
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom cubane-assembled Mn-oxo clusters to monodispersed manganese oxide colloidal nanocrystals†
Yan
He
 a,
Yang
Liu
b,
Huijuan
Zheng
a,
Zhen
Xiang
a,
Zheng
Zhou
*a,
Fengting
Geng
c,
Longlong
Geng
c,
Evgeny V.
Dikarev
*d and
Haixiang
Han
*a
a,
Yang
Liu
b,
Huijuan
Zheng
a,
Zhen
Xiang
a,
Zheng
Zhou
*a,
Fengting
Geng
c,
Longlong
Geng
c,
Evgeny V.
Dikarev
*d and
Haixiang
Han
*a
aInterdisciplinary Materials Research Center, School of Materials Science and Engineering, Tongji University, Shanghai, 201804, China. E-mail: hxhan@tongji.edu.cn
bDepartment of Materials Science, Fudan University, Shanghai, 200433, China
cShandong Provincial Key Laboratory of Monocrystalline Silicon Semiconductor Materials and Technology, College of Chemistry and Chemical Engineering, Dezhou University, Dezhou, 253023, China
dDepartment of Chemistry, University at Albany, State University of New York, Albany, New York 12222, USA
First published on 11th June 2024
Abstract
The assemblies of [M4O4] (M = metal) cubanes represent a fascinating class of materials for a variety of application fields. Although such a structural characteristic is relatively common in small molecules and in extended bulk solids, high nuclearity clusters composed of multiple [M4O4] units as their backbones are rare. In this work, we report two new Mn-oxo clusters, MnII8MnIII10O10(OOCMe)12(OMe)14(py)2 ([Mn18-Ac]) and MnII4MnIII14O14(OOCCMe3)8(OMe)14(MeOH)5(py) ([Mn18-Piv]), whose core structures are assemblies of either 6- or 7-cubanes in different packing patterns, which have been unambiguously revealed by single crystal X-ray diffraction technique. The cubane-assembled structural features can be deemed as the embryonic structures of the bulk manganese oxide. Herein, this report demonstrates the first case study of utilizing Mn-oxo clusters as precursors for the preparation of manganese oxide nanocrystals, which has never been explored before. Through a simple colloidal synthetic approach, high-quality, monodisperse Mn3O4 nanocrystals can be readily prepared by employing both precursors, while their morphologies were found to be quite different. This work confirms that the structural similarity between precursors and nanomaterials is instrumental in affording more kinetically efficient pathways for materials formation, and the structure of the precursor has a significant impact on the morphology of final nanocrystal products.
Introduction
Mn-oxo clusters are well-documented as single-molecule magnets (SMMs) that are capable of functioning as single-domain magnetic particles.1 A particularly intriguing research direction is to explore these molecular structures to achieve new magnetic properties such as high ground state spin number, spin–spin cross relaxation, and quantum tunneling of the magnetization vector (QTM).2–4 Beyond the nanomagnetism field, Mn-oxo clusters are also known to demonstrate promising activities in electrochemistry, catalysis, and electronic transistors.5–7 By virtue of their molecular nature, the cluster structures can be readily elucidated via single-crystal X-ray diffraction at atomic resolution, which provides new perspectives on synthetic strategies, design of analogous assemblies, as well as on the origins of displaying properties.8–10Although a number of Mn-oxo clusters have been obtained to date, it is still challenging to establish a clear relationship between the synthetic pathway and the resultant structure, as many experimental parameters are involved in dictating the final structural topologies. Nevertheless, the introduction of O2− anions to construct a metal–oxygen framework as well as the selection of proper ligands that direct the formation of specific assemblies, have been proven to be critically important tools for the preparation of new Mn-oxo clusters.2,11,12 Practically, the former feature can be readily realized by introducing O2 in synthesis or by employing oxidative reagents like KMnO4 or (Bu4N)MnO4.13 In contrast, the latter tool proved to be quite tricky, and a variety of ligands and possible combinations thereof have been attempted in order to greatly extend the diversity of cluster structures. Among those, synthetic strategies can be principally classified as employing: flexible bridging ligands that impose little morphology constraints (carboxylates),14 rigid bridging ligands that could apply relatively strong constraints to the structural topology (cyanides),15 and ligands with multiple binding groups (tripodal alcohols or dicarboxylates).16,17 In addition to the direct synthetic routes involving simple starting reagents, like MnCl2, Mn(ClO4)2 or Mn(OAc)2,18–21 several “small” Mn-oxo clusters can be used for the preparation of higher nuclearity ones. For example, the largest known {Mn84} cluster was obtained from the smaller {Mn12} unit.22 The above methods have led to an encouraging success in creating a number of unprecedented Mn-oxo clusters.
Based on crystallographic analysis, most of the Mn-oxo clusters are constructed with three types of subunits, namely rhombic [Mn2O2], cubic [Mn4O4] and defective-dicubic [Mn4O6] (ESI, Fig. S5†), providing a significant topological diversity, such as the wheel-like {Mn18},23 {Mn12}24 and {Mn22},25 torus-like {Mn70}26 and {Mn84},22 and cubane-assembled {Mn13} clusters.17 Among different structural topologies, the [M4O4] cubane fragment is especially interesting. Such a unit is built of four metals and four oxygens at alternating vertices of the cube, analogous to the organic cubane (C8H8), and thus being casually described as the [M4O4] cubane.27 It represents an important building unit (denoted as cubane-assembled structure hereafter), which appears as an integral part of metal oxides, such as MnO or Mn3O4.28–31 Even more, cubane-assembled structures, albeit extremely rare, can also be found in some high nuclearity Mn-oxo clusters.17,18,32 Therefore, the cubane-assembled block in molecular clusters can be deemed as an embryonic unit of the bulk oxide nanocrystals, which inspires us to explore if those clusters can serve as precursors for the preparation of high-quality manganese oxide colloidal nanocrystals. During the preparation process of colloidal nanocrystals, the title clusters can undergo partial dissociation, and the structures of the rest species may have a significant impact on the reaction kinetic pathways as well as on the final form of the nanocrystals.33 For example, Brandi and co-workers have successfully employed the [In37P20] cluster to prepare monodisperse InP colloidal nanocrystals. They proposed that small twistane molecular species appeared upon dissociation of [In37P20]. These particular units eventually lead to high-quality InP nanocrystals through a more controllable kinetic pathway.34 However, the utilization of Mn-oxo clusters as precursors for colloidal manganese oxide nanomaterials has never been explored.
Herein, we adopted methoxide and carboxylates to prepare two cubane-assembled Mn-oxo clusters. Upon the exposition to O2, two new MnII/MnIII oxo nanoclusters in about 1.2 nm have been successfully isolated. Their structures were found to be assembled of multiple [Mn4O4] cubanes, decorated with carboxylate and methoxide ligands, bringing about significant differences in their overall structural topologies. Importantly, both clusters are capable of acting as precursors for the preparation of high-quality, monodispersed colloidal Mn3O4 nanocrystals through a conventional colloidal heat-up synthetic approach (Scheme 1).
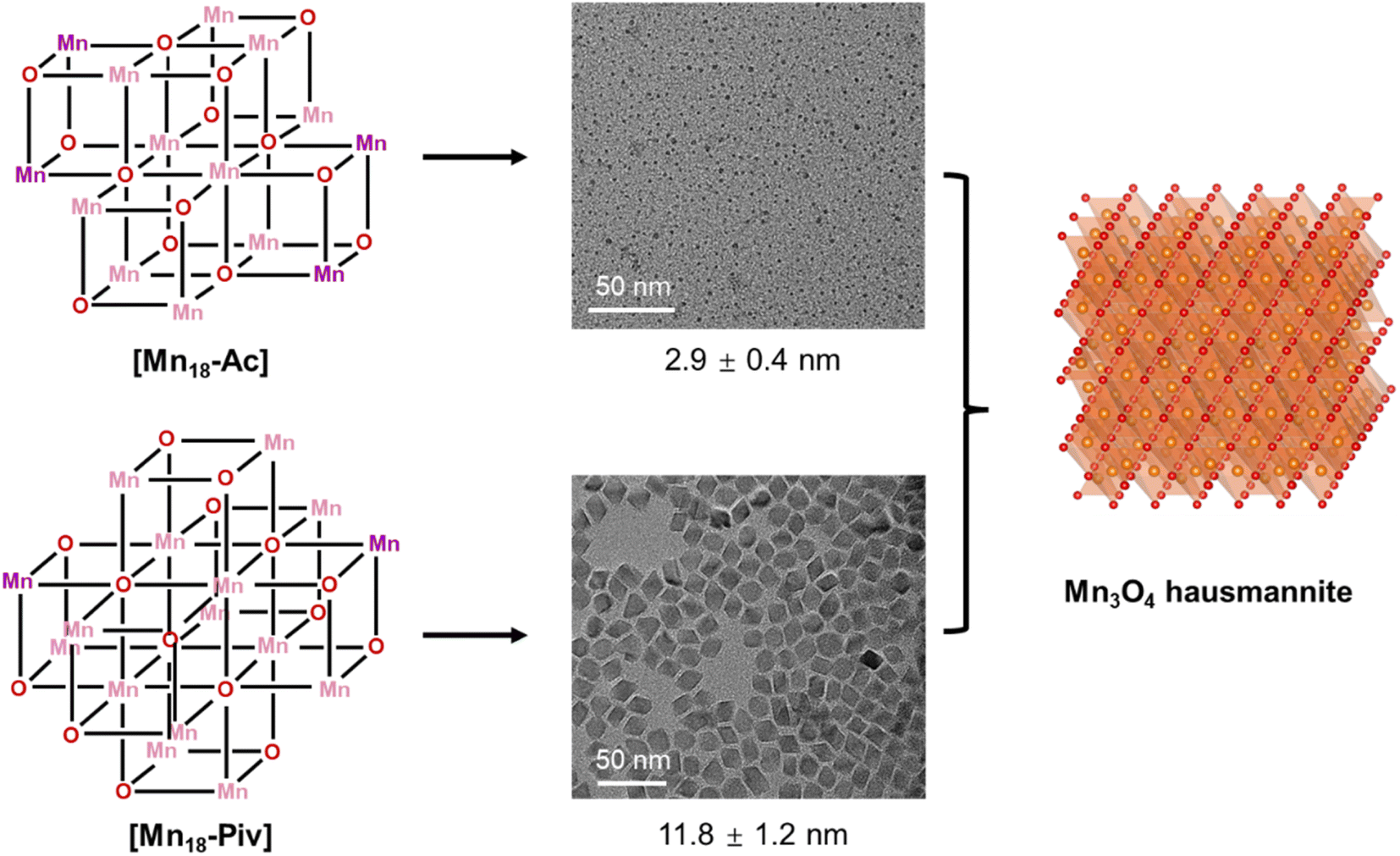 | ||
| Scheme 1 Preparation of Mn3O4 colloidal nanocrystals from Mn-oxo clusters. | ||
Results and discussion
Synthesis of [Mn18-Ac] and [Mn18-Piv]
We have made a thorough analysis of previously reported Mn-oxo clusters, and the ligands used for their decoration (ESI, Table S5†). We first attempted to employ β-diketonates and methanol to prepare high-nuclearity Mn-oxo clusters. Unfortunately, these experiments all failed after a number of attempts. Therefore, our efforts turned to replacement of β-diketonates with more flexible carboxylate ligands (R’COOH), which have been previously used to obtain multinuclear Mn-oxo clusters.14 The better suitability of carboxylates may be ascribed to their flexible coordination behaviors. For example, although β-diketonates represent a class of versatile bidentate ligands by affording 3 electrons,35 the rigid skeleton essentially limits their flexibility in both chelating and bridging connectivity modes.36 The result is that the propensity of forming cubanes or cubane-assembled structures will compromise the coordination of β-diketonates, as well as their spatial arrangement.37 Consequently, the cubic features in the course of structure conversion cannot be retained, and unpredictable structures with indefinite geometries could be obtained.37 In contrast, carboxylate ligands are more flexible and feasible not to impose extra geometry restrictions on Mn centers, which assists in preserving the [Mn4O4] cubane character in the clusters. The latter can be attributed to their multiple binding patterns with metal ions and to the relatively less steric hindrance.38 This is especially important in the synthesis of Mn cubane-assembled structures in order to satisfy the ideal octahedrally-coordinated environment for the metal centers at the vertex sites.39In order to prepare new high-nuclearity Mn-oxo clusters featuring [Mn4O4] cubane-assembled units, we employed two commonly used carboxylate acids, acetate acid (CH3COOH) and pivalate acid (C(CH3)3COOH), which demonstrate different steric hindrance defined by the bulkiness of the respective alkyl groups.40 In addition, it has been revealed in our previous work that the steric effect of alkoxide ligands is also an important factor in defining the cubane-type structures, and there is a trend that the less bulky alkyl groups may appear as more beneficial for the formation of cubane-assembled molecule structures.41 Therefore, methanol was chosen here as the solvent to prepare Mn-oxo clusters. It should be noted here that performing the reaction in strictly anaerobic conditions immediately proved to be unsuccessful, and no products were crystallized out. This is attributed to the lack of oxo-groups for the formation of [–Mn–O–Mn–O–] framework. Furthermore, the hard-donor character of alkoxide can provide more stability to high-valent manganese ions (hard acid).42,43 Therefore, we run the reactions directly in the open air. Under such a scenario, O2 was introduced to partially oxidize divalent Mn to its trivalent state and the reduced oxo-groups were, in turn, engaged in the construction of the extended [–Mn–O–Mn–O–] framework. Eventually, we have successfully obtained (see ESI: Experimental section & General procedures section† for more information) two cubane-assembled, high nuclearity Mn-oxo clusters MnII8MnIII10O10(OOCMe)12(OMe)14(py)2 (abbreviated as [Mn18-Ac] hereafter) and MnII4MnIII14O14(OOCCMe3)8(OMe)14(MeOH)5(py) ([Mn18-Piv]). These two clusters have dimensions of about 1.1–1.2 nm without the organic ligands (Fig. 1). Both clusters appear as brown-colored solids that are stable in the open air. In addition, they are soluble in polar solvents such as methanol or ethanol, while displaying a limited solubility in acetone or acetonitrile, and are insoluble in non-polar solvents.
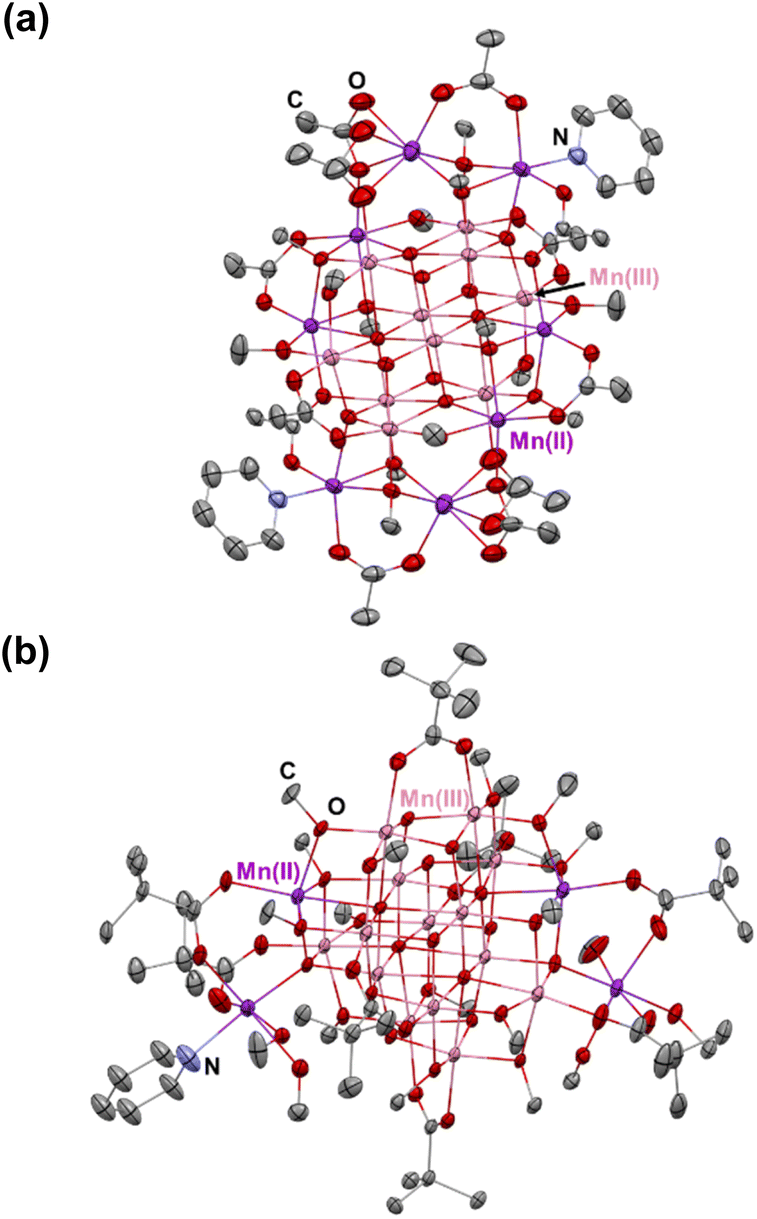 | ||
| Fig. 1 Solid state structures of [Mn18-Ac] (a) and [Mn18-Piv] (b) drawn with thermal ellipsoids at the 30% probability level. Hydrogen atoms are omitted for clarity. Colour code: MnII, purple; MnIII, pink; O, red; N, blue; C, grey. | ||
Single crystal structures of [Mn18-Ac] and [Mn18-Piv]
Single crystal X-ray diffraction analysis reveals that [Mn18-Ac] and [Mn18-Piv] both consist of a [–Mn–O–Mn–] skeleton with 18 Mn atoms, in which the bridging oxygens are either O2− (in inner core) or CH3O− (in outer shell). Regardless of the different topologies, the two clusters can be depicted as the epitaxially grown structures from a central [Mn4O4] cubane, and the main cores of the two clusters are made up of multiple cubane units with nearly identical geometries (Fig. 2a and 3a). Specicially, the structure of [Mn18-Ac] cluster consists of six cubanes, which can be schematically described as a two-step growth from the central cube 1. First, the congruent [Mn4O4] cubane (cubane 2) grows along the +z direction of 1, resulting in an overall cuboidal geometry of packing two cubanes. In the next step, the two cubanes 1 and 2 continue to duplicate and extend in different directions. In particular, cubane 1 replicates cubanes 5 and 6 along its +x and −y directions, and cubane 2 clones cubanes 3 and 4 along its −x and +y directions (Fig. 2c). This eventually brings about a six-cubane assembly in the cluster [Mn18-Ac].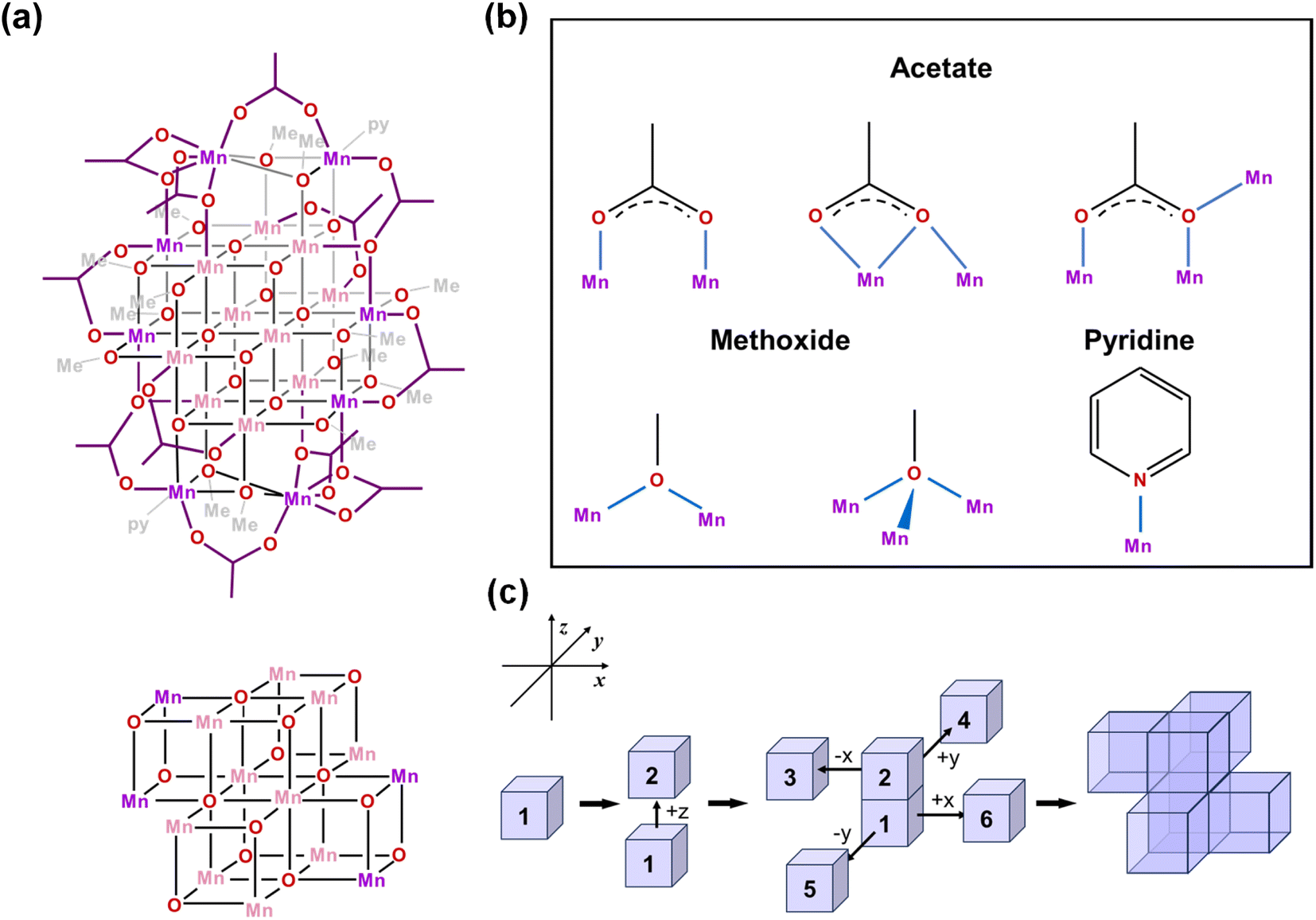 | ||
| Fig. 2 (a) Schematic illustrations of the [Mn18-Ac] cluster and its [Mn4O4]-assembled core. Pink and purple colors designate MnIII and MnII ions, respectively. (b) The coordination modes of acetate, methoxide, and pyridine in [Mn18-Ac]. (c) Schematic representation of the assembling [Mn4O4] cubic units in the core structure of [Mn18-Ac]. | ||
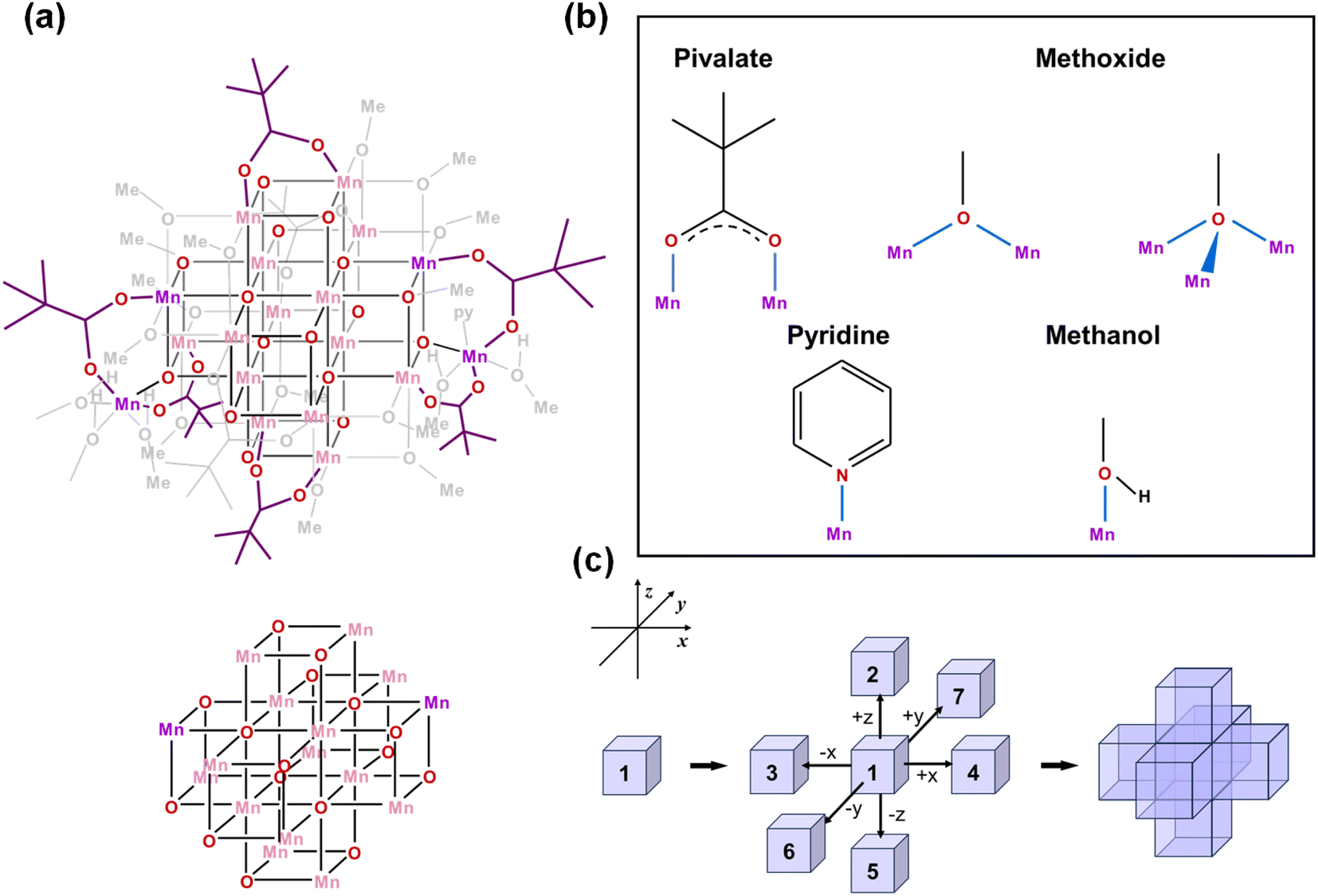 | ||
| Fig. 3 (a) Schematic illustration of the [Mn18-Piv] cluster and its [Mn4O4]-assembled core. Pink and purple colors designate MnIII and MnII ions, respectively. (b) The coordination modes of pivalate, methoxide, and pyridine/methanol ligands in [Mn18-Piv]. (c) Schematic representation of the assembling [Mn4O4] cubic units in the core structure of [Mn18-Piv]. | ||
For the cluster [Mn18-Piv], the growth process is more isotropic and it can be considered as the duplication of the central cubane along all six directions, eventually generating a seven-cubane assembled structure (Fig. 3c). In addition, there are several more Mn atoms continuously growing on the periphery of the cubane-assembled structures for both clusters. Due to the direct binding with the surfactant ligands, those metal centers demonstrate severely distorted coordination environments in order to sustain the intrinsic geometric ligand constraints.
We have carefully inspected possible reasons leading to different topologies of the two clusters. Most likely, those may be attributed to the different binding modes of the carboxylate ligands in each cluster. Detailed structural anatomy reveals that apart from the isolated O2−, oxygens from the methoxide (CH3O−) are also involved in the construction of the [–O–Mn–O–Mn–] framework for both [Mn18-Ac] and [Mn18-Piv] acting in either μ2- or μ3-bridging modes. Neutral pyridine and methanol molecules act as σ-donor ligands that connect to the corner Mn sites. However, the acetate (CH3COO−) in [Mn18-Ac] and pivalate ((CH3)3CCOO−) in [Mn18-Piv] exhibit distinctly different coordination behaviors. In [Mn18-Ac], the acetates act in three different types of coordinating fashions, namely tridentate bridging and chelating/bridging, while the pivalate ligands only display pure bidentate bridging in [Mn18-Piv]. Meanwhile, MeOH molecules can also act as σ-donor ligands binding with the two Mn atoms at the corners of [Mn18-Piv], which cannot be found in [Mn18-Ac] (Fig. 2b and 3b). The disparity of the coordinating patterns for acetate and pivalate is likely the cause for the differences in the overall cluster structure topologies.44
Although [Mn18-Ac] and [Mn18-Piv] both consist of 18 Mn atoms in a mixture of di-/tri-valent oxidation states, they are substantially different in the ratio of MnII to MnIII ions. Specifically, there are 8 MnII and 10 MnIII in [Mn18-Ac], while the [Mn18-Piv] contains 4 MnII and 14 MnIII, resulting in average Mn oxidation states of +2.56 vs. +2.78, respectively. Notably, the trivalent Mn ions are located in the inner section of the cluster assembly in both molecules. The assignment of Mn oxidation states can be reliably made by comparing the Mn–O bond distance due to the valence-trapped nature of Mn-oxo clusters, with electrons are no longer delocalized across the whole network of the Mn–O bonds.45 The Mn ions assigned as divalent exhibit longer averaged Mn–O bond distances than those of trivalent (ESI, Tables S3 and S4†). For instance, the averaged MnII–O bond lengths range from 2.179 to 2.250 Å, and from 2.149 Å to 2.183 Å, for [Mn18-Ac] and [Mn18-Piv], respectively, while MnIII ions exhibit shorter MnIII–O bonds of 2.046 and 2.045 Å (Table 1). The latter is comparable with previously reported clusters displaying MnIII–O bond length of 2.040 Å.46 Moreover, MnIII centers exhibit characteristic Jahn–Teller distortion in either elongated or compressed manner, depending on whether the single eg electron is located at dz2 or dx2−y2 orbital (ESI, Fig. S6†). It is worth mentioning that the total averaged Mn–O bond distance for [Mn18-Ac] is longer than that for [Mn18-Piv], which is in line with the higher averaged oxidation state of Mn than in the latter.
Theoretical calculations
The DFT calculations using PBE/def2-TZVP hybrid functional were performed to reveal the electronic structures of [Mn18-Ac] and [Mn18-Piv] (Fig. 4). The oxidation states of Mn atoms were quantified using the LOBA method (ESI, Tables S6 and S7†),47 and the MnII and MnIII positions in both optimized structures are in good agreement with the crystallographic assignments. As reflected by the partial density of states, the frontier orbitals are predominated by d states of Mn and p states of O in both structures (ESI, Fig. S14 and S15†). To be specific, the highest occupied MOs in [Mn18-Ac] are localized on the exterior MnII ions, which are chelated by the acetate ligands, while the lowest unoccupied MOs are contributed by the internal MnIII ions (ESI, Fig. S16 and Table S8†). In contrast, the frontier molecular orbitals in [Mn18-Piv] are dominated by MnIII ions, except for the HOMO and HOMO-2 that exhibit a larger MO delocalization among the MnIII centers (ESI, Fig. S17, and Table S9†). Overall, the theoretical calculations revealed that the fully structurally relaxed structures and the distributions of MnII and MnIII ions agree well with the data obtained from the X-ray single crystal analysis of clusters [Mn18-Ac] and [Mn18-Piv].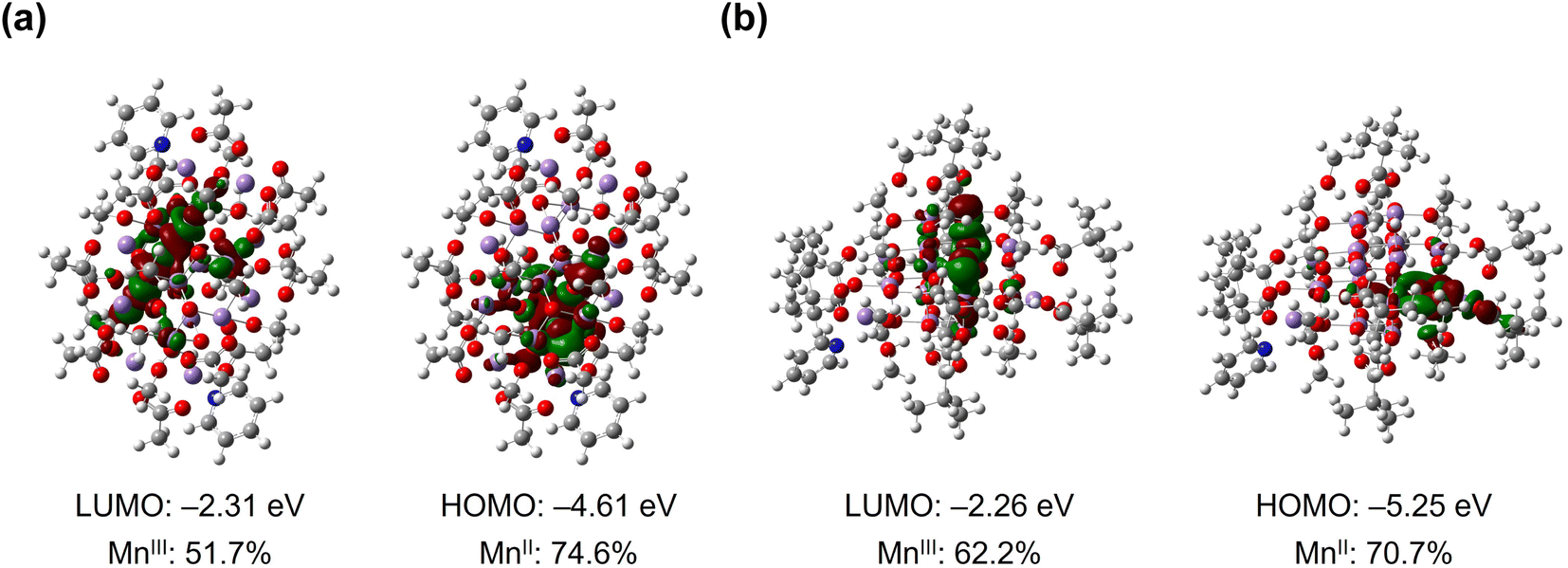 | ||
| Fig. 4 Frontier molecular orbitals (LUMO and HOMO) in [Mn18-Ac] (a) and [Mn18-Piv] (b) with labeled energies and Mn orbital contributions. | ||
Investigations of the oxidation states for Mn in [Mn18-Ac] and [Mn18-Piv]
The oxidation states of metal atoms in nanomaterials can effectively influence the electronic structure of metal-oxo nanoclusters, thereby imposing a significant impact on their chemical and physical properties. In Mn-oxo clusters, the oxidation states for Mn ions define their overall spin state, which is a critical parameter for magnetic properties. For example, [Mn18-Ac] and [Mn18-Piv] display different susceptibility behaviors at room temperature (39.1 and 116.5 cm3 K mol−1, respectively) in the direct-current (DC) magnetic field (ESI, Fig. S7†). To further confirm the average oxidation state in the bulk materials, X-ray photoelectron spectroscopy (XPS) investigation has been carried out to preliminarily determine the oxidation states of Mn for both clusters. The Mn 2p3/2 and Mn 2p1/2 spectra for [Mn18-Ac] and [Mn18-Piv] can be satisfactorily simulated by using the two-phase fitting strategy, suggesting the presence of MnII and MnIII species (see ESI: X-ray photoelectron spectroscopy (XPS) section† for more information). Although the XPS results partially reveal the spectroscopic features of the distinct MnII and MnIII ions, this technique does not provide a strong confirmation of the overall oxidation states of Mn ions in both clusters. Therefore, X-ray absorption near-edge spectroscopy (XANES) has been carried out on cluster samples. Upon the irradiation of X-ray with appropriate energies, the inner shell electrons can be promoted to higher energy orbitals, demonstrating characteristic K-edge absorption signatures, which vary upon the metal charges. Typically, the higher K-edge energy indicates the higher oxidation state of the metal. Here, we chose Mn0 foil, MnIIO, MnIII2O3, and MnIVO2 as the reference samples. It was found that the spectra of [Mn18-Ac] and [Mn18-Piv] both display the typical Mn K-edge features, which can be attributed to the electron transition from 1s to 4p orbitals (Fig. 5a).48 In addition, the K-edge absorption curve for [Mn18-Ac], compared with that for [Mn18-Piv], is obviously left-shifted, indicating the relatively lower averaged oxidation states for Mn in the former. Importantly, the two curves are both located in between the reference samples of MnIIO and MnIII2O3, which agree well with the mixed valences of Mn ions in the title clusters. To quantify the K-edge absorption values for each sample, their first-derivative normalized XANES curves were also plotted (Fig. 5b). It is shown that with the increase of the oxidation states, the four reference samples Mn0 foil, MnIIO, MnIII2O3 and MnIVO2 have the Mn K-edge energies of 6539.0, 6544.5, 6553.0, and 6558.0 eV, respectively. The Mn K-edge energies for [Mn18-Ac] and [Mn18-Piv] fall into the range between the MnIIO and MnIII2O3 standards, being measured at 6548.0 and 6549.0 eV, respectively. It should be mentioned that the K-edge energy generally has a linear relationship with the oxidation state for the same element, however, the significant intrinsic variations of the K-edge energies for Mn usually make the determination of the absolute oxidation state problematic relative to the standards, which is also true for the current work.49 Overall, the XANES investigations unambiguously revealed that [Mn18-Ac] and [Mn18-Piv] both contain a mixture of MnII and MnIII ions, and more MnIII are incorporated in the latter cluster, perfectly in line with the single crystal structure analysis.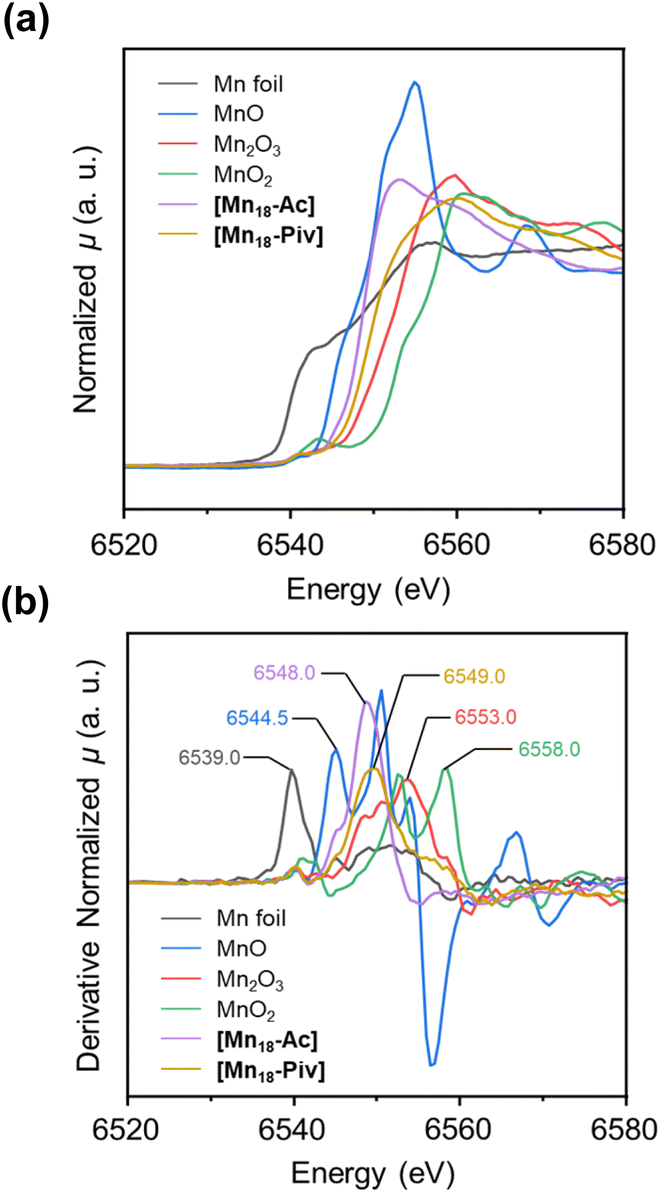 | ||
| Fig. 5 Normalized XANES spectra (a) and the first-derivative normalized XANES spectra (b) for Mn0 foil, MnIIO, MnIII2O3, MnIVO2, [Mn18-Ac], and [Mn18-Piv]. | ||
Photocatalytic properties
The feasible variations of the oxidation states and coordination environments for Mn centers endow Mn-oxo clusters with good catalytic activities in several processes of interest.50,51 Here, we have conducted preliminary evaluation of the photocatalytic performance of [Mn18-Ac] and [Mn18-Piv] by degrading tetracycline (TC) through the advanced oxidation process. With the peroxymonosulfate (PMS) as an activation reagent,52 both [Mn18-Ac] and [Mn18-Piv] demonstrate good catalytic activities in terms of effectiveness and efficiency (Fig. 6a). Overall, a sharp decrease of the characteristic absorption peak for TC can be observed within 5 minutes for both clusters and the irradiation of light can improve the degradation rate of TC. This can be clearly manifested by the pseudo-first-order kinetic plots, as the curve for [Mn18-Ac] demonstrates a steeper slope than that for [Mn18-Piv], and with the induction of light, the slopes for both clusters increase (Fig. 6b). Generally, the [Mn18-Ac] exhibits a slightly better catalytic performance than [Mn18-Piv], and the light irradiation can further increase the TC degradation. From the degradation rate constant (k) histogram (Fig. 6c), [Mn18-Ac] and [Mn18-Piv] demonstrate the k values of about 2.18 × 10−3 s−1 and 1.96 × 10−3 s−1, respectively. Upon the light irradiation, the k values increase 2.4 and 2.1 times, reaching 5.47 × 10−3 s−1 and 3.70 × 10−3 s−1 for [Mn18-Ac] and [Mn18-Piv], respectively. The results imply that the electromagnetic waves within the visible light range can effectively stimulate the transportation of electrons to form strong oxidizing radicals on the cluster surface. Moreover, the photocatalytic performance of the two clusters is more efficient than that of the bulk manganese oxide materials. For example, Tian et al. have recently reported two different microscale MnOx materials (1–2 μm), which require 15 and 45 min to degrade TC to about 20% under the similar conditions.53 The relatively high photocatalytic efficiency of [Mn18-Ac] and [Mn18-Piv] may result from their ultrasmall sizes, which afford large specific surface areas and more active sites on their surfaces.54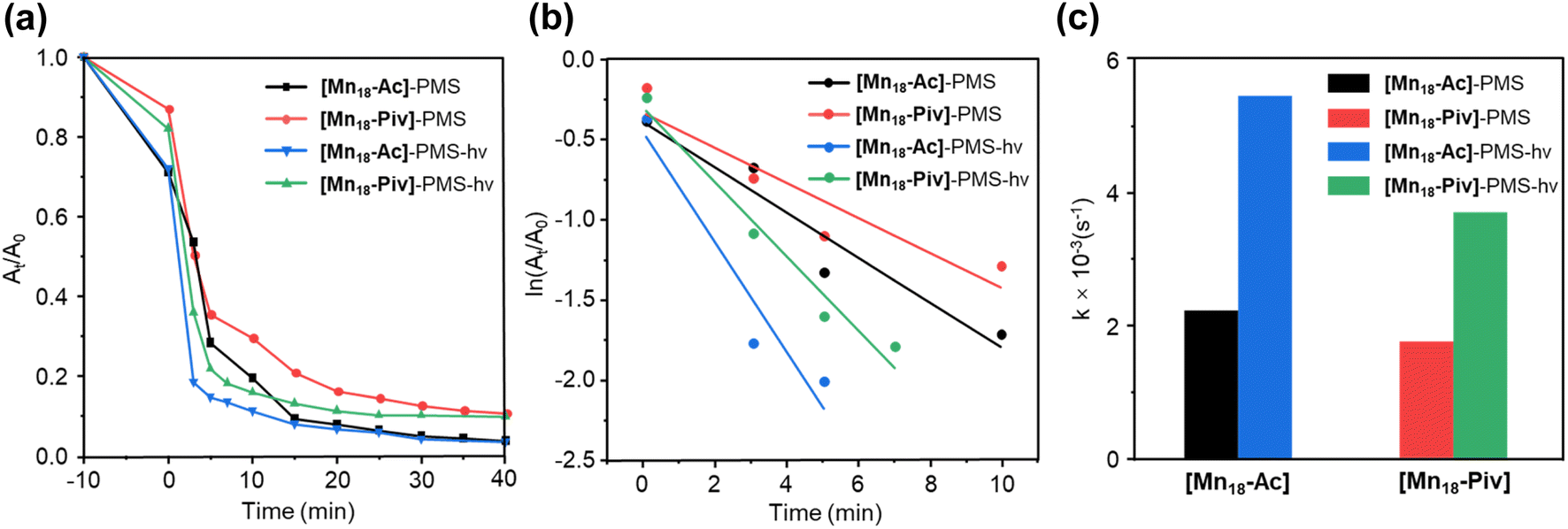 | ||
| Fig. 6 TC degradation rates over reaction time (a) and the pseudo-first-order kinetic modeling (b) for the catalytic behavior of [Mn18-Ac] and [Mn18-Piv] clusters under dark condition and light irradiation; (c) histogram of the degradation rate constant for [Mn18-Ac] and [Mn18-Piv] under dark environment and light irradiation. | ||
Preparation of nanoscale Mn oxide nanocrystals from Mn-oxo cluster precursors
Clusters can be considered as the intermediates to the formation of the corresponding macroscopic nanocrystals, and their innate structural characters may have a significant influence on nanocrystal structure, compositions and morphology.55,56 Here, we have used [Mn18-Ac] and [Mn18-Piv] clusters as precursors to prepare Mn oxide nanocrystals through the conventional colloidal synthetic approach in oleylamine (see ESI: Experimental section & General procedures† for more information).57 The precursors were dispersed in oleylamine and heated to 220 °C for 9 hours to yield the brown precipitates. The nanocrystals were isolated after being centrifuged and washed with DCM/hexane several times. Remarkably, both precursors produced the same hausmannite Mn3O4 phase,28 that was clearly confirmed by the X-ray powder diffraction (Fig. 7a and b); 1-Mn3O4 and 2-Mn3O4 designate the Mn3O4 phases obtained from [Mn18-Ac] and [Mn18-Piv], respectively. Le Bail profile fits show that the two powder patterns can be satisfactorily fitted with the hausmannite Mn3O4 phase crystallized in the tetragonal space group I41/amd. The unit cell parameters refined from the Le Bail fit are very close to those reported in literature.28 However, the FWHM (full width at half maximum) for the two powder patterns are different with the one for 1-Mn3O4 being apparently larger than that for 2-Mn3O4, which indicates that the use of precursor [Mn18-Ac] results in appearance of smaller Mn3O4 nanocrystals under the same conditions. It is worth noting that the structure of Mn3O4 is made of two different subunits: [Mn4O4] cubanes and [MnO4] tetrahedra, which feature six- and four-coordination environments for Mn centers (ESI, Fig. S10†). Therefore, the [Mn4O4] cubane character in clusters can be regarded as one to be retained in larger nanocrystals. The TEM micrographs revealed that both 1-Mn3O4 and 2-Mn3O4 colloidal nanocrystals are monodispersed, with average sizes of 2.9 (±0.4) and 11.8 (±1.2) nm, respectively (Fig. 7c and e and ESI, Fig. S11†). However, due to the small size of 1-Mn3O4, we were unable to clearly recognize its geometry (ESI, Fig. S12†). The careful inspection of the nanocrystals from HRTEM images revealed that most of the exposed crystal planes in 1-Mn3O4 can be indexed as (220) (Fig. 7d). In contrast, the TEM micrographs clearly unveil that the 2-Mn3O4 nanocrystals feature an octahedral geometry (Fig. 7e), with the characteristic crystal planes such as (112), (200), and (204) indexed from the fast Fourier transform (FFT) patterns (Fig. 7f and ESI, Fig. S13†).58 The latter can be better explained from the surface energy point of view. Typically, the formation of octahedral nanocrystals from tetragonal/cubic unit cells requires the exposure of the {101} group of nanocrystal planes. In Mn3O4 unit cell, the surface energy of (101) planes is higher than other low-index crystal planes,59 leading to a naturally adopted octahedral morphology with (101)-terminated surfaces, which are also confirmed by the high-resolution TEM (HRTEM) images (Fig. 7g). From those, most of the exposed surfaces can be reliably indexed as the characteristic (101) planes. Furthermore, the HRTEM images of 2-Mn3O4 (ESI, Fig. S13†) clearly show that the Mn atoms are hexagonally packed, consistent with the atomic arrangement in the Mn3O4 lattice.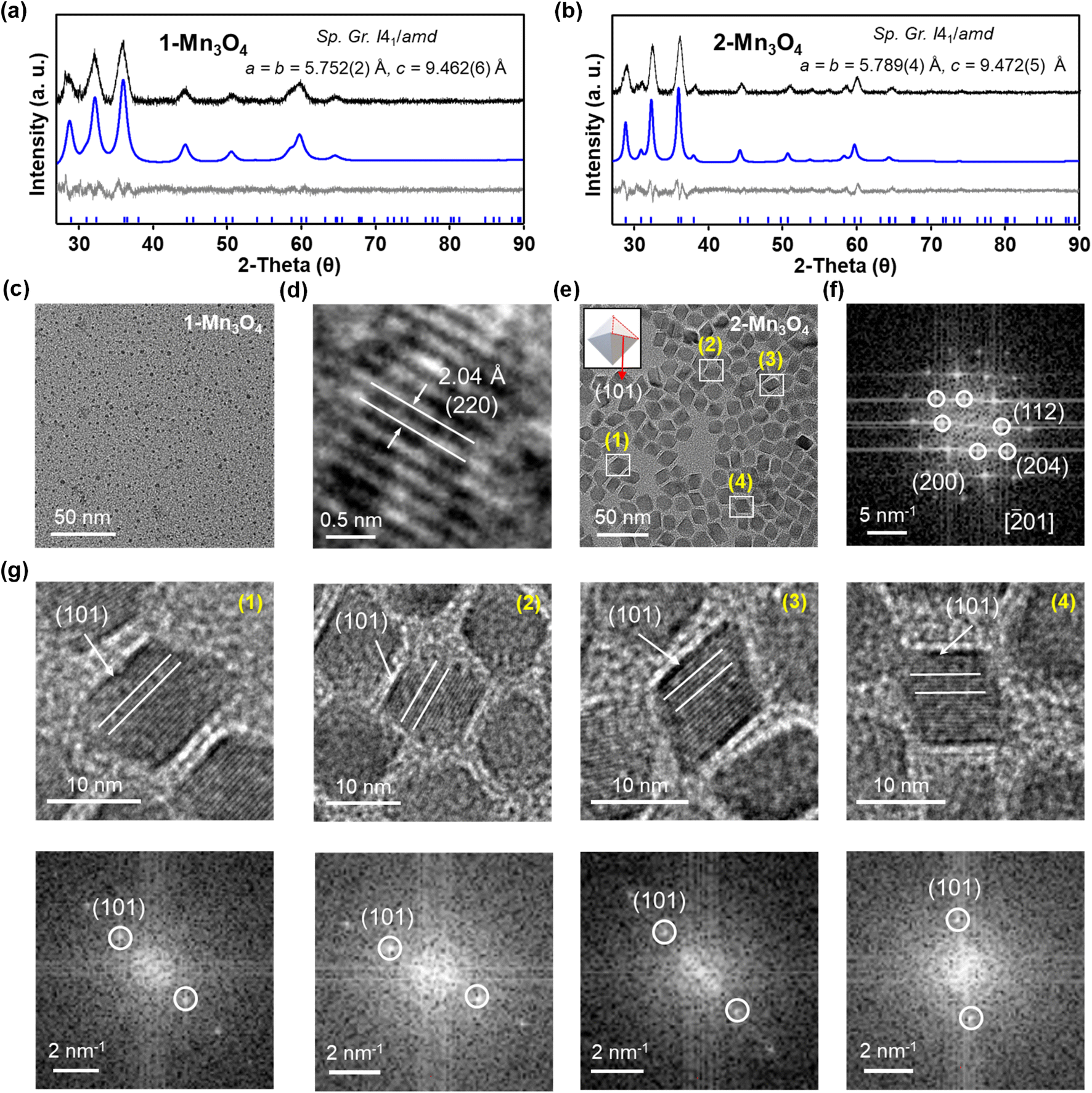 | ||
Fig. 7 (a) and (b) X-ray powder diffraction patterns and Le Bail fit for Mn3O4 nanocrystals obtained from [Mn18-Ac] and [Mn18-Piv]. Black and blue curves are experimental and simulated patterns with theoretical positions shown at the bottom as blue bars. The grey is the difference curve. (c) and (d) TEM micrographs of 1-Mn3O4 nanocrystals obtained from [Mn18-Ac]. (e) TEM micrographs of the Mn3O4 nanocrystals obtained from [Mn18-Piv]. Inset graph is the schematic representation of the octahedron geometry for 2-Mn3O4 nanocrystals. (f) Fast Fourier transform (FFT) pattern for the TEM micrograph of the 2-Mn3O4 nanocrystals from the view of [![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 01] orientation. (g) High-resolution TEM micrographs (top) of selected nanocrystals in (e) and the corresponding fast Fourier transform (FFT) patterns (bottom). 01] orientation. (g) High-resolution TEM micrographs (top) of selected nanocrystals in (e) and the corresponding fast Fourier transform (FFT) patterns (bottom). | ||
It would be interesting to examine possible reasons that cause the disparity in nanocrystal size of 1-Mn3O4 and 2-Mn3O4, especially since they were both obtained under the same preparation conditions. Taking into account that the colloidal Mn3O4 was isolated after a long reaction process (9 hours), it can be rationalized that the nanocrystals are more likely to be thermodynamic rather than kinetic products. A possible mechanism to account for this interesting phenomenon is shown in Fig. 8. At the early stages, the two precursors dispersed in oleylamine first underwent a ligand exchange process, upon which the original alkoxide and carboxylate ligands were replaced with oleylamine. This process is accompanied by the partial structural dissociation leaving only the innermost cubane-assembled base intact.33 Such a phenomenon has also been observed previously in the formation of InP nanocrystals from the In37P20 nanocluster.34 The remaining species then act as either nuclei or foundation blocks for the growth of the nanocrystals. Due to the difference in structures, anisotropies, lattice energies and sizes, of those initial molecules, they eventually go through two different reaction pathways. As a result, the 1-Mn3O4 nanocrystals were the phase that was trapped in the form of relatively small size (∼3 nm) particles, and the applied experimental conditions could not provide sufficient driving force to make nanocrystals grow larger to reach the next thermodynamically stable stage. In contrast, the cubane-assembled core structure of [Mn18-Piv] takes a different transformation route resulting in ∼12 nm Mn3O4 particles being trapped. It has to be admitted that a full mechanism still requires further investigation, but it can be reliably concluded that the innate structural characters of the precursors impose a profound influence on the morphology of the final colloidal nanocrystals.
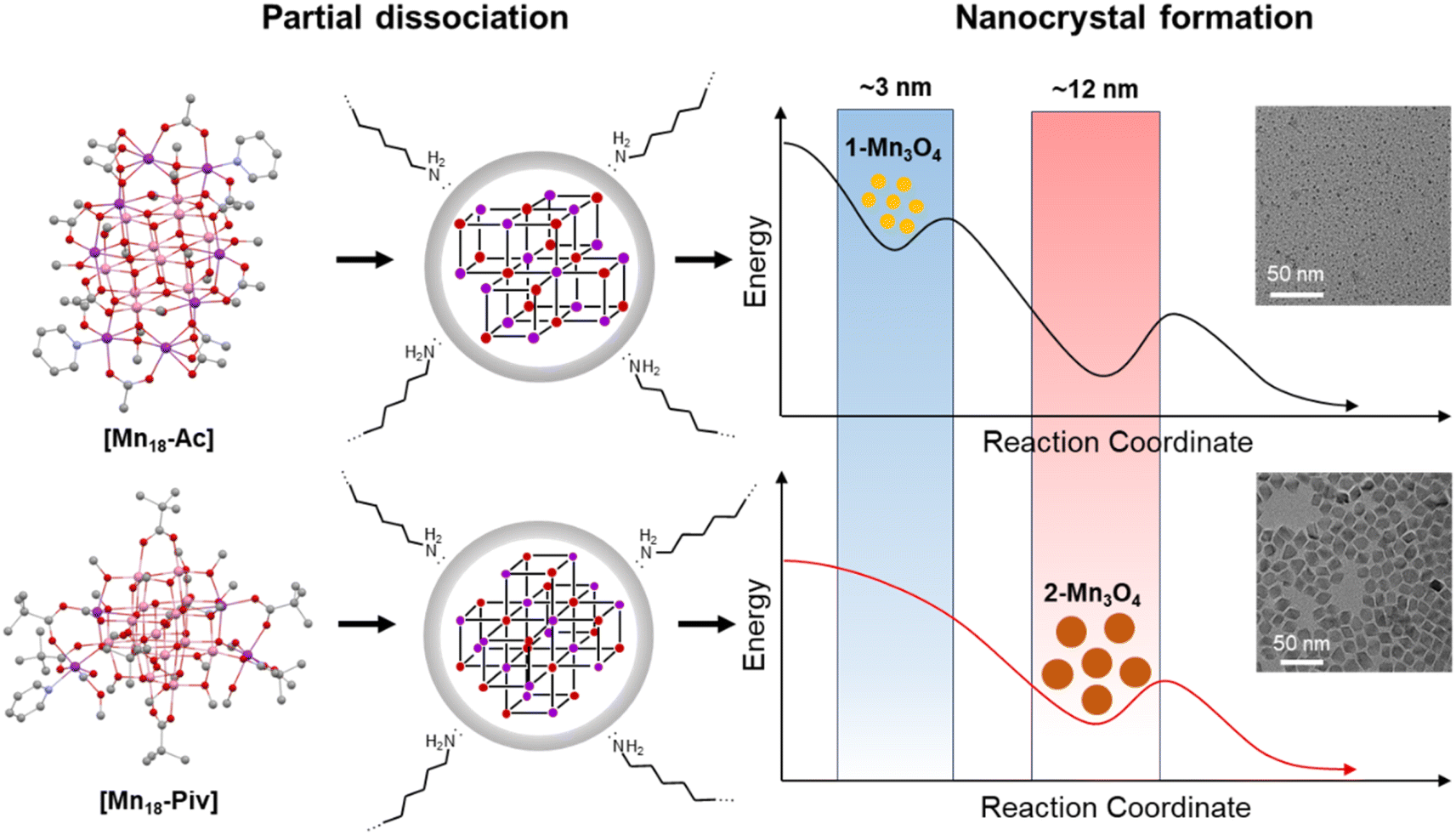 | ||
| Fig. 8 Proposed formation mechanism of Mn3O4 colloidal nanocrystals from [Mn18-Ac] and [Mn18-Piv] precursors. | ||
Conclusions
Two new high nuclearity Mn-oxo clusters [Mn18-Ac] and [Mn18-Piv] have been successfully prepared by employing carboxylate and alkoxide ligands. The clusters exhibit common structural features as being assembled of multiple [Mn4O4] cubanes with different packing patterns, which can be ascribed to the different coordinating modes of the respective carboxylates. The [Mn4O4] cubane-assembled structures are rare in the Mn-oxo clusters family, but ubiquitous in extended manganese oxide solids. The title clusters have been employed as precursors for the preparation of manganese oxide colloidal nanocrystals. X-ray powder diffraction and electron microscopy analysis revealed that both clusters produce Mn3O4 oxide nanocrystals with high monodispersity. The latter may be attributed to the pre-formed cubane-type [–O–Mn–O–Mn–] skeletons in clusters, which afford a more kinetically efficient pathway to the bulk manganese oxide solids. Interestingly, under the same preparation conditions, the dimensions of the Mn3O4 nanocrystals obtained from the two clusters are quite different. This implies that the topologies of the clusters may have a significant impact on the formation of nanocrystals, which should be investigated in future research. To the best of our knowledge, this work represents the first case study to directly prepare high-quality colloidal manganese oxide nanocrystals from the manganese-oxo cluster precursors. It can be envisioned that higher nuclearity metal-oxo clusters with similar structural features to the corresponding bulk solids, can potentially be even better precursor candidates for colloidal synthesis. This comes from the fact that relatively large size of the clusters can effectively reduce the energy of nucleation during the formation of nanocrystals, and the structural similarities between clusters and the final nanocrystals could afford more efficient kinetic routes.60,61Data availability
The crystallographic data was provided in Cambridge Structural Database, and the other necessary data of this study have been provided in the ESI.†Author contributions
H. H. and E. D. conceived this project, supervised and guided the design, analysis, interpretation and wrote the manuscript. Y. H. conducted synthesis of nanoclusters and nanocrystals, performed optical and magnetic investigations. Y. L. and Z. X. contributed to the conduction and analysis of electron microscopy experiments. H. Z. contributed to the analysis and discussion of results. Z. Z. performed theoretical calculations. F. G. and L. G. performed the photocatalytic experiments. All authors contributed to the interpretation of the results and preparation of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported by the Fundamental Research Funds for the Central Universities (22120230060, Z. Z.; 22120220118, H. H.), the National Natural Science Foundation of China (52101229, Z. X.; 22301219, Z. Z.; 22101205, H. H.), and Shandong Provincial Key Laboratory of Monocrystalline Silicon Semiconductor Materials and Technology (2023KFKT009, L. G. and H. H.). L. G. also acknowledges financial support from Qingchuang Science and Technology Plan of Shandong Province (2021KJ054). E. D. thanks the National Science Foundation (CHE-1955585) for support.Notes and references
- L. K. Thompson and L. N. Dawe, Coord. Chem. Rev., 2015, 289–290, 13–31 CrossRef CAS.
- T. C. A. Stamatatos, A. Khalil, W. Wernsdorfer and G. Christou, Angew. Chem., Int. Ed., 2007, 46, 884–888 CrossRef CAS PubMed.
- K. Takeda and K. Awaga, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 14560–14565 CrossRef CAS.
- J. R. Friedman, M. P. Sarachik, J. Tejada and R. Ziolo, Phys. Rev. Lett., 1996, 76, 3830–3833 CrossRef CAS PubMed.
- F. Yang, Y.-K. Deng, L.-Y. Guo, H.-F. Su, Z. Jagličić, Z.-Y. Feng, G.-L. Zhuang, S.-Y. Zeng and D. Sun, CrystEngComm, 2016, 18, 1329–1336 RSC.
- G. Maayan, N. Gluz and G. Christou, Nat. Catal., 2018, 1, 48–54 CAS.
- Ł. Michalak, C. M. Canali, M. R. Pederson, M. Paulsson and V. G. Benza, Phys. Rev. Lett., 2010, 104, 017202 CrossRef.
- C. Xu, Y. Jin, H. Fang, H. Zheng, J. C. Carozza, Y. Pan, P. Wei, Z. Zhang, Z. Wei, Z. Zhou and H. Han, J. Am. Chem. Soc., 2023, 145, 25673–25685 CrossRef CAS PubMed.
- H. Li, P. Wang, C. Zhu, W. Zhang, M. Zhou, S. Zhang, C. Zhang, Y. Yun, X. Kang, Y. Pei and M. Zhu, J. Am. Chem. Soc., 2022, 144, 23205–23213 CrossRef CAS.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- T. C. Stamatatos, K. A. Abboud, W. Wernsdorfer and G. Christou, Angew. Chem., Int. Ed., 2008, 47, 6694–6698 CrossRef CAS.
- M. Riaz, R. K. Gupta, H.-F. Su, Z. Jagličić, M. Kurmoo, C.-H. Tung, D. Sun and L.-S. Zheng, Inorg. Chem., 2019, 58, 14331–14337 CAS.
- T. Sala and M. V. Sargent, J. Chem. Soc., Chem. Commun., 1978, 253–254 RSC.
- G. Arom, S. M. J. Aubin, M. A. Bolcar, G. Christou, H. J. Eppley, K. Folting, D. N. Hendrickson, J. C. Huffman, R. C. Squire, H. L. Tsai, S. Wang and M. W. Wemple, Polyhedron, 1998, 17, 3005–3020 CrossRef.
- B. Sieklucka, J. Szklarzewicz, T. J. Kemp and W. Errington, Inorg. Chem., 2000, 39, 5156–5158 CrossRef CAS PubMed.
- K. L. Taft, A. Caneschi, L. E. Pence, C. D. Delfs, G. C. Papaefthymiou and S. J. Lippard, J. Am. Chem. Soc., 1993, 115, 11753–11766 CrossRef CAS.
- C. Lampropoulos, C. Koo, S. O. Hill, K. Abboud and G. Christou, Inorg. Chem., 2008, 47, 11180–11190 CrossRef CAS PubMed.
- E. K. Brechin, W. Clegg, M. Murrie, S. Parsons, S. J. Teat and R. E. P. Winpenny, J. Am. Chem. Soc., 1998, 120, 7365–7366 CrossRef CAS.
- Y. Deng, Y. Wu, Z. Li, Z. Jagličić, R. K. Gupta, C. Tung and D. Sun, Chin. J. Chem., 2023, 41, 1667–1672 CrossRef CAS.
- B.-Q. Ji, H.-F. Su, M. Jagodič, Z. Jagličić, M. Kurmoo, X.-P. Wang, C.-H. Tung, Z.-Z. Cao and D. Sun, Inorg. Chem., 2019, 58, 3800–3806 CrossRef CAS PubMed.
- K. Sheng, R. Wang, X. Tang, M. Jagodič, Z. Jagličić, L. Pang, J.-M. Dou, Z.-Y. Gao, H.-Y. Feng, C.-H. Tung and D. Sun, Inorg. Chem., 2021, 60, 14866–14871 CrossRef CAS PubMed.
- A. J. Tasiopoulos, A. Vinslava, W. Wernsdorfer, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2004, 43, 2117–2121 CrossRef CAS PubMed.
- M. Coletta, T. G. Tziotzi, M. Gray, G. S. Nichol, M. K. Singh, C. J. Milios and E. K. Brechin, Chem. Commun., 2021, 57, 4122–4125 RSC.
- T. N. Nguyen, W. Wernsdorfer, K. A. Abboud and G. Christou, J. Am. Chem. Soc., 2011, 133, 20688–20691 CrossRef CAS.
- M. Murugesu, J. Raftery, W. Wernsdorfer, G. Christou and E. K. Brechin, Inorg. Chem., 2004, 43, 4203–4209 CrossRef CAS PubMed.
- A. Vinslava, A. J. Tasiopoulos, W. Wernsdorfer, K. A. Abboud and G. Christou, Inorg. Chem., 2016, 55, 3419–3430 CrossRef CAS PubMed.
- K. F. Biegasiewicz, J. R. Griffiths, G. P. Savage, J. Tsanaktsidis and R. Priefer, Chem. Rev., 2015, 115, 6719–6745 CrossRef CAS.
- V. Baron, J. Gutzmer, H. Rundlof and R. Tellgren, Am. Mineral., 1998, 83, 786–793 CrossRef CAS.
- R. E. Pacalo and E. K. Graham, Phys. Chem. Miner., 1991, 18, 69–80 CrossRef CAS.
- Y.-K. Deng, H.-F. Su, J.-H. Xu, W.-G. Wang, M. Kurmoo, S.-C. Lin, Y.-Z. Tan, J. Jia, D. Sun and L.-S. Zheng, J. Am. Chem. Soc., 2016, 138, 1328–1334 CrossRef CAS.
- Y. Chu, L. Guo, B. Xi, Z. Feng, F. Wu, Y. Lin, J. Liu, D. Sun, J. Feng, Y. Qian and S. Xiong, Adv. Mater., 2018, 30, 1704244 CrossRef.
- C.-C. Wu, S. Datta, W. Wernsdorfer, G.-H. Lee, S. Hill and E.-C. Yang, Dalton Trans., 2010, 39, 10160–10168 RSC.
- M. R. Friedfeld, J. L. Stein and B. M. Cossairt, Inorg. Chem., 2017, 56, 8689–8697 CrossRef CAS.
- A. Ritchhart and B. M. Cossairt, Angew. Chem., Int. Ed., 2018, 57, 1908–1912 CAS.
- H. Zhang, B. Li, J. Sun, R. Clérac and E. V. Dikarev, Inorg. Chem., 2008, 47, 10046–10052 CrossRef CAS PubMed.
- Z. Wei, H. Han, A. S. Filatov and E. V. Dikarev, Chem. Sci., 2014, 5, 813–818 RSC.
- S. Petit, F. Baril-Robert, G. Pilet, C. Reber and D. Luneau, Dalton Trans., 2009, 6809–6815 RSC.
- M.-L. Hu, A. Morsali and L. Aboutorabi, Coord. Chem. Rev., 2011, 255, 2821–2859 CrossRef CAS.
- D. Van den Eynden, R. Pokratath and J. De Roo, Chem. Rev., 2022, 122, 10538–10572 CrossRef CAS PubMed.
- T. A. Stromnova, O. N. Shishilov, L. I. Boganova, N. A. Minaeva, A. V. Churakov, L. G. Kuz'mina and J. A. K. Howard, Russ. J. Inorg. Chem., 2005, 50, 179–186 Search PubMed.
- Y. He, Z. Zhou and H. Han, Crystals, 2024, 14, 478–487 CrossRef CAS.
- T. L. Ho, Chem. Rev., 1975, 75, 1–20 CrossRef CAS.
- L.-Y. Guo, H.-F. Su, M. Kurmoo, C.-H. Tung, D. Sun and L.-S. Zheng, J. Am. Chem. Soc., 2017, 139, 14033–14036 CrossRef CAS PubMed.
- R. A. Reynolds and D. Coucouvanis, Inorg. Chem., 1998, 37, 170–171 CrossRef CAS.
- M. T. Caudle, P. Riggs Gelasco, A. K. Gelasco, J. E. Penner Hahn and V. L. Pecoraro, Inorg. Chem., 1996, 35, 3577–3584 CrossRef CAS.
- J. Yoo, A. Yamaguchi, M. Nakano, J. Krzystek, W. E. Streib, L. C. Brunel, H. Ishimoto, G. Christou and D. N. Hendrickson, Inorg. Chem., 2001, 40, 4604–4616 CrossRef CAS PubMed.
- A. J. W. Thom, E. J. Sundstrom and M. Head-Gordon, Phys. Chem. Chem. Phys., 2009, 11, 11297–11304 RSC.
- H. Visser, E. Anxolabéhère Mallart, U. Bergmann, P. Glatzel, J. H. Robblee, S. P. Cramer, J.-J. Girerd, K. Sauer, M. P. Klein and V. K. Yachandra, J. Am. Chem. Soc., 2001, 123, 7031–7039 CrossRef CAS PubMed.
- A. Manceau, M. A. Marcus and S. Grangeon, Am. Mineral., 2012, 97, 816–827 CrossRef CAS.
- E. Saputra, S. Muhammad, H. Sun, H.-M. Ang, M. O. Tadé and S. Wang, Appl. Catal., B, 2013, 142–143, 729–735 CrossRef CAS.
- X. Li, Q. Zhou, S. Wang, Y. Li, Y. Liu, Q. Gao and Q. Wu, J. Phys. Chem. C, 2021, 125, 11963–11974 CrossRef CAS.
- F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 310, 41–62 CrossRef CAS.
- N. Tian, X. Tian, Y. Nie, C. Yang, Z. Zhou and Y. Li, Chem. Eng. J., 2018, 352, 469–476 CrossRef CAS.
- K. Hu, Z. Li, L. Bai, F. Yang, X. Chu, J. Bian, Z. Zhang, H. Xu and L. Jing, Sol. RRL, 2021, 5, 2000472 CrossRef CAS.
- R. S. Dhayal, W. E. van Zyl and C. W. Liu, Acc. Chem. Res., 2016, 49, 86–95 CrossRef CAS.
- C. Xu, Z. Zhou and H. Han, Crystals, 2022, 12, 1236–1246 CrossRef CAS.
- W. S. Seo, H. H. Jo, K. Lee, B. Kim, S. J. Oh and J. T. Park, Angew. Chem., Int. Ed., 2004, 43, 1115–1117 CrossRef CAS PubMed.
- Y. Li, H. Tan, X.-Y. Yang, B. Goris, J. Verbeeck, S. Bals, P. Colson, R. Cloots, G. Van Tendeloo and B.-L. Su, Small, 2011, 7, 475–483 CrossRef CAS.
- P. R. Garcês Gonçalves Jr, H. A. De Abreu and H. A. Duarte, J. Phys. Chem. C, 2018, 122, 20841–20849 CrossRef.
- F. Wang, V. N. Richards, S. P. Shields and W. E. Buhro, Chem. Mater., 2014, 26, 5–21 CrossRef CAS.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, single crystal X-ray data, magnetic measurements, X-ray photoelectron spectra, X-ray absorption near edge spectra, photocatalytic data, transmission electron microscopy and powder X-ray diffraction patterns, theoretical calculations. CCDC 2325203 and 2325204. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01451k |
| This journal is © The Royal Society of Chemistry 2024 |