
Synthesis and evaluation of neuroprotective 4-O-substituted chrysotoxine derivatives as potential multifunctional agents for the treatment of Alzheimer's disease†
Li
Guan
a,
Yanfeng
Hao
a,
Lei
Chen
a,
Meng-Lin
Wei
b,
Qin
Jiang
a,
Wen-Yuan
Liu
b,
Yan-Bo
Zhang
c,
Jie
Zhang
a,
Feng
Feng
*ad and
Wei
Qu
*ad
aDepartment of Natural Medicinal Chemistry, China Pharmaceutical University, Nanjing 210009, China. E-mail: fengfeng@cpu.edu.cn; weiqcpu@126.com
bKey Laboratory of Drug Quality Control and Pharmacovigilance (China Pharmaceutical University), Ministry of Education, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing 210009, China
cSchool of Chinese Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, 10 Sassoon Road, Pokfulam, Hong Kong, China
dKey Laboratory of Biomedical Functional Materials, China Pharmaceutical University, Nanjing 211198, China
First published on 22nd February 2016
Abstract
A series of 4-O-substituted chrysotoxine (CTX) derivatives were designed, synthesized and evaluated as multifunctional agents for the treatment of Alzheimer's disease (AD). In vitro assays indicated that four ring substituted compounds (2a, 2b, 3i and 3j) exhibited significant neuroprotective effects against Aβ25–35-induced toxicity in PC12 cells. The four compounds also inhibited self- and Cu2+-induced Aβ1–42 aggregation and acted as biometal chelators. In particular, compound 2a was a potential lead compound for AD treatment (cell viability up to 100.78% at 50 µM in Aβ25–35-treated PC12 cells, 51.88% and 58.03% inhibition at 25 µM for self- and Cu2+-induced Aβ1–42 aggregation, respectively). A metal chelating experiment showed that compound 2a had a moderate interaction with Cu2+ and Al3+. Moreover, western blot analysis showed that compound 2a attenuated Aβ-induced tau protein hyperphosphorylation at Ser199/202 and Ser396 sites. Furthermore, compound 2a could efficiently cross the blood-brain barrier (BBB) by a parallel artificial membrane permeability assay (PAMPA). In summary, these results suggested that compound 2a was a promising multifunctional compound for AD therapy.
1. Introduction
Alzheimer's disease (AD), characterized by a progressive loss of memory and cognitive impairments, is one of the most common neurodegenerative diseases mainly occurring in the elderly.1 The World Alzheimer Report 2015 showed that 46.8 million people have been suffering from AD and this number is expected to exceed 131.5 million in 2050.2 Current therapeutic drugs for AD include acetylcholinesterase inhibitors (tacrine, donepezil, rivastigmine and galantamine)3 and N-methyl-D-aspartate (NMDA) receptor antagonists (memantine).4 Although these single-target drugs have a modest and transient improvement in memory and cognitive function, they cannot prevent progressive neurodegeneration.3 The etiology of AD still remains elusive, but several factors, such as β-amyloid (Aβ) deposits,5 τ-protein hyperphosphorylation,6 metal dyshomeostasis,7 oxidative stress8 and low levels of acetylcholine,9 have been greatly implicated in AD pathogenesis.Among the multiple factors that induce AD, Aβ plays a primary role.10 The Aβ hypothesis states that the production and accumulation of Aβ peptides in the brain are central events in the AD pathogenesis.11,12 Aβ is a peptide chain cleaved from amyloid precursor protein (APP) ranging in length from 39 to 43 amino acids.13 Aβ1–40 and Aβ1–42 are the main isoforms of Aβ peptides.14 A sustained imbalance between the production and clearance of Aβ40–42 fragments leads to the accumulation of Aβ peptide monomers, oligomers, and finally promotes the large aggregated Aβ plaques, which are thought to initiate the pathogenic cascade, ultimately leading to neuronal loss and dementia.15–17 Another hallmark of AD pathology is the intracellular accumulation of hyperphosphorylated tau protein, which leads to neurofibrillary tangles in the brain.18 Therefore, an inhibition of Aβ aggregation or tau hyperphosphorylation has been recognized as an effective therapeutic strategy for AD.
In addition, recent studies have indicated that excessive biometals (such as Cu2+, Fe2+, Zn2+ and Al3+) exist in the brains of AD patients.19 The abnormal accumulation of biometal ions is closely associated with the formation of Aβ plaques and neurofibrillary tangles, which have been found to induce neurotoxicity.20,21 The modulation of brain biometals represents an additional rational approach for the treatment of AD.
A major impediment to the development of effective compounds for AD therapy is that essentially 100% of large-molecule drugs and >98% of small-molecule drugs fail to cross the blood-brain-barrier (BBB).22 Increasing the lipid solubility of target compounds is regarded as a promising way to improve their BBB permeability.23
Chrysotoxine (CTX), a natural bibenzyl (4-hydroxy-3,5,3′,4′-tetramethoxybibenzyl), has been isolated from plants in genus Dendrobium, the source of a valuable and scarce traditional medicine named “Shihu” in China.24–26 Recent studies have revealed that CTX can inhibit SH-SY5Y cells apoptosis induced by neurotoxins 6-hydroxydopamine and 1-methyl-4-phenyl pyridinium (MPP+) in Parkinson's disease model, which suggested its potential protective effect against neurodegeneration.27,28 Similar to resveratrol, CTX contains two aromatic end groups and a linker region in the middle, which are critical for the efficient bond at the Aβ peptide guided π-stacking.29 However, the scarce source of CTX restrict its further development.
In this paper, we synthesized CTX and its derivatives modified by several series of hydrophobic moieties as potential neuroprotective agents for AD. Then we chose four compounds with significant neuroprotective effects to explore their potential multifunctional properties for AD therapy by the pharmacological evaluations, including self- and Cu2+-induced Aβ aggregation, tau phosphorylation and metal chelation. Meanwhile, theoretical prediction and experimental assay were performed to explore their BBB permeability.
2. Result and discussion
2.1. Chemistry
The natural compound CTX was totally synthesized with Wittig reaction as a key step, which was widely used for the synthesis of bibenzyl skeleton.30 Syringaldehyde (4-hydroxy-3,5-dimethoxylbenzaldehyde, 2) was used as the starting material. Based on the acetylation protection of hydroxyl group, 4-hydroxymethyl-2,6-dimethoxyphenyl acetate (4) was obtained by aldehyde reduction with sodium borohydride. With further hydroxyl chlorination of compound 4, we got the intermediate 4-chloromethyl-2,6-dimethoxyphenyl acetate (5), which was reacted with triphenylphosphine to prepare the phosphonium salt (6), the key material for the Wittig reaction. The phosphonium salt was dissolved completely with 3,4-dimethoxybenzaldehyde in THF and deprotonated with NaH to yield a mixture of Z- and E-stilbenes, which were reduced by treatment with catalytic amounts of 10% Pd–C over H2 (4 atm) to afford bibenzyl skeleton. After deacetylation with sodium methylate, CTX was finally obtained through a total 7-step procedure, and the whole yield of our 7-step reaction was up to 54.1%, which was summarized in Scheme 1.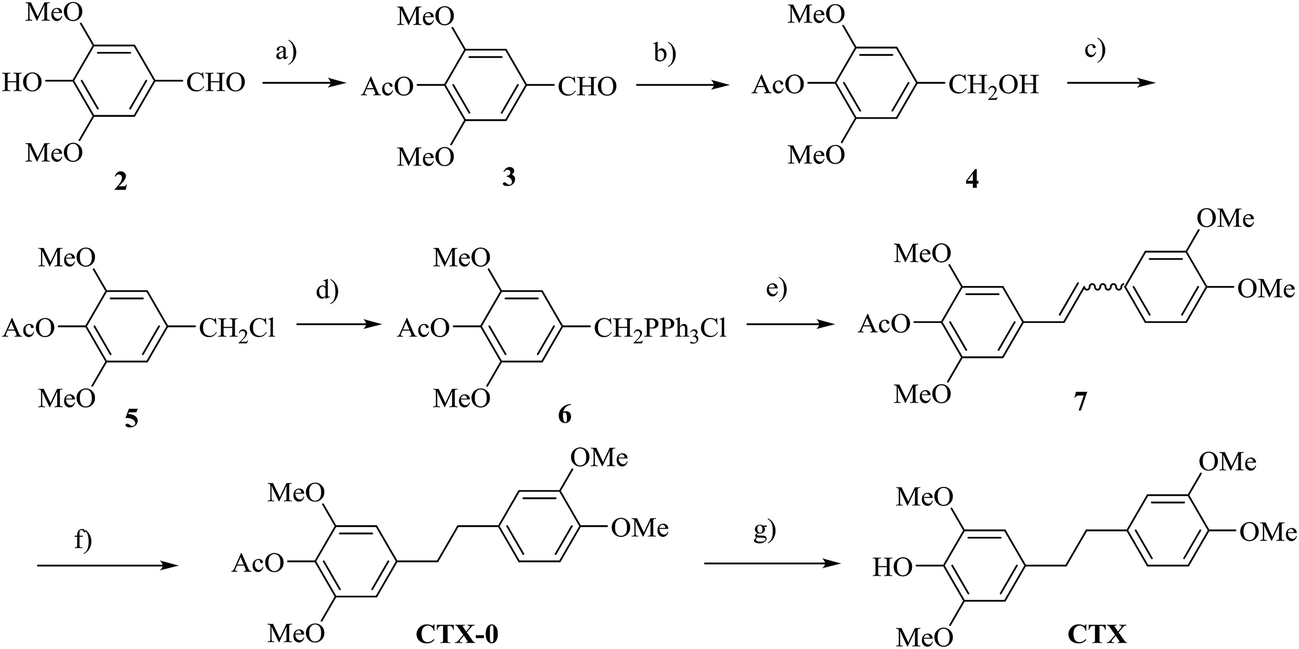 | ||
| Scheme 1 Synthesis steps of lead compound chrysotoxine (CTX). (a) Acetic anhydride, DCM, DMAP, rt, 30 min; (b) NaBH4, EtOH, 0 °C, rt, 30 min; (c) SOCl2, Et3N, DCM, 0 °C, rt, 3 h; (d) PPh3, toluene, reflux, 5 h; (e) 3,4-dimethoxybenzaldehyde, NaH, THF, Ar, 0 °C, rt, overnight; (f) H2, 10% Pd/C, EtOAc, 4 atm; (g) MeONa/MeOH, rt, 30 min. | ||
The chrysotoxine-4-O-substituted esterified derivatives 1a–1f were catalyzed by EDC·HCl in just one step, while compounds 2a–2e were obtained through the esterification with the acid chloride intermediate. These procedures were shown in Scheme 2. The 4-O-substituted chrysotoxine ether derivatives 3a–3j were synthesized by the combination of halon compounds and CTX in DMF, which was described in Scheme 3.
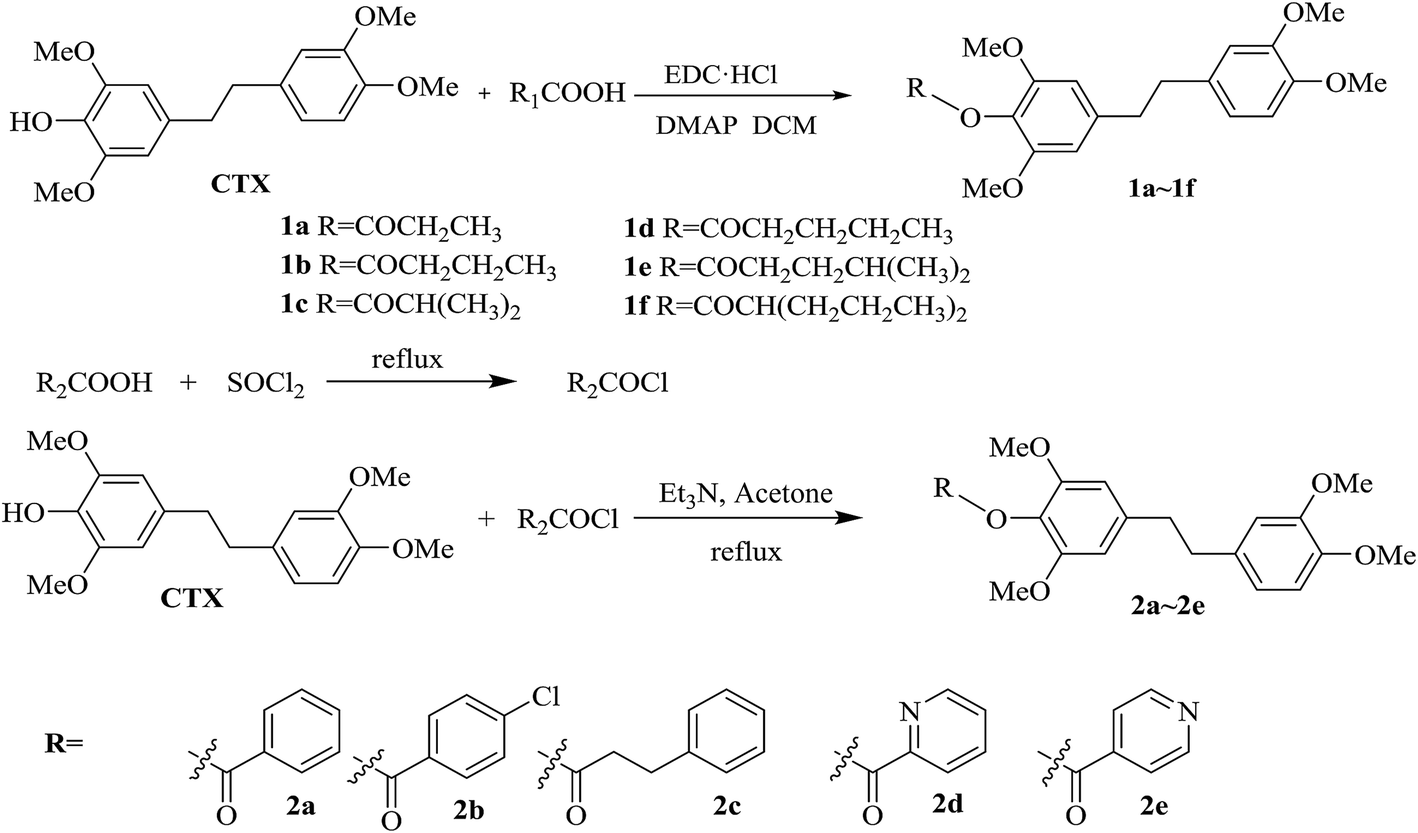 | ||
| Scheme 2 Synthetic procedure of 4-O-ester substituted CTX derivatives. | ||
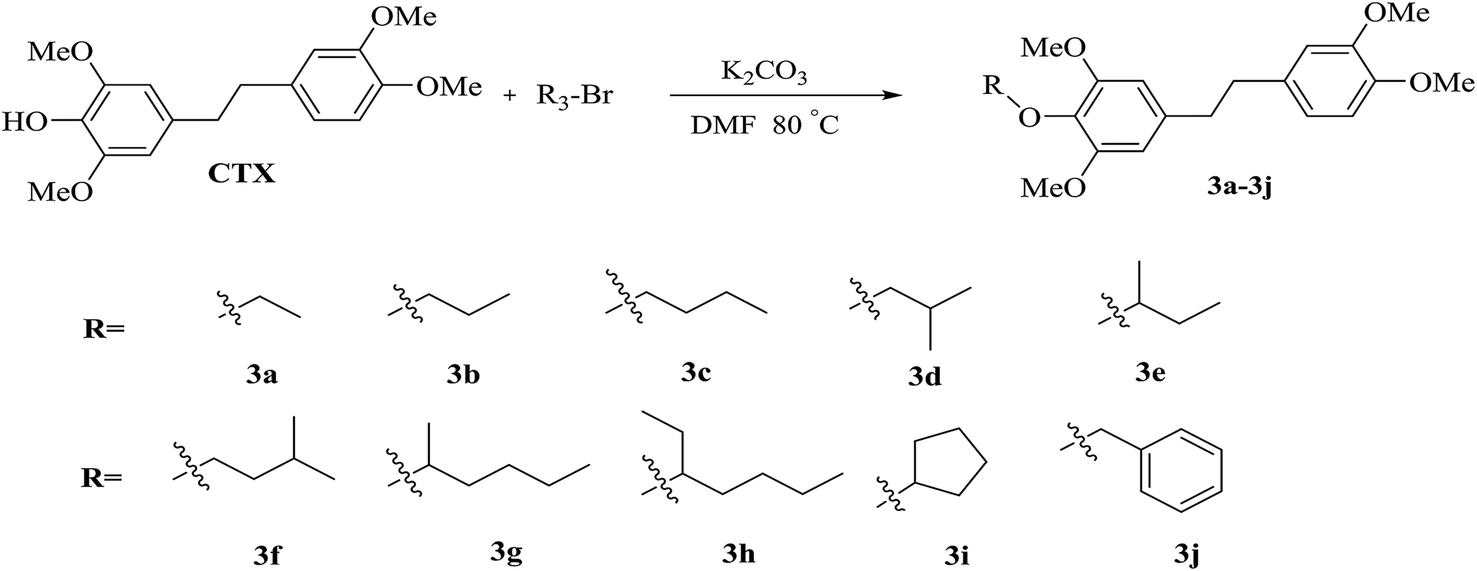 | ||
| Scheme 3 Synthetic procedure of 4-O-ether substituted CTX derivatives. | ||
All compounds were purified by recrystallization or chromatography technology. The analytical and spectroscopic data confirmed their structures, which were shown as details in the Experimental section.
2.2. Pharmacology
| Com. | Cell viabilityb (%) | Com. | Cell viabilityb (%) | ||||
|---|---|---|---|---|---|---|---|
| 50 µM | 25 µM | 10 µM | 50 µM | 25 µM | 10 µM | ||
| a 10 µM Aβ25–35 was added to all the test groups and model group to develop the nerve damage model for 24 h. Compounds were added 0.5 h prior to Aβ25–35 addition. Cell viability was assessed by measuring the MTT reduction. b Data were the mean ± SD expressed as percent of the untreated Aβ25–35 control group. Results are the mean of 3 independent experiments in triplicate. ###p < 0.001 vs. control; ***p < 0.001, **p < 0.01, *p < 0.05 vs. model (ANOVA followed by Newman–Keuls test). | |||||||
| CTX | 58.89 ± 2.47 | 55.02 ± 2.91 | 52.73 ± 4.18 | 2e | 46.56 ± 1.42 | 50.71 ± 2.85 | 53.53 ± 3.51 |
| CTX-0 | 48.87 ± 3.51 | 50.01 ± 4.01 | 48.47 ± 3.93 | 3a | 46.15 ± 3.14 | 50.92 ± 3.42 | 53.84 ± 2.57 |
| 1a | 44.40 ± 6.21 | 44.21 ± 4.30 | 45.31 ± 5.11 | 3b | 48.81 ± 1.55 | 49.11 ± 1.43 | 49.21 ± 2.62 |
| 1b | 49.37 ± 4.02 | 50.12 ± 2.21 | 54.89 ± 3.56 | 3c | 53.44 ± 1.80 | 52.03 ± 3.44 | 51.79 ± 3.09 |
| 1c | 45.91 ± 1.91 | 47.33 ± 3.54 | 49.34 ± 2.81 | 3d | 49.89 ± 2.47 | 50.12 ± 1.96 | 54.53 ± 1.88 |
| 1d | 48.63 ± 3.14 | 41.90 ± 2.91 | 49.73 ± 4.18 | 3e | 48.75 ± 2.02 | 49.73 ± 4.72 | 49.02 ± 3.61 |
| 1e | 49.57 ± 4.15 | 50.40 ± 2.04 | 49.83 ± 1.01 | 3f | 49.48 ± 3.72 | 50.16 ± 4.70 | 50.21 ± 1.51 |
| 1f | 60.02 ± 1.89(*) | 55.21 ± 2.71 | 50.11 ± 1.85 | 3g | 52.18 ± 1.99 | 50.25 ± 3.08 | 50.67 ± 1.40 |
| 2a | 100.78 ± 2.51(***) | 80.12 ± 2.56(***) | 58.18 ± 2.09 | 3h | 58.88 ± 3.51 | 54.78 ± 2.51 | 51.03 ± 2.77 |
| 2b | 95.62 ± 1.22(***) | 69.24 ± 1.52(**) | 56.41 ± 3.04 | 3i | 72.64 ± 5.12(**) | 61.02 ± 1.05(*) | 54.77 ± 2.79 |
| 2c | 49.84 ± 1.09 | 50.22 ± 2.47 | 54.00 ± 1.85 | 3j | 71.00 ± 2.45(***) | 60.89 ± 1.67(*) | 58.22 ± 2.84 |
| 2d | 54.60 ± 3.02 | 50.10 ± 6.07 | 52.97 ± 3.76 | Model | 53.82 ± 2.72(###) | Control | 100.05 ± 3.12 |
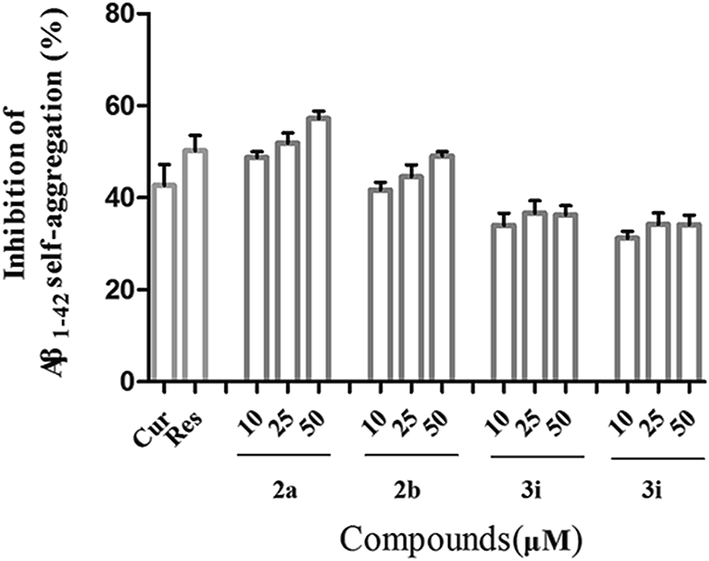 | ||
| Fig. 1 Inhibition of Aβ1–42 self-induced aggregation by compounds 2a, 2b, 3i and 3j. Curcumin (Cur) and resveratrol (Res) were reference compounds. The thioflavin T fluorescence method was used and the measurements were carried out in the presence of test compounds in different concentrations. The mean ± SD values from three independent experiments were shown. | ||
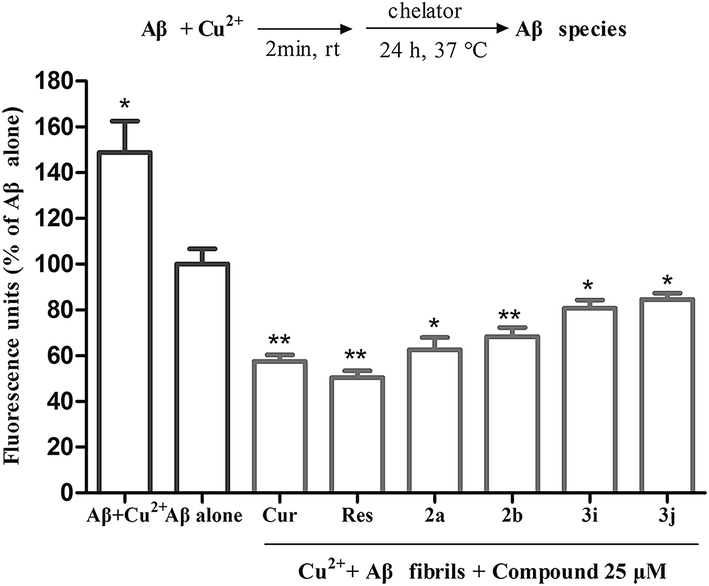 | ||
| Fig. 2 Inhibition of Cu2+-induced Aβ1–42 aggregation by compounds 2a, 2b, 3i and 3j. The final concentration of Aβ1–42, Cu2+ and test compounds were 25 µM. Values are reported as the mean ± SD of three independent experiments.*p < 0.05, **p < 0.01 vs. Aβ alone group. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, which was taken as an indication of the stoichiometry of the complex.
:1 ratio, which was taken as an indication of the stoichiometry of the complex.
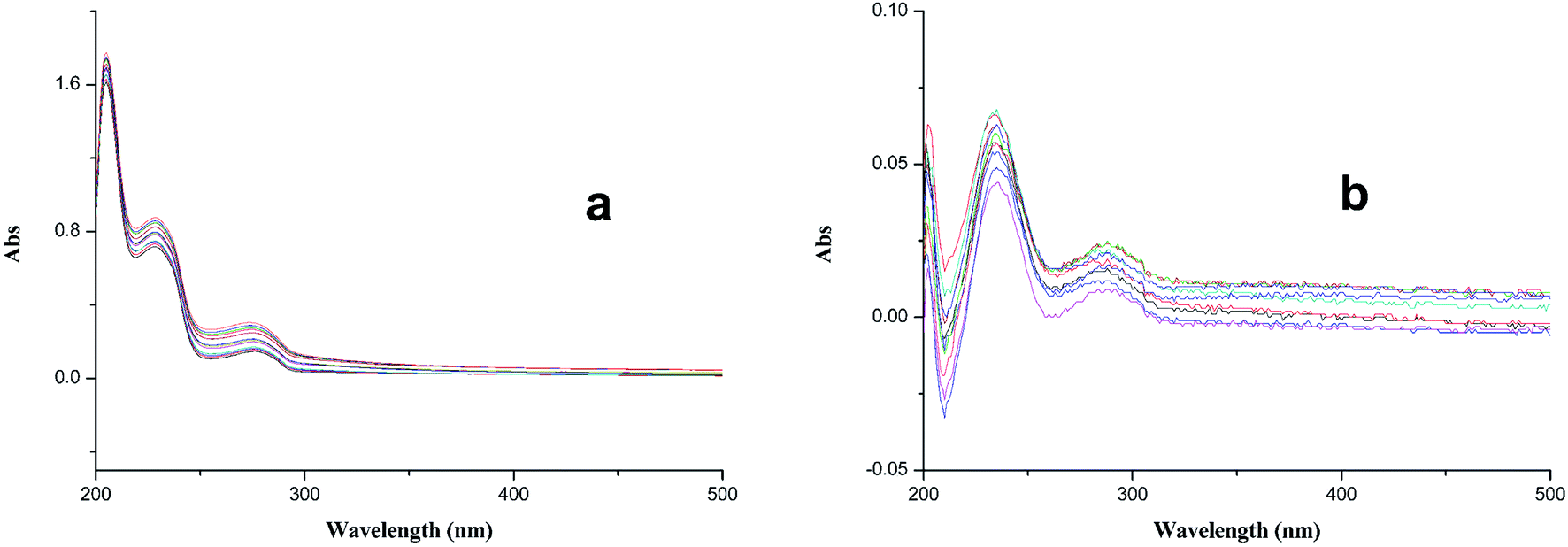 | ||
| Fig. 3 (a) UV-vis (200–500 nm) absorption spectra of compound 2a (25 µM in methanol) alone or in the presence of Cu2+ (2–50 µM in methanol); (b) the differential spectra due to 2a–Cu2+ complex formation obtained by numerical subtraction from the above spectra of those of Cu2+ and 2a at the corresponding concentrations. | ||
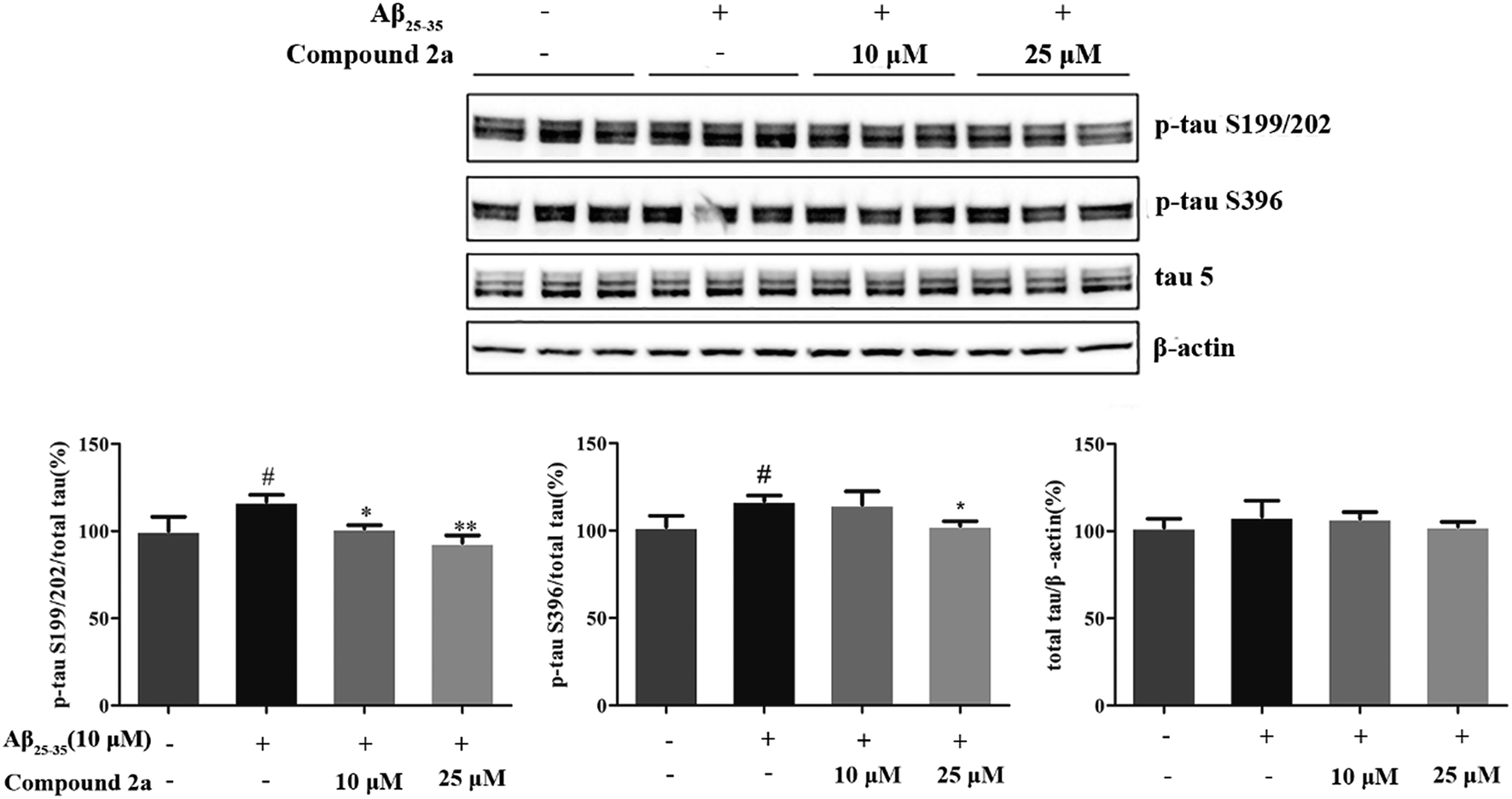 | ||
| Fig. 4 Compound 2a attenuated Aβ25–35-induced tau protein hyperphosphorylation in PC12 cells. PC12 cells were pretreated with compound 2a (10 and 25 µM) for 30 min, and then treated with Aβ25–35 (10 µM) for 6 h. Western blot analysis of phosphorylated tau protein (at Ser199/202 and Ser396 sites) and total tau was performed. Values are mean ± S.D. from triplicate independent experiments. #p < 0.05 vs. control; *p < 0.05, **p < 0.01 vs. Aβ25–35 treatment group. | ||
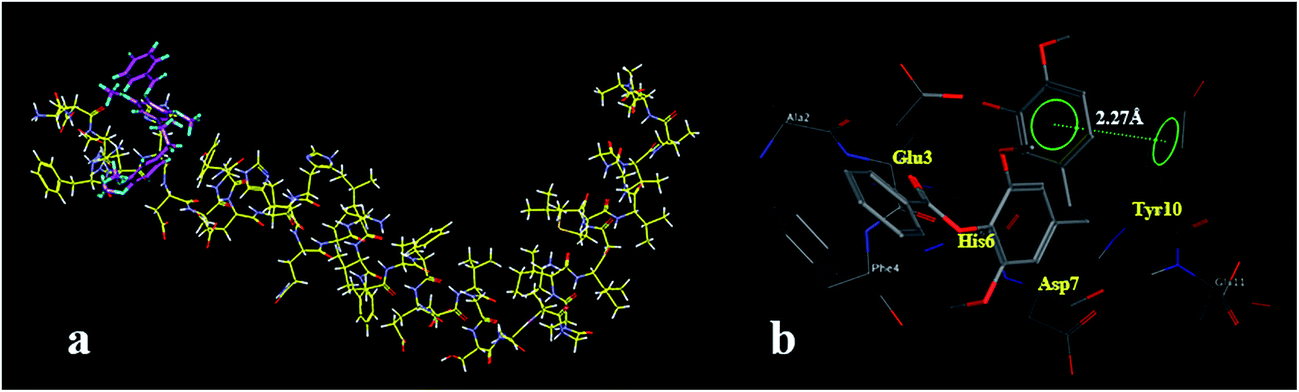 | ||
| Fig. 5 Docking results of compound 2a and Aβ1–42 (PDB code: 1IYT). (a) Cartoon representations of compound 2a (colored purple) interacting with Aβ1–42 (colored yellow); (b) association of compound 2a (colored grey) and Aβ1–42 obtained from docking calculations. The interactions between the ligand and residue Tyr10 are indicated by the green line. | ||
logP) less than 5, the number of hydrogen bond donor atoms (HBD) less than 5, the number of hydrogen bond acceptor atoms (HBA) less than 10 and the small polar surface area less than 90 Å2. Filter for prediction of BBB penetration involves calculation of logBB by means of the equation shown in the footnote of Table 2 and compounds with logBB higher than −1.0 could cross BBB.42 Defined by the terms of Lipinski's rules and calculated logBB for potential applications in brains, compounds 2a, 2b, 3i and 3j fulfill drug-like criteria in general and could possibly penetrate the BBB better than CTX.
| Compounds | MWa |
ClogPa |
HBAa | HBDa | PSAa | logBBa |
|---|---|---|---|---|---|---|
|
a MW, molecular weight; ClogP, calculated logarithm of the octanol–water partition coefficient; HBA, hydrogen-bond acceptor atoms; HBD, hydrogen-bond donor atoms; PSA, polar surface area; logBB = −0.0148 × PSA + 0.152 × ClogP + 0.139.42
|
||||||
| CTX | 318 | 3.211 | 6 | 1 | 57.15 | −0.218 |
| 2a | 422 | 5.105 | 6 | 0 | 63.22 | −0.021 |
| 2b | 456 | 5.818 | 6 | 0 | 63.22 | 0.079 |
| 3i | 386 | 5.018 | 5 | 0 | 46.15 | 0.210 |
| 3j | 408 | 5.314 | 5 | 0 | 46.15 | 0.264 |
| Rules | ≤500 | ≤5.0 | ≤10 | ≤5 | ≤90 | ≥−1.0 |
| Commercial drugs | Bibliographya | Peb (10−6 cm s−1) | Compounds | Peb (10−6 cm s−1) |
|---|---|---|---|---|
|
a Data obtained from ref. 43.
b Experimental data are the mean ± SD of 3 independent experiments, with PBS:EtOH (70:30) as solvent.
|
||||
| Chlorpromazine | 6.5 | 7.1 ± 0.9 | CTX | 4.1 ± 0.5(CNS±) |
| Clonidine | 5.3 | 6.5 ± 1.5 | 2a | 6.9 ± 0.7(CNS+) |
| Desipramine | 12.0 | 14.8 ± 3.3 | 2b | 6.2 ± 0.5(CNS+) |
| Lomefloxacin | 1.1 | 0.9 ± 0.4 | 3i | 7.6 ± 1.1(CNS+) |
| Progesterone | 9.3 | 10.5 ± 1.8 | 3j | 8.2 ± 0.9(CNS+) |
| Dopamine | 0.2 | 0.3 ± 0.1 | — | — |
| Ofloxacin | 0.8 | 1.5 ± 0.5 | — | — |
| Piroxacin | 2.5 | 3.9 ± 1.1 | — | — |
| Testosterone | 17.0 | 18.1 ± 1.9 | — | — |
| Verapamil | 16.0 | 16.5 ± 1.7 | — | — |
3. Conclusion
In summary, a series of 4-O-substituted chrysotoxine (CTX) derivatives were designed, synthesized and evaluated as multifunctional agents for the treatment of Alzheimer's disease (AD). Among the four ring substituted compounds (2a, 2b, 3i and 3j) with good neuroprotective activities, compound 2a exhibited significant inhibition of self- and Cu2+-induced Aβ1–42 aggregation and metal-chelating ability. Moreover, compound 2a was also capable of crossing the BBB in a parallel artificial membrane permeability assay. Furthermore, the result of western blot analysis showed that compound 2a was able to reduce Aβ-induced tau protein hyperphosphorylation. Taken together, these results suggest that compound 2a is a promising lead compound for multifunctional AD treatment. Further investigations of AD therapeutic candidates based on these results are in progress.4. Experimental
4.1. Chemistry
All chemicals (reagent grade) used were purchased from Aladdin Shanghai Biochemical Technology Co., Ltd. or Sinopharm Chemical Reagent Co., Ltd. (China). Reaction progress was monitored using analytical thin layer chromatography (TLC) on precoated silica gel GF254 (Qingdao Haiyang Chemical Plant, Qingdao, China) plates and the spots were detected under UV light (λ = 254 nm). IR spectra were recorded on a Nicolet iS10 spectrophotometer using KBr disks. Melting point was measured on an XT-4 micro melting point apparatus and uncorrected.1H and 13C NMR spectra were measured on a Bruker 300 MHz spectrometer at 298 T and referenced to TMS. Chemical shifts were reported in ppm using the residual solvent line as internal standard. Splitting patterns were designed as s (singlet), d (doublet), t (triplet), m (multiplet).
Mass spectra were obtained on an MS Agilent 1100 Series LC/MSD Trap mass spectrometer (ESI-MS). A Shimadzu LC-2010 series HPLC system (Kyoto, Japan) was used for the purity detection. Samples were separated on a YMC-Pack ODS-A column (50 mm × 4.6 mm, 3 µm, YMC Co. Ltd., Kyoto, Japan) with the column temperature set at 30 °C. The mobile phase consisted of acetonitrile–water (90:10, v/v), delivered at a flow rate of 1 mL min−1. The injection volume was 10 µL. Column chromatography was performed on silica gel (200–300 mesh; Qingdao Marine Chemical Inc.).
4.1.1.1. 4-Formyl-2,6-dimethoxyphenyl acetate (3). Syringaldazine (8.33 g, 0.046 mol) and catalyst DMAP were dissolved in DCM (10 mL), then acetic anhydride (4.75 mL, 0.051 mol) was slowly added. After the mixture was stirred at room temperature for 1 h, the solution was washed with diluted water (10 mL × 3). Organic layer was dried over anhydrous Na2SO4 and concentrated to get white crystals 10.19 g, 99.8% yield; mp 118–119 °C; IR vmax (KBr, cm−1): 3099, 2940, 1759, 1691, 1603, 1499; 1H-NMR (300 MHz, CDCl3) δ 9.93 (1H, s, –CHO), 7.17 (2H, s, H-3, H-5), 3.92 (6H, s, 2 × –OCH3), 2.38 (3H, s, –COCH3); 13C-NMR (75 MHz, CDCl3) δ 191.1, 168.1, 152.8, 134.3, 115.0, 106.0, 56.3, 20.4; ESI-MS m/z: 225.1 [M + H]+.
4.1.1.2. 4-(Hydroxymethyl)-2,6-dimethoxyphenyl acetate (4). To a solution of 3 (8.6 g, 0.038 mol) in EtOH (10 mL), NaBH4 (0.53 g, 0.014 mol) was slowly added under ice bath. After stirred at room temperature for 30 min, 5% HCl was added to adjust pH to 7. Concentrated under vacuum, residue was extracted with DCM (10 mL × 3), then organic layer was dried over anhydrous Na2SO4 and concentrated to get white solid 8.34 g, 96.5% yield; mp 98–99 °C; IR vmax (KBr, cm−1): 3530, 3126, 2944, 1748, 1605, 1508; 1H-NMR (300 MHz, CDCl3) δ 6.66 (1H, s, H-3, H-5), 4.69 (2H, s, Ar-CH2–), 3.84 (6H, s, 2 × –OCH3), 2.36 (3H, s, –COCH3); 13C-NMR (75 MHz, CDCl3) δ 168.8, 152.1, 139.6, 115.3, 103.1, 65.2, 56.1, 20.5; ESI-MS m/z: 227.1 [M + H]+.
4.1.1.3. 4-(Chloromethyl)-2,6-dimethoxyphenyl acetate (5). To a solution of 4 (8.3 g, 0.037 mol) in DCM (10 mL), Et3N (6.2 mL, 0.043 mol), SOCl2 (3.0 mL, 0.043 mol) was slowly dropped under ice salt bath. After stirred at room temperature for 3 h, the solution was washed with diluted water (10 mL × 3). Organic layer was dried over anhydrous Na2SO4 and concentrated to get light yellow solid 7.96 g, 88.7% yield; mp 75–76 °C; IR vmax (KBr, cm−1): 3162, 2971, 1759, 1606, 1508; 1H-NMR (300 MHz, CDCl3) δ 6.85 (1H, s, H-3, H-5), 4.73 (2H, s, Ar-CH2–), 3.76 (6H, s, 2 × –OCH3), 2.24 (3H, s, –COCH3); 13C-NMR (75 MHz, CDCl3) δ 168.3, 152.0, 139.4, 115.0, 103.2, 60.7, 56.1, 20.4; ESI-MS m/z: 227.1 [M + H]+.
4.1.1.4. (4-Acetoxy-3,5-dimethoxybenzyl)triphenylphosphonium chloride (6). 5 (7.8 g, 0.032 mol) and PhP3 (8.4 g, 0.032 mol) were dissolved in toluene (5 mL) under reflux for 6 h. After cooled to temperature, the mixture was filtrated and washed with toluene under vacuum. Dried under infrared light and 14.63 g white powder was obtained, 90.2% yield; 1H-NMR (300 MHz, DMSO-d6) δ 7.92 (3H, m, ArH), 7.76 (6H, dt, J = 3.6 Hz, 8.1 Hz, ArH), 7.69 (6H, m, ArH), 6.30 (2H, d, J = 2.4 Hz, H-3, H-5), 5.09 (2H, J = 15 Hz, Ar-CH2–), 3.37 (6H, s, 2 × –OCH3), 2.21 (3H, s, × –OCH3); 13C-NMR (75 MHz, DMSO-d6) δ 168.4, 152.0, 151.9, 135.5, 134.8, 134.6, 130.6, 130.4, 126.9, 126.7, 118.8, 117.6, 108.5, 108.4, 56.2, 29.4, 28.8, 20.6; ESI-MS m/z: 541.2 [M + Cl]−.
4.1.1.5. 4-[(Z/E)-3,4-Dimethoxystyryl]-2,6-dimethoxyphenyl acetate (7). 6 (3.0 g, 5.920 mmol) and 3,4-dimethoxybenzaldehyde were dissolved in anhydrous THF (3 mL) under Ar protection. NaH (0.15 g, 6.25 mmol) was added slowly under ice salt bath. After overnight reaction in room temperature, the solution was concentrated. Then the mixture of trans- and cis-stilbenes was recrystallized in methanol with 70.4% yield. trans- and cis-Stilbenes could be separated by fast chromatography, and the ratio of trans- and cis- was 3
:1. 4-[(Z)-3,4-Dimethoxystyryl]-2,6-dimethoxyphenyl acetate: 1H-NMR (300 MHz, CDCl3) δ 7.06 (3H, m, ArH), 6.97 (1H, d, J = 8.9 Hz, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 6.89 (1H, d, J = 8.9 Hz, –CHCH–), 6.76 (2H, s, H-3, 5), 3.97 (3H, s, –OCH3), 3.93 (3H, s, –OCH3), 3.90 (6H, s, 2 × –OCH3), 2.37 (3H, s, –COCH3); 13C-NMR (75 MHz, CDCl3) δ 168.7, 151.9, 148.3, 135.8, 130.4, 129.5, 128.5, 122.1, 111.6, 110.7, 106.0, 105.4, 56.0, 55.8, 55.7, 20.5; ESI-MS m/z: 359.1 [M + H]+; 4-[(E)-3,4-dimethoxystyryl]-2,6-dimethoxyphenyl acetate: 1H-NMR (300 MHz, CDCl3) δ 6.87 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.84 (1H, d, J = 1.8 Hz, H-2′), 6.78 (1H, d, J = 8.1 Hz, H-5′), 6.57 (2H, s, H-3, 5), 6.54 (1H, d, J = 12.0 Hz, –CHCH–), 6.49 (1H, d, J = 12.0 Hz, –CHCH–), 3.88 (3H, s, –OCH3), 3.69 (6H, s, 2 × –OCH3), 3.68 (3H, s, –OCH3), 2.34 (3H, s, –COCH3); 13C-NMR (75 MHz, CDCl3) δ 168.9, 152.2, 149.1, 149.0, 136.0, 130.1, 128.8, 126.6, 122.0, 111.2, 108.7, 102.9, 56.1, 56.0, 55.9, 20.5; ESI-MS m/z: 359.1 [M + H]+.
:1) to get a light yellow solid 0.88 g, 99.8% yield. Mp 98–99 °C; IR vmax (KBr, cm−1): 3462, 3133, 2933, 1609, 1517; 1H-NMR (300 MHz, CDCl3) δ 6.81 (1H, d, J = 8.1 Hz, H-5′), 6.72 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.68 (1H, d, J = 1.8 Hz, H-2′), 6.38 (2H, s, H-3, 5), 5.39 (1H, s, –OH), 3.88 (6H, s, 2 × –OCH3), 3.87 (6H, s, 2 × –OCH3), 2.85 (4H, s, –CH2CH2–); 13C-NMR (75 MHz, CDCl3) δ 146.9, 146.3, 140.4, 133.8, 132.3, 127.8, 119.9, 111.4, 110.7, 104.6, 55.7, 55.4, 55.3, 37.9, 37.3; ESI-MS m/z: 319.1 [M + H]+, 336.1 [M + NH4]+; HPLC purity: 100%, tR = 1.71 min.
:1–8:1) as eluent, by which we obtained the corresponding 4-O-modified CTX derivatives.
CH–), 6.89 (1H, d, J = 8.9 Hz, –CHCH–), 6.76 (2H, s, H-3, 5), 3.97 (3H, s, –OCH3), 3.93 (3H, s, –OCH3), 3.90 (6H, s, 2 × –OCH3), 2.37 (3H, s, –COCH3); 13C-NMR (75 MHz, CDCl3) δ 168.7, 151.9, 148.3, 135.8, 130.4, 129.5, 128.5, 122.1, 111.6, 110.7, 106.0, 105.4, 56.0, 55.8, 55.7, 20.5; ESI-MS m/z: 359.1 [M + H]+; 4-[(E)-3,4-dimethoxystyryl]-2,6-dimethoxyphenyl acetate: 1H-NMR (300 MHz, CDCl3) δ 6.87 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.84 (1H, d, J = 1.8 Hz, H-2′), 6.78 (1H, d, J = 8.1 Hz, H-5′), 6.57 (2H, s, H-3, 5), 6.54 (1H, d, J = 12.0 Hz, –CHCH–), 6.49 (1H, d, J = 12.0 Hz, –CHCH–), 3.88 (3H, s, –OCH3), 3.69 (6H, s, 2 × –OCH3), 3.68 (3H, s, –OCH3), 2.34 (3H, s, –COCH3); 13C-NMR (75 MHz, CDCl3) δ 168.9, 152.2, 149.1, 149.0, 136.0, 130.1, 128.8, 126.6, 122.0, 111.2, 108.7, 102.9, 56.1, 56.0, 55.9, 20.5; ESI-MS m/z: 359.1 [M + H]+.
:1) to get a light yellow solid 0.88 g, 99.8% yield. Mp 98–99 °C; IR vmax (KBr, cm−1): 3462, 3133, 2933, 1609, 1517; 1H-NMR (300 MHz, CDCl3) δ 6.81 (1H, d, J = 8.1 Hz, H-5′), 6.72 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.68 (1H, d, J = 1.8 Hz, H-2′), 6.38 (2H, s, H-3, 5), 5.39 (1H, s, –OH), 3.88 (6H, s, 2 × –OCH3), 3.87 (6H, s, 2 × –OCH3), 2.85 (4H, s, –CH2CH2–); 13C-NMR (75 MHz, CDCl3) δ 146.9, 146.3, 140.4, 133.8, 132.3, 127.8, 119.9, 111.4, 110.7, 104.6, 55.7, 55.4, 55.3, 37.9, 37.3; ESI-MS m/z: 319.1 [M + H]+, 336.1 [M + NH4]+; HPLC purity: 100%, tR = 1.71 min.
:1–8:1) as eluent, by which we obtained the corresponding 4-O-modified CTX derivatives.
4.1.4.1. 4-(3,4-Dimethoxyphenethyl)-2,6-dimethoxyphenyl propionate (1a). Yield 63.7%; white solid, mp 101–102 °C; IR vmax (KBr, cm−1): 3422, 2922, 1758, 1604, 1515; 1H-NMR (300 MHz, CDCl3) δ 6.82 (1H, d, J = 8.1 Hz, H-5′), 6.74 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.67 (1H, d, J = 1.8 Hz, H-2′), 6.40 (2H, s, H-3, 5), 3.88 (3H, s, –OCH3), 3.87 (3H, s, –OCH3), 3.79 (6H, s, 2 × –OCH3), 2.88 (4H, s, –CH2CH2–), 2.66 (2H, q, J = 7.8 Hz, –COCH2–), 1.29 (3H, t, J = 7.5 Hz, –CH3); 13C-NMR (75 MHz, CDCl3) δ 172.0, 151.4, 148.3, 146.8, 139.6, 133.7, 119.8, 111.4, 110.7, 104.7, 55.6, 55.5, 55.3, 38.2, 37.0, 26.7, 8.8; ESI-MS m/z: 392.3 [M + NH4]+, 375.2 [M + H]+; HPLC purity: 100%, tR = 2.22 min.
4.1.4.2. 4-(3,4-Dimethoxyphenethyl)-2,6-dimethoxyphenyl butyrate (1b). Yield 42.1%; yellow powder; mp 60–61 °C; IR vmax (KBr, cm−1): 3446, 2953, 1758, 1603, 1508; 1H-NMR (300 MHz, CDCl3) δ 6.82 (1H, d, J = 8.1 Hz, H-5′), 6.74 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.67 (1H, d, J = 1.8 Hz, H-2′), 6.40 (2H, s, H-3, 5), 3.88 (3H, s, –OCH3), 3.86 (3H, s, –OCH3), 3.79 (6H, s, 2 × –OCH3), 2.88 (4H, s, –CH2CH2–), 2.61 (2H, t, J = 7.2 Hz, –COCH2–), 1.82 (2H, m, J = 7.2 Hz, –CH2–), 1.07 (3H, t, J = 7.2 Hz, –CH3); 13C-NMR (75 MHz, CDCl3) δ 171.1, 151.4, 148.5, 139.6, 133.7, 119.8, 114.5, 111.4, 110.7, 104.7, 55.6, 55.5, 55.3, 38.2, 37.0, 35.3, 18.2, 13.1; ESI-MS m/z: 406.3 [M + NH4]+, 389.2 [M + H]+; HPLC purity: 98%, tR = 2.44 min.
4.1.4.3. 4-(3,4-Dimethoxyphenethyl)-2,6-dimethoxyphenyl isobutyrate (1c). Yield 49.2%; yellow powder, mp 80–81 °C; IR vmax (KBr, cm−1): 3433, 2932, 1760, 1601, 1509, 1117; 1H-NMR (300 MHz, CDCl3) δ 6.81 (1H, d, J = 8.1 Hz, H-5′), 6.74 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.67 (1H, d, J = 1.8 Hz, H-2′), 6.40 (2H, s, H-3, 5), 3.88 (3H, s, –OCH3), 3.87 (3H, s, –OCH3), 3.78 (6H, s, 2 × –OCH3), 2.89 (1H, m, –COCH–) 2.88 (4H, s, –CH2CH2–), 1.34 (6H, d, J = 6.9 Hz, 2 × –CH3); 13C-NMR (75 MHz, CDCl3) δ 174.7, 151.4, 148.5, 146.8, 139.4, 133.7, 119.8, 114.5, 111.4, 110.7, 104.8, 55.6, 55.5, 55.3, 38.2, 37.0, 33.4, 18.6; ESI-MS m/z: 406.3 [M + NH4]+, 389.2 [M + H]+; HPLC purity: 100%, tR = 2.47 min.
4.1.4.4. 4-(3,4-Dimethoxyphenethyl)-2,6-dimethoxyphenyl pentanoate (1d). Yield 63.3%; white crystal; mp 77–78 °C; IR vmax (KBr, cm−1): 3448, 2956, 1741, 1601, 1509, 1128; 1H-NMR (300 MHz, CDCl3) δ 6.82 (1H, d, J = 8.1 Hz, H-5′), 6.74 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.67 (1H, d, J = 1.8 Hz, H-2′), 6.40 (2H, s, H-3, 5), 3.88 (3H, s, –OCH3), 3.86 (3H, s, –OCH3), 3.78 (6H, s, 2 × –OCH3), 2.88 (4H, s, –CH2CH2–), 2.62 (2H, t, J = 7.2 Hz, –COCH2–), 1.78 (2H, m, J = 7.2 Hz, –CH2–), 1.48 (2H, m, –CH2–), 0.98 (3H, t, J = 7.2 Hz, –CH3); 13C-NMR (75 MHz, CDCl3) δ 181.1, 151.4, 148.3, 146.8, 139.6, 134.9, 119.8, 111.4, 110.7, 104.7, 55.6, 55.5, 55.3, 38.2, 37.0, 33.1, 26.7, 21.7, 13.3; ESI-MS m/z: 420.3 [M + NH4]+, 403.2 [M + H]+; HPLC purity: 100%, tR = 2.70 min.
4.1.4.5. 4-(3,4-Dimethoxyphenethyl)-2,6-dimethoxyphenyl 3-methylbutanoate (1e). Yield 60.6%; white crystal; mp 102–103 °C; IR vmax (KBr, cm−1): 3422, 2923, 1758, 1607, 1506, 1124; 1H-NMR (300 MHz, CDCl3) δ 6.82 (1H, d, J = 8.1 Hz, H-5′), 6.74 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.67 (1H, d, J = 1.8 Hz, H-2′), 6.40 (2H, s, H-3, 5), 3.88 (3H, s, –OCH3), 3.86 (3H, s, –OCH3), 3.78 (6H, s, 2 × –OCH3), 2.88 (4H, s, –CH2CH2–), 2.49 (2H, d, J = 7.2 Hz, –COCH2–), 2.29 (1H, m, –CH–), 1.09 (6H, d, J = 6.6 Hz, 2 × –CH3); 13C-NMR (75 MHz, CDCl3) δ 170.5, 151.4, 148.2, 146.8, 139.6, 133.65, 119.8, 111.4, 110.7, 104.7, 55.5, 55.4, 55.3, 42.5, 38.2, 37.0, 25.6, 21.8; ESI-MS m/z: 420.3 [M + NH4]+, 403.2 [M + H]+; HPLC purity: 100%, tR = 2.69 min.
4.1.4.6. 4-(3,4-Dimethoxyphenethyl)-2,6-dimethoxyphenyl 2-propylpentanoate (1f). Yield 48.7%; white crystal; mp 63–64 °C; IR vmax (KBr, cm−1): 3417, 2914, 1758, 1607, 1513, 1129; 1H-NMR (300 MHz, CDCl3) δ 6.81 (1H, d, J = 8.1 Hz, H-5′), 6.73 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.66 (1H, d, J = 1.8 Hz, H-2′), 6.38 (2H, s, H-3, 5), 3.88 (3H, s, –OCH3), 3.86 (3H, s, –OCH3), 3.76 (6H, s, 2 × –OCH3), 2.87 (4H, s, –CH2CH2–), 2.67 (1H, m, –COCH–), 1.80 (2H, m, –CH2), 1.56 (2H, m, –CH2), 1.52 (4H, m, 2 × –CH2–), 0.97 (6H, t, J = 8.1 Hz, 2 × –CH3); 13C-NMR (75 MHz, CDCl3) δ 173.8, 151.4, 148.3, 146.8, 139.5, 133.7, 119.8, 111.4, 110.7, 104.7, 55.5, 55.4, 55.3, 44.8, 38.2, 37.0, 34.4, 19.9, 13.6; ESI-MS m/z: 462.3 [M + NH4]+, 445.2 [M + H]+; HPLC purity: 96%, tR = 2.02 min.
:1–3:1).
4.1.5.1. 4-(3,4-Dimethoxyphenethyl)-2,6-dimethoxyphenyl benzoate (2a). Yield 90.3%; white crystal; mp 136–137 °C; IR vmax (KBr, cm−1): 3448, 2935, 1741, 1601, 1509, 1130; 1H-NMR (300 MHz, CDCl3) δ 8.26, 7.61, 7.52 (5H, m, ArH), 6.83 (1H, d, J = 8.1 Hz, H-5′), 6.76 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.70 (1H, d, J = 1.8 Hz, H-2′), 6.46 (2H, s, H-3, 5), 3.89 (6H, s, 2 × –OCH3), 3.79 (6H, s, 2 × –OCH3), 2.91 (4H, s, –CH2CH2–); 13C-NMR (75 MHz, CDCl3) δ 184.1, 151.6, 148.2, 145.7, 139.8, 133.7, 132.8, 131.2, 129.9, 127.9, 119.8, 111.4, 110.7, 104.8, 55.7, 55.5, 55.4, 38.3, 37.1; ESI-MS m/z: 440.3 [M + NH4]+, 423.2 [M + H]+; HPLC purity: 99%, tR = 2.53 min.
4.1.5.2. 4-(3,4-Dimethoxyphenethyl)-2,6-dimethoxyphenyl 4-chlorobenzoate (2b). Yield 87.2%; light yellow powder; mp 130–131 °C; IR vmax (KBr, cm−1): 3447, 2933, 1743, 1601, 1508, 1127; 1H-NMR (300 MHz, CDCl3) δ 8.19 (2H, d, J = 8.4 Hz, ArH), 7.49 (2H, d, J = 8.4 Hz, ArH), 6.83 (1H, d, J = 8.1 Hz, H-5′), 6.76 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.70 (1H, d, J = 1.8 Hz, H-2′), 6.45 (2H, s, H-3, 5), 3.91 (6H, s, 2 × –OCH3), 3.82 (6H, s, 2 × –OCH3), 2.91 (4H, s, –CH2CH2–); 13C-NMR (75 MHz, CDCl3) δ 187.3, 151.5, 149.5, 147.0, 140.0, 135.9, 133.7, 131.3, 128.4, 128.3, 119.8, 111.4, 110.7, 104.7, 55.6, 55.5, 55.4, 42.8, 38.3, 37.0, 33.5; ESI-MS m/z: 474.2 [M + NH4]+, 457.1 [M + H]+; HPLC purity: 95%, tR = 3.06 min.
4.1.5.3. 4-(3,4-Dimethoxyphenethyl)-2,6-dimethoxyphenyl 3-phenylpropanoate (2c). Yield 85.7%; light yellow powder; mp 65–66 °C; IR vmax (KBr, cm−1): 3423, 2923, 1759, 1605, 1508, 1127; 1H-NMR (300 MHz, CDCl3) δ 7.31 (5H, m, ArH), 6.82 (1H, d, J = 8.1 Hz, H-5′), 6.74 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.67 (1H, d, J = 1.8 Hz, H-2′), 6.40 (2H, s, H-3, 5), 3.88 (3H, s, –OCH3), 3.86 (3H, s, –OCH3), 3.76 (6H, s, 2 × –OCH3), 3.12 (2H, t, J = 6.9 Hz, –CH2–), 2.95 (2H, t, J = 6.9 Hz, –CH2–), 2.87 (4H, s, –CH2CH2–); 13C-NMR (75 MHz, CDCl3) δ 170.4, 151.3, 148.5, 146.8, 140.0, 139.7, 133.6, 128.0, 127.9, 125.7, 119.8, 111.4, 110.7, 104.7, 55.6, 55.5, 55.3, 38.3, 37.0, 34.9, 30.5; ESI-MS m/z: 468.2 [M + NH4]+, 451.1 [M + H]+; HPLC purity: 99%, tR = 2.66 min.
4.1.5.4. 4-(3,4-Dimethoxyphenethyl)-2,6-dimethoxyphenyl picolinate (2d). Yield 72.7%; yellow powder; mp 132–133 °C; IR vmax (KBr, cm−1): 3131, 2944, 1757, 1601, 1514; 1H-NMR (300 MHz, CDCl3) δ 8.89, 8.32, 7.95, 7.57 (4H, pyridine), 6.83 (1H, d, J = 8.1 Hz, H-5′), 6.75 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.69 (1H, d, J = 1.8 Hz, H-2′), 6.44 (2H, s, H-3, 5), 3.89 (3H, s, –OCH3), 3.88 (3H, s, –OCH3), 3.78 (6H, s, 2 × –OCH3), 2.91 (4H, s, –CH2CH2–); 13C-NMR (75 MHz, CDCl3) δ 180.1, 151.9, 150.1, 148.9, 146.0, 140.4, 137.0, 134.2, 127.2, 125.9, 120.3, 111.9, 111.2, 105.1, 56.1, 55.9, 55.8, 38.8, 37.5; ESI-MS m/z: 424.1 [M + H]+; HPLC purity: 99%, tR = 2.00 min.
4.1.5.5. 4-(3,4-Dimethoxyphenethyl)-2,6-dimethoxyphenyl isonicotinate (2e). Yield 87.7%; white crystal; mp 118–119 °C; IR vmax (KBr, cm−1): 3131, 2932, 1735, 1601, 1514; 1H-NMR (300 MHz, CDCl3) δ 8.86 (2H, d, J = 5.7 Hz, pyridine), 8.06 (2H, d, J = 5.7 Hz, pyridine), 6.83 (1H, d, J = 8.1 Hz, H-5′), 6.75 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.69 (1H, d, J = 1.8 Hz, H-2′), 6.45 (2H, s, H-3, 5), 3.89 (3H, s, –OCH3), 3.88 (3H, s, –OCH3), 3.78 (6H, s, 2 × –OCH3), 2.91 (4H, s, –CH2CH2–); 13C-NMR (75 MHz, CDCl3) δ 181.5, 155.4, 151.8, 150.6, 141.1, 136.8, 134.1, 123.7, 120.3, 111.9, 111.2, 105.2, 56.1, 56.0, 55.9, 38.8, 37.5; ESI-MS m/z: 424.1 [M + H]+, 406.1 [M − OH]+; HPLC purity: 98%, tR = 2.42 min.
:1–10:1).
4.1.6.1. 5-(3,4-Dimethoxyphenethyl)-2-ethoxy-1,3-dimethoxybenzene (3a). Yield 52.9%; yellow crystal, mp 68–69 °C; IR vmax (KBr, cm−1): 3132, 2957, 1589, 1515; 1H-NMR (300 MHz, CDCl3) δ 6.82 (1H, d, J = 8.1 Hz, H-5′), 6.74 (1H, d, J = 1.8, 8.1 Hz, H-6′), 6.68 (1H, d, J = 1.8 Hz, H-2′), 6.38 (2H, s, H-3, 5), 4.04 (2H, q, J = 7.2 Hz, –OCH2–), 3.88 (3H, s, –OCH3), 3.86 (3H, s, –OCH3), 3.83 (6H, s, 2 × –OCH3), 2.87 (4H, s, –CH2CH2–), 1.37 (3H, t, J = 7.2 Hz, –CH3); 13C-NMR (75 MHz, CDCl3) δ 153.3, 148.7, 146.9, 137.2, 134.3, 120.3, 115.1, 111.9, 111.1, 105.6, 74.2, 56.1, 55.9, 55.8, 38.6, 37.6, 31.1; ESI-MS m/z: 347 [M + H]+, HPLC purity: 99%, tR = 2.38 min.
4.1.6.2. 5-(3,4-Dimethoxyphenethyl)-1,3-dimethoxy-2-propoxybenzene (3b). Yield 61.8%; light yellow oil; IR vmax (KBr, cm−1): 3000, 2958, 2935, 1589, 1513; 1H-NMR (300 MHz, CDCl3) δ 6.81 (1H, d, J = 8.1 Hz, H-5′), 6.73 (1H, d, J = 1.8, 8.1 Hz, H-6′), 6.68 (1H, d, J = 1.8 Hz, H-2′), 6.38 (2H, s, H-3, 5), 3.92 (2H, t, J = 6.9 Hz, –OCH2–), 3.88 (3H, s, –OCH3), 3.86 (3H, s, –OCH3), 3.82 (6H, s, 2 × –OCH3), 2.86 (4H, s, –CH2CH2–), 1.78 (2H, m, –CH2–), 1.01 (3H, t, J = 7.5 Hz, –CH3); 13C-NMR (75 MHz, CDCl3) δ 153.3, 148.7, 146.4, 137.2, 134.3, 120.3, 115.1, 111.9, 111.1, 105.6, 75.1, 56.1, 55.9, 55.8, 38.6, 37.6, 23.3, 10.4; ESI-MS m/z: 378 [M + NH4]+, 361 [M + H]+; HPLC purity: 97%, tR = 2.70 min.
4.1.6.3. 2-Butoxy-5-(3,4-dimethoxyphenethyl)-1,3-dimethoxybenzene (3c). Yield 65.2%; yellow oil; IR vmax (KBr, cm−1): 3132, 2955, 1581, 1514; 1H-NMR (300 MHz, CDCl3) δ 6.81 (1H, d, J = 8.1 Hz, H-5′), 6.73 (1H, d, J = 1.8, 8.1 Hz, H-6′), 6.68 (1H, d, J = 1.8 Hz, H-2′), 6.37 (2H, s, H-3, 5), 3.95 (2H, t, J = 6.9 Hz, –OCH2–), 3.88 (3H, s, –OCH3), 3.86 (3H, s, –OCH3), 3.82 (6H, s, 2 × –OCH3), 2.86 (4H, s, –CH2CH2–), 1.76 (2H, m, –CH2–), 1.49 (2H, m, –CH2–), 0.97 (3H, t, J = 6.5 Hz, –CH3); 13C-NMR (75 MHz, CDCl3) δ 153.3, 148.7, 147.2, 137.2, 134.3, 120.3, 111.9, 111.2, 105.6, 73.2, 56.1, 56.0, 55.8, 38.6, 37.6, 32.2, 19.1, 13.9; ESI-MS m/z: 392.2 [M + NH4]+, 375.2 [M + H]+; HPLC purity: 99%, tR = 3.06 min.
4.1.6.4. 5-(3,4-Dimethoxyphenethyl)-2-isobutoxy-1,3-dimethoxybenzene (3d). Yield 49.8%; yellow oil; IR vmax (KBr, cm−1): 3132, 2935, 1588, 1514; 1H-NMR (300 MHz, CDCl3) δ 6.81 (1H, d, J = 8.1 Hz, H-5′), 6.73 (1H, d, J = 1.8, 8.1 Hz, H-6′), 6.68 (1H, d, J = 1.8 Hz, H-2′), 6.37 (2H, s, H-3, 5), 3.88 (3H, s, –OCH3), 3.86 (3H, s, –OCH3), 3.82 (6H, s, 2 × –OCH3), 3.71 (2H, d, J = 6.9 Hz, –OCH2–), 2.86 (4H, s, –CH2CH2–), 2.07 (1H, m, –CH–), 1.03 (6H, d, 2 × –CH3); 13C-NMR (75 MHz, CDCl3) δ 153.2, 148.8, 147.3, 137.1, 134.3, 120.3, 111.9, 111.1, 105.9, 80.2, 56.2, 56.0, 55.8, 38.5, 37.6, 29.0, 19.2; ESI-MS m/z: 392.2 [M + NH4]+, 375.2 [M + H]+; HPLC purity: 100%, tR = 3.21 min.
4.1.6.5. 2-(sec-Butoxy)-5-(3,4-dimethoxyphenethyl)-1,3-dimethoxybenzene (3e). Yield 42.7%; yellow oil; IR vmax (KBr, cm−1): 3132, 2955, 1584, 1515; 1H-NMR (300 MHz, CDCl3) δ 6.81 (1H, d, J = 8.1 Hz, H-5′), 6.73 (1H, d, J = 1.8, 8.1 Hz, H-6′), 6.67 (1H, d, J = 1.8 Hz, H-2′), 6.37 (2H, s, H-3, 5), 4.11 (1H, m, –CH–), 3.88 (3H, s, –OCH3), 3.85 (3H, s, –OCH3), 3.80 (6H, s, 2 × –OCH3), 2.85 (4H, s, –CH2CH2–), 1.80–161 (2H, m, –OCH2–), 1.23 (3H, d, J = 6.3 Hz, –CH3), 0.99 (3H, t, J = 7.5 Hz, –CH3); 13C-NMR (75 MHz, CDCl3) δ 153.5, 148.7, 142.1, 136.9, 129.3, 120.3, 115.0, 105.6, 80.1, 56.0, 55.9, 55.8, 38.6, 37.6, 29.5, 19.5, 10.0; ESI-MS m/z: 392.2 [M + NH4]+, 375.2 [M + H]+; HPLC purity: 98%, tR = 3.06 min.
4.1.6.6. 5-(3,4-Dimethoxyphenethyl)-2-(isopentyloxy)-1,3-dimethoxybenzene (3f). Yield 44.9%; yellow oil; IR vmax (KBr, cm−1): 3132, 2924, 1589, 1518; 1H-NMR (300 MHz, CDCl3) δ 6.81 (1H, d, J = 8.1 Hz, H-5′), 6.73 (1H, d, J = 1.8, 8.1 Hz, H-6′), 6.68 (1H, d, J = 1.8 Hz, H-2′), 6.37 (2H, s, H-3, 5), 3.97 (2H, t, J = 6.9 Hz, –OCH2–), 3.88 (3H, s, –OCH3), 3.86 (3H, s, –OCH3), 3.82 (6H, s, 2 × –OCH3), 2.86 (4H, s, –CH2CH2–), 1.89 (1H, m, –CH–), 1.67 (2H, t, J = 6.9 Hz, –CH2–), 0.96 (6H, d, J = 6.6 Hz, 2 × –CH3); 13C-NMR (75 MHz, CDCl3) δ 153.3, 148.7, 147.2, 137.2, 134.3, 120.3, 111.9, 111.1, 105.6, 71.9, 56.1, 55.9, 55.8, 38.9, 38.6, 37.6, 24.8, 22.6; ESI-MS m/z: 406.2 [M + NH4]+, 389.2 [M + H]+; HPLC purity: 100%, tR = 3.47 min.
4.1.6.7. 5-(3,4-Dimethoxyphenethyl)-1,3-dimethoxy-2-(pentan-2-yloxy)benzene (3g). Yield 37.4%; yellow oil; IR vmax (KBr, cm−1): 3133, 2935, 1587, 1517; 1H-NMR (300 MHz, CDCl3) δ 6.81 (1H, d, J = 8.1 Hz, H-5′), 6.73 (1H, d, J = 1.8, 8.1 Hz, H-6′), 6.68 (1H, d, J = 1.8 Hz, H-2′), 6.36 (2H, s, H-3, 5), 4.19 (1H, m, –OCH–), 3.88 (3H, s, –OCH3), 3.85 (3H, s, –OCH3), 3.80 (6H, s, 2 × –OCH3), 2.86 (4H, brs, –CH2CH2–), 1.76, 1.51 (4H, m, –CH2CH2–), 1.22 (2H, t, J = 6.3 Hz, –CH2–), 0.95 (3H, t, J = 6.9 Hz, –CH3); 13C-NMR (75 MHz, CDCl3) δ 153.5, 148.6, 147.2, 136.9, 134.4, 120.3, 111.9, 111.1, 105.5, 78.5, 56.0, 55.9, 55.8, 39.1, 38.6, 37.6, 20.0, 18.8, 14.2; ESI-MS m/z: 406.2 [M + NH4]+, 389.2 [M + H]+; HPLC purity: 99%, tR = 3.53 min.
4.1.6.8. 5-(3,4-Dimethoxyphenethyl)-2-[(2-ethylhexyl)oxy]-1,3-dimethoxybenzene (3h). Yield 39.4%; yellow oil; IR vmax (KBr, cm−1): 3133, 2958, 1637, 1513; 1H-NMR (300 MHz, CDCl3) δ 6.81 (1H, d, J = 8.1 Hz, H-5′), 6.72 (1H, dd, J = 1.8, 8.1 Hz, H-6′), 6.68 (1H, d, J = 1.8 Hz, H-2′), 6.36 (2H, s, H-3, 5), 3.88 (3H, s, –OCH3), 3.85 (3H, s, –OCH3), 3.82 (2H, d, J = 6.9 Hz, –OCH2–), 3.80 (6H, s, 2 × –OCH3), 2.86 (4H, brs, –CH2CH2–), 1.40–1.72 (9H, m), 0.95 (6H, t, J = 6.9 Hz, 2 × –CH3); 13C-NMR (75 MHz, CDCl3) δ 153.5, 149.6, 146.9, 137.2, 134.5, 120.3, 111.9, 111.1, 105.8, 75.8, 56.1, 55.9, 55.8, 40.2, 38.5, 37.6, 30.2, 29.0, 23.5, 23.2, 14.2, 10.9; ESI-MS m/z: 448.2 [M + NH4]+, 431.2 [M + H]+; HPLC purity: 98%, tR = 6.65 min.
4.1.6.9. 2-(Cyclopentyloxy)-5-(3,4-dimethoxyphenethyl)-1,3-dimethoxybenzene (3i). Yield 25.8%; yellow crystal, 51–52 °C; IR vmax (KBr, cm−1): 3416, 2361, 1637, 1420, 1231, 1122; 1H-NMR (300 MHz, CDCl3) δ 6.81 (1H, d, J = 8.1 Hz, H-5′), 6.73 (1H, d, J = 1.8, 8.1 Hz, H-6′), 6.67 (1H, d, J = 1.8 Hz, H-2′), 6.37 (2H, s, H-3, 5), 4.76 (1H, t, J = 5.4 Hz, –OCH–), 3.88 (3H, s, –OCH3), 3.85 (3H, s, –OCH3), 3.80 (6H, s, 2 × –OCH3), 2.86 (4H, brs, –CH2CH2–), 1.91, 1.67, 1.58 (8H, m); 13C-NMR (75 MHz, CDCl3) δ 153.8, 149.7, 148.5, 138.3, 137.2, 134.5, 120.3, 111.9, 111.1, 105.7, 84.3, 56.1, 55.9, 55.8, 38.6, 37.5, 32.7, 23.7; ESI-MS m/z: 404.2 [M + NH4]+, 387.2 [M + H]+; HPLC purity: 98%, tR = 3.22 min.
4.1.6.10. 2-(Benzyloxy)-5-(3,4-dimethoxyphenethyl)-1,3-dimethoxybenzene (3j). Yield 96.8%; yellow crystal, 77–78 °C; IR vmax (KBr, cm−1): 3416, 2937, 1589, 1514, 1128, 1028; 1H-NMR (300 MHz, CDCl3) δ 7.52–7.31 (5H, m, ArH), 6.81 (1H, d, J = 8.1 Hz, H-5′), 6.71 (1H, d, J = 1.8, 8.1 Hz, H-6′), 6.67 (1H, d, J = 1.8 Hz, H-2′), 6.37 (2H, s, H-3, 5), 5.00 (2H, s, –CH2–), 3.88 (3H, s, –OCH3), 3.86 (3H, s, –OCH3), 3.81 (6H, s, 2 × –OCH3), 2.87 (4H, s, –CH2CH2–); 13C-NMR (75 MHz, CDCl3) δ 153.3, 148.8, 147.3, 138.0, 137.6, 134.3, 128.5, 128.1, 127.8, 120.4, 111.9, 111.2, 105.6, 75.1, 56.1, 56.0, 55.8, 38.6, 37.6; ESI-MS m/z:426.2 [M + NH4]+, 409.2 [M + H]+; HPLC purity: 100%, tR = 3.53 min.
4.2. Pharmacology
The PC12 cells were purchased from the Institute of Biochemistry and Cell Biology (Shanghai, China) and cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with penicillin (100 U mL−1), streptomycin (100 µg mL−1), and 10% (vol/vol) fetal bovine serum (FBS) at 37 °C in an atmosphere containing 5% CO2. DMEM and FBS were obtained from Gibco (GrandIsland, NY). The other reagents were purchased from Beyotime Institute of Biotechnology (Shanghai, China).
For the experimental studies, the PC12 cells were subcultured in 96 or 6-well plates at a density of 1.5 × 104 or 2 × 105 cells per well and allowed to adhere and grow. When cells reached the required confluence, they were placed in serum-free medium and pretreated with different concentrations of test compounds for 0.5 h,45 and then Aβ25–35 (10 µM) was added to the medium for the corresponding time.
For the inhibition of self-induced aggregation assay, 20 µL Aβ1–42 (25 µM, final concentration) was incubated with 20 µL of test compounds (25 µM, final concentration) in 50 mM phosphate buffer solution (pH 7.4) at 37 °C for 24 h. To minimize evaporation effect the wells were sealed by a transparent heat-resistant plastic film. After incubation, 160 µL of 5 µM thioflavin T (TCI (Shanghai) Development Co., Ltd.) in 50 mM glycine–NaOH buffer (pH 8.5) was added. Fluorescence was measured on a Varioskan Flash Multimode Reader (Thermo Scientific) with excitation and emission wavelengths at 446 nm and 490 nm, respectively. The fluorescence intensities were compared and the percent inhibition due to the presence of the inhibitor was calculated by the following formula: 100 − (IFi/IFc × 100), where IFi and IFc were the fluorescence intensities obtained for Aβ1–42 in the presence and in the absence of inhibitor, respectively. Each assay was run in triplicate.
:1500; p-tau S396 at 1:500; S199/202 at 1:500), the blots were washed in Tween 20-TBS (TBST) (0.02% Tween 20, 100 mM Tris, pH 7.5, 150 nM NaCl) for 20 min and then incubated with secondary antibody for 1 h at room temperature. The blots were washed in TBST for 20 min and incubated with ECL chemiluminescent reagent (Thermo Scientific) for 3 min. Quantification of pixel intensity was done with Image-Pro Plus 6.0 software. β-Actin was used as a control to normalize tau protein.
:3), and the filter membrane was impregnated with 4 µL of PBL in dodecane (20 mg mL−1). Compounds were dissolved in DMSO at 5 mg mL−1 and diluted 50-fold in PBS/EtOH (7:3) to a final concentration of 100 µg mL−1. Then, 200 µL of the solution was added to the donor wells. The acceptor filter plate was carefully placed on the donor plate to form a sandwich, and the plates were incubated undisturbed for 18 h at 25 °C. After incubation, the donor plate was carefully removed, and the concentration of the compounds in the acceptor wells was determined using a UV plate reader (Flexstation® 3). Each sample was analyzed at five wavelengths in four wells and in at least three independent runs. Pe was calculated by the following formula: Pe = −Vd × [Va(Vd + Va)A × t] × ln(1 − drugacceptor/drugequilibrium), where Vd is the volume of the donor well, Va is the volume of the acceptor well, A is the filter area, t is the permeation time, drugacceptor is the absorbance obtained in the acceptor well, and drugequilibrium is the theoretical equilibrium absorbance. The results are given as the mean ± SD. In the experiment, 10 quality control standards with known BBB permeabilities were included to validate the analysis set. A plot of the experimental data versus the literature values gave a strong linear correlation: Pe(exp.) = 1.054Pe(bibl.) + 0.5582 (R2 = 0.987). From this equation and the limit established by Di et al. (Pe(lit.) = 4.0 × 10−6 cm s−1) for BBB permeation, we concluded that compounds with a permeability greater than 4.8 × 10−6 cm s−1 can cross the BBB.
Acknowledgements
This research work was financially supported by the National Natural Science Foundation of China (No. 81373956, No. 81274064, No. 81502950 and No. 81573557), Seed Funding Program for Basic Research from HKU (201111159043), the Priority Academic Program Development of Jiangsu Higher Education Institutions, the Fundamental Research Funds for the Central Universities (2015ZD010), Huahai Graduate Innovation Fund (No. CX14B-007HH), College Students Innovation Project for the R&D of Novel Drugs (J1030830), National Students Innovation and Entrepreneurship Training Program (G15073).References
- H. W. Querfurth and F. M. LaFerla, N. Engl. J. Med., 2010, 362, 329–344 CrossRef CAS PubMed.
- World Alzheimer Report 2015, http://www.alz.co.uk/research/world-report-2015, assessed August 2015.
- G. Pepeu and M. G. Giovannini, Curr. Alzheimer Res., 2009, 6, 86–96 CrossRef CAS PubMed.
- B. Reisberg, R. Doody, A. Stöffler, F. Schmitt, S. Ferris and H. J. Möbius, N. Engl. J. Med., 2003, 348, 1333–1341 CrossRef CAS PubMed.
- K. A. Bates, G. Verdile, Q. X. Li, D. Ames, P. Hudson, C. L. Masters and R. N. Martins, Mol. Psychiatry, 2008, 14, 469–486 CrossRef PubMed.
- A. Boutajangout and T. Wisniewski, Gerontology, 2014, 60, 381–385 CrossRef CAS PubMed.
- A. I. Bush and R. E. Tanzi, Neurotherapeutics, 2008, 5, 421–432 CrossRef CAS PubMed.
- M. A. Ansari and S. W. Scheff, J. Neuropathol. Exp. Neurol., 2010, 69, 155 CrossRef CAS PubMed.
- P. T. Francis, A. M. Palmer, M. Snape and G. K. Wilcock, J. Neurol., Neurosurg. Psychiatry, 1999, 66, 137–147 CrossRef CAS.
- J. Hardy, J. Neurochem., 2009, 110, 1129–1134 CrossRef CAS PubMed.
- M. Jucker and L. C. Walker, Ann. Neurol., 2011, 70, 532–540 CrossRef CAS PubMed.
- M. Hernández-Rodríguez, J. Correa-Basurto, M. I. Nicolás-Vázquez, R. Miranda-Ruvalcaba, C. G. Benítez-Cardoza, A. A. Reséndiz-Albor, J. V. Méndez-Méndez and M. C. Rosales-Hernández, PLoS One, 2015, 10, e0130263 Search PubMed.
- O. Crescenzi, S. Tomaselli, R. Guerrini, S. Salvadori, A. M. D'Ursi, P. A. Temussi and D. Picone, Eur. J. Biochem., 2002, 269, 5642–5648 CrossRef CAS PubMed.
- S. Paul, S. Planque and Y. Nishiyama, Rejuvenation Res., 2010, 13, 179–187 CrossRef CAS PubMed.
- L. Huang, C. Lu, Y. Sun, F. Mao, Z. Luo, T. Su, H. Jiang, W. Shan and X. Li, J. Med. Chem., 2012, 55, 8483–8492 CrossRef CAS PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- T. A. Dineen, K. Chen, A. C. Cheng, K. Derakhchan, O. Epstein, J. Esmay, D. Hickman, C. E. Kreiman, I. E. Marx, R. C. Wahl, P. H. Wen, M. M. Weiss, D. A. Whittington, S. Wood, R. T. Fremeau, R. D. White and V. F. Patel, J. Med. Chem., 2014, 57, 9811–9831 CrossRef CAS PubMed.
- S. Lovestone, C. H. Reynolds, D. Latimer, D. R. Davis, B. H. Anderton, J. M. Gallo, D. Hanger, S. Mulot and B. Marquardt, Curr. Biol., 1994, 4, 1077–1086 CrossRef CAS PubMed.
- A. M. Palmer, Trends Pharmacol. Sci., 2011, 32, 141–147 CrossRef CAS PubMed.
- L. E. Scott and C. Orvig, Chem. Rev., 2009, 109, 4885–4910 CrossRef CAS PubMed.
- S. Boopathi and P. Kolandaivel, RSC Adv., 2014, 4, 38951 RSC.
- D. E. Clark, J. Pharm. Sci., 1999, 88, 815–821 CrossRef CAS PubMed.
- W. M. Pardridge, Alzheimer's Dementia, 2009, 5, 427–432 CrossRef PubMed.
- M. Ono, Y. Ito, C. Masuoka, H. Koga and T. Nohara, Food Sci. Technol. Int., 1995, 1, 115–120 CrossRef CAS.
- J. X. Song, S. C. W. Sze, T. B. Ng, C. K. F. Lee, G. P. H. Leung, P. C. Shaw, Y. Tong and Y. B. Zhang, J. Ethnopharmacol., 2012, 139, 698–711 CrossRef CAS PubMed.
- J. J. Fan, L. Guan, Z. Q. Kou, F. Feng, Y. B. Zhang and W. Y. Liu, J. Chromatogr. B, 2014, 967, 57–62 CrossRef CAS PubMed.
- J. X. Song, P. C. Shaw, C. W. Sze, Y. Tong, X. S. Yao, T. B. Ng and Y. B. Zhang, Neurochem. Int., 2010, 57, 676–689 CrossRef CAS PubMed.
- J. X. Song, P. C. Shaw, N. S. Wong, C. W. Sze, X. S. Yao, C. W. Tang, Y. Tong and Y. B. Zhang, Neurosci. Lett., 2012, 521, 76–81 CrossRef CAS PubMed.
- A. A. Reinke and J. E. Gestwicki, Chem. Biol. Drug Des., 2007, 70, 206–215 CAS.
- Y. Hernández-Romero, J. I. Rojas, R. Castillo, A. Rojas and R. Mata, J. Nat. Prod., 2004, 67, 160–167 CrossRef PubMed.
- Y. W. Lee, D. H. Kim, S. J. Jeon, S. J. Park, J. M. Kim, J. M. Jung, H. E. Lee, S. G. Bae, H. K. Oh, K. H. Ho Son and J. H. Ryu, Eur. J. Pharmacol., 2013, 704, 70–77 CrossRef CAS PubMed.
- A. Diaz, D. Limon, R. Chávez, E. Zenteno and J. Guevara, J. Alzheimer's Dis., 2012, 30, 505–522 CAS.
- Y. G. Kaminsky, M. W. Marlatt, M. A. Smith and E. A. Kosenko, Exp. Neurol., 2010, 221, 26–37 CrossRef CAS PubMed.
- N. Jiang, X. B. Wang, Z. R. Li, S. Y. Li, S. S. Xie, M. Huang and L. Y. Kong, RSC Adv., 2015, 5, 14242–14255 RSC.
- N. Jiang, S. Y. Li, S. S. Xie, Z. R. Li, K. D. G. Wang, X. B. Wang and L. Y. Kong, Eur. J. Med. Chem., 2014, 87, 540–551 CrossRef CAS PubMed.
- S. Y. Li, X. B. Wang and L. Y. Kong, Eur. J. Med. Chem., 2014, 71, 36–45 CrossRef CAS PubMed.
- M. G. Spillantini and M. Goedert, Trends Neurosci., 1998, 21, 428–433 CrossRef CAS PubMed.
- C. Ballatore, V. M. Y. Lee and J. Q. Trojanowski, Nat. Rev. Neurosci., 2007, 8, 663–672 CrossRef CAS PubMed.
- A. A. Reinke and J. E. Gestwicki, Chem. Biol. Drug Des., 2007, 70, 206–215 CAS.
- W. Pardridge, Neurotherapeutics, 2005, 2, 3–14 CrossRef PubMed.
- C. A. Lipinski, Drug Discovery Today, 2004, 1, 337–341 CrossRef CAS PubMed.
- D. E. Clark, J. Pharm. Sci., 1999, 88, 815–821 CrossRef CAS PubMed.
- L. Di, E. H. Kerns, K. Fan, O. J. McConnell and G. T. Carter, Eur. J. Med. Chem., 2003, 38, 223–232 CrossRef CAS PubMed.
- Y. F. Xian, Z. X. Lin, Q. Q. Mao, S. P. Ip, Z. R. Su and X. P. Lai, Cell. Mol. Neurobiol., 2012, 32, 353–360 CrossRef CAS PubMed.
- G. Li, R. Ma, C. Huang, Q. Tang, Q. Fu, H. Liu, B. Hu and J. Xiang, Neurosci. Lett., 2008, 442, 143–147 CrossRef CAS PubMed.
- D. L. Chen, P. Zhang, L. Lin, O. Shuai, H. M. Zhang, S. H. Liu and J. Y. Wang, Cell. Mol. Neurobiol., 2013, 33, 837–850 CrossRef CAS PubMed.
- W. Huang, D. Lv, H. Yu, R. Sheng, S. C. Kim, P. Wu, K. Luo, J. Li and Y. Hu, Bioorg. Med. Chem., 2010, 18, 5610–5615 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra21313d |
| This journal is © The Royal Society of Chemistry 2016 |