
Chromium complexes containing a tetradentate [OSSO]-type bisphenolate ligand as a novel family of catalysts for the copolymerization of carbon dioxide and 4-vinylcyclohexene oxide†
Gaoshan Sib,
Li Zhanga,
Bing Hana,
Hongye Zhanga,
Xiangqing Li*b and
Binyuan Liu*a
aDepartment of Polymer Science and Engineering, Hebei University of Technology, No. 8 Guangrong Road, Tianjin 300130, China. E-mail: byliu@hebut.edu.cn; Tel: +86 22 6020 4305
bSchool of Chemical and Environmental Engineering, Shanghai Institute of Technology, 100 Haiquan Road, Shanghai 201418, China. E-mail: xqli@sit.edu.cn; Tel: +86 21 6087 3061
First published on 22nd February 2016
Abstract
This work reports the synthesis of polycarbonates from 4-vinylcyclohexene oxide (VCHO)/carbon dioxide copolymerization catalyzed by new homogeneous chromium complexes with an [OSSO]-type bis(phenolato) dianionic ligand. Through the structural design, the ethylene-bridged ligands of the chromium complex exhibit the highest activity with the same axial anion Cl. The catalytic activity and product selectivity of these [OSSO]CrIII complexes have been systematically investigated under varied conditions. The 1H NMR spectrum of the obtained copolymers confirmed the resulting polycarbonate with high CO2 incorporation with a carbonate unit above 95%. This is one of the rare examples of a well-defined metal complex with a soft Lewis base as a donor for the efficient copolymerization of carbon dioxide and epoxides.
Introduction
Copolymerization of epoxides and carbon dioxide (CO2) to prepare aliphatic polycarbonates (APCs) has been considered to be an attractive polymerization process for a potential large-scale utilization of greenhouse CO2 in chemical synthesis,1 and the resulting APCs are a promising class of biodegradable polymers for various applications, such as integral components of engineered tissues, medical devices and drug delivery systems.2 Since the pioneering work reported by Inoue in the late 1960s,3 various catalysts have been explored for this reaction, of which particular efforts are devoted to developing structurally well-defined metal complexes supported by different ancillary ligands.1b,4–12 So far, such successful ligands employed dominantly involve hard Lewis base nitrogen and/or oxygen as coordination atoms such as salen4c,d,5 salalen,6 salan,7 porphyrinato,8 corrole,9 β-diketiminates,10 etc.11,12 Compared to considerable reports of the ligands based on the hard donors oxygen and nitrogen, there has been a few examples concerning on the metal complexes chelated with soft atoms for this transformation.13 Duchateau and co-workers found that chromium complex having soft phosphorus atom as one coordinate donor could catalyze the cyclohexene oxide (CHO)–CO2 copolymerization. However, this kind of chromium aminophinate complex shows low activity (turnover frequency (TOF) < 30 h−1) and the product is oligomeric poly(cyclohexene carbonate)s (Mn < 1800 g mol−1) with the appreciable ether linkages (≥6%).13a Capacchione and co-workers have reported that dinuclear Fe(III) complex coordinated by dithioether-triphenolate catalyzed the coupling reaction of CO2 with epoxides, exclusively producing the formation of cyclic carbonate.13bIn our recent work we found that [OSSO]CrX/Lewis base binary catalyst demonstrated efficient activity for the ring opening copolymerization (ROCOP) of epoxides with anhydride.14 Of importance, [OSSO]-type bis(phenolate) dianionic tetradentate ligands are analogous to the [ONNO]-salan ligands, in which the N–Me (H) donors are replaced with soft sulphur donors, i.e. the two phenoxide frameworks are linked to sulfur atom. As revealed by Lu and Darensbourg groups,7 chromium salan complex has shown higher catalytic performance than the corresponding chromium salen complex in catalyzing CO2/epoxides copolymerization, regarding both activity and stereochemistry control. They proposed that such higher catalytic performance should be related to the greater electron-donating feature of amino donor of salan ligand than that of salen counterparts, and cis-configuration structure is also an important contribution factor. Noteworthy, sulphur is a better electron-donating atom than that of nitrogen. Moreover, an characteristics of metal complexes of the [OSSO]-type ligand possess the cis-geometry as similar as complexes bearing [ONNO]-type tetradentate salan ligands.15 Furthermore, some well-defined metal complexes-based efficient catalysts for copolymerization of epoxides with CO2 are often capable of catalyzing the ROCOP of epoxides with anhydride or inverse due to the similarity of copolymerization mechanism.16,17
Inspired by the above-mentioned descriptions, the chromium complex containing a [OSSO]-type dianionic ligands (see Fig. 1) are, therefore, employed to test their catalytic behavior toward CO2/VCHO copolymerization in conjugation with organic onium salts or Lewis base, which is a part of our continuous efforts in search for novel catalyst for the ROCOP of epoxides and CO2.18 The VCHO was selected for this work because VCHO is largely unexplored as substrate for copolymerization with CO2 compared to substantial reports on CHO.19–22 The focuses are paid on the influence of ligand skeleton and reaction parameters on the activity and selectivity.
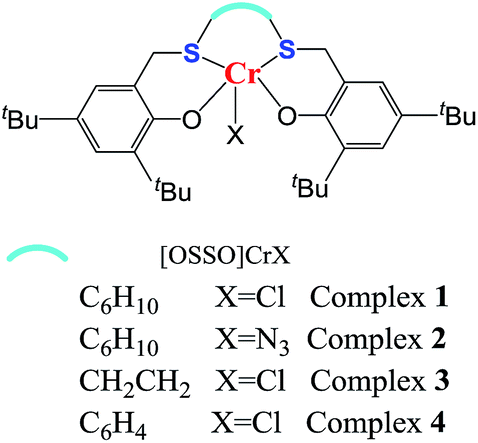 | ||
| Fig. 1 Structures of [OSSO]CrX complexes employed in this work (X = Cl or N3). | ||
Experimental section
Reagents and measurements
Unless otherwise stated, all reagents were purchased from commercial suppliers and used without further purification. All manipulations involving air- and/or water-sensitive compounds were carried out with the standard Schlenk and vacuum line techniques under argon atmosphere. VCHO was stirred over powered CaH2 at room temperature over 48 h and vacuum-distilled before use. All used cocatalysts including bis(triphenylphosphine)iminium chloride (PPNCl), 4-dimethylaminopyridine (DMAP), and tetrabutylammonium chloride (TABCl) were purified by standard methods. Bis(triphenylphosphine)iminium azide (PPNN3), was prepared according to published procedure.23 [OSSO]CrX complexes were synthesized as described in our previous work.14All 1H spectra were recorded on a Bruker-400 spectrometer at frequencies of 400 MHz. Chemical shifts are given in ppm relative to TMS. Infrared (IR) spectra were obtained on a BrukerVector 22 spectrometer at a resolution of 4 cm−1 (16 scans collected). The molecular weight of polymer was determined by using gel permeation chromatography (GPC) on a PL-GPC 220 instrument with a refractive index detector, calibrated with polystyrene standards. The columns used were MIXED-B 300 × 7.5 mm columns held at 40 °C, using THF as eluents at a flow rate of 1.0 mL min−1. The glass transition temperatures (Tg) of polymers were determined at a heating rate of 10 °C min−1 on PerkinElmer Diamond differential scanning calorimetry (DSC) instrument.
General procedure for the copolymerization of epoxides and CO2
The copolymerization of CO2 with VCHO was carried out in a 100 mL autoclave, which was equipped with a magnetic stirrer. Desired amounts of catalyst, cocatalyst and VCHO were transferred into the dried autoclave. The autoclave was heated to the target temperature in a pre-heated oil bath, filled with CO2 to a set pressure and was kept stirring for the desired time. After the reaction, the autoclave was slowly vented CO2, followed addition of mixture was taken for IR spectroscopic analysis. The residual mixture was added excess methanol to precipitate and filtrate. The crude copolymer was washed with methanol for several times. The resulting polymer was dried at 60 °C in vacuo overnight.Results and discussions
To investigate the relationship between complex structure and catalytic performance, four [OSSO]CrX complexes with different skeleton of [OSSO]-type ligands and axial anion (see Fig. 1) were synthesized14 and tested for the copolymerization of VCHO with CO2. As shown in Table 1, all the complexes were active in catalyzing the copolymerization of VCHO with CO2 at a molar ratio of 1000/1/2 ([VCHO]/[[OSSO]CrX]/[PPNCl]) at 90 °C in an efficient way. When X = Cl, complex 3 supported by [OSSO] ligand with ethylene backbone showed the highest activity to poly(vinylcyclohexene carbonate) with TOF up to 78 h−1, followed by the cyclohexylene-bridged chromium complex 1 (entry 1, TOF: 74 h−1, Table 1), and the least phenylene framework complex 4 (entry 4, Table 1). While the axial anion of Cl− was instead of N3− anion, the catalytic activity to the polymer is slightly enhanced by 10% (entry 2 vs. entry 1, Table 1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Complex | TOFb | Selectc | Carbonated | Mne (g mol−1) | PDIe |
a Reaction conditions: [VCHO]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cr:PPNCl = 1000:1:2; VCHO = 12.4 g (0.1 mol); 3.0 MPa, 90 °C, 6 h.b Turnover frequency (TOF) for the copolymer calculated as mole of VCHO consumed per mole of catalyst per hour (h−1).c Selectivity of PVCHC estimated by IR spectroscopy.d Calculated by 1H NMR spectroscopy.e Determined by GPC. :Cr:PPNCl = 1000:1:2; VCHO = 12.4 g (0.1 mol); 3.0 MPa, 90 °C, 6 h.b Turnover frequency (TOF) for the copolymer calculated as mole of VCHO consumed per mole of catalyst per hour (h−1).c Selectivity of PVCHC estimated by IR spectroscopy.d Calculated by 1H NMR spectroscopy.e Determined by GPC. |
||||||
| 1 | 1 | 74 | 73% | 97.5% | 15000 |
1.30 |
| 2 | 2 | 82 | 68% | 98.0% | 20000 |
1.35 |
| 3 | 3 | 78 | 71% | 99.0% | 19000 |
1.27 |
| 4 | 4 | 13 | 44% | 95.5% | 6000 | 1.26 |
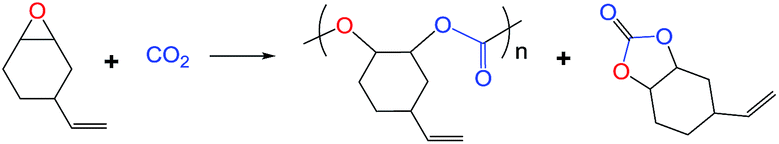
The complex structure also affect the chemoselectivity of the polycarbonate vs. cyclic carbonate. [OSSO] ligand with phenylene bridge complex 4 showed lowest selectivity to the polycarbonate (entry 4, Table 1), whereas complex 1 and 3 gave the comparable selectivity. Not only that, the copolymer obtained from complex 4 possessed lowest molecular weight (MW) and carbonate unit contents in the polymer chain (entry 4, Table 1). As indicated from the 1H NMR spectrum (Fig. S1 in ESI†) and Table 1, the copolymer achieved from complex 3 with ethylene bridge possessed the almost perfectly alternating structure and higher MW. These results indicates that [OSSO]CrX complex with electron-withdrawing framework is unfavorable for the better catalytic performance from the viewpoints of activity and resulting polymer structure. The worse selectivity to polycarbonate vs. cyclic carbonate in the case of [OSSO]CrX-based catalytic system compared to its salanCrX analogue can be attributed to weaker interaction of growing anionic polymer chain with higher electron-rich Cr center as a result of coordination to higher electron-donating soft sulfur atoms in [OSSO]CrX complexes, resulting in a higher propensity to backbite.
Pioneering works revealed that the catalytic activity in the copolymerization of epoxides with CO2 are drastically dependent on the cocatalyst type and loading.24 Thus, the cocatalyst often used in the salenMX catalytic system including DMAP, PPNX (X = Cl, N3), and TABCl were selected to estimate their efficiency for the copolymerization of VCHO and CO2 in neat VCHO at 90 °C within 6 h. It is found that DMAP afforded the copolymer with the lowest yield and the copolymer with lowest MW, while PPNX (X = Cl or N3) onium salt as cocatalyst displayed the highest catalytic activity at same conditions (Table 2). The observation of the catalytic activity consists with the metal salen complexes-based catalyst system for the copolymerization of epoxides and CO2,6a,23,24e,h,25 the stronger interaction between alkylammonium cations with hard anions than their PPN counterparts could account for the less activity for the TBACl than that of PPNCl.6a,24e,h
| Entry | 1/cocatal | TOFb | Selectc (%) | Mnd (g mol−1) | PDId |
|---|---|---|---|---|---|
| a Reaction conditions: [VCHO]:complex 1:cocatalyst = 1000:1:2 (entries 1–4); VCHO = 12.4 g (0.1 mol); 2.0 MPa, 90 °C, 6 h.b Turnover frequency (TOF) for the copolymer calculated as mole of VCHO consumed per mole of catalyst per hour (h−1).c Selectivity of PVCHC estimated by IR spectroscopy.d Determined by GPC. “—” not measured. |
|||||
| 1 | PPNCl(1/2) | 58 | 73 | 9000 | 1.49 |
| 2 | PPNN3(1/2) | 61 | 68 | 12000 |
1.28 |
| 3 | DMAP(1/2) | 15 | 86 | 6000 | 1.43 |
| 4 | TABCl(1/2) | 30 | 67 | 10000 |
1.24 |
| 5 | PPNCl(1/1) | 38 | 74 | — | — |
| 6 | PPNCl(1/3) | 36 | 52 | — | — |
As evidenced from Table 2, an increase in catalytic activity is noted for 2 equiv. of quaternary organic salts over 1 equiv. (entries 1 and 5 in Table 2). This is in contrast to what is observed in previous studies7b utilizing its analogue of salanCrX as catalyst, where 1 equiv. of cocatalyst optimizes the rate of copolymer formation. Further increase the PPNCl/complex 1 ratio to 3 result in the poor selectivity to polycarbonate. This is presumably due to the facile displacement of anionic growing polymer chain from active metal center by competitive coordination of nucleophilic cocatalyst moieties promoted at higher cocatalyst concentration, leading to a higher tendency to form cyclic carbonate via backbiting reaction.8e
The role of cocatalyst in salen or salan complex-based binary catalytic systems for epoxides/CO2 copolymerization have been investigated and found that the Lewis base or anion of onium salt can coordinate to the active metal center and remarkably improve the catalytic activity of the obtained copolymers by weakening the chromium nucleophile bond with the trans effect, which benefits the epoxide coordination and then ring-opening via a nucleophile attack.24e We speculated that cocatalyst employed here play a similar function in a similar manner, that is, which could also coordinate to the central metal site of [OSSO]CrX molecule to form six-coordinated chromium species. This speculation is supported by the shift of azide stretching vibration (ν(N3)) of PPNN3 in the presence of complex 1.
As revealed by Fig. 2, it was found that ν(N3) appeared at 2003 cm−1 for the PPNN3, while the ν(N3) mainly shifted to 2056 cm−1 when one equiv. of complex 1 was added into PPNN3 CH2Cl2 solution, indicating that the N3− coordinate to chromium to generate six-coordinated chromium species. The analogous interaction was occurred between PPNCl and complex 2, where a peak at 2103 cm−1 characteristic of ν(N3) of complex 2 shifted to around 2061 cm−1 for the mixture of one equiv. of PPNCl and complex 2 (see Fig. S2†). A proposed interaction between cocatalyst PPNY (Y = Cl or N3) and [OSSO]CrX (X = Cl or N3) is as shown in Scheme 1. Furthermore, a shift to a lower frequency of the ν(N3) vibration of complex 2 occurred with the addition of DMAP.22 All cases evidenced that both Lewis base and onium salts can coordinate to chromium sites of [OSSO]CrX complexes. Indeed, chromium(III) complexes have a tendency to form six-coordination species as demonstrated by salen,24b,e,26 porphyrin,27 tetramethyltetraazaannulene,9 and N,N′-bis(trifluoroacetylacetone)-1,2-diminine chromium complexes.28 Additionally, blank experiments with complex 1 or PPNCl as the sole initiator for the copolymerization of VCHO and CO2 have been carried out at the VCHO/initiator = 1000:1, 3.0 MPa, and 90 °C for 6 h. The catalytic results show that no polymeric products are produced. Nonetheless, the combination of complex 1 and one of the four co-catalysts (DMAP, PPNCl, PPNN3 and n-Bu4NCl) under the same reaction condition endows the effective copolymerization (entries 1–6 in Table 2). This results further suggest that the real catalytically activity species is the six-coordinate chromium center as proposed in Scheme 1.
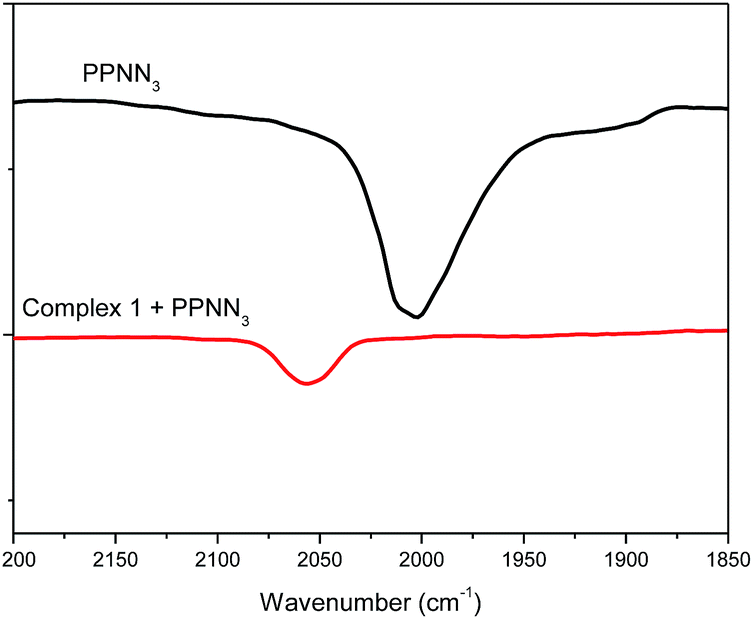 | ||
| Fig. 2 IR spectra of PPNN3 and the mixture of PPNN3/complex (1:1). | ||
 | ||
| Scheme 1 Proposed mechanism of interaction of PPNY/[OSSO]CrX (X, Y = Cl or N3). | ||
According to the observations above-described, a reaction pathway is presumed via a coordination–insertion mechanism as illustrated in Scheme 2. First, the epoxides coordinate to the six-coordinated chromium center and then the metal alkoxide (M–OR) intermediate is formed by ring-opening of the epoxide. Subsequently, CO2 insert into the M–OR bond through a nucleophilic attack to form a metal carbonate intermediate. This metal carbonate intermediate will react with the epoxides to regenerate the M–OR bond again. Therefore, the copolymerization occurs by the continual cycling between the metal alkoxide and carbonate intermediates. If the presence of the active proton compound such as trace H2O in the reaction system, chain transfer reaction will be taken place. This proposal is further evidenced by the presence of weak proton signal at 4.54 ppm and broad signals δ = 3.90 and 3.75 ppm, which indicate chains end-capped with hydroxyl groups,20 arising by chain transfer reaction with the trace water in the reaction system.
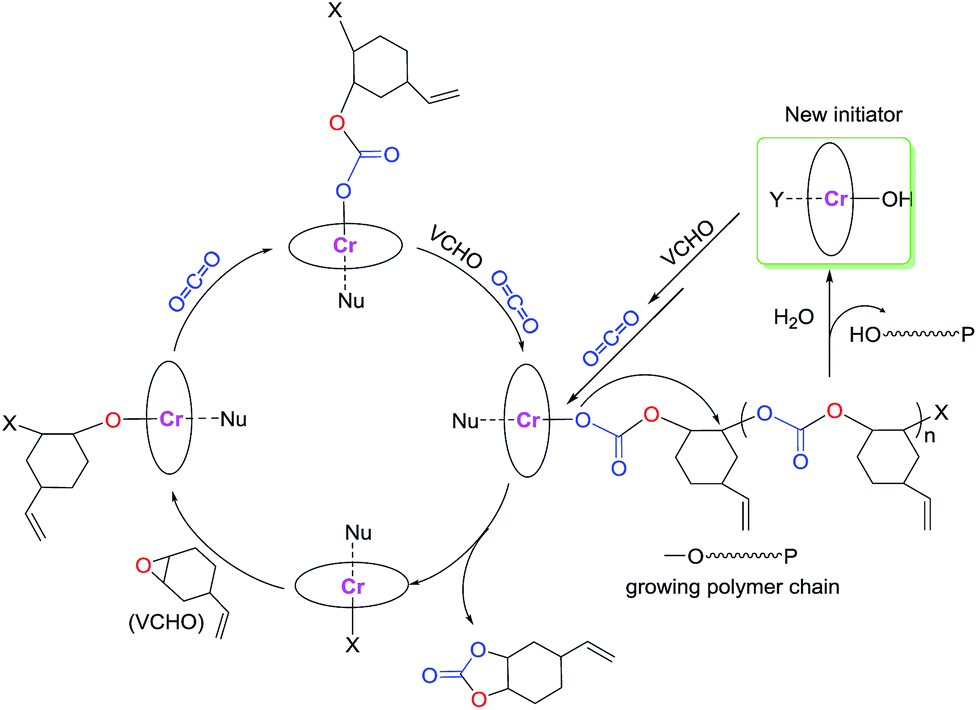 | ||
| Scheme 2 Proposed mechanism for the copolymerization of VCHO with CO2. | ||
To get insight of resulting PVCHC microstructure, the 13C-NMR spectroscopy was recorded. As shown in Fig. 3, the resulting PVCHC presents complex signals in the carbonate region ranging from 153.0 ppm to 153.5 ppm of the 13C NMR spectrum, which is well agreement with the observation of PVCHC prepared by the iron complex-catalyzed the copolymerization of VCHC and CO2.20 As analyzed by Pescarmona et al.,20 the resultant polycarbonate is atactic (Fig. 3). This speculation is further evidenced by the fact of no presence of melting point while only glass transition temperature is present at about 110.6 °C (see Fig. S3†).
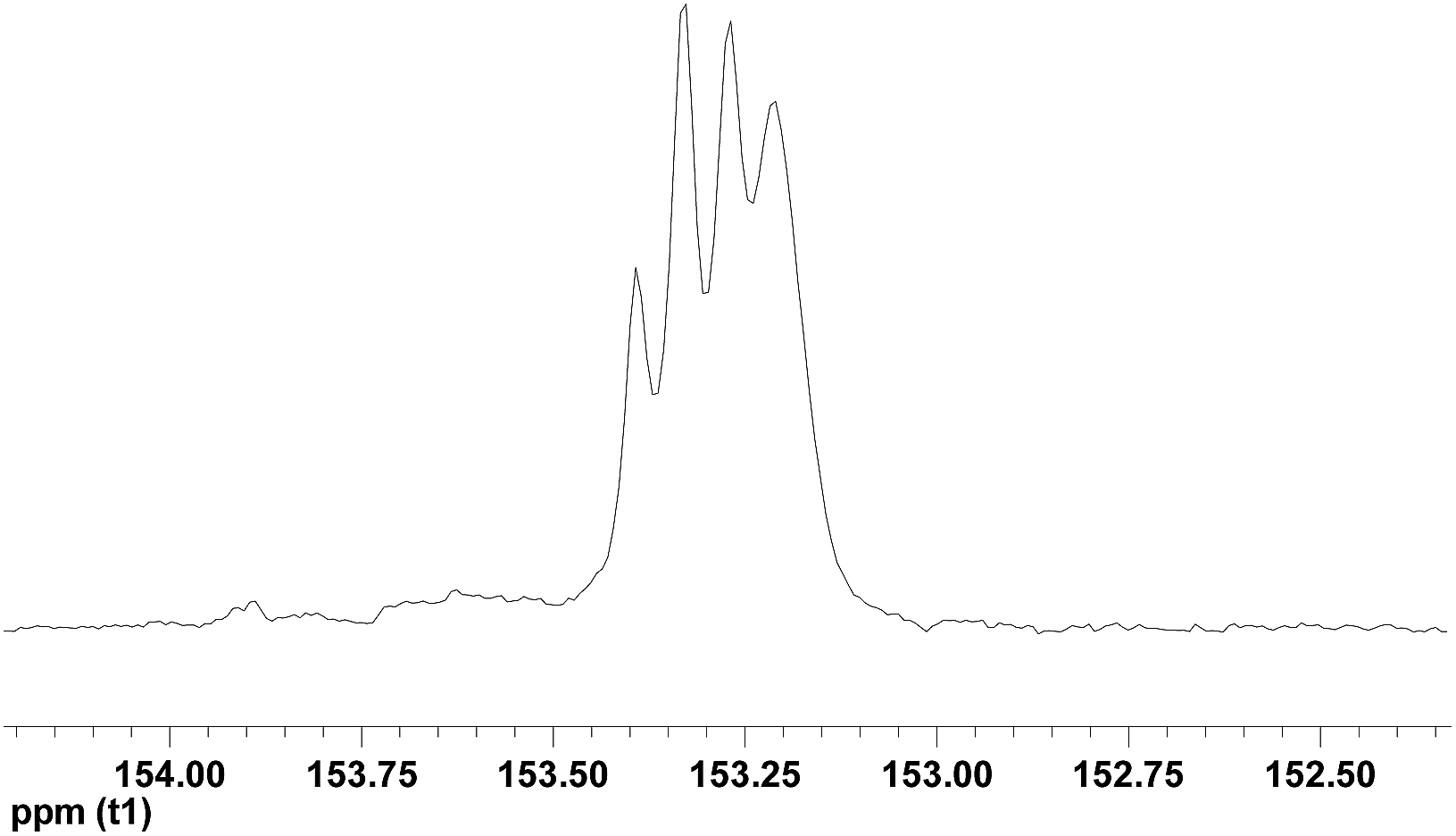 | ||
| Fig. 3 The carbonate region of 13C NMR spectrum of PVCHC (Table 1, entry 1). | ||
The molecular weight (MW) values of resulting copolymers are lower than the theoretical expected MW and GPC curves show a bimodal distribution (see Fig. S4†). This observation is consistent with the above proposal of the chain transfer reaction. Indeed, this phenomenon has been demonstrated to be a general one for a variety of metal complex-catalyzed the copolymerization of CO2 and epoxides.6a,9,24a,29 Although GPC curves show a bimodal distribution, the Mn values display a linear relationship with the VCHO conversion as illustrated in Fig. S5.† The case suggests that the copolymerization proceeds with a controlled manner.
To further optimize the reactivity of this catalyst system, the effect of variables including temperature, CO2 pressure, reaction time, molar ratio of monomer to catalyst on the copolymerization were investigated in the presence of PPNCl as shown in Table 3. It is found that the temperature over or below 90 °C results in the lower polymerization rate (44%, 61% and 56% of VCHO conversion for 80 °C, 90 °C, and 100 °C, respectively). The selectivity to copolymer varies from 74 to 61% with the temperature change from 80 to 100 °C (entries 1–3, Table 3). At the identified conditions, CO2 pressure apparently influences the catalytic activity and reaction rate, the higher pressure is beneficial for the copolymerization process (entries 2, 10, and 11, Table 3), whereas the pressure has a slight effect on the selectivity of the copolymer, especially when the pressure is above 1.5 MPa. The molar ratio of VCHO to [OSSO]CrX complex also considerably affect the copolymerization. It was found that TOF value for copolymers is up to 134 h−1 at a molar ratio of VCHO to complex 1 of 500:1 in 2 h (entry 9, Table 3), even at higher VCHO to complex 1 molar ratio at 1000:1, the TOF for copolymer still reaches to 114 h−1 (entry 8, Table 3) in a same reaction period, further increasing VCHO/complex 1 molar ratio to 1500, the reaction rate decreased (entries 2, 4–6 in Table 3). Whereas the coupling reaction of CO2 with terminal epoxides such as propylene oxide (PO) and epichlorohydrin (ECH), exclusive cyclic carbonates were produced at 80 °C and 3.0 MPa with TOF value of 145 h−1 for PO and 136 h−1 for ECH, respectively.
| Entry | P (MPa) | Temp (°C) | Molar ratiob | T (h) | Select.c (%) | TOFd | Mne (g mol−1) | PDIe |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: VCHO (12.4 g, 0.1 mol); complex 1:PPNCl = 1:2 in neat VCHO.b VCHO]/[complex 1.c Selectivity of PVCHC estimated by IR spectroscopy.d Turnover frequency (TOF) for the copolymer calculated as mole of VCHO consumed per mole of catalyst per hour (h−1).e Determined by GPC. “—” not measured. |
||||||||
| 1 | 3.0 | 80 | 1000:1 |
6 | 74 | 54 | 10000 |
1.26 |
| 2 | 3.0 | 90 | 1000:1 |
6 | 73 | 74 | 15000 |
1.30 |
| 3 | 3.0 | 100 | 1000:1 |
6 | 61 | 57 | 14700 |
1.30 |
| 4 | 3.0 | 90 | 1500:1 |
6 | 70 | 46 | — | — |
| 5 | 3.0 | 90 | 800:1 |
6 | 72 | 69 | — | — |
| 6 | 3.0 | 90 | 500:1 |
6 | 69 | 58 | — | — |
| 7 | 3.0 | 90 | 1000:1 |
3 | 71 | 93 | 9000 | 1.48 |
| 8 | 3.0 | 90 | 1000:1 |
2 | 65 | 114 | 7000 | 1.28 |
| 9 | 3.0 | 90 | 500:1 |
2 | 79 | 134 | 9800 | 1.21 |
| 10 | 1.5 | 90 | 1000:1 |
6 | 73 | 52 | — | — |
| 11 | 1.0 | 90 | 1000:1 |
6 | 68 | 25 | 6000 | 1.26 |
At last, it should be pointed out that [OSSO]CrCl-based catalyst show inferior catalytic activity to the polycarbonate for the copolymerization of CHO with CO2 when compared with its salanCrCl analogue based catalyst. The TOF to copolymer is 39 h−1 for [OSSO]CrCl catalyst, while TOF value for salanCrCl catalyst was up to 240 h−1 and at the same conditions (CHO/catalyst/PPNN3 = 1250/1/2, 60 °C, 3.4 MPa).7b The poorer activity presumedly results from a weaker interaction of the growing anionic polymer chain with a more electron-rich CrIII center arising from the more electron-releasing sulfur donor.
Conclusions
In summary, four chromium complexes with [OSSO] ligands in combination with Lewis base and onium salt have been surveyed as the efficient catalyst for the copolymerization of CO2 and VCHO with TOF up to 134 h−1 and carbonate unit in the polymer chain greater than 95%. Among the catalysts presented in this work, complex supported by [OSSO] ligand with ethylene backbone showed the superior catalytic performance than those of the cyclohexylene- or phenylene-bridged chromium complex. Further investigation are aimed at tailoring the physical and chemical properties of this polymer by free radical-mediated post-polymerization.Acknowledgements
This work was supported by National Natural Science Foundation of China (51373046 and 51473045), Natural Science Foundation of Hebei Province (B2014202013), High-level Excellent Talents in University of Hebei Province. Prof. Xiangqing Li thanks the National Natural Science Foundation of China (21301118) and Innovation Program of Shanghai Municipal Education Commission (No. 15ZZ096) financial supports.Notes and references
- (a) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC; (b) Y. S. Qin and X. H. Wang, Biotechnol. J., 2010, 5, 1164–1180 CrossRef CAS PubMed; (c) D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665–2671 RSC; (d) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed; (e) R.-R. Ang, L. T. Sin, S.-T. Bee, T.-T. Tee, A. A. H. Kadhum, A. R. Rahmat and B. A. Wasmi, J. Cleaner Prod., 2015, 102, 1–17 CrossRef CAS.
- (a) T. Artham and M. Doble, Macromol. Biosci., 2008, 8, 14–24 CrossRef CAS PubMed; (b) J. Xu, E. Feng and J. Song, J. Appl. Polym. Sci., 2014, 5, 39822–39838 Search PubMed; (c) L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS; (d) J. Feng, R.-X. Zhuo and X.-Z. Zhang, Prog. Polym. Sci., 2012, 37, 211–236 CrossRef CAS; (e) S. Tempelaar, L. Mespouille, O. Coulembier, P. Dubois and A. P. Dove, Chem. Soc. Rev., 2013, 42, 1312–1336 RSC.
- (a) S. Inoue, H. Koinuma and T. Tsuruta, Makromol. Chem., 1969, 130, 210–220 CrossRef CAS; (b) S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS.
- (a) G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed; (b) M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC; (c) X.-B. Lu, W.-M. Ren and G.-P. Wu, Acc. Chem. Res., 2012, 45, 1721–1735 CrossRef CAS PubMed; (d) X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC; (e) P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC.
- Y. Wang, Y. Qin, X.-H. Wang and F.-S. Wang, ACS Catal., 2015, 5, 393–396 CrossRef CAS.
- (a) K. Nakano, M. Nakamura and K. Nozaki, Macromolecules, 2009, 42, 6972–6980 CrossRef CAS; (b) Y. Wang, Y. Qin, X.-H. Wang and F.-S. Wang, Catal. Sci. Technol., 2014, 4, 3964–3972 RSC.
- (a) B. Li, G.-P. Wu, W.-M. Ren, Y.-M. Wang, D.-Y. Rao and X.-B. Lu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6102–6113 CrossRef CAS; (b) D. J. Darensbourg, M. Ulusoy, O. Karroonnirum, R. R. Poland, J. H. Reibenspies and B. Çetinkaya, Macromolecules, 2009, 42, 6992–6998 CrossRef CAS.
- (a) H. Sugimoto, H. Ohshima and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3549–3555 CrossRef CAS; (b) W. Wu, Y. S. Qin, X. H. Wang and F. S. Wang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 493–498 CrossRef CAS; (c) H. Sugimoto and K. Kuroda, Macromolecules, 2008, 41, 312–317 CrossRef CAS; (d) X. Jiang, F. L. Gou and H. W. Jing, J. Catal., 2014, 313, 159–167 CrossRef CAS; (e) C. Chatterjee and M. H. Chisholm, Inorg. Chem., 2012, 51, 12041–12051 CrossRef CAS PubMed.
- D. J. Darensbourg and S. B. Fitch, Inorg. Chem., 2007, 46, 5474–5476 CrossRef CAS PubMed.
- (a) M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1998, 120, 11018–11019 CrossRef CAS; (b) D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS PubMed; (c) M. Cheng, D. R. Moore, J. J. Reczek, B. M. Chamberlain, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 8738–8749 CrossRef CAS PubMed; (d) N. M. Rajendran, A. Haleel and N. D. Reddy, Organometallics, 2014, 33, 217–224 CrossRef CAS; (e) M. W. Lehenmeier, S. Kissling, P. T. Altenbuchner, C. Bruckmeier, P. Deglmann, A.-K. Brym and B. Rieger, Angew. Chem., Int. Ed., 2013, 52, 9821–9826 CrossRef CAS PubMed.
- (a) B. Y. Lee, H. Y. Kwon, S. Y. Lee, S. J. Na, S. Han, H. Yun, H. Lee and Y.-W. Park, J. Am. Chem. Soc., 2005, 127, 3031–3037 CrossRef CAS PubMed; (b) T. Bok, H. Yun and B. Y. Lee, Inorg. Chem., 2006, 45, 4228–4237 CrossRef CAS PubMed.
- (a) Y. L. Xiao, Z. Wang and K. Ding, Chem.–Eur. J., 2005, 11, 3668–3678 CrossRef CAS PubMed; (b) Y.-Z. Hua, X.-C. Yang, M.-M. Liu, X. X. Song, M.-C. Wang and J.-B. Chang, Macromolecules, 2015, 48, 1651–1657 CrossRef CAS.
- (a) J. Gurnham, S. Gambarotta, I. Korobkov, L. Jasinska-Walc and R. Duchateau, Organometallics, 2014, 33, 4401–4409 CrossRef CAS; (b) A. Buonerba, A. D. Nisi, A. Grassi, S. Milione, C. Capacchione, S. Vagin and B. Rieger, Catal. Sci. Technol., 2015, 5, 118–123 RSC.
- G. S. Si, L. Zhang, B. Han, Z. Y. Duan, B. Q. Li, J. C. Dong, X. Q. Li and B. Y. Liu, Polym. Chem., 2015, 6, 6372–6377 RSC.
- (a) N. Nakata, T. Toda and A. Ishii, Polym. Chem., 2011, 2, 1597–1610 RSC; (b) J.-C. Buffet, A. N. Martin, M. Kolb and J. Okuda, Polym. Chem., 2011, 2, 2378–2384 RSC; (c) N. Nakata, T. Toda, T. Matsuo and A. Ishii, Inorg. Chem., 2012, 51, 274–281 CrossRef CAS PubMed; (d) C. Costabile, C. Capacchione, D. Saviello and A. Proto, Macromolecules, 2012, 45, 6363–6370 CrossRef CAS; (e) A. Kapelski, J.-C. Buffet, T. P. Spaniol and J. Okuda, Chem.–Asian J., 2012, 7, 1320–1330 CrossRef CAS PubMed; (f) A. Stopper, J. Okuda and M. Kol, Macromolecules, 2012, 45, 698–704 CrossRef CAS; (g) A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC; (h) E. Luciano, F. D. Monica, A. Buonerba, A. Grassi, C. Capacchione and S. Milione, Polym. Chem., 2015, 6, 4657–4668 RSC; (i) A. Ishii, K. Asajima, T. Toda and N. Nakata, Organometallics, 2011, 30, 2947–2956 CrossRef CAS.
- (a) D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS; (b) S. Huijser, E. H. Nejad, R. Sablong, C. de Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS; (c) N. J. V. Zee and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 2665–2668 CrossRef PubMed; (d) R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS PubMed.
- S. Paul, Y. Q. Zhu, C. Romain, R. Brook, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC and references therein.
- (a) B. Y. Liu, C. Y. Tian, L. Zhang, W. D. Yan and W. J. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6243–6251 CrossRef CAS; (b) B. Y. Liu, Y. H. Gao, X. Zhao, W. D. Yan and X. H. Wan, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 359–365 CrossRef CAS; (c) Z. Y. Duan, X. Y. Wang, Q. Gao, L. Zhang, B. Y. Liu and I. Kim, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 789–795 CrossRef CAS; (d) B. Y. Liu, X. Zhao, H. F. Guo, Y. H. Gao, M. Yang and X. H. Wang, Polymer, 2009, 50, 5071–5075 CrossRef CAS; (e) D. Tian, B. Y. Liu, Q. Y. Gan, H. R. Li and D. J. Darensbourg, ACS Catal., 2012, 2, 2029–2035 CrossRef CAS.
- (a) J. F. Zhang, W. M. Ren, X. K. Sun, Y. Meng, B. Y. Du and X. H. Zhang, Macromolecules, 2011, 44, 9882–9886 CrossRef CAS; (b) A. E. Cherian, F. C. Sun, S. S. Sheiko and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11350–21135 CrossRef CAS PubMed.
- M. Taherimehr, J. P. Serta, A. W. Kleij, C. J. Whiteoak and P. P. Pescarmona, ChemSusChem, 2015, 8, 1034–1042 CrossRef CAS PubMed.
- D. J. Darensbourg and S. B. Fitch, Inorg. Chem., 2008, 47, 11868–11878 CrossRef CAS PubMed.
- T.-J. Hsu and C.-S. Tan, Polymer, 2002, 43, 4535–4543 CrossRef CAS.
- K. D. Demadis, T. J. Meyer and P. S. Whitee, Inorg. Chem., 1998, 37, 3610–3619 CrossRef CAS PubMed.
- (a) C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869–10878 CrossRef CAS PubMed; (b) D. J. Darensbourg and A. L. Phelps, Inorg. Chem., 2005, 44, 4622–4629 CrossRef CAS PubMed; (c) X. B. Lu and Y. Wang, Angew. Chem., Int. Ed., 2004, 43, 3574–3577 CrossRef CAS PubMed; (d) D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers and A. L. Phelps, Inorg. Chem., 2004, 43, 1831–1833 CrossRef CAS PubMed; (e) D. J. Darensbourg, R. M. Mackiewicz and J. L. Rodgers, J. Am. Chem. Soc., 2005, 127, 14026–14038 CrossRef CAS PubMed; (f) D. J. Darensbourg and D. R. Billodeaux, Inorg. Chem., 2005, 44, 1433–1442 CrossRef CAS PubMed; (g) Y. S. Niu, W. X. Zhang, X. Pang, X. S. Chen, X. L. Zhuang and X. B. Jing, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5050–5056 CrossRef CAS; (h) X. B. Lu, L. Shi, Y. M. Wang, R. Zhang, Y. J. Zhang, X. J. Peng, Z. C. Zhang and B. Li, J. Am. Chem. Soc., 2006, 128, 1664–1674 CrossRef CAS PubMed.
- C. T. Cohen and G. W. Coates, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5182–5191 CrossRef CAS.
- D. J. Darensbourg and A. I. Moncada, Inorg. Chem., 2008, 47, 10000–10008 CrossRef CAS PubMed.
- C. Chatterjee and M. H. Chisholm, Inorg. Chem., 2012, 51, 12041–12052 CrossRef CAS PubMed.
- (a) D. J. Darensbourg and E. B. Frantz, Inorg. Chem., 2008, 47, 4977–4987 CrossRef CAS PubMed; (b) D. J. Darensbourg and E. B. Frantz, Dalton Trans., 2008, 45, 5031–5036 RSC.
- (a) J. Gurnham, S. Gambarotta, I. Korobkov, L. Jasinska-Walc and R. Duchateau, Organometallics, 2014, 33, 4401–4409 CrossRef CAS; (b) K. Nakano, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2006, 45, 7274–7277 CrossRef CAS PubMed; (c) H. Sugimoto, H. Ohtsuka and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4172–4186 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra27373k |
| This journal is © The Royal Society of Chemistry 2016 |