
Dual-drug delivery of curcumin and platinum drugs in polymeric micelles enhances the synergistic effects: a double act for the treatment of multidrug-resistant cancer†
Wei
Scarano
a,
Paul
de Souza
b and
Martina H.
Stenzel
*a
aCentre for Advanced Macromolecular Design (CAMD), School of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: M.Stenzel@unsw.edu.au
bLiverpool Hospital Clinical School, and Molecular Medicine Research Group, University of Western Sydney, Sydney, NSW 2170, Australia
First published on 18th September 2014
Abstract
Combinational chemotherapy is often used to prevent drug induced resistance in cancer. The aim of this work is to test whether the co-delivery of drugs within one nanoparticle can result in increased synergistic effects of both drugs. Therefore, a micelle system with two different compartments, one for the drug curcumin and one for the conjugation of platinum drugs was designed. A triblock copolymer, based on the biodegradable polycaprolactone PCL, a PEG based shell and an amine bearing polymer as the interphase for the conjugation of platinum drugs was prepared by combination of ring-opening polymerization and RAFT polymerization. Curcumin was incorporated into the self-assembled onion-type micelle by physical encapsulation into the PCL core with an entrapment capacity of 6 wt%. The platinum(IV) drug oxoplatin was reacted with succinic anhydride to yield Pt(NH3)2Cl2[(COOH)2], which acted as the drug and as a crosslinker for the stabilisation of micelles. The size of the dual drug micelles was measured to be 38 nm by DLS, which was confirmed by TEM. The toxicity of the dual drug delivery system was tested against the A2780 human ovarian cancer cell line and compared with the IC50 value of micelles that deliver either curcumin or the platinum drug alone. The results were analysed using the CalcuSyn software. While curcumin and the platinum drug together without a carrier already showed synergy with a combination index ranging from 0.4 to 0.8, the combined delivery in one nanoparticle did enhance the synergistic effects resulting in a combination index of approximately 0.2–0.35. For comparison, a mixture of two nanoparticles, one with curcumin and the other with the platinum drug, was tested revealing a less noticeable synergistic effect compared to the co-delivery of both drugs in one drug carrier.
Introduction
Conventional chemotherapy, if ineffective in eradicating cancer cells, can lead to the development of drug resistance over the course of therapy. The development of drug resistance could be due to two major factors: the inability to deliver drugs to the tumour site and specific genetic alternations in cancer cells.1 Drug delivery to the tumour site can be improved with the use of nanoparticles and delivery systems such as polymeric micelles have been widely used as drug carriers due to their advantageous characteristics such as solubilisation of hydrophobic molecules with high drug loading capacity, low toxicity, high water solubility and high structural stability which ultimately allow prolonged circulation in blood and enhanced accumulation in tumour tissues.2,3To overcome potential drug-induced mutations and drug resistance in tumours, combination chemotherapy may be used. This is where a combination of two or more drugs is employed to disrupt different stages of the cell repair and/or reproduction cycle to enhance the apoptosis of cancer cells. In cancer treatment it is common to utilise a combination of chemotherapeutic agents for treatment,4–8 however it is important to consider their mechanism of action, the dose ratios between the drugs as well as their induced side effects.
Cisplatin is one of the commonly used chemotherapeutic agents today; it disrupts cell repair by cross-linking with DNA strands and ultimately causing apoptosis.9,10 However a potential disadvantage of using platinum-based drugs is that they are required to enter the cell nucleus first, and then attach to DNA before the apoptosis sequence begins.11,12 However, excision repair cross-complementary 1 (ERCC1), a protein involved in the process of nucleotide excision repair, is expressed at high levels in cancer and has been associated with resistance to platinum-based chemotherapy.13 Therefore cisplatin is often used in conjunction with other drugs for cancer treatment in an attempt to circumvent this potential mechanism of platinum resistance.
Curcumin is a natural polyphenolic compound extracted from the root of turmeric plant (curcuma longa) and has been used traditionally for centuries in Asia for medicine, cooking, and other purposes. Curcumin exists in mostly β-diketone tautomer form but also exists between the two equilibrates of the asymmetric keto–enol tautomer,14 and it possesses antioxidant, anti-inflammatory, antibacterial and anti-cancer activities. Curcumin has been shown to exhibit therapeutic potential against various types of cancers15–18 but it has poor bioavailability due to its rapid metabolism in the liver and the intestinal wall. It is understood that the anti-carcinogenic and chemo-preventive effects of curcumin could be due to its action on multiple targets including transcription factors, growth regulators,19 adhesion molecules, apoptotic genes, angiogenesis regulators and cellular signalling molecules.20–22
The synergistic effect of curcumin and various anticancer agents has been explored previously.23–33 However the efficacy of the drug can also be significantly improved with the use of drug delivery systems.34 Drug delivery systems consisting of polymeric materials have been used to deliver a wide range of compounds with the potential of improving the Enhanced Permeability and Retention (EPR) effect on solid tumours.35–37 This is where polymeric macromolecular structures of size above 40 kDa are accumulated in solid tumours due to their pathophysiological uniqueness, unlike low molecular weight compounds.38,39 Therefore delivering curcumin using a polymeric drug delivery system could also improve its activity.40–45 Furthermore, curcumin is able to enhance the antiproliferative properties of platinum complexes.13,46–49 The use of liposomes to deliver both platinum and curcumin can result in a greater growth inhibition and an apoptotic effect with virtually no side effects in animals.50 However, it has never been quantified whether there is an advantage (or a synergistic effect) in co-delivering both drugs.
We hypothesized therefore that the use of a polymeric micelle delivery system to co-deliver curcumin and platinum complexes would result in improved drug efficacy. Both drugs have very different requirements with regard to an optimized drug carrier design. While curcumin is best delivered using a hydrophobic polymer, platinum drugs are typically conjugated to the polymer.51–53 The design of nanocarriers with various compartments of different nature is therefore of paramount importance. Onion-type polymeric micelles based on triblock copolymers with their tuneable characteristics fit the specific requirements. This triblock copolymer needs to satisfy three criteria: a hydrophobic part, which will form the core of the micelle, for the encapsulation of curcumin, a reactive block for the platinum drug conjugation and a water-soluble part that will build up the shell, which determines the interaction with the biological environment. The triblock copolymer will therefore comprise the biodegradable and hydrophobic polymer polycaprolactone as the core, a PEG-based polymer as the shell and amine-bearing polymers in the interphase allowing efficient modification for the attachment of desirable molecules. Primary amine based copolymers have been exploited in numerous applications due to their reactivity towards many functional groups such as acids and isocyanates. Furthermore, we propose to use a platinum-based crosslinker that may shield the compound against deactivation by proteins and peptides, as well as strengthen the structural integrity of the micelle itself (Scheme 1).54 Crosslinking of micelles has been shown to improve the performance of the drug carrier.55,56 The micelles were then tested against the A2780 human ovarian carcinoma cell line. The main focus is the change of the synergistic effects of both drugs when co-delivered in one carrier.
 | ||
| Scheme 1 Synthetic route to the triblock copolymer PCL-b-PABPA-POEGMEA followed by the encapsulation of curcumin and crosslinking using Pt[(COOH)2 for structural stability. | ||
Experimental
Materials
2-Mercapto ethanol (Aldrich, 99%), benzyl bromide (Aldrich, 98%), carbon disulfide (Aldrich, 99%), potassium phosphate, 3-amino-1-propanol (Aldrich, 99%), di-tert-butyl-dicarbonate, acryloyl chloride (Aldrich, 97% with <210 ppm MEHQ as the stabilizer), ε-caprolactone (Aldrich, 97%), lipase acrylic resin (Aldrich, ≥5000 U g−1), fluorescein O-methacrylate (Aldrich, 97%), cis-diammineplatinum(II) dichloride (CDDP; Sigma Aldrich, 99.9%), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC; Aldrich, 98%), N-hydroxysuccinimide (NHS; Aldrich, 98%), succinic anhydride (Fluka, 97%), curcumin (Fluka, analytical standard), L-ascorbic acid (Aldrich, 99%), toluene (Aldrich; purum), N,N-dimethyl acetamide (DMAc; Aldrich, 99.9%), N,N-dimethylformamide (DMF; Aldrich), diethyl ether (Et2O; Ajax Fine Chem, 99%), ethyl acetate (EtOAc; Ajax Finechem, 99.5%), methanol, anhydrous tetrahydrofuran (THF; Aldrich, >99%), n-hexane (Ajax Fine Chem, 95%), hydrochloric acid (HCl; Ajax Fine Chem, 32%), hydrogen peroxide (H2O2; Ajax Fine Chem, 30% w/v), 1,4-dioxane (Ajax Fine Chem, 99%), triethylamine (TEA; Ajax Fine Chem, 99%), and potassium phosphate (K3PO4; Ajax Fine Chem, 99%) were used without any further purification.Oligo(ethylene glycol) methyl ether acrylate (OEGMEA, Mn = 480 g mol−1; Aldrich) was destabilized by passing it over a column of basic alumina and stored at −7 °C. 2,2-Azobisisobutyronitrile (AIBN; Fluka, 98%) was purified by recrystallization from methanol. Deionized (DI) water was produced using the Milli-Q water purification system and has a resistivity of 17.9 mΩ cm−1.
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hexane–ethyl acetate mixture. Yield: 88%. 1H-NMR (CDCl3) δ (ppm): 7.40–7.28 (5H, m, Ph), 4.69 (2H, s, H1), 3.91 (2H, t, H2), 3.66 (2H, t, H3), 2.38 (1H, br, s, H4).
:1). Yield: 50 repeating units of ε-caprolactone. 1.94 g, GPC (THF): Mn = 5300 g mol−1, PDISEC = 1.85.
960 g mol−1 (Mn(SEC DMAc) = 23500 g mol−1) PCL50-b-POEGMEA37 was used for chain extension in subsequent reactions.
054 g mol−1 (Mn(SEC DMAc) = 30000 g mol−1). The polymer PCL50-b-ABPA38-b-POEGMEA37 was stored in the dark and dry area for future reactions.
:1 hexane–ethyl acetate mixture. Yield: 88%. 1H-NMR (CDCl3) δ (ppm): 7.40–7.28 (5H, m, Ph), 4.69 (2H, s, H1), 3.91 (2H, t, H2), 3.66 (2H, t, H3), 2.38 (1H, br, s, H4).
:1). Yield: 50 repeating units of ε-caprolactone. 1.94 g, GPC (THF): Mn = 5300 g mol−1, PDISEC = 1.85.
960 g mol−1 (Mn(SEC DMAc) = 23500 g mol−1) PCL50-b-POEGMEA37 was used for chain extension in subsequent reactions.
054 g mol−1 (Mn(SEC DMAc) = 30000 g mol−1). The polymer PCL50-b-ABPA38-b-POEGMEA37 was stored in the dark and dry area for future reactions.
The percent of curcumin incorporated during nanoparticle preparation was determined by taking 100 μL of the particle solution (1 mg mL−1) and diluting with DMF to give a final volume of 2 mL. The solution was assayed using a UV spectrophotometer at 428 nm and results were obtained using a standard curve. The loading efficiency and entrapment efficiency are calculated by the following formulae:
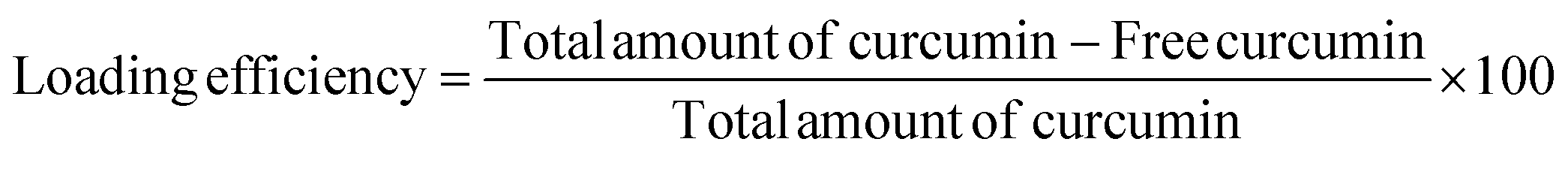
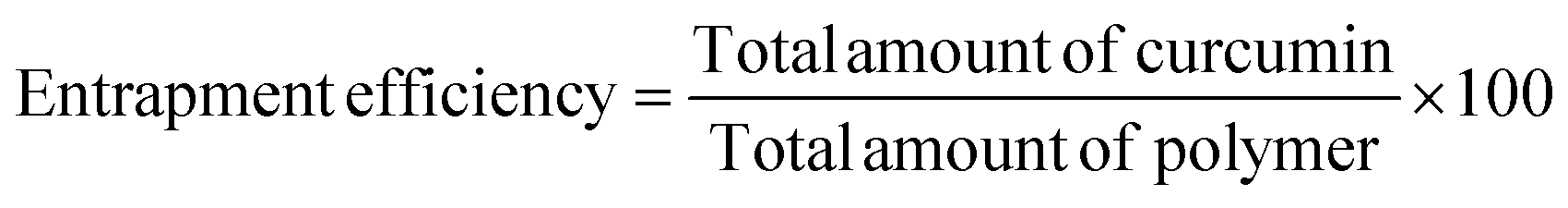 :1.
:1.
Cell culture
The A2780 cell lines were grown in a ventilated tissue culture flask T-75 using Roswell Park Memorial Institute (RPMI-1640) media containing 10% Foetal Bovine Serum (FBS) and antibiotics. The cells were incubated at 37 °C under a 5% CO2 humidified atmosphere and passaged every 2–3 days when monolayers at around 80% confluence were formed. The cell density was determined by counting the number of viable cells using a trypan blue dye (Sigma-Aldrich) exclusion test.Cell viability
The cytotoxicity of PCL-b-PABPA-b-POEGMEA, cisplatin, oxoplatin, Pt IV(COOH)2 and curcumine/platinum(IV) incorporated micelles (M3) was measured by a standard sulforhodamine B colorimetric proliferation assay (SRB assay). The SRB assay was established by the U.S. National Cancer Institute for rapid, sensitive, and inexpensive screening of antitumor drugs in microtiter plates. The cells were seeded at a density of 5000 cells per well in 96-well plates containing 200 μL of growth medium per well and incubated for 24 h. The medium was then replaced with fresh medium (200 μL) containing various concentrations of the material being tested.After 48 h incubation, cells were fixed with trichloroacetic acid 10% w/v (TCA) before washing, incubated at 4 °C for 1 h, and then washed five times with tap water to remove TCA, growth medium, and low molecular weight metabolites. Plates were air dried and then stored until use. TCA-fixed cells were stained for 30 min with 0.4% (w/v) SRB dissolved in 1% acetic acid. At the end of the staining period, SRB was removed and cultures were quickly rinsed five times with 1% acetic acid to remove the unbound dye. Then the cultures were air dried until no conspicuous moisture was visible. The bound dye was shaken for 10 min. The absorbance at 570 nm of each well was measured using a microtiter plate reader scanning spectrophotometer BioTek's PowerWave™ HT Microplate Reader and the KC4™ Software. All experiments were repeated three times. Dose–response curves were plotted (values were expressed as the percentage of control, medium only) and IC50 inhibitory concentrations were obtained using the software Graph Pad PRISM 6.
| Cell viability (%) = (OD490,sample − OD490,blank)/(OD490,control − OD490,blank) × 100 |
Analysis
Results and discussion
Reversible Addition Fragmentation chain Transfer (RAFT) polymerisation is a commonly used technique used for the synthesis of complex polymer architectures. In particular it can be easily combined with other techniques to generate polymer architectures that have degradable properties.58 Several approaches are described in the literature to combine polymers prepared by ring-opening polymerization and RAFT polymerization with the initiation of the ring-opening polymerization using a functional RAFT agent being one of them.58 Polycaprolactone was chosen as one of the materials for drug delivery carriers due to its biodegradability and biocompatibility. This allows the matrix material to be decomposed into non-toxic and low molecular weight molecules and then metabolized or absorbed by the organism.59 Polycaprolactone was synthesized via ring-opening polymerization (ROP) of ε-caprolactone which was carried out according to the literature.60 The reaction was performed under enzymatic conditions at 70 °C in the presence of lipase resin and the benzyl 2-hydroxyethyl carbonotrithioate (BHCT) RAFT agent as the initiator (Scheme 1). The conversion of this reaction was determined by gravimetric analysis and is further confirmed by the 1H NMR spectra (see ESI Fig. S1†) revealing a PCL block with approximately 50 repeating units and is determined by comparing the peak at 4.633 ppm which belongs to the BHCT RAFT and the peak at 4.097–4.053 ppm of the caprolactone repeated units (Table 1, see ESI Fig. S1 and S2†). The results from SEC and the theoretical value obtained from the conversion were in good agreement especially considering that the SEC was calibrated against polystyrene.| Sample | Polymer | Conv. | M Theon (g mol−1) | M SECn (g mol−1) | PDISEC |
|---|---|---|---|---|---|
| PCL | PCL50 | 95% | 5900 | 5200 | 1.85 |
| PCL1 | PCL50-b-POEGMEA37 | 35% | 22960 |
23500 |
1.37 |
| M1 | PCL50-b-PABPA38-b-POEGMEA37 | 65% | 31054 |
30000 |
1.35 |
The newly synthesised PCL macroRAFT agent was used for the polymerisation of OEGMEA. The monomer oligo(ethylene glycol)methyl ether acrylate (OEGMEA) was chosen as the hydrophilic shell of the micelle due to its water solubility and the resemblance to PEG. The monomer OEGMEA had to be polymerized prior to the amine bearing monomer to generate a micelle with a protective water-soluble shell. This was necessary due to the nature of the RAFT agent that carried PCL as part of the Z-group, which led to the monomer insertion close to the PCL block.58 The resulting block polymer, named PCL1, had a molecular weight close to the expected value (Table 1, see ESI Fig. S2†).
For the synthesis of the third block a new amine bearing monomer had to be prepared. The purpose of this block was to act as a functional handle for the conjugation of the platinum(IV) crosslinker while enhancing the structural integrity of the polymeric micelle by crosslinking at the nexus. The amine functionality is not only highly reactive, but the resulting amide bond after reaction with the carboxylate-based Pt(IV) crosslinker is also stable in a typical biological environment preventing premature disassembly of the micelle. The protection of amine functional groups is essential prior to the RAFT polymerisation reaction at neutral pH. This is due to the fact that amines would attack the dithioester functional group of the RAFT agent via nucleophilic substitution reaction, thus destroying or interfering with the functionality of the RAFT agent itself.61,62 Di-tert-butyl dicarbonate was used for the protection of 3-amino-1-propanol and the tert-butyl carbamates can be easily cleaved under an acidic condition. The monomer was synthesised by reacting acryloyl chloride and BOC protected 3-amino-1-propanol in the presence of an auxiliary base, in this case triethylamine, in anhydrous THF solvent (Scheme 2). The final product was purified via gel column chromatography and is soluble in many solvents such as methanol, toluene, and dioxane. Moreover, the monomer was stable, not air sensitive and even after storage in the freezer at −10 °C over several weeks showed no signs of decomposition. The structure of the monomer was confirmed by 1H NMR (see ESI Fig. S3†).
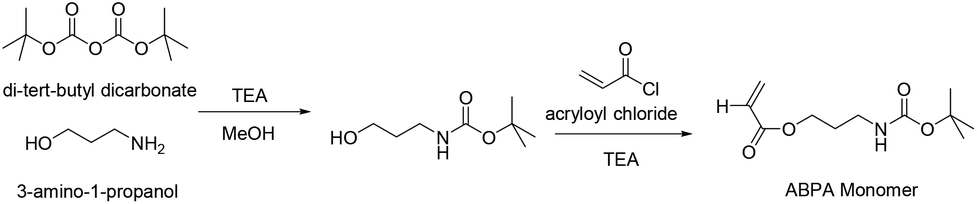 | ||
| Scheme 2 Synthesis route to the ABPA monomer from 3-amino-1-propanol. | ||
The PCL50-b-PEGMEA37 (PCL1) macroRAFT agent was employed for further chain extension with ABPA to yield the triblock copolymer PCL-b-PABPA-b-PEGMEA. The molecular weights of the block copolymers are listed in Table 1 and the overlay of SEC chromatograms is shown in the ESI Fig. S2.† Their measured values are close to the theoretical value. In addition, the molecular weight distribution is narrow.
The subsequent removal of the BOC protective group can be easily achieved using strong acids such as hydrochloric acid or trifluoroacetic acid. The polymer was dissolved in an organic solvent such as DMF or dioxane and HCl (2 M in diethyl ether) was added to the reaction and was left to stir overnight to yield the deprotected PCL50-b-PAPA38-b-POEGMEA37. The polymer was then purified via dialysis against acetone or water to remove unwanted side products. 1H-NMR proves the complete removal of the tert-butyl carbamate protection group (see ESI Fig. S4†) while SEC analysis confirms that during deprotection no side reaction occurred which might have led to crosslinking or loss of PCL (see ESI Fig. S5†).
Two types of block copolymers were used for the synthesis of micelles prior to drug loading: PCL50-b-ABPA38-b-POEGMEA37 (M1) with the BOC protection group and PCL50-b-APA38-b-POEGMEA37 (M2) without the BOC protection group. The size distribution of the nanoparticles is shown in Table 2 with an average hydrodynamic size of about 118 nm with a relative narrow distribution (PDIDLS = 0.104) for PCL50-b-ABPA38-b-POEGMEA37, and an average size of 183 nm for PCL50-b-PAPA38-b-POEGMEA37 with a PDIDLS of 0.117. The obvious size difference between M1 and M2 is due to the removal of the BOC protective group on the polymer backbone to give primary amines. The repulsive forces of the positively charged amino groups lead to swelling and chain stretching and therefore an increase in micelle size.
| Code | Copolymer | D h (d nm−1) | PDIDLS |
|---|---|---|---|
| M1 | PCL50-b-PABPA38-b-POEGMEA37 | 118.2 | 0.104 |
| M2 | PCL50-b-PAPA38-b-POEGMEA37 | 183.4 | 0.117 |
| M3 | PCL50-b-PAPA38-b-POEGMEA37 cuc + Pt | 28.7 | 0.389 |
| M4 | PCL50-b-PAPA38-b-POEGMEA37 cuc + Pt (freeze dried and redissolved in DMAc) | 68.8 | 0.386 |
| M1 cuc | PCL50-b-PABPA38-b-POEGMEA37 cuc | 78.5 | 0.242 |
| M2 Pt | PCL50-b-PAPA38-b-POEGMEA37 Pt | 155 | 0.399 |
A series of curcumin loaded micelles formulated with different amounts of curcumin (w/w, % of polymer) was prepared to study the curcumin encapsulation efficiency. The successful encapsulation was made visible by the clear yellow solution of curcumin loaded into micelles compared to free curcumin (Fig. 1). The encapsulation of curcumin is determined using a UV-VIS spectrophotometer where 100 μL of the 1 mg mL−1 solution was diluted to 2 mL in DMF and a standard curve was set up as a guideline. The encapsulation efficiency of curcumin is shown in Table 3. The results showed that with a constant amount of polymer used the maximum entrapment efficiency was limited between 6 and 7 wt%. Hence, more curcumin led to a decrease in encapsulation efficiency.
 | ||
| Fig. 1 Photograph showing the solubility of curcumin loaded M1 micelles (1 mg mL−1) and the insolubility of curcumin itself in aqueous solution. | ||
| Polymer used (mg) | Curcumin added (mg) | Encapsulation efficiency (%) | Entrapment efficiency (%) |
|---|---|---|---|
| 15 | 1 | 87 | 5.8 |
| 15 | 2 | 53 | 6.9 |
| 15 | 5.5 | 19 | 6.3 |
At this stage, the Pt(IV) drug for conjugation to the polymer was prepared. Various deactivation pathways of Pt(II) anticancer drugs can be avoided using Pt(IV) compounds as prodrugs.63,64 The Pt(IV) complex used in this project is based on the idea that the drug itself can be used as a crosslinker to strengthen the micelle's integrity preventing premature breakdown and reducing burst release of curcumin. In addition, this dual drug carrier was designed to sufficiently carry both drugs through the bloodstream and release a cytotoxic dose of cisplatin upon intracellular reduction once inside a tumour cell; at the same time curcumin would be released with the breakdown of the micelle. The synthesis of the platinum(IV) crosslinker requires a two-step reaction. The first step involves the oxidation of cisplatin using hydrogen peroxide solution (30%) and heat, followed by the ring opening reaction with succinic anhydride. The final product was readily soluble in water as well as DMF, therefore the unreacted complex can be easily removed by dialysis.
The attachment of the platinum(IV) crosslinker was performed in conjunction with the incorporation of curcumin. This minimises the loss of curcumin via diffusion during dialysis with water. The conjugation reaction was carried out at room temperature for 48 hours, and EDC and NHS were used for the esterification as the coupling agent and catalyst, respectively. The reason EDC was chosen as opposed to DCC is due to the solubility of EDC in water and can be easily removed by dialysis.
Once the curcumin was encapsulated and the micelles were crosslinked with the platinum(IV) compound, the size of the micelles (M3) were measured by DLS and results showed that the average size of these micelles has dropped dramatically to 38 nm and the size is confirmed by TEM (Fig. 1). The significant reduction in size can be explained by the loss of cationic charges during crosslinking and the force that contracts the micelle when crosslinking.
The solution was then freeze dried and redissolved in DMAc in order to evaluate the stability of these micelles in a good solvent that can dissolve all blocks. Structural integrity under these conditions confirms the success of the crosslinking process. The hydrodynamic diameter (M4) measured in DMAc is about 68.8 nm. The increase in size is expected since the micelle would start swelling under these conditions, but not disintegrate, which is proof of evidence that crosslinking indeed occurred (Fig. 2). The overlay of the average size distribution of micelles as measured by DLS can be found in ESI Fig. S6.†
 | ||
| Fig. 2 TEM image of M3 micelles (loaded with cuc and crosslinked with Pt) (concentration of micelles 1 mg mL−1 in water): A (scale bar = 200 nm) and B (scale bar = 50 nm). | ||
To determine the amount of platinum crosslinker conjugated to the micelle PCL50-b-APA38-b-POEGMEA37 was used to synthesis blank micelles (concentration = 1 mg mL−1) without the encapsulation of curcumin. The ratio between the Pt(IV)[(COOH2)] carboxyl and the polymer amine functional group was set to 2:1, which would in theory lead to polymers with pendant, non-crosslinked platinum drugs as depicted in Scheme 3 on the right. The ratio was chosen to ensure that all amine groups have the possibility to react to eliminate potentially toxic cationic charges. After reaction, the polymer was dialysed to remove the unreacted Pt(IV)[(COOH2)]. ICP-MS was used to determine the amount of platinum attached revealing a consumption of 79% of the initial drug. Assuming all amine functional groups have reacted, further calculations disclose that out of the 38 repeating units of the amine containing monomer 41% are crosslinked and that 59% are conjugated to a platinum unit but not crosslinked (Scheme 3).
 | ||
| Scheme 3 Conjugation of Pt(IV)[(COOH)2]as a crosslinker for the amine bearing monomers. | ||
The release of curcumin was performed in three different scenarios to evaluate the rate of release with and without the crosslinking as well as compared with the decomposition of the platinum crosslinker. The amount of platinum released was determined by UV-Vis spectroscopy since the absorbance is not affected by the presence of platinum drugs. Firstly, M1 was used for the encapsulation of curcumin and the micelles were dialysed against water to measure the release of curcumin in the absence of the platinated crosslinker. In another experiment, two M3 solutions (5 mL) were placed in two separate dialysis bags, one is placed in a 0.1 M PBS solution at pH 7.4 and the other in a 5 mM ascorbic acid solution. Ascorbic acid is a reducing agent which reduces the Pt(IV) complexes to Pt(II) where the compound loses its axial ligands. Studies have shown that the reduction rate of Pt(IV) complexes strongly depends on the electron-withdrawing power and the steric hindrance of the axial ligands. The rate of reduction increases in the order of axial ligands: OH < OCOCH3 < Cl < OCOCF3.65 Samples were taken from the dialysis bag and were measured with UV-VIS spectroscopy at 428 nm in DMF to quantify the amount of curcumin remaining.
From the results obtained, the fastest release of curcumin is from the uncrosslinked micelles indicating that the flexible structure did not delay the release. After 8 hours 40% of the curcumin is released and the rate of release has slowed down for the next 2 days, reaching a maximum release of 63% in 5 days. Comparing the platinum crosslinked micelles, a faster release rate of curcumin could be observed with the reduction of the platinum crosslinker in the presence of ascorbic acid within the first 20 hours but slowly levels off to similar values of crosslinked micelles in 0.1 M PBS solution as shown in Fig. 3. In summary, crosslinking of the micelles at the interphase of hydrophilic and hydrophobic sections slows down the leaching of the encapsulated curcumin avoiding the initial burst release of the drug. Although crosslinking of micelles can result in a much more noticeable delay of the release rate,66 the measure rate here is not a sign that crosslinking has not occurred, but more a sign that the crosslinking density is not high.
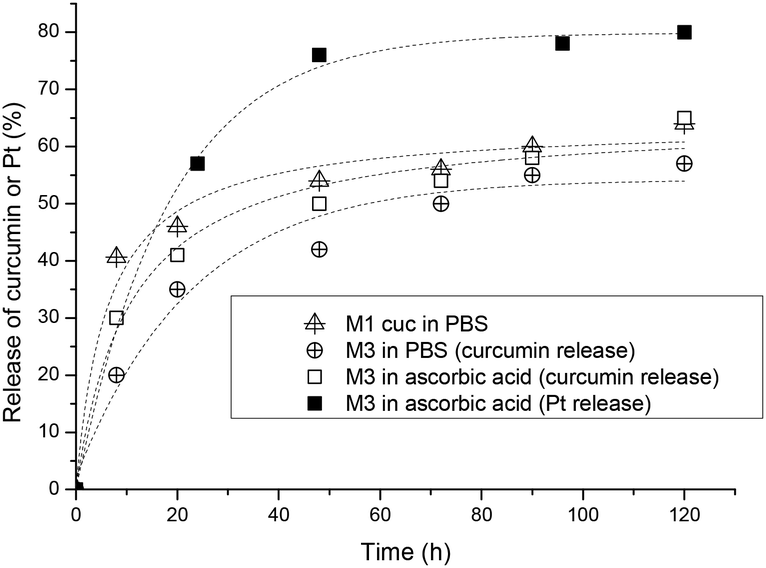 | ||
| Fig. 3 The release of curcumin (open symbol) and platinum (closed symbol) from the platinum crosslinked PCL50-b-PAPA38-b-POEGMEA37 M3 micelle in both PBS (0.1 M) and ascorbic acid (5 mM) over 150 h as well as the release of curcumin from PCL50-b-PABPA38-b-POEGMEA37 M1 in PBS. | ||
The release of platinum was measured by ICP-MS in a reductive environment of ascorbic acid and the results show that more than 50% of the platinum was released within the first 24 h and levels off to about 75% after 2 days (Fig. 3). The micelles in the absence of ascorbic acid did not release Pt-drugs.
The cytotoxicity effect of the curcumin and platinum incorporated micelle and the free drugs (curcumin, cisplatin, oxoplatin and platinum-diacid) was studied against A2780 cells using the standard SRB assay. First the toxicity of the polymer at various concentrations was investigated prior to drug incorporation and it can be deduced that the naked polymer itself has little or no effect on the growth of A2780 cells at low concentrations however concentrations higher than 250 μg mL−1 show a slight cell growth inhibition effect (Fig. 4).
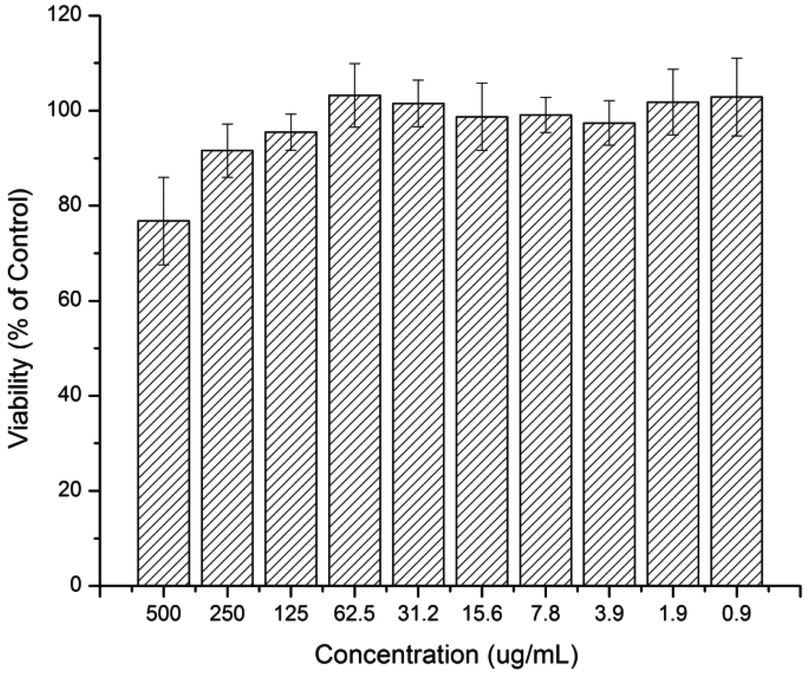 | ||
| Fig. 4 Human Ovarian Carcinoma A2780 viability after being exposed to a solution containing M1 after 48 h. | ||
Subsequently the cytotoxicity of free drugs oxoplatin, cisplatin, Pt(IV)[(COOH)2] and curcumin was tested against A2780 (Fig. 5). The results were compared with the drug incorporated micelles; the comparison allows the determination of the efficacy of the drug carriers themselves. As expected, cisplatin is more potent than oxoplatin as oxoplatin is a platinum(IV) compound, which requires an extra reduction step to form the active diaquo–diamino platinate(II) active species in order to bind to DNA to cause cell apoptosis. Furthermore, cisplatin is more potent towards the A2780 cells than curcumin, Pt[(COOH)2] and oxoplatin, where A2780 cells are least susceptible to curcumin, giving the highest IC50 value. IC50 values of all compounds tested are shown in Table 4.
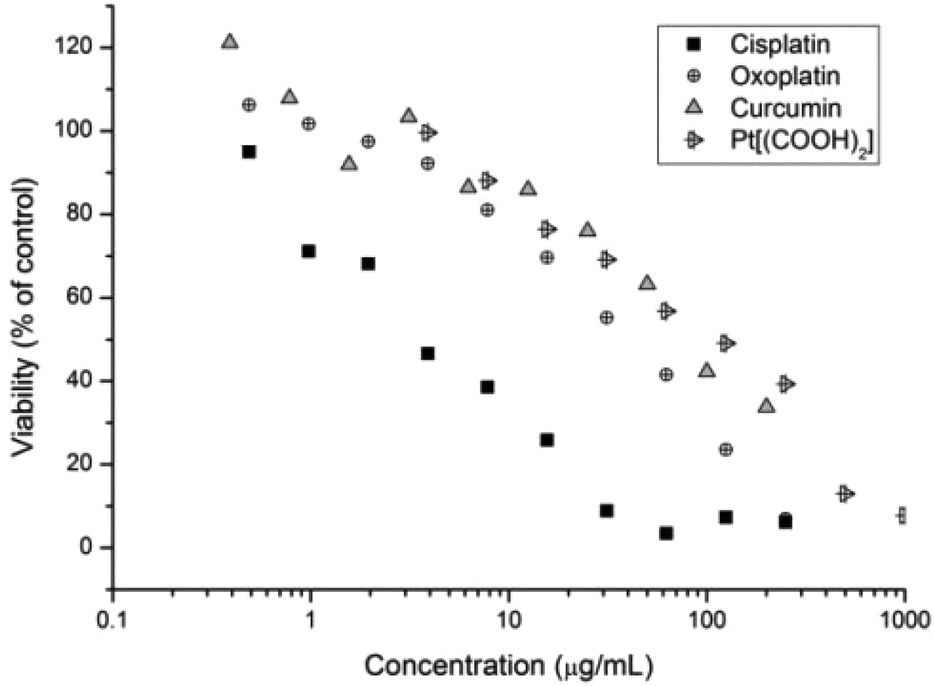 | ||
| Fig. 5 Cell growth inhibition study of curcumin, Pt-diacid, cisplatin and oxoplatin against A2780 human ovarian carcinoma cancer cells for 48 h. | ||
| Compound | IC50 (μg mL−1 of the drug) | IC50 μM |
|---|---|---|
| Oxoplatin | 38.22 | 114.8 |
| Cisplatin | 3.99 | 13.3 |
| Pt(IV)[(COOH)2] | 97.38 | 172.0 |
| Curcumin | 83.72 | 227 |
| M1 cuc | 26.24 | 71.3 |
| M2 Pt | 16.47 | 29.1 |
| M3 | Pt: 12.08 | Pt: 21.3 |
| Cuc: 8.68 | Cuc: 23.5 |
Drug incorporated micelles were also tested against the A2780 human ovarian cancer cell line. The purpose of this experiment was to demonstrate the importance of drug delivery systems for the enhanced cytotoxicity compared to free drugs. Three micelles were compared: a micelle loaded with curcumin only using PCL50-b-PAPA38-b-POEGMEA37 (M1 cuc), a micelle crosslinked with Pt(IV)[(COOH)2] (M2 Pt micelle) and a micelle loaded with both drugs (M3). The molar ratio of curcumin and Pt in the latter micelle was measured by UV-Vis spectroscopy and ICP-MS to be 1:0.95 indicating that roughly the same molar amounts of both drugs are present in the system.
The IC50 of free curcumin was compared with curcumin incorporated micelles in the same way as curcumin encapsulated platinum crosslinked micelles were compared with each other (Fig. 6). The results show that with the aid of the polymeric drug delivery system, the cytotoxicity of curcumin is increased compared with the free drug. The decrease in IC50 from 83.72 μg mL−1 to 26.24 μg mL−1 with the use of polymeric micelles most likely indicates the efficiency of transporting larger amounts of curcumin into the cell by endocytosis rather than through ion channels. PEG-based micelles are known to be efficiently taken up by various cells while simultaneously transporting large amounts of drug across the cell membrane.67–69 In addition, studies have shown that when curcumin accumulates in the endoplasmic reticulum (ER) membrane, the intracellular iron pool is reduced. This may result in lengthening of the G1 phase of the cell cycle, thereby slowing growth in vitro.70
 | ||
| Fig. 6 Cell growth inhibition study of free curcumin, curcumin incorporated micelles and curcumin platinum crosslinked micelles against A2780 human ovarian cancer cells for 48 h. | ||
Similar results were found when investigating the platinum drug loaded micelle with the drug being less toxic compared to platinum crosslinked micelles (Fig. 7). The reason is that micelles are internalised by endocytosis and the platinum complex attached to the polymer backbone will undergo a reduction process in the cell cytoplasm and thus platinum(II) will be released. Once enough platinum(IV) has been cleaved off the backbone the structural integrity of the micelle becomes unstable and it eventually falls apart. This in turn gives the lower IC50 value compared to the free Pt[(COOH)2] complex. With the aid of curcumin the IC50 value of micelles containing both drugs is again lower than the micelles containing only Pt[(COOH)2].
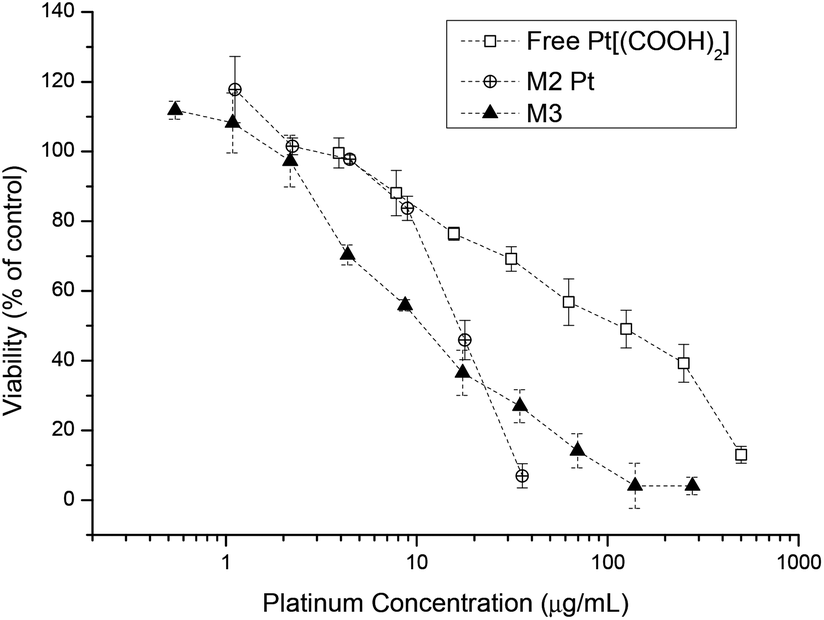 | ||
| Fig. 7 Cell growth inhibition study of free Pt[(COOH)2], M2-Pt micelles and curcumin platinum crosslinked micelles M3 against A2780 human ovarian cancer cells for 48 h. | ||
The computer software CalcuSyn for Windows (Biosoft, UK) was employed to evaluate the synergistic effect between curcumin and Pt[(COOH)2] as free drugs as well as micelle-incorporated moieties. In this system, synergism, additivity, or antagonism is defined by the combination index; a CI value <1 indicates the synergistic effect, a CI value of 1 indicates an additive effect and a CI value >1 indicates an antagonistic effect.
The experimental set up consisted of constant concentration ratios between curcumin and Pt[(COOH)2] against A2780 cells individually and combined as free drugs, mixed micelles and as well being incorporated in micelles. The results are represented in an Isobologram as well as a Median effect analysis plot (Fig. 8). Details of the analysis can be found in the ESI.†
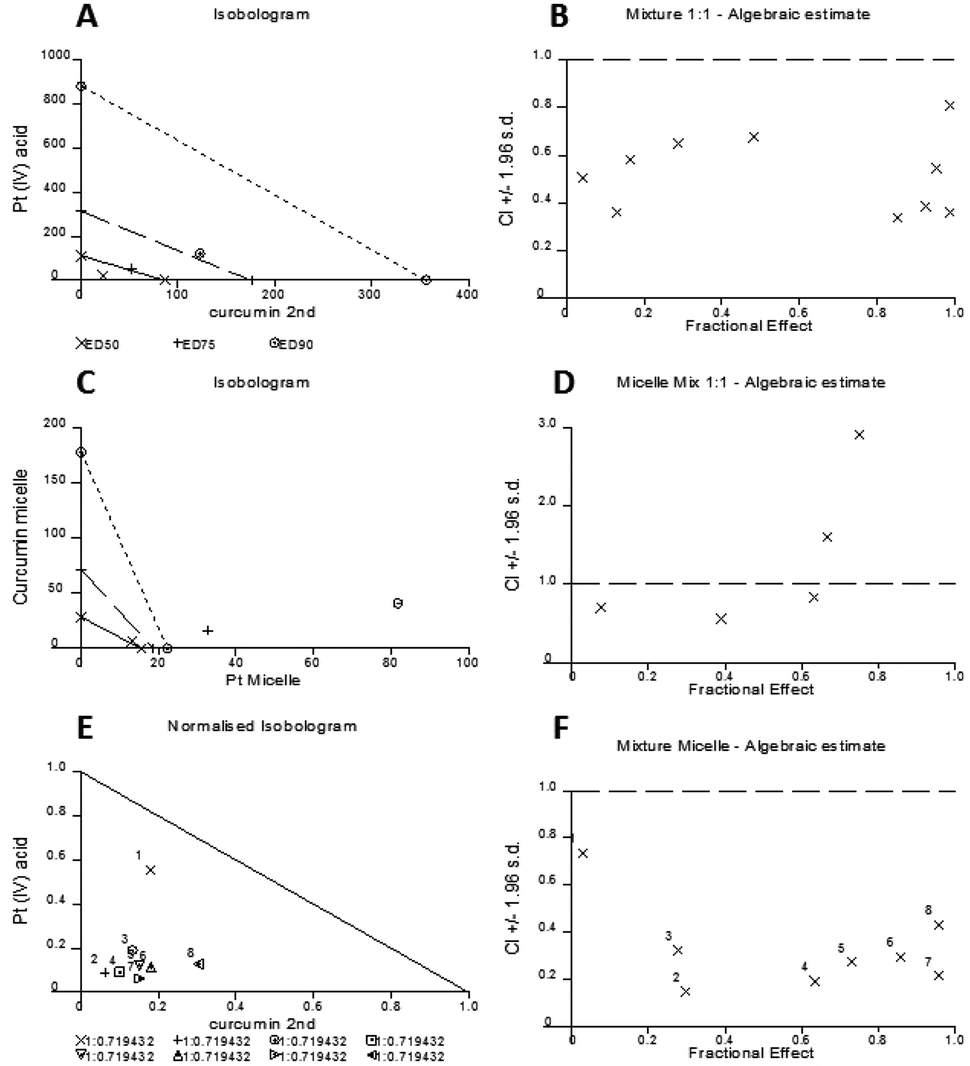 | ||
| Fig. 8 CalcuSyn software evaluation of the synergistic effect of free curcumin and free Pt(IV) acid complex as an isobologram (A) and Median effect analysis plot (B), the mixture effect of two separate micelle solutions as an isobologram (C) and Median effect analysis plot (D), the effect of both drugs in one micelle as an isobologram (E) and Median effect plot (F). | ||
The experimental data are represented in dots and data points that are located below the line in the median effect analysis indicating their synergism within the fractional effect between zero and one. The fractional effect on cell death is calculated by the formula: (100-alive cells)/100. The calculation using the computer software CalcuSyn of the in vitro experimental results showed the synergistic effect of curcumin and Pt[(COOH)2] in 1:1 ratio as free drugs (Fig. 8A and B). This can also be seen for the combination of the two drugs in one micelle in the same ratio of curcumin to Pt[(COOH)2] (Fig. 8E and F). However, it appears that the synergistic effect of the mixture of two individual drugs encapsulated in micelles is prominent at low fractional effects. From the refined values from the combination index method described by Chou and Talalay it can be seen that the combination of curcumin and Pt[(COOH)2] as free drugs (CI values between 0.3 and 0.7) is less synergistic compared to the micelle form (CI values between 0.1 and 0.4).
Here, it can be seen that the combination of free Pt[(COOH)2] and curcumin results in marked synergy (Fig. 8B), where the combination indices all range between 0.4 and 0.8. When the same drugs are incorporated into micelles in the same concentration ratios, the combination indices are mostly less than 0.4 and reach 0.2 (Fig. 8D), indicating highly synergistic combinations. These data confirm that the combination of cisplatin and curcumin is synergistic, but also that the incorporation of both drugs in micelles increases the synergy considerably. At this point, it is not clear why the behaviour is different between the micelle loaded with both drugs and two micelles loaded with curcumin and platinum drug respectively. It is possible that different uptake pathways of the two different micelles can result in this outcome while drugs loaded into one carrier will have the same uptake pathway together.
Conclusion
This project demonstrated the enhanced cytotoxicity of micelles incorporating two drugs as combinational chemotherapy against a resistant cancer cell line. A triblock copolymer was synthesised with amphiphilic properties to allow the physical encapsulation of curcumin and it also allows for the crosslinking using Pt(NH3)2Cl2[(COOH)2] at the interphase which strengthens the structural integrity of the micelle to ensure a slower release of curcumin and to prevent premature disassociation of the delivery vehicle. The synergistic effect of both drugs as free drugs as well as in the micelle form is calculated using the CalcuSyn Biosoft software which indicates that the use of both drugs is a highly synergistic combination.Acknowledgements
The authors would like to thank the Australian Research Council (ARC) for funding. M. H. Stenzel and W. Scarano acknowledge the Centre for Advanced Macromolecular Design (CAMD), the St George Research Education Centre (CPT), the UNSW Analytical Centre for support and the UNSW Electron Microscope Unit for support.References
- M. M. Gottesman, Annu. Rev. Med., 2002, 53, 615–627 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2012, 64(Supplement), 37–48 CrossRef PubMed.
- R. Haag, Angew. Chem., Int. Ed., 2004, 43, 278–282 CrossRef CAS PubMed.
- E. M. McKelvey, J. A. Gottlieb, H. E. Wilson, A. Haut, R. W. Talley, R. Stephens, M. Lane, J. F. Gamble, S. E. Jones, P. N. Grozea, J. Gutterman, C. Coltman and T. E. Moon, Cancer, 1976, 38, 1484–1493 CrossRef CAS.
- T. Tippayamontri, R. Kotb, B. Paquette and L. Sanche, Anticancer Res., 2013, 33, 3005–3014 CAS.
- R. Yang, B. Wang, Y.-J. Chen, H.-B. Li, J.-B. Hu and S.-Q. Zou, Anti-Cancer Drugs, 2013, 24, 871–877 CrossRef CAS PubMed.
- Z. H. Lu, J. Li, M. Lu, X. T. Zhang, J. Zhou, X. C. Wang, J. F. Gong, J. Gao, Y. Li and L. Shen, Med. Oncol., 2013, 30, 664–664 CrossRef CAS PubMed.
- P. Prasad, A. Shuhendler, P. Cai, A. M. Rauth and X. Y. Wu, Cancer Lett., 2013, 334, 263–273 CrossRef CAS PubMed.
- V. Brabec and J. Kasparkova, Drug Resist. Updates, 2005, 8, 131–146 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- R. J. Knox, F. Friedlos, D. A. Lydall and J. J. Roberts, Cancer Res., 1986, 46, 1972–1979 CAS.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed.
- Y.-W. Lin, M.-S. Tsai, S.-H. Weng, Y.-H. Kuo and Y.-F. Chiu, Mol. Pharm., 2011, 255(3), 327–338 Search PubMed.
- F. Payton, P. Sandusky and W. L. Alworth, J. Nat. Prod., 2007, 70, 143–146 CrossRef CAS PubMed.
- P. Anand, C. Sundaram, S. Jhurani, A. B. Kunnumakkara and B. B. Aggarwal, Cancer Lett., 2008, 267, 133–164 CrossRef CAS PubMed.
- M. Hollborn, R. Chen, P. Wiedemann, A. Reichenbach, A. Bringmann and L. Kohen, PLoS One, 2013, 8 Search PubMed.
- P. Kuppusamy, K. Hideg and H. Kalman, Univ Ohio State Res Found.
- G. R. Pillai, A. S. Srivastava, T. I. Hassanein, D. P. Chauhan and E. Carrier, Cancer Lett., 2004, 208, 163–170 CrossRef PubMed.
- A. Saha, T. Kuzuhara, N. Echigo, A. Fujii, M. Suganuma and H. Fujiki, Biol. Pharm. Bull., 2010, 33, 1291–1299 CAS.
- R. K. Maheshwari, A. K. Singh, J. Gaddipati and R. C. Srimal, Life Sci., 2006, 78, 2081–2087 CrossRef CAS PubMed.
- Q. Chen, Y. Wang, K. Xu, G. Lu, Z. Ying, L. Wu, J. Zhan, R. Fang, Y. Wu and J. Zhou, Oncol. Rep., 2010, 23, 397–403 CAS.
- Q. Y. Chen, G. H. Lu, Y. Q. Wu, Y. Zheng, K. Xu, L. J. Wu, Z. Y. Jiang, R. Feng and J. Y. Zhou, Oncol. Rep., 2010, 23, 1285–1292 CAS.
- A. O. Boztas, O. Karakuzu, G. Galante, Z. Ugur, F. Kocabas, C. Z. Altuntas and A. O. Yazaydin, Mol. Pharm., 2013, 10, 2676–2683 CrossRef CAS PubMed.
- Q. Du, B. Hu, H.-M. An, K.-P. Shen, L. Xu, S. Deng and M.-M. Wei, Oncol. Rep., 2013, 29, 1851–1858 CAS.
- M. M. Hossain, N. L. Banik and S. K. Ray, Neurochem. Int., 2012, 61, 1102–1113 CrossRef CAS PubMed.
- D. Sutaria, B. K. Grandhi, A. Thakkar, J. Wang and S. Prabhu, Int. J. Oncol., 2012, 41, 2260–2268 CAS.
- A. Cort, M. Timur, E. Ozdemir, E. Kucuksayan and T. Ozben, Mol. Med. Rep., 2012, 5, 1481–1486 CAS.
- R. Manikandan, M. Beulaja, C. Arulvasu, S. Sellamuthu, D. Dinesh, D. Prabhu, G. Babu, B. Vaseeharan and N. M. Prabhu, Microsc. Res. Tech., 2012, 75, 112–116 CrossRef CAS PubMed.
- H. Yin, R. Guo, Y. Xu, Y. Zheng, Z. Hou, X. Dai, Z. Zhang, D. Zheng and H. e. Xu, Acta Biochim. Biophys. Sin., 2012, 44, 147–153 CrossRef CAS PubMed.
- M. A. Parasramka and S. V. Gupta, J. Oncol., 2012, 2012, 709739–709739 CrossRef PubMed.
- R. A. Siddiqui, J. D. Altenburg, A. A. Bieberich, K. A. Harvey, Z. Xu and V. J. Davisson, FASEB J., 2011, 25 Search PubMed.
- B. B. Patel and A. P. N. Majumdar, Nutr. Cancer, 2009, 61, 842–846 CrossRef CAS PubMed.
- T. M. Elattar and A. S. Virji, Anticancer Res., 2000, 20, 1733–1738 CAS.
- M. Sun, X. Su, B. Ding, X. He, X. Liu, A. Yu, H. Lou and G. Zhai, Nanomedicine, 2012, 7, 1085–1100 CrossRef CAS PubMed.
- H. Maeda, J. Controlled Release, 2012, 164, 138–144 CrossRef CAS PubMed.
- H. Maeda, Cancer Sci., 2013, 104, 779–789 CrossRef CAS PubMed.
- H. Maeda, H. Nakamura and J. Fang, Adv. Drug Delivery Rev., 2013, 65, 71–79 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- H. Maeda, G. Y. Bharate and J. Daruwalla, Eur. J. Pharm. Biopharm., 2009, 71, 409–419 CrossRef CAS PubMed.
- J.-C. Tang, H.-S. Shi, L.-Q. Wan, Y.-S. Wang and Y.-Q. Wei, Asian Pacific J. Cancer Prev., 2013, 14, 2307–2310 CrossRef.
- M. Gou, K. Men, H. Shi, M. Xiang, J. Zhang, J. Song, J. Long, Y. Wan, F. Luo, X. Zhao and Z. Qian, Nanoscale, 2011, 3, 1558–1567 RSC.
- L. Liu, L. Sun, Q. Wu, W. Guo, L. Li, Y. Chen, Y. Li, C. Gong, Z. Qian and Y. Wei, Int. J. Pharm., 2013, 443, 175–182 CrossRef CAS PubMed.
- Z. Ma, A. Haddadi, O. Molavi, A. Lavasanifar, R. Lai and J. Samuel, J. Biomed. Mater. Res., Part A, 2008, 86A, 300–310 CrossRef CAS PubMed.
- K. Wang, T. Zhang, L. Liu, X. Wang, P. Wu, Z. Chen, C. Ni, J. Zhang, F. Hu and J. Huang, Int. J. Nanomed., 2012, 7, 4487–4497 CAS.
- S. Manju and K. Sreenivasan, J. Colloid Interface Sci., 2012, 368, 144–151 CrossRef CAS PubMed.
- L. M. Howells, S. Sale, S. N. Sriramareddy, G. R. B. Irving, D. J. L. Jones, C. J. Ottley, D. G. Pearson, C. D. Mann, M. M. Manson, D. P. Berry, A. Gescher, W. P. Steward and K. Brown, Int. J. Cancer, 2011, 129, 476–486 CrossRef CAS PubMed.
- M. Al Moundhri, S. Al-Salam, A. Al Mahrouqee, S. Beegam and B. Ali, J. Med. Toxicol., 2013, 9, 25–33 CrossRef CAS PubMed.
- M. U. Nessa, P. Beale, C. Chan, J. Q. Yu and F. Huq, Anticancer Res., 2012, 32, 4843–4850 CAS.
- M. Notarbartolo, P. Poma, D. Perri, L. Dusonchet, M. Cervello and N. D'Alessandro, Cancer Lett., 2005, 224, 53–65 CrossRef CAS PubMed.
- L. Li, B. Ahmed, K. Mehta and R. Kurzrock, Mol. Cancer Ther., 2007, 6, 1276–1282 CrossRef CAS PubMed.
- V. T. Huynh, S. Pearson, J. M. Noy, A. Abboud, R. H. Utama, H. X. Lu and M. H. Stenzel, ACS Macro Lett., 2013, 2, 246–250 CrossRef CAS.
- V. T. Huynh, J. Y. Quek, P. L. de Souza and M. H. Stenzel, Biomacromolecules, 2012, 13, 1010–1023 CrossRef CAS PubMed.
- M. Callari, J. Aldrich-Wright, P. L. de Souza and M. H. Stenzel, Prog. Polym. Sci., 2014, 39(9), 1614–1643 CrossRef CAS PubMed.
- H. T. T. Duong, V. T. Huynh, P. de Souza and M. H. Stenzel, Biomacromolecules, 2010, 11, 2290–2299 CrossRef CAS PubMed.
- Y. Kim, M. H. Pourgholami, D. L. Morris, H. Lu and M. H. Stenzel, Biomater. Sci., 2013, 1, 265–275 RSC.
- V. T. Huynh, S. Binauld, P. L. de Souza and M. H. Stenzel, Chem. Mater., 2012, 24, 3197–3211 CrossRef CAS.
- J. Skey and R. K. O'Reilly, Chem. Commun., 2008, 4183–4185 RSC.
- A. Gregory and M. H. Stenzel, Prog. Polym. Sci., 2012, 37, 38–105 CrossRef CAS PubMed.
- D. R. Chen, J. Z. Bei and S. G. Wang, Polym. Degrad. Stab., 2000, 67, 455–459 CrossRef CAS.
- S. Sinnwell, A. J. Inglis, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Chem. Commun., 2008, 2052–2054 RSC.
- J. Xu, J. He, D. Fan, X. Wang and Y. Yang, Macromolecules, 2006, 39, 8616–8624 CrossRef CAS.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS.
- S. v. Zutphen and J. Reedijk, Coord. Chem. Rev., 2005, 249, 2845–2853 CrossRef PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- S. Choi, C. Filotto, M. Bisanzo, S. Delaney, D. Lagasee, J. L. Whitworth, A. Jusko, C. Li, N. A. Wood, J. Willingham, A. Schwenker and K. Spaulding, Inorg. Chem., 1998, 37, 2500–2504 CrossRef CAS.
- Y. Kim, M. H. Pourgholami, D. L. Morris and M. H. Stenzel, Biomacromolecules, 2012, 13, 814–825 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- A. Mero, O. Schiavon, G. Pasut, F. M. Veronese, E. Emilitri and P. Ferruti, J. Bioact. Compat. Polym., 2009, 24, 220–234 CrossRef CAS PubMed.
- S. Minear, A. F. O'Donnell, A. Ballew, G. Giaever, C. Nislow, T. Stearns and M. S. Cyert, Eukaryotic Cell, 2011, 10, 1574–1581 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, GPC and DLS of the block copolymers. See DOI: 10.1039/c4bm00272e |
| This journal is © The Royal Society of Chemistry 2015 |