
Capacitive deionization for nutrient recovery from wastewater with disinfection capability†
Zheng
Ge
a,
Xi
Chen
a,
Xia
Huang
b and
Zhiyong Jason
Ren
 *a
*a
aDepartment of Civil, Environmental, and Architectural Engineering, University of Colorado Boulder, Boulder, Colorado 80309, USA. E-mail: zhiyong.ren@colorado.edu; Fax: +1 303 492 7317; Tel: +1 303 492 4137
bState Key Joint Laboratory of Environment Simulation and Pollution Control, School of Environment, Tsinghua University, Beijing, 100084, P. R. China
First published on 17th October 2017
Abstract
This study demonstrates that capacitive deionization can be effectively used for the removal and recovery of dominant nitrogen (ammonium) and phosphorus (phosphate salts) species present in wastewater. Moreover, low concentrations of chlorine and other oxidants can be generated in situ for disinfection. With an applied voltage from 1.2 V to 3.0 V, salts, ammonium, and phosphorus can be quickly removed from water and adsorbed on the electrodes due to the formation of an electrical double layer, and results show that the removal efficiency was 77.5–91.2% for salts, 60.5–95.7% for ammonium, and 46.4–80.7% for phosphorus, respectively. In addition, most of the adsorbed ions are released back to the concentrate during regeneration, so high nutrient recovery can be accomplished as well. Such a simple electrochemical process can be promising to solve both nutrient and salinity problems after biological treatments for discharge and reuse, and the additional disinfection function adds further benefits to improve water quality and safety with low cost.
Water impactNutrient removal and recovery is an emerging need for wastewater treatment, but current biological processes to convert nitrogen and phosphorus into resources can be expensive, time consuming, and energy intensive. Using abiotic electrochemical methods may provide an efficient alternative for nutrient recovery from wastewater and alleviate salinity problems in water reuse. |
Introduction
Nutrient removal and recovery is becoming a major task beyond organic removal for wastewater treatment facilities.1–3 Traditional biological nutrient removal (BNR) processes consume large amounts of energy and chemicals, but most of them only remove nutrients from wastewater without the capability of recovering such valuable resources. Emerging anaerobic treatment processes such as anaerobic membrane bioreactors (AnMBRs) and microbial electrochemical systems (MESs) can recover energy during organic degradation but still have limited capability for nutrient removal.4–6 In fact, nutrients remaining in the anaerobic effluent have become a major challenge for these technologies due to the difficulties in meeting discharge permit limits. The major nutrient species after anaerobic biological treatment are ammonium and phosphate salts, which are charged ion species under neutral conditions.7 Therefore, in this study, we hypothesize that an electrochemical process such as capacitive deionization or electrosorption may be effective in removing low concentrations of nutrients from wastewater and the anaerobic treatment effluent and recovering them in the concentrate during regeneration.Capacitive deionization is a technology that has been increasingly used in water desalination and softening.8–10 Because the main goal of such applications has been salt removal, the process has been focused on deionizing saline water to generate fresh water. The goal of this study, however, includes both removing charged salt and nutrient species from wastewater and recovering them from concentrates. By applying an electric field between two closely placed porous electrodes, ions and charged particles in water can be separated. Cations/positively charged species move toward the negatively biased electrode (cathode), while anions/negatively charged species move toward the positively biased electrode (anode). This electrochemical process shares a similar principle to supercapacitors but is modified for salt removal from aqueous solution rather than storing charge on electrodes. Because this process moves ions rather than water, it can be more efficient under low- to moderate-salinity water conditions (TDS < 5000 mg L−1).10–13 In general, the total dissolved solid (TDS) in the wastewater effluent is around 1000 mg L−1, so CDI can be an ideal polishing process to remove charged nutrient compounds after mainstream biological carbon removal. For example, by coupling membrane capacitive deionization with ion exchange, more than 65% of the ammonium was recovered from low strength wastewater.14 Moreover, such a process can effectively reduce salinity in the effluent and therefore minimize salt accumulation in reclaimed water.15,16 Voltage is a crucial parameter to control the formation of an electrical double layer (EDL), which thermodynamically affects the system performance.17 In theory, a higher voltage promotes faster ion movement toward the counter electrode as well as the formation of a thicker EDL for ion adsorption. However, the typical potential window in CDI is controlled below 1.5 V to restrict faradaic current generation such as water splitting.12,18,19 Low voltage input is an advantage because the energy consumption is very low compared with membrane-based desalination technologies.20
Since CDI has been shown to be effective in low salinity salt removal, we hypothesize that similar functions may be obtained for charged nutrient species removal and recovery as well. This approach can be especially interesting when deployed after biological treatment of municipal wastewater, because such water contains high concentrations of nutrient species yet low concentrations of other ions. In this study, we tested this hypothesis by investigating the influences of low voltage on the removal of dominant nitrogen (ammonium) and phosphorus (phosphate salts) species in CDI setups as well as their recoveries during reactor regeneration. Moreover, we characterized the feasibility of generating chlorine and other oxidants during the process to achieve disinfection in the effluent. Such a simple electrochemical process can be quite promising to solve both nutrient and salinity problems after biological treatments for both discharge and reuse, and the additional disinfection function adds further benefits to improve water quality and safety with low cost.
Materials and methods
Four identical CDI cells were fabricated by using activated carbon cloth (ACC, Chemviron Carbon, UK) as the electrodes (Fig. S1†). Each electrode consisted of 14 cm2 (2 × 7 cm2, 3 g) ACC, which was soaked in deionized water at 60 °C overnight followed by thorough rinsing to ensure the absence of dissolved compounds.21,22 A titanium mesh was used as the current collector, and plastic meshes (2 mm thick) were placed between two electrodes as spacers to prevent short-circuiting. Each reactor has an effective liquid volume of 40 mL, and the pH-neutral electrolyte was a synthetic solution containing (per liter in deionized water): 68.8 mg NH4Cl, 8.9 mg NaH2PO4·H2O, 8.6 mg Na2HPO4·12H2O, 8 mg NaCl, and 21 mg Na2SO4. The measured conductivity, NH4+-N, and PO43−-P were 260 ± 11 μs cm−1, 18 ± 1 mg L−1, and 3.2 ± 0.3 mg L−1, respectively. A potentiostat (PC4/300, Gamry Instruments, NJ) was used to provide DC voltage and monitor the current flow. The operation voltage input ranged from 1.2 to 3.0 V.For the dynamic tests of salinity reduction and nutrient removal, different voltages ranging from 1.2–3.0 V were applied for each 12 h cycle. Conductivity, ammonium nitrogen (NH4+-N), total phosphorus (PO43−-P), and chlorine content were analyzed using a spectrophotometer following the protocols provided by HACH (Sension 5, HACH, CO, TNT 830, TNT 843, and TNT 866).23 The concentrations of ions (Na+, NH4+, Cl−, SO42−, and PO43−) were also analyzed by ion chromatography (IC) for charge efficiency calculation. Because charged species can be adsorbed on the porous electrode surface via both physical and electrical processes, to differentiate electrical sorption from physical sorption, additional tests were performed by soaking electrodes into a concentrated nutrient solution to saturate physical sorption. After soaking all electrodes in the concentrated solution for 24 h, charge–discharge experiments were conducted to investigate nutrient recovery under the electrical potential. The charging voltage started at 1.2 V for 12 h to allow electrosorption, then two electrodes were short-circuited for ion discharge until the difference in electrical potential between the electrodes dropped to ∼100 mV.24 The charge–discharge cycles were repeated at least 4 times to ensure repeatability before new cycles were performed with another applied voltage. After 1.2 V, the applied voltage was increased sequentially to 1.8 V, 2.4 V, and 3.0 V, with each condition repeated for 4 cycles. During the discharge steps, DI water was used as the initial recovery solution.
The ion removal performance was evaluated by a specific rate based on the unit amount of electrode materials as calculated from eqn (1)25
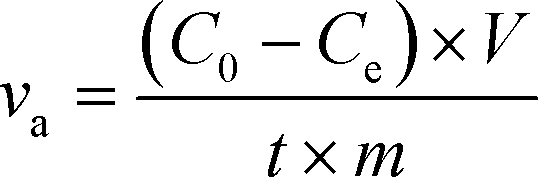 | (1) |
The charge efficiency was calculated to evaluate the electron utilization on ion removal based on eqn (2).
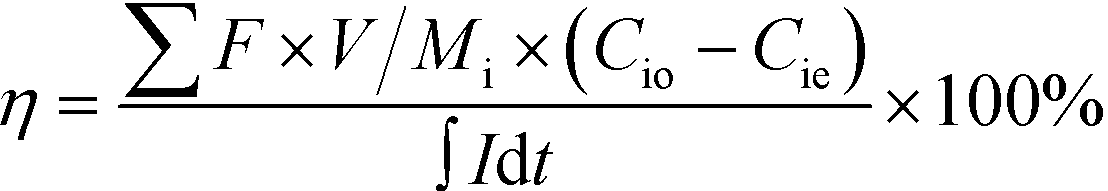 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1); and I is the current generated (A).
485 C mol−1); and I is the current generated (A).
Results and discussion
Electrochemical salt and nutrient removal
Fig. 1A shows that the solution conductivity gradually dropped when an external voltage was applied, while the conductivity remained stable under non-voltage control. When 1.2 V was applied, the conductivity decreased by 78% during a 12 hour batch cycle from 267 ± 8 to 59 ± 1 μs cm−1, and faster removal was accomplished with higher voltages. For example, to achieve 83.1% salt removal, 8 hours were needed under 1.8 V while only 4 hours were needed under 3.0 V. The conductivity reduction rate during the first 4 hours increased from 37.8 ± 3.1 to 54.2 ± 3.0 μs cm−1 h−1 when the voltage was increased from 1.2 to 3.0 V (Fig. 2). With a higher voltage input, salts transported faster and more ions were adsorbed onto the electrodes with the formation of an EDL.17 In addition, the transport rate in bulk solution between electrodes was also accelerated according to the Poisson–Nernst–Planck (PNP) model.26 In the meantime, limited water electrolysis was observed due to the low conductivity in the solution.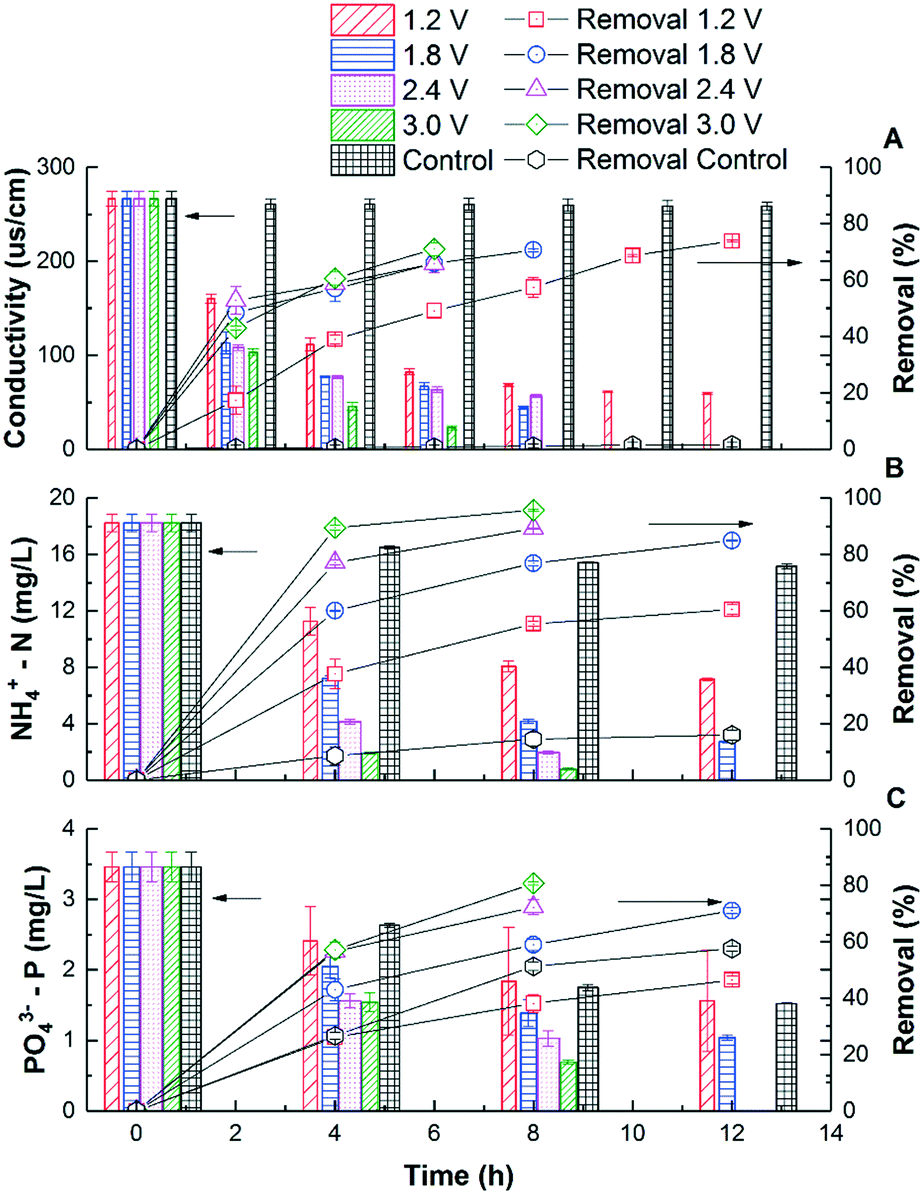 | ||
| Fig. 1 The removal kinetics and percentage of conductivity (A), NH4+-N (B), and PO43−-P (C) in CDI reactors under different voltages. | ||
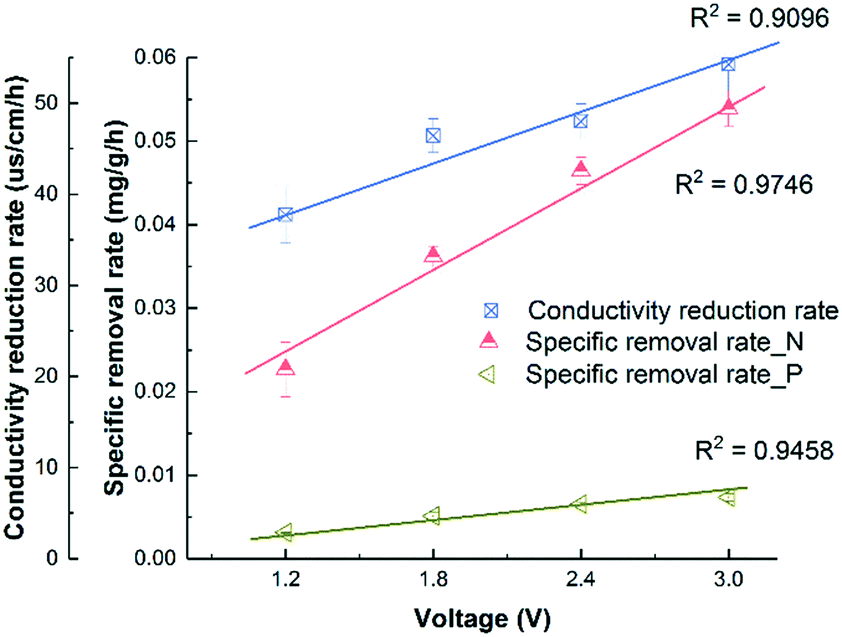 | ||
| Fig. 2 The correlation between the conductivity reduction rate or specific removal rate of NH4+-N and PO43−-P and the external applied voltage. | ||
A similar profile was observed in ammonium removal (Fig. 1B), where the removal rate was positively correlated with the applied voltage. Fig. 2 shows that the NH4+-N adsorption rate increased linearly from 0.023 ± 0.003 mg g−1 h−1 at 1.2 V to 0.054 ± 0.002 mg g−1 h−1 at 3.0 V, which led to the decrease of NH4+-N concentration from 18.2 ± 0.6 to 7.1 ± 0.1 mg L−1. Starting from the same initial NH4+-N concentration, 61% removal was observed in 12 hours under 1.2 V, while 95.7% removal was observed in 8 hours under 3.0 V (Fig. 1B). In other words, to reduce NH4+-N from 18.2 ± 0.6 to 7.1 ± 0.1 mg L−1, 83.3% time can be saved when increasing the voltage from 1.2 V to 3.0 V. The non-voltage control shows that 16.7% of the ammonium was physically adsorbed in 12 hours, much less than those reactors with applied voltages.
The profile of phosphate removal is more complicated with the applied voltage. While the concentration of phosphate dropped from 3.46 ± 0.21 to 1.56 ± 0.72 mg L−1 (46% removal) within 12 hours under 1.2 V, the non-voltage control shows a comparable removal (55%) during the same period. When the voltage was increased to 1.8 V and 3.0 V, the removal rate increased to 0.0051 ± 0.0005 and 0.0073 ± 0.0004 mg g−1 h−1, respectively. As a result, the effluent phosphate concentration dropped to 1.04 ± 0.04 mg L−1 and 0.69 ± 0.03 mg L−1 from 3.46 ± 0.21 mg L−1 within 12 hours, respectively.
The non-voltage control tests show that 24% of the phosphorus was physically adsorbed onto the electrode in 4 h, and this value increased to 56% after 12 h. This is comparable with the removal under 1.2 V, indicating that a lower voltage is not effective in driving phosphate removal via electrosorption, even though it is effective for other ions. It is known that the total electrosorption capacity consists of the capacity of the outer Helmholtz plane (OHP) and the capacity of the truly diffuse charge. The former is independent of the difference in electrical potential between the electrodes. However, when a larger potential difference is created via a higher applied voltage, the charge storage capacitance of electrosorption increases, especially the capacitance of the truly diffuse charge according to the Gouy–Chapman–Stern (GCS) model,17 which means that more ions can be adsorbed as an EDL. Compared with CDI used in seawater or brackish water desalination, which limits the applied voltage between 1.2–1.5 V to avoid electrolysis and energy loss from faradaic current generation, the 3 V used in this study for phosphate removal is high and will lead to potential water splitting and chlorine production.18 However, this may not be a big issue but can rather be a benefit as discussed later in this study. Compared with the hydrated radius of ammonium (0.331 nm), the hydrated radius of phosphate ions (H2PO4−/HPO42−) ranges from 0.375 to 0.391 nm, which requires higher driving force to move, that's why a higher voltage is required for phosphate electrosorption.27,28 Also, due to the low chloride concentration and low conductivity in municipal wastewater (∼1000 μs cm−1), energy loss caused by side reactions in saline water could be negligible. The concentration of phosphate ions (0.08 mmol L−1) was lower than that of ammonium (1.29 mmol L−1), so they were less influenced by the electric field. Overall, in order to effectively remove phosphates from water, a higher voltage is preferred due to their larger molecular size, hydrated radius, and low molar concentration.29,30
Recovery of N and P nutrients in concentrates
While traditionally the goal of CDI is to remove charged ions from water to achieve desalination, this study further investigated the benefits of nutrient recovery during CDI regeneration. The adsorbed nutrient ions can desorb from the electrodes back to the electrolyte, so nutrient concentrates can be recovered. This regeneration process is similar to capacitor discharge and can be achieved by either short circuiting the two electrodes or applying a reverse electrical potential.31 Preliminary tests showed that 90.9 ± 17.8% of the salts that was adsorbed on the electrodes was recovered in concentrates, and the recovery of NH4+-N was 81.7 ± 4.0%. However, the desorption of P was limited when physical and electrical adsorptions were not separated. Different from salts and ammonium, limited phosphate salts were released back when low voltage (1.2–1.8 V) was applied. This is understandable as physical adsorption plays an important role in phosphate removal which is not subject to electrical potential difference (Fig. 3C). To understand the distribution between physical and electrical adsorptions, all electrodes were soaked in a concentrated solution with nitrogen (3600 mg NH4+-N/L) and phosphorus (700 mg PO43−-P/L) for 24 h to saturate the electrodes with physical adsorption first. After that, all CDI cells were pre-operated at 1.2 V for over 20 24 h cycles so that the adsorption/desorption status approaches equilibrium conditions. The next step was that sixteen 24 h charging–discharging cycles were continuously conducted with the charging voltage increased from 1.2 to 3.0 V as shown in Fig. 3. The results show the conductivity and concentrations of N and P after charging (adsorbing) and discharging (desorbing) steps. The removal of conductivity, ammonium and phosphorus has no significant difference from previous test results as shown in Fig. 1. The discharge rate of conductivity increased from 16.5 ± 1.2 to 19.2 ± 2.6 μs cm−1 h−1 when the voltage increased from 1.2 V to 2.4 V, while the discharge rate of NH4+ increased from 0.42 ± 0.04 to 0.49 ± 0.02 mg g−1 h−1. During regeneration, the conductivity in the concentrate reached 231 ± 37 μs cm−1, and the NH4+ concentration reached 17.5 ± 1.8 mg L−1 under 2.4 V. The amount of P released after discharging under low voltage operation was similar to that after charging steps. At 1.2 V, 1.42 ± 1.0 mg L−1 PO43−-P was removed, while 1.60 ± 0.03 mg L−1 was collected in the recovery solution, indicating that almost all of the electrically adsorbed P along with a small fraction of the physically adsorbed P was released from saturated electrodes. When the voltage was increased to 1.8 V, a balance was observed between adsorbed and desorbed P as concentrations of both species were about 1.60 mg L−1 on average. With voltages further increased to 2.4 and 3.0 V, 2.15 ± 0.52 and 2.60 ± 0.16 mg L−1 P were observed, respectively. In the meantime, phosphate salt recovery was also increased as shown in Fig. 3C, indicating that the physically adsorbed P may be partially released back to the concentrate solution under high voltage.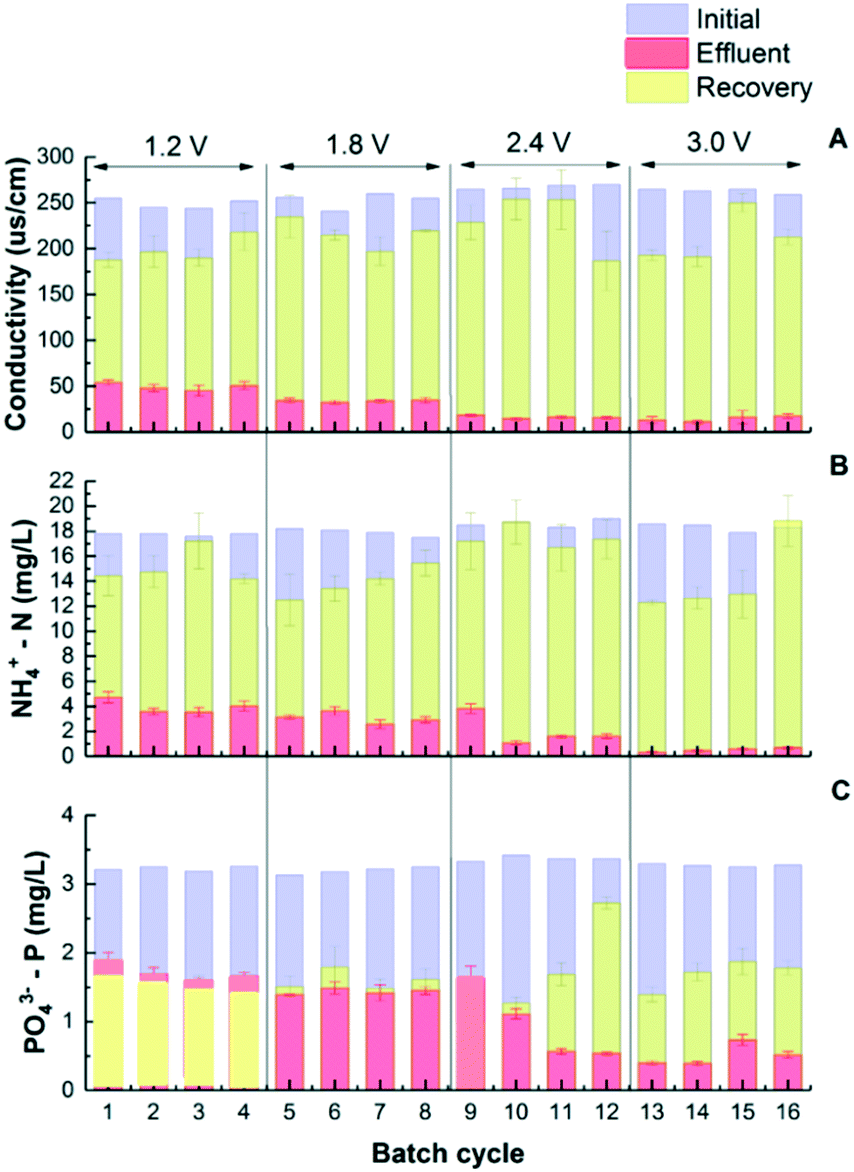 | ||
| Fig. 3 The removal and recovery of conductivity (A), NH4+-N (B), and PO43−-P (C) between electrosorption and regeneration under different voltages. | ||
Wastewater contains different types of anions and cations, so the electrosorption of these ions is a competitive process that is related to the ion selectivity, valence, size, and corresponding hydrated radius. For example, Gabelich et al. reported that the capacity of a carbon electrode for monovalent ions (Na+) was higher than that for divalent ions (Ca2+).9,12 For carbon-based materials, poor affinity for cations was reported but the difference in sorption capacity maybe more related to their hydrated radius, especially for ions with similar sizes. Previous work also found that ions with small hydrated radii exhibited higher selectivity over those with larger hydrated radii due to size affinity.29,30 Therefore, NH4+ ions were found to be easily adsorbed on negatively charged electrodes due to their small size (0.145 nm) and hydrated radius (0.331 nm).32 However, phosphate salts exhibit a larger size/radius (0.24/0.38 nm)27 which slowed down their migration and adsorption. The valence of specific ions also plays an important role in electrosorption but for large compounds, the molecular size and hydrated radius were found to be more important in migration and adsorption.30 We acknowledge that the CDI process does not have high selectivity for targeted nutrient removal and recovery at the present stage, but with future material and process development, such a function will be available in the near future.
Disinfection benefits of CDI with high voltage
Most CDI studies apply a voltage lower than 1.5 V to restrict faradaic current generation caused by water redox.12,19,33 As Porada et al. summarized, most common water redox reactions that occur in CDI/electrosorption include water splitting (producing H2 on the cathode and O2 on the anode), oxygen reduction on the cathode, and chlorination on the anode.19 As a result, charge efficiency could also decrease as expected since more electrons were taken for electrochemical reactions. Fig. 4 shows that in a 6 h operation, low voltage operation (1.2 V) achieved a high efficiency of 83.1 ± 11.7%, but this value decreases along with the increase of the applied voltage. When 3.0 V was used, the charge efficiency dropped to 18.1 ± 0.5% for adsorption. However, it was not all waste of electron flow. To take advantage of such electrochemical reactions, disinfection is achievable during regeneration by producing chlorine,34 peroxide,33 and other coating materials.35 The idea of using electrosorption as post treatment after anaerobic treatment could be employed not only to remove and recover charged nutrients and dissolved salts, but also to disinfect the effluent by chlorine or other oxidants generated when higher voltage is applied. For example, if dissolved oxygen is present in the water effluent, oxygen reduction reaction may happen to generate peroxide via a two-electron pathway:| O2 + 2H+ + 2e− → H2O2; E0 = 0.67 V vs. NHE | (3) |
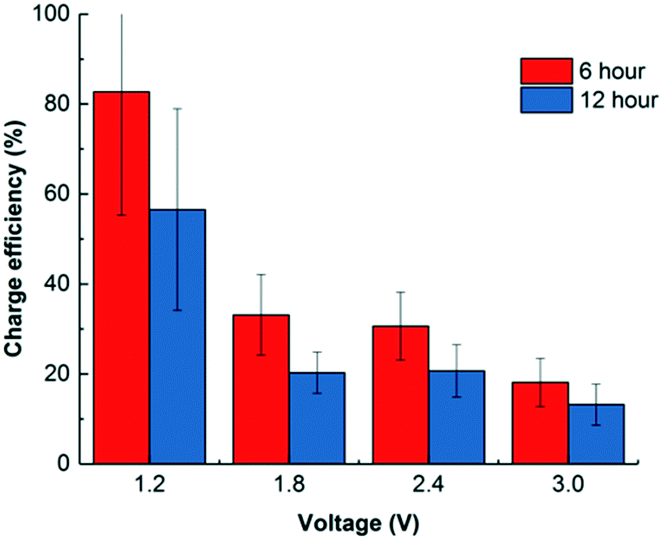 | ||
| Fig. 4 Charge efficiency of CDI operated in 6 and 12 hours under various voltage inputs. | ||
On the other hand, for an anaerobic effluent containing no oxygen but some amount of chloride, chlorine generation and chlorination will occur as follows:
| 2Cl− + Cl2 + 2e−; E0 = −1.3583 V vs. NHE(anode) | (4) |
| Cl2 + H2O ↔ HOCl + HCl | (5) |
Meanwhile, carbon oxidation may occur on the anode as shown in eqn (6) when a high voltage is applied:36
| C + H2O → CO2 + 4H+ + 4e− | (6) |
During the whole experiment, no obvious carbon electrode oxidation was observed after >100 cycles under different operating conditions. This shows the stability of the high-surface area activated carbon electrode used under low conductivity water conditions. However, the effects of faradaic reactions under high voltages need to be investigated to understand how to minimize electrode performance loss. pH also dropped under these conditions from neutral to ∼5.5 due to the reduction of buffer capacity (phosphate salt removal) and partial faradaic reactions.37,38
In this study, chlorination was observed when the input voltage was higher than 2 V. Due to the high overpotential created by the electrode and synthetic wastewater solution, low concentrations of total chlorine and free chlorine were generated at 0.10 ± 0.02 mg L−1 total Cl2 and 0.08 ± 0.01 mg L−1 free Cl2, respectively. This amount of chlorine can be sufficient to keep the electrodes clean from biofouling and satisfy the compliance requirements of the effluent discharge limit (0.25 mg L−1).39 If electrosorption is used to polish treated wastewater for salinity reduction and nutrient removal, biofouling on carbon-based porous materials can be a potential issue which may significantly decrease the system performance. On the other hand, energy consumption based on the unit volume of treated water was calculated as shown in Table S2.† The energy used ranged from 0.045 to 0.873 kW h m−3, with higher consumption associated with higher voltage and longer operation. While this study only shows the potential of such a disinfection process, it is not the focus of the study, so further investigations are needed to characterize the disinfectant production during high voltage CDI/electrosorption and its efficacy in effluent disinfection.
Outlook
This study demonstrates for the first time that CDI and electrosorption can be effective in both ammonium and phosphate removal and recovery. This approach not only expands the areas of applications for the technology but also provides a new method for advanced wastewater treatment and reuse. Compared with traditional complex and energy intensive nutrient recovery processes, this electrochemical approach can be an efficient post-treatment for biological processes, and the generation of disinfectants offers additional benefits to improve effluent safety.Although electrosorption is not the only electrochemical process that can be used for N and P removal, the unique advantage of electrosorption over other methods such as electrocoagulation, electrooxidation, and electrodialysis is its non-faradaic nature. Nitrogen, phosphorus, and other organics can be removed through electrocoagulation/oxidation assisted by electrode reactions.40 Electrodialysis also relies on electrode reactions but the ionic ammonium and phosphates in the liquid phase are separated by faradaic current generation from electrode reactions.41 Comparably speaking, electrosorption with a non-faradaic process is simple with low energy consumption, because N and P are simply stored in the pores of carbon materials and can be recovered by neutralizing the electrodes.
This study shows that salts and ammonium can be easily removed and recovered under an externally applied potential but phosphate recovery requires a higher voltage to be desorbed from the electrode due to its high affinity and physical adsorption. However, for targeted nutrient recovery, present CDI systems have limitations due to their poor selectivity for ion removal and recovery. This is a universal challenge for CDI reactors, and the development of functional electrode materials or coatings has good potential for targeted ion removal/recovery. Luckily, the conductivity of traditional municipal wastewater is low, so it is possible to separate nutrient ions by post treatment such as tuning the pH and chemistry for struvite precipitation or ammonium stripping. Moreover, implementing such electrochemical units before water reuse systems can reduce salt build-up in those water lines. Further investigations should also focus on improving P recovery especially from physical adsorption, and strategies may include developing functionalized electrode materials for P adsorption, staged operation for physical and electrical adsorption and desorption, and selective membrane electrosorption.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the US Office of Naval Research under the Award N000141612210.References
- K. S. Le Corre, E. Valsami-Jones, P. Hobbs and S. A. Parsons, Crit. Rev. Environ. Sci. Technol., 2009, 39, 433–477 CrossRef CAS.
- Z. J. Ren and A. K. Umble, Nature, 2016, 529, 25–25 CrossRef CAS PubMed.
- W. Mo and Q. Zhang, J. Environ. Manage., 2013, 127, 255–267 CrossRef CAS PubMed.
- D. Hou, L. Lu, D. Sun, Z. Ge, X. Huang, T. Y. Cath and Z. J. Ren, Water Res., 2017, 114, 181–188 CrossRef CAS PubMed.
- P. T. Kelly and Z. He, Bioresour. Technol., 2014, 153, 351–360 CrossRef CAS PubMed.
- L. Lu and Z. J. Ren, Bioresour. Technol., 2016, 215, 254–264 CrossRef CAS PubMed.
- B. K. Mayer, L. A. Baker, T. H. Boyer, P. Drechsel, M. Gifford, M. A. Hanjra, P. Parameswaran, J. Stoltzfus, P. Westerhoff and B. E. Rittmann, Environ. Sci. Technol., 2016, 50, 6606–6620 CrossRef CAS PubMed.
- M. E. Suss, S. Porada, X. Sun, P. M. Biesheuvel, J. Yoon and V. Presser, Energy Environ. Sci., 2015, 8, 2296–2319 CAS.
- Z. Huang, L. Lu, Z. Cai and Z. J. Ren, J. Hazard. Mater., 2016, 302, 323–331 CrossRef CAS PubMed.
- C. Forrestal, P. Xu and Z. Ren, Energy Environ. Sci., 2012, 5, 7161–7167 CAS.
- M. A. Anderson, A. L. Cudero and J. Palma, Electrochim. Acta, 2010, 55, 3845–3856 CrossRef CAS.
- C. J. Gabelich, T. D. Tran and I. H. M. Suffet, Environ. Sci. Technol., 2002, 36, 3010–3019 CrossRef CAS PubMed.
- C. Forrestal, Z. Stoll, P. Xu and Z. J. Ren, Environ. Sci.: Water Res. Technol., 2015, 1, 47–55 CAS.
- Z. Wang, H. Gong, Y. Zhang, P. Liang and K. Wang, Chem. Eng. J., 2017, 316, 1–6 CrossRef CAS.
- J.-B. Lee, K.-K. Park, H.-M. Eum and C.-W. Lee, Desalination, 2006, 196, 125–134 CrossRef CAS.
- J. Kang, T. Kim, H. Shin, J. Lee, J.-I. Ha and J. Yoon, Desalination, 2016, 398, 144–150 CrossRef CAS.
- A. J. B. A. L. R. Faulkner, Electrochemical Methods Fundamentals and Applications, JOHN WILEY & SONS, INC, New York, 2nd edn, 2001 Search PubMed.
- D. He, C. E. Wong, W. Tang, P. Kovalsky and T. D. Waite, Environ. Sci. Technol. Lett., 2016, 3, 222–226 CrossRef CAS.
- S. Porada, R. Zhao, A. van der Wal, V. Presser and P. M. Biesheuvel, Prog. Mater. Sci., 2013, 58, 1388–1442 CrossRef CAS.
- Y. Qu, P. G. Campbell, L. Gu, J. M. Knipe, E. Dzenitis, J. G. Santiago and M. Stadermann, Desalination, 2016, 400, 18–24 CrossRef CAS.
- C. Forrestal, A. Haeger, L. Dankovich, T. Y. Cath and Z. J. Ren, Environ. Sci.: Water Res. Technol., 2016, 2, 353–361 CAS.
- N. Li, J. An, X. Wang, H. Wang, L. Lu and Z. J. Ren, Desalination, 2017, 419, 20–28 CrossRef CAS.
- L. Lu, D. Xing and Z. J. Ren, Bioresour. Technol., 2015, 195, 115–121 CrossRef CAS PubMed.
- Z. A. Stoll, C. Forrestal, Z. J. Ren and P. Xu, J. Hazard. Mater., 2015, 283, 847–855 CrossRef CAS PubMed.
- P. Liang, X. Sun, Y. Bian, H. Zhang, X. Yang, Y. Jiang, P. Liu and X. Huang, Desalination, 2017, 420, 63–69 CrossRef CAS.
- P. M. Biesheuvel, B. van Limpt and A. van der Wal, J. Phys. Chem. C, 2009, 113, 5636–5640 CAS.
- Y. Marcus, Chem. Rev., 1988, 88, 1475–1498 CrossRef CAS.
- C. A. Pasternak, Monovalent Cations in Biological Systems, CRC Press, 1990 Search PubMed.
- C.-H. Hou and C.-Y. Huang, Desalination, 2013, 314, 124–129 CrossRef CAS.
- Y. Li, C. Zhang, Y. Jiang, T.-J. Wang and H. Wang, Desalination, 2016, 399, 171–177 CrossRef CAS.
- E. García-Quismondo, C. Santos, J. Soria, J. Palma and M. A. Anderson, Environ. Sci. Technol., 2016, 50, 6053–6060 CrossRef PubMed.
- E. R. Nightingale, J. Phys. Chem., 1959, 63, 1381–1387 CrossRef CAS.
- B. Shapira, E. Avraham and D. Aurbach, Electrochim. Acta, 2016, 220, 285–295 CrossRef CAS.
- D. Liu, X. Wang, Y. F. Xie and H. L. Tang, Sci. Total Environ., 2016, 568, 19–25 CrossRef CAS PubMed.
- Y. Wang, A. G. El-Deen, P. Li, B. H. L. Oh, Z. Guo, M. M. Khin, Y. S. Vikhe, J. Wang, R. G. Hu, R. M. Boom, K. A. Kline, D. L. Becker, H. Duan and M. B. Chan-Park, ACS Nano, 2015, 9, 10142–10157 CrossRef CAS PubMed.
- J. E. Dykstra, K. J. Keesman, P. M. Biesheuvel and A. van der Wal, Water Res., 2017, 119, 178–186 CrossRef CAS PubMed.
- J.-H. Lee, W.-S. Bae and J.-H. Choi, Desalination, 2010, 258, 159–163 CrossRef CAS.
- X. Gao, A. Omosebi, J. Landon and K. Liu, Energy Environ. Sci., 2015, 8, 897–909 CAS.
- Wastewater Technology Fact Sheet Chlorine Disinfection, https://www3.epa.gov/npdes/pubs/chlo.pdf, 1999 Search PubMed.
- C. Feng, N. Sugiura, S. Shimada and T. Maekawa, J. Hazard. Mater., 2003, 103, 65–78 CrossRef CAS PubMed.
- X. Wang, X. Zhang, Y. Wang, Y. Du, H. Feng and T. Xu, J. Membr. Sci., 2015, 490, 65–71 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ew00350a |
| This journal is © The Royal Society of Chemistry 2018 |