynylated meroterpenoids have expanded the enzyme inventory and offered a potential route for the engineered biosynthesis of alkyne-tagged compounds.21–23 These findings have increased our understanding of the enzymatic machineries participating in acetylenic natural product generation. Previously, Minto et al.24 and Haritos et al.25 summarized the biosynthesis of polyacetylenes and allied natural products. Herein, we update the recent research progress in the biosynthesis of alkyne-containing natural products and introduce de novo biosynthesis for in situ generating alkyne-tagged natural products.
2. Biosynthesis of acetylenic natural products
2.1 Fatty acids
Acetylenic fatty acids containing one or more C–C triple bonds are widespread metabolites in plants, fungi, marine sponges, and insects.26–28 Crepenynic acid (1), stearolic acid (2), and tariric acid (3) are general precursors to many acetylenic fatty acid-derived metabolites (Fig. 1A).29,30 Biosynthetic studies of crepenynic acid from Crepis alpina seed oil led to the characterization of the bifunctional enzyme Crep1, responsible for the double bond and subsequent acetylenic bond formation.31 The microsomal fraction from developing C. alpina seeds converted [14C]linoleic acid into [14C]crepenynic acid in the presence of NADH, clearly establishing that the Δ12 triple bond formation employs an acyl chain with a double bond at Δ12 position as a precursor (Fig. 1B). Furthermore, the in vivo heterologous expression of the crep1 gene in Saccharomyces cerevisiae (YN94-1 strain) afforded results consistent with those from the in vitro experiment. The results reflected the recognition of a special fatty acid desaturase (FAD), designated as an acetylenase, which is capable of catalyzing the conversion of olefinic bonds to acetylenic bonds and is distinct from enzymes that only convert single bonds to double bonds.28,31,32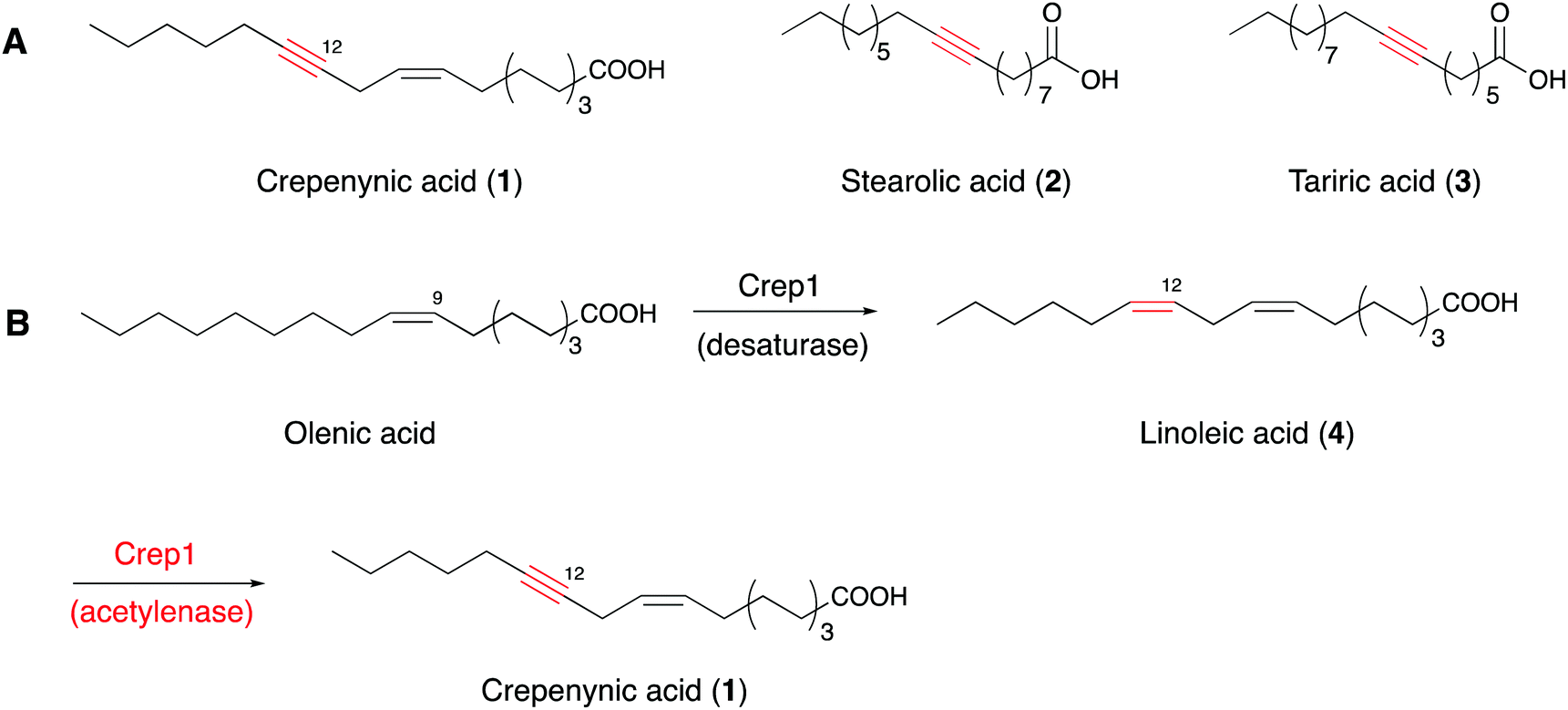 | ||
| Fig. 1 (A) Structures of 1–3. (B) Reactions catalyzed by the bifunctional Crep1. | ||
Although the exact triple bond-forming mechanism by the acetylenase remains unclear due to the lack of a membrane desaturase crystal structure, the desaturase-mediated formation of a double bond in a fatty acid is reportedly diiron-dependent,33–36 thus providing an important reference for understanding the triple C–C bond-forming reaction catalyzed by Crep1. Crep1 is composed of 375 amino acid residues and shares high sequence similarity to a widely distributed group of membrane-bound, non-heme diiron enzymes in living organisms.35 As shown in Fig. 2, the selected sequences include mouse stearoyl-CoA desaturases 1 and 3, and other fatty acid desaturases from mammals, yeast, Arabidopsis thaliana, etc. Among them, the crystal structure of mouse stearoyl-CoA desaturase 1 (SCD1) (protein data bank ID 4YMK), an integral membrane protein, clearly shows the dizinc-center with longer distance between the two metals which is different from previously solved structures of soluble di-iron enzymes.33,37 The other obvious difference is the absence of glutamate residue in SCD1 which serves as bridging ligand of the two iron ions in the reported soluble enzymes.33 As Zn2+ has an ionic radius of 0.88 Å, it may serve as a reasonable substitute for Fe2+ (0.92 Å) and didn’t alter the structure of SCD1.33 From the sequence alignment results (Fig. 2), the histidine motifs for di-iron center ligation in SCD1 are consistent with the reported desaturases. In addition to the two HxxHH and HxxxxH motifs (marked in red frames) that are also conserved in Crep1, there are two other potential histidine-rich motifs (HHTH and HVMHH, shadowed in red), indicating that Crep1 possesses a similar manner of metal chelation.
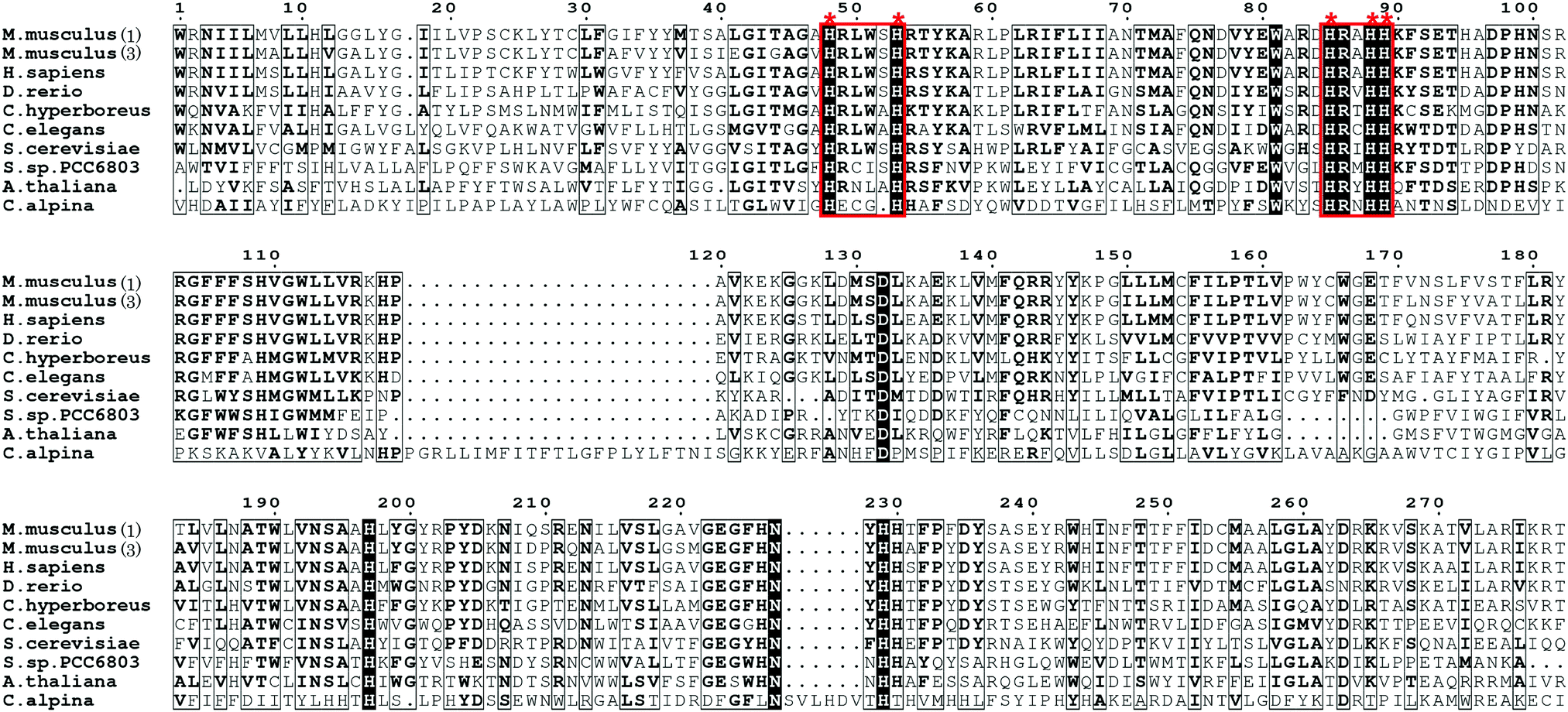 | ||
| Fig. 2 Partial sequence alignment of Crep1 with other integral membrane desaturases [The accession numbers for sequences included in the alignment are: mouse SCD1 (GI: 31543675), mouse SCD3 (GI: 13277368), human SCD1 (GI: 53759151), zebrafish SCD1 (GI: 28394115), D. melanogaster desat2 (GI: 24646295), C. hyperboreus ChDes9-1 (GI: 589834955), C. elegans FAT5 (GI: 544604099), delta-9 desaturase from Synechocystis sp. PCC 6803 (GI: 339274799), delta-9 desaturase from A. thaliana (GI: 18402641), yeast OLE1 (GI: 1322552), and Crep1 from C. alpine (Genbank accession No. CAA76158.2)]. (This alignment was generated by ESPript 3.0.40) | ||
SCD1 catalyzes the C9–C10 cis-double bond formation from steroyl-CoA to oleyl-CoA.33 This step requires oxygen, which is activated by the metal center. The electrons needed for the desaturation reaction are obtained from cytochrome b5, which in turn gained electrons from NAD(P)H via cytochrome b5 reductase.33 Fatty acid desaturation in plants also relies on cytochrome b5 reductase to transfer electrons.38 Crep1 may undergo two rounds of oxidation to catalyze the triple-bond formation in a saturated fatty acid. The kinetic isotope effect (KIE) study, using Crep1 expressed in yeast, supported a conventional two-step mechanism from alkene to alkyne.39 The large primary deuterium isotope effect (kH/kD = 14.6 ± 3.0) for C12–H bond cleavage, and the minimal isotope effect of the C13–H bond breaking step (kH/kD = 1.25 ± 0.08), suggested that crepenynic acid is produced via an initial H-atom abstraction at C12 of a linoleoyl substrate (Fig. 3).39
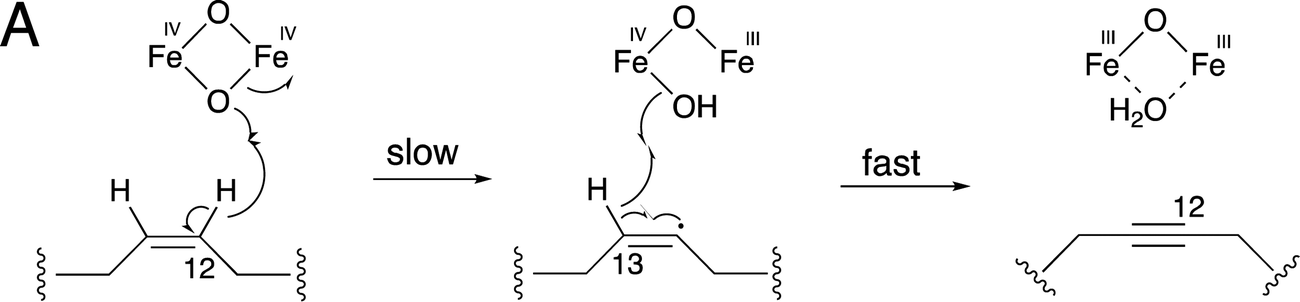 | ||
| Fig. 3 Proposed mechanism of non-heme diiron fatty acid desaturases that catalyze the oxidation from alkene to alkyne. | ||
Intriguingly, a homolog of Crep1 has been discovered in the biosynthetic gene cluster of falcarindiol (5),41 a dietary metabolite from edible crops such as carrots, tomatoes and celery, that exhibits antifungal activity and cytotoxicity against several human cancer cell lines. The conversion of linoleic acid (4) to crepenynic acid (5) by a Crep1-like acetylenase was proposed to be involved in an early catalytic step in the falcarindiol biosynthetic pathway in tomato (Solanum lycopersicum) (Fig. 4).41 As only a few amino acid changes distinguish acetylenases from other desaturases, it is difficult to find orthologs of Crep1 by a homology search of genome databases. Instead, researchers correlated the expression levels of 40 corresponding transcripts from their RNA sequencing datasets with the falcarindiol accumulation in tomato leaves, to prioritize the putative acetylenases responsible for falcarindiol alkyne installation. Four sequentially located genes, with expression levels showing the best correlations with falcarindiol accumulation, were predicted to encode a cluster of enzymes for falcarindiol alkyne formation. Two genes (ACET1a and ACET1b) from the candidate cluster were determined to possess acetylenase activity by analyses of their overexpression in the heterologous host Nicotiana benthamiana. The results revealed that ACET1a and ACET1b encode identical acetylenases that catalyze the conversion of linoleic acid to crepenynic acid in the first step of falcarindiol biosynthesis in tomato. In addition to ACET1a and ACET1b, Solyc12g100250 and Solyc12g100270, which respectively encode a desaturase and a putative decarboxylase, are also involved in the biosynthesis of falcarindiol (Fig. 4).41
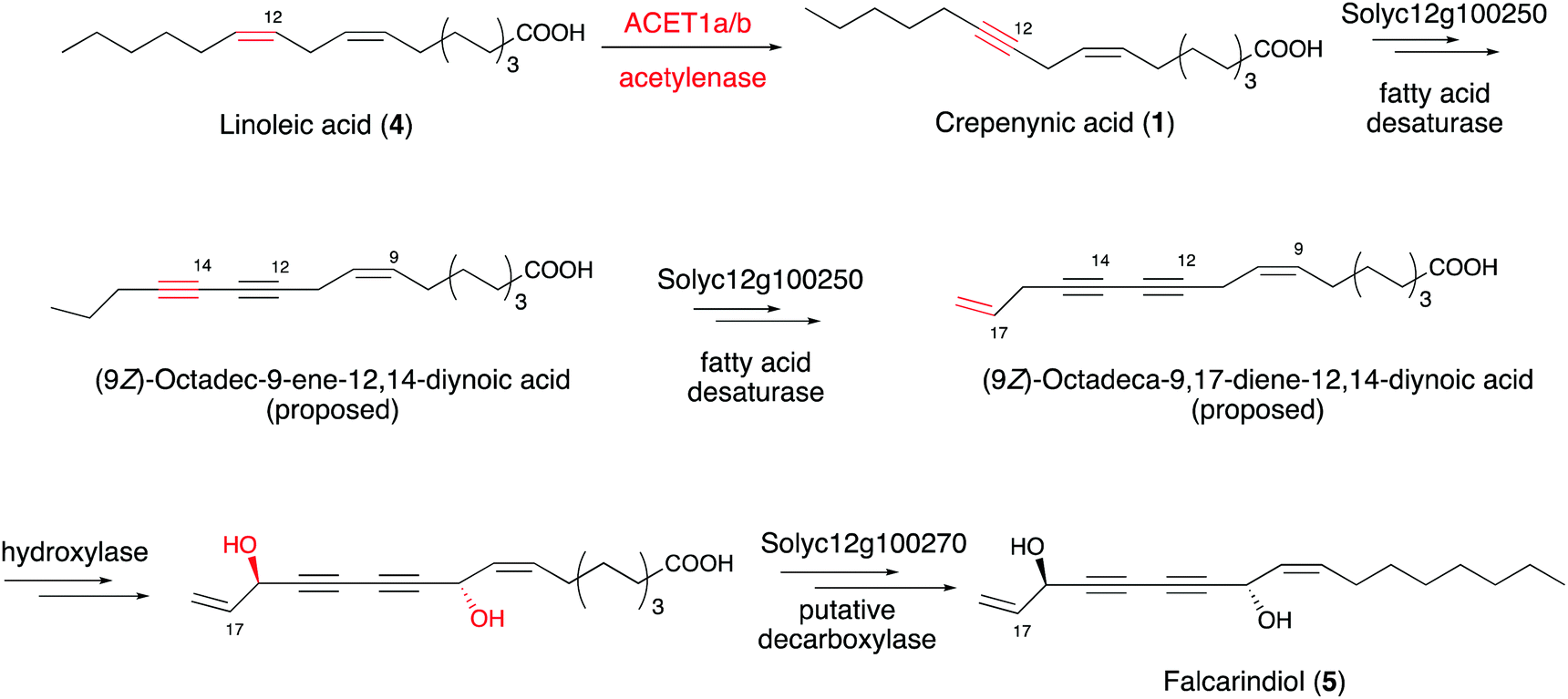 | ||
| Fig. 4 Proposed pathway from linoleic acid to falcarindiol. | ||
2.2 Polyketides
Many microbial and marine polyacetylenes are formed from polyketides or peptides. Enediyne natural products, mainly produced by soil and marine microorganisms, have an unusual bicyclo[7.3.0]-dodecadienediyne core that contains the 1,5-diyne-3-ene unit embedded in a 9-membered ring or the bicyclo[7.3.1]-tridecadiynene core in a 10-membered ring.42,43 These kinds of natural products feature special molecular architectures with promising antibiotic and antitumor activities that are attracting considerable attention.44,45 The subunit six-carbon enediyne was investigated to determine whether it is the key structural element responsible for the biological activity. The enediyne moiety can form a reactive diradical intermediate by Bergman cyclisation. The highly reactive diradical abstracts hydrogen atoms from the DNA and generates a carbon-centered radical, leading to double stranded DNA cleavage in the presence of O2.45,46Early experiments feeding 9-membered neocarzinostatin47 and 10-membered esperamicin48 with 13C-labeled sodium acetate to their producers revealed that each was formed by the head-to-tail assembly of acetate units. In 2002, Shen and co-workers identified the biosynthetic gene cluster of C-1027 (6), a chromoprotein antibiotic isolated from Streptomyces globisporus, by chromosomal walking from the sgcAB locus, which is essential for 6 production in S. globisporus.49,50 At the same time, Thorson and co-workers identified the biosynthetic gene locus for calicheamicin γ (7) by screening a genomic library for cosmids that confer calicheamicin resistance to Micromonospora echinospora.51 Both gene clusters contain the five-gene cassette (the minimal enediyne PKS gene cassette or warhead gene cassette) proposed to be responsible for enediyne core formation (Fig. 5A). Genome mining of the gene clusters producing other enediyne natural products and sequencing of some microbial genomes in the following years further confirmed the conservation of the warhead gene cassettes.52–56
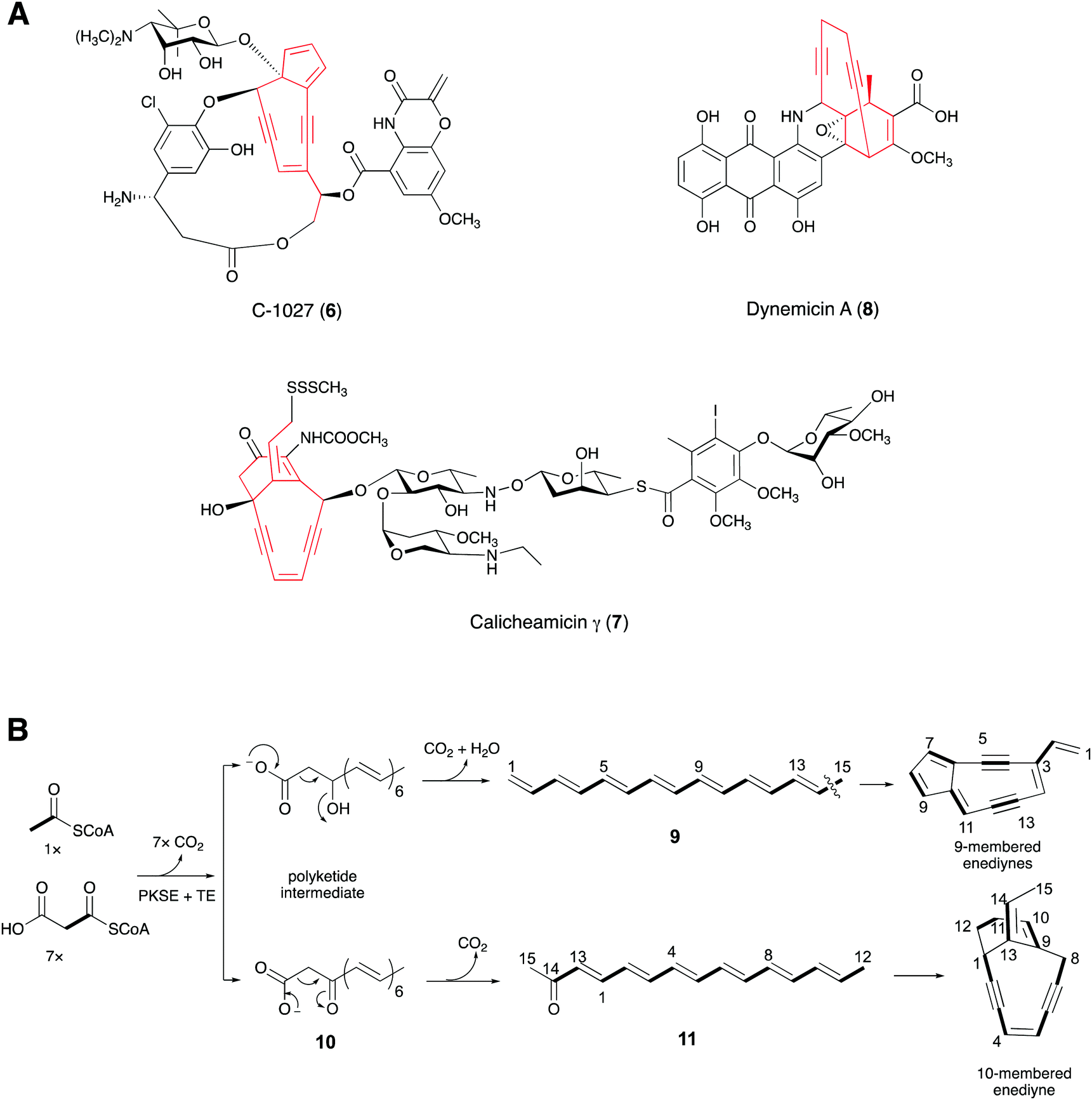 | ||
| Fig. 5 (A) Structures of 6–8. (B) Postulated biosynthetic routes to 9- and 10-membered enediynes. | ||
Shen's group co-expressed the PKS (SgcE) for C-1027 with its cognate thioesterase TE (SgcE10) in E. coli, which produced a 15-carbon polyene (9).57 Further in vivo experiments showed that SgcE can be incorporated into the biosynthetic pathways of other 9-membered enediynes, supporting the idea that 9 is the precursor to the 9-membered enediyne cores. Meanwhile, Liang and co-workers obtained a linear carbonyl-conjugated polyene (11), from the enzymatic assay of the PKS (CalE8) of 7 with the assistance of its TE (CalE7).58,59 Using mass spectrometry, Townsend and co-workers proposed that 11 was derived from a β-keto carboxylic intermediate (10) (Fig. 5B).60 The enediyne core and the other polyketide moieties of the enediyne-containing compounds were considered to have different biosynthetic origins.51,52 Interestingly, by gene deletion and 18O-labeling experiments, Townsend's group recently demonstrated that the enediyne PKS, DynE8, plays a dual role in the biosyntheses of the enediyne core and the anthraquinone scaffold in dynemicin A (8).61,62 Furthermore, Liang proposed that CalU15, encoded by a gene in the warhead gene cassette of 7 and sharing minimal sequence identity with the families of diiron containing desaturases or acetylenases, catalyzes the acetylenic bond formation in the 1,5-diyne-3-ene unit.58 However, the enzymatic activity of the protein must still be proved.
2.3 Polyketide/non-ribosomal peptide hybrids
The first gene clusters for terminal alkyne biosynthesis were identified from jamaicamide B (12) and carmabin A (13), which are terminal alkyne-bearing polyketide–nonribosomal peptide (PK–NRP) hybrids produced by the marine cyanobacteria Moorea producens JHB and M. producens 3L, respectively.20,63 A bioinformatics analysis revealed the conserved trigene cassettes, jamABC and camABC, with high amino acid sequence identities of 95.7–97.4%.64 JamA is a fatty acyl-CoA ligase, while JamB is a membrane associated fatty acid desaturase with strict substrate specificity for 5-hexanoic acid, and together with JamC, an acyl carrier protein (ACP), they are responsible for alkyne formation in a PKS–NRPS system.20 In addition, genome mining revealed more than 80 gene operons homologous to jamABC across diverse bacterial genera, indicating that this gene operon is likely to be employed in the synthesis of a variety of natural products containing an alkyne motif.19 Further experimental studies determined the biosynthetic pathway of the terminal alkyne generated by the three proteins. Hexenoic acid was first activated with ATP by JamA, and then loaded onto the ACP JamC. Subsequent dehydrogenation of the terminal alkene moiety was accomplished by the membrane associated fatty acid desaturase/acetylenase JamB to generate the terminal alkyne (Fig. 6B).65,66 Zhang's group further demonstrated that desaturase/acetylenase (JamB) has a more stringent interaction with ACP than the acyl-ACP ligase (JamA) by both in vitro and in vivo assays, which provided new insights into the role of ACP in alkyne biosynthesis.67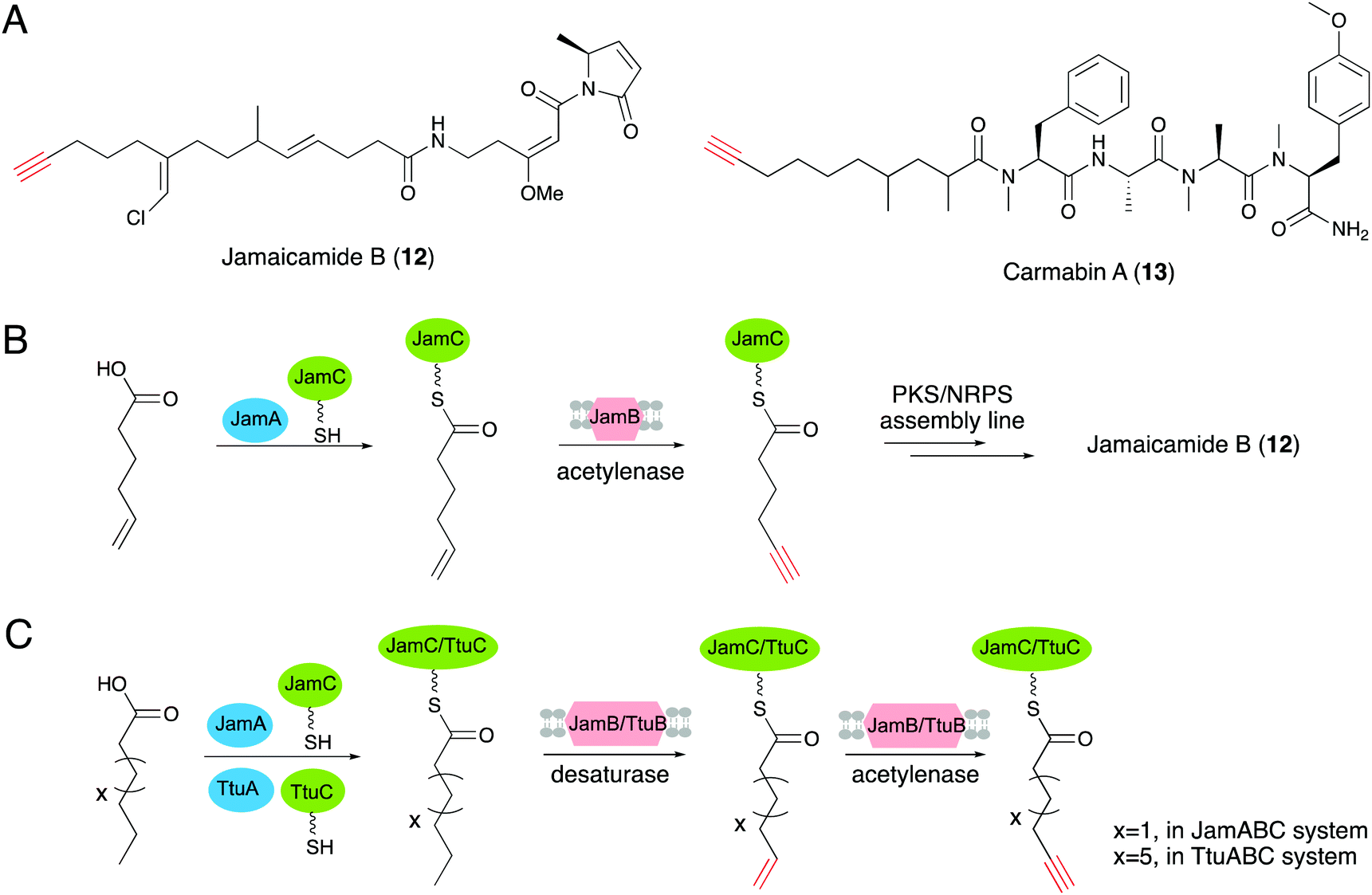 | ||
| Fig. 6 (A) Structures of 12 and 13. (B) The biosynthetic pathway for the C6 alkynoic starter unit generation and incorporation into jamaicamide B. (C) Terminal alkyne biosynthetic pathways of JamABC and TtuABC that modify C6 and C10 fatty acids, respectively. | ||
The discovery of the first terminal alkyne biosynthetic pathway has inspired further characterizations and applications of this unique biosynthetic route. Genome mining of JamABC homologs led to the discovery of the new terminal alkyne biosynthetic cassette ttuABC, from Teredinibacter turnerae T7901.19 TtuABC shows an analogous mode of action with JamABC to form a terminal alkyne moiety. The medium-chain fatty acid is activated by acyl-CoA ligase TtuA and load onto carrier protein TtuC. One obvious difference between operons TtuABC and JamABC is that the desaturase/acetylenase TtuB is specific for decanoic acid (C10 chain length) rather than hexanoic acid (C6 chain length) for substrate selection (Fig. 6C).19
Gerwick and co-workers discovered vatiamides A–D (14–17) from Moorea producens, which feature a fatty acid tail harboring a terminal alkyne or bromoalkyne that is identical to the moiety found in jamaicamides.20,68 Genome mining of jamABC homologs identified the PKS/NRPS gene cluster vatA-O responsible for the formation of vatiamides A and B. Efforts toward finding the genes for the biosynthesis of vatiamides C and D illuminated a 40 kb extension of NRPS and PKS modules downstream of vatO in the original 50 kb cluster. This research characterized the first example of a special combinatorial non-collinear PKS/NRPS system in which the upstream PKS cassette interacts with three separate NRPS partners (Fig. 7B). Terminal alkynes can undergo a copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction to allow sensitive alkyne identification in a crude extract.28 The researchers synthesized a fluorogenic “click”-based probe that exhibited a potent fluorescence increase at 490 nm in the triazole adduct formation. Three alkynylated lipopeptides and their brominated analogues vatiamides A–F (14–19), which are predicted to be produced by the unique non-collinear PKS/NRPS system, were discovered with the use of this fluorogenic azide-based click probe (Fig. 7A).68
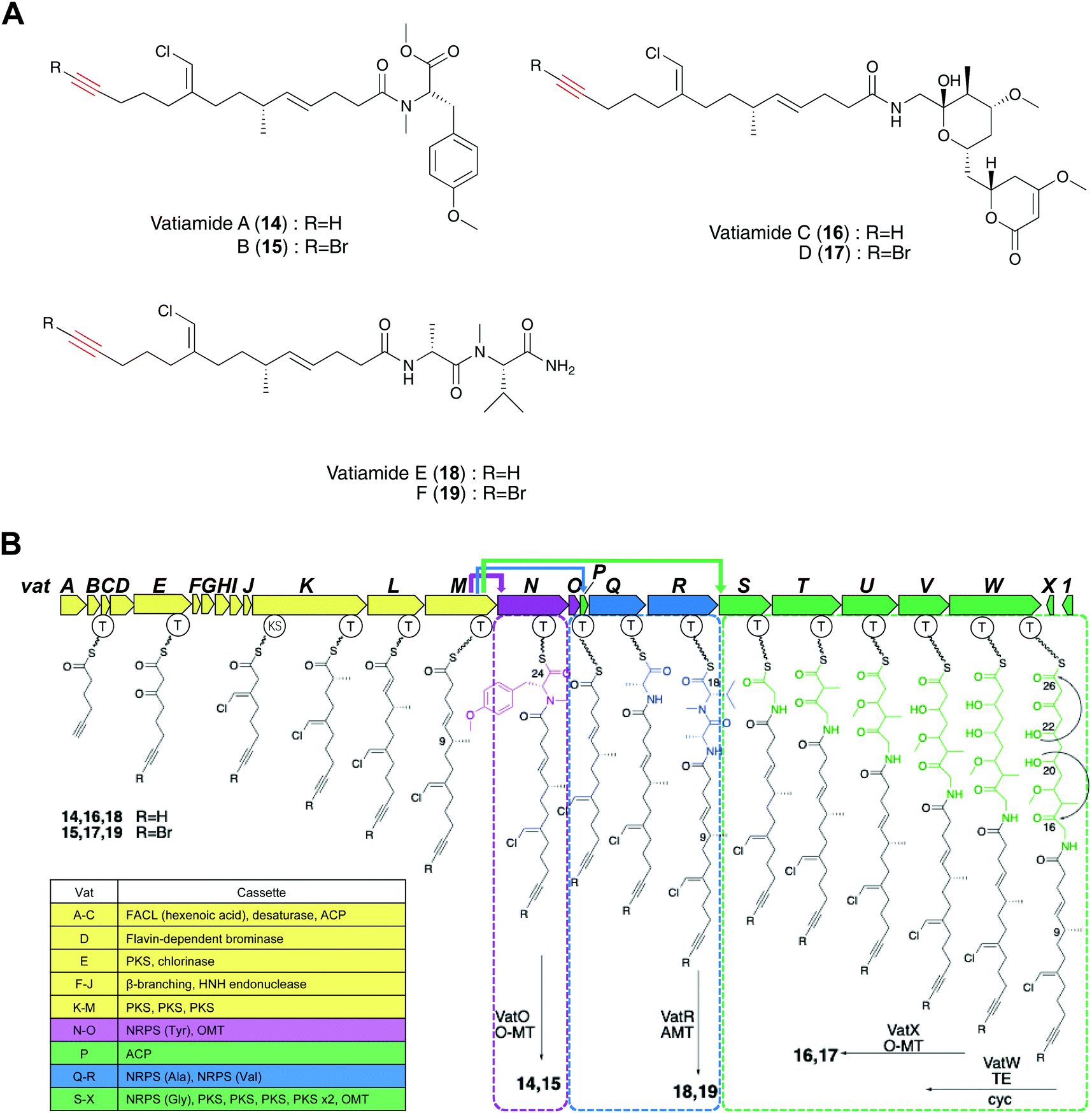 | ||
| Fig. 7 (A) Structures of 14–19. (B) Biosynthetic pathway of vatiamides in Moorea producens. | ||
2.4 Amino acids
A series of terminal alkyne-containing amino acids are reportedly produced by fungi and bacteria.69,70 Early studies revealed that some terminal alkynes originate from fatty acid or polyketide biosynthetic pathways.24,65 Recently, Chang and co-workers characterized a unique pathway to produce a terminal alkyne-containing amino acid, β-ethynylserine (βes) (20), from Streptomyces cattleya.21 Knock outs of three fatty acid desaturases (SCAT_4823, SCAT_p1525 and SCAT_0184) did not disrupt βes production, thus suggesting a different route than canonical desaturation for target compound production. The putative biosynthetic gene cluster (BesA-F), related to amino acid metabolism, was identified by a genomic comparison of two Streptomyces spp. that produce acetylenic amino acids (Fig. 8A) with 26 other control Streptomyces spp. genomes.21 In contrast to jamaicamide B, terminal alkyne installation in βes is catalyzed by three crucial enzymes, including BesB, a pyridoxal-5′-phosphate (PLP)-dependent cystathionine-β-lyase homolog, BesC, which shares homology with the non-heme iron oxidase HemeO superfamily, and BesD, a non-heme Fe/α-ketoglutarate (αKG)-dependent oxygenase.21 The pathway involves multiple unique functional groups to yield the terminal alkyne, including the BesD-mediated 4-chlorination of L-lysine (23), followed by oxidative cleavage via BesC to produce a chlorinated terminal alkene (24), with ammonia and formaldehyde as co-products. BesB catalyzes the conversion of 24 to 21 with the aid of a PLP cofactor. To probe the mechanism of BesB, 24 was used as substrate in D2O and lead to the formation of [M + 2D]+ Pra which suggested at least two deprotonation–protonation events occurring during the BesB catalysis. The Cα-deprotonation giving the 4-Cl-Alg-ketimine intermediate is consistent with the cystathionine γ-synthase mechanism.71 Further deprotonation at the β position initiated the elimination of chloride to form an allene intermediate (27). The terminal alkyne was formed by subsequent isomerization of the allene intermediate and release from PLP (Fig. 8B and C). Pra is then ligated to glutamate to form the γ-glutamyl-L-dipeptide (25), which is the substrate of the BesE hydroxylase to produce the βes dipeptide (26). Notably, the glutamylation step is considered to be a physiological step that reduces side reactions and facilitates transport. The amide hydrolase is responsible for the release of free Pra (21) and βes (20).21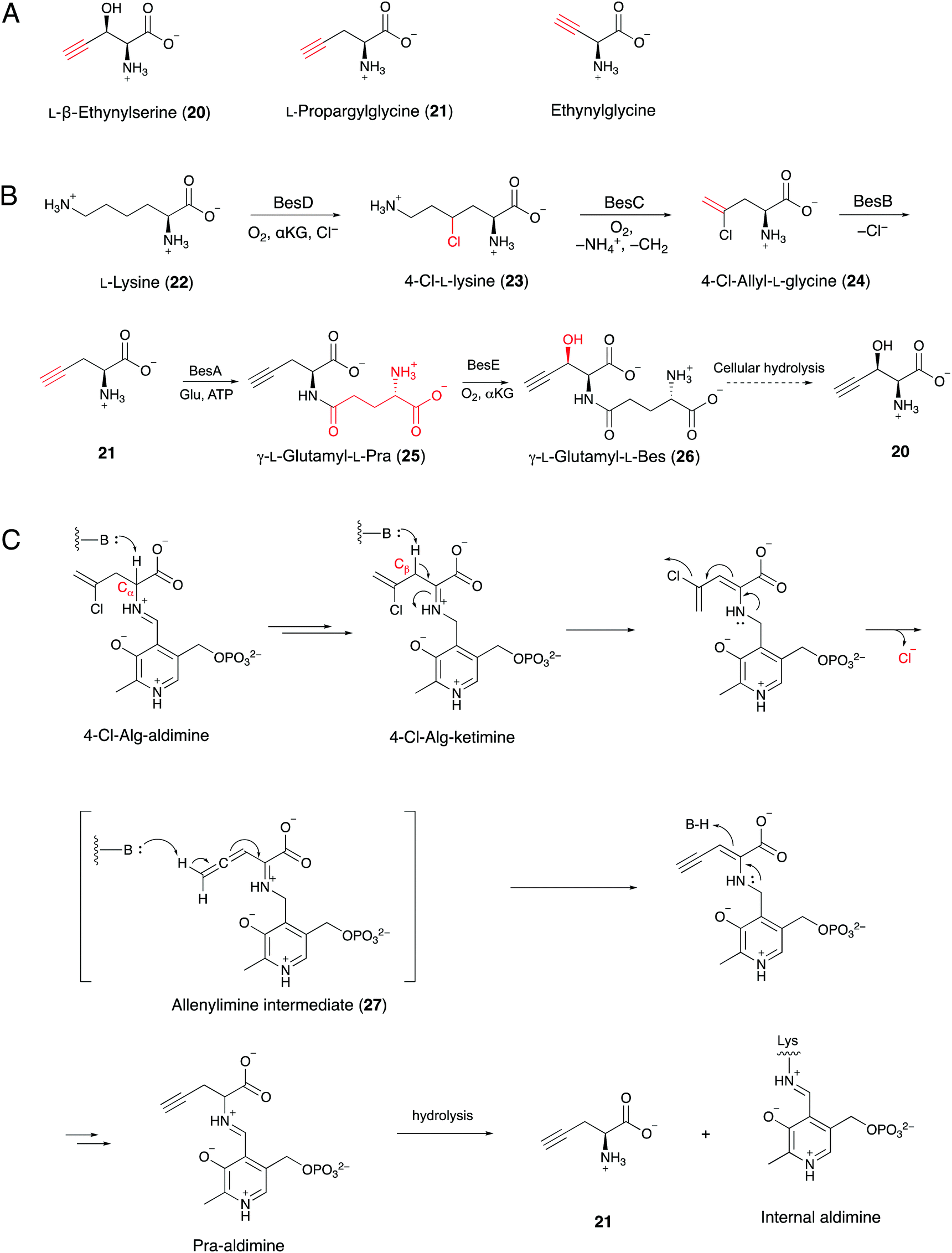 | ||
| Fig. 8 (A) Terminal-alkyne amino acids from Streptomyces spp. (B) Proposed biosynthetic pathway of βes from L-lysine. (C) Putative mechanism for BesB-catalyzed formation of Pra (21) from 4-Cl-allylglycine (24). | ||
Searches for BesA-D homologs revealed five additional Streptomyces spp. with the genes encoding the proteins, but their abilities to produce terminal alkyne containing amino acids have not been reported.21 Subsequently, the authors found that three of the strains could produce either Pra or βes. Their discovery will lead to the identification of enzyme homologs responsible for the formation of a wider range of terminal alkyne amino acid products. In addition, characterization of the βes biosynthetic pathway will provide an opportunity for the production of non-proteinogenic amino acids, such as halo-, terminal-alkene and terminal-alkyne amino acids, that can be used for downstream applications.21
2.5 Meroterpenoids
Acetylenic meroterpenoids are structurally unique compounds with an alkynyl moiety in prenyl chains. These compounds display a wide range of properties, such as cytotoxic, antifungal and antibacterial bioactivities.72,73 Although some chemical approaches have been developed to synthesize these acetylenic compounds, little is known about the biosynthesis and molecular basis for the acetylenic prenyl chain formation.74,75 Recently, two research groups simultaneously reported that the alkynyl moieties in the prenyl chains of biscognienyne B (28) and asperpentyn (29) are generated by cytochrome P450 enzymes, as determined via heterologous expression, feeding experiments and in vitro enzyme reactions.22,23 Furthermore, the two groups demonstrated that the stereochemistry of the double bond in the intermediates is important for substrate recognition.Gao and co-workers previously isolated 28 and several other acetylenic meroterpenoids from an endolichenic fungus Biscogniauxia sp. (71-10-1-1).76 Based on the previously reported prenyltransferase IacE and the cupin protein IacF, which are reportedly associated with the biosynthesis of biscognienyne B,77 they identified the putative gene cluster consisting of 11 genes. Through heterologous expression in Aspergillus oryzae NSAR1, feeding experiments and in vitro assays, they demonstrated that 28 is synthesized from 4-hydroxybenzoic acid (30) by six enzymes (Fig. 9A).22 Particularly, they found that the alkyne in 28 was installed by the cytochrome P450 enzyme BisI, which is different from the other reported enzymes dedicated to acetylenic bond biosynthesis. In order to explore the mechanism and substrate scope of the BisI-mediated biosynthesis of the alkynyl moiety, the authors chemically prepared the 1-OH methylated form (31) of 4-hydroxy-3-prenylbenzoic acid and performed feeding experiments (Fig. 9B). The results indicated that BisI can convert 31 to the dehydrogenated product (7E)-diene (32). However, further feeding experiments and in vitro assays revealed that BisI cannot accept 32 or its demethylated derivative 33 as a substrate to yield the corresponding acetylenic products, but it shows promiscuous activities towards C5 and C15 prenyl chains.22
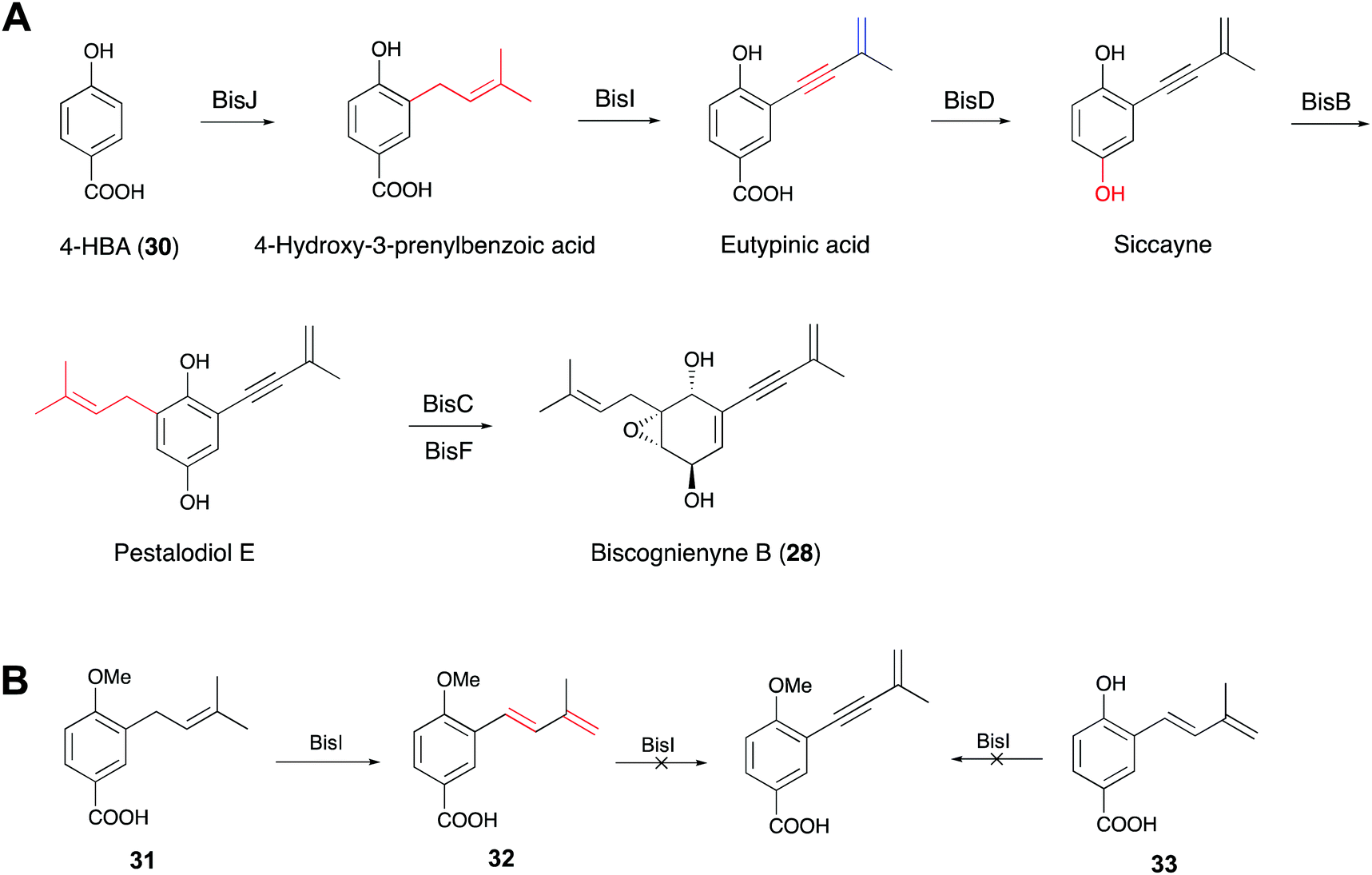 | ||
| Fig. 9 (A) Proposed biosynthetic pathway for biscognienyne B. (B) The substrate scope of BisI. | ||
Meanwhile, Lin and co-workers identified the similar cytochrome P450 enzyme AtyI, responsible for the 1,3-enyne formation in asperpentyn (29) (Fig. 10B).23 To investigate the mechanism of the enyne moiety formation by AtyI, a series of substrate derivatives were synthesized and fed to Saccharomyces cerevisiae expressing atyI. Feeding (7Z)-alkene (34) to S. cerevisiae expressing atyI led to the production of compound 35, while (7E)-alkene (37) could not be converted (Fig. 10A). Further experiments feeding 35 and its 4-OH methylated derivative 36 to S. cerevisiae expressing atyI showed no conversion to the enyne, suggesting that 7-alkyne containing intermediates were not involved in the enyne formation by AtyI. In contrast, when S. cerevisiae expressing atyI was fed 38, compound 39 was detected (Fig. 10A). The above results indicated that the AtyI-catalyzed formation of the 1,3-enyne moiety occurs through a (7Z)-diene intermediate. This P450 monooxygenase was thus verified to possess dual functions. It catalyzes prenyl chain dehydrogenation to yield a (7Z)-diene from 40, followed by further dehydrogenation to form the 1,3-enyne moiety (Fig. 10B).23 The formation of the (7Z)-diene intermediate might be conducive to the abstraction of the two hydrogen atoms on the same side, by the active ferryl-oxo complex of the P450 enzyme. Particularly, Compound I sequentially abstracts the 7-H and 8-H to give the enyne moiety. This mechanism is similar to that of a fatty acid desaturase; however, the iron cofactors are different. This hypothesis also suggests opportunities for discovering novel acetylenic meroterpenoids and expanding the enzymatic tools for the de novo biosynthesis of alkyne-tagged compounds.
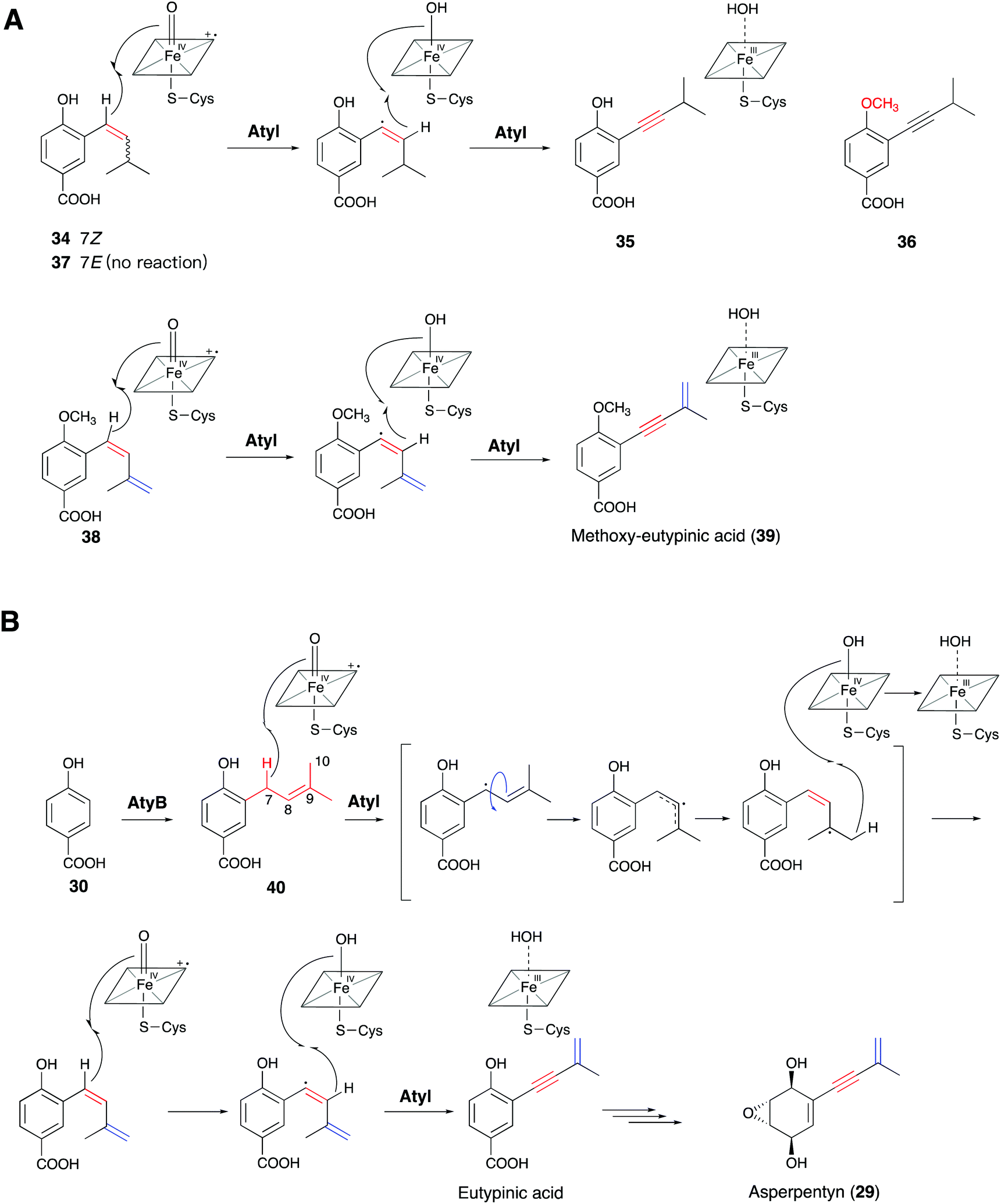 | ||
| Fig. 10 Proposed mechanisms of (A) AtyI-catalyzed synthetic substrate analogs 34 and 38 by feeding experiments, and (B) the natural AtyI-catalyzed pathway from 40 to asperpentyn (29). | ||
3. Engineered biosynthesis of alkyne-tagged products
Tagging natural products with a unique chemical handle facilitates the stabilization, quantification, visualization, and functional analysis of natural products through bioorthogonal chemistry.78–80 In particular, the terminal alkyne can be widely employed through the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) ‘click’ reaction, which is one of the most powerful tools in chemical biology.68,81–83 In addition to tagging natural products by semi-synthesis14 and total synthesis,15 precursor-directed biosynthesis, which is a more facile and gentle way of chemical synthesis, has been used for azide- or alkyne-tag installation.16,84,85 However, for precursor-directed biosynthesis, a major limitation is the low conversion efficiency of precursor into product, which in turn causes the problem of the coexistence of precursors and products with the same chemical handle.18 Given the progress achieved toward understanding alkyne biosynthesis, de novo biosynthesis, which is employed for the creation of alkyne-labeled molecules through biosynthetic pathway engineering, has drawn keen attention. This strategy avoids the feeding of alkynoic precursors and overcomes the limitation of precursor-directed biosynthesis.65The function of the trigene cassette jamABC for terminal alkyne formation in jamaicamide B was first biochemically characterized by Zhang et al.65 Based on this result, they generated a JamABC in situ system for introducing terminal alkyne tags into natural products. JamB has strict substrate specificity for 5-hexanoic acid and the acyl carrier protein (JamC, CamC). Therefore, only a 5-hexynoyl moiety is incorporated into alkyne-tagged natural products by the JamABC in situ system. The terminal alkyne biosynthetic cassette was first co-expressed with a type III PKS (HsPKS1) system in a starter unit, with malonyl-CoA as the extension unit, in the E. coli BAP1 strain. After induction by IPTG, the strain was grown for 2 days in F1 minimal medium supplemented with 5-hexanoic acid. Analysis of the culture extracts revealed the production of the terminal alkyne-tagged PK compound 41 (compound 3 in ref. 65) (Fig. 11A). However, the low activity of JamB leads outcompeted 5-hexenoic acid supplied than the target terminal alkyne. Zhang and co-workers improved the activity of JamB up to 20-fold to convert hexanoic acid to the terminal alkyne in E. coli and developed a rapid and sensitive platform through fluorogenic click reaction for detecting extracellular alkene-tagged metabolites directly in crude cultures.86
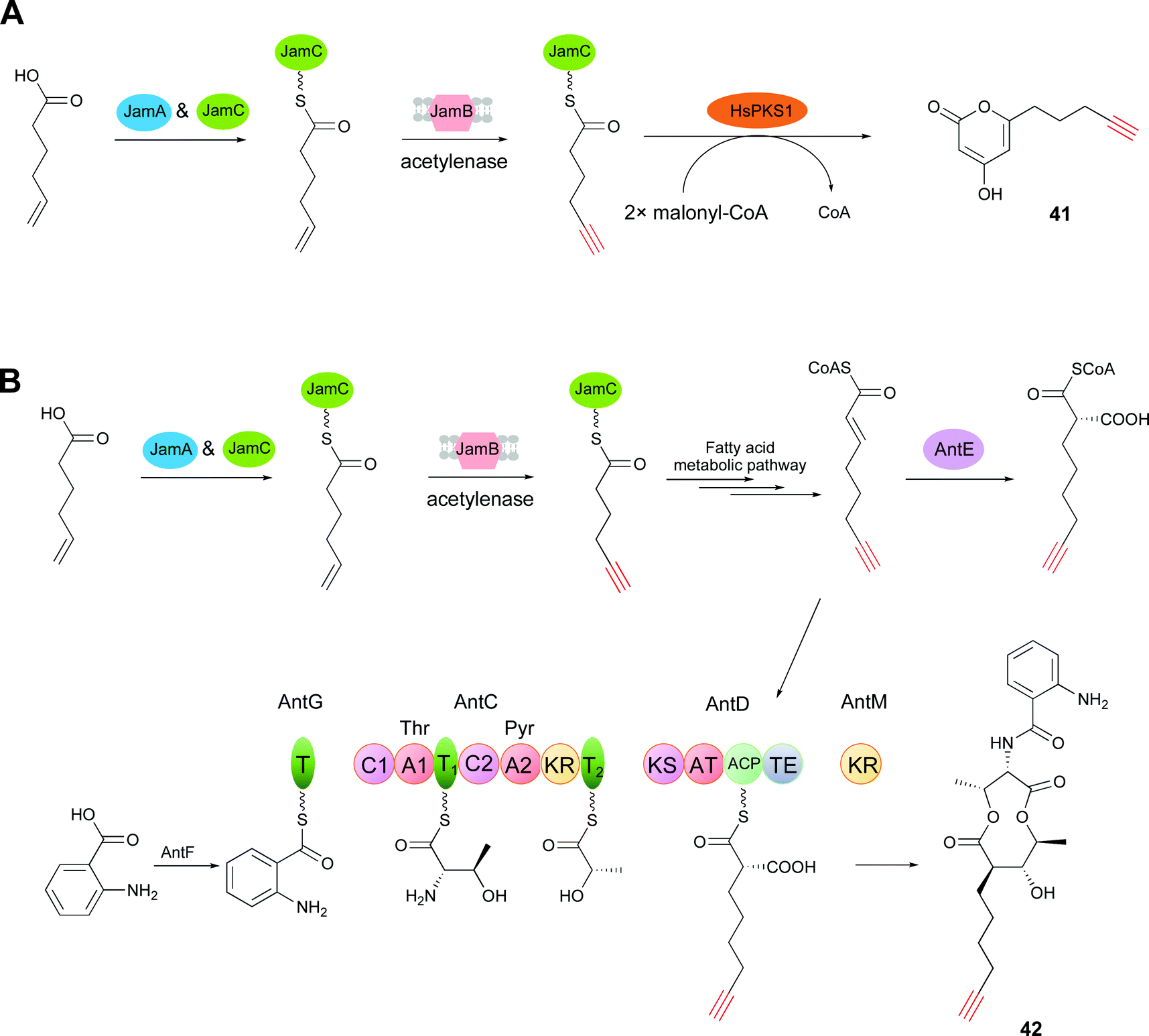 | ||
| Fig. 11 (A) Biosynthesis of 41via starter unit engineering. (B) Biosynthesis of 42via extender unit engineering. | ||
Further explorations for incorporating an alkynoic extender unit into the molecular scaffolds of PKs employed the gene cassette antCDEFGM, required for the generation of the antimycin dilactone scaffold,87 and the jamABC cassette. Liu and co-workers demonstrated that an alkynoic extender unit can be accepted by the promiscuous AntE (crotonyl-CoA reductase/carboxylase (CCR)-like protein) and the acyltransferase (AT) domain embedded in AntD to synthesize an alkyne-labeled antimycin by the precursor directed biosynthesis in Streptomyces.88 To avoid feeding alkynoic precursors, co-expression of the two gene cassettes, antCDEFGM and jamABC, in E. coli BAP1 led to the production of the alkyne-bearing PK/NRP hybrid antimycin analog 42 (compound 5 in ref. 65) (Fig. 11B).65
TtuABC, encoded from the genome of T. turnerae T7901, shows a similar mode of function to JamABC in forming a terminal alkyne moiety. TtuABC has different substrate specificity towards fatty acyl moieties with a C10 chain length, instead of hexanoic acid (C6 chain length). Zhang's group employed the type III PKS (HsPKS1) in a TtuABC in situ system that specifically recognized the C10 fatty acid of the starter unit to generate the novel terminal alkyne-tagged polyketide compound 43 (compound 1 in ref. 19) in E. coli (Fig. 12A).19 Based on these results, they proved that PKS starter unit engineering is a viable way to install the alkynyl moiety from JamABC onto a polyketide to generate terminal alkyne-tagged products. They further tried to generalize this strategy to other kinds of PKSs by employing two well studied type I PKSs, LipPKS1 and DEBSM6, to produce the target alkyne-tagged polyketides.89 LipPKS1 is the first module in lipomycin biosynthesis and DEBSM6 is the last PKS module in the erythromycin biosynthetic pathway.90,91 Engineered LipPKS1 and DEBSM6 have been constructed to utilize malonyl-CoA instead of methylmalonyl-CoA as the extender unit, with the promiscuous DEBS thioesterase, to promote the release of a carboxylic acid product.92 Zhang and co-workers further removed the AT and ACP loading domains of the engineered LipPKS1, to create a truncated version that facilitates the alternative starter unit incorporation. The two engineered modules (LipPKS1* and DEBSM6*), in combination with JamABC working as a trans loading system, were employed to produce the alkyne-tagged polyketide 3-hydroxydec-9-ynoic acid (44) (compound 1 in ref. 89) (Fig. 12B).89 These findings have shed light on the discovery and development of synthetic tools for the de novo biosynthesis of various acetylenic products.
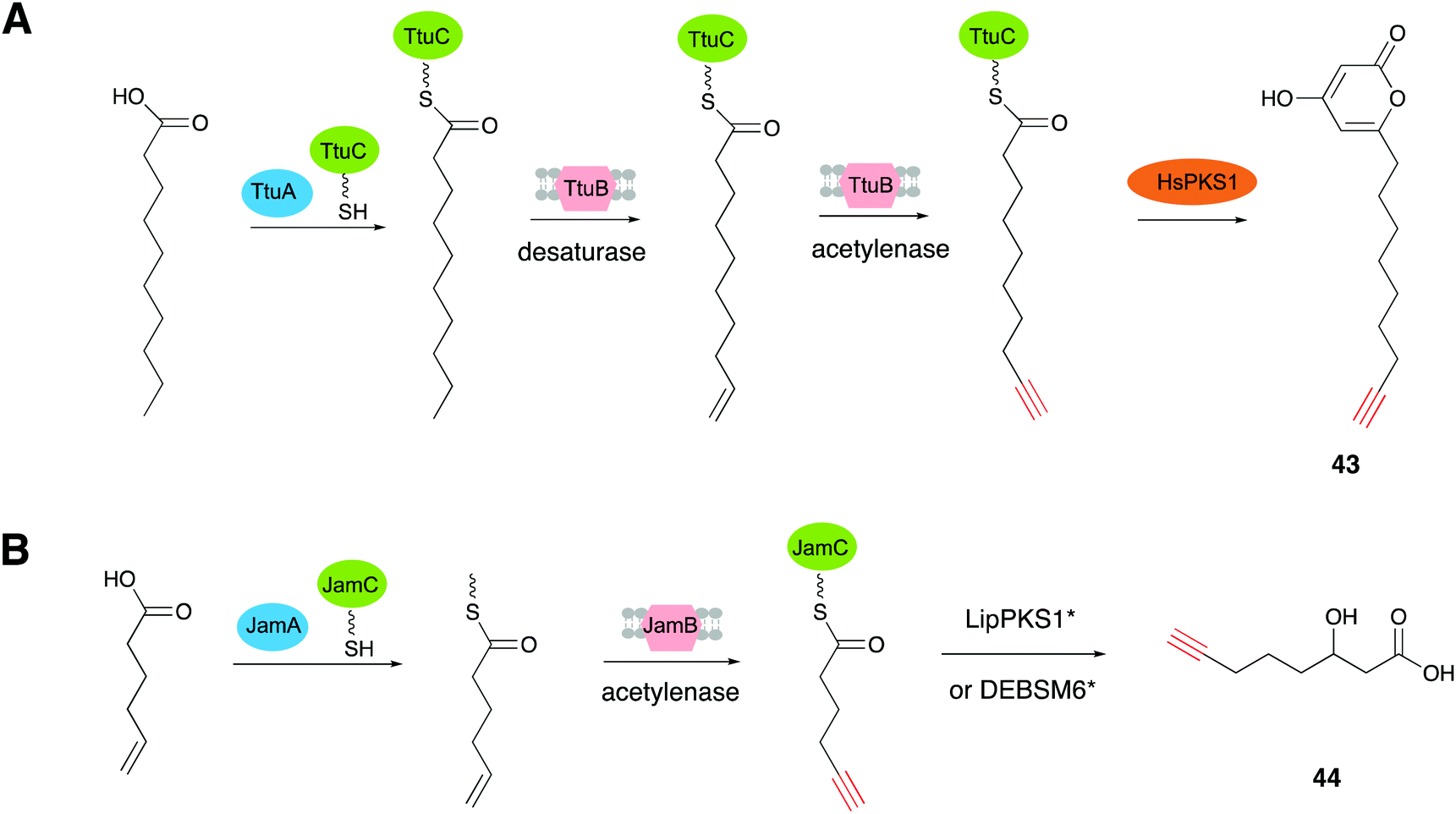 | ||
| Fig. 12 (A) Biosynthesis of 43 using the in vivo TtuABC system. (B) Scheme of the in vivo (or in vitro) reactions between JamABC and engineering type I PKSs, LipPKS1* and DEBSM*, to produce 44. | ||
4. Conclusion
The alkyne is a biological and chemical functional moiety found in numerous natural products. Hitherto, a few types of gene cassettes were identified in the biosynthetic pathways of alkyne-containing natural products. The recent progress in identifying the biosynthetic enzymes and pathways of acetylenic products offers a potential route for the de novo biosynthesis of alkyne-labeled products, for use as powerful tools for diverse downstream applications in drug discovery and chemical biology. Meanwhile, with these enzymes in hand, finding more homologs will lead to an expanded alkyne biosynthetic enzyme inventory capable of efficiently generating acetylenic products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express sincere appreciation to an excellent group of coworkers whose contributions are cited in the text. This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (JSPS KAKENHI Grant No. JP16H06443, JP20H00490, and JP20KK013), and Japan Science and Technology Agency (JST SICORP Grant No. JPMJSC1701).References
- Q. Y. Chai, Z. Yang, H. W. Lin and B. N. Han, Mar. Drugs, 2016, 14, 216 CrossRef PubMed.
- K. C. Nicolaou, W. M. Dai, S. C. Tsay, V. A. Estevez and W. Wrasidlo, Science, 1992, 256, 1172–1178 CrossRef CAS PubMed.
- H. Kim, H. Lee, D. Lee, S. Kim and D. Kim, J. Am. Chem. Soc., 2007, 129, 2269–2274 CrossRef CAS PubMed.
- P. Wessig and G. Müller, Chem. Rev., 2008, 108, 2051–2063 CrossRef CAS PubMed.
- K. Campbell, C. J. Kuehl, M. J. Ferguson, P. J. Stang and R. R. Tykwinski, J. Am. Chem. Soc., 2002, 124, 7266–7267 CrossRef CAS PubMed.
- G. Chen, L. Wang, D. W. Thompson and Y. Zhao, Org. Lett., 2008, 10, 657–660 CrossRef CAS PubMed.
- B. M. Trost and J. T. Masters, Chem. Soc. Rev., 2016, 45, 2212–2238 RSC.
- Y. Jiang, J. Chen, C. Deng, E. J. Suuronen and Z. Zhong, Biomaterials, 2014, 35, 4969–4985 CrossRef CAS PubMed.
- C. G. Liu, Y. Wang, P. Liu, Q. L. Yao, Y. Y. Zhou, C. F. Li, Q. Zhao, G. H. Liu and X. L. Zhang, ACS Chem. Biol., 2020, 15, 1554–1565 CrossRef CAS PubMed.
- Z. Hao, S. Hong, X. Chen and P. R. Chen, Acc. Chem. Res., 2011, 44, 742–751 CrossRef CAS PubMed.
- M. Grammel and H. C. Hang, Nat. Chem. Biol., 2013, 9, 475–484 CrossRef CAS PubMed.
- P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 10, 4905–4979 CrossRef PubMed.
- L. Wei, F. Hu, Y. Shen, Z. Chen, Y. Yu, C. C. Lin, M. C. Wang and W. Min, Nat. Methods, 2014, 11, 410–412 CrossRef CAS PubMed.
- J. Seidel, Y. Miao, W. Porterfield, W. Cai, X. Zhu, S. J. Kim, F. Hu, S. Bhattarai-Kline, W. Min and W. Zhang, Chem. Commun., 2019, 55, 9379–9382 RSC.
- I. Staub and S. A. Sieber, J. Am. Chem. Soc., 2008, 130, 13400–13409 CrossRef CAS PubMed.
- C. J. Harvey, J. D. Puglisi, V. S. Pande, D. E. Cane and C. Khosla, J. Am. Chem. Soc., 2012, 134, 12259–12265 CrossRef CAS PubMed.
- K. Bravo-Rodriguez, S. Klopries, K. R. M. Koopmans, U. Sundermann, S. Yahiaoui, J. Arens, S. Kushnir, F. Schulz and E. Sanchez-Garcia, Chem. Biol., 2015, 19, 1425–1430 CrossRef PubMed.
- J. F. Martin, J. Casqueiro and P. Liras, Curr. Opin. Microbiol., 2005, 8, 282–293 CrossRef CAS PubMed.
- X. Zhu, M. Su, K. Manickam and W. Zhang, ACS Chem. Biol., 2015, 10, 2785–2793 CrossRef CAS PubMed.
- D. J. Edwards, B. L. Marquez, L. M. Nogle, K. McPhail, D. E. Goeger, M. A. Roberts and W. H. Gerwick, Chem. Biol., 2004, 11, 817–833 CrossRef CAS PubMed.
- J. A. Marchand, M. E. Neugebauer, M. C. Ing, C. I. Lin, J. G. Pelton and M. C. Y. Chang, Nature, 2019, 567, 420–424 CrossRef CAS PubMed.
- J. M. Lv, Y. H. Gao, H. Zhao, T. Awakawa, L. Liu, G. D. Chen, X. S. Yao, D. Hu, I. Abe and H. Gao, Angew. Chem., Int. Ed., 2020, 132, 13633–13638 CrossRef.
- Y. R. Chen, A. Naresh, S. Y. Liang, C. H. Lin, R. J. Chein and H. C. Lin, Angew. Chem., Int. Ed., 2020, 59, 13537–13541 CrossRef CAS PubMed.
- R. E. Minto and B. J. Blacklock, Prog. Lipid Res., 2008, 47, 233–306 CrossRef CAS PubMed.
- V. S. Haritos, Nat. Chem. Biol., 2015, 11, 98–99 CrossRef CAS PubMed.
- B. J. Blacklock, B. E. Scheffler, M. R. Shepard, N. Jayasuriya and R. E. Minto, J. Biol. Chem., 2010, 285, 28442–28449 CrossRef CAS PubMed.
- V. S. Haritos, I. Horne, K. Damcevski, K. Glover, N. Gibb, S. Okada and M. Hamberg, Nat. Commun., 2012, 3, 1150 CrossRef PubMed.
- C. Ross, K. Scherlach, F. Kloss and C. Hertweck, Angew. Chem., Int. Ed., 2014, 53, 7794–7798 CrossRef CAS PubMed.
- X. Bao, S. Katz, M. Pollard and J. Ohlrogge, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7172–7177 CrossRef CAS PubMed.
- N. El-Jaber, A. Estevez-Braun, A. G. Ravelo, O. Munoz-Munoz, A. Rodriguez-Afonso and J. R. Murguia, J. Nat. Prod., 2003, 66, 722–724 CrossRef CAS PubMed.
- M. Lee, M. Lenman, A. Banaś, M. Bafor, S. Singh, M. Schweizer, R. Nilsson, C. Liljenberg, A. Dahlqvist, P.-O. Gummeson, S. Sjödahl, A. Green and S. Stymne, Science, 1998, 280, 915–918 CrossRef CAS PubMed.
- J. Shanklin, J. E. Guy, G. Mishra and Y. Lindqvist, J. Biol. Chem., 2009, 284, 18559–18563 CrossRef CAS PubMed.
- Y. Bai, J. G. McCoy, E. J. Levin, P. Sobrado, K. R. Rajashankar, B. G. Fox and M. Zhou, Nature, 2015, 524, 252–256 CrossRef CAS PubMed.
- H. G. Enoch, A. Catalá and P. Strittmatter, J. Biol. Chem., 1976, 251, 5095–5103 CrossRef CAS.
- J. Shanklin, E. Whittle and B. G. Fox, Biochemistry, 1994, 33, 12787–12794 CrossRef CAS PubMed.
- P. H. Buist and B. Behrouzian, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2003, 68, 107–112 CrossRef.
- M. Högbom, Y. Huque, B.-M. Sjöberg and P. Nordlund, Biochemistry, 2002, 41, 1381–1389 CrossRef PubMed.
- L. L. Wayne, J. G. Wallis, R. Kumar, J. E. Markham and J. Browse, Plant Cell, 2013, 25, 3052–3066 CrossRef CAS PubMed.
- D. W. Reed, D. R. Polichuk, P. H. Buist, S. J. Ambrose, R. J. Sasata, C. K. Savile and P. S. Covello, J. Am. Chem. Soc., 2003, 125, 10635–10640 CrossRef CAS PubMed.
- X. Robert and P. Gouet, Nucleic Acids Res., 2014, 42, W320–W324 CrossRef CAS PubMed.
- J. E. Jeon, J. G. Kim, C. R. Fischer, N. Mehta, C. Dufour-Schroif, K. Wemmer, M. B. Mudgett and E. Sattely, Cell, 2020, 180, 176–187.e19 CrossRef CAS PubMed.
- K. Edo, M. Mizugaki, Y. Koide, H. Seto, K. Furihata, N. Ōtake and N. Ishida, Tetrahedron Lett., 1985, 26, 331–334 CrossRef CAS.
- M. D. Lee, T. S. Dunne, C. C. Chang, G. A. Ellestad, M. M. Siegel, G. O. Morton and D. B. Borders, J. Am. Chem. Soc., 1987, 109, 3466–3468 CrossRef CAS.
- J. Thorson, E. Sievers, J. Ahlert, E. Shepard, R. Whitwam, K. Onwueme and M. Ruppen, Curr. Pharm. Des., 2000, 6, 1841–1879 CrossRef CAS PubMed.
- A. L. Smith and K. C. Nicolaou, J. Med. Chem., 1996, 39, 2103–2117 CrossRef CAS PubMed.
- J. B. Biggins, J. R. Prudent, D. J. Marshall, M. Ruppen and J. S. Thorson, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13537–13542 CrossRef CAS PubMed.
- O. D. Hensens, J. L. Giner and I. H. Goldberg, J. Am. Chem. Soc., 1989, 111, 3295–3299 CrossRef CAS.
- K. S. Lam, J. A. Veitch, J. Golik, B. Krishnan, S. E. Klohr, K. J. Volk, S. Forenza and T. W. Doyle, J. Am. Chem. Soc., 1993, 115, 12340–12345 CrossRef CAS.
- W. Liu and B. Shen, Antimicrob. Agents Chemother., 2000, 44, 382–392 CrossRef CAS PubMed.
- W. Liu, S. D. Christenson, S. Standage and B. Shen, Science, 2002, 297, 1170–1173 CrossRef CAS PubMed.
- J. Ahlert, E. Shepard, N. Lomovskaya, E. Zazopoulos, A. Staffa, B. O. Bachmann, K. Huang, L. Fonstein, A. Czisny, R. E. Whitwam, C. M. Farnet and J. S. Thorson, Science, 2002, 297, 1173–1176 CrossRef CAS PubMed.
- W. Liu, K. Nonaka, L. P. Nie, J. Zhang, S. D. Christenson, J. Bae, S. G. Van Lanen, E. Zazopoulos, C. M. Farnet, C. F. Yang and B. Shen, Chem. Biol., 2005, 12, 293–302 CrossRef CAS PubMed.
- Q. Gao and J. S. Thorson, FEMS Microbiol. Lett., 2008, 282, 105–114 CrossRef CAS PubMed.
- S. G. Van Lanen, T. J. Oh, W. Liu, E. Wendt-Pienkowski and B. Shen, J. Am. Chem. Soc., 2007, 129, 13082–13094 CrossRef CAS PubMed.
- D. W. Udwary, L. Zeigler, R. N. Asolkar, V. Singan, A. Lapidus, W. Fenical, P. R. Jensenand and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10376–10381 CrossRef CAS PubMed.
- Z. J. Low, G. L. Ma, H. T. Tran, Y. Zou, J. Xiong, L. Pang, S. Nuryyeva, H. Ye, J. F. Hu, K. N. Houk and Z. X. Liang, J. Am. Chem. Soc., 2020, 142, 1673–1679 CrossRef CAS PubMed.
- J. Zhang, S. G. V. Lanen, J. Ju, W. Liu, P. C. Dorrestein, W. Li, N. L. Kelleher and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1461–1465 Search PubMed.
- R. Kong, L. P. Goh, C. W. Liew, Q. S. Ho, E. Murugan, B. Li and Z. X. Liang, J. Am. Chem. Soc., 2008, 130, 8142–8143 CrossRef CAS PubMed.
- M. Kotada, R. Kong, I. Qureshi, Q. S. Ho, H. Sun, C. W. Liew, L. P. Goh, P. Cheung, Y. Mu, J. Lescar and Z. X. Liang, J. Biol. Chem., 2009, 284, 15739–15749 CrossRef PubMed.
- K. Belecki, J. M. Crawford and C. A. Townsend, J. Am. Chem. Soc., 2009, 131, 12564–12566 CrossRef CAS PubMed.
- D. R. Cohen and C. A. Townsend, Nat. Chem., 2018, 10, 231–236 CrossRef CAS PubMed.
- D. R. Cohen and C. A. Townsend, Angew. Chem., Int. Ed., 2018, 57, 5650–5654 CrossRef CAS.
- K. L. McPhail, J. Correa, R. G. Linington, J. González, E. Ortega-Barría, T. L. Capson and W. H. Gerwick, J. Nat. Prod., 2007, 70, 984–988 CrossRef CAS PubMed.
- A. C. Jones, E. A. Monroe, S. Podell, W. R. Hess, S. Klages, E. Esquenazi, S. Niessen, H. Hoover, M. Rothmann, R. S. Lasken, J. R. Yates III, R. Reinhardt, M. Kube, M. D. Burkart, E. E. Allen, P. C. Dorrestein, W. H. Gerwick and L. Gerwick, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8815–8820 CrossRef CAS PubMed.
- X. Zhu, J. Liu and W. Zhang, Nat. Chem. Biol., 2015, 11, 115–120 CrossRef CAS PubMed.
- P. C. Dorrestein, J. Blackhall, P. D. Straight, M. A. Fischbach, S. Garneau-Tsodikova, D. J. Edwards, S. McLaughlin, M. Lin, W. H. Gerwick, R. Kolter, C. T. Walsh and N. L. Kelleher, Biochemistry, 2006, 45, 1537–1546 CrossRef CAS PubMed.
- M. Su, X. Zhu and W. Zhang, AIChE J., 2018, 64, 4255–4262 CrossRef CAS PubMed.
- N. A. Moss, G. Seiler, T. F. Leao, G. Castro-Falcon, L. Gerwick, C. C. Hughes and W. H. Gerwick, Angew. Chem., Int. Ed., 2019, 58, 9027–9031 CrossRef CAS PubMed.
- M. Sanada, T. Miyano and S. Iwadare, J. Antibiot., 1986, 39, 304–305 CrossRef CAS PubMed.
- H. C. Potgieter, N. M. J. Vermeulen and D. J. J. Potgieter, Phytochemistry, 1977, 16, 1757–1759 CrossRef CAS.
- P. Brzović, E. L. Holbrook, R. C. Greene and M. F. Dunn, Biochemistry, 1990, 29, 442–451 CrossRef PubMed.
- D. V. Kuklev, A. J. Domb and V. M. Dembitsky, Phytomedicine, 2013, 20, 1145–1159 CrossRef CAS PubMed.
- K. Sakai, T. Hirose, M. Iwatsuki, T. Chinen, T. Kimura, T. Suga, K. Nonaka, T. Nakashima, T. Sunazuka, T. Usui, Y. Asami, S. Ōmura and K. Shiomi, J. Nat. Prod., 2018, 81, 1604–1609 CrossRef CAS PubMed.
- P. Lan, L. E. White, E. S. Taher, P. E. Guest, M. G. Banwell and A. C. Willis, J. Nat. Prod., 2015, 78, 1963–1968 CrossRef CAS PubMed.
- J. Li, S. Park, R. L. Miller and D. Lee, Org. Lett., 2009, 11, 571–574 CrossRef CAS PubMed.
- H. Zhao, G. D. Chen, J. Zou, R. R. He, S. Y. Qin, D. Hu, G. Q. Li, L. D. Guo, X. S. Yao and H. Gao, Org. Lett., 2017, 19, 38–41 CrossRef CAS PubMed.
- Y. Pan, L. Liu, F. Guan, E. Li, J. Jin, J. Li, Y. Che and G. Liu, ACS Chem. Biol., 2018, 13, 703–711 CrossRef CAS PubMed.
- D. O’Hagan, C. Schaffrath, S. L. Cobb, J. T. G. Hamilton and C. D. Murphy, Nature, 2002, 416, 279 CrossRef PubMed.
- V. Agarwal, Z. D. Miles, J. M. Winter, A. S. Eustaquio, A. A. El Gamal and B. S. Moore, Chem. Rev., 2017, 117, 5619–5674 CrossRef CAS PubMed.
- Y. Sugai, Y. Katsuyama and Y. Ohnishi, Nat. Chem. Biol., 2016, 12, 73–75 CrossRef CAS PubMed.
- F. Amblard, J. H. Cho and R. F. Schinazi, Chem. Rev., 2009, 109, 4207–4220 CrossRef CAS PubMed.
- J. Lehmann, M. H. Wright and S. A. Sieber, Chemistry, 2016, 22, 4666–4678 CrossRef CAS PubMed.
- E. S. Hems, B. A. Wagstaff and G. Saalbach, Chem. Commun., 2018, 54, 12234–12237 RSC.
- I. Koryakina, C. Kasey, J. B. McArthur, A. N. Lowell, J. A. Chemler, S. Li, D. A. Hansen, D. H. Sherman and G. J. Williams, ACS Chem. Biol., 2017, 12, 114–123 CrossRef CAS PubMed.
- X. Zhu and W. Zhang, Front. Chem., 2015, 3, 11 Search PubMed.
- X. Zhu, P. Shieh, M. Su, C. R. Bertozzi and W. Zhang, Chem. Commun., 2016, 52, 11239–11242 RSC.
- M. Sandy, Z. Rui, J. Gallagher and W. Zhang, ACS Chem. Biol., 2012, 7, 1956–1961 CrossRef CAS PubMed.
- Y. Yan, J. Chen, L. Zhang, Q. Zheng, Y. Han, H. Zhang, D. Zhang, T. Awakawa, I. Abe and W. Liu, Angew. Chem., Int. Ed., 2013, 52, 12308–12312 CrossRef CAS PubMed.
- W. B. Porterfield, N. Poenateetai and W. Zhang, iScience, 2020, 23, 100938 CrossRef CAS PubMed.
- C. Bihlmaier, E. Welle, C. Hofmann, K. Welzel, A. Vente, E. Breitling, M. Muller, S. Glaser and A. Bechthold, Antimicrob. Agents Chemother., 2006, 50, 2113–2121 CrossRef CAS PubMed.
- B. J. Rawlings, Nat. Prod. Rep., 2001, 18, 190–227 RSC.
- S. Yuzawa, K. Deng, G. Wang, E. E. Baidoo, T. R. Northen, P. D. Adams, L. Katz and J. D. Keasling, ACS Synth. Biol., 2017, 6, 139–147 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |