
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed phosphonyldifluoromethylation of alkenes with bromodifluoromethylphosphonate†
Zhang
Feng
,
Yu-Lan
Xiao
and
Xingang
Zhang
*
Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: xgzhang@mail.sioc.ac.cn; Fax: (+86)-21-6416-6128; Tel: (+86)-21-5492-5333
First published on 29th January 2016
Abstract
An efficient palladium-catalyzed phosphonyldifluoromethylation of alkenes with bromodifluoromethylphosophonate is described. The method provides a facile access to a series of phosphonyldifluoromethylated alkenes that are of interest in medicinal chemistry. Mechanistic studies reveal that a phosphonyl difluouromethyl radical is involved in the reaction.
 Xingang Zhang | Xingang Zhang is a research professor in the Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences (CAS). He graduated in 1998 from Sichuan University and received a Ph.D. in 2003 at SIOC, CAS. After his postdoctoral work at the University of Illinois at Urbana Champaign (UIUC), he joined the faculty of SIOC as a research associate professor in 2008, and became a research professor in 2012. His current research interests are focused on organofluorine chemistry. He received the Thieme Chemistry Journal Award 2014, the National Science Fund for Distinguished Young Scholars 2014, the Fifth Chinese Chemical Society-Royal Society of Chemistry Award for Outstanding Young Chemist (2015), and the 2015 RSC Fluorine Chemistry Prize. |
Efficient and straightforward methods for the synthesis of fluorinated compounds are increasingly important in the discovery of new biologically active molecules and advanced functional materials. Over the past few years, considerable efforts have been made in the fluoroalkylation of arenes,1 however efficient strategies for the synthesis of alkenes bearing fluorinated functional groups are less studied.2 Very recently, we developed an efficient and straightforward method for the preparation of fluoroalkylated alkenes through palladium-catalyzed Heck-type reaction of fluoroalkyl halides.3 However, the reaction of bromodifluoromethylphosphonate (BrCF2PO(OEt)2) 1avia such a strategy remains a synthetic challenge because of the significant hydrodebromination of 1a. In this study, we describe the discovery and development of the reaction that meet this challenge.
On the other hand, organic molecules containing the CF2PO(OR)2 group constitute a distinct class of fluorinated compounds due to their important applications in medicinal chemistry.4 For instance, difluoromethylphosphonates in which the hydrolysable P–O bonds are replaced by a stable P–CF2 bond are the best mimics of the phosphonate esters, thus providing a useful tool for the drug discovery and development.5 Conceptually, incorporation of CF2PO(OR)2 into alkenes could lead to the development of interesting molecules in life sciences because of the unique properties of the CF2PO(OR)2 group and the versatile synthetic utility of the carbon![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) carbon double bond. Unfortunately, efficient methods to access phosphonyldifluoromethylated alkenes are very limited. Previously, such a structural motif can be prepared through copper catalyzed cross-coupling between [(di-ethoxyphosphinyl)difluoromethyl]zinc bromides and alkenyl halides.6 However, the requirement of multi-steps to prepare alkenyl halides and the use of the shelf-unstable difluoroalkylated zinc reagent restrict its wide-spread synthetic applications. The radical addition of alkynes with a phosphonyldifluoromethyl radical is an alternative strategy to produce phosphonyldifluoromethylated alkenes.6 But a Z/E mixture was obtained. Therefore, it is of great interest to develop a new and straightforward method to prepare such a valuable structural motif.
carbon double bond. Unfortunately, efficient methods to access phosphonyldifluoromethylated alkenes are very limited. Previously, such a structural motif can be prepared through copper catalyzed cross-coupling between [(di-ethoxyphosphinyl)difluoromethyl]zinc bromides and alkenyl halides.6 However, the requirement of multi-steps to prepare alkenyl halides and the use of the shelf-unstable difluoroalkylated zinc reagent restrict its wide-spread synthetic applications. The radical addition of alkynes with a phosphonyldifluoromethyl radical is an alternative strategy to produce phosphonyldifluoromethylated alkenes.6 But a Z/E mixture was obtained. Therefore, it is of great interest to develop a new and straightforward method to prepare such a valuable structural motif.
We began this study by choosing diethyl (bromodifluoromethyl)phosphonate 1a and styrene 2a as model substrates (Table 1). Initially, the reaction carried out in the presence of Pd(MeCN)2Cl2 (10 mol%), Xantphos (20 mol%), and K2CO3 (2.0 equiv.) in DCE at 80 °C failed to provide product 3aa (entry 1). Instead, a significant amount of hydrodebrominated HCF2PO(OEt)2 and other uncertain byproducts were formed. Further optimization of reaction conditions through a survey of different reaction parameters, such as palladium salts, ligands, and solvents, also led to negative results (for details, see the ESI†). Taking into account the fact that a fluoroalkyl radical via a single-electron-transfer (SET) pathway is involved in the palladium-catalyzed reaction of fluoroalkyl halides with alkenes,3 we envisioned that these negative results may arise from the formation of a difluoromethylene phosphonate radical in situ through a Pd(0) induced SET pathway, and such a difluoroalkylated radical is too reactive and prone to generation of the above by-products. Thus, if the resulting difluoroalkyl radical can be stabilized by the steric effect7 through the replacement of the ethyl group of 1a with a bulky group, it will facilitate the production of the desired product 3a. Accordingly, sterically hindered substrates, such as dibutyl (bromodifluoromethyl)phosphonate 1b and diisopropyl (bromodifluoromethyl)phosphonate 1c were synthesized. To our delight, a 12% yield of 3ab was obtained when 1b was used (entry 2). However, the hydrodebrominated HCF2PO(OEt)2 and other side products were still observed. Switching 1b with a more sterically hindered substrate 1c led to an increased yield (21%) (entry 3). Encouraged by these results, a survey of the reaction parameters, such as palladium sources, bases, solvents, and ligands, was conducted. Among the tested palladium salts (entries 4–9), Pd(PhCN)2Cl2 was the best catalyst and provided 3ac in 41% yield (entry 4). Switching the base from K2CO3 to K3PO4 increased the yield of 3ac to 50% (entry 10). But other bases led to lower yield or no product (entries 11 and 12). The reaction was very sensitive to the solvent and ligand, and the combination of DCE and Xantphos was still the best choice (entry 10, for details, see the ESI†). Finally, the optimal reaction conditions were identified by increasing the reaction temperature to 120 °C with a utility of 5 mol% of Pd(PhCN)2Cl2, providing 3ac in 92% yield upon isolation (entry 14). No product was observed in the absence of the palladium catalyst or Xantphos (entries 15 and 16), thus demonstrating the pivotal role of Pd/Xantphos in promotion of the reaction.
|
|
||||
|---|---|---|---|---|
| Entry | 1 | [Pd] (X) | Base | 3a, yieldb (%) |
| a Reaction conditions (unless otherwise specified): 1 (2.0 equiv.), 2a (0.3 mmol, 1.0 equiv.), base (2.0 equiv.), DCE (2 mL), 24 h. b Determined by 19F NMR using fluorobenzene as an internal standard (isolated yield in parentheses). c Reaction ran at 120 °C for 12 h. | ||||
| 1 | 1a | PdCl2(MeCN)2 (10) | K2CO3 | 3aa, trace |
| 2 | 1b | PdCl2(MeCN)2 (10) | K2CO3 | 3ab, 12 |
| 3 | 1c | PdCl2(MeCN)2 (10) | K2CO3 | 3ac, 21 |
| 4 | 1c | PdCl2(PhCN)2 (10) | K2CO3 | 3ac, 41 |
| 5 | 1c | PdCl2 (10) | K2CO3 | 3ac, 39 |
| 6 | 1c | PdCl2(PPh3)2 (10) | K2CO3 | 3ac, 28 |
| 7 | 1c | PdCl2(dppf) (10) | K2CO3 | 3ac, trace |
| 8 | 1c | [PdCl(C3H5)]2 (5) | K2CO3 | 3ac, nd |
| 9 | 1c | Pd(PPh3)4 (10) | K2CO3 | 3ac, 31 |
| 10 | 1c | PdCl2(PhCN)2 (10) | K3PO4 | 3ac, 50 |
| 11 | 1c | PdCl2(PhCN)2 (10) | Cs2CO3 | 3ac, nd |
| 12 | 1c | PdCl2(PhCN)2 (10) | Na2CO3 | 3ac, 44 |
| 13c | 1c | PdCl2(PhCN)2 (10) | K3PO4 | 3ac, (94) |
| 14c | 1c | PdCl2(PhCN)2 (5) | K3PO4 | 3ac, (92) |
| 15c | 1c | None | K3PO4 | 3ac, nd |
| 16c | 1c | PdCl2(PhCN)2 (5) | K3PO4 | 3ac, nd |
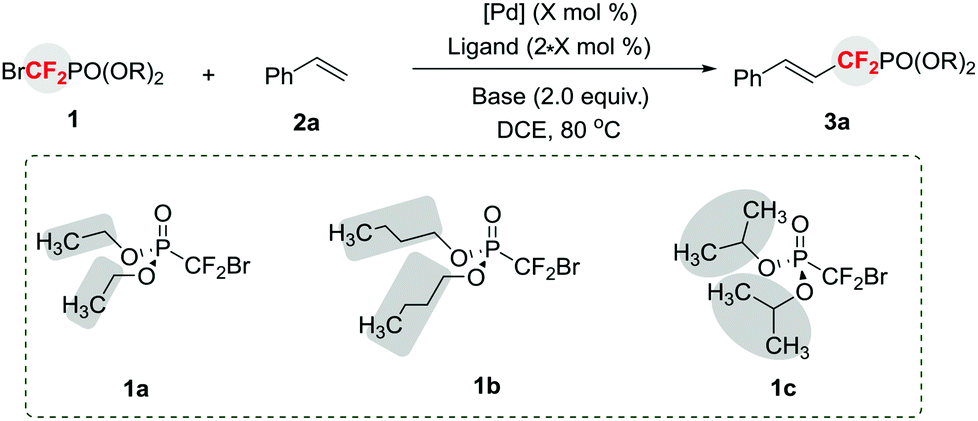
To demonstrate the substrate scope of this method, reactions of 1c with a variety of alkenes were performed and provided 3 in moderate to high yields (Table 2). Generally, aromatic alkenes bearing electron-rich groups afforded higher yields than those substrates bearing electron-deficient groups. Particularly, excellent yields of 3c and 3d were obtained when 4-tert-butylstyrene or 4-methoxystyrene was employed. A variety of versatile functional groups, including base or nucleophile sensitive functionalities, such as cyano, nitro, alkoxycarbonyl, and formyl were compatible with the reaction (3f–3i). Interestingly, a terminally branched alkene bearing an alkyl group solely afforded the double bond migrated product in good yield (61%) (3j), thus providing an efficient way to prepare phosphonyldifluoromethylated allylic compounds. Other branched alkenes, such as cyclic aromatic alkenes, were also applicable to the reaction and provided the corresponding products 3k and 3l in good to excellent yields. It should be mentioned that the conjugated alkene underwent the reaction smoothly without observation of other by-products (3m). In particular, a heterocycle 4-methyl-5-vinylthiazole was also a suitable substrate (3n). The current process can also be extended to enamide and furnish 3o in good yield. Most importantly, coumarin and quinolinone, the important scaffolds of biologically active molecules,8 afforded the corresponding products 3p and 3q in synthetically useful yields, thus providing a straightforward strategy for the discovery of new biologically interesting molecules. However, aliphatic alkenes, such as allyl ethers, cyclic aliphatic alkenes, and acrylate, were not suitable substrates.
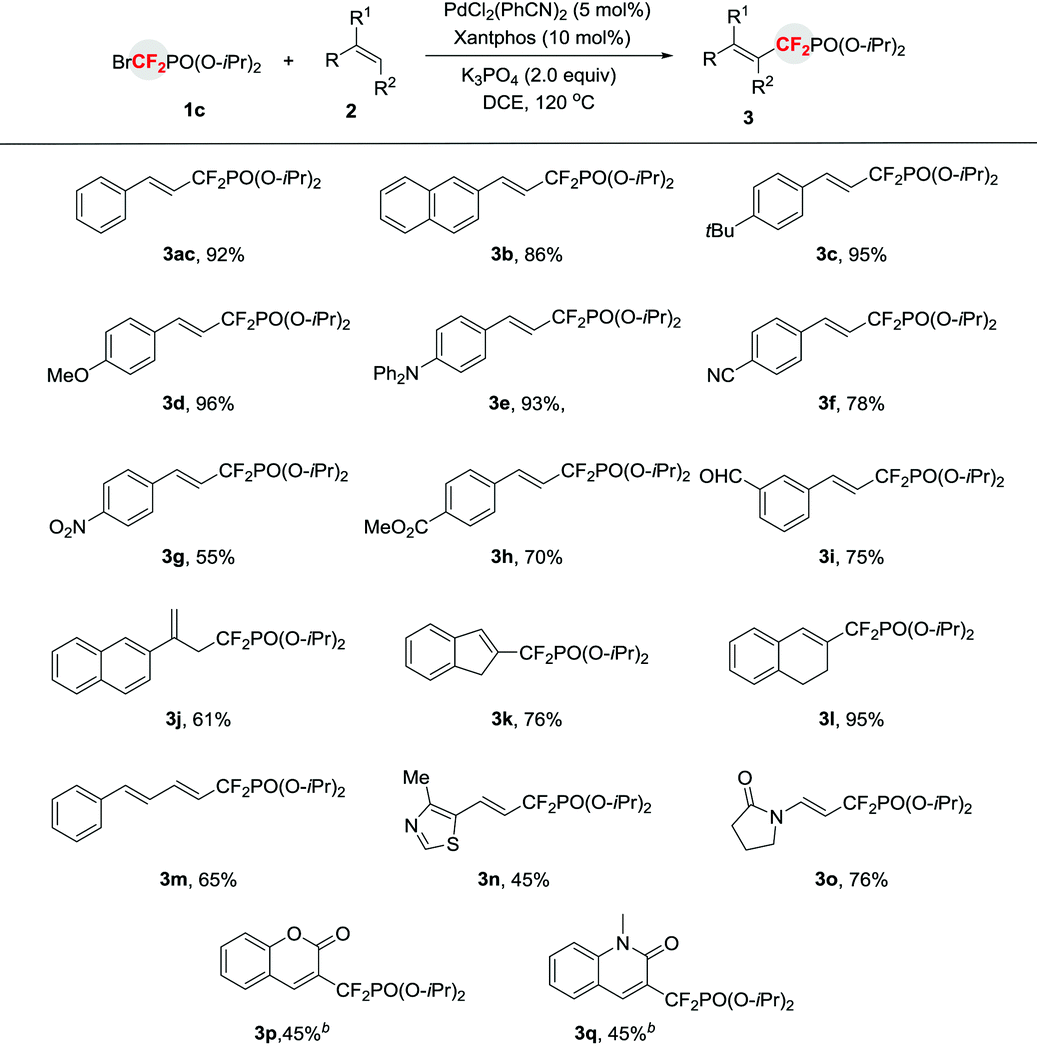
The utility of this reaction can also be demonstrated by the late stage functionalization for the synthesis of phosphonyldifluoromethylated biologically active molecule. As shown in Scheme 1, treatment of the estrone-derived alkene 4 with diisopropyl (bromodifluoromethyl)phosphonate 1c afforded the corresponding phosphonyldifluoromethylated compound 5 in good yield, thus providing an efficient access to fluorinated bioactive molecules.
 | ||
| Scheme 1 Late-stage phosphonyldifluoromethylation in the synthesis of biologically active molecules. | ||
To gain some mechanistic insight into the present reaction, radical inhibition experiments were performed (Scheme 2). When a reaction of 1c with 2a was carried out in the presence of PdCl2(PhCN)2Cl2 (5 mol%), Xantphos (10 mol%) and K3PO4 in DCE, the addition of a ET scavenger 1,4-dinitrobenzene3 or a radical inhibitor hydroquinone dramatically diminished the yield of 3ac, thus implying that a SET pathway via phosphonyldifluoromethyl radical may be involved in the catalytic cycle.
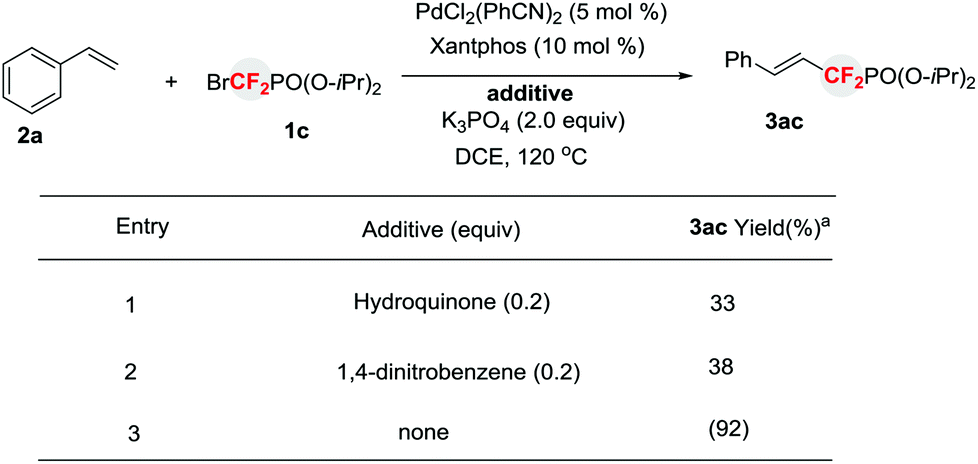 | ||
| Scheme 2 Inhibition experiments for palladium-catalyzed Heck-type reaction of diisopropyl (bromodifluoromethyl)phosphonate 1c with styrene. aDetermined by 19F NMR using fluorobenzene as an internal standard (isolated yield in parentheses). | ||
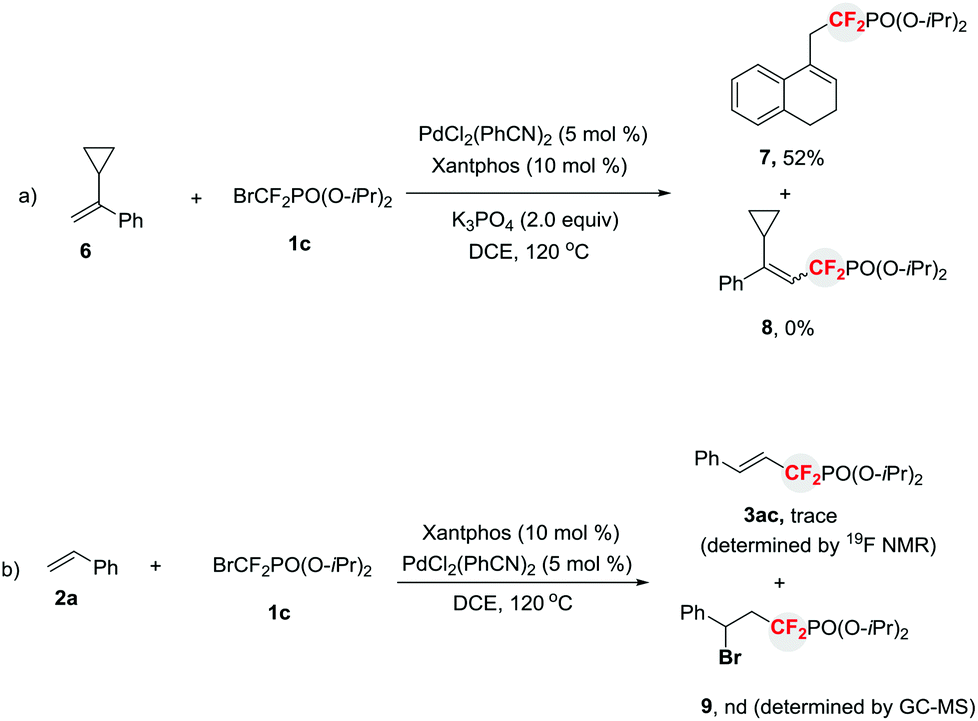
To further confirm that a free difluoroalkyl radical existed in the reaction, a radical clock experiment was conducted. As illustrated in Scheme 3a, a ring-opened compound 7 instead of the Heck-type product 8 was afforded when α-cyclopropylstyrene 69 was treated with 1c under standard reaction conditions. Thus this finding clearly demonstrates that a difluoroalkyl radical is indeed involved in the reaction. In addition, to rule out the possibility that the formation of compounds 3via sequential bromine atom transfer radical addition to alkenes,10 followed by base-assisted elimination of the resulting benzyl bromide, the reaction of 2a with 1c in the presence of PdCl2(PhCN)2 and Xantphos without base was conducted (Scheme 3b). It was found that no benzyl bromide 9 was observed. This result suggests that the base-assisted two-step process for the formation of phosphonyldifluoromethylated alkenes is less likely. This finding is consistent with our previous results, in which a Pd-initiated SET pathway via a Heck-type catalytic cycle is involved in the reaction.3
 | ||
| Scheme 3 Experiments for mechanistic studies. | ||
Conclusions
In conclusion, we have disclosed an efficient method for the preparation of phosphonyldifluoromethylated alkenes through palladium-catalyzed Heck-type reaction with bromodifluoromethylphosphonate. The steric effect of the diisopropyl (bromodifluoromethyl)phosphonate is critical for the reaction efficiency. The significant advantage of this protocol is the synthetic simplicity, thus providing a facile route for application in the discovery of biologically interesting molecules.Acknowledgements
The National Basic Research Program of China (973 Program (no. 2012CB821600 and 2015CB931900)), the NSFC (no. 21425208, 21421002, and 21332010), and SIOC are greatly acknowledged for funding this work.Notes and references
- For selected reviews, see: (a) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (b) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (c) C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929 RSC; (d) T. Besset, C. Schneider and D. Cahard, Angew. Chem., Int. Ed., 2012, 51, 5048 CrossRef CAS PubMed; (e) F.-L. Qing, Chin. J. Org. Chem., 2012, 32, 815 CrossRef CAS.
- T. Besset, T. Poisson and X. Pannecoucke, Chem. – Eur. J., 2014, 20, 16830 CrossRef CAS PubMed.
- Z. Feng, Q.-Q. Min, H.-Y. Zhao, J.-W. Gu and X. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1270 CrossRef CAS PubMed.
- (a) T. R. Burke and K. Lee, Acc. Chem. Res., 2003, 36, 426 CrossRef CAS PubMed; (b) V. D. Romanenko and V. P. Kukhar, Chem. Rev., 2006, 106, 3868 CrossRef CAS PubMed.
- (a) G. M. Blackbum, Chem. Ind., 1981, 134 Search PubMed; (b) G. M. Blackhum, D. E. Kent and F. Kolkmann, J. Chem. Soc., Perkin Trans. I, 1984, 1119 Search PubMed; (c) Z.-Y. Zhang, Acc. Chem. Res., 2003, 36, 385 CrossRef CAS PubMed; (d) S. Mitra and A. M. Barrios, ChemBioChem, 2008, 9, 1216 CrossRef CAS PubMed; (e) P. K. Mandal, W. S.-L. Liao and J. S. McMurray, Org. Lett., 2009, 11, 3394 CrossRef CAS PubMed; (f) Y. Xu and G. D. Prestwich, Org. Lett., 2002, 4, 4021 CrossRef CAS PubMed; (g) G. G. Durgam, T. Virag, M. D. Walker, R. Tsukahara, S. Yasuda, K. Liliom, L. A. van Meeteren, W. H. Moolenaar, N. Wilke, W. Siess, G. Tigyi and D. D. Miller, J. Med. Chem., 2005, 48, 4919 CrossRef CAS PubMed; (h) Y. Xiao and P. Liu, Angew. Chem., Int. Ed., 2008, 47, 9722 CrossRef CAS PubMed; (i) K. Panigrahi, G. A. Applegate, G. Malik and D. B. Berkowitz, J. Am. Chem. Soc., 2015, 137, 3600 CrossRef CAS PubMed; (j) N. Krishnan, K. Krishnan, C. R. Connors, M. S. Choy, R. Page, W. Peti, L. V. Aelst, S. D. Shea and N. K. Tonks, J. Clin. Invest., 2015, 125, 3163 Search PubMed.
- T. Yokomatsu, K. Suemune, T. Murano and S. Shibuya, J. Org. Chem., 1996, 61, 7207 CrossRef CAS PubMed.
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 317 CrossRef CAS.
- (a) P. O. Patil, S. B. Bari, S. D. Firke, P. K. Deshmukh and S. T. Donda, Bioorg. Med. Chem., 2013, 21, 2434 CrossRef CAS PubMed; (b) C. B. M. Poulie and L. Bunch, ChemMedChem, 2013, 8, 205 CrossRef CAS PubMed; (c) J. M. Dickinson, Nat. Prod. Rep., 1993, 10, 71 RSC.
- J. E. Baldwin, Chem. Rev., 2003, 103, 1197 CrossRef CAS PubMed.
- Q.-Y. Chen, Z.-Y. Yang, C.-X. Zhao and Z.-M. Qiu, J. Chem. Soc., Perkin Trans. 1, 1988, 563 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00005c |
| This journal is © the Partner Organisations 2016 |