
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable luminescence of hyperbranched polysiloxanes by nonsaturation-induced electrostatic potential polarization for activatable fluorescent theranostics†
Yan
Zhao
 ,
Zhixuan
Feng
,
Miaomiao
He
,
Xiangyi
Wang
,
Weixu
Feng
,
Wei
Tian
and
Hongxia
Yan
*
,
Zhixuan
Feng
,
Miaomiao
He
,
Xiangyi
Wang
,
Weixu
Feng
,
Wei
Tian
and
Hongxia
Yan
*
Xi'an Key Laboratory of Hybrid Luminescent Materials and Photonic Device, School of Chemistry and Chemical Engineering, Northwestern Polytechnical University, Xi'an 710129, Shaanxi, China. E-mail: hongxiayan@nwpu.edu.cn
First published on 12th May 2025
Abstract
Developing high-efficiency luminescent polysiloxanes with activatable fluorescent theranostics is highly desirable in biomedicine. In this work, we compared hyperbranched polysiloxanes (HBPSis) with diverse nonsaturation and built an intelligent chemodynamic therapy (CDT) system with activatable fluorescence imaging. Experimental characterization and theoretical calculations reveal that increased nonsaturation offers numerous delocalized π electrons to modulate the polarization of electrostatic potential, which further drives the clusterization of carbonyl groups. Consequently, through–space interactions are enhanced, leading to a red-shifted emission and improved quantum yield. Furthermore, an intelligent CDT system, HBPSi–Fe3+@AT, is fabricated. Both the CDT process and fluorescence imaging can be activated in the presence of overexpressed glutathione. This work provides fresh insight into nonsaturation-induced electrostatic potential polarization and presents an activatable CDT system for versatile fluorescence imaging-guided theranostics.
Introduction
Activatable real-time fluorescence (FL) theranostics have become one of the most valuable approaches toward cancer diagnosis and therapy.1–3 Compared with the most commonly reported “always on” mode, activatable FL materials could be triggered by stimuli in the tumor microenvironment, such as pH,4,5 redox,6,7 enzymes or proteins,8,9etc. Such specifically designed activatable materials remain in the “off” state at normal tissues, while precisely switching to the “on” state with a specific stimulus in the tumor microenvironment, resulting in high sensitivity, a satisfactory signal-to-background ratio and minimal nonspecific damages.10 In this regard, activatable FL materials have been promising candidates due to their intrinsic features in cancer diagnosis and therapy, such as chemotherapy, photodynamic therapy, photothermal therapy, chemodynamic therapy, gene therapy, etc.11–14Unconventional polysiloxanes have been rising stars in activatable FL theranostics due to their negligible connate toxicity, tunable water-solubility and degradable features.15–17 Compared with traditional fluorescent polymers with large π conjugates, unconventional polysiloxanes contain electron-rich heteroatoms and Si atoms.18 The unique combination of inorganic Si–O bonds and organic C–O segments results in outstanding FL performance benefiting from the syncretic flexibility and rigidity. Generally, most polysiloxanes are limited by short-wavelength emission, low quantum yield (QY), and insufficient understanding of the structure–property relationships, especially for polysiloxanes with topological structures. Electrostatic potential (ESP) is a powerful tool that is derived from a reliable quantum chemical method and has been used in exploring the electronic features of molecules and identifying possible interaction sites.19–22 Since ESP is experimentally observable, its topological characteristics have been used as valuable electronic descriptors for localized/delocalized electron distributions.23–26 Additionally, ESP distributions have proven to be an effective method for the investigation of non-covalent interactions.27 Despite great progress, the deep understanding of how to accelerate electrostatic potential polarization in unconventional polysiloxanes and the role of ESP in the luminescence process still remains a puzzle.
In this work, we compared four HBPSis containing three diols with diverse nonsaturation and systematically studied their FL properties. A photophysical study indicates a red-shifted emission and enhanced QY with an increased degree of unsaturation. Experimental and theoretical results demonstrate that the delocalized π electrons generated by the increased degree of unsaturation can significantly regulate the ESP polarization and promote the electronic enrichment near O atoms in carbonyl groups, further leading to the clusterization of carbonyl groups (Scheme 1). Then, through–space interactions (TSIs), especially n⋯π TSI, are enhanced in the clusters, achieving a red-shifted emission at 512 nm with a QY of 29.6%. Therefore, we propose that nonsaturation of the segments can induce ESP polarization, which achieves tunability of the luminescence behaviors. In addition, we fabricated an intelligent CDT system with activatable FL imaging via the loading of 3-amino-1,2,4-triazole and coordination of Fe3+ with HBPSi-BYD. The fabricated HBPSi–Fe3+@AT exhibits a dual-promoted CDT effect with activatable FL imaging triggered by intracellular glutathione. This work contributes to a deeper understanding of nonsaturation-induced ESP polarization and provides a new CDT biomaterial for FL imaging-guided cancer therapy.
 | ||
| Scheme 1 Illustration of nonsaturation-induced ESP polarization and TSIs in HBPSis. | ||
Results and discussion
Studies towards the FL properties
In this work, HBPSi-BYD and HBPSi-MTMS were synthesized according to a reported transesterification polycondensation reaction (Fig. S1†),28 and their FL properties are compared with those of HBPSi-BDO and HBPSi-CBD. HBPSi-BYD is synthesized with 1-[3-(trimethoxysilyl)propyl]urea and 2-butyne-1,4-diol, which contain ureido and alkynyl groups. As a comparison, HBPSi-MTMS contains methyl and alkynyl groups, HBPSi-BDO contains ureido and alkyl groups, and HBPSi-CBD contains ureido and alkenyl groups. The chemical structures of HBPSi-BYD and HBPSi-MTMS were characterized by FTIR, 1H NMR, and gel permeation chromatography, and the characterization data are depicted in the ESI.† The characterization data of HBPSi-BDO and HBPSi-CBD can be found in ref. 29. The alkynyl groups alter the electronic distribution of polysiloxanes, thus influencing the FL features. According to the UV absorption spectra in Fig. 1B, HBPSi-BYD has three absorption peaks, located at 216 nm, 250 nm and ∼290 nm, respectively. The peak at 216 nm is caused by the n–σ* transition of free amine and hydroxyl groups, and that at 250 nm belongs to the n–σ* transition of the –Si(O)3 group. The maximum absorption peak appears at ∼290 nm, and an obvious absorption intensity increase was observed with the rise of HBPSi-BYD concentration. According to our previous research on unconventional polysiloxanes, the newly emerging long-wavelength λab should originate from the forbidden n–π* transition of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– and –C
C– and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.15,23 HBPSi-BDO and HBPSi-CBD also have absorption peaks around 220 nm, 260 nm and 300 nm.29 However, their λab at ∼300 nm is weak, and the strongest absorption appeared at ∼220 nm. Compared with HBPSi-BDO and HBPSi-CBD, the λab at ∼290 nm is dramatically enhanced in HBPSi-BYD, indicating that the alkynyl groups can promote the forbidden n–π* transition significantly.
O.15,23 HBPSi-BDO and HBPSi-CBD also have absorption peaks around 220 nm, 260 nm and 300 nm.29 However, their λab at ∼300 nm is weak, and the strongest absorption appeared at ∼220 nm. Compared with HBPSi-BDO and HBPSi-CBD, the λab at ∼290 nm is dramatically enhanced in HBPSi-BYD, indicating that the alkynyl groups can promote the forbidden n–π* transition significantly.
 | ||
| Fig. 1 (A) Chemical structures of HBPSi-BYD. (B) UV absorption spectra of HBPSi-BYD. (C) FL spectra of HBPSi-BYD (λex = 270 nm). (D) Plots of FL intensity versus concentration for HBPSi-BYD. (E) Emission spectra of HBPSi-BYD with various excitation wavelengths. (F) Corresponding Commission Internationale de L'Eclairage plots in (E). | ||
The FL spectra of HBPSi-BYD with different concentrations were further tested to kinetically monitor the emission variation during aggregation. Our preliminary studies have proved that the free electron-rich groups can form small clusters to generate emission peaks in the short-wavelength region (<400 nm).30,31 In Fig. 1C, as the concentration increases, the intensity at 300 nm weakened, and a new long-wavelength emission at ∼460 nm appeared (Fig. 1C and S7A†), the intensity of which gradually strengthened with the increase of concentrations. The decreased FL at 300 nm and the increased FL at ∼460 nm demonstrate the formation of stabilized larger clusters. The emission at ∼350 nm slightly strengthened with the rise in concentrations, suggesting the existence of moderate clusters. For a better explanation of the FL intensity variation in HBPSi-BYD, the emission intensity with varying concentrations was explored in detail. In Fig. 1D and S7B,† the FL intensity of HBPSi-BYD exhibited distinct stages. The turning point represents the critical cluster concentration (CCC), demonstrating the generation of stabilized clusters.32,33 The CCC values for HBPSi-BYD were found to be 0.67 mg mL−1 (300 nm), 1.20 mg mL−1 (350 nm) and 0.90 mg mL−1 (460 nm). As the concentration increases beyond CCC, the clusters accumulate and the molecular motions are restricted. Consequently, the FL intensity is dramatically enhanced.34,35 In the presence of alkynyl groups, the formation of clusters is obviously promoted, leading to a red-shifted emission at 512 nm and a high QY of 29.6%, as well as multicolor emissions. In comparison, HBPSi-BDO and HBPSi-CBD, which contain different non-saturations, exhibit the highest QY of only 16.1% with an FL emission at 484 nm.
Contrary to the above HBPSis, HBPSi-MTMS is a kind of polysiloxane that possesses minimum O/N atoms and abundant π electrons. It shows distinct absorption at 250 nm and ∼280 nm (Fig. S9A†) and weak λab at 216 nm due to the limited electron-rich atoms. Similarly, distinct FL stages were also observed in HBPSi-MTMS. According to Fig. S11C and D,† the CCC values for HBPSi-MTMS were 18.7 mg mL−1 (303 nm), 17.0 mg mL−1 (350 nm) and 18.7 mg mL−1 (451 nm), which were larger than that of HBPSi-BYD. The larger CCC values correspond to the emergence of clusters at higher concentrations, indicating that the formation of clusters in HBPSi-MTMS is limited by the deficient O/N atoms. As a result, the FL intensities in HBPSi-MTMS are weaker, and the QY is 4.9% with a maximal emission at 458 nm. The photoluminescence results reveal that the alkynyl groups are conducive to the formation of stabilized clusters.
Quantum chemical calculations on the FL mechanism
Next, the photoluminescence mechanism was further explored by density functional theory at the B3LYP/6-31G(d,p) level.36–38 To investigate the ESP in manipulating the FL performance of unconventional polysiloxanes, we compared the ESP of three HBPSis that contain ureido groups in silane and diols with different degree of nonsaturation. As presented in Fig. 2, the ESP diagrams of the three HBPSis reflect uniform to uneven distributions with an increase in the degree of unsaturation. In the ESP map of HBPSi-BYD, the positive charge is localized on the H atoms of hydroxyl and amino groups, and the negative charge is mainly distributed on the O atoms of carbonyl groups. An obvious electronic enrichment near O atoms in carbonyl groups was observed, which further led to the clusterization of carbonyl groups. Such results indicate that the delocalized π electrons in alkynyl groups can regulate the ESP polarization and facilitate the formation of carbonyl clusters, which is conducive to enhancing the TSIs in HBPSis. | ||
| Fig. 2 ESP maps and NTOs for HBPSi-BDO, HBPSi-CBD, and HBPSi-BYD, and their corresponding emission diagrams. | ||
Then, the TSIs in HBPSis were validated via the natural transition orbitals (NTOs). HBPSi-BDO is the polysiloxane with alkyl groups. According to their NTOs in Fig. 2, the hole is mainly localized on the ureido group, and the electron is distributed on the hydroxyl and amino groups, suggesting that the TSIs in HBPSi-BDO are mainly inter/intramolecular n⋯n TSIs. In the NTOs in HBPSi-CBD with alkenyl groups, both the hole and electron are distributed on the alkenyl and hydroxyl groups within the same molecule, indicating the existence of intramolecular n⋯n/n⋯π TSIs. The NTOs of HBPSi-BYD demonstrate different distributions. The hole is mainly distributed on the ureido group, and the electron is localized on alkynyl and hydroxyl groups. The orbital distributions in HBPSi-BYD further substantiate the inter/intramolecular n⋯n/n⋯π TSI. With the strengthened TSI, the energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) decreased. According to Fig. 2, the calculated ΔE gradually decreased from 7.32 eV to 5.29 eV with an increase in the degree of unsaturation.29 The lowering energy gap is closely related to the red shift in the corresponding emission. Consequently, the longest emission wavelength (512 nm) and the highest QY (29.6%) were observed in HBPSi-BYD.
The TSIs were also confirmed by the distance between electron-rich atoms and π units. According to Fig. S13,† the inter/intramolecular dO⋯O (2.94, 2.59, 2.80, 2.77, and 2.77 Å), dO⋯N (2.99, 2.92, 2.93, 2.96, 3.04, and 2.97 Å), dO⋯CO (2.92, 3.06, and 3.13 Å), dO⋯CC (2.41, 2.41, 2.91, 3.14, 3.11, 3.21, and 3.15 Å), and dN⋯CC (3.23 Å) in 4 HBPSi-BYD molecules are shorter than the sum of the van der Waals radii of the atoms, revealing the existence of inter/intramolecular n⋯n/n⋯π TSIs.39,40 With the increased molecular numbers, HBPSi-BYD becomes more compact due to the clusterization of carbonyl groups, and the n⋯n/n⋯π TSIs are enhanced. For example, 1, 2, 3 and 4 molecules of HBPSi-BYD show 1, 6, 9, and 11 pairs of n⋯n TSIs and 0, 1, 6, and 11 pairs of n⋯π TSIs. Interestingly, no π⋯π interactions are observed in Fig. S13,† indicating that the π–π stacking is negligible in HBPSi-BYD.
Different from the above polysiloxanes, HBPSi-MTMS is a kind of polysiloxane that possesses minimum O/N atoms and abundant π electrons. Its ESP map demonstrates a uniform distribution and the NTO calculation reveals that the hole and electron are localized on alkynyl and hydroxyl groups within the same molecule, suggesting intramolecular n⋯n/n⋯π TSIs (Fig. S16†). The uniform ESP distribution and weak TSIs are caused by the limited number of electron-rich atoms in HBPSi-MTMS. Consequently, a larger CCC value was observed, accompanied by a blue emission at 458 nm with a low QY of 4.9%. The calculations of HBPSi-MTMS reveal that the abundant n electrons are the prerequisite to enhance the FL performance.
According to the above results, a schematic could be summarized. For polysiloxanes with complex structures, the abundant electron-rich groups with n electrons are the prerequisite to induce electron delocalization. With the increased degree of unsaturation, more π electrons that participate in the electron delocalization are generated to induce ESP polarization. The electronic enrichment near O atoms in carbonyl groups further drives the clusterization of carbonyl groups, resulting in enhanced TSIs and a lowered energy gap, accompanied by red-shifted emission and enhanced FL efficiency. These findings confirm that nonsaturation of the segments can induce ESP polarization of unconventional polysiloxanes, which achieves tunability of the luminescence behaviors.
CDT performance of HBPSi–Fe3+@AT
Since HBPSi-BYD is a kind of electron-rich polymer with abundant O and N atoms, it generally coordinates with metal ions. As revealed in Fig. 3C, the FL intensity of HBPSi-BYD greatly weakens in the presence of Fe3+, but it is almost unchanged with Fe2+. As the Fe3+ concentration increased from 0 to 1 mM, the FL intensity progressively quenched. A linear relationship was observed in the range of 1–100 (R2 = 0.9902) μM, and the calculated limit of detection was 0.94 μM (Fig. 3D, inset). Such a phenomenon could be caused by the electron transfer between HBPSi-BYD and metal ions. Among various metal ions, Fe3+ has the largest charge radius ratio to coordinate with the O and N atoms in HBPSi-BYD to form a stabilized HBPSi–Fe3+ nanocomplex.41 Then, its strongest oxidizing ability quenches the FL of HBPSi-BYD via the electron transfer process, as evidenced by the FL lifetime tests in Fig. S17.† Consequently, Fe3+ quenched the FL the most while Fe2+ had negligible influence on HBPSi-BYD. Such fascinating behavior makes HBPSi-BYD a visual tracker for the transformation of Fe3+ and Fe2+.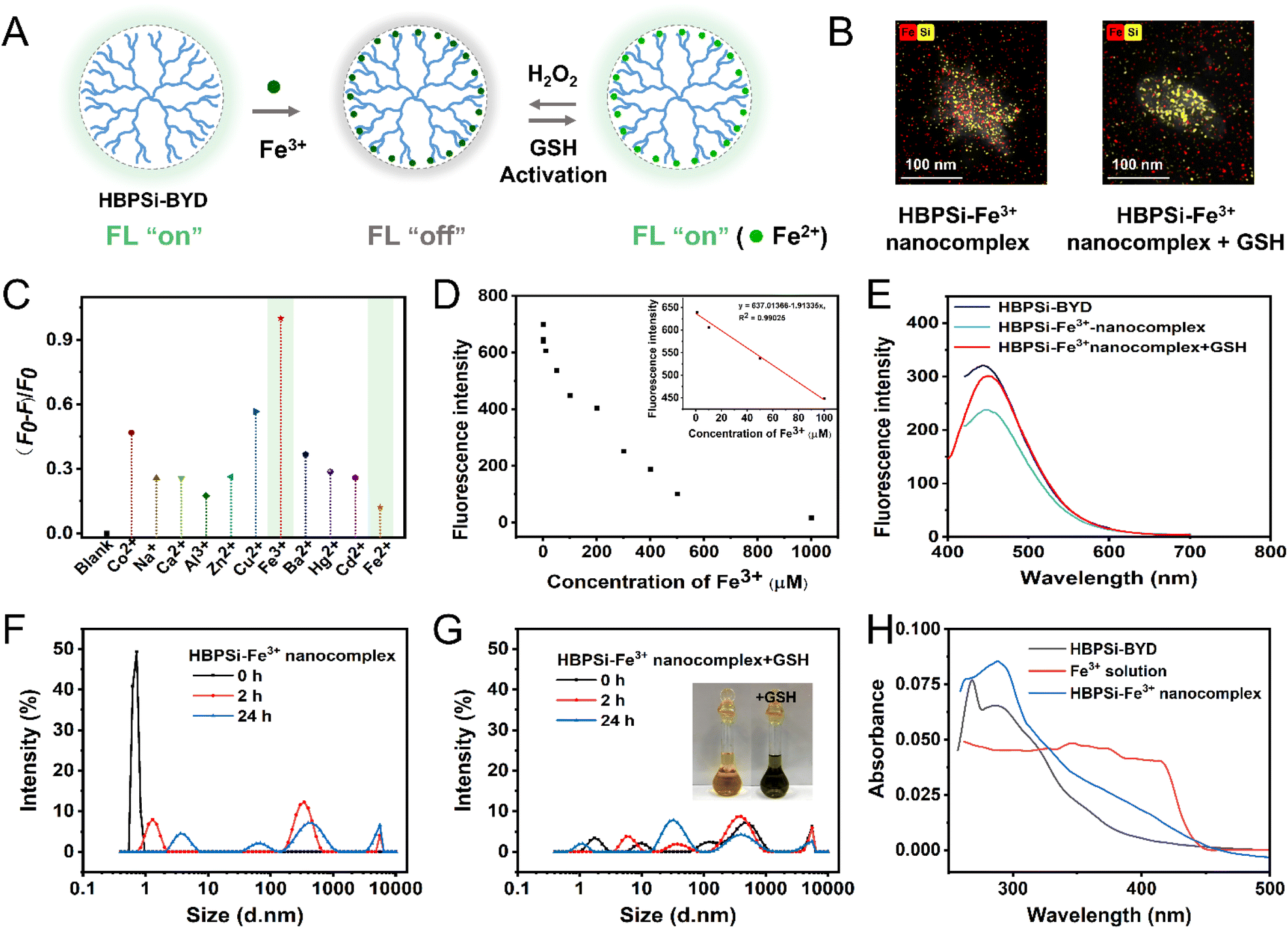 | ||
| Fig. 3 (A) Schematic illustration of the FL variation of HBPSi-BYD. (B) Elemental mapping images of the HBPSi–Fe3+ nanocomplex and GSH-treated HBPSi–Fe3+ nanocomplex. (C) FL intensity of HBPSi-BYD with metal ions. (D) FL intensity of HBPSi-BYD with various Fe3+ concentrations. Inset: linear relationship between FL intensity and Fe3+ concentrations. (E) FL intensity of HBPSi-BYD, the HBPSi–Fe3+ nanocomplex and the GSH-treated HBPSi–Fe3+ nanocomplex. (F) Size distributions of the HBPSi–Fe3+ nanocomplex. (G) Size distributions of the GSH-treated HBPSi–Fe3+ nanocomplex. (H) UV absorption spectra of HBPSi-BYD, Fe3+ solution and the HBPSi–Fe3+ nanocomplex. | ||
In addition, glutathione (GSH), a novel intracellular antioxidant, shows a satisfactory reducing ability towards Fe3+. As depicted in Fig. 3E, the addition of 1 mM Fe3+ leads to an obvious FL drop, and the subsequent addition of 10 mM GSH restores the FL, suggesting that GSH could reduce Fe3+ to Fe2+. Further, the formation of the HBPSi–Fe3+ nanocomplex and the GSH reduction process were confirmed by dynamic light scattering (DLS), UV absorption spectrophotometry and transmission electron microscopy (TEM). In Fig. 3F, the DLS data at 0 h revealed the existence of small aggregates with diameters of about 0.8 nm, indicating that the coordination between HBPSi-BYD and Fe3+ has not occurred yet with an incubation time of 0 h. With prolonged incubation time, larger aggregates appeared, mainly ascribed to the coordination between O/N atoms and Fe3+. Compared with Fe3+, Fe2+ has a weak coordination ability with HBPSi-BYD. As a result, Fig. 3G suggests decreased sizes after adding GSH. The formation of the HBPSi–Fe3+ nanocomplex was confirmed by UV absorption spectra. According to Fig. 3H, the absorption spectrum of the HBPSi–Fe3+ nanocomplex shows strong absorption at ∼288 nm, which is generated by the forbidden n–π* transition of –CC– and –CO in HBPSi-BYD. The strengthened absorption intensity at ∼288 nm suggests that HBPSi-BYD became more clustered in the presence of Fe3+. Moreover, the absorption spectrum of the HBPSi–Fe3+ nanocomplex also exhibits enhanced absorption at 340–430 nm compared to that of HBPSi-BYD, which may be caused by Fe3+. The UV absorption spectra reveal that Fe3+ could coordinate with HBPSi-BYD and lead to its aggregation. The TEM images also demonstrate similar trends to the DLS data. In Fig. S19B,† HBPSi–Fe3+ nanocomplexes exhibit an average diameter of 232 nm, which is larger than that of HBPSi-BYD aggregates (∼160 nm). The enlarged sizes are caused by the Fe3+ coordination-induced HBPSi-BYD aggregation. In the presence of GSH, the sizes of HBPSi–Fe3+ nanocomplexes decreased to ∼171 nm (Fig. S19C†), indicating that the larger aggregates dissociated into smaller ones in response to the GSH reduction within 15 min. The aggregate sizes of HBPSi-BYD, the HBPSi–Fe3+ nanocomplex and the GSH-treated HBPSi–Fe3+ nanocomplex are beneficial to endocytosis. To further confirm the coordination process between Fe3+ and HBPSi-BYD, we recorded element mapping images. According to Fig. 3B, the HBPSi–Fe3+ nanocomplex shows a compact structure with an obvious enrichment of Fe and Si elements, confirming the coordination of Fe3+ and HBPSi-BYD. The GSH-treated HBPSi–Fe3+ nanocomplex displays a smaller and looser structure with different element distributions. In the element mapping image of the GSH-treated HBPSi–Fe3+ nanocomplex in Fig. 3B, the Si element is mainly located inside the aggregate, and the Fe element shows a uniform distribution without local enrichment, suggesting that Fe3+ has been reduced to Fe2+ by GSH and Fe2+ did not coordinate with HBPSi-BYD. The reduction process was further confirmed by the color change. As shown in Fig. 3G (inset), the color of the HBPSi–Fe3+ nanocomplex changed from yellow to green after incubation with GSH, further confirming the reduction ability of GSH. Moreover, by alternately adding GSH and H2O2 to the HBPSi–Fe3+ nanocomplex, the FL switched between “off” and “on” states (Fig. 3A and S20B†). The above results demonstrated that GSH could restore the FL of HBPSi-BYD by reducing Fe3+ to Fe2+, and H2O2 could oxidize Fe2+ to Fe3+, accompanied by a switched FL readout.
Taking advantage of the unique FL performance of HBPSi-BYD and the reductive nature of GSH, we propose a GSH-triggered CDT system with activatable FL imaging based on the HBPSi–Fe3+ nanocomplex (Fig. 4). CDT is a tumor-specific therapeutic approach that generates highly toxic hydroxyl radicals (˙OH) via the Fenton or Fenton-like reaction with H2O2 in the tumor microenvironment and has been a rising star in selective tumor therapy.42–45 Moreover, to maintain a high level of H2O2, 3-amino-1,2,4-triazole (AT), a specific catalase inhibitor, was introduced into the cavities of HBPSi-BYD to fabricate HBPSi–Fe3+@AT. HBPSi–Fe3+@AT exhibits quenched FL and repressed CDT due to Fe3+. In the tumor microenvironment, the CDT function is triggered once Fe3+ is reduced to Fe2+ by GSH, accompanied by the recovered FL of HBPSi-BYD. Fe2+ catalyzes the Fenton-like reaction through the following reactions:
| 2Fe3+ + 2GSH → 2Fe2+ + GSSG + 2H+ | (1) |
| Fe2+ + H2O2 → Fe3+ + ˙OH + OH− | (2) |
 | ||
| Fig. 4 Illustration of the fabrication of HBPSi–Fe3+@AT and the dual-promoted CDT. | ||
Additionally, the released AT can suppress the catalase activity for maintaining H2O2 content, which is conducive to the generation of ˙OH. As a result, the introduction of Fe3+ provides extra Fe to disrupt iron metabolism, and the GSH depletion and AT-induced overexpressed H2O2 can disturb the intracellular redox homeostasis, leading to dual-promoted CDT with activatable FL imaging.
For in vitro CDT tests, salicylic acid was chosen as the trapping agent to evaluate the ˙OH generation.46,47 In the presence of ˙OH, the colorless salicylic acid can be oxidized to a colored compound with strong absorption at ∼510 nm. As shown in Fig. 5A, the HBPSi–Fe2+ nanocomplex shows a significant increase in the signal of oxidized salicylic acid, revealing the burst generation of ˙OH and confirming that Fe2+ works as a catalyst to initiate the Fenton-like reaction. In contrast, the absorbance of HBPSi–Fe3+ nanocomplex solution is weak, indicating that less ˙OH is generated by Fe3+. After co-incubation with GSH and the HBPSi–Fe3+ nanocomplex, the absorbance at ∼510 nm is almost the same as that of HBPSi–Fe2+ nanocomplex solution, confirming the proposed CDT strategy. These results demonstrated that HBPSi–Fe3+@AT is a potential candidate for GSH-triggered CDT.
 | ||
| Fig. 5 (A) UV absorption spectra of the HBPSi–Fe3+ nanocomplex, HBPSi–Fe2+ nanocomplex, or HBPSi–Fe3+ nanocomplex and GSH in salicylic acid solution. (B) UV absorption spectra of a mixture of HBPSi–Fe3+@AT and catalase, HBPSi–Fe3+@AT, or a mixture of the HBPSi–Fe3+ nanocomplex and catalase in salicylic acid solution in the presence of GSH. (C) Cytotoxicity of HBPSi-BYD, the HBPSi–Fe3+ nanocomplex and HBPSi–Fe3+@AT. The error bar suggests the standard deviation of 6 measurements. (D) Mean FL intensity of ROS in different cell groups. (E–H) Flow cytometric dot plots of CoLo 205 cells pre-incubated with PBS (E), HBPSi-BYD (F), the HBPSi–Fe3+ nanocomplex (G), and HBPSi–Fe3+@AT (H). (I–L) Representative fluorescence images of live/dead staining of CoLo 205 cells pre-incubated with PBS (I), HBPSi-BYD (J), the HBPSi–Fe3+ nanocomplex (K), and HBPSi–Fe3+@AT (L). (M–P) Confocal laser scanning microscope images of Hepa1-6 cells pre-incubated with PBS (M), HBPSi-BYD (N), the HBPSi–Fe3+ nanocomplex (O), and HBPSi–Fe3+@AT (P). | ||
Then, the in vitro catalase inhibition performance of AT was also tested with salicylic acid. In the UV absorption spectra in Fig. 5B, the presence of catalase can degrade H2O2 and produce an absorption of 2.54 L g−1 cm−1 in HBPSi–Fe3+ nanocomplex solution. As expected, the content of ˙OH was notably amplified after co-incubation with catalase and HBPSi–Fe3+@AT and showed a negligible difference with HBPSi–Fe3+@AT-treated solution (3.71 L g−1 cm−1), revealing that the loading of AT could effectively disrupt H2O2 metabolism to promote CDT.
Before antitumor tests, the cytotoxicity of HBPSi-BYD, the HBPSi–Fe3+ nanocomplex and HBPSi–Fe3+@AT against CoLo 205 cells was studied via a CCK-8 assay.48 As depicted in Fig. 5C, the cell viability slightly decreased after incubation with 100 μg mL−1 HBPSi-BYD for 24 h, which could be caused by the terminal amino groups that may damage cell membranes. In contrast, high cytotoxicity is observed in the HBPSi–Fe3+ nanocomplex or HBPSi–Fe3+@AT groups. Approximately 67% or 45% of cells remained alive when their equivalent concentrations reached 100 μg mL−1. Additionally, HBPSi–Fe3+@AT presents higher cytotoxicity than the HBPSi–Fe3+ nanocomplex with a significant difference (P < 0.01), confirming that AT is conducive to ˙OH generation via suppressing catalase activity. The cytotoxicity results are consistent with in vitro tests, suggesting that both the HBPSi–Fe3+ nanocomplex and HBPSi–Fe3+@AT can generate ˙OH, and HBPSi–Fe3+@AT exhibits a better cancer cell-killing effect.
Next, the cancer cell-killing effects of HBPSi-BYD, the HBPSi–Fe3+ nanocomplex and HBPSi–Fe3+@AT were investigated by the calcein AM and propidium iodide co-staining assay and flow cytometry analysis.49,50 In the calcein AM and propidium iodide co-staining assay, red and green FL represented dead cells and live cells, respectively. According to Fig. 5J, most cells remain alive after incubation with HBPSi-BYD. After being co-cultured with the HBPSi–Fe3+ nanocomplex or HBPSi–Fe3+@AT, more dead cells were observed, and HBPSi–Fe3+@AT shows the strongest cancer cell-killing ability (Fig. 5L). To quantitatively assess the CDT performance, flow cytometry was used to evaluate the cellular ˙OH content and monitor cell apoptosis. Fig. 5D depicts the detection of cellular ˙OH. Compared with untreated cells, FL intensity slightly increased after incubation with HBPSi-BYD. In contrast, the HBPSi–Fe3+ nanocomplex group and HBPSi–Fe3+@AT group showed enhanced FL intensity, and the highest signal was observed in the HBPSi–Fe3+@AT group, confirming that more ˙OH is generated by HBPSi–Fe3+@AT. Then, cells were double stained with Annexin V-FITC and propidium iodide.51 According to Fig. 5E, approximately 91.0% of untreated CoLo 205 cells remain alive. After incubation with HBPSi-BYD, the proportion of live cells is 75.6%, revealing the slight cytotoxicity of HBPSi-BYD. In addition, the proportion of live cells dramatically decreased to 53.5% in the HBPSi–Fe3+@AT group (Fig. 5H), showing a distinctly enhanced CDT effect. Based on the cellular test results, we believe that the designed HBPSi–Fe3+@AT can significantly dual-promote the CDT effect by disrupting the cellular iron metabolism and regulating redox homeostasis for amplified ˙OH production.
Besides the GSH-triggered CDT, the FL nature of HBPSi-BYD makes it a visual tracker for activatable FL imaging. The FL images of HBPSi-BYD, the HBPSi–Fe3+ nanocomplex or HBPSi–Fe3+@AT towards tumor cells (Hepa1-6) are recorded in Fig. 5N–P. In Fig. 5N, the HBPSi-BYD-treated Hepa1-6 cells exhibited bright FL emissions. In contrast, weak FL was observed in the HBPSi–Fe3+ nanocomplex group in Fig. 5O. Furthermore, obvious FL can be distinguished from HBPSi–Fe3+@AT treated cells accompanied by distinct karyopyknosis (Fig. 5P). The enhanced FL confirms that more Fe2+ is generated in the presence of HBPSi–Fe3+@AT, thus leading to improved CDT efficiency. However, negligible FL was observed in HBPSi–Fe3+@AT treated normal cells (Fig. S23†), revealing that HBPSi–Fe3+@AT was able to distinguish cancer cells from normal cells due to the different GSH contents. The FL imaging tests confirmed that HBPSi–Fe3+@AT can realize stimuli-triggered CDT with activatable FL imaging in cancer cells, which is a versatile system among theranostic platforms (Table S3†).
Conclusions
In summary, we systematically studied nonsaturation-induced ESP polarization and its influence on FL performance and constructed a stimuli-triggered CDT system with activatable FL imaging. Photoluminescence analysis emphasizes that the increased degree of unsaturation offers delocalized π electrons, which participate in electron delocalization with abundant n electrons, which leads to modulated ESP polarization with an obvious electronic enrichment near O atoms in carbonyl groups. The clusterization of carbonyl groups driven by ESP further enhances the TSIs and lowers the energy gap to promote the red-shifted emission at 512 nm with an enhanced QY of 29.6%. Combining the experimental and theoretical calculation results, we proposed a new mechanism that nonsaturation of the segments can induce ESP polarization, which further achieves tunability of the luminescence behaviors. In addition, HBPSi–Fe3+@AT, an intelligent stimuli-triggered CDT system with activatable FL imaging, has been successfully constructed. With the reduction of Fe3+ to Fe2+ by GSH, the CDT was triggered via the Fe2+ catalyzed Fenton-like reaction, accompanied by the recovered FL of HBPSi-BYD. The coordination of Fe3+ with HBPSi-BYD disrupts the intracellular iron metabolism, and the depleting GSH and AT-induced overexpressed H2O2 regulate the redox homeostasis, resulting in dual-promoted CDT. This work discovers a new strategy to regulate the FL behaviors based on nonsaturation-induced ESP polarization and offers new insights into understanding the luminescence process, as well as the development of a dual-promoted CDT system with activatable FL imaging.Data availability
The authors confirm that the data of this study are available within the article or its ESI.†Author contributions
Yan Zhao: methodology, validation, investigation, and writing – original draft. Zhixuan Feng: investigation and visualization. Miaomiao He: investigation. Xiangyi Wang: investigation. Weixu Feng: supervision. Wei Tian: supervision. Hongxia Yan: supervision and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22175143), Shaanxi Fundamental Science Research Project for Chemistry and Biology (23JHQ087) and the Innovation Capability Support Program of Shaanxi (2025RS-CXTD-022).Notes and references
- X. F. Wu, R. Wang, N. Kwon, H. M. Ma and J. Yoon, Chem. Soc. Rev., 2022, 51, 450–463 RSC.
- M. Zhao, B. Li, H. Zhang and F. Zhang, Chem. Sci., 2021, 12, 3448–3459 RSC.
- Y. Zhang, S. S. He, C. Xu, Y. Jiang, Q. Q. Miao and K. Y. Pu, Angew. Chem., Int. Ed., 2022, 61, e202203184 CrossRef CAS PubMed.
- X. Zhao, K. C. Zhao, L. J. Chen, Y. S. Liu, J. L. Liu and X. P. Yan, Chem. Sci., 2021, 12, 442–452 RSC.
- C. Qi, J. He, L. H. Fu, T. He, N. T. Blum, X. K. Yao, J. Lin and P. Huang, ACS Nano, 2021, 15, 1627–1639 CrossRef CAS PubMed.
- S. Sun, Q. Chen, Z. D. Tang, C. Liu, Z. J. Li, A. G. Wu and H. W. Lin, Angew. Chem., Int. Ed., 2020, 59, 21041–21048 CrossRef CAS PubMed.
- W. Wei, Y. Zhang, F. Yang, L. Zhou, Y. Zhang, Y. Wang, S. Yang, J. Li and H. Dong, Chem. Sci., 2023, 14, 5503–5509 RSC.
- S. Yan, P. Sun, N. Niu, Z. Zhang, W. Xu, S. Zhao, L. Wang, D. Wang and B. Z. Tang, ACS Nano, 2022, 16, 9785–9798 CrossRef CAS PubMed.
- Y. Yao, Y. Zhang, C. Yan, W. H. Zhu and Z. Guo, Chem. Sci., 2021, 12, 9885–9894 RSC.
- A. Mukherjee, P. C. Saha, R. S. Das, T. Bera and S. Guha, ACS Sens., 2021, 6, 2141–2146 CrossRef CAS PubMed.
- C. H. Zhang, K. Cai, P. G. Zhang, Z. Wu, M. Ma and B. Chen, Biosens. Bioelectron., 2022, 195, 113654 CrossRef CAS PubMed.
- S. Diao, Z. Zhang, S. Zhao, Q. Li, X. Zhang, X. Yang, Z. Xu, M. Liu, W. Zhou, R. Li, C. Xie and Q. Fan, Adv. Sci., 2024, 11, 2409833 CrossRef CAS PubMed.
- P. Chen, R. Wang, K. Wang, J. N. Han, S. Kuang, Z. Nie and Y. Huang, Chem. Sci., 2022, 13, 12187–12197 RSC.
- Y. Lai, Y. Dang, F. Li, C. Ding, H. Yu, W. Zhang and Z. Xu, Adv. Funct. Mater., 2022, 32, 2200016 CrossRef CAS.
- Y. Zhao, L. Xu, Y. He, Z. Feng, W. Feng and H. Yan, Aggregate, 2023, 5, e471 CrossRef.
- S. Wang, X. Wang, S. Feng, W. Lv, M. Lin, Q. Ling and Z. Lin, Inorg. Chem. Front., 2022, 9, 3619–3626 Search PubMed.
- D. A. Tomalia, B. Klajnert-Maculewicz, K. A. M. Johnson, H. F. Brinkman, A. Janaszewska and D. M. Hedstrand, Prog. Polym. Sci., 2019, 90, 35–117 CrossRef CAS.
- Y. Zuo, Z. Guo, W. Quan and W. Lin, Coord. Chem. Rev., 2021, 438, 213887 CrossRef CAS.
- C. H. Suresh and S. Anila, Acc. Chem. Res., 2023, 56, 1884–1895 CrossRef CAS PubMed.
- P. B. Torres, S. Baldor, E. Quiroga, A. J. Ramirez-Pastor, D. Spelzini, V. Boeris and C. F. Narambuena, Soft Matter, 2024, 20, 2100–2112 Search PubMed.
- X. J. Xie, Y. Wang, Q. Y. Cao, R. Krishna, H. Zeng, W. Lu and D. Li, Chem. Sci., 2023, 14, 11890–11895 RSC.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- Y. He, Y. Qiao, Z. Li, W. Feng, Y. Zhao, W. Tian, B. Z. Tang and H. Yan, Angew. Chem., Int. Ed., 2024, 63, e202413425 CrossRef CAS PubMed.
- H. Ji, Z. Wang, S. Wang, C. Wang, Y. Chu, H. Liu, Y. Zhang and L. Han, Adv. Funct. Mater., 2023, 33, 2213277 CrossRef CAS.
- H. Wang, Z. Zhu, L. L. Droitte, W. Liao, O. Cador, B. L. Guennic and J. Tang, Chem. Sci., 2023, 14, 7208–7214 RSC.
- A. Yahagh, R. R. Kaswan, S. Kazemi, P. A. Karrb and F. D'Souza, Chem. Sci., 2024, 15, 906–913 RSC.
- S. Jena, J. Dutta, K. D. Tulsiyan, A. K. Sahu, S. S. Choudhury and H. S. Biswal, Chem. Soc. Rev., 2022, 51, 4261–4286 RSC.
- Y. Zhao, M. He, L. Xu, C. Zhang, L. Guo, W. Feng and H. Yan, Biomacromolecules, 2023, 24, 1888–1900 CrossRef CAS PubMed.
- Y. Zhao, L. Xu, Z. Feng, S. Yin, W. Feng and H. Yan, Biomacromolecules, 2024, 25, 2635–2644 CrossRef CAS PubMed.
- Y. Zhao, B. Liu, R. Lou, Y. Qi, M. He, S. Long, W. Feng and H. Yan, Biomater. Adv., 2022, 17, 212848 CrossRef PubMed.
- Y. He, F. Ding, Y. Zhao, W. Tian, W. Feng and H. Yan, Polym. Chem., 2022, 13, 6534–6542 RSC.
- B. He, J. Zhang, J. Zhang, H. Zhang, X. Wu, X. Chen, K. H. S. Kei, A. Qin, H. H. Y. Sung, J. W. Y. Lam and B. Z. Tang, Adv. Sci., 2021, 8, 2004299 CrossRef CAS PubMed.
- Z. Zhang, H. Zhang, M. Kang, N. Li, D. Wang and B. Z. Tang, Sci. China: Chem., 2021, 64, 1990–1998 CrossRef CAS.
- Y. He, W. Feng, Y. Qiao, Z. Tian, B. Z. Tang and H. Yan, Angew. Chem., Int. Ed., 2023, 62, e202312571 CrossRef CAS PubMed.
- S. Yang, Y. Qu, L. Liao, Z. Jiang and S. Lee, Adv. Mater., 2022, 34, 2104125 CrossRef CAS PubMed.
- W. Feng, D. Chen, Y. Zhao, B. Mu, H. Yan and M. Barboiu, J. Am. Chem. Soc., 2024, 146, 2484–2493 Search PubMed.
- J. E. Barker, G. W. Richings, Y. Xie, J. Y. Rho, C. T. J. Ferguson, R. K. O'Reilly and S. Habershon, Chem. Sci., 2024, 15, 19400–19410 RSC.
- A. Wakamiya, T. Murakamib and S. Yamaguchi, Chem. Sci., 2013, 4, 1002–1007 RSC.
- Q. Xu, J. Zhang, J. Sun, H. Zhang and B. Z. Tang, Nat. Photonics, 2024, 18, 1185–1194 Search PubMed.
- B. Chu, H. Zhang, K. Chen, B. Liu, Q. Yu, C. Zhang, J. Sun, Q. Yang, X. Zhang and B. Z. Tang, J. Am. Chem. Soc., 2022, 144, 15286–15294 CrossRef CAS PubMed.
- T. Bai, H. Yan, Y. Zhang, L. Guo and Y. Zhao, J. Phys. Chem. B, 2021, 125, 4321–4329 CrossRef CAS PubMed.
- Y. Zhu, P. Gong, J. Wang, J. Cheng, W. Wang, H. Cai, R. Ao, H. Huang, M. Yu, L. Lin and X. Chen, Angew. Chem., Int. Ed., 2023, 62, e202218407 CrossRef CAS PubMed.
- J. Wang, Y. Liu, T. Cui, H. Yang and L. Lin, Chem. Sci., 2024, 15, 9915–9926 Search PubMed.
- J. Shen, H. Yu, Y. Shu, M. Ma and H. Chen, Adv. Funct. Mater., 2021, 31, 2106106 CrossRef CAS.
- S. Zhang, C. Cao, X. Lv, H. Dai, Z. Zhong, C. Liang, W. Wang, W. Huang, X. Song and X. Dong, Chem. Sci., 2020, 11, 1926–1934 Search PubMed.
- S. He, C. Chen, F. Li, W. Xu, D. Li, M. Liang and X. Yang, Nano Lett., 2023, 23, 8978–8987 Search PubMed.
- R. S. Lankone, A. R. Deline, M. Barclay and D. H. Fairbrother, Talanta, 2020, 218, 121148 Search PubMed.
- Y. Liu, D. Zhu, X. Zhu, G. Cai, J. Wu, M. Chen, P. Du, Y. Chen, W. Liu and S. Yang, Chem. Sci., 2020, 11, 11435–11442 Search PubMed.
- P. Hu, S. Zhao, J. Shi, F. Li, S. Wang, Y. Gan, L. Liu and S. Yu, Nanoscale, 2022, 14, 12219–12231 RSC.
- Y. Teng, D. Wang, Z. Yang, R. Wang, S. Ning, R. Zhang, H. Yang, X. Feng, J. Liu, L. Yang and Y. Tian, Biomaterials, 2025, 317, 123092 CrossRef CAS PubMed.
- Y. N. Zheng, C. Y. Xiong, Y. Zhuo, Y. Q. Chai, W. B. Liang and R. Yuan, Chem. Commun., 2020, 56, 8488–8491 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, characterization data and ESI figures. See DOI: https://doi.org/10.1039/d5sc01009h |
| This journal is © The Royal Society of Chemistry 2025 |