
Recent advances of polymer acceptors for high-performance organic solar cells
Congcong
Zhao
,
Jiuxing
Wang
 *,
Jiqing
Jiao
,
Linjun
Huang
and
Jianguo
Tang
*
*,
Jiqing
Jiao
,
Linjun
Huang
and
Jianguo
Tang
*
Institute of Hybrid Materials, National Center of International Joint Research for Hybrid Materials Technology, National Base of International Sci. & Tech. Cooperation on Hybrid Materials, School of Materials Science and Engineering, Qingdao University, 308 Ningxia Road, Qingdao 266071, China. E-mail: jiuxingwang@qdu.edu.cn; tang@qdu.edu.cn
First published on 20th November 2019
Abstract
In the past few decades, polymer solar cells (PSCs) have been intensively investigated in academic fields. The study of non-fullerene polymer acceptors has become a hot research focus due to their excellent opto-electronic properties such as wide light-absorbing ability, appropriate molecular energy levels, and easy chemical modifications. The much higher power conversion efficiencies (PCEs) of non-fullerene PSCs relative to fullerene PSCs revealed the significant potential of non-fullerene acceptors in PSCs. This review systematically summarizes the recent advancements of efficient polymer acceptors, including perylene diimide-based, naphthalene diimide-based, diketopyrrolopyrrole-based, double B←N bridged bipyridyl-based, and other polymer acceptors. Their structure–property relationships were thoroughly analyzed and summarized, which may provide new guidance for the rational structural design of high-performance photovoltaic materials.
 Congcong Zhao | Congcong Zhao received his BS degree in Materials Science and Engineering from Liaocheng University in 2018. He is currently pursuing a MS degree at Qingdao University under the supervision of Dr Jiuxing Wang. His research interests are the design and synthesis of efficient organic and polymer materials in solar cells. |
 Jiuxing Wang | Jiuxing Wang received his BS degree in Polymer Materials and Engineering from Sichuan University in 2011 and PhD degree in Chemical Engineering with Prof. Renqiang Yang from University of Chinese Academy of Sciences in 2016. He has been an Assistant Professor at Qingdao University since 2016. His research interests focus on organic and polymer materials for photovoltaic applications. |
 Jianguo Tang | Jianguo Tang received his PhD degree from Shanghai Jiaotong University in 2000, after which, he worked at University Bayreuth as a Visiting Professor (2001). Then, he joined Prof. L. A. Belfiore's group at Colorado State University as a research fellow (2002–2003). Currently, he is the director of National Center of International Joint Research for Hybrid Materials at Qingdao University. His research interests include hybrid organic–inorganic luminescent materials and solar cells. |
1. Introduction
With the rapid economic growth, the consumption of fossil energy sources has greatly increased, which has resulted in critical energy crises and severe environmental problems. Therefore, it is of great importance to develop renewable and clean energy resources to alleviate energy crises and environmental problems. Solar energy has attracted a great deal of attention due to its clean nature and large capacity to meet the whole world's energy demands.1 Photovoltaics is a convenient way to utilize solar energy. Among the various kinds of solar cells, polymer solar cells (PSCs) have emerged during the past decades2–10 because of the following intrinsic advantages compared to its counterparts. (1) The excellent solution processability of organic active layer materials makes it possible to realize roll-to-roll processing as well as large-area industrial production.11 (2) It is easy to fine-tune energy levels of both donors and acceptors.12 (3) The mechanical flexibility contributes to a wide range of applications.13The device structures used for organic solar cells have experienced the revolution of single layer, bilayer, bulk heterojunction (BHJ), and tandem structures. The very first solar cell device structure was a single layer configuration (a, Fig. 1), which possessed a poor power conversion efficiency (PCE) of less than 0.5% due to the low charge mobility of the organic materials.14 The representative structure of the bilayer heterojunction (b, Fig. 1) contained a p-type semiconductor and n-type semiconductor. This structure dramatically facilitated exciton dissociation and charge transport and thus improved the PCE to 1%.15 Later, a BHJ device structure (c, Fig. 1) was invented to increase the D/A interfacial area by blending donor materials and acceptor materials. The PCEs of single junction devices range from 13–15%.16,17 To further enhance the PCEs of PSCs, tandem solar cells were fabricated to more efficiently utilize the sunlight. The highest PCE of tandem devices has exceeded 17%.18,19 To date, BHJ and tandem solar cells are widely-used strategies to improve PCEs. Moreover, the incorporation of hole and electron transport materials (e.g., PEDOT:PSS, MoO3, and TiOx) further enhanced the PCEs.20,21
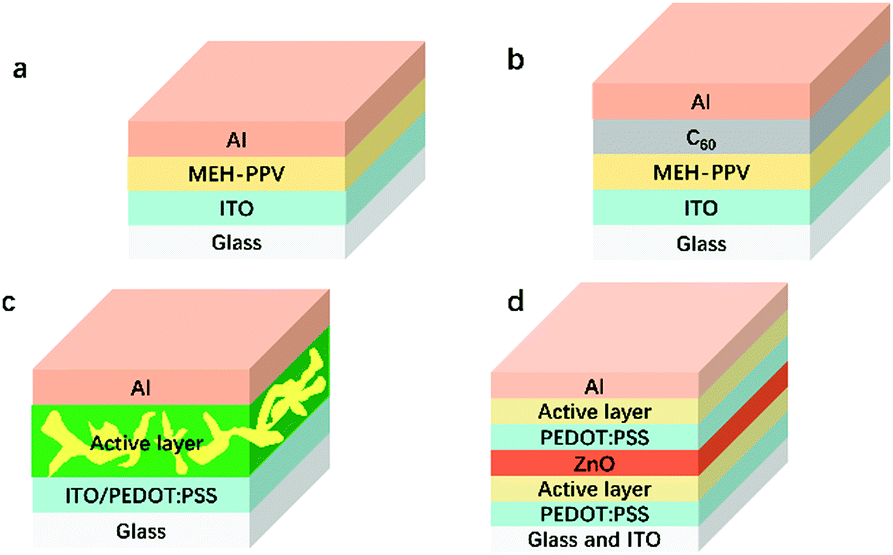 | ||
| Fig. 1 Evolution of the solar cell device structures. | ||
The development of active layer materials plays an important role in improving PCEs. In the early phases, acceptors were mainly fullerene derivatives (Fig. 2). The enhanced PCEs were mostly ascribed to the development of efficient donors.22–25 In recent years, the rapid development of non-fullerene acceptors has greatly promoted PCEs from ∼12% to over 16% (Fig. 2).6,26,27
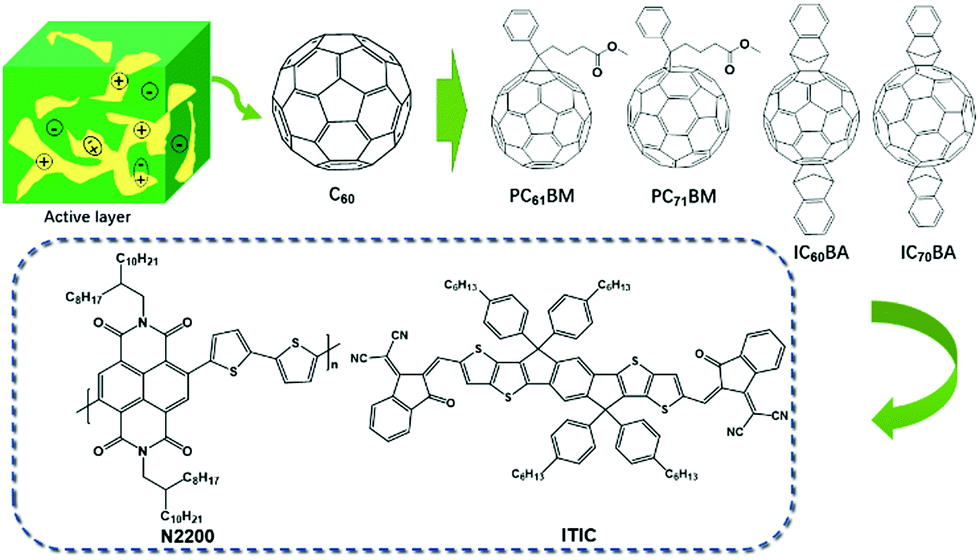 | ||
| Fig. 2 Evolution of the acceptor structures. | ||
In the early stages, the most efficient acceptors were fullerene derivatives. The photo-induced ultrafast electron transfer between a conjugated polymer donor MEH-PPV and acceptor C60 was discovered by Sariciftci et al. in 1993.28 It was the first time that C60 was selected as an electron acceptor. However, the PCE of this double layered device was low because the short diffusion length (usually ca. 10 nm) of the excitons resulted in the recombination of excitons, leading to a low charge separation efficiency. To overcome this difficulty, BHJ was introduced in PSCs by Heeger and coworkers in 1995.29 In this type of device, a blend of conjugated polymer donors and C60 derivative acceptor PC61BM was used as the active layer. By controlling the phase separation into the interpenetrating network, a much larger interfacial D/A contacting area was achieved, which allowed for the collection and dissociation of a larger number of excitons and thus improved the PCE. However, a drawback of PC61BM is its very weak absorption in the visible region due to the high degree of symmetry of C60. Therefore, C70 derivative PC71BM, which possesses a stronger visible absorption than PC61BM was widely used in the fabrication of BHJ PSCs. In addition, to further improve the lowest unoccupied molecular orbital (LUMO) energy level, indene-C60 bisadduct (IC60BA) and indene-C70 bisadduct (IC70BA) were synthesized.30,31 The LUMO energy level of ICBA was increased to −3.74 eV, which was 0.17 eV higher than that of PCBM, and thus an open-circuit voltage (Voc) of 0.82 V was achieved. After selecting P3HT as the donor, the PCE of the ICBA-based PSC was 6.2% by optimizing the device structure, which was a 40% increase in comparison with the PCE of the PCBM-based PSC (4.4%).32
As mentioned above, fullerene and its derivatives as electron acceptors have dominated the field of PSCs for a long time because they exhibited a good solubility in organic solvents, had high electron mobilities, and possessed electron-affinity values that were suitable for achieving charge separation with various donors.33–35 Nevertheless, fullerene and its derivatives have notable drawbacks such as weak absorption, high cost, limited energy level variability, thermal instability, and inferior mechanical flexibility.36–38 Because of these shortcomings, the highest PCE of fullerene-based PSCs was restricted to about 12%.
Compared with traditional fullerene acceptors, non-fullerene acceptors possess superior properties such as easier tunable energy levels, greater light absorption, and higher stability (Fig. 3). Recently, a breakthrough in the material design and synthesis of non-fullerene acceptors opened up a new landscape for PSCs.39–50 Non-fullerene acceptors include small molecules and polymers.
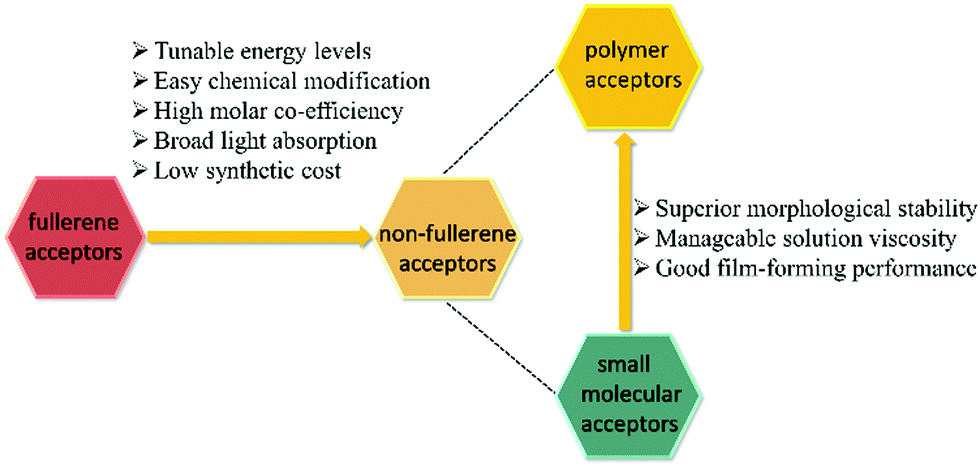 | ||
| Fig. 3 Advantages of polymer acceptors. | ||
1.1 Small molecule acceptors
Owing to the rigid and planar structure, high photochemical stability, easy synthesis, and modification, non-fullerene small molecule acceptors are widely employed as electron-deficient materials. Among various small molecule acceptors, functionalizations on indacenodithiophene (IDT) or indacenodithienothiophene (IDTT) cores with various electron-deficient terminal groups have been extensively studied. For example, the star molecule ITIC developed by Zhan et al. exhibited a broad absorption, low LUMO and HOMO energy levels, good electron transport ability, and good miscibility with polymer donors.51 They continued to modify the structure of ITIC, including extending the fused-ring cores, modulating the end-capping units, and tailoring the side chains.52–56 The corresponding acceptors showed improved properties and thus PCEs over 12% were achieved. In recent years, due to major advancements in the ITIC family, the PCEs of single-junction PSCs have realized over 15%, which has inaugurated a new era in the OPV field.16,57,58 Also, aromatic diimides (PDI and NDI) were widely used as the electron-deficient core or terminal units due to their easy synthesis and high electron mobility.27 More specifically, the trimeric PDI derivatives showed a superior PCE of about 9%. Different numbers of PDI molecules may form a twisted 3D-structure with a certain moiety as the core, which not only inhibits the self-aggregation of a mixed membrane, but also promotes the formation of phase separation on the nanometer scale.59–61 In addition, diketopyrrolopyrrole (DPP) units were widely investigated owing to their excellent photochemical stability and strong absorption in the visible and near-infrared region.1.2 Polymer acceptors
Polymer acceptors are another kind of non-fullerene acceptors. Compared with small molecular acceptors, n-type polymer acceptors have the following advantages. (a) Polymer acceptors have a higher morphological stability and better compatibility with various donor materials. Polymers are more inherently ductile than small molecular acceptors, leading to a sufficient entanglement with other polymer chains.62 (b) Polymer acceptors have lower costs in the same design rules due to an easier synthesis and modification of the molecular structures.63 (c) After blending with polymer donor materials, the solution viscosity is more easily controlled, which is beneficial for a large-scale production through the solution process.64 (d) Polymer acceptors exhibit an excellent mechanical stability because of the greater intrinsic flexibility and the strengthened donor/acceptor interfaces by the entanglements among the polymer chains.65 Due to these merits, polymers have a great potential as non-fullerene acceptors and have attracted the increasing attention of researchers. They have become a new hot-spot topic in solar cell research.In this work, we summarized recently reported high-performance polymer acceptors and classified them into five groups according to their structural features, including perylene diimide (PDI)-based, naphthalene diimide (NDI)-based, diketopyrrolopyrrole (DPP)-based, double B←N bridged bipyridyl (BNBP)-based, and other polymer acceptors. The design strategies and structure–property relationships were also discussed.
2. Recent progress of polymer acceptors in photovoltaic applications
2.1 Perylene diimide (PDI)-based polymer acceptors
Perylene diimide (PDI, Fig. 4) and its derivatives, which have outstanding chemical and physical properties such as high electron mobility and high molar absorption coefficients represent one of the most promising acceptors for PSCs.66–68 There are various positions for functionalization in a PDI molecular structure, including bay positions, ortho positions, and imide positions.69–71 The modifications in the molecular structures can be easily accomplished by introducing other moieties at these positions to disturb the planarity and construct twisted configurations.72 For instance, the bay positions are easy to modify to regulate optical and electronic properties and have a great influence on the absorption in the visible to near-infrared spectral region, which is considered to be the most common and efficient method. Substitutions at the imide positions are chemically simple but less effective in changing the planarity of the molecular backbone.73 In addition, the substitutions could alter the solubility of the molecules as well as the film morphology, thereby affecting the efficiency of the devices. Functionalization of ortho positions can also effectively improve the performance of the devices, but it is synthetically difficult to accomplish to some degree. For conjugated PDI molecules fused at the bay positions, the extended fused unit is believed to have a large π-electron delocalization, relatively high electron mobilities, and a good electron accepting ability.74 However, the steric hindrance of the substituent groups affects the π–π intermolecular stacking of the PDI core, thus impacting the solubility, aggregation, and electron mobility of the material in the solid state. How to design rational PDI structures that suppress aggregation while retaining a high electron mobility is an important topic. The optoelectronic properties and photovoltaic characteristics of PDI-based polymer acceptors are summarized in Table 1.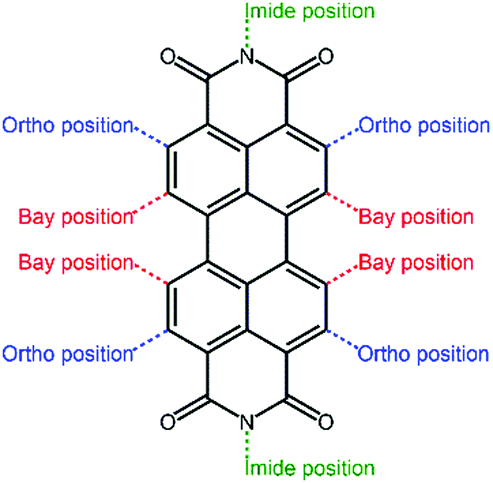 | ||
| Fig. 4 Core structure of the PDI unit. | ||
| Acceptors | HOMO (eV) | LUMO (eV) | E g (eV) | Donors | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a1 | — | — | — | Polythiophene derivative | 0.63 | 4.2 | 39 | 1.5 | 75 |
| a2 | −6.10 | −4.30 | 1.80 | PTB7-Th | 0.70 | 9.98 | 51.0 | 3.58 | 76 |
| a3 | −5.77 | −4.07 | 1.70 | PTB7-Th | 0.75 | 11.2 | 54 | 4.5 | 77 |
| a4 | −5.76 | −4.11 | 1.65 | PTB7-Th | 0.85 | 7.2 | 42 | 2.5 | 77 |
| a5 | −5.46 | −3.87 | 1.59 | PTB7-Th | 0.78 | 10.65 | 49 | 4.07 | 78 |
| a6 | −5.75 | −4.04 | 1.71 | PTB7-Th | 0.76 | 14.13 | 58 | 6.23 | 78 |
| a7 | −5.63 | −3.87 | 1.76 | PTB7-Th | 0.81 | 11.8 | 65 | 6.2 | 79 |
| a8 | −5.65 | −3.86 | 1.79 | PTB7-Th | 0.80 | 10.6 | 63 | 5.3 | 79 |
| a9 | −5.82 | −4.12 | 1.70 | PTB7-Th | 0.73 | 13.47 | 65 | 6.39 | 80 |
| a10 | −5.94 | −4.15 | 1.79 | PTB7-Th | 0.67 | 13.31 | 60 | 5.35 | 80 |
| a11 | −5.97 | −4.11 | 1.86 | PTB7-Th | 0.77 | 12.74 | 64 | 6.28 | 81 |
| a12 | −6.02 | −4.11 | 1.91 | PTB7-Th | 0.76 | 14.00 | 61 | 6.49 | 81 |
| a13 | −6.03 | −4.12 | 1.91 | PTB7-Th | 0.76 | 13.96 | 62 | 6.58 | 81 |
| a14 | −6.05 | −3.95 | 2.10 | PTB7-Th | 0.74 | 12.98 | 54.0 | 5.20 | 82 |
In 2007, Zhan et al. first reported a PDI-based polymer acceptor (a1, Fig. 5) consisting of alternating PDI and dithienothiophene units.75 A high electron mobility and broad absorption that extended from the visible to near-infrared region (ca. 850 nm) were observed in e1. After selecting a bi(thienylenevinylene)-substituted polythiophene as the donor, the corresponding solar cell exhibited a Voc of 0.63 V, a short-circuit current density (Jsc) of 4.2 mA cm−2, and a fill factor (FF) of 39%. The average PCE of the device ranged from 1% to 2%. Although the PCEs were not very high, the pioneering work drew wide attention in studying PDI-based polymer acceptors. Kim et al. synthesized a triple bond-linked PDI conjugated polymer (a2, Fig. 5).76 After selecting PTB7-Th as the donor, the blend film showed a very small domain size (ca. 20 nm) because of the ability of a2 to self-assemble highly ordered structures. Li and coworkers reported two co-polymers (a3 and a4, Fig. 5) consisting of a perylene bisimide dimer or single perylene bisimide as the acceptor and bithiophene as the donor unit attached to the imide position.77 They found that PTB7-Th:a3 cells showed a significantly high photocurrent and PCEs of 4.5% compared to that of PTB7-Th:a4 cells (2.5%). Atomic force microscopy (AFM) showed that the a3-based thin films had a better mixed morphology with a small domain size, which contributed to an efficient charge separation. It was found that a3 had a poor crystalline tendency because of the highly twisted backbone, leading to a better miscibility with PTB7-Th. Zhao et al. synthesized two novel acceptor–acceptor type polymer acceptors (a5 and a6, Fig. 5) containing PDI and electron deficient 4,7-dithienyl-2,1,3-benzothiadiazole (DTBT) units.78 With PTB7-Th as the polymer donor, a5-based PSC showed a dramatic PCE of 4.07%, whereas the photovoltaic performance of the a6-based device was further improved with a PCE of 6.23%. They also optimized the device performance by adding different additives into the polymer/polymer blends. The results showed that the device performance of PTB7-Th:a5 was improved with an enhanced Jsc (10.65 mA cm−2) and FF (49%) upon adding 3% (v/v) 1-chloronaphthalene (CN). Similarly, an enhanced performance was observed with 0.5% (v/v) 1,8-diiodooctane (DIO) for the PTB7-Th:a6 device (Jsc = 14.13 mA cm−2, FF = 58%).
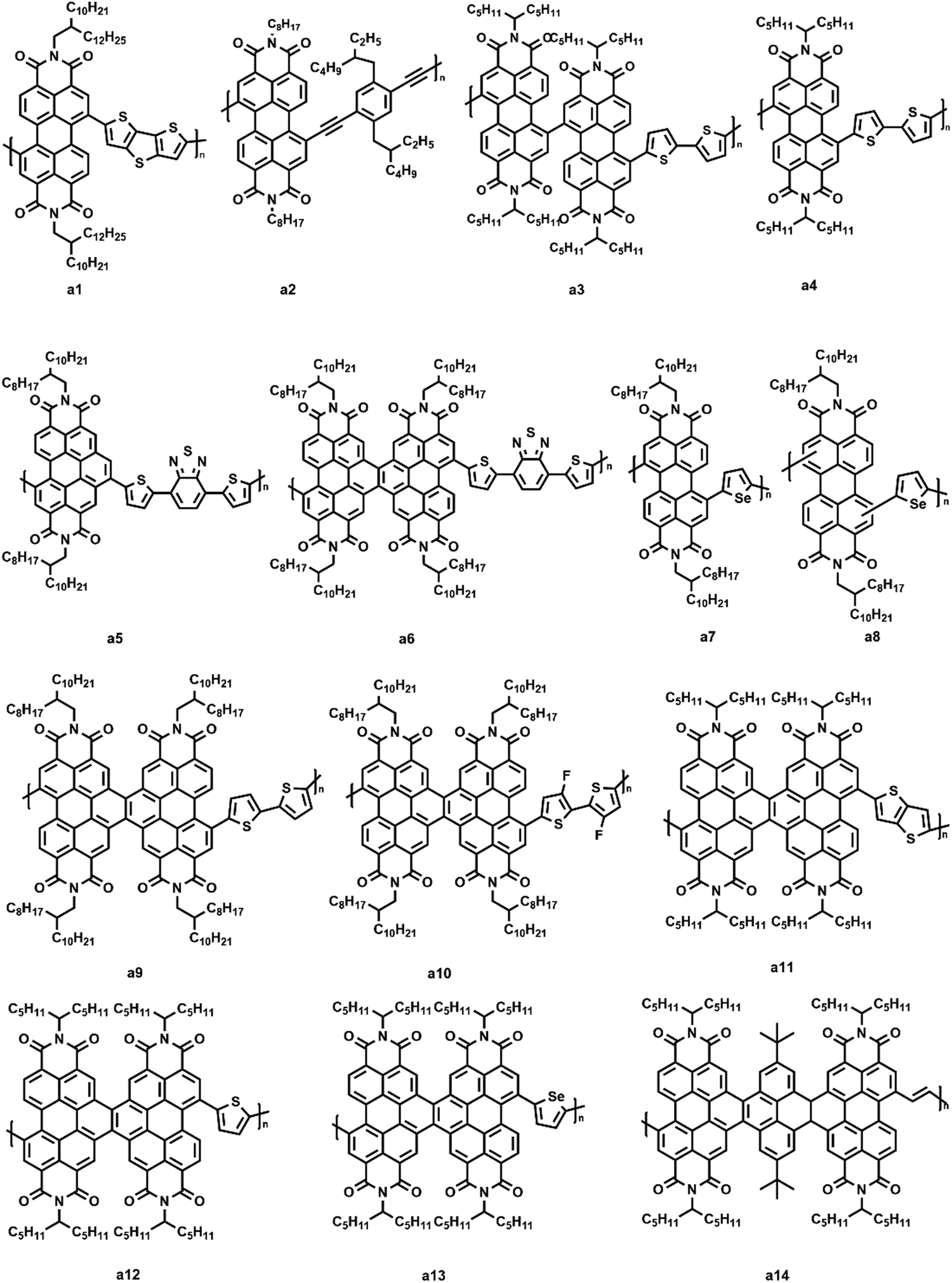 | ||
| Fig. 5 The structures of PDI-based polymer acceptors. | ||
It is a common phenomenon that researchers focus largely on employing various π-linkers and/or extending π-conjugated PDI cores to build objective acceptor polymers. However, the regioregularity of polymers strongly influences the conjugation length and crystalline order of the polymers. To study how regioregularity impacts the properties of corresponding polymers, Zhan and his group reported two new regioregular and regioirregular model copolymer acceptors based on selenophene and perylenetetracarboxylic diimide moieties, respectively.79 They investigated the photovoltaic properties of PSCs that contained blends of the PTB7-Th donor material with two polymer acceptors (a7 and a8, Fig. 5). The regioirregular a8-based PSCs displayed promising average PCEs of about 5.3%, whereas the regioregular a7 based-PSCs displayed a significantly improved average PCE of up to 6.2%. Zhao and coworkers synthesized two polymeric electron acceptors (a9 and a10, Fig. 5) based on the fused PDI and bithiophene or difluorobithiophene units in their previous work.80 When fabricated with PTB7-Th as the polymeric electron donor, a PCE as high as 6.39% based on PTB7-Th:a9 was achieved, which was significantly higher than that of the all-PSC based non-fused PDI counterparts. Recently, their group reported three n-type polymer acceptors (a11–a13, Fig. 5) based on fused PDI and thieno[3,2-b]thiophene, thiophene, or selenophene units for PSCs.81 The PCEs of PSCs based on three polymers as acceptors and PTB7-Th as a polymer donor achieved an exceeding 6%. They also demonstrated that the photovoltaic performances of the corresponding devices were determined by a fused PDI with a small effect from the comonomers. A twisted ladder-type polymer (a14, Fig. 5) with a bearing PDI linked by pyrene units was synthesized by Ba et al.82 The highly twisted structure was considered to prevent the formation of π–π stacking and crystallization in the aggregated states. Then, the PTB7-Th:a14-based device showed a high PCE of 5.20%.
In brief, the photovoltaic performance of the PDI-based PSCs can be improved by the following methods. (1) Judiciously selecting proper donor units to fine-tune the absorption ability and LUMO energy levels. (2) Employing small π linkers such as vinylene or ethynylene into copolymers relieved the steric hindrance, thus facilitating π–π stacking and electron mobility. (3) Extending the fused PDI units resulted in torsional configurations and prevented the strong aggregation of the PDI units, which was helpful for improving the photovoltaic performance.
2.2 Naphthalene diimide (NDI)-based polymer acceptors
In addition to PDI and its derivatives, its counterpart, naphthalene diimide (NDI, Fig. 6), was also employed to construct non-fullerene acceptors. Similar to PDI, the imide positions were easily modified with various substituents to improve the solubility as well as adjust the polymer crystallinity, which affected the morphological control and phase separation in the blend polymer film.83 Moreover, owing to the high electron affinity as well as good chemical and photochemical stabilities, NDI was regarded as an ideal candidate.84 However, compared to PDI, NDI-based polymer acceptors exhibited a lower molar absorption coefficient and lower electron mobilities due to the shortened conjugated backbone. Therefore, NDI was often used as the electron-withdrawing moiety to design polymer acceptors. The optoelectronic properties and photovoltaic characteristics of NDI-based polymer acceptors are illustrated in Table 2.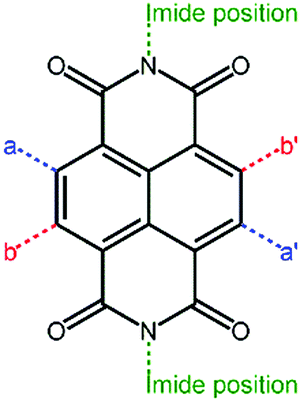 | ||
| Fig. 6 Core structure of the NDI unit. | ||
| Acceptors | HOMO (eV) | LUMO (eV) | E g (eV) | Donors | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| b1 | — | — | — | PTzBI-Si | 0.86 | 16.8 | 78.8 | 11.0 | 90 |
| b2 | −5.82 | −3.95 | 1.87 | PBDB-T | 0.85 | 18.32 | 57 | 9.38 | 91 |
| b3 | −5.91 | −3.91 | 2.00 | PBDB-T | 0.85 | 14.83 | 64.32 | 8.13 | 92 |
| b4 | −5.59 | −3.95 | 1.64 | PBDTTTPD | 0.94 | 12.15 | 65 | 7.4 | 93 |
| b5 | −5.59 | -3.95 | 1.64 | PBDTTTPD | 0.95 | 10.41 | 66 | 6.5 | 93 |
| b6 | −6.36 | −4.05 | 2.31 | PTB7-Th | 0.83 | 12.9 | 71 | 7.6 | 94 |
| b7 | −6.37 | −4.05 | 2.32 | PTB7-Th | 0.83 | 9.7 | 52 | 4.2 | 94 |
| b8 | −6.38 | −4.00 | 2.38 | PTB7-Th | 0.83 | 5.8 | 48 | 2.3 | 94 |
| b9 | −5.37 | −3.94 | 1.43 | PTB7-Th | 0.74 | 9.42 | 39.2 | 2.74 | 96 |
| b10 | −5.36 | −3.93 | 1.43 | PTB7-Th | 0.77 | 11.4 | 54.1 | 4.75 | 96 |
| b11 | −6.08 | −3.82 | 2.26 | PBDB-T | 0.85 | 12.8 | 68 | 7.4 | 97 |
| b12 | −6.02 | −3.78 | 2.24 | PBDB-T | 0.86 | 12.6 | 65 | 7.0 | 97 |
| b13 | −5.78 | −3.90 | 1.88 | PBDT-TAZ | 0.86 | 11.4 | 69 | 6.8 | 98 |
| b14 | −5.81 | −3.91 | 1.90 | PBDT-TAZ | 0.84 | 12.9 | 75 | 8.1 | 98 |
| b15 | −5.82 | −3.98 | 1.84 | PBDT-TAZ | 0.83 | 11.9 | 73 | 7.3 | 98 |
| b16 | −5.83 | −4.07 | 1.76 | PBDT-TAZ | 0.82 | 10.7 | 69 | 6.1 | 98 |
| b17 | −5.65 | −3.82 | 1.83 | PBDTTTPD | 1.04 | 9.55 | 61.2 | 6.1 | 99 |
| b18 | −5.59 | −3.82 | 1.77 | PBDTTTPD | 1.04 | 10.23 | 61.2 | 6.5 | 99 |
| b19 | −5.56 | −3.85 | 1.71 | PBDTTTPD | 1.04 | 10.96 | 62.5 | 7.1 | 99 |
| b20 | −5.59 | −3.86 | 1.73 | PBDTTTPD | 1.03 | 10.67 | 62.0 | 6.8 | 99 |
Recently, a NDI–bithiophene copolymer P(NDI2OD-T2), also named N2200, was extensively studied, from which, a promising PSC performance with a PCE of 8% or higher was achieved.85–89 In particular, Cao et al. achieved a high photovoltaic performance for all-polymer solar cells (all-PSCs) using a green solvent system based on cyclopentyl methyl ether (CPME) to manipulate the BHJ morphology.90 After processing with CPME, the PTzBI-Si:N2200 (b1, Fig. 7) blend film exhibited an enhanced absorption coefficient in the range of 300–480 nm and a small phase separation with improved polymer mixing. Thus, the highest PCE of 11.0% was obtained. Similarly, Jenekhe and coworkers synthesized an NDI–biselenophene copolymer (b2, Fig. 7), which was a selenophene analogue of N2200.91 After using PBDB-T as the polymer donor, the all-PSCs achieved up to a 9.38% PCE, high Jsc (18.32 mA cm−2), and low optical band-gap energy loss (0.53 eV). The high molecular weight and short alkyl side chains of b2 were helpful in enhancing the electron mobility and face-on orientations. An additive-free acceptor (b3, Fig. 7) was synthesized by Li et al. by introducing a small amount (molar percentage: 5%) of rhodanine-based dye groups into the NDI-based polymer acceptor to partly replace the NDI blocks in N2200.92 The incorporation of the dye groups into the backbones dramatically improved the absorption coefficient and LUMO energy level as well as reduced crystallization. Consequently, the PBDBT:b3 based device achieved a high PCE of 8.13%. Two NDI-based copolymers (b4 and b5, Fig. 7) that incorporated electron-withdrawing cyanovinylene groups into the backbone with 2-hexyldecyl and 2-octyldodecyl side chains were synthesized by Kim et al.93 There was an enhancement in the dipole moment change (Δμge) and delocalization of the LUMO energy level upon the incorporation of the cyanovinylene groups. The PSCs fabricated from these polymer acceptors and polymer donor (PBDTTTPD) exhibited a maximum PCE of 7.4% and a high FF of 65%. To modulate the crystallinity of N2200, Wang et al. introduced a certain number of single thiophene units to replace the bithiophene units in the N2200 backbone and synthesized a series of random copolymers (b6–b8, Fig. 7).94 These copolymers were expected to have a more flexible main chain and lower crystallinity since the regularity of the random copolymers was reduced. Their photovoltaic performance was studied using PTB7-Th as the donor in all-PSCs. The highest PCE of 7.6% with a high Jsc of 12.9 mA cm−2 and a record FF of 71% were achieved due to an optimal crystallinity and miscibility of b6 with PTB7-Th, which resulted in balanced hole and electron mobilities and reduced bimolecular recombination. Their group also obtained high-performance all-PSCs based on PTB7-Th and b6 by roll-to-roll printing techniques.95 They found that the addition of 0.4 vol% DIO in the BHJ solution prolonged the drying time from 127 to 1066 s, resulting in a better morphology and a higher crystallinity. The FF was boosted to 71% and the PCE up to 8.61%. Moreover, the flexible ITO-free all-PSCs with a large-area active layer (2.03 cm2) gave an excellent PCE of 6.65%, indicating the bright future for the practical applications of all-PSC devices. Side chain engineering is an effective way to modulate the conformation and ordering of the polymer chains in the films. Chen et al. designed and synthesized two novel polymer acceptors (b9 and b10, Fig. 7) with thiophene groups in the side chains attached to NDI units.96 When paired with PTB7-Th as the donor, the b10-based PSC device demonstrated a PCE of 4.75%, which was higher than that of the b9-based PSC (2.74%) because the shorter alkyl side chains of b9 led to its relatively poor solubility and unbalanced charge transport. Chen and coworkers reported two novel acceptor polymers (b11 and b12, Fig. 7) containing siloxane-terminated side chains.97 By modulating the proper loadings of the siloxane-terminated side chains on the acceptor polymers, the PBDB-T:b11 all-PSC obtained a maximum PCE of 7.4% with an outstanding FF of 68%. Similarly, the Cao group designed a series of polymer acceptors (b13–b16, Fig. 7) by introducing a fraction of linear oligoethylene oxide side chains to replace the branched alkyl chains on the NDI units.98 After blending with PBDT-TAZ, the b14-based all-PSC offered a much higher PCE of 8.1% with an FF of 75%. Kim et al. reported a series of NDI-based polymers (b17–b20, Fig. 7) with different branching points. They systematically studied the effect of the branching point of the side chain in the NDI-based polymers.99 It was found that when the branching point was far away from the conjugated backbone, the steric hindrance was relaxed and the intermolecular interactions were stronger, thus facilitating the formation of crystalline structures in the thin film state. When these polymers were applied in the all-PSCs as electron acceptors, a high PCE of 7.1% was achieved in the b19-based all-PSC.
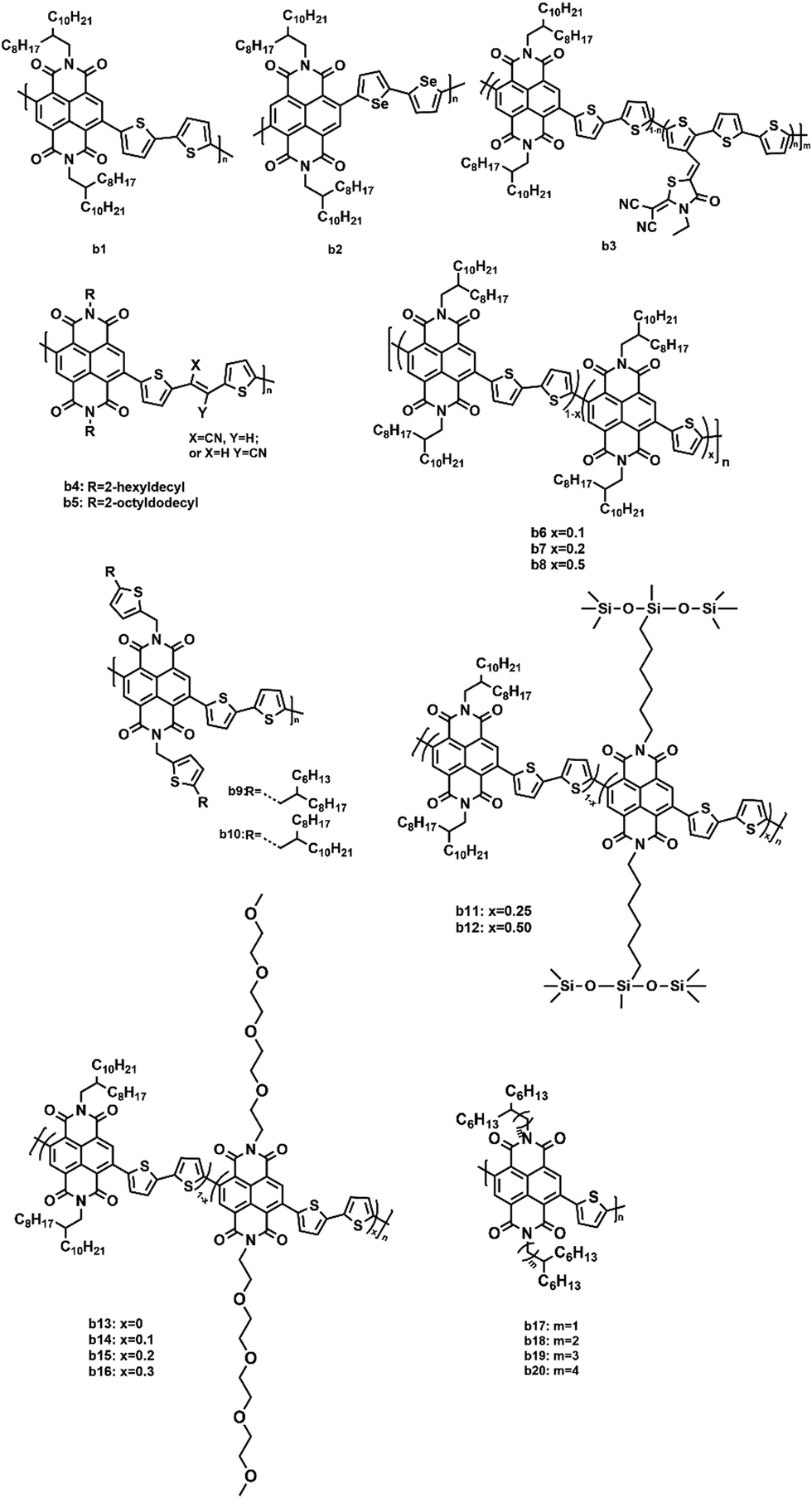 | ||
| Fig. 7 The structures of NDI-based polymer acceptors. | ||
As discussed above, the optimization strategies to improve the photovoltaic performance of NDI-based polymers focused on a rational molecular design and BHJ morphology control. The use of a proper solvent system plays an important role in mixing NDI-based polymers and donor materials, which is desirable for the molecular orientation and nanoscale phase separation. From the perspective of the molecular design, introducing dye groups into the backbone of NDI-based polymers is conductive to improving the absorption coefficient and LUMO energy levels. Moreover, the insertion of a flexible side chain is an efficient way to moderate the crystallinity.
2.3 Diketopyrrolopyrrole (DPP)-based polymer acceptors
Diketopyrrolopyrrole (DPP, Fig. 8) has now become one of the most favored electron deficient units in constructing narrow band gap conjugated polymers. In general, DPP polymers consist of four primary building blocks: a DPP unit, aromatic substituent, π-conjugated segment, and alkyl side chains. The aromatic substituent connects the DPP unit to the conjugated segment that is comprised of electron rich or electron deficient units or combinations. The optical and electronic properties of the DPP polymers can be tuned by changing the aromatic substituents or π-conjugated segments.100,101 The alkyl side chains linked to the DPP unit promote its solubility as well as adjust the aggregation and crystallization of the polymer.102 The optoelectronic properties and photovoltaic characteristics are summarized in Table 3.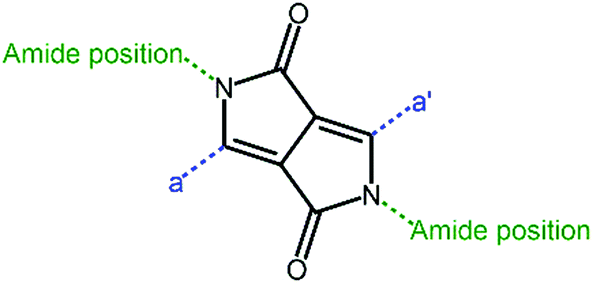 | ||
| Fig. 8 Core structure of the DPP unit. | ||
| Acceptors | HOMO (eV) | LUMO (eV) | E g (eV) | Donors | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| c1 | −5.8 | −4.1 | 1.7 | PTB7-Th | 1.04 | 6.57 | 40 | 2.72 | 103 |
| c2 | −6.19 | −3.98 | 2.21 | PTB7-Th | 1.02 | 5.9 | 39 | 2.3 | 104 |
| c2 | −6.19 | −3.98 | 2.21 | PBDTTS-FTAZ | 1.07 | 9.1 | 43 | 4.2 | 104 |
| c3 | −5.70 | −4.26 | 1.44 | PTB7-Th | 0.71 | 0.66 | 29 | 0.14 | 105 |
| c4 | −5.71 | −4.18 | 1.53 | PTB7-Th | 0.95 | 3.2 | 36 | 1.1 | 105 |
| c5 | −5.69 | −4.16 | 1.53 | PTB7-Th | 0.99 | 6.4 | 37 | 2.4 | 105 |
| c6 | −5.67 | −4.09 | 1.58 | PTB7-Th | 0.94 | 7.5 | 45 | 3.1 | 105 |
| c7 | −5.47 | −4.03 | 1.44 | PDPP5T | 0.81 | 7.1 | 49 | 2.9 | 106 |
| c8 | −5.85 | −4.39 | 1.46 | PDPP5T | 0.79 | 5.7 | 49 | 2.4 | 106 |
| c9 | −5.78 | −4.27 | 1.51 | PDPP5T | 0.75 | 4.1 | 46 | 1.7 | 106 |
| c10 | −5.79 | −4.30 | 1.49 | PDPP5T | 0.75 | 1.6 | 43 | 0.68 | 106 |
| c11 | −5.75 | −4.24 | 1.51 | PDPP5T | 0.76 | 1.8 | 42 | 0.54 | 106 |
| c12 | −6.14 | −4.56 | 1.58 | PDPP5T | 0.67 | 7.4 | 41 | 2.1 | 106 |
McCulloch et al. synthesized novel thieno[2,3-b]pyridine-flanked DPP polymers (c1, Fig. 9).103 When copolymerized with 3,4-difluorothiophene, c1 possessed a highly planar backbone due to the fused thieno[2,3-b]pyridine flanking unit that effectively alleviated the steric hindrance with both the central DPP core and the 3,4-difluorothiophene repeat unit. A PCE of 2.72% with a high Voc of 1.04 V was achieved for the all-PSC using PTB7-Th as the polymer donor. Wang et al. synthesized an acceptor (c2, Fig. 9) containing pyridine-flanked diketopyrrolopyrrole and isoindigo units.104 They selected two donors (PTB7-Th, PBDTTS-FTAZ) to blend with c2. As a result, the PBDTTS-FTAZ based all-PSC attained a higher PCE of 4.2% due to the lower lying HOMO energy level (−6.19 eV) and enhanced absorption coefficient (8.1 × 104 cm−1) of c2. Li and coworkers designed several DPP-based polymers containing different-sized aromatic units from small thiophene units to benzodithiophene and large alkylthio-benzodithiophene units (c3–c6, Fig. 9).105 When blended with PTB7-Th as the donor, c6 with large-sized aromatic units exhibited a maximum PCE of 3.1%. However, the introduction of bulky linkers led to a blue-shifted absorption. Li et al. designed a series of polymers (c7–c12, Fig. 9) consisting of thiazole-flanked DPP units with a varying ratio of thiophene and perfluoroalkyl benzodithiophene units.106 The polymers were found to have small optical band gaps and high crystalline properties, depending on the ratio of the linked units. The optimized PCE of PSC was up to 2.9%.
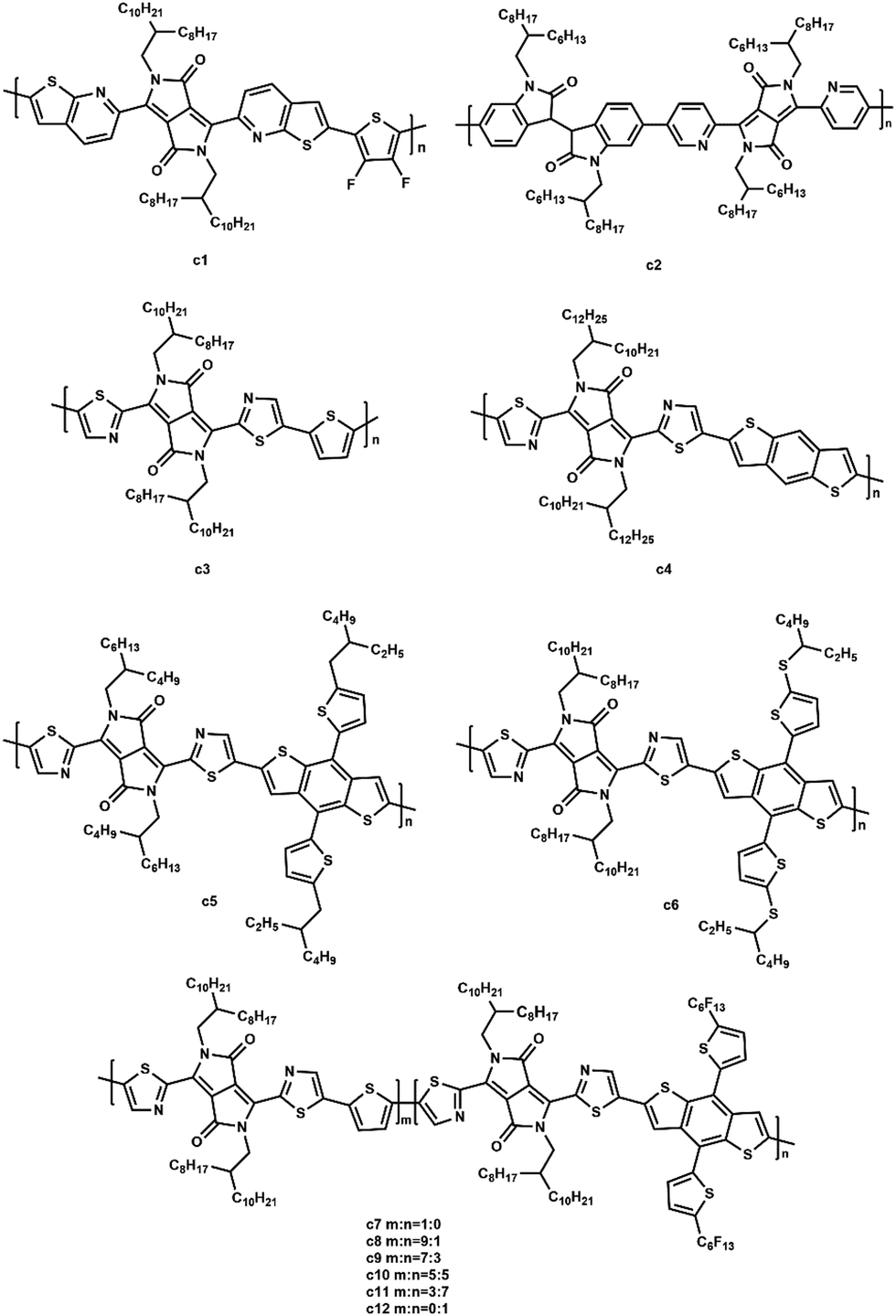 | ||
| Fig. 9 The structures of DPP-based polymer acceptors. | ||
In conclusion, modifications on the aromatic substituent, π-conjugated segment, and alkyl side chains are three pathways to enhance the photovoltaic performance of DPP-based polymers. A planar backbone can be obtained by using π-conjugated moieties as the aromatic substituent due to a reduced steric hindrance. In addition, the introduction of isoindigo units contributed to a lower HOMO energy level and enhanced absorption coefficient.
2.4 Double B←N bridged bipyridyl (BNBP)-based polymer acceptors
The double B←N bridged bipyridyl (BNBP, Fig. 10) is a novel electron-deficient building block for polymer acceptors as a consequence of a fixed planar configuration and low-lying LUMO/HOMO energy levels.107–110 The optoelectronic properties and photovoltaic characteristics of the BNBP-based polymer acceptors are listed in Table 4.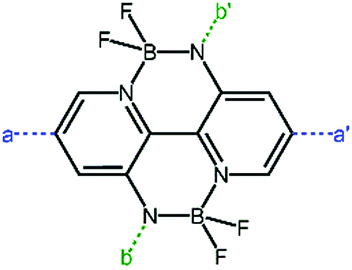 | ||
| Fig. 10 Core structure of BNBP unit. | ||
| Acceptors | HOMO (eV) | LUMO (eV) | E g (eV) | Donors | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| d1 | −5.64 | −3.45 | 2.19 | P3HT | 1.01 | 4.98 | 35 | 1.76 | 111 |
| d2 | −5.89 | −3.60 | 2.29 | PTB7-Th | 0.99 | 8.78 | 44.06 | 3.83 | 112 |
| d3 | −5.77 | −3.50 | 2.27 | PTB7-Th | 1.12 | 5.24 | 39 | 2.27 | 113 |
| d4 | −5.84 | −3.66 | 2.18 | PTB7-Th | 1.03 | 10.02 | 42 | 4.26 | 113 |
| d5 | −5.82 | −3.63 | 2.19 | PTB7 | 1.04 | 6.21 | 38 | 2.47 | 114 |
| d6 | −5.45 | −3.87 | 1.58 | PTB7 | 0.88 | 7.54 | 41 | 2.69 | 115 |
| d7 | −5.81 | −3.42 | 2.39 | J61 | 1.27 | 8.61 | 50 | 5.46 | 116 |
| d7 | −5.81 | −3.42 | 2.39 | PBDTTT-E-T | 1.10 | 7.56 | 42 | 3.51 | 116 |
| d8 | −5.96 | −3.46 | 2.50 | J61 | 1.24 | 2.71 | 39 | 1.30 | 116 |
| d8 | −5.96 | −3.46 | 2.50 | PBDTTT-E-T | 1.11 | 1.18 | 28 | 0.36 | 116 |
| d9 | −5.74 | −3.72 | 2.02 | PTB7-Th | 1.11 | 7.58 | 45 | 3.77 | 117 |
| d10 | −5.75 | −3.73 | 2.19 | PTB7-Th | 1.07 | 9.21 | 45 | 4.46 | 117 |
Wang and coworkers made efforts to design a series of polymers based on BNBP units. They synthesized a polymer acceptor (d1, Fig. 11) composed of an alternating BNBP unit and a cyclopenta-[2,1-b:3,4-b′]-dithiophene (CDT) unit to research the impact of the donor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acceptor blend ratio.111 The highest PCE of 1.76% was achieved in the device with a P3HT:d1 ratio of 5:1.
:acceptor blend ratio.111 The highest PCE of 1.76% was achieved in the device with a P3HT:d1 ratio of 5:1.
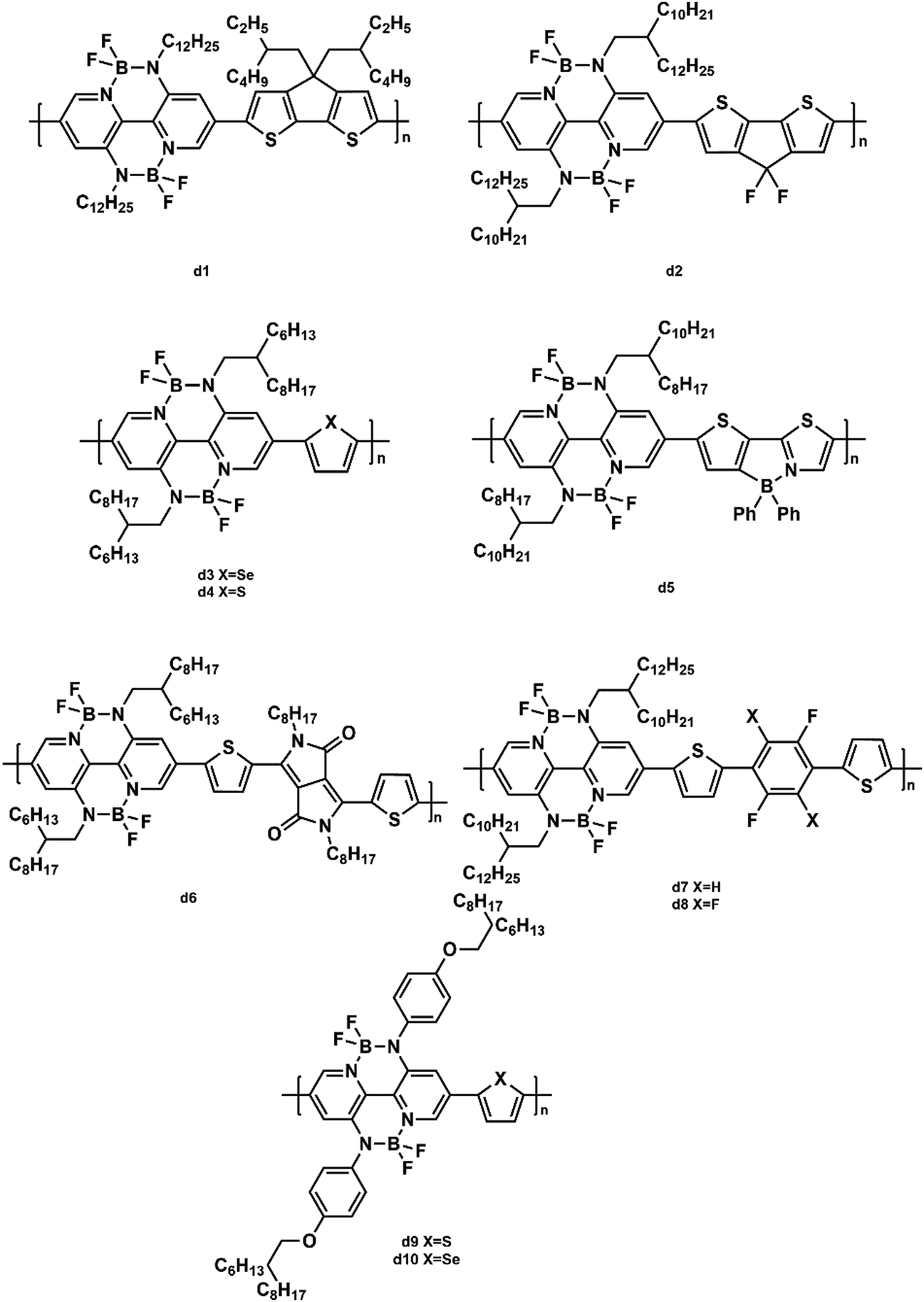 | ||
| Fig. 11 The structures of BNBP-based polymer acceptors. | ||
Wang et al. also introduced fluoro-substitutes into CDT to combine with the BNBP unit.112 The generated polymer acceptor (d2, Fig. 11) exhibited a down-shifted LUMO energy level, a diminished steric hindrance effect, and strong intermolecular interaction, leading to a PCE of 3.83% in the PSC with PTB7-Th as the donor. In addition, their group synthesized two copolymers (d3 and d4, Fig. 11) based on BNBP units and thiophene/selenophene units.113 The introduction of selenophene decreased the Voc because of its lower LUMO energy level, but increased the electron mobility (2.07 × 10−4 cm2 V−1 s−1) of the corresponding blend film. As a result, when d4 blended with PTB7-Th, the device showed a PCE of 4.26%. A new alternating polymer (d5, Fig. 11) based on BNBP and B←N bridged thienylthiazole units exhibited a strong light absorption in the visible region, low-lying LUMO/HOMO energy levels, and a moderate electron mobility.114 The corresponding device based on the PTB7:d5 blend gave a PCE of 2.47%. Their group copolymerized the BNBP unit and DPP unit to develop a new polymer acceptor (d6, Fig. 7) with a small band gap of 1.56 eV.115 The optimal PTB7:d6 (w:w, 2:1) device achieved an enhanced PCE of 2.69% and an increased Jsc of 7.54 mA cm−2 after optimization. To improve the electron mobility of the polymer acceptors, the same group synthesized two polymer acceptors (d7 and d8, Fig. 11) containing BNBP units and fluorinated moieties.116 Fluorination on the polymer backbone could improve the charge carrier mobility by enhancing the main chain planarity and impacting the molecular energy levels. Due to the high electron mobility (5.40 × 104 cm2 V−1 s−1), high-lying LUMO/HOMO energy levels (−3.42/−5.81 eV) of d7, the all-PSC with d7 as the acceptor demonstrated a PCE of 5.46%. They also designed two polymers (d9 and d10, Fig. 11) by incorporating conjugated alkoxyphenyl side chains into the BNBP unit.117 The introduction of the alkoxyphenyl side chain endowed the polymer acceptors with low-lying LUMO energy levels, an enhanced π–π stacking, and high electron mobilities. When blended with PTB7-Th, the resulting all-PSCs exhibited an optimal PCE of 4.46%.
For the BNBP-based polymers, fluorination of the backbones and the introduction of the alkoxyphenyl side chains both alleviated the steric hindrance and decreased the π–π stacking distances (dπ–π). Consequently, the small dπ–π led to strong intermolecular interactions and facilitated the electron mobilities of the resulting polymers.
2.5 Other polymer acceptors
Besides the above-discussed polymer acceptors, there were still many miscellaneous acceptors reported. Their optoelectronic properties and photovoltaic characteristics are illustrated in Table 5.| Acceptors | HOMO (eV) | LUMO (eV) | E g (eV) | Donors | V oc (V) | J sc (mA cm−2) | FF (%) | PCE (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| e1 | −5.78 | −3.85 | 1.93 | PM6 | 0.96 | 15.27 | 68 | 10.3 | 118 |
| e2 | −5.69 | −3.77 | 1.92 | PBDT-TS1 | 1.1 | 6.0 | 41 | 2.6 | 119 |
| e3 | −5.62 | −3.86 | 1.76 | PBDT-TS1 | 1.0 | 11.0 | 44 | 4.8 | 119 |
| e4 | — | — | — | P3HT | 1.26 | 3.88 | 55 | 2.70 | 120 |
| e5 | −5.59 | −3.75 | 1.43 | PBDB-T | 0.90 | 14.20 | 65 | 8.32 | 121 |
| e6 | −5.33 | −3.64 | 1.69 | P3HT | 0.84 | 2.75 | 49.28 | 1.09 | 122 |
| e7 | −6.08 | −3.50 | 1.96 | PTB7-Th | 1.04 | 13.82 | 45.0 | 6.50 | 123 |
| e8 | −6.13 | −3.42 | 1.86 | PTB7-Th | 1.04 | 12.72 | 56.25 | 7.42 | 124 |
| e9 | −6.15 | −3.41 | 1.87 | PTB7-Th | 1.05 | 13.56 | 58.25 | 8.28 | 124 |
| e10 | −6.16 | −3.40 | 1.87 | PTB7-Th | 1.07 | 12.23 | 49.95 | 6.54 | 124 |
| e11 | −5.95 | −3.72 | 2.23 | PBFTAZ | 0.99 | 9.3 | 50 | 4.6 | 125 |
| e12 | −5.98 | −3.75 | 2.23 | PBFTAZ | 1.00 | 8.2 | 42 | 3.7 | 125 |
| e13 | −5.98 | −3.75 | 2.23 | PBFTAZ | 0.97 | 13.2 | 55 | 7.3 | 125 |
| e14 | −5.23 | −3.46 | 1.77 | PTB7-Th | 1.03 | 14.88 | 58.46 | 8.98 | 126 |
| e15 | −5.69 | −3.69 | 1.61 | p-DTS(FBTTh2)2 | 0.97 | 2.91 | 53.2 | 1.50 | 127 |
| e16 | −5.58 | −3.69 | 1.55 | p-DTS(FBTTh2)2 | 0.94 | 2.12 | 50.0 | 1.00 | 127 |
| e17 | −5.84 | −3.80 | 2.04 | PTB7-Th | 0.92 | 11.37 | 48 | 5.04 | 128 |
| e17 | −5.84 | −3.80 | 2.04 | PTB7 | 0.93 | 9.05 | 45 | 3.80 | 128 |
| e18 | −5.85 | −3.73 | 2.12 | PTB7-Th | 1.08 | 0.51 | 22 | 0.12 | 128 |
| e18 | −5.85 | −3.73 | 2.12 | PTB7 | 1.00 | 2.48 | 30 | 0.70 | 128 |
| e19 | −5.81 | −3.71 | 2.1 | PTB7-Th | 0.89 | 14.24 | 52 | 6.60 | 129 |
| e20 | — | −3.75 | 2.16 | PTB7 | 0.39 | 0.90 | 33 | 0.11 | 130 |
| e21 | — | −4.35 | 2.07 | PTB7 | 0.41 | 1.55 | 38 | 0.22 | 130 |
| e22 | −5.04 | −3.18 | 1.53 | — | — | — | — | — | 131 |
| e23 | −5.06 | −3.17 | 1.54 | — | — | — | — | — | 131 |
| e24 | −5.88 | −3.72 | 1.80 | PTB7-Th | 1.00 | 6.51 | 43.4 | 2.83 | 132 |
| e25 | −5.76 | −3.58 | 1.83 | PTB7-Th | 1.08 | 4.47 | 32.4 | 1.56 | 132 |
| e26 | −5.76 | −3.72 | 2.04 | p-DTS(FBTTh2)2 | 1.03 | 1.71 | 36.3 | 0.64 | 133 |
| e27 | −5.80 | −3.50 | 1.77 | P3HT | 0.90 | 0.52 | 57.14 | 0.27 | 134 |
| e28 | −6.12 | −3.69 | 1.86 | P3HT | 0.87 | 0.26 | 44.97 | 0.10 | 134 |
| e29 | −6.21 | −3.62 | 2.20 | P3HT | 0.88 | 0.26 | 45.90 | 0.11 | 134 |
Yan et al. synthesized a high-performing polymer acceptor (e1, Fig. 12) with a maximum absorption coefficient of 2.74 × 105 cm−1.118 By pairing with PM6, a PCE of 10.3% was achieved with a high Voc of 0.97 V. They also found that the addition of 1-chloronaphthalene (CN) improved the domain purity and enhanced the crystallization of the polymers, which was responsible for the dramatic increase in the PCE. The strong electron-withdrawing 2,1,3-benzothiadiazole (BT) unit was used in the polymer acceptors because of its high electron mobility and low-optical-bandgap advantages. Beaujuge et al. reported two polymer acceptors (e2 and e3, Fig. 12) based on BT units.119 The introduction of the BT unit narrowed the optical bandgap from 1.9 eV to 1.7 eV and also extended the optical absorption to longer wavelengths (by ∼70 nm). Optimized devices based on e3 achieved a large Voc of 1.0 V, a high Jsc of 11.0 mA cm−2 as well as a PCE of 4.8%. Miyake and coworkers reported a copolymer (e4, Fig. 12) consisting of BT and fluorene units.120 They investigated the impacts of molecular weight on the morphology of the polymer blends. Even under an annealing temperature of 140 °C, a small domain size and interconnected networks were observed in the high molecular weight (Mn = 28000 g mol−1) polymer (e4) blends, owing to the high glass transition temperature and reduced chain mobility of e4. This blend morphology accounted for a PCE of 2.7% with a high Voc of 1.26 V, a Jsc of 3.88 mA cm−2, and an FF of 55%. Guo et al. reported a polymer acceptor (e5, Fig. 12) based on dicyanobenzothiadiazole (DCNBT) and IDT units.121 The strong electron-withdrawing cyano unit enabled e5 with a n-type character. Compared with N2200, e5 showed a high-lying LUMO energy level (−3.75 eV), narrow bandgap (1.43 eV), and high absorption coefficient (6.15 × 104 cm−1). When e5 was blended with PBDB-T, the all-PSCs exhibited a large Voc of 0.90 V, a high PCE of 8.32%, and a small energy loss of 0.53 V. A new polymer acceptor (e6, Fig. 12) based on a thiophene-fused benzoxadizole unit was synthesized by Chen et al.122 It showed low HOMO–LUMO energy levels of −5.33 eV and −3.64 eV, which matched well with P3HT as a donor for all-PSCs. Consequently, a PCE of 1.09% with a high Voc of 0.84 V was achieved.
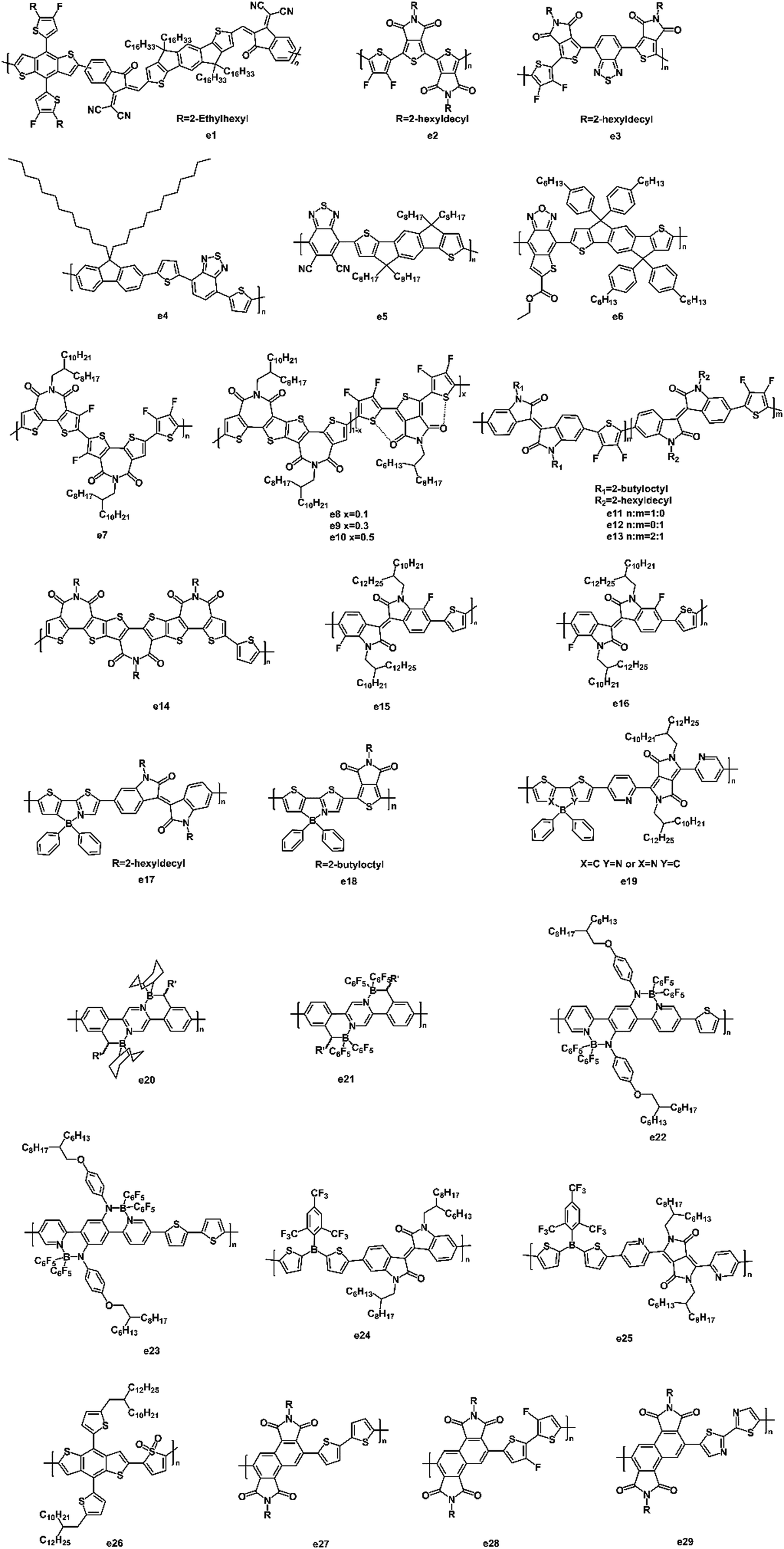 | ||
| Fig. 12 The structures of other polymer acceptors. | ||
Guo et al. synthesized a fluorinated bithiophene imide (BTI)-based polymer acceptor (e7, Fig. 12).123 The introduction of F atoms on the BTI core induced intra- and intermolecular non-covalent interactions, which were beneficial for charge transport. When e7 was blended with PTB7-Th, the PSC device achieved a high Voc of 1.04 V with a PCE of 6.50%. In 2019, Guo et al. first incorporated the noncovalent interactions into terpolymer acceptors (e8–e10, Fig. 12) to tune the crystallinity and miscibility.124 The introduction of the TPD content caused noncovalent S–O interactions as well as increased the face-on polymer packing and in-chain charge transport. By blending with PTB7-Th, the root-mean-square (RMS) roughness was reduced from 3.75 nm to 1.16 nm as the TPD content increased from 10% to 50% in the polymer acceptor. As a result, a remarkable PCE of 8.28% with a small energy loss of 0.53 eV was achieved for the PTB7-Th:e9 based solar cells. Beaujuge and coworkers synthesized a series of branched alkyl-substituted polymer acceptors (e11–e13, Fig. 12) containing alternating isoindigo and difluorothiophene units.125 Due to the long alkyl side chains, the morphology of the active layer showed a high degree of donor–acceptor mixing with small domain sizes. The optimized devices exhibited a maximum PCE of 7.3% with a Jsc as high as 13.2 mA cm−2. Guo et al. synthesized a triimide-functionalized polymer (e14, Fig. 12), thus enabling an all-PSC with a high PCE of 8.98%.126 The increased imide number on e14 resulted in a slightly wider absorption and narrower bandgap. By blending e14 with PTB7-Th, a well-mixed film morphology with the desired polymer chain orientation was observed, yielding a larger Jsc (14.88 mA cm−2) and FF (58%). In 2018, Liu et al. reported two fluorinated isoindigo-based polymer acceptors (e15 and e16, Fig. 12).127 The two fluorine substituents enabled a close stacking of polymer chains through a noncovalent F–H and F–S interaction. When e15 and e16 blended with p-DTS(FBTTh2)2, respectively, the e15-based solar cell showed a PCE up to 1.5% with a Voc of 0.97 V, a Jsc of 2.91 mA cm−2, and an FF of 53.2%.
Recently, many polymers based on the B←N unit were reported, indicating the great potential of this class of compounds as acceptors for PSCs. Wang and coworkers synthesized two polymer acceptors (e17 and e18, Fig. 12) containing a thieno [3,4-c]pyrrole-4,6-dione (TPD) unit.128 They extended the length of the repeating units to alleviate the steric hindrance and promote π–π stacking of the polymer backbones. As a result, the all-PSCs with e17 as the acceptor and PTB7-Th as the donor exhibited a PCE of 5.04% because of the enhanced electron mobility (1.88 × 10−4 cm2 V−1 s−1) in the well-mixed active blend. Liu et al. synthesized an amorphous polymer acceptor (e19, Fig. 12) based on B←N units to study the active layer morphologies of the devices.129 When PTB7-Th was used as the donor, the blends showed uniform morphologies and a smooth surface with a RMS roughness of 1.67 nm. The electron mobility was as high as 2.20 × 10−4 cm2 V−1 s−1. As a result, a PCE of 6.6% with a Jsc of 14.24 mA cm−2 and an FF of 52% were achieved. Pammer and coworkers reported two ladder-type polymer acceptors (e20 and e21, Fig. 12) containing B←N units by postfunctionalization.130 The increased electron affinities and decreased optical bandgaps of these polymers were achieved by the borylation method. The two polymers showed an ambipolar charge transport with hole and electron mobilities on the order of 2 × 10−5 cm2 V−1 s−1. A new building block with an intramolecular D–A character was designed by fusing two B←N-linked pyridyl rings (acceptor unit) and a diamine-substituted phenylene ring (donor unit) together.131 The new building block exhibited a narrow bandgap and red-shifted absorption spectrum. Consequently, the LUMO/HOMO energy levels were −3.18 eV/−5.04 eV for e22 and −3.17 eV/−5.06 eV for e23. The two polymers (e22 and e23, Fig. 12) exhibited strong absorption bands with a peak at 698 nm/422 nm for e22 and 691 nm/451 nm for e23.
Liu et al. synthesized two conjugated polymers (e24 and e25, Fig. 12) based on triarylborane units.132 The two polymers exhibited low-lying LUMO/HOMO energy levels (−3.72/−5.88 eV) due to the p–π conjugation of the triarylborane unit and electron-deficient properties of the isoindigo and diketopyrrolopyrrole units. After selecting PTB7-Th as the donor, the e24:PTB7-Th blend showed a smaller phase separation than that of the e25:PTB7-Th blends. As a result, the e24-based PSC device achieved a PCE of 2.83% with a Jsc of 6.51 mA cm−2. To study a new strategy using the thiophene-S,S-dioxide (TDO) unit to design the polymer acceptor, Liu et al. synthesized a polymer acceptor (e26, Fig. 12) consisting of an alternating TDO unit and BDT unit.133 The introduction of the TDO unit led to a downshift in the LUMO/HOMO energy level by 0.9/0.4 eV and a red-shift in the absorption spectra by ∼110 nm. As a result, the PSC device with e26 as the acceptor showed a high Voc of 1.03 V, a Jsc of 1.71 mA cm−2, an FF of 36.3%, and a PCE of 0.64%. To raise the LUMO energy levels of the polymer acceptors, Liu et al. used a 1,2,5,6-naphthalenediimide (iso-NDI) unit to design the three polymers (e27–e29, Fig. 12).134 The results indicated that the iso-NDI-based polymers had higher LUMO energy levels by ∼0.30 V compared to that for the NDI-based polymer (N2200) due to it having less electron-deficient properties than the NDI unit. Therefore, all three iso-NDI-based polymers exhibited a high Voc in the range of 0.88–0.90 V, which was higher than that of N2200 (0.60 V) by ∼0.30 V. Among the three polymers, e27 exhibited the best device performance: Voc = 0.90 V, Jsc = 0.52 mA cm−2, FF = 57%, and PCE = 0.27%.
The impressive performance of these polymers was achieved by e1 with a PCE of 10.3%. It is worth noting that this polymer consisted of an IDT-based unit, which is often used in small molecule acceptors. The efforts for getting strong absorption coefficients, a high electron mobility, and appropriate energy levels for the polymer acceptors are ongoing.
3. Conclusions and future prospects
In recent years, tremendous progress has been made in the field of non-fullerene polymer acceptors, which possess an overwhelming potential to replace fullerene and its derivatives. Due to advantages such as high absorption coefficients, a good film-forming performance, and tunable energy levels, polymer acceptors show a great application prospect. The design of D–A type conjugated polymer acceptors has become one of the most effective strategies to acquire highly-efficient all-PSCs. However, there are still some challenges in designing rational all-PSCs.First, electron mobility plays an important role in the active layer. How to enhance the electron mobility has become an essential issue that needs to be addressed. The molecular orientation of conjugated polymers is a crucial factor. Unlike fullerene and its derivatives, polymers acceptors possess anisotropic charge transport properties due to the two-dimensional structures. In addition, extending fused units is conductive to enhance the crystallinity and thus improve electron mobility.
Secondly, how to judiciously control the size of the phase separation is of equal importance to achieve a high PCE. An appropriate phase separation is required for suppressing the bimolecular recombination between charge carriers and facilitating charge transport. However, the large-scale phase separation often observed is attributed to an undesired mixing of the polymer donors and acceptors of all-PSCs.
Third, it's a big challenge to obtain a homogeneous molecular weight of polymers. In general, the use of higher molecular weight polymers often leads to better miscibility between donor and acceptor materials due to a reduced domain size of the blends. Consequently, a more efficient exciton dissociation and better charge transport are achieved. Methodically modulating the molecular weight of conjugated polymers could be an effective strategy for optimizing the morphology of all-PSCs.
Other challenges also deserve attention in order to obtain high-performance PSCs. Long-term stability against ambient conditions is a crucial factor impacting device performance, especially ensuring that the thermal stability of PSCs is indispensable for their manufactures and applications. For further commercialization, efforts should also focus on fabricating large-area printed all-PSCs transferring from the lab scale to industry scale without sacrificing performance.
Overall, this review summarizes and analyses, in detail, the high-performance polymer acceptors in recent years and puts forward challenges and solutions. Many strategies for boosting the device performance have been employed, including structure modification, morphology optimization, and interfacial engineering. Among these developments, designing novel polymer acceptors plays an important role. Lots of polymer acceptors have been systematically studied to date. PDI and NDI based polymer acceptors account for a large proportion due to easy modifications and other outstanding chemical properties. However, other polymer acceptors such as DPP and BNBP-based polymers have also attracted much attention. The photovoltaic properties of polymer acceptor-based PSCs are continually improving. Although there are some key issues that need to be settled, polymer acceptors demonstrate a very bright future for commercial applications.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by (1) the National Natural Science Foundation of China (51703104), the Shandong Provincial Natural Science Foundation (ZR2017BEM035, BS2014CL025), and the China Postdoctoral Science Foundation (2017M612198, 2014M56053), (2) the State Key Project of International Cooperation Research (2016YFE0110800, 2017YFE0108300), (3) the National Natural Science Foundation of China (51403114, 51473082, 51878361), and (4) the National Program for Introducing Talents of Discipline to Universities (“111” plan), the 1st class discipline program of Materials Science of Shandong Province, and the Double-Hundred Foreign Expert Program of Shandong Province (2019–2021).Notes and references
- Z. Genene, W. Mammo, E. Wang and M. R. Andersson, Adv. Mater., 2019, 31, 1807275 CrossRef.
- J. Wang, M. Xiao, W. Chen, M. Qiu, Z. Du, W. Zhu, S. Wen, N. Wang and R. Yang, Macromolecules, 2014, 47, 7823–7830 CrossRef CAS.
- C. Lee, S. Lee, G. U. Kim, W. Lee and B. J. Kim, Chem. Rev., 2019, 119, 8028–8086 CrossRef CAS.
- H. Sun, Y. Tang, C. W. Koh, S. Ling, R. Wang, K. Yang, J. Yu, Y. Shi, Y. Wang, H. Y. Woo and X. Guo, Adv. Mater., 2019, 31, 1807220 CrossRef.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- P. Cheng, G. Li, X. W. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- H. Xu, Y. Yang, C. Zhong, X. Zhan and X. Chen, J. Mater. Chem. A, 2018, 6, 6393–6401 RSC.
- J. Hou, O. Inganas, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS.
- X. Zhan and S. R. Marder, Mater. Chem. Front., 2019, 3, 180 RSC.
- J. Wang, X. Bao, D. Ding, M. Qiu, Z. Du, J. Wang, J. Liu, M. Sun and R. Yang, J. Mater. Chem. A, 2016, 4, 11729–11737 RSC.
- S. Zhang, Y. Qin, J. Zhu and J. Hou, Adv. Mater., 2018, 30, 1800868 CrossRef.
- L. Meng, Y. Q. Yi, X. Wan, Y. Zhang, X. Ke, B. Kan, Y. Wang, R. Xia, H. L. Yip, C. Li and Y. Chen, Adv. Mater., 2019, 31, 1804723 CrossRef.
- X. e. Jia, Z. Chen, C. Duan, Z. Wang, Q. Yin, F. Huang and Y. Cao, J. Mater. Chem. C, 2019, 7, 314–323 RSC.
- D. Wöhrle and D. Meissner, Adv. Mater., 1991, 3, 129–138 CrossRef.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- J. Yuan, Y. Q. Zhang, L. Y. Zhou, G. C. Zhang, H. L. Yip, T. K. Lau, X. H. Lu, C. Zhu, H. J. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. F. Li and Y. P. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- W. P. Wang, B. F. Zhao, Z. Y. Cong, Y. Xie, H. M. Wu, Q. B. Liang, S. Liu, F. Liu, C. Gao, H. B. Wu and Y. Cao, ACS Energy Lett., 2018, 3, 1499–1507 CrossRef CAS.
- Y. Zhang, B. Kan, Y. Sun, Y. Wang, R. Xia, X. Ke, Y. Q. Yi, C. Li, H. L. Yip, X. Wan, Y. Cao and Y. Chen, Adv. Mater., 2018, 30, 1707508 CrossRef.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H. L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094–1098 CrossRef CAS.
- B. Zhang, S. Yi, G. Chen, Z. He, H.-B. Wu, W. Yang, F. Niu, J. Qu, P. Zeng and Y. Cao, J. Mater. Chem. C, 2017, 5, 4858–4866 RSC.
- Y. F. Wang, Z. Z. Liang, X. M. Li, J. C. Qin, M. L. Ren, C. Y. Yang, X. C. Bao, Y. J. Xia and J. F. Li, J. Mater. Chem. C, 2019, 7, 11152–11159 RSC.
- H. Shang, H. Fan, Y. Liu, W. Hu, Y. Li and X. Zhan, Adv. Mater., 2011, 23, 1554–1557 CrossRef CAS.
- M. Zhang, X. Guo, S. Zhang and J. Hou, Adv. Mater., 2014, 26, 1118–1123 CrossRef CAS.
- D. Ding, J. Wang, Z. Du, F. Li, W. Chen, F. Liu, H. Li, M. Sun and R. Yang, J. Mater. Chem. A, 2017, 5, 10430–10436 RSC.
- S. Dai, S. Chandrabose, J. Xin, T. Li, K. Chen, P. Xue, K. Liu, K. Zhou, W. Ma, J. M. Hodgkiss and X. Zhan, J. Mater. Chem. A, 2019, 7, 2268–2274 RSC.
- Y. Cui, H. Yao, J. Zhang, T. Zhang, Y. Wang, L. Hong, K. Xian, B. Xu, S. Zhang, J. Peng, Z. Wei, F. Gao and J. Hou, Nat. Commun., 2019, 10, 2515 CrossRef PubMed.
- H. Li, J. Wang, Y. Wang, F. Bu, W. Shen, J. Liu, L. Huang, W. Wang, L. A. Belfiore and J. Tang, Sol. Energy Mater. Sol. Cells, 2019, 190, 83–97 CrossRef CAS.
- N. S. Sariciftci, D. Braun, C. Zhang, V. I. Srdanov, A. J. Heeger, G. Stucky and F. Wudl, Appl. Phys. Lett., 1993, 62, 585–587 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- Y. He, H. Y. Chen, J. Hou and Y. Li, J. Am. Chem. Soc., 2010, 132, 1377–1382 CrossRef CAS PubMed.
- G. Zhao, Y. He and Y. Li, Adv. Mater., 2010, 22, 4355–4358 CrossRef CAS PubMed.
- Y. J. Cheng, C. H. Hsieh, Y. He, C. S. Hsu and Y. Li, J. Am. Chem. Soc., 2010, 132, 17381–17383 CrossRef CAS PubMed.
- T. Liu and A. Troisi, Adv. Mater., 2013, 25, 1038–1041 CrossRef CAS.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS.
- Z. Xiao, X. J. Geng, D. He, X. Jia and L. M. Ding, Energy Environ. Sci., 2016, 9, 2114–2121 RSC.
- Z. Liu and N. Wang, J. Mater. Chem. C, 2019, 7, 10039–10048 RSC.
- D. Baran, T. Kirchartz, S. Wheeler, S. Dimitrov, M. Abdelsamie, J. Gorman, R. S. Ashraf, S. Holliday, A. Wadsworth, N. Gasparini, P. Kaienburg, H. Yan, A. Amassian, C. J. Brabec, J. R. Durrant and I. McCulloch, Energy Environ. Sci., 2016, 9, 3783–3793 RSC.
- Z. Liang, J. Tong, H. Li, Y. Wang, N. Wang, J. Li, C. Yang and Y. Xia, J. Mater. Chem. A, 2019, 7, 15841–15850 RSC.
- B. He, Q. Yin, X. Yang, L. Liu, X.-F. Jiang, J. Zhang, F. Huang and Y. Cao, J. Mater. Chem. C, 2017, 5, 8774–8781 RSC.
- G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS.
- B. Guo, W. B. Li, G. P. Luo, X. Guo, H. F. Yao, M. J. Zhang, J. H. Hou, Y. F. Li and W. Y. Wong, ACS Energy Lett., 2018, 3, 2566–2572 CrossRef CAS.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423–9429 CrossRef CAS.
- J. Sun, X. Ma, Z. Zhang, J. Yu, J. Zhou, X. Yin, L. Yang, R. Geng, R. Zhu, F. Zhang and W. Tang, Adv. Mater., 2018, 30, 1707150 CrossRef.
- C. Cao, M. Xiao, X. Yang, J. Zhang, F. Huang and Y. Cao, J. Mater. Chem. C, 2018, 6, 8020–8027 RSC.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS.
- F. Zhao, S. Dai, Y. Wu, Q. Zhang, J. Wang, L. Jiang, Q. Ling, Z. Wei, W. Ma, W. You, C. Wang and X. Zhan, Adv. Mater., 2017, 29, 1700144 CrossRef.
- H. Zhang, H. Yao, J. Hou, J. Zhu, J. Zhang, W. Li, R. Yu, B. Gao, S. Zhang and J. Hou, Adv. Mater., 2018, 30, 1800613 CrossRef.
- S. S. Chen, H. T. Yao, B. Hu, G. Y. Zhang, L. Arunagiri, L. K. Ma, J. C. Huang, J. Q. Zhang, Z. L. Zhu, F. J. Bai, W. Ma and H. Yan, Adv. Energy Mater., 2018, 8, 1800529 CrossRef.
- W. Li, L. Ye, S. Li, H. Yao, H. Ade and J. Hou, Adv. Mater., 2018, 30, 1707170 CrossRef.
- H. Benten, D. Mori, H. Ohkita and S. Ito, J. Mater. Chem. A, 2016, 4, 5340–5365 RSC.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed.
- Y. Yang, Z. G. Zhang, H. Bin, S. Chen, L. Gao, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 15011–15018 CrossRef CAS PubMed.
- G. Cai, J. Zhu, Y. Xiao, M. Li, K. Liu, J. Wang, W. Wang, X. Lu, Z. Tang, J. Lian, P. Zeng, Y. Wang and X. Zhan, J. Mater. Chem. A, 2019, 7, 21432–21437 RSC.
- C. Yan, W. Wang, T.-K. Lau, K. Li, J. Wang, K. Liu, X. Lu and X. Zhan, J. Mater. Chem. A, 2018, 6, 16638–16644 RSC.
- J. Zhu, Y. Wu, J. Rech, J. Wang, K. Liu, T. Li, Y. Lin, W. Ma, W. You and X. Zhan, J. Mater. Chem. C, 2018, 6, 66–71 RSC.
- Z. Li, S. Dai, J. Xin, L. Zhang, Y. Wu, J. Rech, F. Zhao, T. Li, K. Liu, Q. Liu, W. Ma, W. You, C. Wang and X. Zhan, Mater. Chem. Front., 2018, 2, 537–543 RSC.
- Y. Cui, H. Yao, L. Hong, T. Zhang, Y. Xu, K. Xian, B. Gao, J. Qin, J. Zhang, Z. Wei and J. Hou, Adv. Mater., 2019, 31, 1808356 CrossRef.
- B. Fan, D. Zhang, M. Li, W. Zhong, Z. Zeng, L. Ying, F. Huang and Y. Cao, Sci. China: Chem., 2019, 62, 746–752 CrossRef CAS.
- X. Zhang, C. Zhan and J. Yao, Chem. Mater., 2014, 27, 166–173 CrossRef.
- S. M. McAfee, A. J. Payne, S. V. Dayneko, G. P. Kini, C. E. Song, J. C. Lee and G. C. Welch, J. Mater. Chem. A, 2017, 5, 16907–16913 RSC.
- S. Li, H. Zhang, W. Zhao, L. Ye, H. Yao, B. Yang, S. Zhang and J. Hou, Adv. Energy Mater., 2016, 6, 1501991 CrossRef.
- Z. G. Zhang, Y. Yang, J. Yao, L. Xue, S. Chen, X. Li, W. Morrison, C. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 13503–13507 CrossRef CAS.
- N. J. Zhou and A. Facchetti, Mater. Today, 2018, 21, 377–390 CrossRef CAS.
- A. Facchetti, Mater. Today, 2013, 16, 123–132 CrossRef CAS.
- T. Kim, J. H. Kim, T. E. Kang, C. Lee, H. Kang, M. Shin, C. Wang, B. Ma, U. Jeong, T. S. Kim and B. J. Kim, Nat. Commun., 2015, 6, 8547 CrossRef CAS PubMed.
- P. Cheng, C. Yan, Y. Li, W. Ma and X. Zhan, Energy Environ. Sci., 2015, 8, 2357–2364 RSC.
- P. Cheng, L. Ye, X. Zhao, J. Hou, Y. Li and X. Zhan, Energy Environ. Sci., 2014, 7, 1351–1356 RSC.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS.
- Y. H. Cai, L. J. Huo, X. B. Sun, D. H. Wei, M. S. Tang and Y. M. Sun, Adv. Energy Mater., 2015, 5, 1500032 CrossRef.
- N. Liang, K. Sun, Z. Zheng, H. Yao, G. Gao, X. Meng, Z. Wang, W. Ma and J. Hou, Adv. Energy Mater., 2016, 6, 1600060 CrossRef.
- Y. Guo, Y. Li, O. Awartani, J. Zhao, H. Han, H. Ade, D. Zhao and H. Yan, Adv. Mater., 2016, 28, 8483–8489 CrossRef CAS PubMed.
- Z. T. Liu, Y. Wu, Q. Zhang and X. Gao, J. Mater. Chem. A, 2016, 4, 17604–17622 RSC.
- F. Fernandez-Lazaro, N. Zink-Lorre and A. Sastre-Santos, J. Mater. Chem. A, 2016, 4, 9336–9346 RSC.
- J. Yang, B. Xiao, A. Tang, J. Li, X. Wang and E. Zhou, Adv. Mater., 2019, 31, 1804699 CrossRef CAS PubMed.
- X. W. Zhan, Z. A. Tan, B. Domercq, Z. S. An, X. Zhang, S. Barlow, Y. F. Li, D. B. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246–7247 CrossRef CAS PubMed.
- S. Nam, S. G. Hahm, H. Han, J. Seo, C. Kim, H. Kim, S. R. Marder, M. Ree and Y. Kim, ACS Sustainable Chem. Eng., 2015, 4, 767–774 CrossRef.
- F. Yang, C. Li, G.-T. Feng, X.-D. Jiang, A.-D. Zhang and W.-W. Li, Chin. J. Polym. Sci., 2016, 35, 239–248 CrossRef.
- M. Liu, J. Yang, Y. L. Yin, Y. Zhang, E. J. Zhou, F. Y. Guo and L. C. Zhao, J. Mater. Chem. A, 2018, 6, 414–422 RSC.
- Y. Liang, S. Lan, P. Deng, D. Zhou, Z. Guo, H. Chen and H. Zhan, ACS Appl. Mater. Interfaces, 2018, 10, 32397–32403 CrossRef CAS PubMed.
- M. Liu, J. Yang, C. L. Lang, Y. Zhang, E. J. Zhou, Z. T. Liu, F. Y. Guo and L. C. Zhao, Macromolecules, 2017, 50, 7559–7566 CrossRef CAS.
- Y. Yin, J. Yang, F. Guo, E. Zhou, L. Zhao and Y. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 15962–15970 CrossRef CAS PubMed.
- J. R. Cheng, B. Li, X. J. Ren, F. Liu, H. C. Zhao, H. J. Wang, Y. G. Wu, W. P. Chen and X. W. Ba, Dyes Pigm., 2019, 161, 221–226 CrossRef CAS.
- Z. J. Li, W. Zhang, X. F. Xu, Z. Genene, D. D. Rasi, W. Mammo, A. Yartsev, M. R. Andersson, R. A. J. Janssen and E. G. Wang, Adv. Energy Mater., 2017, 7, 1602722 CrossRef.
- J. Yang, B. Xiao, K. Tajima, M. Nakano, K. Takimiya, A. L. Tang and E. J. Zhou, Macromolecules, 2017, 50, 3179–3185 CrossRef CAS.
- B. Fan, L. Ying, P. Zhu, F. Pan, F. Liu, J. Chen, F. Huang and Y. Cao, Adv. Mater., 2017, 29, 1703906 CrossRef PubMed.
- L. Gao, Z. G. Zhang, L. Xue, J. Min, J. Zhang, Z. Wei and Y. Li, Adv. Mater., 2016, 28, 1884–1890 CrossRef CAS PubMed.
- B. B. Fan, L. Ying, Z. F. Wang, B. T. He, X. F. Jiang, F. Huang and Y. Cao, Energy Environ. Sci., 2017, 10, 1243–1251 RSC.
- X. Liu, Y. Zou, H. Q. Wang, L. Wang, J. Fang and C. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 38302–38309 CrossRef CAS PubMed.
- J. Jung, W. Lee, C. Lee, H. Ahn and B. J. Kim, Adv. Energy Mater., 2016, 6, 1600504 CrossRef.
- Z. Y. Li, L. Ying, P. Zhu, W. K. Zhong, N. Li, F. Liu, F. Huang and Y. Cao, Energy Environ. Sci., 2019, 12, 157–163 RSC.
- N. B. Kolhe, H. Lee, D. Kuzuhara, N. Yoshimoto, T. Koganezawa and S. A. Jenekhe, Chem. Mater., 2018, 30, 6540–6548 CrossRef CAS.
- D. Chen, J. Yao, L. Chen, J. Yin, R. Lv, B. Huang, S. Liu, Z. G. Zhang, C. Yang, Y. Chen and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 4580–4584 CrossRef CAS PubMed.
- H. H. Cho, S. Kim, T. Kim, V. G. Sree, S. H. Jin, F. S. Kim and B. J. Kim, Adv. Energy Mater., 2018, 8, 1701436 CrossRef.
- Z. Li, X. Xu, W. Zhang, X. Meng, W. Ma, A. Yartsev, O. Inganas, M. R. Andersson, R. A. Janssen and E. Wang, J. Am. Chem. Soc., 2016, 138, 10935–10944 CrossRef CAS PubMed.
- Y. Lin, S. Dong, Z. Li, W. Zheng, J. Yang, A. Liu, W. Cai, F. Liu, Y. Jiang, T. P. Russell, F. Huang, E. Wang and L. Hou, Nano Energy, 2018, 46, 428–435 CrossRef CAS.
- X. Wu, Y. M. Tang, Y. X. Wang, X. Y. Liu, C. M. Liu, X. N. Zhang, Y. G. Yang, X. Y. Gao, F. Chen, X. G. Guo and Z. K. Chen, J. Polym. Sci. Part A: Polym. Chem., 2017, 55, 3679–3689 CrossRef CAS.
- S. Z. Feng, C. Liu, X. F. Xu, X. C. Liu, L. J. Zhang, Y. W. Nian, Y. Cao and J. W. Chen, ACS Macro Lett., 2017, 6, 1310–1314 CrossRef CAS.
- X. Liu, C. Zhang, C. Duan, M. Li, Z. Hu, J. Wang, F. Liu, N. Li, C. J. Brabec, R. A. J. Janssen, G. C. Bazan, F. Huang and Y. Cao, J. Am. Chem. Soc., 2018, 140, 8934–8943 CrossRef CAS PubMed.
- H. You, D. Kim, H. H. Cho, C. Lee, S. Chong, N. Y. Ahn, M. Seo, J. Kim, F. S. Kim and B. J. Kim, Adv. Funct. Mater., 2018, 28, 1803613 CrossRef.
- Y. Ji, C. Xiao, Q. Wang, J. Zhang, C. Li, Y. Wu, Z. Wei, X. Zhan, W. Hu, Z. Wang, R. A. Janssen and W. Li, Adv. Mater., 2016, 28, 943–950 CrossRef CAS PubMed.
- A. Tang, C. Zhan, J. Yao and E. Zhou, Adv. Mater., 2017, 29, 1600013 CrossRef PubMed.
- W. Li, K. H. Hendriks, M. M. Wienk and R. A. Janssen, Acc. Chem. Res., 2016, 49, 78–85 CrossRef CAS PubMed.
- H.-Y. Chen, M. Nikolka, A. Wadsworth, W. Yue, A. Onwubiko, M. Xiao, A. J. P. White, D. Baran, H. Sirringhaus and I. McCulloch, Macromolecules, 2017, 51, 71–79 CrossRef.
- Z. Li, X. Xu, W. Zhang, Z. Genene, W. Mammo, A. Yartsev, M. R. Andersson, R. A. J. Janssen and E. Wang, J. Mater. Chem. A, 2017, 5, 11693–11700 RSC.
- Y. P. Yu, S. C. Zhou, X. H. Wang, C. Li, G. T. Feng, Y. G. Wu, W. Ma and W. W. Li, Org. Electron., 2017, 47, 133–138 CrossRef CAS.
- A. D. Zhang, Q. Wang, R. A. A. Bovee, C. Li, J. Q. Zhang, Y. Zhou, Z. X. Wei, Y. F. Li, R. A. J. Janssen, Z. H. Wang and W. W. Li, J. Mater. Chem. A, 2016, 4, 7736–7745 RSC.
- Z. Ding, X. Long, B. Meng, K. Bai, C. Dou, J. Liu and L. Wang, Nano Energy, 2017, 32, 216–224 CrossRef CAS.
- C. Dou, X. Long, Z. Ding, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 1436–1440 CrossRef CAS PubMed.
- X. Long, Z. Ding, C. Dou, J. Zhang, J. Liu and L. Wang, Adv. Mater., 2016, 28, 6504–6508 CrossRef CAS PubMed.
- Z. J. Zhang, Z. C. Ding, X. J. Long, C. D. Dou, J. Liu and L. X. Wang, J. Mater. Chem. C, 2017, 5, 6812–6819 RSC.
- X. Long, Z. Ding, C. Dou, J. Liu and L. Wang, Mater. Chem. Front., 2017, 1, 852–858 RSC.
- R. Zhao, Y. Min, C. Dou, J. Liu and L. Wang, Chem. – Eur. J., 2017, 23, 9486–9490 CrossRef CAS PubMed.
- Z. Ding, X. Long, C. Dou, J. Liu and L. Wang, Chem. Sci., 2016, 7, 6197–6202 RSC.
- R.-Y. Zhao, C.-D. Dou, J. Liu and L.-X. Wang, Chin. J. Polym. Sci., 2016, 35, 198–206 CrossRef.
- X. Long, N. Wang, Z. Ding, C. Dou, J. Liu and L. Wang, J. Mater. Chem. C, 2016, 4, 9961–9967 RSC.
- X. Long, J. Yao, F. Cheng, C. Dou and Y. Xia, Mater. Chem. Front., 2019, 3, 70–77 RSC.
- R. Y. Zhao, Z. Z. Bi, C. D. Dou, W. Ma, Y. C. Han, J. Liu and L. X. Wang, Macromolecules, 2017, 50, 3171–3178 CrossRef CAS.
- H. T. Yao, F. J. Bai, H. W. Hu, L. Arunagiri, J. Q. Zhang, Y. Z. Chen, H. Yu, S. S. Chen, T. Liu, J. Y. L. Lai, Y. P. Zou, H. Ade and H. Yan, ACS Energy Lett., 2019, 4, 417–422 CrossRef CAS.
- S. J. Liu, X. Song, S. Thomas, Z. P. Kan, F. Cruciani, F. Laquai, J. L. Bredas and P. M. Beaujuge, Adv. Energy Mater., 2017, 7, 1602574 CrossRef.
- D. Mori, H. Benten, H. Ohkita, S. Ito and K. Miyake, ACS Appl. Mater. Interfaces, 2012, 4, 3325–3329 CrossRef CAS PubMed.
- S. Shi, P. Chen, Y. Chen, K. Feng, B. Liu, J. Chen, Q. Liao, B. Tu, J. Luo, M. Su, H. Guo, M. G. Kim, A. Facchetti and X. Guo, Adv. Mater., 2019, 31, 1905161 CrossRef CAS PubMed.
- Y. Yang, J. C. Wang, X. W. Zhan and X. G. Chen, RSC Adv., 2017, 7, 19990–19995 RSC.
- H. Sun, Y. Tang, H. Guo, M. A. Uddin, S. Ling, R. Wang, Y. Wang, X. Zhou, H. Y. Woo and X. Guo, Sol. RRL, 2019, 3, 1800265 CrossRef.
- H. Sun, B. Liu, C. W. Koh, Y. Zhang, J. Chen, Y. Wang, P. Chen, B. Tu, M. Su, H. Wang, Y. Tang, Y. Shi, H. Y. Woo and X. Guo, Adv. Funct. Mater., 2019, 29, 1903970 CrossRef CAS.
- S. Liu, Y. Firdaus, S. Thomas, Z. Kan, F. Cruciani, S. Lopatin, J. L. Bredas and P. M. Beaujuge, Angew. Chem., Int. Ed., 2018, 57, 531–535 CrossRef CAS PubMed.
- Y. F. Wang, Z. L. Yan, M. A. Uddin, X. Zhou, K. Yang, Y. M. Tang, B. Liu, Y. Q. Shi, H. L. Sun, A. Y. Deng, J. F. Dai, H. Y. Woo and X. G. Guo, Sol. RRL, 2019, 3, 1900107 CrossRef.
- J. Miao, H. Xu, B. Meng, J. Liu and L. Wang, Chin. J. Chem., 2018, 36, 411–416 CrossRef CAS.
- R. Zhao, C. Dou, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 5313–5317 CrossRef CAS PubMed.
- R. Zhao, B. Lin, J. Feng, C. Dou, Z. Ding, W. Ma, J. Liu and L. Wang, Macromolecules, 2019, 52, 7081–7088 CrossRef CAS.
- M. Grandl, J. Schepper, S. Maity, A. Peukert, E. von Hauff and F. Pammer, Macromolecules, 2019, 52, 1013–1024 CrossRef.
- X. Shao, C. Dou, J. Liu and L. Wang, Sci. China: Chem., 2019, 62, 1387–1392 CrossRef CAS.
- B. Meng, Y. Ren, J. Liu, F. Jakle and L. Wang, Angew. Chem., Int. Ed., 2018, 57, 2183–2187 CrossRef CAS PubMed.
- B. Meng, J. Miao, J. Liu and L. Wang, Macromol. Rapid Commun., 2018, 39, 1700505 CrossRef PubMed.
- S. Zhang, J. Liu, Y. Han and L. Wang, Macromol. Chem. Phys., 2017, 218, 1600606 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |