
Thiol–ene immobilisation of carbohydrates onto glass slides as a simple alternative to gold–thiol monolayers, amines or lipid binding†
Caroline I.
Biggs
a,
Steve
Edmondson
b and
Matthew I.
Gibson
 *ac
*ac
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: m.i.gibson@warwick.ac.uk
bSchool of Materials, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cWarwick Medical School, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK
First published on 18th September 2014
Abstract
Carbohydrate arrays are a vital tool in studying infection, probing the mechanisms of bacterial, viral and toxin adhesion and the development of new treatments, by mimicking the structure of the glycocalyx. Current methods rely on the formation of monolayers of carbohydrates that have been chemically modified with a linker to enable interaction with a functionalised surface. This includes amines, biotin, lipids or thiols. Thiol-addition to gold to form self-assembled monolayers is perhaps the simplest method for immobilisation as thiolated glycans are readily accessible from reducing carbohydrates in a single step, but are limited to gold surfaces. Here we have developed a quick and versatile methodology which enables the use of thiolated carbohydrates to be immobilised as monolayers directly onto acrylate-functional glass slides via a ‘thiol–ene’/Michael-type reaction. By combining the ease of thiol chemistry with glass slides, which are compatible with microarray scanners this offers a cost effective, but also useful method to assemble arrays.
Introduction
Due to a combination of the global decrease in the discovery of new antibiotics and the increase in antibiotic resistance,1 the need for new technologies to investigate bacterial infection and allow rapid diagnoses are more important than ever. Prior to the occurrence of infection, pathogens typically must adhere onto the host cells, which is commonly through protein–carbohydrate interactions.2 This is a key step in the infection process for pathogens such as cholera toxin, human immunodeficiency virus (HIV) or Escherichia coli (E. coli) Probing these interactions can be used to detect the bacteria, provide structural information on their adhesion proteins (lectins) and carbohydrate specificities.3 Furthermore, understanding these interactions is assisting the development of anti-adhesion therapy, which is a promising prophylactic alternative to conventional antibiotics.4 These glycan–lectin interactions are also crucial in virus biology and can be potential targets for antiviral strategies.5However, glycans (carbohydrates) are inherently complex and dynamic and do not display specific receptor–ligand interactions in the same manner as e.g. biotin/streptavidin, but it is actually observed that lectins are capable of binding a range of related carbohydrate structures, to varying extents. This promiscuity underlines the challenge of glycomics and assigning protein–carbohydrate interactions. To overcome this inherently complex problem, carbohydrate microarrays, whereby 10's or 100's of carbohydrates are immobilised onto a solid substrate have been developed.6
Carbohydrate arrays have transformed a number of medical and biological research areas, and can be considered as simplified biomimics of the cell glycocalyx.7 The screening of human sera using microarrays has been used to identify antibodies for the malaria toxin8 serological markers for Crohn's disease9 and glycoprotein arrays have been developed to study HIV glycobiology.10
There are two general immobilisation strategies employed to assemble carbohydrate arrays: either based on covalent interactions or based on non-covalent interactions.11 Non covalent methods include hydrophobic binding of lipids or non-specific absorption onto nitrocellulose-coated surfaces12 or black polystyrene.13 Whilst straightforward these non-covalent methods can be liable to washing-off effects.14 Covalent immobilisation can be achieved in both a site specific and a non-specific manner. For example, cyanuric chloride surfaces react with hydroxyl groups, as do boronic acids,15 phtalimides16 and azidoaryl17 coated surfaces. Their advantage of requiring no chemical modification is limited by the multiple orientations in which the glycans can be presented. Hydrazine or amino-oxy functionalities can react specific with the reducing termini of sugars, to give predominantly ring closed structures, but again this does give a mixture. For these reasons, site specific covalent attachment using orthogonal linkers incorporated into the saccharide are widely employed,11 including the Consortium for Functional Glycomics which employed amino linkers to react with succinimidyl ester glass slides.18 ‘Click’ chemistry inspired routes have also been employed, utilising alkyne or azido-sugars with the complementary surfaces. More recently, thiol–ene type click reactions have gained attention in both their radical and nucleophilic (Michael addition) formats,19 particularly in materials chemistry for polymer and surface modification.
We therefore rationalised that thiol–ene type chemistries would be the ideal method to install carbohydrates onto array-type surfaces as thiolylated carbohydrates are readily accessible and orthogonal to most functional groups (amines, sulphates, phosphates, alcohols). Thiolylated carbohydrates have also been explored for assembly onto gold surfaces as self-assembled monolayers.20 In this work, we demonstrate that thiolated monosaccharides can be conveniently attached to glass slides which have been simply coated with an acrylated silane, forming a SAM (self-assembled monolayer)-type monolayer which is suitable for probing carbohydrate–lectin interactions.
Results and discussion
Glass slides are desirable, versatile, surfaces for covalent carbohydrate immobilisation due to their availability, low cost and compatibility with most array scanners. However, they are not intrinsically reactive towards thiols, our desired bio-orthogonal group to introduce carbohydrates, and is summarised against current technologies in Fig. 1. To incorporate thiol-reactive acrylate functionality onto the glass (for ‘thiol–ene click’) 3-(trimethoxysilyl)propyl acrylate was chosen. Silanes are widely employed for producing ultra-thin coatings on inorganic substrates. Following cleaning with ‘piranha solution’ (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2SO4:H2O2), the silane was added to the surface by solution phase deposition from toluene. Silicon wafers were employed in addition to glass slides due to their very similar silicon dioxide surfaces and to enable chemical characterisation by XPS (X-ray photoelectron spectroscopy) and ellipsometry, which are both more challenging to conduct on glass. Following cleaning, the static water contact angle of the glass and silicon surfaces was ∼20° which increased to 50° (glass) and 70° (silicon) following addition of the silane, indicating successful surface functionalization, with the changes in surface energy in line with literature reports21,22 (Fig. 2). To probe the surface functionality and to demonstrate that the surface acrylate is available for further reactions, a range of thiols were added to the surface and changes in contact angles measured (Fig. 2). On glass surfaces, addition of dodecane-thiol lead to an increase in contact angle to 65° due to the addition of hydrophobic alkyl chains, and addition of thioglycerol lead to a lower value indicating hydrophilicity consistent with the presence of hydroxyl groups. Addition of thio-glucose (Glc-SH) gave rise to an unexpectedly high contact angle, but this is in line with other reports for the comparison of glucose and galactose on glass.23 The role of an amine catalyst on the thiol–ene functionalization was also probed on the silicon surface, ethanolamine was chosen as a suitable primary amine. Amines have been reported to accelerate the thiol–ene reaction in solution, by catalysing the Michael addition process,24 but surprisingly no significant differences were observed here in terms of the contact angle. This observation does not rule out amine catalysis (vide infra), but rather suggests that the degree of functionalization does not have a linear correlation with the observed contact angle or that the functionalization is essentially complete without catalysis. The differences in absolute contact angles between glass and silicon are likely due to the complex dependence of contact angle on the roughness of the substrate, especially for static angles.25
:1 H2SO4:H2O2), the silane was added to the surface by solution phase deposition from toluene. Silicon wafers were employed in addition to glass slides due to their very similar silicon dioxide surfaces and to enable chemical characterisation by XPS (X-ray photoelectron spectroscopy) and ellipsometry, which are both more challenging to conduct on glass. Following cleaning, the static water contact angle of the glass and silicon surfaces was ∼20° which increased to 50° (glass) and 70° (silicon) following addition of the silane, indicating successful surface functionalization, with the changes in surface energy in line with literature reports21,22 (Fig. 2). To probe the surface functionality and to demonstrate that the surface acrylate is available for further reactions, a range of thiols were added to the surface and changes in contact angles measured (Fig. 2). On glass surfaces, addition of dodecane-thiol lead to an increase in contact angle to 65° due to the addition of hydrophobic alkyl chains, and addition of thioglycerol lead to a lower value indicating hydrophilicity consistent with the presence of hydroxyl groups. Addition of thio-glucose (Glc-SH) gave rise to an unexpectedly high contact angle, but this is in line with other reports for the comparison of glucose and galactose on glass.23 The role of an amine catalyst on the thiol–ene functionalization was also probed on the silicon surface, ethanolamine was chosen as a suitable primary amine. Amines have been reported to accelerate the thiol–ene reaction in solution, by catalysing the Michael addition process,24 but surprisingly no significant differences were observed here in terms of the contact angle. This observation does not rule out amine catalysis (vide infra), but rather suggests that the degree of functionalization does not have a linear correlation with the observed contact angle or that the functionalization is essentially complete without catalysis. The differences in absolute contact angles between glass and silicon are likely due to the complex dependence of contact angle on the roughness of the substrate, especially for static angles.25
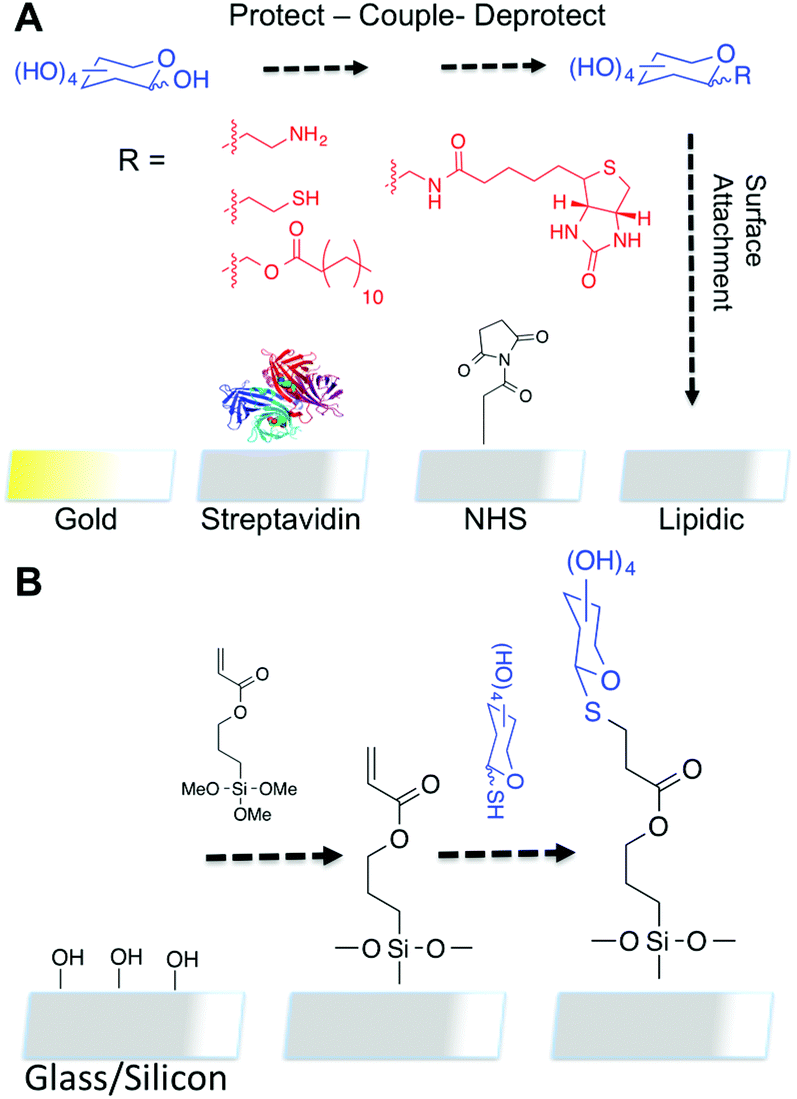 | ||
| Fig. 1 (A) Summary of some current technologies requiring specific surfaces and linkers; (B) Proposed thiol–ene route using acrylate glass/silicon as template surface for forming covalently tethered monolayers. | ||
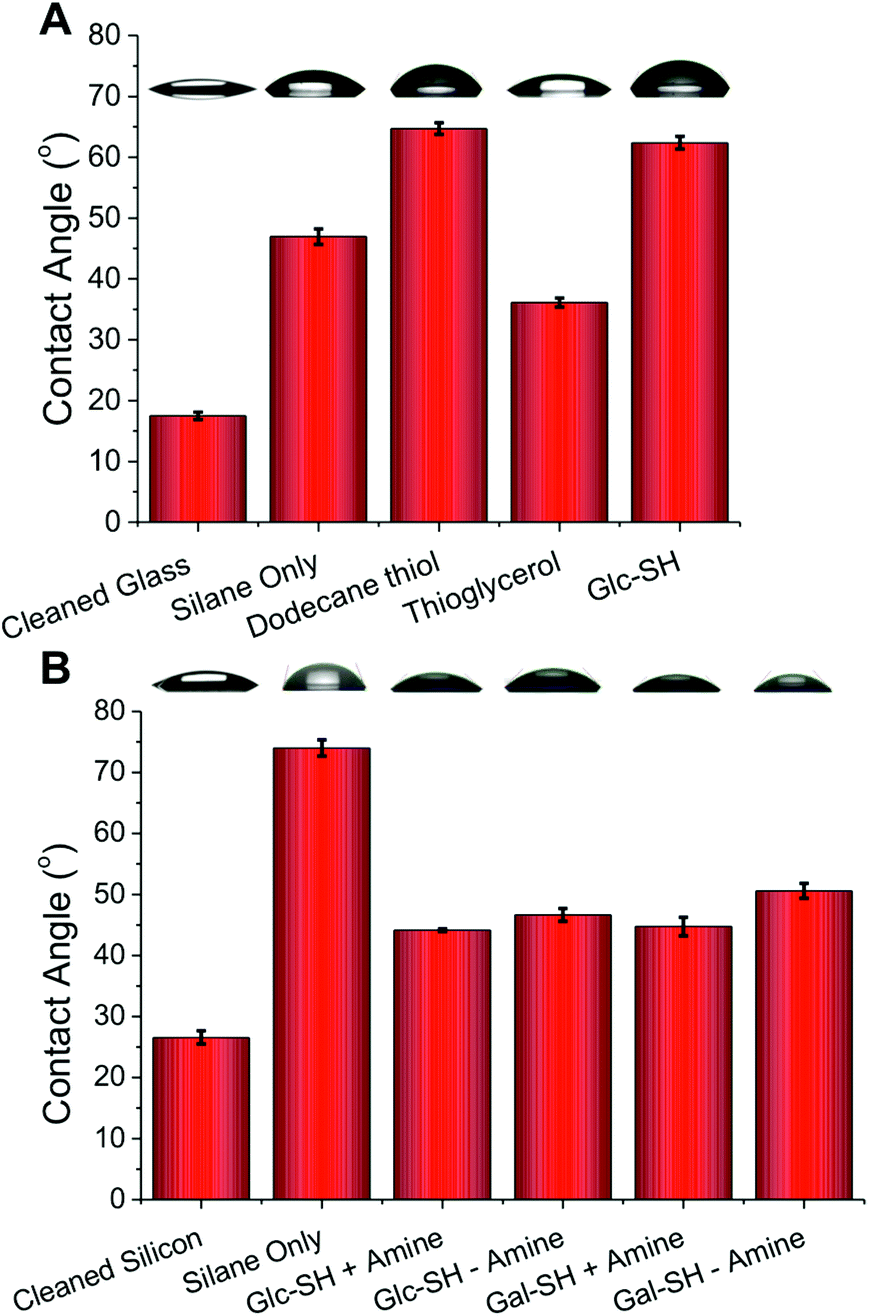 | ||
| Fig. 2 Contact angle measurements on functionalised surfaces; (A) Glass slides; (B) Silicon wafers ± amine indicates if catalytic ethanolamine was added. Error bars represent ± standard deviation from a minimum of 3 measurements. Representative image of the water droplets used to obtained measurements are shown above each bar. | ||
To further probe the surface coating, ellipsometry was employed on the silicon chips, and the measured thickness of each individual layer is shown in Fig. 3. The initial silane layer was found to be 0.6 nm thick suggesting that a monolayer (rather than thick multilayers which can form during solution-phase deposition due to siloxane polycondensation) was formed. Subsequent addition of thiols resulted in an increase in thickness of 0.4–0.5 nm for all thiols with or without addition of amine, which supports the contact angle evidence that amine addition does not change the surface properties of the films. The total layer thickness is also consistent with the formation of monolayers of thiols (as hoped for), rather than uncontrolled deposition.
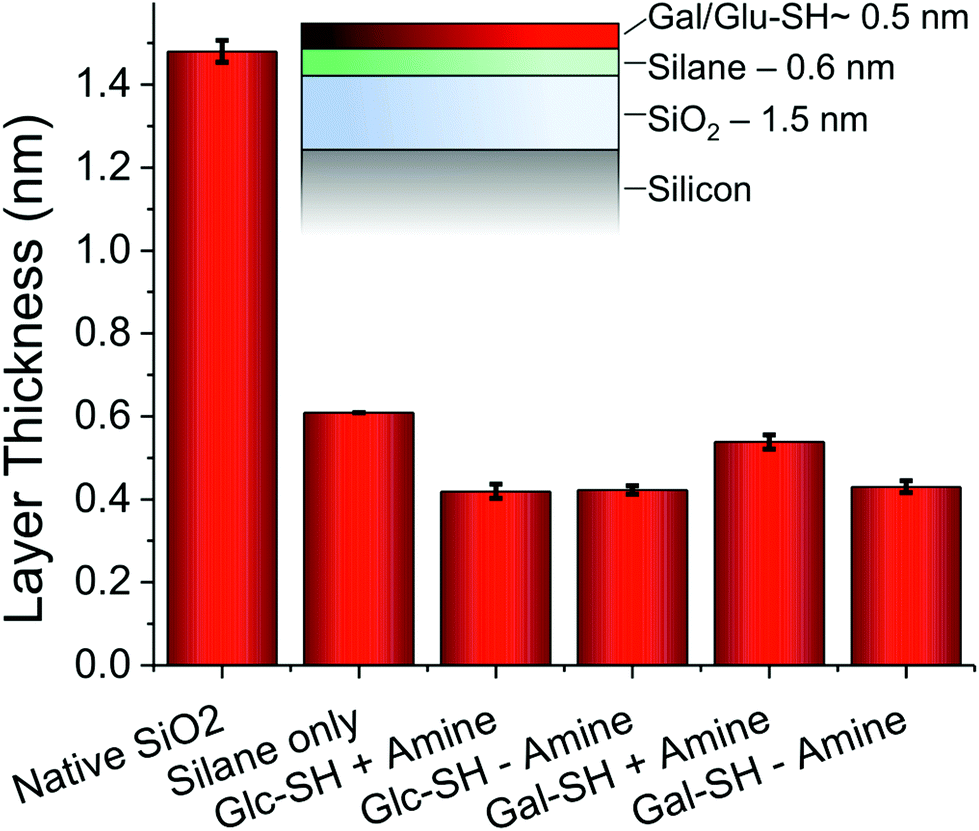 | ||
| Fig. 3 Layer thicknesses determined by ellipsometry on silicon wafers. Indicated thickness is of each individual layer, not cumulative thickness. Error bars represent standard deviation from minimum of 3 independent measurements. | ||
As a final analytical method to determine the chemical composition of the surface, XPS was employed. Fig. 4A shows the carbon: silicon ratio upon addition of the thiols/amines. The clear increase in the relative carbon concentration following the addition of the glycosyl-thiols is indicative of successful thiol–ene coupling. Since the total layer thickness increases after coupling, as confirmed by ellipsometry, the limited penetration depth of XPS (typically 5–10 nm) results in less of the underlying silicon being detected. For both Gal-SH and Glc-SH there was a significant increase in the carbon:silicon ratio upon addition of the amine catalyst. This is in contrast to the results of contact angle and ellipsometry that could not resolve these differences in grafting density. A representative C 1s region of the spectra for Gal-SH is shown in Fig. 4B to demonstrate that the increased carbon signal is attributable to functionality consistent with a carbohydrate being present (e.g. C–C, C–O).26 This analysis demonstrates the need for both molecular and surface property measurements for new array surfaces, otherwise the role of added amine would not have been elucidated from contact-angle/ellipsometry measurements alone.
 | ||
| Fig. 4 X-ray photoelectron spectroscopy analysis of carbohydrate functionalised surfaces; (A) Change in carbon:silicon ratio upon addition of different sugars; (B) Representative high-resolution XPS spectrum of C 1s region before and after addition of thio-galactose with amine catalyst. | ||
Having confirmed the successful, and facile, thio–ene glycosylation of the glass slides and silicon wafers, it was possible to evaluate their use for probing carbohydrate–protein interactions under array-type conditions. A series of surfaces were prepared bearing either silane alone or Gal-SH/Glc-SH. These surfaces were exposed to fluorescent fluorescein-labelled lectins; PNA (peanut agglutinin) and Con A (Concanavalin A). PNA has preference for galactose resides and Con A for glucose/mannose. The lectins were added to the surface at a concentration of 0.1 mg mL−1 and incubated with the surface for 30 minutes at 37 °C before being extensively washed and their fluorescence measured. As would be expected at this lectin concentration, the galactose surface had more PNA bound than Con A, and the glucose surface had more Con A than PNA.
As mentioned in the introduction, carbohydrate–lectin interactions tend to be non-specific, and are better described as being preferential. Multivalent presentation has also been reported to affect specificity, which must be considered when applying arrays27 along with considerations of the relative fluorescent labelling density of different proteins in comparative glycomics. Hence, there was still some binding of each lectin to their non-native carbohydrate target. Quantitative evaluation is shown in Fig. 5B.
 | ||
| Fig. 5 Lectin binding onto differentially functionalised surfaces using 0.1 mg mL−1 of FITC-lectin. (A) Collated array-scanner fluorescence micrographs (green/red channels); (B) Quantitative analysis of total green fluorescence (excitation 480 nm, emission 520 nm). Error bars show standard deviation from minimum of 3 independent measurements. | ||
A commonly employed array format is based on the SP8 linker ((CH2)3NH2), which is used to immobilise amino sugars onto NHS-functional surfaces.28Fig. 6 shows a comparison of our linkage method relative to SP-8 linked carbohydrates, presenting a similar alkyl chain length (which is required to ensure the lectin can access the carbohydrate on the surface) and hence should enable comparisons to be made in the future between these surfaces, and those already used elsewhere.
 | ||
| Fig. 6 Comparison of (A) thiol–ene immobilisation versus (B) NHS/SP8 immobilisation. | ||
Conclusions
This work has demonstrated the feasibility, and simplicity of thiol–gold self-assembled monolayers can be extended to cheaper and more readily available glass substrates, through the use of silane linkers. The addition of an acrylate-functional silane onto the glass or silicon surface provides an orthogonally reactive layer for the subsequent covalent immobilisation of thiolated carbohydrates. The characterisation, by drop shape analysis, ellipsometry, XPS and fluorescence binding assay, demonstrated the successful modulation of surface properties. Importantly, the role of the amine catalyst was studied and shown to increase binding density, which was only revealed by elemental XPS analysis rather than surface properties only. Using fluorescence binding, the selectivity of the carbohydrates towards two model lectins was measured. This methodologies main strength lies in its simplicity, and versatility and can be extended to other biomolecules and surfaces.Experimental
Materials and methods
All reagents and solvents were used as received from the supplier. Laboratory solvents were purchased from Fisher Scientific, 3-(trimethoxysilyl)propyl acrylate and (3-glycidyloxypropyl)-trimethoxysilane from Sigma-Aldrich. Microscope slides were purchased from Fisher Scientific (ground edges, plain glass, product code: 12383118) and silicon wafers from IDB Technologies, with a resistivity of 1–10 Ω. Phosphate-buffered saline (PBS) solution was prepared by dissolving a pre-formulated tablet (Sigma-Aldrich) in 200 mL ultra-high quality water. The resulting PBS solution has a composition of 0.01 M phosphate, 0.0027 M potassium chloride and 0.138 M sodium chloride, pH 7.4. 10 mmol HEPES buffer, containing 0.1 mmol CaCl2, pH 6.5, was prepared in 250 mL ultra-high quality water. Ethanolamine was purchased from Sigma-Aldrich, thiosugars (1-thio-β-D-glucose sodium salt and 1-thio-β-D-galactose sodium salt) from Carbosynth and fluorescently labelled lectins (PNA, Con A) from Vector Labs (Fluorescein FLK-2100 labelled).Contact angle measurements
The water contact angle measurements were conducted at room temperature using a Krüss drop shape analysis system DSA100 equipped with a movable sample table and microliter syringe. Deionized water was used as the wetting liquid and the drop size was set to 10 μL. Samples were placed onto the sample table, using tweezers, and aligned within the field of view of the camera. The microliter syringe was advanced until a drop of 10 μL was formed and suspended at the end of the syringe needle. The sample table was then elevated, until the sample touched the bottom of the drop, causing it to detach from the end of the needle and form on the surface. The sample table was the moved back to the original position and an image immediately recorded. The baseline and contact advancing angles were then computed from the image. This process was repeated five times for each sample and the reported values are the average taken from the repeat measurements.Ellipsometery
Ellipsometry measurements were carried out on a Nanofilm auto-nulling imaging ellipsometer with a resolution of 0.001° (delta and psi). A 550 nm wavelength light source was used and all measurements were taken using an angle of incidence scan at 50, 60 and 70° using four zone nulling. Firstly a cleaned silicon surface was measured. It was placed upon the sample table, under the alignment laser dot and automatically aligned to be level. Following the set up procedure (setting of stage-height to find maximum intensity, rotating the polariser to find the brightest image and calibrating the focus based on a small contaminant, such as a speck of dust) the an angle of incidence scan was performed. The values of psi and delta for each angle were recorded and inputted into the WVase software. The thickness of the oxide layer present on the silicon was calculated by adding a “SiO2” layer29 into the model and fitting the data to the model, to obtain the thickness. The sample could then be silanated and remeasured, with the organic layer modelling the layer using a Cauchy layer (i.e. with optical constants approximated using the Cauchy dispersion equation) on top of the existing SiO2 layer. Further layers added on top of the silane were modelled using the same single Cauchy layer, the thickness of such layers being calculated by subtracting the value previously obtained for the silane away from the total thickness of the Cauchy layer. Three values for each sample were recorded and the values stated are the average of these repeats and the error bars represent the standard deviation within the measurements. Each value obtained from the software also has an experimental error associated with it and a mean-square error between fitted and measured delta and psi values.X-ray photoelectron spectroscopy
The samples were mounted on to Omicron-style sample plates using electrically-conductive carbon tape and loaded in to the fast-entry chamber. Once the fast-entry chamber had been evacuated to an appropriate pressure, the samples were transferred in to the 12-stage carousel for storage at pressures of less than 1 × 10−10 mbar. XPS data were acquired in the main analysis chamber using an Omicron SPHERA analyser (Omicron Nanotechnology, UK). Core level XPS spectra were recorded using a pass energy of 10 eV (0.47 eV resolution), with the sample illuminated using an Omicron XM1000 Al Kα X-ray source (hν = 1486.6 eV). Analysis of the XPS data was carried out using the CasaXPS software, using mixed Gaussian–Lorentzian (Voigt) lineshapes. The transmission function of the analyser has been carefully determined using clean Au, Ag and Cu foils, whilst the work function of the analyser was determined using the Fermi edge of a polycrystalline Ag sample at regular intervals throughout the experiment, thereby allowing accurate composition and binding energy shifts to be determined. All binding energies have been referenced to the C 1s peak arising from adventitious carbon at 284.6 eV, a necessary correction due to the insulating nature of the oxide termination of the Si substrate.Microarray scanner
The fluorescence images were obtained using an Agilent G2565CA Scanner with a 2 μm resolution. Standard two colour scanning protocols were used with a SHG-YAG laser (532 nm) and a helium–neon laser (633 nm). The top left hand corner of each slide was marked with permanent pen, away from the lectin spots, and the slides were each placed into a sample holder; orientated so that the resulting image would correspond to the original slide orientation. The samples were loaded and the standard two colour scan was run, producing the data as a Tagged Image File (TIF). The resulting image files were analysed using Agilent Feature Extraction Software. The average fluorescent intensity was calculated for the sample area of interest (the lectin spot) by taking the average output value for the green channel for that set area. The background fluorescence (the average output value for the green channel for all of the areas of the sample without a lectin spot or pen mark) was calculated manually and subtracted.Experimental procedures
Surface cleaning
The solid surfaces used in this work (glass slides and silicon wafers) were cleaned using piranha solution [caution – reacts violently with organic material]. The surfaces were placed into a 3:1 (v/v) mixture of 98% sulphuric acid and 30% hydrogen peroxide, on ice, for 20 minutes, then rinsed with deionized water and dried in a gentle stream of dry nitrogen.
Silanization and control surfaces
Immediately following the cleaning process, the samples were immersed into a solution of 3-(trimethyoxysilyl)propyl acrylate (5 mL, 2% v/v in toluene, 2 hours, RT), washed with toluene (5 × 2 mL) and water (5 × 2 mL), then blown under a stream of nitrogen until dry. This process applied to the glass slides and the silicon wafers.Carbohydrate functionalisation of silane coated surfaces
Following silanization, the samples were immersed into the chosen thiolated sugar solution (2 mg mL−1 in water) for 2 hours (RT) then washed with distilled water (3 × 2 mL) and dried under a stream of nitrogen. For the samples that were functionalised in the presence of amine, ethanolamine (0.1 mL) was added into the thiolated sugar solution prior to addition of the sample.Lectin binding studies
Samples were subjected to spots (20 μL) of each of the fluorescently labelled lectins at intervals of 20 mm: Con A (0.1 mg mL−1 in HEPES) and PNA (0.1 mg mL−1 in HEPEs, diluted from PBS stock) for 30 minutes (RT, dark). The protein solutions were then removed by pipette and the surface was washed (5 mL appropriate buffer, 2 × 5 mL deionised water) and dried under a stream of nitrogen. Samples were stored in the dark until tested on the fluorescence scanner.Acknowledgements
Equipment used was supported by the Birmingham Science City (SC) Advanced Materials project, with support from Advantage West Midlands and part funded by the European Regional Development Fund. MIG was a Science City Research Fellow, supported HEFCE. CIB has a PhD Scholarship from the BBSRC-funded Life Science Training Centre. Dr Marc Walker is thanked for performing XPS analysis on the UoW Science-City XPS facility.Notes and references
- H. C. Neu, Science, 1992, 257, 1064–1073 CAS.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS.
- R. J. Pieters, Med. Res. Rev., 2007, 27, 796–816 CrossRef CAS PubMed.
- N. Sharon, Biochim. Biophys. Acta, Gen. Subj., 2006, 1760, 527–537 CrossRef CAS PubMed.
- W. Van Breedam, S. Pöhlmann, H. W. Favoreel, R. J. de Groot and H. J. Nauwynck, FEMS Microbiol. Rev., 2014, 38, 598–632 CrossRef CAS PubMed.
- J. Hirabayashi, M. Yamada, A. Kuno and H. Tateno, Chem. Soc. Rev., 2013, 42, 4443–4458 RSC.
- P. H. Seeberger and D. B. Werz, Nature, 2007, 446, 1046–1051 CrossRef CAS PubMed.
- L. Schofield, M. C. Hewitt, K. Evans, M. A. Siomos and P. H. Seeberger, Nature, 2002, 418, 785–789 CrossRef CAS PubMed.
- I. Dotan, S. Fishman, Y. Dgani, M. Schwartz, A. Karban, A. Lerner, O. Weishauss, L. Spector, A. Shtevi, R. T. Altstock, N. Dotan and Z. Halpern, Gastroenterology, 2006, 131, 366–378 CrossRef CAS PubMed.
- D. M. Ratner and P. H. Seeberger, Curr. Pharm. Des., 2007, 13, 173–183 CrossRef CAS.
- N. Laurent, J. Voglmeir and S. L. Flitsch, Chem. Commun., 2008, 4400–4412 RSC.
- D. N. Wang, S. Y. Liu, B. J. Trummer, C. Deng and A. L. Wang, Nat. Biotechnol., 2002, 20, 275–281 CrossRef CAS PubMed.
- W. G. T. Willats, S. E. Rasmussen, T. Kristensen, J. D. Mikkelsen and J. P. Knox, Proteomics, 2002, 2, 1666–1671 CrossRef CAS.
- S. Park, J. C. Gildersleeve, O. Blixt and I. Shin, Chem. Soc. Rev., 2013, 42, 4310–4326 RSC.
- H.-Y. Hsiao, M.-L. Chen, H.-T. Wu, L.-D. Huang, W.-T. Chien, C.-C. Yu, F.-D. Jan, S. Sahabuddin, T.-C. Chang and C.-C. Lin, Chem. Commun., 2011, 47, 1187–1189 RSC.
- D. Wang, G. T. Carroll, N. J. Turro, J. T. Koberstein, P. Kovac, R. Saksena, R. Adamo, L. A. Herzenberg, L. A. Herzenberg and L. Steinman, Proteomics, 2007, 7, 180–184 CrossRef CAS PubMed.
- Z. Pei, H. Yu, M. Theurer, A. Walden, P. Nilsson, M. Yan and O. Ramstrom, ChemBioChem, 2007, 8, 166–168 CrossRef CAS PubMed.
- M. D. Disney and P. H. Seeberger, Chem. Biol., 2004, 11, 1701–1707 CrossRef CAS PubMed.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- B. T. Houseman and M. Mrksich, Chem. Biol., 2002, 9, 443–454 CrossRef CAS.
- J. Wang, M. I. Gibson, R. Barbey, S. J. Xiao and H. A. Klok, Macromol. Rapid Commun., 2009, 30, 845–850 CrossRef CAS PubMed.
- J. W. Grate, M. G. Warner, J. W. Pittman, K. J. Dehoff, T. W. Wietsma, C. Zhang and M. Oostrom, Water Resour. Res., 2013, 49, 4724–4729 CrossRef CAS.
- M. Janado and Y. Yano, J. Solution Chem., 1985, 14, 891–902 CrossRef CAS.
- G. Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196–1204 RSC.
- D. Quere, Physica a-Statistical Mechanics and Its Applications, 2002, 313, 32–46 CrossRef CAS.
- NIST, NIST X-ray Photoelectron Spectroscopy Database, Version 4.1, National Institute of Standards and Technology, Gaithersburg, 2012, http://srdata.nist.gov/xps/ Search PubMed.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2755–2794 CrossRef CAS.
- O. C. Grant, H. M. K. Smith, D. Firsova, E. Fadda and R. J. Woods, Glycobiology, 2014, 24, 17–25 CrossRef CAS PubMed.
- x, SiO2 DATA, from Palik Handbook of optical constants of solids Vol. 1, p. 759.
Footnote |
| † Electronic supplementary information (ESI) available: Full XPS and ellipsometry data. See DOI: 10.1039/c4bm00176a |
| This journal is © The Royal Society of Chemistry 2015 |