
Organocatalytic asymmetric conjugate addition of diaryloxazolidin-2,4-diones to nitroolefins: an efficient approach to chiral α-aryl-α-hydroxy carboxylic acids†
Lihui
Jiao‡
a,
Xiaowei
Zhao‡
a,
Huixin
Liu
bc,
Xinyi
Ye
b,
Yun
Li
a and
Zhiyong
Jiang
*a
aKey Laboratory of Natural Medicine and Immuno-Engineering of Henan Province, Henan University, Kaifeng, Henan 475004, People's Republic of China. E-mail: chmjzy@henu.edu.cn
bDivision of Chemistry and Biological Chemistry, Nanyang Technological University, 21 Nanyang Link, 637371, Singapore
cDepartment of Chemistry, Tsinghua University, Beijing, 100084, People's Republic of China
First published on 3rd February 2016
Abstract
The first catalytic asymmetric conjugate addition of diaryloxazolidin-2,4-diones to nitroolefins is described. By employing an L-threonine-based tertiary amine-urea as the catalyst, the reaction proceeded in an excellent enantio- and diastereoselective manner (up to >99% ee and >19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). The adducts could be conveniently transformed to valuable α-aryl-α-hydroxy carboxylic acids, which structurally feature two adjacent hetero-quaternary and tertiary stereogenic centers.
:1 dr). The adducts could be conveniently transformed to valuable α-aryl-α-hydroxy carboxylic acids, which structurally feature two adjacent hetero-quaternary and tertiary stereogenic centers.
Introduction
Enantiomeric pure α-aryl-α-hydroxy carboxylic acids, which feature a tertiary alcohol and an aryl group on the α-position of the carboxylic acid, are privileged structural scaffolds in many natural and medicinally important agents.1 For instance (Fig. 1a), chimonamidine is one of the isolated alkaloids from the seeds of Chimonanthus praecox Link that are employed as folk medicine for the therapy of rheumatoid arthritis in China.1a Convolutamides A–F are γ-lactam alkaloids from the marine bryozoan Amathia convoluta with cytotoxicity against L1210 murine leukemia and KB human epidermoid carcinoma cells.1b Other examples include tribulusamide C,1c mandelamide derivative [inhibitor of hepatitis C virus (HCV) NS5A],1d IQNP [ligand for the muscarinic acetylcholinergic receptor (mAChR)],1e and so on.1f–h Accordingly, the development of synthetic methodologies for the preparation of these valuable entities has attracted great interest of chemists during the past few decades.2–5 To date, great improvements have been made in the addition of nucleophiles to α-ketoesters.2 In addition, the aldol reaction to isatins3 can produce the precursors of α-aryl-α-hydroxy carboxylic acids, namely 3-hydroxy-2-oxindoles; the desired carboxylic acids could be obtained after hydrolysis, but their ortho-substituted amines on the aromatic rings are unavoidable and difficult to modify.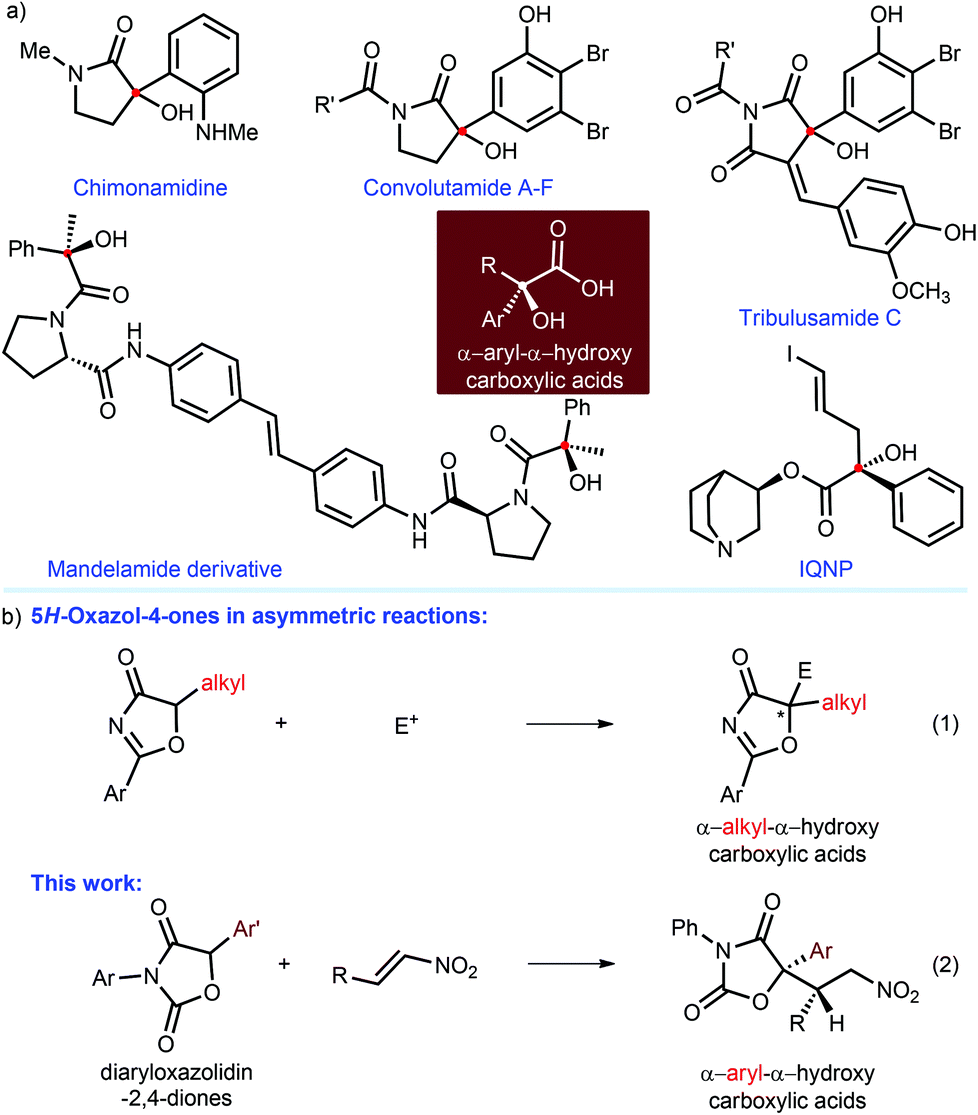 | ||
| Fig. 1 (a) Representative natural and non-natural products. (b) 5H-Oxazol-4-ones in the build of α-alkyl-α-hydroxy carboxylic acids (eqn (1)) and this work (eqn (2)). | ||
In 2004, Trost and co-workers6 introduced 5H-oxazol-4-ones as α-alkyl-α-hydroxy ester surrogates in a highly enantioselective allylic alkylation, thus providing a kind of significant α-alkyl-α-hydroxy carboxylic acids (Fig. 1b, eqn (1)). Since then, catalytic asymmetric reaction of 5H-oxazol-4-ones has been demonstrated as one of the most efficient strategies to build α-alkyl-α-hydroxy carboxylic acids.7,8 Our group have also successively developed a series of organocatalytic asymmetric reactions of 5H-oxazol-4-ones, including conjugate addition to nitroolefins8a and vinyl sulfones,8b Mannich reaction,8c sulfenylation,8d alkylation,8e conjugate addition–protonation8f and [4 + 2] cycloaddition.8f All of these works have demonstrated that 5H-oxazol-4-ones cannot be used to construct α-aryl-α-hydroxy carboxylic acids since 5-aryl-substituted 5H-oxazol-4-ones are very unstable and thus inaccessible. Nonetheless, employing feasible surrogates to access the desired α-aryl-α-tertiary hydroxy carboxylic acids could be recognized as plausible tactics in terms of these contributions.
In 2006, the Maruoka group4 reported a highly enantioselective phase-transfer-catalyzed alkylation of diaryloxazolidin-2,4-diones, in which the alkylated adducts could be conveniently transformed to α-benzyl-α-aryl-α-hydroxy carboxylic acids with satisfactory results. This pioneering work indicated the possibility of diaryloxazolidin-2,4-diones as the surrogates of α-aryl-α-hydroxy carboxylic esters. However, no other example has yet been reported. It is thus highly desirable to develop new reaction patterns involving diaryloxazolidin-2,4-diones, to afford diverse chiral α-aryl-α-hydroxy carboxylic acids with biological targets. Certainly, the unforeseen reactivity and stereoselectivity of diaryloxazolidin-2,4-diones in the unmet reactions should be the two key challenges. In recent years, we were keen on developing asymmetric organocatalytic reactions to build multitudinous chiral tertiary alcohols.3d,8,9 As an extension of these ongoing research efforts, herein, we report a highly enantio- and diastereoselective conjugate addition reaction of diaryloxazolidin-2,4-diones to nitroolefins via a hydrogen-bonding catalysis, leading to an efficient approach to a series of valuable α-aryl-α-hydroxy carboxylic acid derivatives, which contain two adjacent hetero-quaternary and tertiary stereogenic centers (Fig. 1b, eqn (2)).
Results and discussion
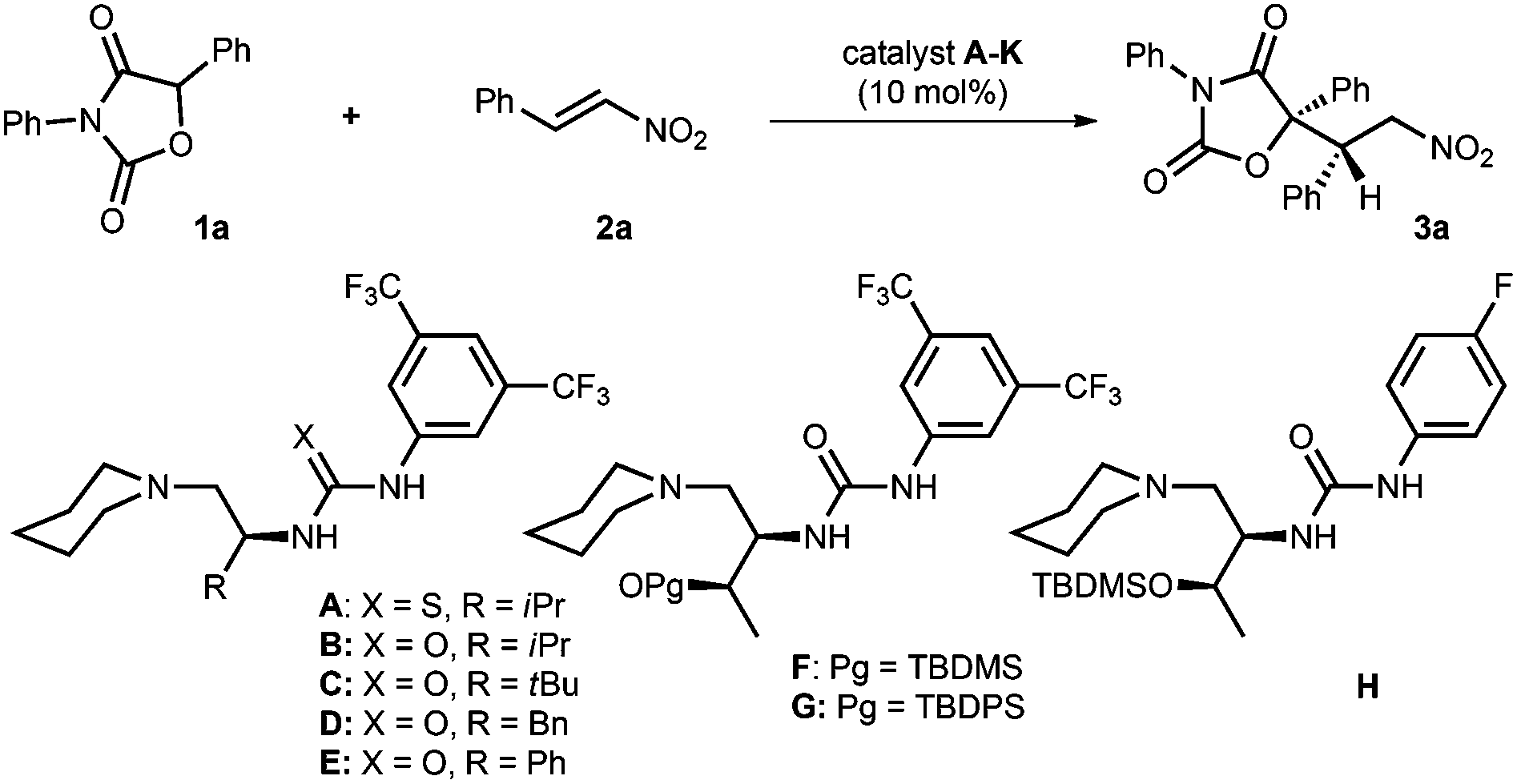
At the outset, the conjugate addition of diphenyloxazolidin-2,4-dione 1a to nitroolefin 2a was chosen as the model reaction to investigate the reaction conditions (Table 1). The reaction was first conducted in CH2Cl2 at 25 °C and in the presence of 10 mol% of L-valine-based tertiary amine-thiourea A as the bifunctional catalyst.10 It was found that the desired adduct 3a could be obtained in 76% yield within 18 hours, but the enantio- and diastereoselectivity were very poor (entry 1). The reactivity (89% yield) and enantioselectivity (31% ee) were improved by utilizing catalyst B which contains urea instead of thiourea as the H-bonding donor, indicating the feasibility of this bifunctional combination in the stereoselective control (entry 2). Accordingly, a series of L-amino acid-based tertiary amine-urea catalysts (C–G) prepared from diverse L-amino acids were subjected to the reactions (entries 3–7); catalyst F with L-threonine as the chiral skeleton and tert-butyldimethylsilyl (TBDMS) as the protective group of alcohol was found to present the best results, in which 3a was obtained in 92% yield with 66% ee and 60:40 dr (entry 6). Noteworthy is that no reaction was detected when catalyst G with a bulkier tert-butyldiphenylsilyl (TBDPS) as the protective group was used (entry 7). Catalyst H bearing 4-fluorophenyl instead of 3,5-di(trifluoromethyl)phenyl as the substituent of urea could slightly increase both the enantioselectivity and diastereoselectivity (68% ee, 68:32 dr, entry 8). Afterwards, a range of solvents, including THF, ether, toluene and m-xylene, were screened in succession (entries 9–12). It was found that m-xylene presented the best results, providing 3a in 99% yield with 71% ee and 74:26 dr (entry 12). The enantioselectivity was further improved to 87% ee by performing the reaction at −20 °C (entry 14). The additive effect was then evaluated (entries 15–17). 4 Å molecular sieves were found to slightly increase the ee and dr values (entry 15). Similar results were obtained when 10 mol% of NaCl was used (entry 16). Finally, 3a with 94% ee and 92:8 dr was attainable when a set of 25 mg of 4 Å molecular sieves and 10 mol% of NaCl were subjected to the reaction (entry 17).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat. | Solvent | T (°C) | t (h) | Yieldb (%) | eec (%) | drc |
| a The reaction was carried out with 0.05 mmol of 1a, 0.06 mmol of 2a and 0.005 mmol of catalyst in 0.5 mL solvent. b Isolated yield. c ee value of major diastereomer determined by HPLC methods. d 10 mg 4 Å molecular sieves (MS) were used. e 0.005 mmol of NaCl were used. f 25 mg 4 Å molecular sieves and 0.005 mmol of NaCl was used. | |||||||
| 1 | A | CH2Cl2 | 25 | 18 | 76 | 2 | 56:44 |
| 2 | B | CH2Cl2 | 25 | 18 | 89 | 31 | 60:40 |
| 3 | C | CH2Cl2 | 25 | 18 | 93 | 20 | 65:35 |
| 4 | D | CH2Cl2 | 25 | 18 | 76 | 19 | 62:38 |
| 5 | E | CH2Cl2 | 25 | 18 | 72 | 28 | 58:42 |
| 6 | F | CH2Cl2 | 25 | 18 | 92 | 66 | 60:40 |
| 7 | G | CH2Cl2 | 25 | 30 | N.R. | N.A. | N.A. |
| 8 | H | CH2Cl2 | 25 | 18 | 95 | 68 | 68:32 |
| 9 | H | THF | 25 | 18 | 85 | 61 | 64:36 |
| 10 | H | Et2O | 25 | 18 | 87 | 64 | 65:35 |
| 11 | H | Toluene | 25 | 12 | 99 | 65 | 71:29 |
| 12 | H | m-Xylene | 25 | 12 | 99 | 71 | 74:26 |
| 13 | H | m-Xylene | 0 | 32 | 64 | 81 | 78:22 |
| 14 | H | m-Xylene | −20 | 60 | 92 | 89 | 89:11 |
| 15d | H | m-Xylene | −20 | 60 | 91 | 92 | 90:10 |
| 16e | H | m-Xylene | −20 | 55 | 91 | 92 | 92:8 |
| 17f | H | m-Xylene | −20 | 55 | 91 | 94 | 92:8 |
With the optimal reaction conditions in hand, we explored the substrate scope involving both diaryloxazolidin-2,4-diones and nitroolefins (Table 2). First, we examined the viability of aryl nitroolefins (2a–m) with diverse steric and electronic properties by using diphenyloxazolidin-2,4-dione 1a as the nucleophile (entries 1–13). The reactions were completed within 96 hours and gave the corresponding adducts 3a–3m in 88–98% yield with 86–96% ee and 8:1 to 19:1 dr. The studies showed that the introduction of methoxy as the electron-donating group on the para-, meta- and ortho-position presented a slightly decreased diastereoselectivity (8:1 to 16:1 dr, entries 8–10). Analogous results were observed for those containing 2-naphthyl (3k) and 2-thienyl (3m) as the aryl groups (7:1 and 8:1 dr, entries 11 and 13). It was found that the α,β,γ,δ-unsaturated nitroolefin 2n was also suitable for the reaction conditions, providing adduct 3n in good enantio- and diastereoselectivity; after a single recrystallization, 99% ee and 99:1 dr of 3n was attainable (entry 14). Nitroolefin 2o containing a cyclohexyl gave the corresponding adduct 3o with a modest ee value yet good dr (50% ee, 15:1 dr, entry 15). Furthermore, other diaryloxazolidin-2,4-diones 1 with different aryl groups on the 5-position provided the corresponding adducts 3p–s in 85 − 91 yields with 85–98% ee and 11:1 to 14:1 dr (entries 16–18). The absolute configurations of the conjugate addition products were assigned based on X-ray crystallographic analysis of a single crystal of 3a.11
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1, Ar | 2, R | 3 | Yieldb (%) | eec (%) | drd |
| a The reaction was carried out with 0.1 mmol of 1, 0.12 mmol of 2a, 0.01 mmol of catalyst F, 0.01 mmol of NaCl and 50 mg 4 Å MS in 1.0 mL solvent. b Isolated yield. c Determined by HPLC methods; ee in parentheses was obtained after single recrystallization. d Determined by 1H NMR analysis. | ||||||
| 1 | 1a, Ph | 2a, Ph | 3a | 98 | 94 (99) | 19:1 |
| 2 | 1a, Ph | 2b, 4-ClPh | 3b | 92 | 94 | 19:1 |
| 3 | 1a, Ph | 2c, 3-ClPh | 3c | 96 | 95 | 10:1 |
| 4 | 1a, Ph | 2d, 2-ClPh | 3d | 92 | 92 | 19:1 |
| 5 | 1a, Ph | 2e, 4-MePh | 3e | 88 | 94 | 18:1 |
| 6 | 1a, Ph | 2f, 3-MePh | 3f | 96 | 90 | 19:1 |
| 7 | 1a, Ph | 2g, 2-MePh | 3g | 95 | 96 | 19:1 |
| 8 | 1a, Ph | 2h, 4-MeOPh | 3h | 96 | 91 | 16:1 |
| 9 | 1a, Ph | 2i, 3-MeOPh | 3i | 98 | 91 | 14:1 |
| 10 | 1a, Ph | 2j, 2-MeOPh | 3j | 95 | 91 | 8:1 |
| 11 | 1a, Ph | 2k, 2-naphthyl | 3k | 95 | 86 | 7:1 |
| 12 | 1a, Ph | 2l, 2-furyl | 3l | 90 | 90 | 16:1 |
| 13 | 1a, Ph | 2m, 2-thienyl | 3m | 92 | 91 | 8:1 |
| 14 | 1a, Ph |
2n, PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– CH– |
3n | 90 | 78 (>99) | 19:1 |
| 15 | 1a, Ph | 2o, Cy | 3o | 59 | 50 | 15:1 |
| 16 | 1b, 4-FPh | 2a, Ph | 3p | 90 | 98 | 14:1 |
| 17 | 1c, 4-ClPh | 2a, Ph | 3q | 89 | 91 | 12:1 |
| 18 | 1d, 4-MeOPh | 2a, Ph | 3r | 91 | 85 | 11:1 |
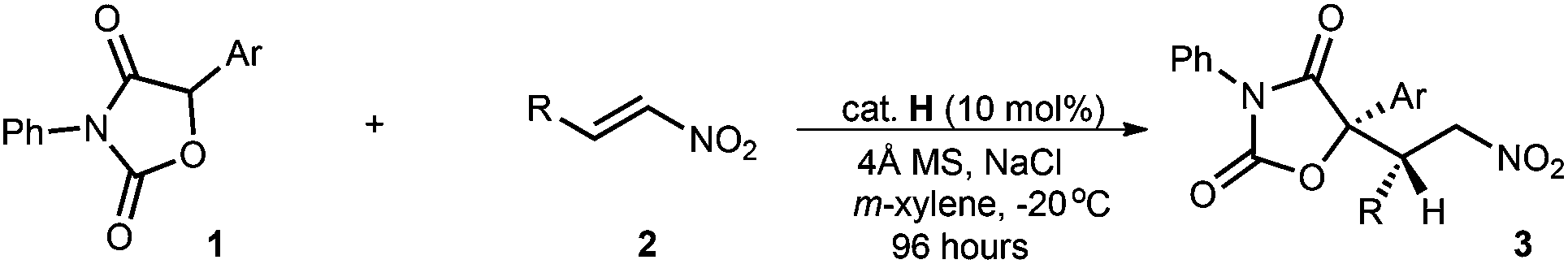
To demonstrate the utility of this methodology, synthetic transformations of conjugate addition products 3 were subsequently processed (Scheme 1a). Using KOH as a base and EtOH/H2O (3:7) as a solvent, the adduct 3a could be readily converted into α-tertiary hydroxyl amide 4 through hydrolysis in 85% yield. Then, the reduction of 4 in the presence of zinc powder and HCl afforded the corresponding amine 5 in good yield and without compromising the ee value. It was found that the amine 5 was able to easily transform to the valuable γ-lactam 65 that is a key chiral scaffold existing in many biologically important molecules, such as chimonamidine,1a convolutamide A–F,1b and tribulusamide C1c (Fig. 1).
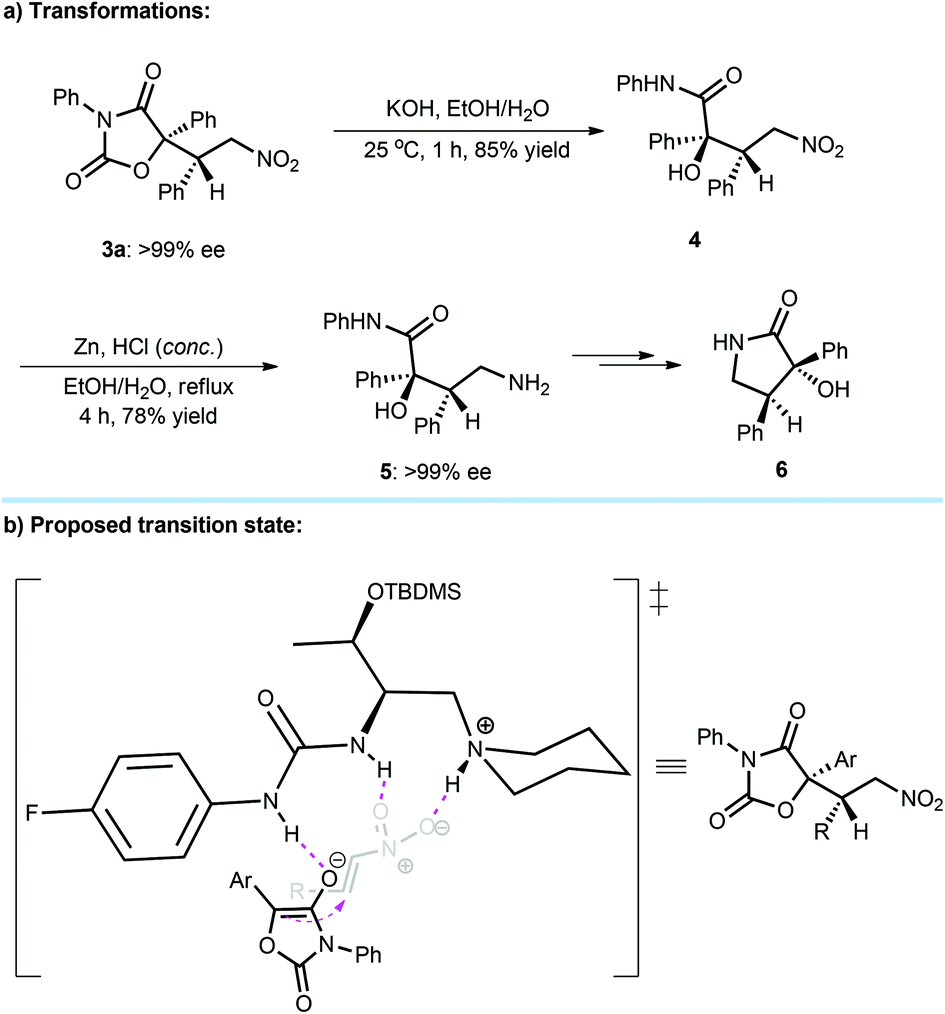 | ||
| Scheme 1 (a) Transformations of the Michael adduct 3a. (b) Plausible transition state. | ||
Although the mechanism towards L-amino acid-based tertiary amine-urea as a catalyst remains to be clarified, a plausible transition-state model is proposed (Scheme 1b), which is different from L-amino acid-based tertiary amine-thiourea7a due to the entirely distinct enantioselectivity (Table 1, entries 1 and 2). Noteworthy is that similar enantioselectivities were observed when catalysts F (Table 1, entry 6) and H (Table 1, entry 8) respectively with 3,5-bis(trifluoromethyl)phenyl and 4-fluoro-phenyl groups as the urea substituents were used. Therefore, the generated enolate of diaryloxazolidin-2,4-dione after deprotonation would bind to the outside N–H bond of the urea unit, and the ortho C–H bond12 of the aryl group of urea may not participate in the interaction with the enolate. The R3NH+ arm of the catalyst and another N–H bond of urea can contribute a dual H-bonding mode to interact with nitroolefin. After nucleophilic addition, the conjugate adducts were attainable with the observed stereoselective results.
Conclusions
In summary, we have developed the first catalytic asymmetric conjugate addition of diaryloxazolidin-2,4-diones to nitroolefins. In the presence of an L-threonine-based tertiary amine-urea as the bifunctional catalyst, a series of conjugate adducts were obtained with good to excellent enantio- and diastereoselectivities (up to >99% ee and >19:1 dr). This strategy provides an efficient approach to valuable α-aryl-α-hydroxy carboxylic acids which structurally feature two adjacent hetero-quaternary and tertiary stereogenic centers. Further investigation to extend diaryloxazolidin-2,4-diones in novel asymmetric patterns is currently underway.
Acknowledgements
Financial support from the National Science Foundation of China (no. 21072044), Henan Province (14IRTSTHN006, 152300410057) and Henan University is greatly appreciated.Notes and references
- (a) H. Takayama, Y. Matsuda, K. Masubuchi, A. Ishida, M. Kitajima and N. Aimi, Tetrahedron, 2004, 60, 893 CrossRef CAS; (b) H.-P. Zhang, H. Shigemori, M. Ishibashi, T. Kosaka, G. R. Pettit, Y. Kamano and J. Kobayashi, Tetrahedron, 1994, 50, 10201 CrossRef CAS; (c) X. Zhang, N. Wei, J. Huang, Y. Tan and D. Jin, Nat. Prod. Res., 2012, 26, 1922 CrossRef CAS PubMed; (d) M. Belema, V. N. Nguyen, D. R. St Laurent, O. D. Lopez, U. Qiu, A. C. Good, P. T. Nower, L. Valera, D. R. O'Boyle II, J.-H. Sun, M. Liu, R. A. Fridell, J. A. Lemm, M. Gao, J. O. Knipe, N. A. Meanwell and L. B. Snyder, Bioorg. Med. Chem. Lett., 2013, 23, 4428 CrossRef CAS PubMed; (e) D. W. McPherson, C. R. Lambert, K. Jahn, V. Sood, R. C. McRee, B. Zeeberg, R. C. Reba and F. F. Knapp Jr., J. Med. Chem., 1995, 38, 3908 CrossRef CAS PubMed; (f) H. B. Rasmussen and J. K. MacLeod, J. Nat. Prod., 1997, 60, 1152 CrossRef CAS; (g) K. A. Bergström, C. Halldin, J. Hiltunen, C. G. Swahn, H. Ito, N. Ginovart, H. Hall, D. W. McPherson, F. F. Knapp Jr., S. Larsson, P.-O. Schnell and L. Farde, Nucl. Med. Biol., 1998, 25, 209 CrossRef; (h) R. P. Hof and R. M. Kellogg, J. Org. Chem., 1996, 61, 3423 CrossRef CAS.
- For selected examples of catalytic asymmetric aldol reaction to α-ketoesters, see: (a) E. F. DiMauro and M. C. Kozlowski, J. Am. Chem. Soc., 2002, 124, 12668 CrossRef CAS PubMed; (b) E. F. DiMauro and M. C. Kozlowski, Org. Lett., 2002, 4, 3781 CrossRef CAS PubMed; (c) K. Funabashi, M. Jachmann, M. Kanai and M. Shibasaki, Angew. Chem., Int. Ed., 2003, 42, 5489 CrossRef CAS PubMed; (d) H. Li, B. Wang and L. Deng, J. Am. Chem. Soc., 2006, 128, 732 CrossRef CAS PubMed; (e) G. Blay, I. Fernández, A. Marco-Aleixandre and J. R. Pedro, Org. Lett., 2006, 8, 1287 CrossRef CAS PubMed; (f) K. Zheng, B. Qin, X. Liu and X. Feng, J. Org. Chem., 2007, 72, 8478 CrossRef CAS PubMed; (g) F. Wang, Y. Xiong, X. Liu and X. Feng, Adv. Synth. Catal., 2007, 349, 2665 CrossRef CAS; (h) H.-L. Wu, P.-Y. Wu, Y.-Y. Shen and B.-J. Uang, J. Org. Chem., 2008, 73, 6445 CrossRef CAS PubMed; (i) H.-F. Duan, J.-H. Xie, X.-C. Qiao, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2008, 47, 4351 CrossRef CAS PubMed; (j) X. Zhu, A. Lin, L. Fang, W. Li, C. Zhu and Y. Cheng, Chem. – Eur. J., 2011, 17, 8281 CrossRef CAS PubMed; (k) J. Luo, H. Wang, X. Han, L.-W. Xu, J. Kwiatkowski, K.-W. Huang and Y. Lu, Angew. Chem., Int. Ed., 2011, 50, 1861 CrossRef CAS PubMed; (l) F. Cai, X. Pu, X. Qi, V. Lynch, A. Redha and J. M. Ready, J. Am. Chem. Soc., 2011, 133, 18066 CrossRef CAS PubMed; (m) T.-S. Zhu, S.-S. Jin and M.-H. Xu, Angew. Chem., Int. Ed., 2012, 51, 780 CrossRef CAS PubMed; (n) A. Crespo-Peña, D. Monge, E. Martín-Zamora, E. Álvarez, R. Fernández and J. M. Lassaletta, J. Am. Chem. Soc., 2012, 134, 12912 CrossRef PubMed; (o) R. Infante, J. Nieto and C. Andrés, Chem. – Eur. J., 2012, 18, 4375 CrossRef CAS PubMed; (p) B. L. Kohn, N. Ichiishi and E. R. Jarvo, Angew. Chem., Int. Ed., 2013, 52, 4414 CrossRef CAS PubMed.
- For selected reviews, see: see: (a) F. Zhou, Y.-L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 CrossRef CAS; (b) R. Dalpozzo, G. Bartoli and G. Bencivenni, Chem. Soc. Rev., 2012, 41, 7247 RSC. For selected examples, see: (c) W.-B. Chen, X.-L. Du, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Tetrahedron, 2010, 66, 1441 CrossRef CAS; (d) B. Zhu, W. Zhang, R. Lee, Z. Han, W. Yang, D. Tan, K.-W. Huang and Z. Jiang, Angew. Chem., Int. Ed., 2013, 52, 6666 CrossRef CAS PubMed.
- T. Ooi, K. Fukumoto and K. Maruoka, Angew. Chem., Int. Ed., 2006, 45, 3839 CrossRef CAS PubMed.
- P. S. Hynes, D. Stranges, P. A. Stupple, A. Guarna and D. J. Dixon, Org. Lett., 2007, 9, 2107 CrossRef CAS PubMed.
- B. M. Trost, K. Dogra and M. Franzini M, J. Am. Chem. Soc., 2004, 126, 1944 CrossRef CAS PubMed.
- (a) T. Misaki, G. Takimoto and T. Sugimura, J. Am. Chem. Soc., 2010, 132, 6286 CrossRef CAS PubMed; (b) T. Misaki, K. Kawano and T. Sugimura, J. Am. Chem. Soc., 2011, 133, 5695 CrossRef CAS PubMed; (c) H. Huang, K. Zhu, W. Wu, Z. Jin and J. Ye, Chem. Commun., 2012, 48, 461 RSC; (d) D. Zhao, L. Wang, D. Yang, Y. Zhang and R. Wang, Angew. Chem., Int. Ed., 2012, 51, 7523 CrossRef CAS PubMed; (e) B. M. Trost and K. Hirano, Angew. Chem., Int. Ed., 2012, 51, 6480 CrossRef CAS PubMed; (f) E. Badiola, B. Fiser, E. Gómez-Bengoa, A. Mielgo, I. Olaizola, I. Urruzuno, J. M. García, J. M. Odriozola, J. Razkin, M. Oiarbide and C. Palomo, J. Am. Chem. Soc., 2014, 136, 17869 CrossRef CAS PubMed; (g) W. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 377 CrossRef CAS PubMed; (h) T. Wang, Z. Yu, D. L. Hoon, K.-W. Huang, Y. Lan and Y. Lu, Chem. Sci., 2015, 6, 4912 RSC.
- (a) B. Qiao, Y. An, Q. Liu, W. Yang, H. Liu, J. Shen, L. Yan and Z. Jiang, Org. Lett., 2013, 15, 2358 CrossRef CAS PubMed; (b) Q. Liu, B. Qiao, K. F. Chin, C.-H. Tan and Z. Jiang, Adv. Synth. Catal., 2014, 356, 3777 CrossRef CAS; (c) Z. Han, W. Yang, C.-H. Tan and Z. Jiang, Adv. Synth. Catal., 2013, 355, 1505 CrossRef CAS; (d) M. Xu, B. Qiao, S. Duan, H. Liu and Z. Jiang, Tetrahedron, 2014, 70, 8696 CrossRef CAS; (e) S. Duan, S. Li, X. Ye, N.-N. Du, C.-H. Tan and Z. Jiang, J. Org. Chem., 2015, 80, 7770 CrossRef CAS PubMed; (f) B. Zhu, R. Lee, J. Li, X. Ye, S.-N. Hong, S. Qiu, M. L. Coote and Z. Jiang, Angew. Chem., Int. Ed., 2016, 55, 1299 CrossRef CAS PubMed.
- (a) W. Zhang, D. Tan, R. Lee, G. Tong, W. Chen, B. Qi, K.-W. Huang, C.-H. Tan and Z. Jiang, Angew. Chem., Int. Ed., 2012, 51, 10069 CrossRef CAS PubMed; (b) Y. Yang, F. Moinodeen, W. Chin, T. Ma, Z. Jiang and C.-H. Tan, Org. Lett., 2012, 14, 4762 CrossRef CAS PubMed; (c) X. Bai, Z. Jing, Q. Liu, X. Ye, G. Zhang, X. Zhao and Z. Jiang, J. Org. Chem., 2015, 80, 12686 CrossRef CAS PubMed; (d) Z. Jing, X. Bai, W. Chen, G. Zhang, B. Zhu and Z. Jiang, Org. Lett., 2016, 18, 260 CrossRef CAS PubMed.
- For a selected review, see: (a) Z. Chai and G. Zhao, Catal. Sci. Technol., 2012, 2, 29 RSC; (b) O. V. Serdyuk, C. M. Heckel and S. B. Tsogoeva, Org. Biomol. Chem., 2013, 11, 7051 RSC; (c) X. Zhao, B. Zhu and Z. Jiang, Synlett, 2015, 2216 CAS.
- CCDC 1417451 (3a) contains the supplementary crystallographic data for this paper.
- K. M. Lippert, K. Hof, D. Gerbig, D. Ley, H. Hausmann, S. Guenther and P. R. Schreiner, Eur. J. Org. Chem., 2012, 5919 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1417451. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00428d |
| ‡ L. J. and X. Z. made equal contributions to this work. |
| This journal is © the Partner Organisations 2016 |