
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering of soluble bacteriorhodopsin†
Andrey
Nikolaev
,
Yaroslav
Orlov
,
Fedor
Tsybrov
 ,
Elizaveta
Kuznetsova
,
Pavel
Shishkin
,
Alexander
Kuzmin
,
Anatolii
Mikhailov
,
Yulia S.
Nikolaeva
,
Arina
Anuchina
,
Igor
Chizhov
,
Oleg
Semenov
,
Ivan
Kapranov
,
Valentin
Borshchevskiy
,
Alina
Remeeva
and
Ivan
Gushchin
*
,
Elizaveta
Kuznetsova
,
Pavel
Shishkin
,
Alexander
Kuzmin
,
Anatolii
Mikhailov
,
Yulia S.
Nikolaeva
,
Arina
Anuchina
,
Igor
Chizhov
,
Oleg
Semenov
,
Ivan
Kapranov
,
Valentin
Borshchevskiy
,
Alina
Remeeva
and
Ivan
Gushchin
*
Research Center for Molecular Mechanisms of Aging and Age-Related Diseases, Moscow Institute of Physics and Technology, Dolgoprudny, Russia. E-mail: ivan.gushchin@phystech.edu
First published on 13th May 2025
Abstract
Studies and applications of membrane proteins remain challenging due to the requirement of maintaining them in a lipid membrane or a membrane mimic. Modern machine learning-based protein engineering methods offer a possibility of generating soluble analogs of membrane proteins that retain the active site structure and ligand-binding properties; however, clear examples are currently missing. Here, we report successful engineering of proteins dubbed NeuroBRs that mimic the active site (retinal-binding pocket) of bacteriorhodopsin, a light-driven proton pump and well-studied model membrane protein. NeuroBRs are soluble and stable, bind retinal and exhibit photocycles under illumination. The crystallographic structure of NeuroBR_A, determined at anisotropic resolution reaching 1.76 Å, reveals an excellently conserved chromophore binding pocket and tertiary structure. Thus, NeuroBRs are promising microbial rhodopsin mimics for studying retinal photochemistry and potential soluble effector modules for optogenetic tools. Overall, our results highlight the power of modern protein engineering approaches and pave the way towards wider development of molecular tools derived from membrane proteins.
Introduction
Membrane proteins are notoriously difficult objects for study. Their function is strongly intertwined with composition and physico-chemical properties of the membrane or membrane-like media they are embedded into.1 For isolation and purification, membrane proteins need to be transferred to membrane mimics such as detergents, amphiphilic polymers or nanodiscs, yet identification of the best approach can be laborious, and the studied protein may become unstable.2–5 Consequently, significant effort was devoted to develop efficient solubilization methods.5,6 Alternatively, several protein engineering approaches were devised to engineer soluble analogues of membrane proteins.6 In two cases, those of tetrameric potassium channel KcsA and pentameric cation channel, nicotinic acetylcholine receptor nAChR, successful design of soluble variants was reaffirmed by obtaining NMR structures.7–9 Later, a simplistic QTY methodology was introduced, which involves replacing hydrophobic amino acids in transmembrane regions with polar ones, namely leucine with glutamine (Q), isoleucine and valine with threonine (T), and phenylalanine with tyrosine (Y).10 The QTY approach allowed generation of soluble forms of GPCRs able to bind their expected ligands,10 although respective proteins have so far resisted structure determination efforts.6More recently, a plethora of easy-to-use machine learning-based protein engineering techniques were developed displaying high success rates. ProteinMPNN, utilizing message-passing neural networks, predicts amino acid sequences that would fold into a desired shape, taking the protein backbone coordinates as an input.11 Proteins engineered with ProteinMPNN are produced by cells in greater amounts and display higher solubility, stability and, in some cases, higher activity.12 Recently, a modified version of ProteinMPNN, dubbed SolubleMPNN, was used to generate soluble proteins with folds closely matching those of claudin, rhomboid protease and GPCRs, as confirmed using X-ray crystallography.13 Whereas the interior of these proteins was completely redesigned, grafting of functional residues on the protein surface endowed them with the ability to bind natural partners of their prototypes.13
Bacteriorhodopsin (BR), a protein from the archaeon Halobacterium salinarum discovered in 1971,14 is a prototypical member of the microbial rhodopsin family and probably the best studied membrane protein overall.15,16 It consists of seven α-helices (labeled A to G) and covalently binds cofactor retinal, which undergoes isomerisation upon illumination. Photoactive proteins from this family are ubiquitous in the biosphere17 and are being used extensively in optogenetics.18,19 Several unsuccessful attempts to engineer soluble BR analogues were previously reported.6 On the other hand, protein engineering approaches advanced rapidly, allowing, for example, design of α-helical proteins that bound retinal analogs and could be used as near infrared fluorescent proteins.20 Here, we describe engineering of soluble BR analogs using modern neural network-based approaches. We started with a crystallographic structure of BR21 as a template, used SolubleMPNN13 to sample the amino acids outside the retinal-binding pocket and AlphaFold2 (ref. 22) to identify sequences with the best structure prediction metrics, and subjected the selected designs to molecular dynamics simulations. We then tested the artificial constructs experimentally and found that they bound the retinal and retained the basic functionality of bacteriorhodopsin.
Results and discussion
Here, we applied SolubleMPNN13 to engineer soluble variants of bacteriorhodopsin. In line with previous work applying neural networks to ligand-binding proteins,12,23,24 we fixed the retinal-binding pocket residues (Fig. 1a), while allowing all other residues to be mutated. We generated 52 sequences using SolubleMPNN and used the ColabFold implementation of AlphaFold2 (ref. 22 and 25) to select three sequences, dubbed NeuroBR_A, B, and C, for which general folds and retinal binding pockets were confidently predicted to be close to those of the wild type protein (Fig. 1b). Sequence identities between WT BR and NeuroBR_A, B, and C are 41.2, 43.4, and 44.7%, respectively. We then conducted molecular dynamics simulations of the three designs, which revealed potential stability of the developed variants (Fig. 1c). Phylogenetic analysis places NeuroBRs in the same branch as WT BR, but much further from the origin (Fig. 1d). While the exterior of the 7-helical bundle is mostly hydrophobic in the WT BR, predicted structures show a lot of polar or potentially charged residues on the protein surface (Fig. 1e). | ||
| Fig. 1 Sequence design and initial characterisation of soluble BR variants. (a) Chromophore-binding pocket in WT BR. (b) Backbone RMSD, pocket RMSD and pLDDT of AlphaFold-predicted models for WT BR and SolubleMPNN-generated sequences. (c) Amplitude of thermal fluctuations of Cα atoms of engineered BR variants in MD simulations. (d) Phylogenetic tree for NeuroBRs and representative microbial rhodopsins (based on the dataset by Rozenberg et al.17). (e) Surface amino acid properties for WT and engineered BR variants. Histidines and cysteines are absent in WT and engineered variants, except for 6×His tags added to NeuroBRs for metal affinity purification (not shown). (f and g) SEC profiles for NeuroBRs reconstituted with retinal and for monomeric fractions of NeuroBR_A and C after one week of incubation at 4 °C, respectively. Chromatography was performed using a Superdex 75 Increase 10/300 GL column with a void volume V0 of approximately 8.2 ml. (h) Visual appearance of WT BR in purple membranes and NeuroBR_A and C in detergent-free buffer. (i) Absorbance spectra of trimeric WT BR in purple membranes and engineered monomeric BR in solution. (j) Comparison of absorption spectra of dark-adapted and illuminated samples of NeuroBR_A and C. (k) Thermal denaturation of engineered BR variants. | ||
Next, we proceeded with experimental characterization of NeuroBRs. Usually, during heterologous expression of retinal-binding proteins in Escherichia coli, the retinal is provided exogenously as it is not synthesized by the bacterium. Free all-trans retinal in solution is colored yellow, with the absorption maximum at 380 nm. In the correctly folded complex, the retinal is covalently bound to a lysine in the retinal-binding pocket, forming a protonated Schiff base. Retinal-loaded BR is expected to be colored purple in the absence of illumination, with the absorption maximum in the range of 548–568 nm depending on the oligomeric state.14,26 Upon illumination, BR and other microbial rhodopsins undergo conformational changes; intermediate states with deprotonated retinal Schiff base display absorption maxima in the range of 360–410 nm.27
Despite retinal supplementation, all three artificially designed proteins were expressed and purified (Fig. S1†) in colorless apo-forms. We believe this to be the consequence of the cell wall being impermeable to the hydrophobic retinal, originally added as an ethanol solution. Consequently, we incubated the purified proteins in yellow-colored retinal solution overnight and observed that the samples turned pink, indicating formation of retinal-protein complexes. Denaturation by heating or excessive illumination converted the samples into an orange form (absorption maximum at ∼400 nm), with NeuroBR_B converting much faster compared to NeuroBR_A and NeuroBR_C. Such absorption presumably corresponds to the deprotonated retinal Schiff base, which is not expected to be found in correctly folded proteins under physiological conditions.
To further characterize the samples, we subjected them to size exclusion chromatography (SEC), while recording the absorption at three different characteristic wavelengths: 280 nm, which corresponds to aromatic amino acids and allows us to track the overall protein in the sample; 375 nm, roughly corresponding either to free retinal or the orange form of the proteins; and 550 nm, roughly corresponding to the expected absorption maximum of solubilized bacteriorhodopsin in monomeric form.26 SEC revealed that after the incubation with retinal the holo-proteins were present as mixtures of the proteins in oligomeric orange and monomeric pink forms (Fig. 1f). Separated pink forms of NeuroBR_A and NeuroBR_C remained stable and monomeric for at least a week (Fig. 1g and h).
NeuroBR_B was discarded from further study as its pink form was unstable, converting with time into an orange form. Judged by the ratio of orange (absorbing at ∼400 nm) and pink (absorbing at ∼530 nm) forms, the protein was not fully correctly folded at any temperature, with the maximum amount of pink form (∼50%) observed between 20 and 30 °C, both in the PBS buffer, pH = 7.5, and in the low salt buffer containing 10 mM NaPi, pH = 8. Increasing or decreasing pH from the range of 7 to 8 causes the protein to transition to the orange form. Although no quantitative measurements were made, we note that NeuroBR_B had significantly weaker photo-stability compared to NeuroBR_A and C, quickly converting into the orange form under daylight. Being unable to obtain a stable pink form of NeuroBR_B and having much better variants A and C, we did not pursue further characterization of NeuroBR_B.
Absorption maxima of purified monomeric NeuroBR_A and C were observed at 523–526 nm (Fig. 1i), shifted from 548 to 568 nm (depending on oligomerization and conditions) observed for WT BR. Whereas WT BR displays dark adaptation (blue shift of the absorption maximum), no alterations in NeuroBR spectra were observed after storing in darkness for 12 hours (Fig. 1j). The proteins were stable with thermally induced transitions to the orange form observed at 72 and 65 °C (Fig. 1k).
Stability of NeuroBR_A and NeuroBR_C allowed us to collect transient absorption spectroscopy data at different buffer pH values. Both proteins were observed to undergo a photocycle (Fig. 2, 3 and S2†). Three major distinct intermediates were clearly resolved: an early red-shifted K535 intermediate, a strongly blue-shifted M400 intermediate (presumably corresponding to the deprotonated form of the Schiff base), and a late red-shifted O585 intermediate. Absorption maxima of all of these intermediates were shifted to shorter wavelengths compared to those of WT protein (K590, M410, and O640),28,29 mirroring the shift of the absorption of the ground state (Fig. 1i). The three intermediates were the most prominent at acidic pH. At neutral pH, decay of the M state, likely linked to reprotonation of the Schiff base, was slowed down significantly, probably due to relative scarcity of protons in the solution compared to acidic conditions, and consequently the O state was not efficiently accumulated. Above pH 8.0, NeuroBR_A converted into a long-lived orange state upon illumination, partially recovering to the pink form over several hours in darkness. Slowdown of the photocycle of NeuroBR variants to fractions of a second compared to 1–10 ms for the WT BR28,29 primarily stems from slow reprotonation and probably follows from the lack of the proton donor residue in engineered proteins, as observed in the D96N BR variant.30,31
 | ||
| Fig. 2 Photocycle and structure of soluble BR variant NeuroBR_A. (a) Changes in absorbance of NeuroBR_A in solution after flash illumination. M and O correspond to areas, where absorbance raises due to formation of putative M and O intermediates. GSB is the ground state bleaching area, where absorbance corresponding to the ground state is diminished due to formation of photocycle intermediates. (b) Model of the NeuroBR_A photocycle at pH 4.0, where the different photocycle intermediates are clearly distinguishable. (c) Recovered absorption spectra of NeuroBR_A photocycle intermediates. (d) Dependence of NeuroBR_A photocycle intermediate half-lives on pH. (e) Overlay of the backbone and retinal structures for WT BR (purple) and NeuroBR_A (yellow and green). (f) Weighted 2Fo − Fc electron density map around the retinal contoured at the 2σ level. (g) Comparison of the retinal Schiff base environment in WT BR and NeuroBR_A. w406 is absent in NeuroBR_A and R82 changes its conformation. | ||
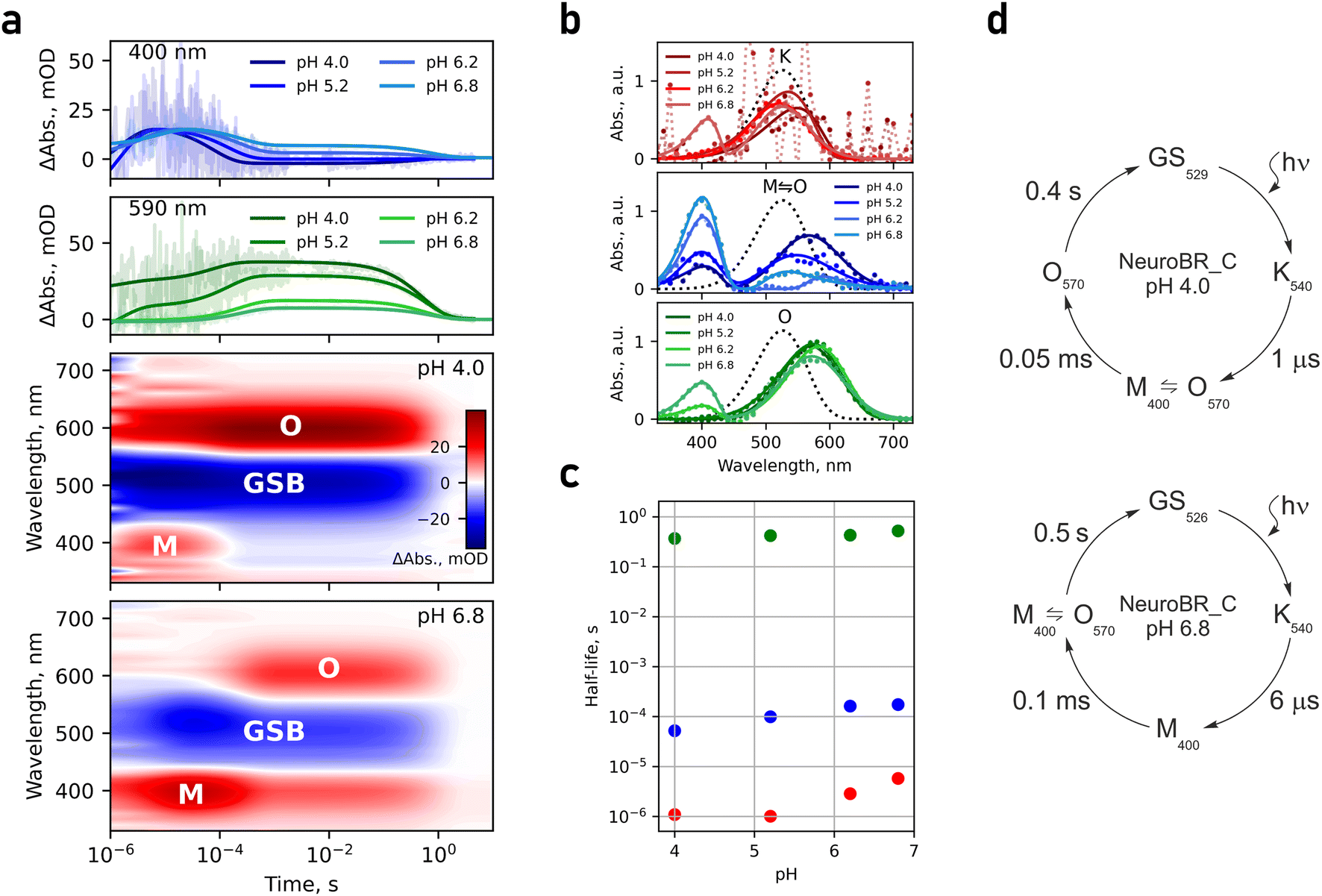 | ||
| Fig. 3 Photocycle of soluble BR variant NeuroBR_C. (a) Changes in absorbance of NeuroBR_C in solution after flash illumination. M and O correspond to areas where absorbance raises due to formation of putative M and O photocycle intermediates. GSB is the ground state bleaching area where absorbance corresponding to the ground state is diminished due to formation of photocycle intermediates. (b) Recovered absorption spectra of NeuroBR_C photocycle intermediates at different buffer pH values. The intermediates are designated in accordance with the photocycle at low pH. Dotted spectra correspond to the ground state. (c) Dependence of NeuroBR_C photocycle intermediate half-lives on pH. (d) Model of NeuroBR_C photocycles at pH 4.0 and pH 6.8. | ||
Following photophysical characterization, we attempted crystallization of NeuroBR_A and NeuroBR_C. We observed formation of box-shaped crystals of NeuroBR_A in 2 days (Fig. S3†). The crystals diffracted anisotropically, with resolution cut-offs for the best single crystal dataset of 1.76/1.78/2.21 Å. The structure was solved using molecular replacement with an AlphaFold-generated model, with 4 molecules in the asymmetric unit. No apparent oligomerization interfaces were observed; the proteins pack in layers in the crystal (Fig. S4†).
Whereas NeuroBR sequences correspond to amino acids 5–231 of WT BR, below we use WT BR numbering of amino acids for NeuroBR for clarity and for correspondence with earlier literature. Electron density allowed clear assignment of most amino acids, with the exception of flexible N- and C-termini and the amino acid 161 in the chain D; this amino acid is located in the E–F loop that is also disordered in many WT BR structures. The overall fold is extremely well conserved (Fig. 2e) with a root mean square deviation (RMSD) of all heavy atom positions from those in WT BR of 1.43–1.55 Å and RMSD of retinal-binding pocket atom positions of 0.69–0.77 Å. Within the four copies of NeuroBR_A in the crystals, RMSD of heavy atom positions ranges from 0.59 Å for chains C and D to 0.81 Å for chains A and C, highlighting rigidity of the structure.
In all four molecules, the retinal is in apparent all-trans conformation and slightly more straightened compared to the WT structure. The conserved water molecule W402 is clearly resolved near the retinal Schiff base (Fig. 2f and S5†). Interestingly, Arg82 is reoriented away from the Schiff base compared to its position in the ground state of WT BR (Fig. 2g). The only notable deviations of the obtained structure from the design are 2.7 Å displacement of the beta-hairpin element (B–C loop) and rearrangement of the E–F loop (disordered in most WT BR structures; Fig. 2e). We thus conclude that the employed engineering strategy was efficient at maintaining the protein structure and overall at obtaining a soluble analogue of a membrane protein.
Previously, denaturation and renaturation of native BR as well as reassembly from fragments were extensively studied32,33 and provided early understanding of membrane protein folding mechanisms.34 The protein was shown to be very stable and tolerant to a wide variety of mutations.35 Yet, soluble BR analogues were not previously reported. Solubilization of native BR with artificial “peptitergents” was possible,36 yet screening of BR analogue libraries, where 41 surface-exposed hydrophobic amino acids were randomly replaced with a hydrophilic one didn't result in purple-colored variants, although one tested construct yielded a soluble protein.37 Interestingly, the latter artificial variant and its K216C mutant covalently linked to a retinal analogue by a single carbon–nitrogen bond both displayed absorption peaks at ∼415 nm, similarly to the orange species reported here.37 Earlier computation-based potentially soluble variants of BR were not tested experimentally.38 Later, computation-based replacement of surface hydrophobic amino acids with charged and polar ones worked for helical peptide phospholamban39,40 and oligomeric channels KcsA and nAChR,7,9 but a similar approach resulted in water-insoluble variants of BR (up to 24.3% surface amino acids altered) that did not bind retinal.35 We assume that successful engineering of soluble BR reported here may be ascribed to higher predictive power of modern machine learning and neural network-based approaches and to stronger modification of the protein, because not only the surface-exposed amino acids were mutated, but also some of the internally facing ones distant from the retinal-binding pocket.
Historically, working with membrane proteins presented researchers with numerous challenges at every step of investigation. The proteins of interest are usually scarce in the native environment, and their host cells are not suitable for overexpression. Consequently, heterologous expression systems are often employed,4 yet they may lack the correct machinery needed for recognizing the particular protein for insertion into membrane and for inserting and folding it.41 Once produced, membrane proteins need to be extracted from the membrane for purification and handling; this was done initially using detergents2 and later also using amphiphilic polymers or nanodiscs.3,5 Transfer to such membrane mimics often destabilizes membrane proteins and affects their structure and function. Consequently, emergence of easy-to-use computational approaches for development of soluble analogues of membrane proteins would be beneficial for the field. Given that the engineering strategy presented here relies on publicly available computational tools (SolubleMPNN,13 ColabFold,25 and GROMACS42), it should be easy to replicate for other membrane proteins. However, we must note that the soluble analogues of membrane proteins would also have some limitations as study objects. Regulation of their function by the membrane and its constituents would be lost, and allosteric effects would likely be altered due to loss of interactions with lipids. Thus, this engineering approach is likely to be more suitable for development of high-throughput platforms for screening of binding of potential ligands, for conversion of membrane-embedded enzymes to soluble ones and for engineering of novel soluble molecular tools based on membrane proteins, and less suitable for studies aiming at direct and quantitative understanding of native membrane systems and their regulation by membrane-specific signals.
Conclusions
Using modern protein engineering approaches, we obtained three artificial sequences of soluble bacteriorhodopsin analogues with 41–45% sequence identity to the original protein. All three constructs were expressed in E. coli and formed a complex with retinal, the natural chromophore of bacteriorhodopsin. Whereas NeuroBR_B was unstable, NeuroBR_A and NeuroBR_C remained folded up to 72 and 65 °C, respectively, and exhibited retinal Schiff base deprotonation and reprotonation upon absorption of photons. The crystal structure of NeuroBR_A revealed a conserved overall fold and a retinal binding pocket in the artificial construct. Engineering of NeuroBR_A presents a clear-cut and unambiguous example of converting a membrane protein into a soluble one, while retaining the ligand-binding ability of its intramembrane part and its core function. This soluble bacteriorhodopsin analogue may be used for probing the retinal photophysics and photochemistry in a unique environment of an α-helical soluble protein, complementing as a model system for the soluble rhodopsin mimic based on the human cellular retinol binding protein II.43 It may also give rise to a new class of soluble optogenetic tools, where the effector domain activity is controlled by the conformation of the retinal-binding domain. Given the excellent conservation of the active site structure demonstrated here, a similar engineering methodology may probably be used to generate soluble analogues of drug target membrane proteins for easier screening of potential binder molecules. Overall, our results further highlight the power of modern protein engineering approaches and pave the way towards wider development of molecular tools derived from membrane proteins.Methods
Computational protein engineering
To preserve the natural ligand-binding ability of BR, we fixed the binding site residues during re-engineering.12,23,24 Using the high resolution structure of ground state BR (PDB ID 7Z09 (ref. 21)) as a reference, we fixed residues with side chain atoms within 5 Å of the retinal and glycines with heavy atoms within the same distance of the retinal. We also fixed the identity of Y57 and R82 for proper coordination of D212 and fixed T178 for proper coordination of W182. Finally, we fixed the region of the G helix containing residues 212–220 around the retinal-binding K216. In total, 34 residues were fixed: 20, 49, 50, 53, 57, 82, 83, 85, 86, 89, 90, 93, 118, 119, 122, 138, 141, 142, 145, 178, 182, 185, 186, 189, 208, 212, 213, 214, 215, 216, 217, 218, 219, 220.We applied ProteinMPNN11 retrained only on structures of soluble proteins13 to coordinates of the residues 5–231 of the WT BR structure. We chose a SolubleMPNN model that predicts amino acid identity considering 48 neighboring amino acids and was trained using Gaussian noise with an SD of 0.2 Å, with a sampling temperature of 0.1. In total, we generated 52 sequences and predicted the corresponding atomistic models using AlphaFold2-based ColabFold v. 1.5.5 with an alphafold2_ptm model,22,25 with the number of recycles set to 6. For each sequence, 5 models were obtained. The best model for consequent analysis was selected using average pLDDT. No AMBER relaxation was performed afterwards. Next, three sequences were selected for further characterization using ColabFold-predicted metrics: average pLDDT, root mean squared deviation (RMSD) of all Cα atoms and RMSD of heavy atoms of fixed residues. The sequences were dubbed A, B, and C in descending order of average pLDDT. Metal affinity tags consisting of six histidines were added via a GSG linker at the C-terminus of each sequence. Final amino acid and nucleotide sequences are available in Data S1 and S2.† Properties of respective proteins are listed in Table S1.†
Molecular dynamics simulations
NeuroBR_A, B, and C were simulated in the monomeric form. Initial models were obtained by combining the previously obtained models and positions of internal water molecules and retinal from the crystallographic structure of WT BR in the ground state (PDB ID 7Z09) using PyMOL.44 The dodecahedron unit cell contained 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 455 water molecules, 50 Na+ and 38 Cl− ions for NeuroBR_A; 13367 water molecules, 48 Na+ and 40 Cl− ions for NeuroBR_B; 12472 water molecules, and 54 Na+ and 38 Cl− ions for NeuroBR_C. All atomistic systems were prepared using the tools of GROMACS 2022.42 Protonation states of all titratable residues were assigned in accordance with a pH of 7.0 using PROPKA 3.5.1;45 the retinal Schiff base was protonated.
455 water molecules, 50 Na+ and 38 Cl− ions for NeuroBR_A; 13367 water molecules, 48 Na+ and 40 Cl− ions for NeuroBR_B; 12472 water molecules, and 54 Na+ and 38 Cl− ions for NeuroBR_C. All atomistic systems were prepared using the tools of GROMACS 2022.42 Protonation states of all titratable residues were assigned in accordance with a pH of 7.0 using PROPKA 3.5.1;45 the retinal Schiff base was protonated.
MD simulations were conducted using GROMACS 2022, with the CHARMM36 force field,46 and TIP3P water model.47 Parameters for the retinal bound to lysine were adapted from ref. 48. Systems were energy minimized using the steepest descent method, thermalized, equilibrated and simulated for 200 ns using the leapfrog integrator with a time step of 2 fs, at a reference temperature of 303.15 K and at a reference pressure of 1 bar. Temperature was coupled using Nosé–Hoover thermostat49 with a coupling constant of 1 ps−1. Pressure was coupled with isotropic Parrinello–Rahman barostat50 with a relaxation time of 5 ps and compressibility of 4.5·10−5 bar−1. Coordinates of the systems were collected every 20 ps.
The simulations were performed using periodic boundary conditions. The covalent bonds to hydrogens were constrained using the LINCS algorithm.51 The nonbonded pair list was updated every 20 steps with a cutoff of 1.2 nm. A force based switching function with a switching range of 1.0–1.2 nm and the particle mesh Ewald (PME) method52 with 0.12 nm Fourier grid spacing and 1.2 nm cutoff were used for treatment of the van der Waals and electrostatics interactions. The Python library MDAnalysis53 was used for analyses.
Phylogenetic analysis
We used MAFFT 7.520 (ref. 54) to align sequences of designed proteins NeuroBR_A, B, and C to the trimmed alignment of 315 sequences from the review by Rozenberg et al.17 as available via GitHub (https://github.com/BejaLab/RhodopsinsReview/blob/main/data/). A phylogenetic tree was constructed using IQ-TREE 1.6.10 (ref. 55) with additional options that correspond to those used by Rozenberg et al.17 (-m TEST -bb 1000 -pers 0.2 -nstop 500). The tree image was generated using TreeViewer 2.2.0.56Protein expression and purification
Protein-coding sequences were synthesized de novo and cloned into pET-28a(+) plasmids via XbaI (TCTAGA) and BamHI (GGATCC) restriction sites. E. coli strain C41 (DE3) cells transformed with protein-encoding plasmids pET-28a(+) were cultured in shaking flasks in 400 ml TBP-5052, one liter of which was prepared by mixing 930 ml of TB (12 g per L tryptone and 24 g per L yeast extract), 1 ml of 1 M MgSO4 solution, 20 ml of 50 × 5052 (250 g per L glycerol, 25 g per L glucose, and 100 g per L α-lactose) and finally 50 ml of 20xNPS (66 g per L (NH4)2SO4, 136 g per L NaH2PO4, and 142 g per L Na2HPO4). Kanamycin was added to a concentration of 50 mg L−1. Cell cultures were incubated at 37 °C until reaching an optical density of 0.5–0.7. Protein expression was induced by addition of 1 mM IPTG, followed by incubation for 20 h at 20 °C. Harvested cells were resuspended in lysis buffer containing 300 mM NaCl and 50 mM Tris–HCl, pH 8.0, and were disrupted in an M-110P lab homogenizer (Microfluidics, USA). The cell membrane fraction was removed from the lysate by ultracentrifugation at 100 kg for 45 minutes at 10 °C. Clarified supernatants were incubated overnight with Ni–nitrilotriacetic acid (Ni–NTA) resin (Qiagen, Germany) with constant stirring at 4 °C. Supernatants with Ni–NTA resin were loaded on a gravity flow column and washed with the PBS buffer. The proteins were eluted in the PBS buffer additionally containing 200 mM imidazole, 30 mM NaCl and 5 mM Tris–HCl, pH 8.0. All of the following steps were performed under red light to avoid photodamage. Retinal was added to the eluted proteins in approximately two-fold molar excess. Resulting mixtures were dialysed overnight against the PBS buffer. During the dialysis, the color of the samples changed from orange to pinkish red, indicating retinal binding. After the dialysis, samples were again incubated overnight with Ni–NTA resin with constant stirring at 4 °C. Resulting solutions with Ni–NTA resin were loaded on a gravity flow column and washed with the PBS buffer to get rid of excess retinal. The protein was eluted in the PBS buffer additionally containing 200 mM imidazole, 30 mM NaCl and 5 mM Tris–HCl, pH 8.0. Resulting solution contained the protein in two stable soluble forms – the orange (presumably misfolded) oligomeric state and pink monomeric state. We performed size exclusion chromatography (SEC) using a Superdex 75 Increase 10/300 GL column (Cytiva, USA) to separate the two forms. Sample purity was assessed by SDS-PAGE (Fig. S1†) and SEC (Fig. 1f and g).For pH sensitivity analyses and transient absorption spectroscopy, a series of buffers was prepared, each containing 75 mM NaCl and 10 mM of MES, MOPS, HEPES, CAPS, and TRIS. pH values were adjusted by incrementally adding HCl or NaOH until reaching the desirable values. The concentrated protein solution in the PBS buffer was mixed in a 1:7 volume ratio with the prepared buffers; the resulting pH value was determined by measuring separately the pH value of a large volume 1:7 mixture of the buffers without the protein.
Thermal stability
For determination of thermal stability of NeuroBR_A and C, protein solutions in the PBS buffer were heated up from 10 to 85 °C at a 1 °C per minute rate. Absorption spectra were recorded using an AvaSpec-2048L spectrometer (Avantes). Samples were illuminated using an AvaLight-DHc full-range light source (Avantes). The proportion of the orange misfolded form was determined based on the ratio of the absorption peaks observed at 523 and 359 nm.Dark adaptation
NeuroBR_A and C in PBS buffer pH 7.5 were incubated overnight and transferred to a cuvette holder in complete darkness at 4 °C. Absorption spectra were measured as described above. After the measurement, the samples were intensely illuminated and returned back to the cuvette holder for subsequent measurement.Transient absorption spectroscopy
The transient absorption changes were measured using a custom-built setup.57 In brief, protein activation was achieved with a Brilliant BRILL/IR-10 Nd:YAG laser coupled with a Rainbow OPO (420–680 nm, Quantel, France), delivering near-nanosecond pulses at 530 nm wavelength with an energy of approximately 1 mJ per pulse. A 75 W Xe-arc lamp (Hamamatsu, Japan) served as the probing light source. The samples were positioned between two monochromators (LSH-150, LOT, Germany) controlled by stepper motors. A photomultiplier tube (R12829, Hamamatsu Photonics, Japan) detected the light. Data were collected from 330 to 730 nm with a 10 nm step size, with each wavelength measurement averaged over 10 repetitions. The sample temperature was maintained at 20 °C using a temperature-controlled cuvette holder. Data acquisition was carried out using two digital oscilloscopes (Keysight DSO-X 4022A) in overlapping time windows.Transient absorption changes were fitted with a multiexponential global fit with MEXFIT.58 Both for NeuroBR_A and NeuroBR_C, 3 time constants were used. After that, the model of irreversible sequential transitions was used to obtain absorption spectra of intermediates.28 Absorption spectra of intermediates and ground states were fitted with skewed Gaussian functions28 for absorption peaks and a power function with a negative exponent for background scattering.
Crystallization, data collection, and structure determination
The monomeric fractions of NeuroBR_A,C were concentrated and crystallized by a sitting drop vapor diffusion approach using an NT8 robotic system (Formulatrix, USA). The drops contained 150 nL concentrated protein solution and 150 nL reservoir solution. Crystallization plates were stored at 20 °C in the dark. The rectangular crystals of NeuroBR_A appeared after 2 days in the probes containing 0.2 M ammonium sulfate and 30% w/v PEG 4000 or PEG 8000 as precipitant solutions. The crystals were harvested using micromounts under red light illumination, cryoprotected by immersion into 15% glycerol, flash-cooled and stored in liquid nitrogen. Diffraction data were collected at the BL02U1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF). Data integration was performed using XIA2 software,59–63 and scaling was completed with the STARANISO web server,64 revealing anisotropic diffraction limits of 1.76, 1.78, and 2.21 Å (Table S2†). Elliptically truncated data were subsequently utilized for molecular replacement using Phaser,65 using the AF2-predicted model as the template. Structure refinement was conducted using Refmac5,66,67 with related statistics provided in Table S3.†Data availability
All generated nucleotide and amino acid sequences are available in the ESI.† Molecular dynamics trajectories were deposited to Zenodo and are available using the following link: https://zenodo.org/doi/10.5281/zenodo.14194414. Crystallographic structure factors and coordinates for NeuroBR_A were deposited into the Protein Data Bank (PDB) under accession code 9KME.Author contributions
I. G. and A. N. conceived the project. I. G. supervised the project. A. N. developed the computational pipeline and obtained NeuroBR sequences. P. S. and A. K. conducted the molecular dynamics simulations. A. A. prepared the phylogenetic tree. A. N. expressed, purified and characterized the proteins. Y. O., Y. S. N., E. K., A. M. and O. S. helped with experiments. F. T. measured the photocycle data. I. C. supervised the photocycle data processing. A. R. obtained the crystals. V. B. and I. K. collected and processed the diffraction data. I. G. and A. N. solved and refined the structure. A. N. and I. G. prepared the original draft of the manuscript with contributions from all coauthors. All authors contributed to the preparation of the final version and reviewed it.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We are grateful to Valentin Gordeliy for inspiration to study bacteriorhodopsin and for advice. Computational parts of the work were supported by the Ministry of Science and Higher Education of the Russian Federation, agreement 075-03-2025-662 (project FSMG-2025-0003, to I. G.). Functional studies were supported by the Russian Science Foundation (21-64-00018, to V. B.). Synchrotron data collection was supported by the Ministry of Science and Higher Education of the Russian Federation (grant no. 075-15-2021-1354, to I. K.). We thank SSRF for providing the opportunity to collect crystallographic data at the beamline BL02U1.References
- I. Levental and E. Lyman, Nat. Rev. Mol. Cell Biol., 2023, 24, 107–122 CrossRef CAS PubMed.
- G. G. Privé, Methods, 2007, 41, 388–397 CrossRef PubMed.
- E. Serebryany, G. A. Zhu and E. C. Y. Yan, Biochim. Biophys. Acta, 2012, 1818, 225–233 CrossRef CAS PubMed.
- A. Pandey, K. Shin, R. E. Patterson, X.-Q. Liu and J. K. Rainey, Biochem. Cell Biol., 2016, 94, 507–527 Search PubMed.
- Q. Zhang and V. Cherezov, Curr. Opin. Struct. Biol., 2019, 58, 278–285 CrossRef CAS PubMed.
- R. Qing, S. Hao, E. Smorodina, D. Jin, A. Zalevsky and S. Zhang, Chem. Rev., 2022, 122, 14085–14179 CrossRef CAS PubMed.
- A. M. Slovic, H. Kono, J. D. Lear, J. G. Saven and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1828–1833 CrossRef CAS PubMed.
- D. Ma, T. S. Tillman, P. Tang, E. Meirovitch, R. Eckenhoff, A. Carnini and Y. Xu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16537–16542 CrossRef CAS PubMed.
- T. Cui, D. Mowrey, V. Bondarenko, T. Tillman, D. Ma, E. Landrum, J. M. Perez-Aguilar, J. He, W. Wang, J. G. Saven, R. G. Eckenhoff, P. Tang and Y. Xu, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 617–626 CrossRef CAS PubMed.
- S. Zhang, F. Tao, R. Qing, H. Tang, M. Skuhersky, K. Corin, L. Tegler, A. Wassie, B. Wassie, Y. Kwon, B. Suter, C. Entzian, T. Schubert, G. Yang, J. Labahn, J. Kubicek and B. Maertens, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E8652–E8659 CrossRef CAS PubMed.
- J. Dauparas, I. Anishchenko, N. Bennett, H. Bai, R. J. Ragotte, L. F. Milles, B. I. M. Wicky, A. Courbet, R. J. de Haas, N. Bethel, P. J. Y. Leung, T. F. Huddy, S. Pellock, D. Tischer, F. Chan, B. Koepnick, H. Nguyen, A. Kang, B. Sankaran, A. K. Bera, N. P. King and D. Baker, Science, 2022, 378, 49–56 CrossRef CAS PubMed.
- K. H. Sumida, R. Núñez-Franco, I. Kalvet, S. J. Pellock, B. I. M. Wicky, L. F. Milles, J. Dauparas, J. Wang, Y. Kipnis, N. Jameson, A. Kang, J. De La Cruz, B. Sankaran, A. K. Bera, G. Jiménez-Osés and D. Baker, J. Am. Chem. Soc., 2024, 146, 2054–2061 CrossRef CAS PubMed.
- C. A. Goverde, M. Pacesa, N. Goldbach, L. J. Dornfeld, P. E. M. Balbi, S. Georgeon, S. Rosset, S. Kapoor, J. Choudhury, J. Dauparas, C. Schellhaas, S. Kozlov, D. Baker, S. Ovchinnikov, A. J. Vecchio and B. E. Correia, Nature, 2024, 631, 449–458 CrossRef CAS PubMed.
- D. Oesterhelt and W. Stoeckenius, Nature, 1971, 233, 149–152 CAS.
- M. Grote, M. Engelhard and P. Hegemann, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 533–545 CrossRef CAS PubMed.
- V. Gordeliy, K. Kovalev, E. Bamberg, F. Rodriguez-Valera, E. Zinovev, D. Zabelskii, A. Alekseev, R. Rosselli, I. Gushchin and I. Okhrimenko, Methods Mol. Biol., 2022, 2501, 1–52 Search PubMed.
- A. Rozenberg, K. Inoue, H. Kandori and O. Béjà, Annu. Rev. Microbiol., 2021, 75, 427–447 Search PubMed.
- K. Kojima, A. Shibukawa and Y. Sudo, Biochemistry, 2020, 59, 218–229 CrossRef CAS PubMed.
- A. Alekseev, V. Gordeliy and E. Bamberg, in Rhodopsin: Methods and Protocols, ed. V. Gordeliy, Springer US, New York, NY, 2022, pp. 71–100 Search PubMed.
- D. Baker, C. Xu, Y. Liu, B. Arús, K. Mishra, M. Luciano, V. Bandi, A. Kumar, Z. Guo, M. Bick, M. Xu, K. Zhang, J. Lingg, J. Bae, A. Kang, S. Gerben, A. Bera, J. Vaughan, J. Manton, E. Derivery, M. Schnermann, A. Stiel, O. Bruns and Y. Guan, Research Square, 2024 DOI:10.21203/rs.3.rs-4652998/v1.
- V. Borshchevskiy, K. Kovalev, E. Round, R. Efremov, R. Astashkin, G. Bourenkov, D. Bratanov, T. Balandin, I. Chizhov, C. Baeken, I. Gushchin, A. Kuzmin, A. Alekseev, A. Rogachev, D. Willbold, M. Engelhard, E. Bamberg, G. Büldt and V. Gordeliy, Nat. Struct. Mol. Biol., 2022, 29, 440–450 Search PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 Search PubMed.
- A. Nikolaev, A. Kuzmin, E. Markeeva, E. Kuznetsova, Y. L. Ryzhykau, O. Semenov, A. Anuchina, A. Remeeva and I. Gushchin, Protein Sci., 2024, 33, e4958 CrossRef CAS PubMed.
- A. S. Nikolaev, D. A. Lunegova, R. I. Raevskii, P. E. Shishkin, A. A. Remeeva, B. Ge, E. G. Maksimov, I. Y. Gushchin and N. N. Sluchanko, Protein Sci., 2024, 33, e5216 CrossRef CAS PubMed.
- M. Mirdita, K. Schütze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed.
- N. A. Dencher and M. P. Heyn, FEBS Lett., 1978, 96, 322–326 CrossRef CAS PubMed.
- O. P. Ernst, D. T. Lodowski, M. Elstner, P. Hegemann, L. S. Brown and H. Kandori, Chem. Rev., 2014, 114, 126–163 CrossRef CAS PubMed.
- I. Chizhov, D. S. Chernavskii, M. Engelhard, K. H. Mueller, B. V. Zubov and B. Hess, Biophys. J., 1996, 71, 2329–2345 CrossRef CAS PubMed.
- C. Wickstrand, P. Nogly, E. Nango, S. Iwata, J. Standfuss and R. Neutze, Annu. Rev. Biochem., 2019, 88, 59–83 Search PubMed.
- H. J. Butt, K. Fendler, E. Bamberg, J. Tittor and D. Oesterhelt, EMBO J., 1989, 8, 1657–1663 CrossRef CAS PubMed.
- M. Holz, L. A. Drachev, T. Mogi, H. Otto, A. D. Kaulen, M. P. Heyn, V. P. Skulachev and H. G. Khorana, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 2167–2171 CrossRef CAS PubMed.
- M. J. Liao, K. S. Huang and H. G. Khorana, J. Biol. Chem., 1984, 259, 4200–4204 CrossRef CAS PubMed.
- T. W. Kahn and D. M. Engelman, Biochemistry, 1992, 31, 6144–6151 CrossRef CAS PubMed.
- J. L. Popot and D. M. Engelman, Annu. Rev. Biochem., 2000, 69, 881–922 CrossRef CAS PubMed.
- K. Mitra, T. A. Steitz and D. M. Engelman, Protein Eng., 2002, 15, 485–492 CrossRef CAS PubMed.
- C. E. Schafmeister, L. J. Miercke and R. M. Stroud, Science, 1993, 262, 734–738 Search PubMed.
- C. E. Schafmeister, PhD thesis, University of California, San Francisco, 1997.
- C. Gibas and S. Subramaniam, Protein Eng., 1997, 10, 1175–1190 Search PubMed.
- S. Frank, R. A. Kammerer, S. Hellstern, S. Pegoraro, J. Stetefeld, A. Lustig, L. Moroder and J. Engel, Biochemistry, 2000, 39, 6825–6831 CrossRef CAS PubMed.
- H. Li, M. J. Cocco, T. A. Steitz and D. M. Engelman, Biochemistry, 2001, 40, 6636–6645 CrossRef CAS PubMed.
- S. A. Kim, H. G. Kim, W. C. B. Wijesinghe, D. Min and T.-Y. Yoon, Annu. Rev. Biophys., 2025, 54, 141–162 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. Wang, Z. Nossoni, T. Berbasova, C. T. Watson, I. Yapici, K. S. S. Lee, C. Vasileiou, J. H. Geiger and B. Borhan, Science, 2012, 338, 1340–1343 CrossRef CAS PubMed.
- W. L. DeLano, San Carlos: DeLano Scientific, 2012 Search PubMed.
- M. H. M. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 Search PubMed.
- R. B. Best, X. Zhu, J. Shim, P. E. M. Lopes, J. Mittal, M. Feig and A. D. Jr. MacKerell, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 Search PubMed.
- S. Zhu, M. F. Brown and S. E. Feller, J. Am. Chem. Soc., 2013, 135, 9391–9398 CrossRef CAS PubMed.
- D. J. Evans and B. L. Holian, J. Chem. Phys., 1985, 83, 4069–4074 Search PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 Search PubMed.
- B. Hess, H. Bekker, H. Berendsen and J. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
- K. Katoh and D. M. Standley, Mol. Biol. Evol., 2013, 30, 772–780 Search PubMed.
- L.-T. Nguyen, H. A. Schmidt, A. von Haeseler and B. Q. Minh, Mol. Biol. Evol., 2015, 32, 268–274 Search PubMed.
- G. Bianchini and P. Sánchez-Baracaldo, Ecol. Evol., 2024, 14, e10873 CrossRef PubMed.
- D. Soloviov, V. Borshchevskiy and I. Chizhov, in Rhodopsin: Methods and Protocols, ed. V. Gordeliy, Springer US, New York, NY, 2022, pp. 169–179 Search PubMed.
- K.-H. Müller and Th. Plesser, Eur. Biophys. J., 1991, 19, 231–240 CrossRef.
- P. Evans, Acta Crystallogr., Sect. D:Struct. Biol., 2006, 62, 72–82 CrossRef PubMed.
- G. Winter, J. Appl. Crystallogr., 2010, 43, 186–190 CrossRef CAS.
- G. Winter, D. G. Waterman, J. M. Parkhurst, A. S. Brewster, R. J. Gildea, M. Gerstel, L. Fuentes-Montero, M. Vollmar, T. Michels-Clark, I. D. Young, N. K. Sauter and G. Evans, Acta Crystallogr., Sect. D:Struct. Biol., 2018, 74, 85–97 CrossRef CAS PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Acta Crystallogr., Sect. D:Struct. Biol., 2011, 67, 235–242 CrossRef CAS PubMed.
- P. R. Evans and G. N. Murshudov, Acta Crystallogr., Sect. D:Struct. Biol., 2013, 69, 1204–1214 CrossRef CAS PubMed.
- I. J. Tickle, C. Flensburg, P. Keller, W. Paciorek, A. Sharff, C. Vonrhein and G. Bricogne, STARANISO Global Phasing Ltd, Cambridge, United Kingdom, 2016 Search PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 Search PubMed.
- A. A. Vagin, R. A. Steiner, A. A. Lebedev, L. Potterton, S. McNicholas, F. Long and G. N. Murshudov, Acta Crystallogr., Sect. D:Struct. Biol., 2004, 60, 2184–2195 CrossRef PubMed.
- J. Agirre, M. Atanasova, H. Bagdonas, C. B. Ballard, A. Baslé, J. Beilsten-Edmands, R. J. Borges, D. G. Brown, J. J. Burgos-Mármol, J. M. Berrisford, P. S. Bond, I. Caballero, L. Catapano, G. Chojnowski, A. G. Cook, K. D. Cowtan, T. I. Croll, J. É. Debreczeni, N. E. Devenish, E. J. Dodson, T. R. Drevon, P. Emsley, G. Evans, P. R. Evans, M. Fando, J. Foadi, L. Fuentes-Montero, E. F. Garman, M. Gerstel, R. J. Gildea, K. Hatti, M. L. Hekkelman, P. Heuser, S. W. Hoh, M. A. Hough, H. T. Jenkins, E. Jiménez, R. P. Joosten, R. M. Keegan, N. Keep, E. B. Krissinel, P. Kolenko, O. Kovalevskiy, V. S. Lamzin, D. M. Lawson, A. A. Lebedev, A. G. W. Leslie, B. Lohkamp, F. Long, M. Malý, A. J. McCoy, S. J. McNicholas, A. Medina, C. Millán, J. W. Murray, G. N. Murshudov, R. A. Nicholls, M. E. M. Noble, R. Oeffner, N. S. Pannu, J. M. Parkhurst, N. Pearce, J. Pereira, A. Perrakis, H. R. Powell, R. J. Read, D. J. Rigden, W. Rochira, M. Sammito, F. Sánchez Rodríguez, G. M. Sheldrick, K. L. Shelley, F. Simkovic, A. J. Simpkin, P. Skubak, E. Sobolev, R. A. Steiner, K. Stevenson, I. Tews, J. M. H. Thomas, A. Thorn, J. T. Valls, V. Uski, I. Usón, A. Vagin, S. Velankar, M. Vollmar, H. Walden, D. Waterman, K. S. Wilson, M. D. Winn, G. Winter, M. Wojdyr and K. Yamashita, Acta Crystallogr., Sect. D:Struct. Biol., 2023, 79, 449–461 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Data S1 and S2, Tables S1–S3, and Fig. S1–S5. See DOI: https://doi.org/10.1039/d5sc02453f |
| This journal is © The Royal Society of Chemistry 2025 |