
A facile approach to high-performance trifunctional electrocatalysts by substrate-enhanced electroless deposition of Pt/NiO/Ni on carbon nanotubes†
Yaru
Bian
a,
Hao
Wang
a,
Zhi
Gao
a,
Jintang
Hu
a,
Dong
Liu
 *a and
Liming
Dai
*b
*a and
Liming
Dai
*b
aState Key Laboratory of Organic–Inorganic Composites, Beijing Advanced Innovation Center for Soft Matter Science and Engineering, College of Chemical Engineering, Beijing University of Chemical Technology, Beijing, China. E-mail: liudong@mail.buct.edu.cn
bSchool of Chemical Engineering, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: l.dai@unsw.edu.au
First published on 10th June 2020
Abstract
Trifunctional electrocatalysts for the oxygen reduction reaction (ORR), oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) are crucial for many electrochemical devices. Herein, novel trifunctional electrocatalysts of a hierarchically-structured Pt/NiO/Ni/CNT with ultrasmall Pt nanoparticles (∼2 nm) were synthesized via substrate-enhanced electroless deposition. The resulting catalysts exhibited a higher ORR activity (half-wave potential at 0.942 V) than that of the benchmark commercial Pt/C (20 wt%) and a similar OER activity (overpotential of 350 mV at 10 mA cm−2) to that of RuO2 in alkaline media. Moreover, the newly-developed Pt/NiO/Ni/CNT catalysts also showed a high mass activity superior to Pt/C towards the HER in both acid and alkaline electrolytes. The Pt/NiO/Ni/CNT catalysts, when used for overall water splitting, self-powered by two Zn–air batteries with the Pt/NiO/Ni/CNT air electrodes connected into series, displayed superb performance with 1.61 V at 10 mA cm−2. This work represents a breakthrough in the development of facile approaches to trifunctional catalysts from low-cost Earth-abundant materials for efficient energy conversion and storage.
Introduction
Renewable energy technologies, including metal–air batteries,1–3 water splitting cells4,5 and fuel cells,6 are essential to mitigate the current energy and environmental crises.7–11 The oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) are the heart of Zn–air batteries while the OER and hydrogen evolution reaction (HER) are necessary for water splitting.12–14 However, all of the three aforementioned reactions require catalysts. Commercial Pt/C is highly efficient for the ORR and HER,15,16 and Ru- or Ir-based catalysts (RuO2, IrO2, Ir/C, etc.) are typically used for the OER. However, the scarce and high cost of noble metals have prevented these electrocatalysts from large-scale applications.17,18 To facilitate the development of renewable energy devices and their integrated systems, it is highly desirable to devise ORR/OER/HER trifunctional electrocatalysts from Earth-abundant materials at low cost.Platinum (Pt) has long been known as the best ORR catalyst,19 though expensive. In order to reduce the usage of Pt, the PtNi catalyst was identified as a promising alternative for the ORR,20–22 which has attracted a great deal of interest.23–25 Fortunately, certain PtNi-based catalysts were also demonstrated to exhibit considerable OER activities. Specifically, Ding et al.26 reported an onset potential (Eonset) of ∼1.52 V and an overpotential at 10 mA cm−2 (η10) of 0.4 V for Ni23.6Pt wires while the observed outstanding OER activity was attributed to the synergetic effect between the small content of the Pt dopant and the nickel matrix, coupled with the rough and tip-like structures. On the other hand, Zhang et al.27 reported an ORR and OER bifunctional electrocatalyst based on PtNi on carbon (PtNi/C), and attributed the improved OER activity to a synergistic effect between the intrinsic activity of Ni and excellent conductivity of Pt. HER electrocatalysts based on NiPt hybrids have also been reported. Examples include Ni(OH)2 on Pt28 and NiO on PtNi–O.29 However, as far as we are aware, the ORR/OER/HER trifunctional electrocatalyst based on the NiPt hybrid has not been reported so far.
In this study, we have, for the first time, synthesized carbon nanotube (CNT)-supported Pt/NiO/Ni (Pt/NiO/Ni/CNT) hybrid electrocatalysts by inter-dispersed NiO and Ni onto the CNT through substrate-enhanced electroless deposition (SEED),30,31 followed by calcination under an Ar/H2 (v/v 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) atmosphere, and then decorated NiO/Ni/CNTs with ultrasmall Pt nanoparticles (NPs, ∼2 nm) by a galvanic displacement reaction. The hierarchically structured Pt/NiO/Ni/CNT electrocatalysts thus prepared possessed a large number of active sites, including Pt–Ni interfaces, NiO/Ni surfaces and selectively exposed Pt (111) facets, for the ORR, OER and HER. Consequently, the resulting Pt/NiO/Ni/CNT hybrid electrocatalysts exhibited excellent performance for the ORR, OER and HER – similar to the benchmark RuO2 for the OER, and even superior to the commercial Pt/C for the ORR and HER in an alkaline medium. Furthermore, two Zn–air batteries based on the Pt/NiO/Ni/CNT air electrode for the ORR and OER could successfully self-power an overall water splitting process using the same Pt/NiO/Ni/CNT electrocatalyst for the OER and HER, demonstrating great potential for practical applications. This work reports the first NiPt hybrid with a low Pt loading for ORR/OER/HER trifunctional electrolysis in Zn–air batteries and water splitting processes, providing a facile approach to trifunctional catalysts from low-cost earth-abundant materials for efficient energy conversion and storage.
:10) atmosphere, and then decorated NiO/Ni/CNTs with ultrasmall Pt nanoparticles (NPs, ∼2 nm) by a galvanic displacement reaction. The hierarchically structured Pt/NiO/Ni/CNT electrocatalysts thus prepared possessed a large number of active sites, including Pt–Ni interfaces, NiO/Ni surfaces and selectively exposed Pt (111) facets, for the ORR, OER and HER. Consequently, the resulting Pt/NiO/Ni/CNT hybrid electrocatalysts exhibited excellent performance for the ORR, OER and HER – similar to the benchmark RuO2 for the OER, and even superior to the commercial Pt/C for the ORR and HER in an alkaline medium. Furthermore, two Zn–air batteries based on the Pt/NiO/Ni/CNT air electrode for the ORR and OER could successfully self-power an overall water splitting process using the same Pt/NiO/Ni/CNT electrocatalyst for the OER and HER, demonstrating great potential for practical applications. This work reports the first NiPt hybrid with a low Pt loading for ORR/OER/HER trifunctional electrolysis in Zn–air batteries and water splitting processes, providing a facile approach to trifunctional catalysts from low-cost earth-abundant materials for efficient energy conversion and storage.
Results and discussion
Synthesis and characterization
Fig. 1 schematically shows the procedure for preparing Pt/NiO/Ni/CNT catalysts: Ni was first deposited on CNTs through a galvanic displacement reaction between Ni2+ and Zn substrates by SEED.30,31 During the SEED process, Ni(OH)2 also formed due to a pH increase caused by the galvanic displacement between H+ and Zn (ESI, Fig. S1†). After sonication and drying in air at 70 °C, we then obtained the Ni(OH)2/Ni/CNT powder with Ni and Ni(OH)2 particles supported by CNTs, along with some ZnO impurities (Fig. 1a). Thereafter, the Ni(OH)2/Ni/CNT powder was converted into NiO/Ni/CNTs by annealing at 950 °C under an Ar/H2 atmosphere, which was accompanied by the removal of ZnO (Fig. 1b and c). By stirring the powdered NiO/Ni/CNTs in aqueous solutions of K2PtCl6 with different concentrations of 15.4, 30.8 and 61.5 μmol L−1, we finally obtained Pt/NiO/Ni/CNT-1, Pt/NiO/Ni/CNT-2 and Pt/NiO/Ni/CNT-3 (Fig. 1d), respectively, for subsequent electrocatalysis (Fig. 1e). Details for all the reactions involved in this process could be found in the ESI.†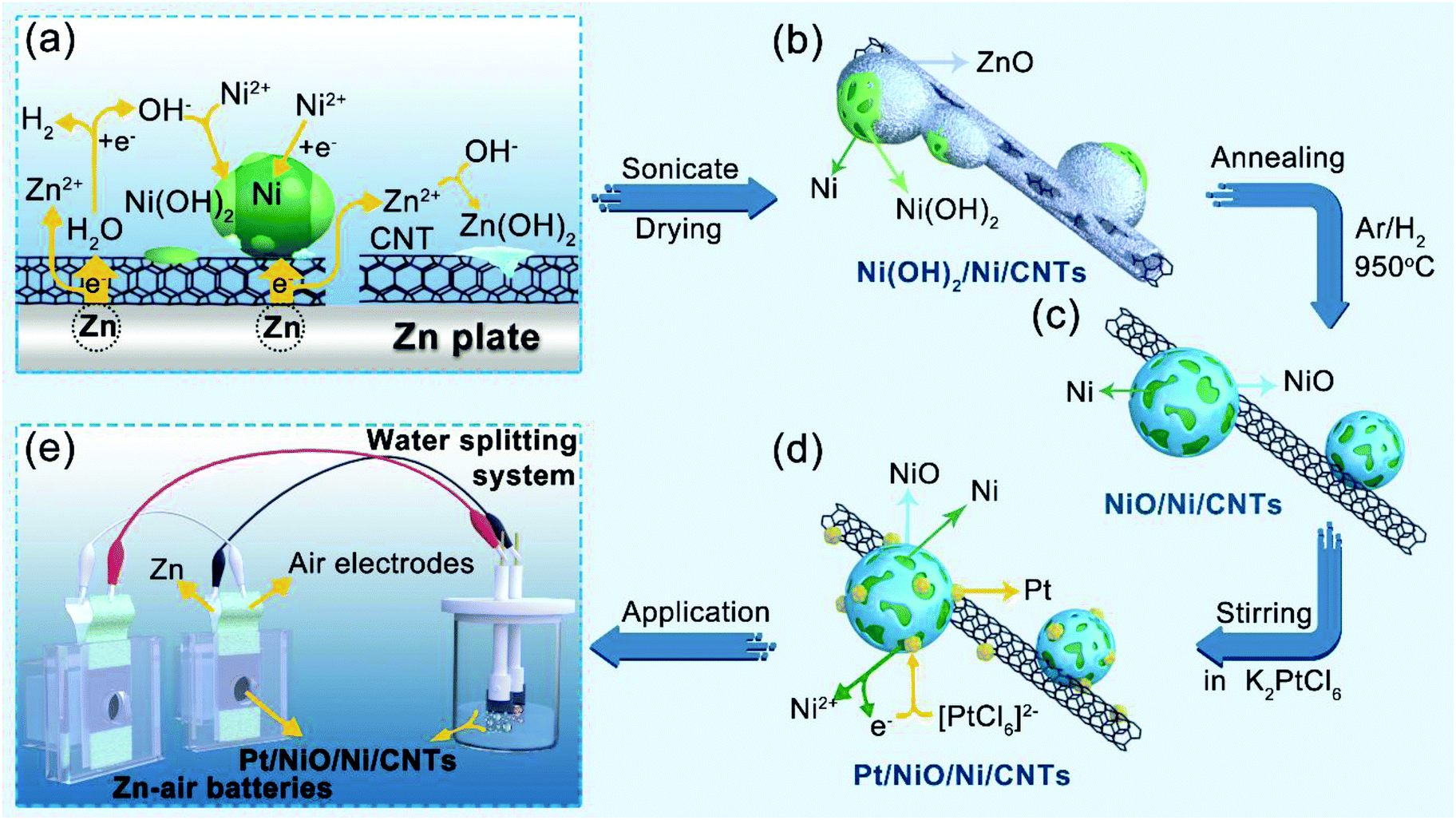 | ||
| Fig. 1 Schematic representation of the synthetic process. (a) Deposition of Ni and Ni(OH)2 onto CNTs supported by a Zn plate via SEED. (b) Powdered Ni(OH)2/Ni/CNTs were obtained after sonication and drying. (c) Ni(OH)2/Ni/CNTs were converted into NiO/Ni/CNTs by annealing in Ar/H2 at 950 °C. (d) Ultrasmall Pt NPs were decorated on the NiO/Ni/CNTs by the galvanic displacement reaction to obtain the Pt/NiO/Ni/CNT catalyst. (e) The resulting Pt/NiO/Ni/CNT catalyst was used as an OER/HER bifunctional catalyst for overall water splitting, which was powered by two Zn–air batteries using the same Pt/NiO/Ni/CNT catalyst as the air electrode for the ORR and OER. | ||
As shown in Fig. 2a, the X-ray diffraction (XRD) patterns of Ni(OH)2/Ni/CNTs exhibited a typical face-centered cubic structure (fcc) of Ni (JCPDS no. 04-0850), indicating that Ni was successfully deposited on CNTs. Some weak peaks from Ni(OH)2/Ni/CNTs indicated the presence of ZnO (JCPDS no. 36-1451) and Ni(OH)2·0.75H2O (JCPDS no. 38-0715). The peaks of ZnO disappeared in NiO/Ni/CNTs due to the removal of ZnO after thermal annealing. It was worth noting that no NiO peak was observed, suggesting the amorphous state of NiO in NiO/Ni/CNTs, as also evidenced by a high-resolution transmission electron microscopy (HRTEM) image and X-ray photoelectron spectra (XPS) – see below. Although no distinct Pt peak was observed in the XRD patterns for the Pt/NiO/Ni/CNT catalysts (Fig. 2a and S2a†), the Ni (111) peaks shifted towards Pt (111) by 0.014°, 0.028° and 0.042° in Pt/NiO/Ni/CNT-1, Pt/NiO/Ni/CNT-2 and Pt/NiO/Ni/CNT-3 (ESI, Fig. S2b†), respectively. These much weaker shifts than those for previously reported PtNi alloys23,32,33 indicate a relatively weak alloying effect between Pt and Ni in this particular case.
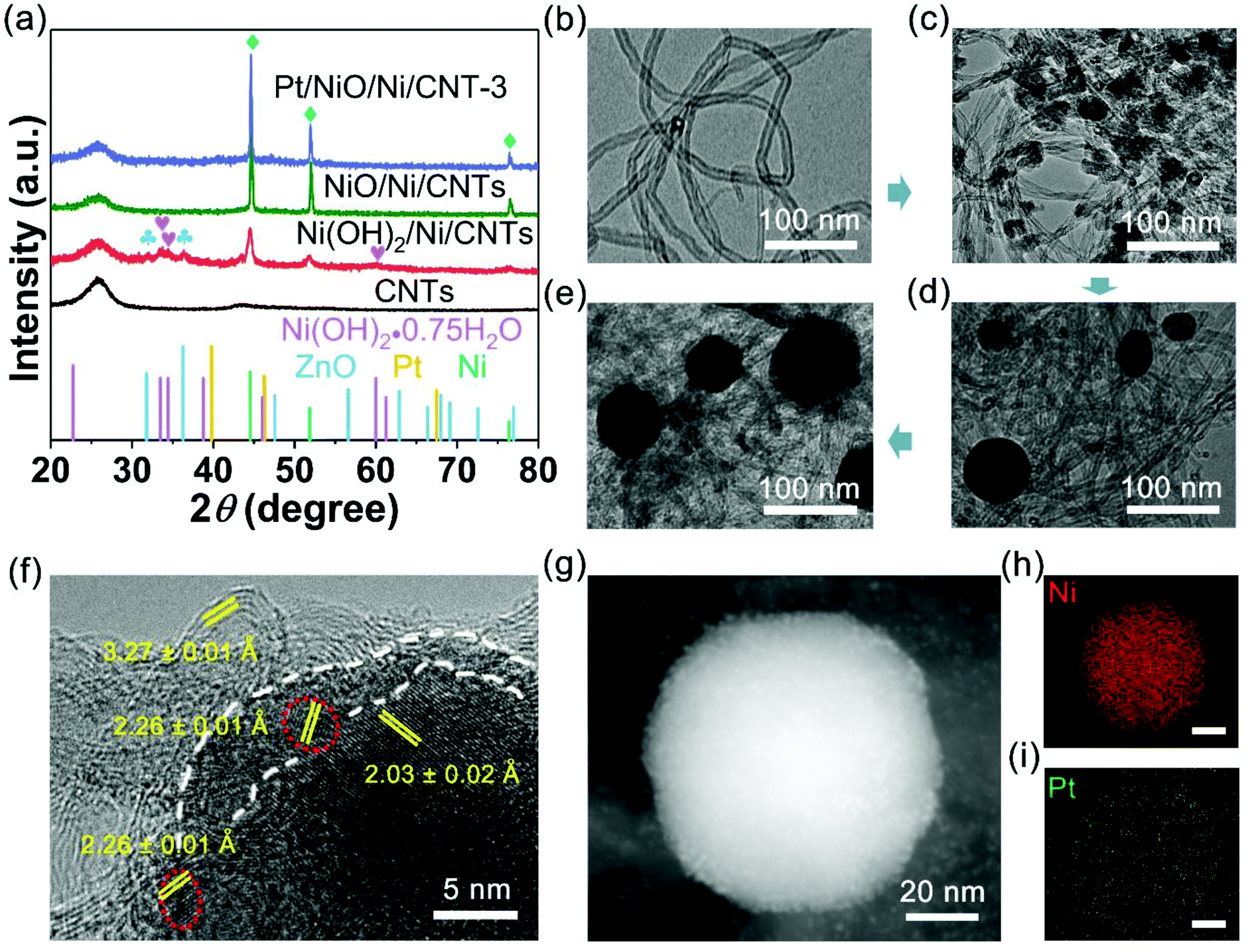 | ||
Fig. 2 (a) XRD patterns of CNTs, Ni(OH)2/Ni/CNTs, NiO/Ni/CNTs and Pt/NiO/Ni/CNT-3 (the peaks for Ni(OH)2·0.75H2O ( ), ZnO ( ), ZnO ( ) and Ni ( ) and Ni ( ) were marked). TEM images of (b) CNTs, (c) Ni(OH)2/Ni/CNTs, (d) NiO/Ni/CNTs and (e) Pt/NiO/Ni/CNT-3. (f) Representative HRTEM image of Pt/NiO/Ni/CNT-3, and the typical Pt NPs are highlighted with red dotted circles. (g) HAADF-STEM image of a single NP of Pt/NiO/Ni/CNT-3. STEM-elemental mappings of (h) Ni and (i) Pt corresponding to (g). The scale bars in (h) and (i) are 20 nm. ) were marked). TEM images of (b) CNTs, (c) Ni(OH)2/Ni/CNTs, (d) NiO/Ni/CNTs and (e) Pt/NiO/Ni/CNT-3. (f) Representative HRTEM image of Pt/NiO/Ni/CNT-3, and the typical Pt NPs are highlighted with red dotted circles. (g) HAADF-STEM image of a single NP of Pt/NiO/Ni/CNT-3. STEM-elemental mappings of (h) Ni and (i) Pt corresponding to (g). The scale bars in (h) and (i) are 20 nm. | ||
Fig. 2b shows a typical transmission electron microscopy (TEM) image for the CNTs. The Raman spectra of the acid-treated CNTs exhibit a stronger D band than G band (ESI, Fig. S3†), indicating some defects induced by an acid. Upon performing the SEED of Ni and Ni(OH)2, NPs formed on CNTs (Fig. 2c). After thermal annealing, some relatively big nanospheres formed on CNTs, as shown in the TEM (Fig. 2d) and scanning electron microscopy (SEM) images of NiO/Ni/CNTs (ESI, Fig. S4a†). Pt decoration did not cause any obvious morphological change (Fig. 2e and S3 in the ESI†), and no obvious particle size change either (ESI, Fig. S4†), suggesting a rather limited Pt loading as small NPs. As shown in HRTEM (Fig. 2f), the lattices with a d-spacing of 3.27 ± 0.01 Å and 2.03 ± 0.02 Å are associated with CNTs and Ni (111) facets, respectively. The amorphous scattered fringes around the Ni nanocrystal (i.e., enclosed by the white dashed line) indicate the formation of amorphous NiO, which was also verified by XPS below. The presence of scattered amorphous NiO on the Ni crystal surface could accelerate the OER and HER in alkaline (vide infra) media. The ultrasmall NPs (∼2 nm) highlighted by red dotted circles are typical Pt (111) facets with a spacing of 2.26 ± 0.01 Å, which have been proved to be more active than the (100) facet34–36 and are attractive for the ORR and HER. The smaller size of Pt NPs in Pt/NiO/Ni/CNT-3 than that in commercial Pt/C (20 wt%, Fig. S5 in the ESI†) is beneficial for exposing more Pt active sites for the ORR and HER at a constant Pt loading. The precise loading of Pt in Pt/NiO/Ni/CNTs determined using an inductively coupled plasma-optical emission spectrometer (ICP-OES, Table S1 in the ESI†) is lower than 4 wt%. As expected, the corresponding high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and STEM-elemental mappings (Fig. 2g–i) for Pt/NiO/Ni/CNT-3 show the co-existence of Pt and Ni.
The elemental compositions and surface chemical states of NiO/Ni/CNTs and Pt/NiO/Ni/CNTs were investigated by XPS. Fig. 3a reproduces the XPS survey spectra, which show the presence of Ni, O and C in NiO/Ni/CNTs, and Ni, Pt, O and C in Pt/NiO/Ni/CNTs. Compared with the commercial Pt/C, Pt/NiO/Ni/CNTs exhibited a much weaker Pt peak, relative to the corresponding C peak (Fig. 3a). Due to the limited detection depth of XPS, the mass contents of all the elements calculated from XPS (ESI, Table S2†) show some differences from the ICP-OES results (ESI, Table S1†). Nevertheless, Tables S1 and S2† show that the mass contents of Pt in Pt/NiO/Ni/CNTs are much lower than that in the commercial Pt/C. The high-resolution XPS Ni 2p spectra (Fig. 3b) show peaks at 870.2 eV and 852.8 eV corresponding to Ni 2p1/2 and Ni 2p3/2, respectively.37 The peaks at 873.7 eV (2p1/2) and 855.6 eV (2p3/2)38,39 are arising from NiO. The high-resolution XPS O 1s spectra (Fig. 3c) display the peak for the Ni–O bond (529.9 eV)40 associated with NiO in NiO/Ni/CNTs and Pt/NiO/Ni/CNTs, along with other peaks for the C–O bond (O-2 at 532.4 ± 0.2 eV: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in esters, amides, anhydrides and lactones, and C–O in phenols and esters; O-3 at 533.5 ± 0.1 eV: ester C–O in COOR/(CO)–O–(CO))41–43 attributable to CNTs (ESI, Fig. S6†). The high-resolution XPS Pt 4f spectra in Fig. 3d show that the Pt0 peaks for Pt/NiO/Ni/CNTs are at much higher binding energies than those of the commercial Pt/C (Pt04f7/2 (70.9 eV) and Pt04f5/2 (74.2 eV)),44 and increasingly shift to higher binding energies (ESI, Table S3†) with increasing Pt loading. These modifications in the Pt electronic structure are beneficial for desorbing oxygen intermediates on Pt to minimize the ORR overpotential.45,46
O in esters, amides, anhydrides and lactones, and C–O in phenols and esters; O-3 at 533.5 ± 0.1 eV: ester C–O in COOR/(CO)–O–(CO))41–43 attributable to CNTs (ESI, Fig. S6†). The high-resolution XPS Pt 4f spectra in Fig. 3d show that the Pt0 peaks for Pt/NiO/Ni/CNTs are at much higher binding energies than those of the commercial Pt/C (Pt04f7/2 (70.9 eV) and Pt04f5/2 (74.2 eV)),44 and increasingly shift to higher binding energies (ESI, Table S3†) with increasing Pt loading. These modifications in the Pt electronic structure are beneficial for desorbing oxygen intermediates on Pt to minimize the ORR overpotential.45,46
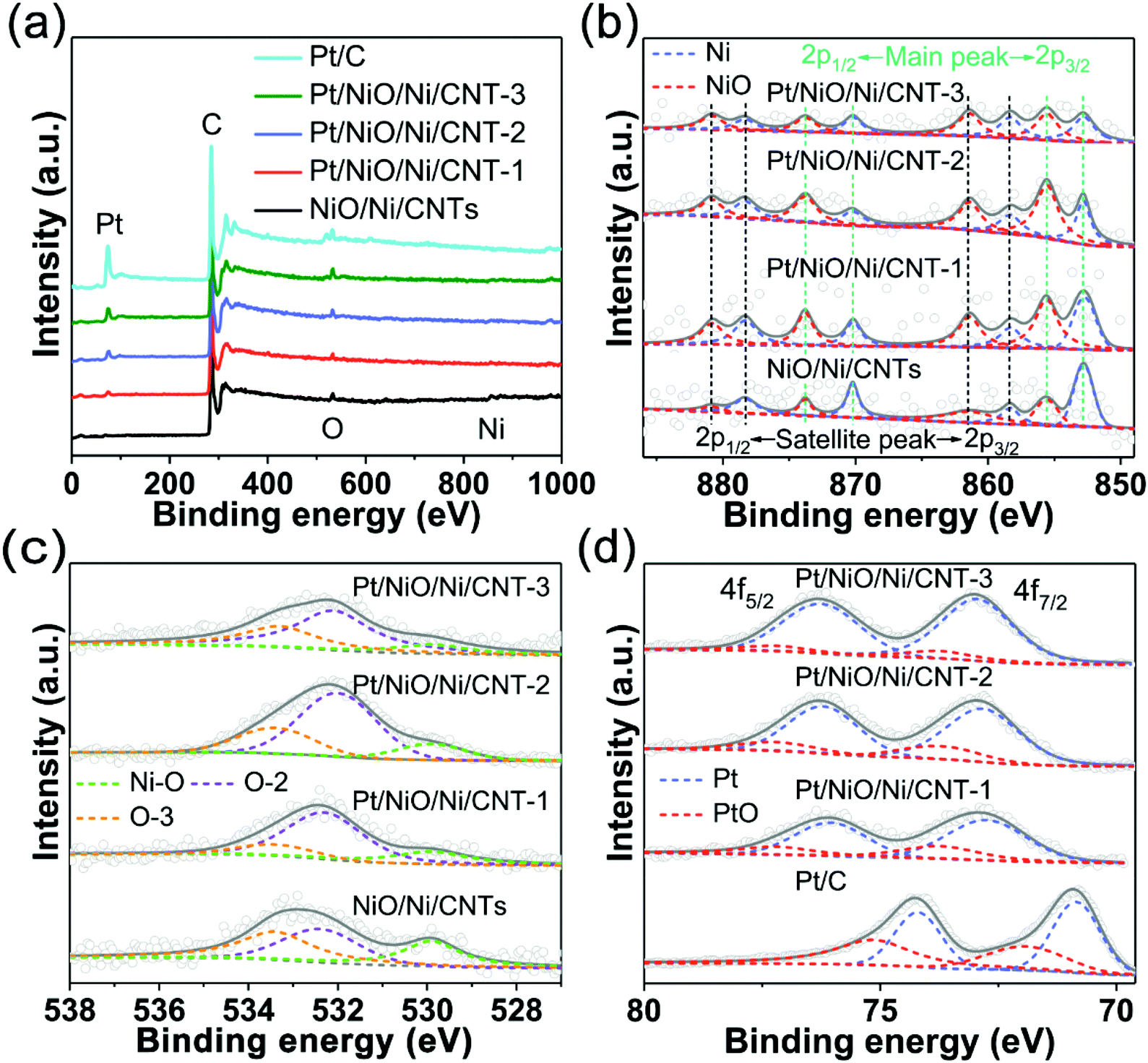 | ||
| Fig. 3 (a) XPS spectra of NiO/Ni/CNTs, Pt/NiO/Ni/CNTs and commercial Pt/C. High-resolution XPS spectra of (b) Ni 2p, (c) O 1s, and (d) Pt 4f. | ||
ORR and OER performance
To investigate the well-characterized Pt/NiO/Ni/CNTs for electrocatalytic energy conversion and storage, we performed the linear sweep voltammetry (LSV) study. Fig. 4a shows LSV curves obtained in O2-saturated 0.1 M KOH. As can be seen, Pt/NiO/Ni/CNT-3 with a low Pt loading (8.15 μg cm−2) exhibited an Eonset of 1.06 V and a half-wave potential (E1/2) of 0.942 V, outperforming commercial 20 wt% Pt/C (50.96 μg cm−2) by 30 and 64 mV, respectively. For comparison, the Eonset, E1/2 and limiting diffusion current density (JL) for all catalysts tested in this study are shown in Fig. S7a (ESI†). To better understand the ORR kinetics of Pt/NiO/Ni/CNTs, mass-corrected Tafel plots were obtained, as shown in Fig. S7b (ESI†). The Tafel plots of the Pt/NiO/Ni/CNTs show slightly lower slopes (72–74 mV dec−1) than that of Pt/C (78 mV dec−1), verifying that Pt/NiO/Ni/CNTs have good kinetics for the ORR due possibly to the unique electronic and surface structures of the Pt nanoparticles in Pt/NiO/Ni/CNTs to facilitate the adsorption/desorption of oxygen intermediates. Fig. S8 (ESI†) shows the corresponding CV curves, which are closely related to electrochemical surface areas (ECSA), determined by hydrogen underpotential deposition (HUPD) in N2-saturated 0.1 M HClO4.47 Due to the low Pt loading and the weak alloying effect between the Ni and Pt (111) facets, Pt/NiO/Ni/CNTs exhibited a weaker intensity and smoother Pt–H oxidation bump (marked with the grey dashed rectangle in Fig. S8†)47 than those of Pt/C.45 The ECSAs and area-specific kinetic current densities (Jarea) of the ORR for Pt/NiO/Ni/CNTs and Pt/C are listed in Table S4 (ESI†), in which the Jarea of Pt/NiO/Ni/CNT-3 exceeds that of the commercial Pt/C by a factor of 4.75. Also, the ORR activities are normalized by Pt mass to obtain the mass-specific ORR kinetic current density (Jmass) at 0.9 V (ESI, Fig. S9†). The Jmass of Pt/NiO/Ni/CNT-3 manifests an enhancement factor of 36.9 compared with Pt/C. The observed ORR performance for Pt/NiO/Ni/CNT-3 is also superior to most reported catalysts, as shown in Table S5 (ESI†). The LSV measurements were performed at the rotating speeds ranging from 1600 rpm to 400 rpm (ESI, Fig. S10a–d†), from which the linear Koutecy–Levich plots were obtained, indicating the first-order reaction kinetics in terms of the dissolved O2.48 The rotating ring-disk electrode (RRDE) measurements (Fig. 4b) were also carried out to reveal a transferred electron number of 3.93–3.95 and a peroxide yield of 2.5–3.4% at 0.6 V for Pt/NiO/Ni/CNTs, demonstrating a high ORR selectivity via a four-electron pathway. Fig. 4c shows chronoamperometric curves for Pt/NiO/Ni/CNT-1, Pt/NiO/Ni/CNT-2, and Pt/NiO/Ni/CNT-3, which reveal excellent durabilities with a 95.0%, 93.8% and 95.3%, respectively, retention of the initial current densities after 10000 s. These values are higher than that of the commercial Pt/C (83.0%), indicating an enhanced stability for Pt in the presence of NiO/Ni due possibly to the aforementioned optimization of the electronic structure and adsorption energy. The decent ORR performance of Pt/NiO/Ni/CNTs is attributable to the following three factors: (1) the proper electronic structure of Pt modified by Ni; (2) the preferential exposure of the Pt (111) facet; and (3) the good electric conductivity of CNTs.
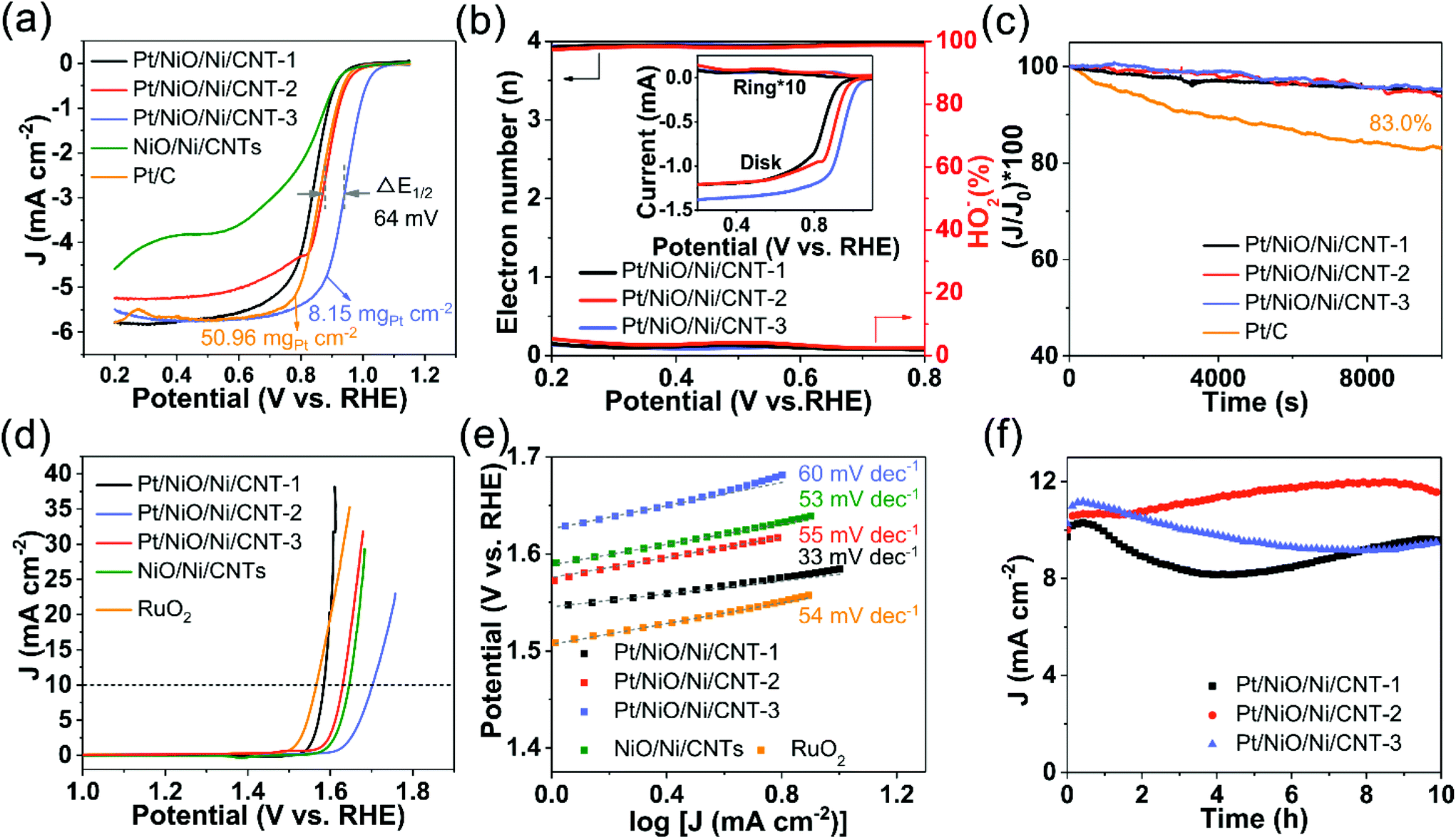 | ||
| Fig. 4 (a) LSV curves of Pt/NiO/Ni/CNTs, NiO/Ni/CNTs and the commercial Pt/C (scan rate 10 mV s−1) for the ORR at 1600 rpm in O2-saturated 0.1 M KOH. The potential at 0.1 mA cm−2 was defined as Eonset. (b) Transferred electron number per oxygen molecule and H2O2 yield calculated from the RRDE curves for Pt/NiO/Ni/CNTs. The inset shows the corresponding RRDE curves. (c) ORR durability curves of Pt/NiO/Ni/CNTs and commercial Pt/C for 10000 s at 0.6 V. (d) LSV curves of Pt/NiO/Ni/CNTs and RuO2 (scan rate of 10 mV s−1) with iR-compensation for the OER at 1600 rpm in O2-saturated 0.1 M KOH. The potential at 1 mA cm−2 was defined as Eonset. (e) Tafel plots corresponding to (d). (f) Chronoamperometric curves (i–t) of Pt/NiO/Ni/CNTs for the OER at the potential where the initial current density is 10 mA cm−2. | ||
Fig. 4d shows LSV curves of Pt/NiO/Ni/CNTs for the OER in 0.1 M KOH. As can be seen, both Pt/NiO/Ni/CNT-1 and Pt/NiO/Ni/CNT-2 exhibited a lower Eonset (1.55 V and 1.57 V, respectively) than that of NiO/Ni/CNTs (Eonset of 1.59 V). Furthermore, Pt/NiO/Ni/CNT-1 exhibited an overpotential at 10 mA cm−2 (η10 = 350 mV) close to that of the benchmark RuO2 (193.5 μgRu cm−2, η10 = 340 mV), along with a low Tafel slope of 33 mV dec−1 (Fig. 4e) compared to RuO2 (54 mV dec−1) and NiO/Ni/CNTs (53 mV dec−1). The high OER activity for Pt/NiO/Ni/CNT-1 is also evidenced by its high turnover frequency for Ni (TOFNi) and Pt (TOFPt). As shown in Fig. S11 (ESI†), the TOFNi value of 0.06 O2 s−1 for Pt/NiO/Ni/CNT-1 is superior to those of Pt/NiO/Ni/CNT-2 (0.01 O2 s−1), Pt/NiO/Ni/CNT-3 (0.002 O2 s−1) and NiO/Ni/CNTs (0.004 O2 s−1); while the TOFPt value of 0.02 O2 s−1 for Pt/NiO/Ni/CNT-1 is also higher than those of Pt/NiO/Ni/CNT-2 (0.003 O2 s−1) and Pt/NiO/Ni/CNT-3 (2.7 × 10−4 O2 s−1). These results indicate that a low content of Pt is beneficial for improving OER kinetics for both Ni and Pt active sites, due probably to more balanced energies for the adsorption/desorption of oxygen. Fig. 4f shows good durabilities for Pt/NiO/Ni/CNTs with >95% current retentions after 10 h.
HER performance in acid and alkaline media
Using a graphite rod as the counter electrode (CE), we have also evaluated the HER activities for the Pt/NiO/Ni/CNTs, NiO/Ni/CNTs, and commercial Pt/C in acid (N2-saturated 0.5 M H2SO4) and alkaline (N2-saturated 0.1 M KOH) media. As shown in Fig. 5a, Pt/NiO/Ni/CNTs exhibited a similar or even more positive Eonset compared to commercial Pt/C in the acid medium. The Tafel slope of Pt/C (31 mV dec−1, Fig. 5b) is in good agreement with that previously reported.49,50 Pt/NiO/Ni/CNTs showed Tafel slopes similar to that of Pt/C, indicating that the Tafel step (2M − Hads → 2 M + H2) is the rate-determining one for these catalysts.51,52 Compared to Pt/C, Pt/NiO/Ni/CNT-1 and Pt/NiO/Ni/CNT-3 showed a factor of 3.95 and 1.08, respectively, improved Jmass at the overpotential of 50 mV (Jη=50) (Fig. 5c). However, Fig. S12a (ESI†) shows a better charge transfer capability for Pt/C than Pt/NiO/Ni/CNTs. Thus, the high Jmass observed for Pt/NiO/Ni/CNTs with a low Pt loading is mainly attributable to the ultrasmall size and well dispersion of the Pt NPs to provide numerous active sites for hydrogen intermediate adsorption.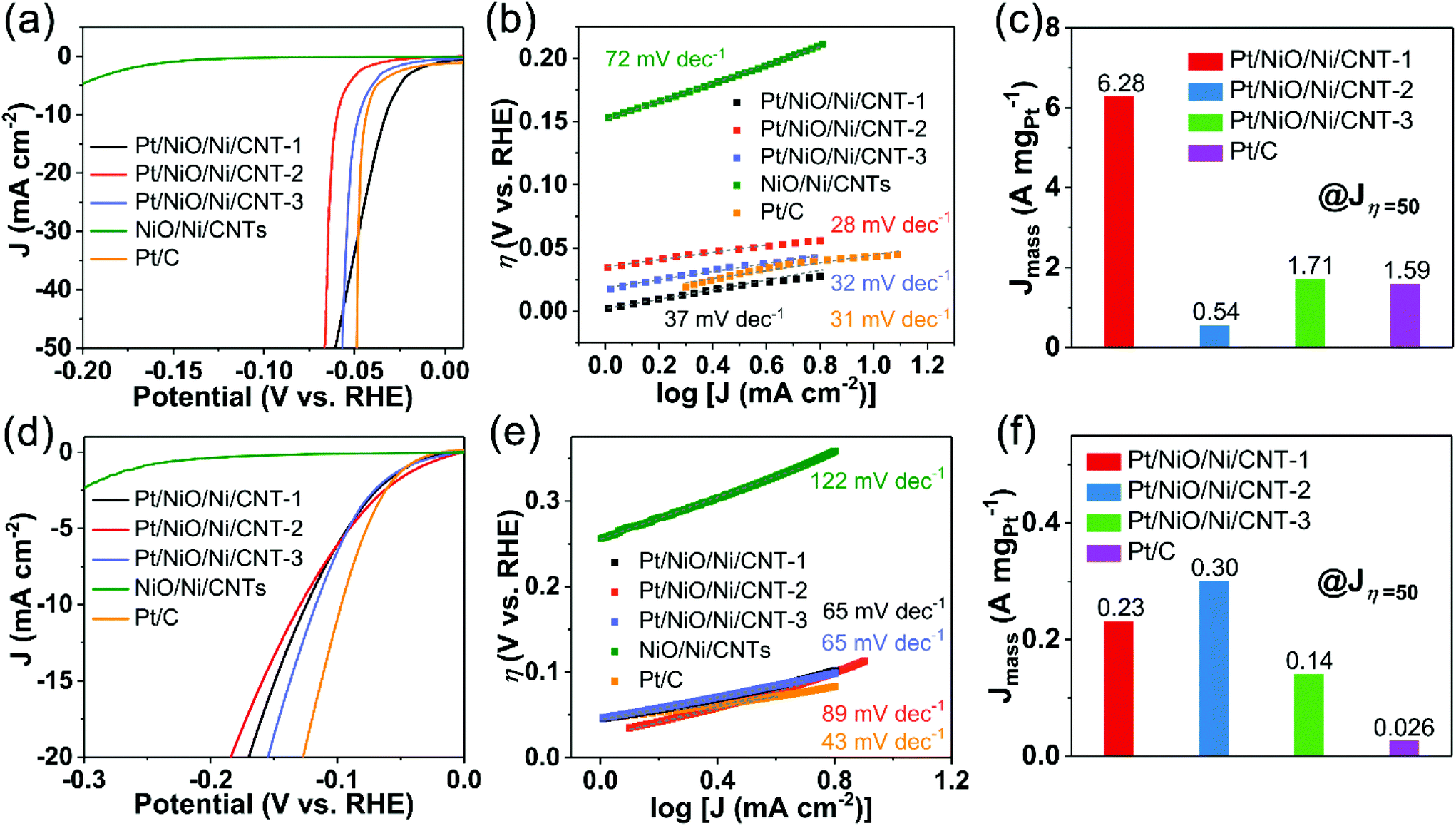 | ||
| Fig. 5 (a) LSV curves and (b) the corresponding Tafel plots of Pt/NiO/Ni/CNTs, NiO/Ni/CNTs and commercial Pt/C for the HER with iR-compensation in N2-saturated 0.5 M H2SO4 at 1600 rpm. (c) Jmass at Jη=50 in N2-saturated 0.5 M H2SO4. (d) LSV curves and (e) Tafel plots of Pt/NiO/Ni/CNTs, NiO/Ni/CNTs and commercial Pt/C for the HER with iR-compensation in N2-saturated 0.1 M KOH at 1600 rpm. (f) Jmass at Jη=50 in N2-saturated 0.1 M KOH. | ||
Fig. 5d shows the LSV curves measured in the alkaline medium, in which η10 for Pt/NiO/Ni/CNT-1, Pt/NiO/Ni/CNT-2, Pt/NiO/Ni/CNT-3, and commercial Pt/C are 125 mV, 129 mV, 117 mV, and 97 mV, respectively. The good HER kinetics are also indicated by the low Tafel slopes for Pt/NiO/Ni/CNTs. Jmass at the overpotential of Jη=50 for Pt/NiO/Ni/CNTs and Pt/C are given in Fig. 5f, which shows about 8.8-, 11.5- and 5.4-fold enhancements for Pt/NiO/Ni/CNT-1, Pt/NiO/Ni/CNT-2 and Pt/NiO/Ni/CNT-3, respectively, compared with that of Pt/C. However, Fig. S12b (ESI†) shows a weaker electron transfer capability for Pt/NiO/Ni/CNTs than that of Pt/C. Even in the alkaline medium, the excellent HER activities of Pt/NiO/Ni/CNTs with a low content of Pt can be attributed, once again, to the unique electronic structure and balanced adsorption/desorption energies. Specifically, the homogeneous dispersion of Pt NPs on Ni nanospheres created numerous active sites to effectively adsorb the hydrogen intermediate while the large-scale exposure of the NiO/Ni surface facilitated the capture of OH− from H2O to accelerate the dissociation of H2O.
The above results indicate that Pt/NiO/Ni/CNTs are highly active electrocatalysts for the HER in both acid and alkaline media. Table S6 (ESI†) summarizes the ORR, OER and HER performance for Pt/NiO/Ni/CNTs with a low Pt loading in comparison with various trifunctional electrocatalysts previously reported. Unlike single active sites in other electrocatalysts, multiple active sites (Pt, NiO and Ni) are efficiently exposed and utilized in Pt/NiO/Ni/CNTs. Moreover, these active sites have synergistic effects to further promote electrocatalytic performance. Also, the use of CNTs as the substrate can ensure efficient electron transport. The electrocatalytic performance indicates that the Pt/NiO/Ni/CNTs are a class of highly efficient trifunctional electrocatalysts of practical significance.
Zn–air batteries and water splitting performance
To demonstrate potential applications of Pt/NiO/Ni/CNTs as ORR/OER/HER trifunctional electrocatalysts, we devised an integrated water splitting system with the Pt/NiO/Ni/CNT as a bifunctional electrocatalyst for the OER and HER self-powered by Zn–air batteries based on the same Pt/NiO/Ni/CNT as the air cathode for the ORR and OER. Fig. 6a shows the cell structure of a primary Zn–air battery with 6 M KOH as the electrolyte. As can be seen in Fig. 6b, all the Zn–air batteries composed of the Pt/NiO/Ni/CNT cathode exhibited a higher peak power density than that of its counterpart with the Pt/C + RuO2 (mass ratio of 1:3, 90 mW cm−2) mixture cathode. Specifically, Zn–air batteries based on the Pt/NiO/Ni/CNT-1, Pt/NiO/Ni/CNT-2 and Pt/NiO/Ni/CNT-3 air cathodes showed power densities of 128.3, 158.6 and 166.9 mW cm−2, respectively, with an open circuit voltage (OCV) of 1.52 V (Fig. 6b and c). After normalizing to the weight of the consumed zinc electrode (ESI, Fig. S13†), the Pt/NiO/Ni/CNT-3 based Zn–air battery showed specific capacities of 708.2 mA h g−1 at 10 mA cm−2, even higher than that of its counterpart based on the Pt/C + RuO2 cathode. Furthermore, no obvious voltage drop was observed over 40 h continuous operation (Fig. 6d), indicating a remarkable durability for Pt/NiO/Ni/CNTs toward the ORR in the primary Zn–air battery.
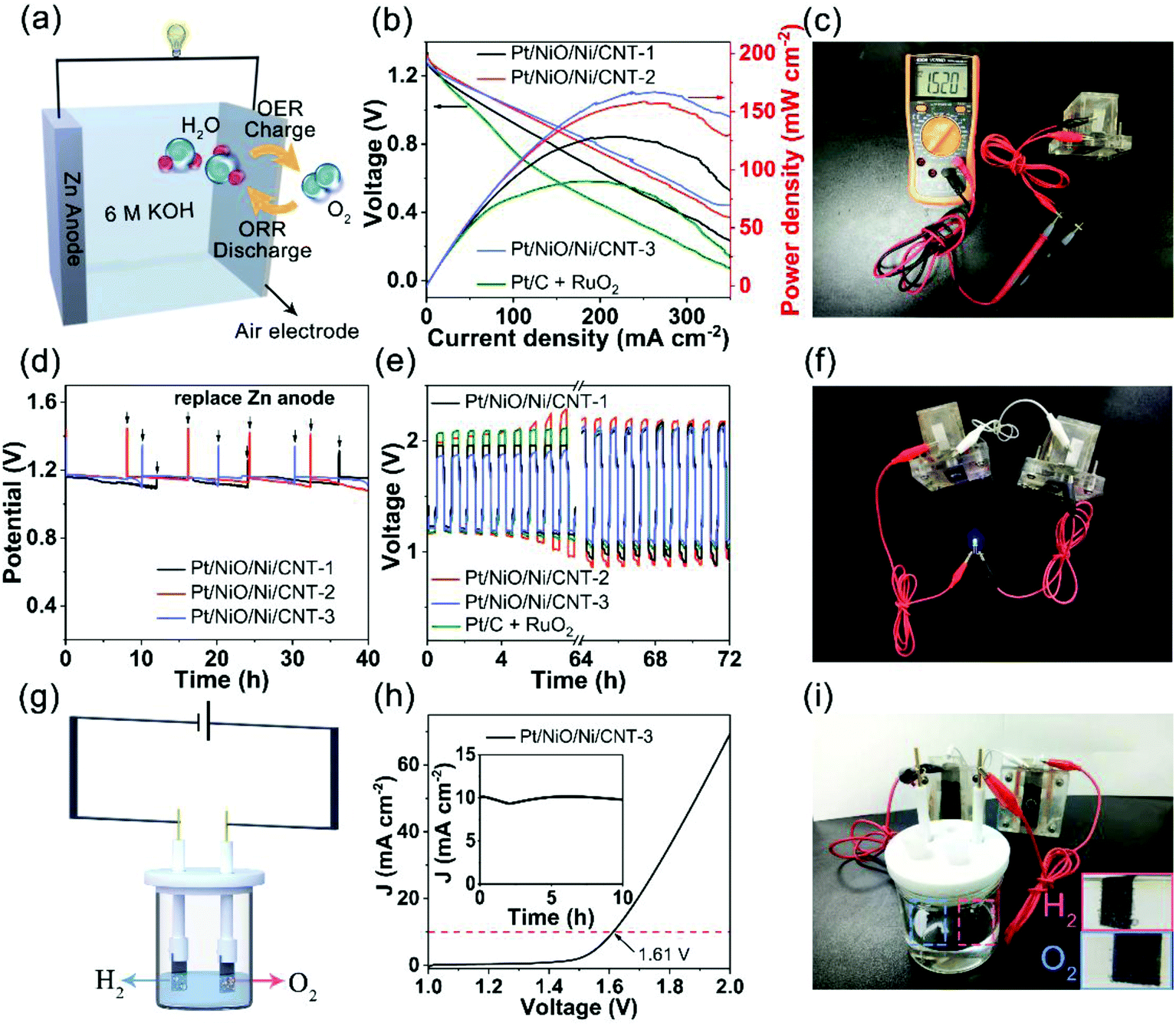 | ||
| Fig. 6 (a) Schematic illustration of a primary Zn–air battery. (b) Discharge polarization curves and power density curves from the primary Zn–air batteries with Pt/NiO/Ni/CNTs or Pt/C + RuO2 as the air catalyst. (c) The photograph for the OCV of the primary Zn–air battery measured using a multimeter. (d) Discharge curves of the primary Zn–air battery with Pt/NiO/Ni/CNTs as the air electrode at 10 mA cm−2. The pulse-like voltage changes correspond to the replacement of the Zn plate. (e) Galvanostatic discharge–charge cycling performance of the rechargeable Zn–air batteries using Pt/NiO/Ni/CNTs or Pt/C + RuO2 as the air electrode at 10 mA cm−2. (f) A typical photograph for a blue LED light (∼1.8 V) being lighted up by two Zn–air batteries in series. (g) Schematic illustration of an electrochemical cell for overall water splitting. (h) The LSV curve and corresponding stability profile at 1.61 V for 10 h (inset) for overall water splitting catalyzed by Pt/NiO/Ni/CNT-3 in 1 M KOH. (i) A typical photograph of the Pt/NiO/Ni/CNT-3∥Pt/NiO/Ni/CNT-3 water-splitting cell powered by two Zn–air batteries in series using Pt/NiO/Ni/CNT-3 as the air electrode (the insets show H2 and O2 generated on the Pt/NiO/Ni/CNT-3 cathode and anode, respectively). | ||
To evaluate the feasibility of Pt/NiO/Ni/CNTs as an ORR/OER bifunctional air-electrode, a rechargeable Zn–air battery based on the Pt/NiO/Ni/CNT-3 cathode was cycled at 10 mA cm−2 for 72 h, which showed an initial charge potential of 1.86 V and a discharge potential of 1.21 V with a smaller voltage gap of 0.65 V and a higher round-trip efficiency of 73.3% than those of its counterpart based on the Pt/C + RuO2 cathode (Fig. 6e). As can also be seen in Fig. 6e, Zn–air batteries based on all the Pt/NiO/Ni/CNT cathodes showed an increase in the charge potential with increasing cycle number due, most probably, to the formation of Ni(OH)2 or NiOOH during prolonged cycling.29,53,54 Compared with the Pt/C + RuO2 cathode, the discharge potential for the Pt/NiO/Ni/CNTs was relatively stable and remained to be 1.10 V over 72 h cycling, indicating a good cycling stability. As shown in Fig. 6f, a blue light emitting diode (LED, ∼1.8 V) was lighted up by two rechargeable Zn–air batteries based on the Pt/NiO/Ni/CNT-3 cathode connected in series, demonstrating its potential for practical applications.
Fig. 6g schematically shows a water splitting system with both the cathode and anode based on Pt/NiO/Ni/CNT-3 as an OER/HER bifunctional electrocatalyst. As can be seen in Fig. 6h, the Pt/NiO/Ni/CNT-3∥Pt/NiO/Ni/CNT-3 water splitting cell displayed a potential of 1.61 V at 10 mA cm−2, which is more active than most other water-splitting electrocatalysts recently reported (ESI, Table S7†). The inset of Fig. 6h shows the chronoamperometric curve at 1.61 V, indicating a good stability with a current retention of 97.98% after 10 h continuous operation. Finally, Fig. 6i shows a photograph of an integrated overall water splitting system self-powered by two Zn–air batteries in series, in which Pt/NiO/Ni/CNT-3 was used as a trifunctional electrocatalyst for the ORR and OER in the Zn–air batteries as well as the OER and HER for water splitting. As shown in the inset of Fig. 6i and Movie S1 (ESI†), H2 and O2 bubbles are obviously generated on the two electrodes, indicating the successful operation of this integrated and self-powered overall water splitting system.
Conclusions
In summary, we have used the SEED method to successfully synthesize the first hierarchically structured Pt/NiO/Ni/CNTs with a low loading of ultrasmall Pt NPs (∼2 nm) and preferentially exposed (111) facets for the efficient ORR, OER and HER. The resulting catalysts also exhibited a higher ORR activity (half-wave potential at 0.942 V) than that of the benchmark commercial Pt/C (20 wt%) and a similar OER activity (overpotential of 350 mV at 10 mA cm−2) to that of RuO2 in alkaline media. Moreover, the newly-developed Pt/NiO/Ni/CNT catalysts also showed a high mass activity superior to Pt/C towards the HER in both acid and alkaline electrolytes. A rechargeable Zn–air battery composed of the Pt/NiO/Ni/CNT-3 air cathode delivered a high OCV of 1.52 V. The overall water splitting unit with a Pt/NiO/Ni/CNT-3∥Pt/NiO/Ni/CNT-3 cell structure exhibited a potential of 1.61 V at 10 mA cm−2 and good stability, which could be powered by two Zn–air batteries in series using the same Pt/NiO/Ni/CNT-3 as a bifunctional electrocatalyst for the ORR and OER. This work provides a facile approach to high-performance low-cost trifunctional catalysts from Earth-abundant materials for efficient energy conversion and storage.Experimental
Chemicals and materials
NiCl2·6H2O was purchased from Shanghai Macklin Biochemical Co., Ltd. Zinc acetate and K2PtCl6 (Pt content 40.1 wt%) were purchased from Tianjin Heowns Biochemical Technology Co., Ltd. Multiwalled carbon nanotubes (CNTs) were purchased from Nanjing XFNANO Materials Technology Co., Ltd. HClO4 (70–72%) and KOH (≥99.99%) were purchased from Aladdin Reagent Co., Ltd. H2SO4 (95–98%), HCl (36–38%), HNO3 (65–68%), acetone (≥99.5%) and anhydrous ethanol (EtOH, ≥99.7%) were purchased from Beijing Chemical Reagent Company. Nafion solution (5 wt%), Zn plate (thickness of 0.25 mm, diameter <8 nm, length of 10–30 μm) and Pt/C (20 wt% on carbon black) were purchased from Alfa Aesar Co., Ltd. RuO2 was purchased from Beijing Innochem Technology Co., Ltd. The polytetrafluoroethylene (PTFE) emulsion (60 wt%) was purchased from Beijing J&K Scientific Co., Ltd. Ni foam was purchased from Shenzhen Yongxingye Co., Ltd. All reagents, except CNTs, were used as received.Materials synthesis
:H2 (v/v) = 50:10) at 950 °C for 2 h with the ramp rate of 5 °C min−1 to obtain NiO/Ni/CNTs.
Materials characterization
SEM images were recorded using a Hitachi S-4700 scanning electron microscope operated at 20 kV. TEM imaging was conducted on a Hitachi 7700 transmission electron microscope operated at 100 kV. HRTEM, HAADF-STEM and EDS were performed using a JEM-2200FS microscope with an accelerating voltage of 200 kV. STEM-elemental mapping was performed on an Oxford X-Max 80T microscope. Compositions of the NiO/Ni/CNTs and Pt/NiO/Ni/CNTs were quantified using ICP-OES on an Agilent 725-ES instrument. The XRD patterns were obtained using a Bruker D2 Phaser with a Cu Kα radiation source (λ = 1.54 Å) at a scan rate of 5° min−1. XPS measurements were carried out on an ESCALAB 250 spectrometer (ThermoFisher) using 150 W Al Kα radiation (1486.6 eV) with the binding energies calibrated by the C 1s line at 284.8 eV. Raman spectra were recorded at a wavelength of 532 nm on a Raman spectrometer (Horiba Lab RAM HR Evolution). The pH values of a blank NiCl2 solution and the NiCl2 solution near the Zn plate were measured using a pH meter (Five Easy Plus, METTLER TOLEDO).Electrochemical measurements
All electrochemical measurements were conducted on a 760E electrochemical workstation (Shanghai CH Instruments). The trifunctional electrocatalyses for the ORR, OER and HER were measured in three-electrode cells. A glassy carbon rotating disk electrode (RDE, PINE Research Instrumentation, USA) with a diameter of 5 mm was used as the substrate of the working electrode (WE). A Pt wire was used for the ORR and OER and a graphite rod was used for the HER as a CE. Ag/AgCl was used as a reference electrode (RE) for all the electrochemical measurements.The catalyst ink was typically prepared by dispersing 5 mg catalyst in 490 μL EtOH and 10 μL Nafion (5 wt%) under ultrasonication for 30 min. 5 μL catalyst ink was dropped onto a freshly polished RDE and dried (catalyst loading of 0.255 mg cm−2). The benchmark commercial Pt/C and RuO2 were prepared into catalyst ink in the same way as for the Pt/NiO/Ni/CNTs.
ORR activity testing was performed in O2-saturated 0.1 M KOH. The CV curves were firstly recorded at a scan rate of 50 mV s−1 for 20 cycles. The ORR activity was then evaluated with the LSV technique on a RDE at 1600 rpm. The calculation details for the transferred electron number, H2O2 yield (HO2−%), ECSA, Jarea and Jmass are shown in the ESI.†
The OER was measured in O2-saturated 0.1 M KOH and the polarization curves were conducted by the LSV technique to determine the OER activity. The CV technique was firstly used to activate the catalysts for 20 cycles until the curves were stabilized. Then, the LSV curves were collected at 10 mV s−1. For the durability test, the chronoamperometric technique was performed at the potential, where the initial current density was 10 mA cm−2 for 10 h without iR-compensation. The calculation details for TOFNi and TOFPt are shown in the ESI.†
HER activity was measured in N2-saturated 0.5 M H2SO4 or 0.1 M KOH. At first, CV testing was conducted 15 times to activate the electrocatalysts. Then, LSV curves were obtained at the scan rate of 10 mV s−1. EIS was measured with frequencies from 105 to 0.1 Hz, and the Jmass of Pt was calculated from the following equation:
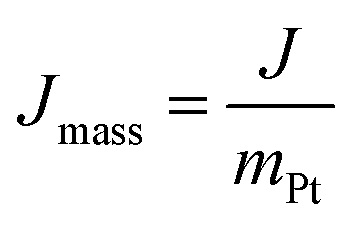
All the potentials were converted to a reversible hydrogen electrode (RHE) according to the equation: Evs. RHE = Evs. Ag/AgCl + EAg/AgCl + 0.05915 pH. EAg/AgCl was calibrated vs. RHE according to the CV curve measured in H2-saturated 0.5 M H2SO4 with the Pt wire as both the WE and CE, and the Ag/AgCl electrode as the RE. As shown in Fig. S14 (ESI†), the average value of the two potentials, where the currents are zero, is determined to be EAg/AgCl (0.213 V).
Liquid Zn–air battery assembly
The air electrodes in liquid Zn–air batteries were prepared according to a previous report43 using Ni foam as a current collector of the air electrodes. The resulting catalyst loading was 2 mg cm−2 with an efficient circle area (0.785 cm2) exposed to the electrolyte. In the control experiment, the catalyst ink containing commercial Pt/C and RuO2 with a mass ratio of 1:3 was used. A polished Zn plate was used as the anode. 6 M KOH served as the electrolyte for primary Zn–air batteries, while 6 M KOH containing 0.2 M zinc acetate served as the electrolyte for rechargeable Zn–air batteries.
Liquid Zn–air battery testing
All the Zn–air batteries were tested under an ambient atmosphere with the reactor volume of 80 mL electrolyte. The polarization curves were measured with the LSV technique at a scan rate of 5 mV s−1 in a primary Zn–air battery. The current density and power density of all the catalysts were normalized to the effective surface area of the air electrode. The power density and specific capacity density were calculated as follows: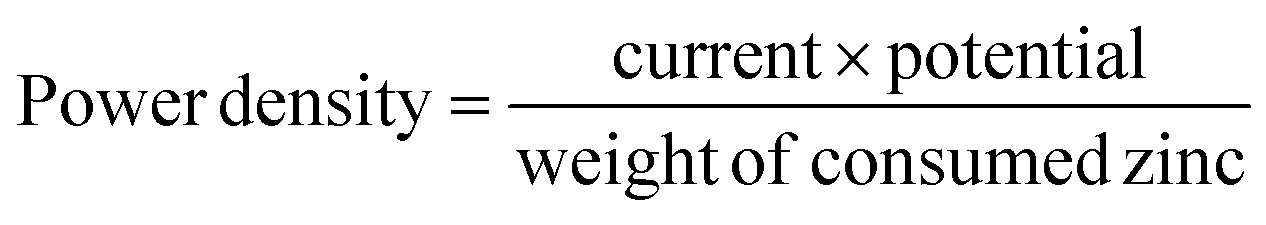
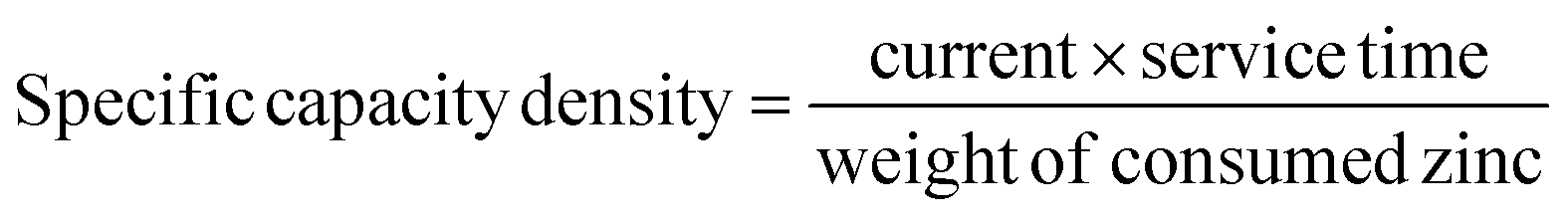
For a primary Zn–air battery, the discharge durability was also measured for 40 h, during which the Zn plate was replaced several times. The charge–discharge cycling stability for a rechargeable Zn–air battery with a cycling interval of 40 min (20 min for charge and 20 min for discharge) at 10 mA cm−2 was carried out using a LAND-CT2001A testing system.
Overall water splitting measurements
For water splitting measurements, the catalyst ink of Pt/NiO/Ni/CNT-3 was dropped onto 1 cm2 carbon paper with a catalyst loading of 2 mg cm−2. Then, this carbon paper was dried at 60 °C for 4 h and used as both the cathode and anode for water splitting. The polarization curve and durability were tested in N2-saturated 1 M KOH without iR-compensation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by The National Key Research and Development Program of China (2017YFA0206500), the Key Program of National Natural Science Foundation of China (51732002), the Distinguished Scientist Program at BUCT (buctylkxj02), and the Australian Research Council (ARC, DP 190103881 and FL 190100126). The authors also thank the financial support from the Beijing Advanced Innovation Center for Soft Matter Science and Engineering, and BUCT.Notes and references
- L. An, Y. Li, M. Luo, J. Yin, Y.-Q. Zhao, C. Xu, F. Cheng, Y. Yang, P. Xi and S. Guo, Adv. Funct. Mater., 2017, 27, 1703779 CrossRef.
- Z. Wang, H. Jin, T. Meng, K. Liao, W. Meng, J. Yang, D. He, Y. Xiong and S. Mu, Adv. Funct. Mater., 2018, 28, 1802596 CrossRef.
- C. Tang, B. Wang, H.-F. Wang and Q. Zhang, Adv. Mater., 2017, 29, 1703185 CrossRef PubMed.
- T. Tang, W.-J. Jiang, S. Niu, N. Liu, H. Luo, Q. Zhang, W. Wen, Y.-Y. Chen, L.-B. Huang, F. Gao and J.-S. Hu, Adv. Funct. Mater., 2018, 28, 1704594 CrossRef.
- Q. Hu, X. Liu, B. Zhu, L. Fan, X. Chai, Q. Zhang, J. Liu, C. He and Z. Lin, Nano Energy, 2018, 50, 212–219 CrossRef CAS.
- R. Gao, Q. Dai, F. Du, D. Yan and L. Dai, J. Am. Chem. Soc., 2019, 141, 11658–11666 CrossRef CAS PubMed.
- J. Zhang, L. Qu, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., Int. Ed., 2016, 128, 2270–2274 CrossRef.
- C. N. R. Rao and K. Pramoda, Bull. Chem. Soc. Jpn., 2019, 92, 441–468 CrossRef CAS.
- H. Jiang, J. Gu, X. Zheng, M. Liu, X. Qiu, L. Wang, W. Li, Z. Chen, X. Ji and J. Li, Energy Environ. Sci., 2019, 12, 322–333 RSC.
- Q. Shi, Q. Liu, Y. Ma, Z. Fang, Z. Liang, G. Shao, B. Tang, W. Yang, L. Qin and X. Fang, Adv. Energy Mater., 2020, 10, 1903854 CrossRef CAS.
- T. Oh, K. Kim and J. Kim, J. Energy Chem., 2019, 38, 60–67 CrossRef.
- Q. Hu, G. Li, G. Li, X. Liu, B. Zhu, X. Chai, Q. Zhang, J. Liu and C. He, Adv. Energy Mater., 2019, 9, 1803867 CrossRef.
- S. Wang, J. Qin, T. Meng and M. Cao, Nano Energy, 2017, 39, 626–638 CrossRef CAS.
- Y. Wang, C. Xie, Z. Zhang, D. Liu, R. Chen and S. Wang, Adv. Funct. Mater., 2018, 28, 1703363 CrossRef.
- J. Greeley and N. M. Markovic, Energy Environ. Sci., 2012, 5, 9246–9256 RSC.
- L. Zhang, L. Han, H. Liu, X. Liu and J. Luo, Angew. Chem., Int. Ed., 2017, 56, 13694–13698 CrossRef CAS PubMed.
- Y. P. Zhu, C. Guo, Y. Zheng and S.-Z. Qiao, Acc. Chem. Res., 2017, 50, 915–923 CrossRef CAS PubMed.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- Q. Chang, Y. Xu, Z. Duan, F. Xiao, F. Fu, Y. Hong, J. Kim, S.-I. Choi, D. Su and M. Shao, Nano Lett., 2017, 17, 3926–3931 CrossRef CAS.
- R. Kavian, S.-I. Choi, J. Park, T. Liu, H.-C. Peng, N. Lu, J. Wang, M. J. Kim, Y. Xia and S. W. Lee, J. Mater. Chem. A, 2016, 4, 12392–12397 RSC.
- S. Kühl, M. Gocyla, H. Heyen, S. Selve, M. Heggen, R. E. Dunin-Borkowski and P. Strasser, J. Mater. Chem. A, 2019, 7, 1149–1159 RSC.
- H. Fan, M. Cheng, Z. Wang and R. Wang, Nano Res., 2017, 10, 187–198 CrossRef CAS.
- L. Gan, M. Heggen, S. Rudi and P. Strasser, Nano Lett., 2012, 12, 5423–5430 CrossRef CAS.
- T. Ding, W. Wang, X. Zhou, L. Zhang, C. Wang, J. Jiang, W. Yang and Q. Yang, APL Mater., 2015, 4, 015702 CrossRef.
- G.-R. Zhang and S. Wöllner, Appl. Catal., B, 2018, 222, 26–34 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- Z. Zhao, H. Liu, W. Gao, W. Xue, Z. Liu, J. Huang, X. Pan and Y. Huang, J. Am. Chem. Soc., 2018, 140, 9046–9050 CrossRef CAS PubMed.
- L. Qu and L. Dai, J. Am. Chem. Soc., 2005, 127, 10806–10807 CrossRef CAS PubMed.
- L. Qu, L. Dai and E. Osawa, J. Am. Chem. Soc., 2006, 128, 5523–5532 CrossRef CAS PubMed.
- J. Choi, Y. Lee, J. Kim and H. Lee, J. Power Sources, 2016, 307, 883–890 CrossRef CAS.
- N. N. Kariuki, M. F. Cansizoglu, M. Begum, M. Yurukcu, F. M. Yurtsever, T. Karabacak and D. J. Myers, ACS Catal., 2016, 6, 3478–3485 CrossRef CAS.
- V. Viswanathan, H. A. Hansen, J. Rossmeisl and J. K. Nørskov, ACS Catal., 2012, 2, 1654–1660 CrossRef CAS.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- J. Chen, L. Fang, S. Luo, Y. Liu and S. Chen, J. Phys. Chem. C, 2017, 121, 6209–6217 CrossRef CAS.
- A. N. Mansour, Surf. Sci. Spectra, 1994, 3, 221–230 CrossRef CAS.
- A. N. Mansour, Surf. Sci. Spectra, 1994, 3, 231–238 CrossRef CAS.
- T. Xia, J. Liu, S. Wang, C. Wang, Y. Sun, L. Gu and R. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 10841–10849 CrossRef CAS PubMed.
- X. Liu, W. Liu, M. Ko, M. Park, M. G. Kim, P. Oh, S. Chae, S. Park, A. Casimir, G. Wu and J. Cho, Adv. Funct. Mater., 2015, 25, 5799–5808 CrossRef CAS.
- S. Sun, F. Jaouen and J.-P. Dodelet, Adv. Mater., 2008, 20, 3900–3904 CrossRef CAS.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- Q. Liu, Y. Wang, L. Dai and J. Yao, Adv. Mater., 2016, 28, 3000–3006 CrossRef CAS PubMed.
- S. Wang, L. Xiong, J. Bi, X. Zhang, G. Yang and S. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 27009–27018 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- N. K. Chaudhari, J. Joo, B. Kim, B. Ruqia, S.-I. Choi and K. Lee, Nanoscale, 2018, 10, 20073–20088 RSC.
- Y. Garsany, O. A. Baturina, K. E. Swider-Lyons and S. S. Kocha, Anal. Chem., 2010, 82, 6321–6328 CrossRef CAS PubMed.
- Y. Guo, P. Yuan, J. Zhang, H. Xia, F. Cheng, M. Zhou, J. Li, Y. Qiao, S. Mu and Q. Xu, Adv. Funct. Mater., 2018, 28, 1805641 CrossRef.
- B. E. Conway and B. V. Tilak, Electrochim. Acta, 2002, 47, 3571–3594 CrossRef CAS.
- X. Cheng, Y. Li, L. Zheng, Y. Yan, Y. Zhang, G. Chen, S. Sun and J. Zhang, Energy Environ. Sci., 2017, 10, 2450–2458 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 RSC.
- S. L. Medway, C. A. Lucas, A. Kowal, R. J. Nichols and D. Johnson, J. Electroanal. Chem., 2006, 587, 172–181 CrossRef CAS.
- P. Oliva, J. Leonardi, J. F. Laurent, C. Delmas, J. J. Braconnier, M. Figlarz, F. Fievet and A. D. Guibert, J. Power Sources, 1982, 8, 229–255 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr03378b |
| This journal is © The Royal Society of Chemistry 2020 |