
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRobust battery interphases from dilute fluorinated cations†
Chulgi Nathan
Hong
a,
Mengwen
Yan
b,
Oleg
Borodin
 c,
Travis P.
Pollard
c,
Langyuan
Wu
d,
Manuel
Reiter
a,
Dario Gomez
Vazquez
a,
Katharina
Trapp
a,
Ji Mun
Yoo
a,
Netanel
Shpigel
d,
Jeremy I.
Feldblyum
b and
Maria R.
Lukatskaya
*a
c,
Travis P.
Pollard
c,
Langyuan
Wu
d,
Manuel
Reiter
a,
Dario Gomez
Vazquez
a,
Katharina
Trapp
a,
Ji Mun
Yoo
a,
Netanel
Shpigel
d,
Jeremy I.
Feldblyum
b and
Maria R.
Lukatskaya
*a
aElectrochemical Energy Systems Laboratory, Department of Mechanical and Process Engineering, ETH Zurich, Zürich 8092, Switzerland. E-mail: mlukatskaya@ethz.ch
bDepartment of Chemistry, The University at Albany, State University of New York, Albany, NY 12222, USA
cBattery Science Branch, DEVCOM Army Research Laboratory, Adelphi, MD 20783, USA
dDepartment of Chemical Sciences, Ariel University, Ariel 40700, Israel
First published on 2nd May 2024
Abstract
Controlling solid electrolyte interphase (SEI) in batteries is crucial for their efficient cycling. Herein, we demonstrate an approach to enable robust battery performance that does not rely on high fractions of fluorinated species in electrolytes, thus substantially decreasing the environmental footprint and cost of high-energy batteries. In this approach, we use very low fractions of readily reducible fluorinated cations in electrolyte (∼0.1 wt%) and employ electrostatic attraction to generate a substantial population of these cations at the anode surface. As a result, we can form a robust fluorine-rich SEI that allows for dendrite-free deposition of dense Li and stable cycling of Li-metal full cells with high-voltage cathodes. Our approach represents a general strategy for delivering desired chemical species to battery anodes through electrostatic attraction while using minute amounts of additive.
Broader contextStable and cost-effective Li-metal batteries (LiMBs) are necessary for non-incremental improvement of energy density in commercial batteries. However, the implementation of Li-metal anodes is impeded by an unacceptably low cycle life and safety concerns when conventional electrolytes are used. In particular, the formation of electronically inactive “dead” lithium and dendrites takes place during cycling. Prior research suggests that fluorine-rich interfacial layer chemistry is important for the stabilization of Li-metal anodes, which can be achieved when electrolytes with a high fraction of fluorinated solvents and/or salts are used. Herein, we introduce an alternative approach that leverages electrostatic attraction between positively charged fluorinated cations and the negatively charged Li-metal anode, generating a significant population of fluorinated species near the electrode surface with a very low fraction of additive in the electrolyte (∼0.1 wt%). As a result, a robust fluorine-rich interfacial layer is formed, enabling dendrite-free deposition of dense Li metal. In general, we present a strategy for delivering desired chemical species to the battery anodes through electrostatic attraction while using minute amounts of additive and therefore can notably reduce costs and environmental footprint of implementing high energy batteries. |
Introduction
Replacement of traditional intercalation anodes such as graphite (372 mA h g−1) or lithium titanium oxide (175 mA h g−1) with Li metal (3860 mA h g−1) can enable high-energy battery cells,1 providing a solution to the current energy density bottlenecks for batteries.2–4 However, Li metal anodes are prone to developing dendritic and porous (mossy) deposits during repetitive charge/discharge cycling. This leads to an unacceptably low cycle life and introduces serious safety concerns, prohibiting practical implementation of lithium metal batteries (LiMBs).2–5 The Li plating/stripping process and cycle life of LiMBs are strongly affected by the properties of the so-called solid electrolyte interphase (SEI). An SEI is a layer formed on the electrode surface as a result of the decomposition of electrolyte components such as solvent, salt, and molecular additives.3,6 Electrolyte composition is known to dramatically affect the properties of the resulting SEI (e.g. structure, chemistry, and homogeneity) and lithium plating/stripping (coulombic) efficiency, morphology and cycle life.4 Commercial Li-ion electrolyte chemistries are based on carbonate solvents and Li-ion salts that were originally developed and optimized for graphite anodes. However, when these conventional carbonate-based electrolytes are used in combination with Li-metal anodes, they yield highly unsatisfactory cycle life due to unfavorable SEI chemistry at the Li-metal surface.2–5 Specifically, insufficient electronic insulation, structural instability and heterogeneity of the SEI result in uncontrolled SEI growth and the formation of electronically disconnected “dead lithium” and/or dendrites.5,7 This results in low coulombic efficiencies (well below 99% that is required7 for every cycle in long-life LiMBs). Therefore, the development of new electrolytes that can yield favorable SEI on Li-metal anodes constitutes a critical research direction.7Recent research suggests that fluorine-rich SEI yields superior performance compared to fluorine-free SEI.3,8,9 To achieve fluorine-rich SEI, state-of-the-art approaches rely on large volume fractions of fluorinated species in the electrolyte that have a statistically higher probability of being reduced at the electrode surface and, therefore, generate fluorine-rich SEI.10 For example, solvent-in-salt electrolytes with a high concentration of fluorine-containing anions yield fluorine-rich SEIs and much improved coulombic efficiencies compared to their dilute counterparts.11 Recently, another way to generate a fluorine enriched SEI from fluorine-containing salts at intermediate concentrations was reported by employing a modified solvent with siloxane groups12 or through usage of surface-modified separators.13 Also, fluorinated solvents themselves can be used to form high-quality SEIs.8,14 The implementation of a fluorinated ionic liquid as a battery solvent is another way to create a fluorine-rich SEI that can suppress formation of dead lithium.15,16
Yet, despite enabling promising performance, these approaches have notable drawbacks when it comes to their practical implementation. The high cost of Li salts and increased solution viscosity at high salt concentration makes highly concentrated electrolytes challenging to implement in commercial batteries. Similarly, the replacement of conventional solvents with heavily fluorinated ones can lead to substantial increases in battery costs and environmental footprint.
Herein, we establish an alternative approach that relies on the electrostatic attraction of fluorinated cations to a negatively charged anode. Through this approach, a significant population of fluorinated species can reach the electrode surface even when the overall additive concentration in the bulk electrolyte is in the millimolar range. Importantly, to ensure the predominant contribution of fluorinated cations to SEI formation, their reduction potential should be significantly higher than that of solvent molecules or anions in electrolyte (as schematically depicted in Fig. 1a). To this end, we selected fluorinated methylpyridinium cations (Fig. 1a) that can offer early decomposition potentials of ∼2 V vs. Li/Li+ (at least ∼1.5 V before solvent decomposition starts). First, we show that the addition of fluorinated methylpyridinium cations even in millimolar amounts (0.08–0.14 wt%) to a conventional electrolyte based on 1,2-dimethoxyethane (DME) enables F-enriched SEI and dense Li plating with an increased coulombic efficiency of 99.6% (compared to 96.4% obtained in additive-free electrolytes). Second, we demonstrate prolonged cycling of a full cell with Li metal anode and Ni-rich high-voltage cathode in ether-based electrolytes with 99.6% average coulombic efficiency, which is achieved by suppressing oxidative decomposition of DME upon addition of small amounts of fluorinated cations. Third, the fluorinated cations also suppress corrosion of the Al current collector caused by chlorine impurities in the electrolyte, thus assisting with prolonged cycling of the full cell.
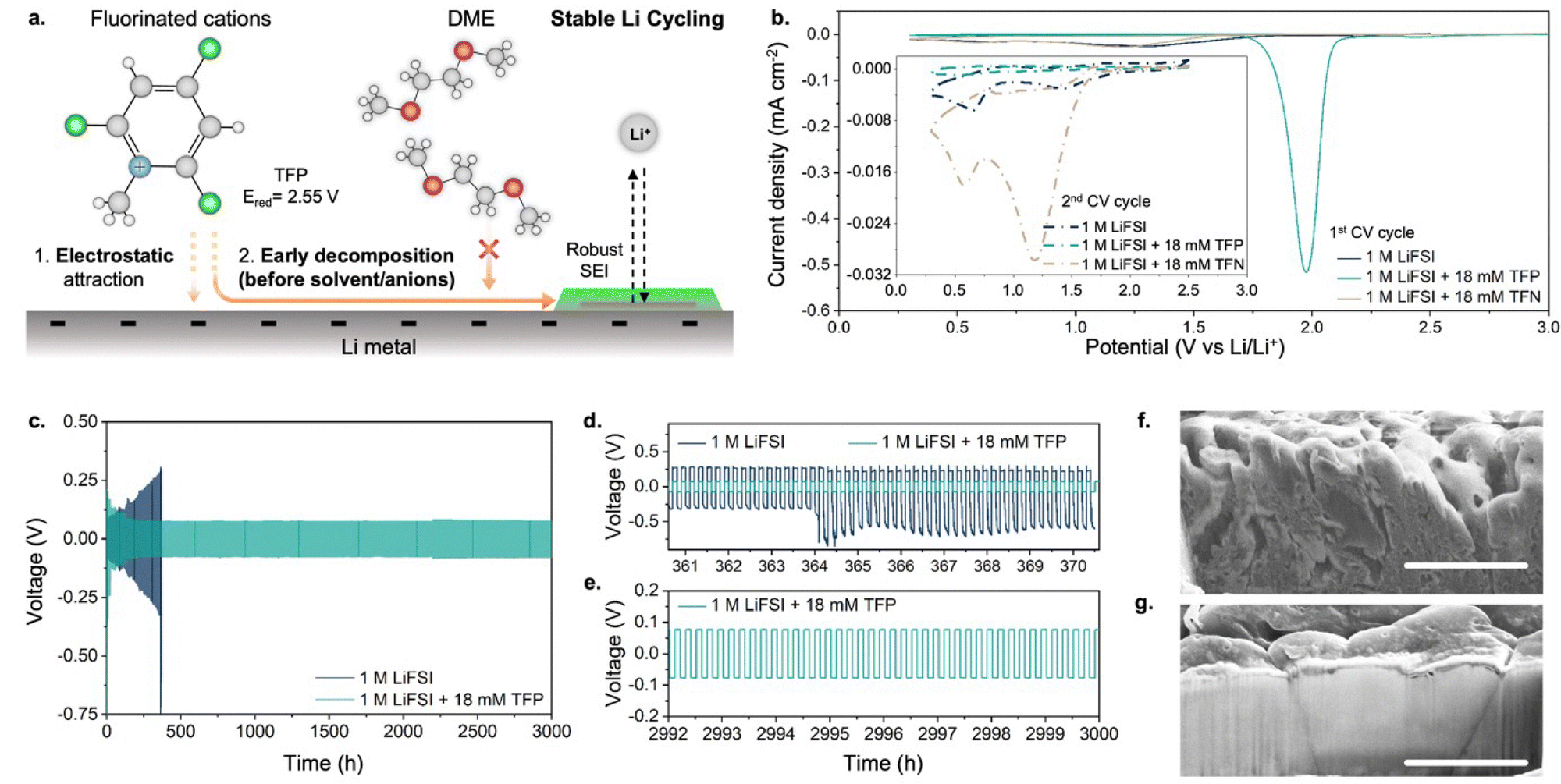 | ||
| Fig. 1 (a) Schematic diagram of SEI formation from a fluorinated cationic additive on a Li-metal anode. (b) First and second (inset) cycle CV profiles collected in DME + 1 M LiFSI electrolytes with and without fluorinated cations (TFP) and fluorinated neutral analogue (TFN) using a 1 mm Cu disk working electrode at a 0.5 mV s−1 scan rate and a voltage window of 0.3–2.5 V vs. Li/Li+. (c) Galvanostatic cycling of a Li0–Li0 symmetric cell at 10 mA cm−2 using DME + 1 M LiFSI as an electrolyte with and without fluorinated cations (TFP). (d) and (e) Zoomed-in voltage profile of (c). (f) and (g) Cross-sectional SEM images of the cycled Li metal (f – after 1852 cycles in DME + 1 M LiFSI, g – after 2000 cycles in DME + 1 M LiFSI + 12 mM TFP) showing Li metal deposit morphologies. Scale bar: 5 μm. Before SEM, a lamella (5 μm deep) was cut out using cryo-FIB. | ||
Results and discussion
Effect of fluorinated cations on cycling of lithium metal anodes
The choice of fluorinated methylpyridinium cations was dictated by the following considerations: (1) previous experiments showed pyridinium-based cations undergo reductive decomposition at ∼1.75 V vs. Li/Li+,17,18 which is well above the decomposition potentials of non-aqueous solvents and anions; (2) quantum-chemical calculations suggest that fluorination of methylpyridinium cations would further shift the reduction potential to even higher values19 (Fig. S1, ESI†), potentially providing a simple pathway for the formation of fluorine-rich SEI. In our experiments, we used N-methyl-2,4,6-trifluoropyridinium (TFP) as a fluorinated cationic additive in a form of perchlorate salt. As the baseline electrolyte we used 1 M lithium bis(fluorosulfonyl)imide (LiFSI) in DME because it shows one of the highest coulombic efficiencies (96.4%) for Li plating/stripping among additive-free electrolyte formulations.8Cyclic voltammetry (CV) profiles collected in additive-containing electrolytes show a pronounced reduction peak at ∼1.98 V vs. Li/Li+ (Fig. 1b) during the 1st cycle, which is absent in the additive-free electrolyte. The current associated with the reduction peak scales with the concentration of TFP (Fig. S2, ESI†). Furthermore, an identical position of TFP-related reduction peak is observed regardless of whether TFP additive is used in the form of perchlorate or triflate salt (Fig. S3, ESI†). The reduction peak disappears in the 2nd cycle, consistent with passivation of the anode surface, preventing further reduction of fluorinated cations. These experiments confirm that the reduction of fluorinated cations occurs at potentials nearly ∼1.5 V higher than the onset of decomposition of the additive-free electrolyte.20 In contrast, no distinct reduction CV peaks or subsequent passivation was observed upon addition of the same concentrations of the neutral analogue, 2,4,6-trifluoropyridine (TFN), indicating importance of the additive charge in enabling its efficiency (Fig. 1b). These results are also in agreement with density functional theory (DFT) calculations that predict TFN's low reduction potential of 0.23 V vs. Li/Li+ (Fig. S1a, ESI†).
To quantify how the addition of fluorinated cations affects the coulombic efficiency of Li plating/stripping, we used a modified Aurbach protocol21 (Fig. S4a, ESI†). The test revealed a substantial improvement of average coulombic efficiency from 96.4% for the TFP-free electrolyte to 99.6% for the cells containing TFP (∼0.1 wt%, Fig. S4b, ESI†). This efficiency is comparable to those reported for the best-performing electrolytes containing fluorinated solvent8,22 or salts in high concentrations,11,23 demonstrating that large fractions of fluorinated species in electrolytes can be avoided when aiming for high coulombic efficiencies.
Next, we performed long-term galvanostatic cycling at 10 mA cm−2 using a Li0–Li0 symmetric cell configuration (Fig. 1c). A dramatic difference was observed between the cells with and without TFP. For cells with additive-free electrolyte, a continuous increase in overpotential due to the formation of dead lithium was observed with cycling (similar to previous reports24,25), resulting in cell failure after 400 h (Fig. 1d). In contrast, addition of millimolar amounts of fluorinated cations enabled outstanding cycling stability and, after initial stabilization, the overpotential remained almost unchanged for at least 3000 h of cycling (Fig. 1e). Moreover, the cycling stability increases with the concentration of fluorinated cations (Fig. S5, ESI†), showing the greatest stability for the 18 mM concentration (Fig. 1c).
Importantly, the morphology of Li metal correlated with the evolution of overpotential with cycling. Initially, comparable overpotentials and similar Li morphology were observed for both electrolytes (Fig. S6, ESI†). After 371 hours of cell cycling, for the additive-free electrolyte a rough Li surface with multiple cracks can be seen by scanning electron microscopy (SEM; Fig. 1f and Fig. S7a, ESI†), with highly porous Li deposits and in agreement with prior studies.26 In contrast, a smooth surface with large and dense Li grains was observed for Li metal that was cycled in the electrolyte containing TFP (Fig. 1g and Fig. S7b, ESI†).
Solid electrolyte interphase
To understand the origin of the superior cycling performance of the Li-metal anode in TFP-containing electrolytes, we performed studies of the SEI layers formed in the presence of TFP using X-ray photoelectron spectroscopy (XPS), electrochemical quartz crystal microbalance with dissipation monitoring (EQCM-D), and quantum chemistry calculations (Fig. 2). The XPS data collected from cycled Cu electrodes revealed a high fluorine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :carbon (F:C) atomic ratio for the SEI formed in the presence of cationic additives (F:C = 3.1–4.5), whereas the F:C ratio of 0.17–0.24 was observed for the SEI generated in reference electrolyte (Fig. 2a). This indicates a dramatically decreased relative contribution of the solvent decomposition products. Furthermore, analysis of F 1s and Li 1s spectra revealed LiF as a dominant species for SEI samples obtained in the presence of fluorinated cations (Fig. 2b and Fig. S8 and Table S1, ESI†). Similarly, a more than three-fold enrichment of SEI with fluorine was observed for a Li metal electrode cycled in the presence of TFP (Li0‖Li0 symmetric cell, one week of galvanostatic cycling at 10 mA cm−2, Fig. S9, ESI†). This indicates that even millimolar addition of the fluorinated cations can yield favorable F-rich SEI.
:carbon (F:C) atomic ratio for the SEI formed in the presence of cationic additives (F:C = 3.1–4.5), whereas the F:C ratio of 0.17–0.24 was observed for the SEI generated in reference electrolyte (Fig. 2a). This indicates a dramatically decreased relative contribution of the solvent decomposition products. Furthermore, analysis of F 1s and Li 1s spectra revealed LiF as a dominant species for SEI samples obtained in the presence of fluorinated cations (Fig. 2b and Fig. S8 and Table S1, ESI†). Similarly, a more than three-fold enrichment of SEI with fluorine was observed for a Li metal electrode cycled in the presence of TFP (Li0‖Li0 symmetric cell, one week of galvanostatic cycling at 10 mA cm−2, Fig. S9, ESI†). This indicates that even millimolar addition of the fluorinated cations can yield favorable F-rich SEI.
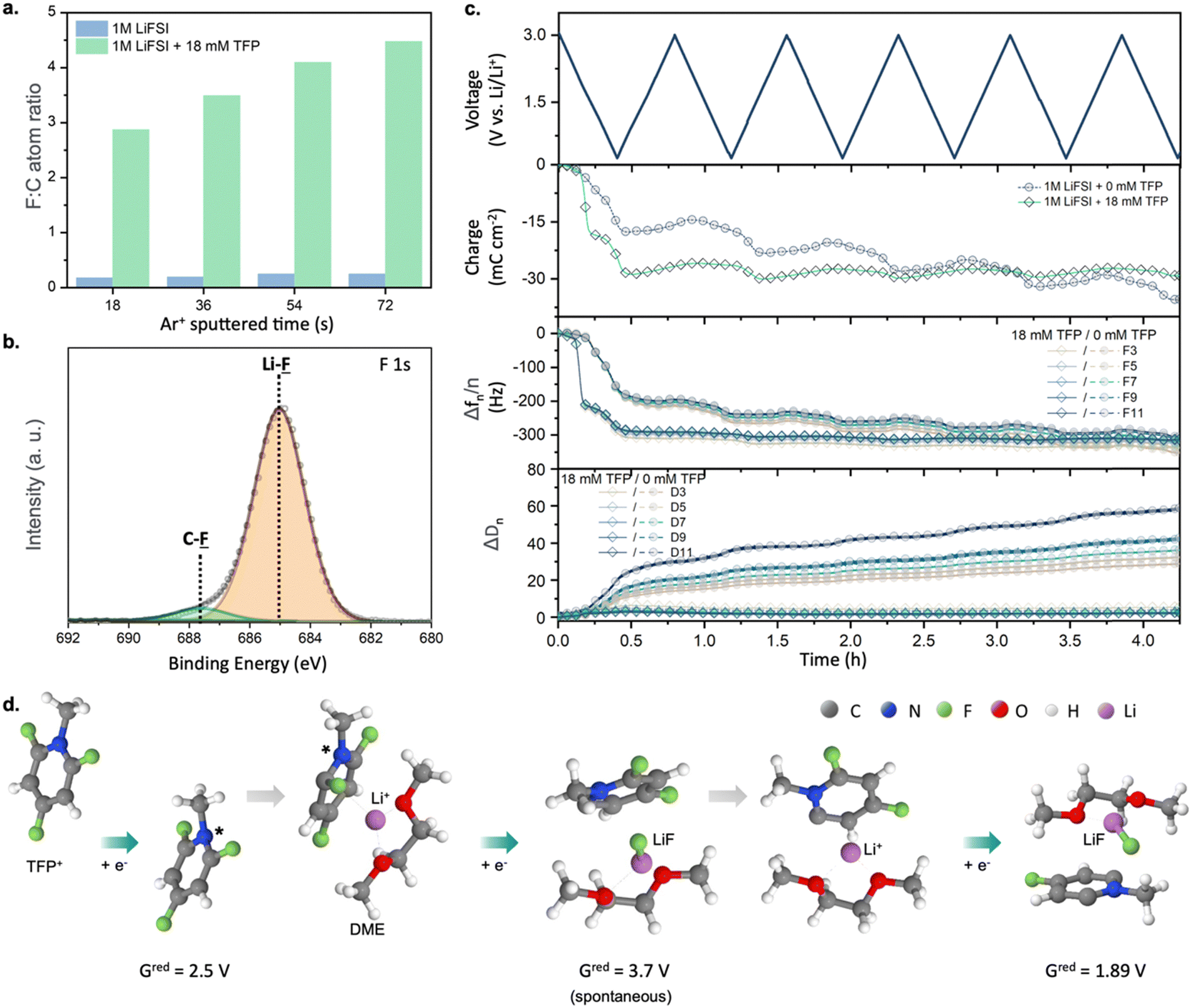 | ||
| Fig. 2 (a) F:C atomic ratio in the SEI layer formed on a Cu electrode as a function of depth. (b) XPS F 1s spectrum of a SEI layer formed on a Cu electrode after cycling in DME + 1 M LiFSI + 18 mM TFP with Ar+ sputtered time of 72 s (estimated depth: 2.0 nm). (c) Voltage, charge, frequency, and dissipation change versus time using EQCM-D analysis for DME + 1 M LiFSI and DME + 1 M LiFSI + 18 mM TFP. (d) Products of reduction at the negative electrode obtained from DFT calculations, see Fig. S11 (ESI†) for further details. | ||
Next, EQCM-D measurements show that the SEI formed in electrolytes with cationic additive is highly rigid since the change of the overtone-normalized resonant frequency (Δfn/n) and energy dissipation (ΔDn) are independent27 of overtone order (n) (Fig. 2c). Therefore, we can apply the Sauerbrey equation to obtain gravimetric information for the formed SEI. By comparing the theoretical and experimental frequencies,28 we attribute the main frequency decrease at ∼2 V to the formation of LiF, which is in good agreement with the XPS results (Fig. S10, ESI† and Fig. 2b). Moreover, no further mass gain was observed after the first CV cycle, indicating the highly desirable robust and passivating nature of the formed SEI. In contrast, the SEI formed in the additive-free electrolyte displays viscoelastic behavior: ΔDn depends on overtone order and larger values of dissipation modulus are observed.27–29 Upon cycling, a continuous increase in charge, Δfn/n and ΔDn can be seen, indicative of uncontrolled SEI growth due to the non-compact and porous nature of the formed interfacial layers.27
Since both XPS and EQCM-D suggest that SEI formed in the presence of TFP is mainly LiF, we further investigated the detailed SEI formation pathway using DFT calculations and MD simulations. The DFT calculations suggest that TFP reduction is the dominant process due to the electrostatic attraction of TFP to the negative electrode and its higher reduction potential (2.1–2.5 V vs. Li/Li+) compared to FSI− (Fig. 2d and Fig. S11, ESI†). Meanwhile, FSI− reduction is unlikely here due to the low affinity of FSI− to the negative electrode30 and low fraction of contact ion pairs (CIPs) or aggregates (AGGs) of LiFSI in DME at this salt concentration (<30%) observed in MD simulations (see Supplementary discussion, ESI†) and previous work.31,32 Reduced TFP* radicals at the anode surface undergo a second reduction and release of F− that results in the formation of LiF when TFP* is in close proximity to Li+ (Fig. 2d and Fig. S11, ESI†). A subsequent reduction and defluorination is also likely when the Li+/TFP−(−F) complex is exposed to potentials below 1.9 V vs. Li/Li+. This allows the additive to deliver multiple F− to form LiF-rich SEI. Alternatively, the reduced TFP* radicals can dimerize after their 1st reduction to form a fluorinated viologen species which undergoes spontaneous defluorination in proximity of Li+ across the newly formed C–C bond (left panel of Fig. S12, ESI†). The viologen species can form an anion (2nd reduction) at ∼0.6 V with subsequent release of another LiF (3rd reduction; in the box in Fig. S12, ESI†).
Full cell performance
Improvement of battery energy density requires coupling of Li-metal anodes with high-voltage cathodes.11,33 Previous studies suggest that high-voltage cathode materials cannot be used in non-concentrated DME-based electrolytes due to the oxidative decomposition of DME solvent above 4 V vs. Li/Li+.8,22,34 When we studied the oxidative stability of DME-based electrolytes with fluorinated cations, we found that the potential for the oxidative decomposition is shifted by approximately +200 mV (4.76 V to 4.95 V at 0.5 mA cm−2) when TFP+ is added to the electrolyte (Fig. S13, ESI†). Because we use perchlorate as a counter anion, we also explored its effect on the stability of the electrolyte. By performing cyclic voltammetry tests in solutions containing 10 mM LiClO4 and 10 mM TFP-ClO4 (in DME + 1 M LiFSI), we observed that the addition of 10 mM LiClO4 results in lower decomposition currents at 5 V compared to a pure DME + 1 M LiFSI solution. This observation suggests that perchlorate anions contribute to enhancing the oxidation stability of DME-based electrolytes (as shown in Fig. S13c, ESI†). Notably, the electrolyte containing both TFP+ and ClO4− demonstrates even greater stability, with oxidative currents reduced by half, emphasizing the advantageous role of TFP+.Such an increase in the oxidation stability of the DME-based electrolyte with TFP cations implies that these electrolytes can be used with high voltage cathodes. To study this, we first performed galvanostatic cycling using a LiNi0.8Co0.1Mn0.1O2 (NCM811) cathode and the Li metal anode (NCM811‖Li metal) in a coin cell configuration with different concentrations of TFP additive to find the optimal one (Fig. S14a, ESI†). Without TFP, the cells show severe capacity fading and a decrease in coulombic efficiency around the 30th cycle, in agreement with previous reports (Fig. S14 and S15, ESI†).8 In contrast, cells with TFP demonstrate a dramatic improvement in cycling stability even for the TFP concentrations as low as 4 mM (∼0.03 wt%; 250 μm Li full cells; Fig. S14a, ESI†), with the optimal concentration being 12 mM (0.1 wt%). The NCM811‖50 μm Li metal full cells with the electrolyte containing 12 mM TFP maintained 94% of its discharge capacity even after 275 cycles (as measured at 0.1C) and an average coulombic efficiency of 99.6% (1C, Fig. 3a). In comparison, for the full cells containing 12 mM LiClO4 (Fig. S15, ESI†), we observed a much faster capacity loss than for 12 mM TFP-ClO4.
 | ||
| Fig. 3 NCM811‖Li full-cell performance. (a) Long-term cycling of NCM811‖50 μm Li full cells in DME + 0.96 M LiFSI + 12 mM TFP ClO4 (0.1C-rate for three cycles and 1C-rate for 50 cycles in a loop). (b) Voltage profile of NCM811‖Li full-cells at 0.1C-rate (top) and 1C-rate (bottom). (c) XPS F 1s spectra of CEI layer formed on NCM811 electrodes cycled in DME + 0.96 M LiFSI + 12 mM TFP ClO4. (d) DFT results predicting H-transfer from DME to LiNiO2 positive electrode followed by DME*(−H) reaction with TFP+ that leads to the F-enriched CEI formation in the presence of additive. | ||
The charge/discharge voltage profiles (Fig. 3b) show that the cells containing fluorinated cations can be successfully and repetitively charged up to 4.2 V. The differential capacity profiles (dQ/dV, Fig. S16, ESI†) for the cells with TFP showed both phase transitions expected for the NCM811 cathode during charging/discharging [from hexagonal (H1) to monoclinic (M) occurring between 3.6 V and 3.8 V and from monoclinic to hexagonal (H2) at ∼4.0 V].35,36 This is in contrast to the cells without additive that showed severe discharge capacity fading when charging up to 4.2 V due to electrolyte decomposition (Fig. S14, ESI†).8 The electrochemical impedance spectroscopy data also show a minimal increase in impedance with cycling for the full cells with TFP (Fig. S17, ESI†), in good agreement with the cells’ dQ/dV profiles.
In addition, we found that the presence of TFP in the electrolyte helps to suppress current collector corrosion. SEM of the aluminum current collectors that supported the NCM811 cathode showed that after 160 cycles in the electrolyte with TFP+, the current collector had no signs of corrosion, while in the additive-free electrolyte a significant roughening of the Al current collector was observed already after 20 cycles (Fig. S18, ESI†). Based on the DFT calculations, we suggest that TFP+ may act as a scavenger of chloride ions and chlorine radicals that are often present in commercial LiFSI, resulting in decreased corrosion of aluminum and cathode surfaces (Fig. S19, ESI†).
Oxidative decomposition and solvation structure of electrolytes
We hypothesize that the improved oxidation stability of the electrolyte and stable cycling of NCM811‖Li cells with fluorinated cations are due to the formation of a favorable cathode electrolyte interphase (CEI). We probe this hypothesis using XPS (Fig. 3c) and quantum chemistry calculations (Fig. 3d). XPS of cycled NCM811 shows that the CEI has a substantial amount of fluorine in the TFP-containing electrolyte, indicating a possible role of fluorinated cations in CEI formation (Fig. 3c and Fig. S18–S20 and Table S1, ESI†). According to DFT calculations, direct oxidation of isolated TFP cation is unlikely due to its high oxidation potential and electrostatic repulsion (Fig. S1a, ESI†). Next, we considered participation of TFP+–FSI− and TFP+–(FSI−)2 aggregates in CEI formation. Although these aggregates would not be electrostatically repelled from the cathode surface, direct oxidation of these complexes is also unlikely within our operating potential window of 3.0–4.2 V vs. Li/Li+ (Fig. S1c, ESI†). Therefore, we considered a different mechanism that involves reaction of TFP cations with DME radicals [C4H9O2]* that are formed at the cathode surface (Fig. 3d).37–39 DFT calculations show that this process has a low energy barrier indicative of fast kinetics (Fig. 3d, central panel). Further H-transfer35 from the ether tail of the [C4H9O2]*–TFP+ to the cathode surface leads to the formation of radicals that are scavenged by TFP cations (Fig. 3d, left panel). After that, the formed molecular aggregates precipitate at the cathode surface (Fig. 3d, right panel), resulting in the protective CEI that is rich in C–F containing species which agrees well with the XPS results for the surface layer formed in the presence of TFP (Fig. 3c and Fig. S20, ESI†). Apart from the CEI mechanism associated with TFP+ forming aggregates with DME, we cannot rule out the possibility that TFP-based products that form at the anode can diffuse to the cathode and contribute to the CEI formation (i.e. cross-talk). Although DFT calculations shed light on the interplay between TFP+ and DME, additional in situ experiments are required to unveil how TFP+ participates in the CEI formation. Also, our results show that the counter-ions for TFP+ play an important role in stabilizing the cathode–electrolyte interphase.Next, we performed molecular dynamics simulations (MD) to provide insights into electrolyte structure and the solvation of the TFP+ additive (Fig. 4a and Fig. S21, ESI†). Higher TFP+ concentrations were used in MD simulations to ensure that at least 12 TFP+ cations are present in a simulation cell. Two TFP+ concentrations (0.27 M and 0.14 M TFP) were examined to understand concentration dependence of additive solvation (Fig. S22, ESI†). A snapshot of the MD simulation cell is shown in Fig. 4 together with the representative Li+ solvates (Fig. S23, ESI†). At room temperature, the Li+(DME)3 solvates are prevalent: 75% of Li+ are not coordinated by either ClO4− or FSI− anions. The rest of Li+ are part of the Li+/FSI− and Li+/ClO4− contact ion pairs (CIPs) shown in Fig. 4c and Fig. S23a–d (ESI†).
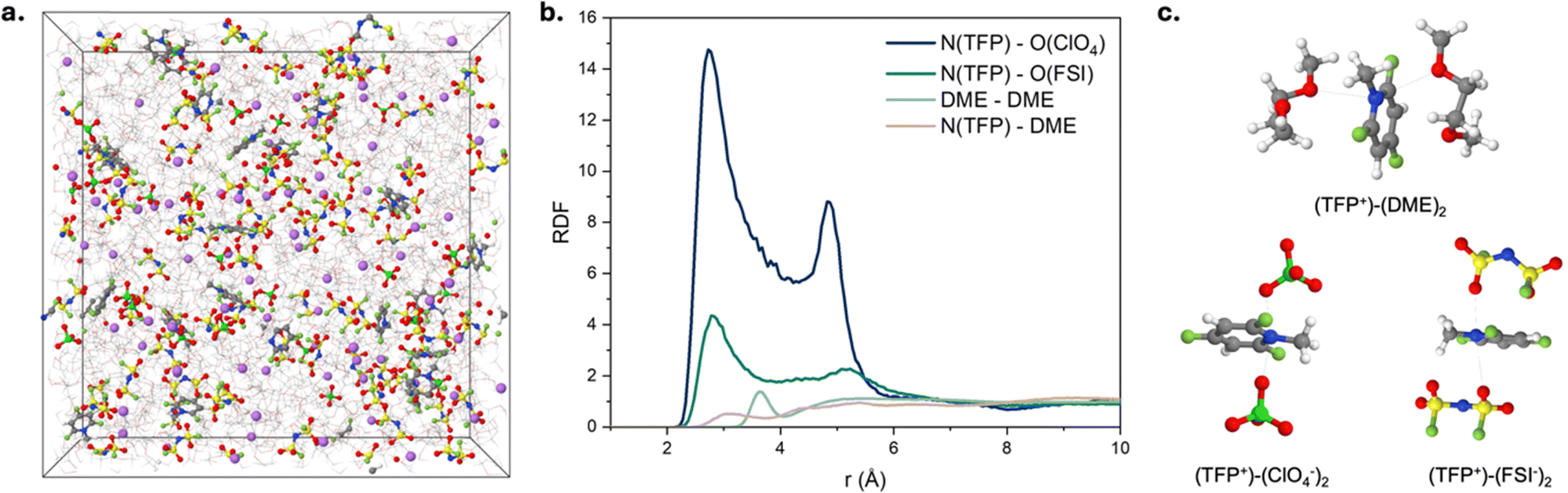 | ||
| Fig. 4 (a) A snapshot of the MD simulation cell for 0.91 M LiFSI and 0.27 M [TFP][ClO4] in DME. (b) Radial distribution functions for 0.91 M LiFSI and 0.27 M [TFP+][ClO4−] in DME at 298 K and (c) representative cation solvates for 0.91 M LiFSI and 0.27 M [TFP+][ClO4−] in DME at 298 K. | ||
Fig. 4b and Fig. S24 (ESI†) show the radial distribution functions (RDFs) for the respective electrolytes. We observe similar magnitudes of the first peak for Li+ with ether oxygen atoms of DME [EO(DME)] and O(ClO4−), while Li–O(FSI) peak is significantly smaller indicating strong affinity of Li+ to DME and ClO4− anions compared to FSI− (Fig. S24, ESI†). The TFP+–ClO4− RDF also shows a much higher first peak than that for TFP+–FSI− (Fig. 4b). Interestingly, both magnitude and widths of the first TFP+/ClO4− peak (Fig. 4b) are larger than the first peak in the Li+/ClO4− RDF (Fig. S24, ESI†), indicating significantly stronger predisposition of the TFP+ cation to form CIPs and aggregates than the corresponding lithium salts. We observe that while most of Li+ (75%) exist as free ions in agreement with the previous reports for 1 M LiFSI in DME,40 a significant fraction of TFP+ cations (∼80%) participate in CIPs and aggregates. Negatively charged aggregates such as TFP+(ClO4−)2 are expected to be found at the positive electrode surface, while free TFP+ and positive aggregates such as (TFP+)2(Anion−) are expected to be found at the negative electrode surface.
We observe the following concentration dependence of the TFP+ environments that is summarized in Table S2 (ESI†) for both [TFP+][ClO4−] concentrations. MD simulations also reveal an increase in “free” (not coordinated by either anion) TFP+ cations from 21% to 28% as the concentration of TFP+ decreases from 0.27 M to 0.14 M. This indicates a greater fraction of free TFP+ at lower concentrations. It is important to emphasize that our electrochemical experiments have TFP+ concentrations that are ∼10–25 times lower than those in the simulated electrolytes. Therefore, we expect an even higher fraction of free TFP+ in the 10–20 mM TFP solutions yielding a sufficient amount of free TFP+ to be electrostatically attracted to anode surface to participate in formation of LiF-rich SEI (in agreement with our EQCM-D results, Fig. 2c).
Conclusions
In summary, we demonstrate that high fractions of fluorinated species in electrolytes are not necessary to generate robust battery interphases. Instead, a very low fraction of readily reducible fluorinated cations can be used to form a favorable fluorine-rich SEI and allow for the deposition of dense Li. Moreover, we show that fluorinated cations, when coupled with perchlorate anions, can dramatically suppress the oxidative decomposition of ether-based electrolytes and corrosion of the aluminum current collectors, enabling stable cycling of full cells with nickel-rich high-voltage cathodes with 99.6% coulombic efficiency. In general, we present a strategy for delivering desired chemical species to the battery anodes through electrostatic attraction while using minute amounts of additive and therefore can notably reduce costs and environmental footprint of implementing high energy batteries.Materials and methods
Materials
2,4,6-Trifluoropyridine (98%, Matrix Scientific), methyl trifluoromethanesulfonate (97%, Matrix Scientific), ion exchange resin Amberlyst® A26 (hydroxide form, Acros Organics), perchloric acid (70%, VWR BDH Chemicals), acetonitrile (ACS reagent, ≥99.5%, EMD Millipore), dichloroethane (≥99.8%, Spectrum Chemical), methanol (≥99.8%, VWR BDH Chemicals) and anhydrous DME (99.5%, Sigma Aldrich) were used as received without further purification. LiFSI salt (98.0%, TCI Europe) was dried under vacuum at 110 °C for 24 hours before use. Unless otherwise stated, this LiFSI salt (98.0%, TCI Europe) was used to prepare electrolytes for all measurements. High-purity LiFSI (99.9%, Solvionic) was used only for measurements presented in Fig. S14 and S18 (ESI†).Preparation of ion exchange column in CIO4− form
The desired amount of Amberlyst® A26 ion exchange resin in OH− form (approximately 20 g of Amberlyst per 2 mmol of ionic starting material) was washed with deionized water twice before loading into a glass column (1.5 cm in diameter). The column was then flushed with methanol. Perchloric acid solution (2% in methanol) was passed through the column until the eluted solution possessed the same pH value as the original acid solution; the color of the column also changed from pink to light yellow during this process. Pure methanol was used to thoroughly remove acid residue from the column, which was then washed with acetonitrile to replace the methanol for further ion exchange. We note that all compounds passed through the ion exchange column in this work are sensitive to base and alcohol. Hence, the complete removal of methanol is imperative for successfully obtaining the desired compounds.Synthesis of N-methyl-2,4,6-trifluoropyridinium (TFP) perchlorate
N-Methyl-2,4,6-trifluoro-pyridinium triflate (0.5 g, 1.68 mmol) was synthesized first as reported previously with slight modification.41 Briefly, 2,4,6-trifluoropyridine (0.45 mL, 5 mmol) and methyl triflate (0.7 mL, 6.4 mmol) were mixed in a 25 mL degassed Schlenk tube and stirred at 50 °C for 4 h, leading to precipitation of a white solid. This solid was collected by filtration and washed with ether (3 × 15 mL). The solid was then recrystallized in an acetonitrile:dichloroethane 1:3 (v:v) mixture at 60 °C to obtain the final product (1.4 g, 4.7 mmol, 94%). Approximately 0.5 g of pure N-methyl-2,4,6-trifluoropyridinium triflate was then dissolved in acetonitrile (2 mL) and passed through the CIO4− ion exchange column with a constant flow of acetonitrile. The eluted solution (ca. 75 mL) was subject to rotary evaporation to afford the crude N-methyl-2,4,6-trifluoropyridinium perchlorate solid salt. The compound was then recrystallized by allowing an acetonitrile:dichloroethane 1:3 (v:v) solution to cool from 40 °C to 0 °C to obtain the final product (0.24 g, 0.97 mmol, 48%).
IR: νmax 3070 cm−1, 2161 cm−1, 2034 cm−1, 1675 cm−1, 1597 cm−1, 1496 cm−1, 1167 cm−1, 1077 cm−1, 877 cm−1, 622 cm−1.
1H NMR (500 MHz, acetonitrile-d3) δ 1.94 (p, J = 2.5 Hz, 1H), 4.02 (s, 1H), 7.64 (dd, J = 7.0, 2.4 Hz, 1H). (Fig. S25a, ESI.†)
13C NMR (126 MHz, acetonitrile-d3) δ 1.31, 35.56, 100.99, 118.26, 159.01, 161.09, 176.58, 178.69. (Fig. S25b, ESI.†)
19F NMR (471 MHz, acetonitrile-d3) δ −75.32 (t, J = 28.6 Hz), −69.33 (s). (Fig. S25c, ESI.†)
HRMS (m/z) N-methyl-2,4,6-trifluoropyridinium cation [M + H]+: Calcd: 148.0368, found: 148.0370; perchlorate anion [M − H]−: Calcd: 98.9410, found: 98.8446.
Density functional theory (DFT) calculations
DFT calculations of the additive reduction, oxidation and reactivity were performed using Gaussian 16 package, revision C.01.42 All complexes were immersed in implicit solvent represented using polarized continuum model (PCM) with ether parameters with the exception of the molecules shown in Fig. S1 (ESI†). Computationally expedient wB97XD DFT calculations with a compact 6-31+G(d,p) basis set yielded oxidation and reduction potentials for additives in good agreement with the more computationally expensive and reliable hybrid DFT/wave-function G4MP2 methodology (Fig. S1, ESI†) and were used throughout the paper. All optimized geometries were confirmed to be minima without imaginary frequencies. Intrinsic reaction coordinate calculations (IRC) are shown in Fig. 3d.Reduction potential for the complexes containing an additive, solvent, Li+ denoted as a complex A was calculated as the negative of the free energy of formation of the reduced species A− in solution [ΔGS298(A → A−) = GS298(A−) − GS298(A)] divided by Faraday's constant as given by:
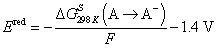
The difference between the Li/Li+ and absolute reduction potential of 1.4 V was subtracted to convert results to Li/Li+ scale as discussed extensively elsewhere.43 Oxidation potential for a complex A was calculated as the free energy of formation of the oxidized specie A+ in solution [ΔGS298(A → A+) = GS298(A+) − GS298(A)] divided by Faraday's constant as given by:
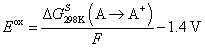
The H-transfer reaction from DME to LiNiO2 cathode surface was adapted from previous work35 and is shown in Fig. 3d. Details and discussion of the molecular dynamics simulations is expanded upon in the ESI.†
Electrochemical characterization
CV profiles were collected using 3-electrode electrochemical cell with a Cu disc working electrode (∅ = 1 mm, embedded in PEEK, eDAQ), Au disc working electrode (∅ = 2 mm, embedded in Kel-F, CH Instrument), Glassy carbon disc working electrode (∅ = 3 mm, embedded in Kel-F, CH Instrument), a Li chip (∅ = 4 mm, 250 μm, Xiamen Tmax Battery Equipments Limited) physically attached to stainless steel rod (McMaster Carr) was used as a counter electrode, leakless Ag/AgCl was used as a reference electrode (∅ = 5 mm, eDAQ). The 3-electrode CV data were collected using a VSP-300 Biologic potentiostat.Li‖Li symmetric cell tests were performed using CR2032 coin cell parts (Hohsen Corp), 250 μm Li chips (∅ = 11 mm, Xiamen Tmax Battery Equipments Limited), and 8 μm Cu current collectors (∅ = 13 mm, Xiamen Tmax Battery Equipment Limited). Two layers of Celgard 2500 were used as a separator (∅ = 19 mm). The amount of electrolyte used for every coin cell was ∼40–75 μl. The cells were aged at room temperature for at least 5 hours before operation. Galvanostatic cycling was performed for the symmetric cell tests with a current density of 10 mA cm−2 (6 min charge and 6 min discharge, specific areal capacity of 1 mA h cm−2).44 Biologic MPG-200 potentiostat and Arbin battery cycler (LBT21084UC) were used for the data collection.
The coulombic efficiency was obtained by averaging results of at least 5 independent measurements that were generated using a modified Aurbach protocol (described below). The modified Aurbach protocol was performed using asymmetric Cu‖Li coin cells, assembled with 75 μl of electrolyte. The detailed experimental sequence was the following: (1) 10 CV cycles were performed to pre-form SEI on a Cu electrode with a scan rate of 5 mV s−1 within the voltage range of 0 V to 2.5 V; (2) a deposition, stripping and re-deposition of excess amount of Li was performed (25 mA h cm−2 with a current density of 2.5 mA cm−2); (3) 100 galvanostatic Li plating and stripping cycles were performed at current density of 10 mA cm−2 and a specific areal capacity of 1 mA h cm−2; (4) final Li stripping was carried out with current density of 2.5 mA cm−2 with a cut off voltage of 1 V.
For full cell tests, a single-sided NCM811 electrodes (2 mA h cm−2 areal loading on 16 μm thick Al current collector, 99.6%, NEI Corp) were used as a cathode and Li metal chips (50 μm and 250 μm, Xiamen Tmax Battery Equipments Limited) were used as anodes. 50 μm thick Li metal anodes were prepared by thinning 250 μm thick Li foil using a roll press. 20 μm thick Li metal anodes were purchased from China Energy Lithium Co., Ltd. For full cell cycling tests a constant current (CC) step was initially used for all charging steps with a cut-off voltage of 4.2 V followed by a constant current constant voltage (CCCV) step with the terminating conditions of either 5% of the 1C-rate current or 30 minutes of the constant voltage step. A CC step was applied for all discharging steps with a cut-off voltage of 3 V. A sequence of three cycles at 0.1C-rate and 50 cycles at 1C-rate was repeated during full cell testing. EIS data were acquired using a VSP-300 (Biologic) in a frequency range from 200 kHz to 100 mHz, with a sinus amplitude of 10.0 mV.
EQCM-D measurements
The frequency and dissipation change for 3–11 overtones were collected by QCM-D instrument (Biolin Scientific AB for SEI studies and AWSensors for CEI studies). The measurements were conducted in a sealed homemade cell connected to a potentiostat (BioLogic VSP-300). For SEI studies, a 5 MHz Cu-coated quartz sensor (QC) was served as a working electrode and Li foil as both counter and reference electrodes. For CEI studies, a 5 MHz Au-coated QC was served as a working electrode and Li foil as both counter and reference electrodes. The gravimetric information was obtained by comparing the theoretical frequency Ftheo calculated from the faradaic mass using Sauerbrey equation to the experimental frequency as given by:27where Cm is the mass sensitivity constant of 5 MHz quartz sensor, Q is the obtained charge, Mw is the atomic mass of the inserted cation in its desolvated form, n is the number of electrons, and F is Faraday's constant.
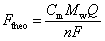
Preparation of the cycled Li anode cross-section using cryogenic focused-ion beam
Sample transfer to the cryo-sample holder was performed within 3 seconds of air exposure. After sample transfer, the sample was cooled to below −140 °C and subject to reduced pressure (ca. 10−6 mbar). Continuous sample cooling was performed using nitrogen gas during the FIB operation. First, a preliminary Ga+ beam cross-sectional cut was performed with a current of 5 nA followed by a cleaning cut with a 0.5 nA beam current.X-Ray photoelectron spectroscopy (XPS)
The cycled electrodes were rinsed 3 times with DME prior to analysis. XPS analysis was performed on a PHI Quantum 2000 using a monochromated Al Kα X-ray source (1486.6 eV) with a pass energy of 30 eV. Sample charging was minimized by charge compensation with a low energy electron and an Ar ion gun. XPS data were processed with the CasaXPS software. The corrected relative sensitivity factors (RSFs) were derived from in-house Matlab code. The corrected RSF values for Li 1s, C 1s, N 1s, O 1s, F 1s and S 2p are 0.625, 7.388, 12.075, 18.301, 25.924 and 18.715, respectively. Shirley background subtraction was applied before peak fitting. Gaussian/Lorentzian product line functions with 30% Lorentzian and 70% Gaussian contribution were used for fitting each XPS spectrum. The spectra were calibrated by assigning the Li–F component of the F 1s spectra to a 685.0 eV binding energy. Samples for XPS were prepared in three-electrode cell with fixed working electrode area: Cu (≥99.8%, NEI Corporation) or Al metal foils (99.6%, NEI Corporation) were used as a working electrode (geometrical area of 0.126 cm−2) and 250 μm Li metal (∅ = 11 mm, Xiamen Tmax Battery Equipments Limited) as a counter and Ag/AgCl leakless electrode (eDAQ) as a reference electrode. The cycled NCM811 and Li metal samples were retrieved from 2032-type coin cell samples. The cells were disassembled, and the retrieved electrodes were washed three times with fresh DME prior to analysis. A custom-made transfer vessel was used for air-free transfer of the samples to the XPS machine.FTIR measurements
Ex situ ATR-FTIR spectra were obtained by averaging 128 scans using a Vertex-70v spectrometer equipped with a high-resolution MCT detector and a Specac Golden Gate Diamond ATR accessory.Raman measurements
The ex situ Raman spectra were obtained with 0.5% laser power (785 nm laser) and 20× magnification using a Renishaw InVia Raman microscope.NMR spectroscopy
1H-, 13C- and 19F-solution nuclear magnetic resonance (NMR) spectra were recorded at room temperature on a Bruker ASCEND 400 spectrometer. Acetonitrile-d3 was used as solvent for all measurements. Spectra were analyzed using MestReNova. For indirect referencing, the magnet was locked and shimmed using a sample containing 0.4 mL DME and 0.1 mL acetonitrile-d3 (referenced to residual solvent signals of acetonitrile-d3), and subsequent samples were measured without locking/shimming.Author contributions
M. R. L. planned the study and supervised the research. C. N. H. performed all electrochemical studies and conducted detailed XPS analysis. J. Y. contributed to interpretation of the data and research supervision. D. G. V. performed SEM and NMR measurements. M. Y. synthesized and characterized the fluorinated cationic additive under supervision of J. I. F. M. R. performed synthesis of additives following procedure from M. Y. and contributed to electrochemical testing to ensure reproducibility and NMR studies. T. P. P. and O. B. performed DFT calculations and MD simulations. N. S. and L. W. performed EQCM-D measurements and relevant data analysis. K. T. performed Raman und FTIR measurements and assisted in CEI characterization. All authors discussed the results and contributed to the writing of the manuscript.Conflicts of interest
The authors have a patent (US patent provisional application number 63398320) related to the electrolytes described in this article.Acknowledgements
M. R. L. and C. N. H. acknowledge support from Swiss National Science Foundation (starting Grant TMSGI_218234). J. I. F. acknowledges the support of startup funds from the University at Albany, State University of New York. L. W. acknowledges financial support from the program of China Scholarships Council (202106830054). O. B. and T. P. P. acknowledge support from the US Army and DEVCOM Army Research Laboratory as well as support under the Department of Energy interagency agreement 89243322SEE000018. We thank Jan Bosse, Dr Zbynek Novotny and Dr Andrei Akbasheu (Paul Scherrer Institute) and Dr Andrea Arcifa (Swiss Federal Laboratories for Materials Science and Technology (EMPA)) for helpful discussions on XPS experiments and analysis. We also thank Dr Rabeb Grissa, Dr Corsin Battaglia (EMPA) for providing XPS transfer vessel for air-free sample transfer.References
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- D. Lin, J. Zhao, J. Sun, H. Yao, Y. Liu, K. Yan and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4613–4618 CrossRef CAS PubMed.
- A. J. Louli, A. Eldesoky, R. Weber, M. Genovese, M. Coon, J. deGooyer, Z. Deng, R. T. White, J. Lee, T. Rodgers, R. Petibon, S. Hy, S. J. H. Cheng and J. R. Dahn, Nat. Energy, 2020, 5, 693–702 CrossRef CAS.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- K.-H. Chen, K. N. Wood, E. Kazyak, W. S. LePage, A. L. Davis, A. J. Sanchez and N. P. Dasgupta, J. Mater. Chem. A, 2017, 5, 11671–11681 RSC.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- G. M. Hobold, J. Lopez, R. Guo, N. Minafra, A. Banerjee, Y. Shirley Meng, Y. Shao-Horn and B. M. Gallant, Nat. Energy, 2021, 6, 951–960 CrossRef CAS.
- Z. Yu, H. Wang, X. Kong, W. Huang, Y. Tsao, D. G. Mackanic, K. Wang, X. Wang, W. Huang, S. Choudhury, Y. Zheng, C. V. Amanchukwu, S. T. Hung, Y. Ma, E. G. Lomeli, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2020, 5, 526–533 CrossRef CAS.
- T. Fan, Y. Wang, V. K. Harika, A. Nimkar, K. Wang, X. Liu, M. Wang, L. Xu, Y. Elias, H. Sclar, M. S. Chae, Y. Min, Y. Lu, N. Shpigel and D. Aurbach, Adv. Sci., 2022, 9, 2202627 CrossRef CAS PubMed.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- X. Fan, L. Chen, X. Ji, T. Deng, S. Hou, J. Chen, J. Zheng, F. Wang, J. Jiang, K. Xu and C. Wang, Chem, 2018, 4, 174–185 CAS.
- Y. Huang, R. Li, S. Weng, H. Zhang, C. Zhu, D. Lu, C. Sun, X. Huang, T. Deng, L. Fan, L. Chen, X. Wang and X. Fan, Energy Environ. Sci., 2022, 15, 4349–4361 RSC.
- Y. Liu, X. Tao, Y. Wang, C. Jiang, C. Ma, O. Sheng, G. Lu and X. W. Lou, Science, 2022, 375, 739–745 CrossRef CAS PubMed.
- Z. Yu, P. E. Rudnicki, Z. Zhang, Z. Huang, H. Celik, S. T. Oyakhire, Y. Chen, X. Kong, S. C. Kim, X. Xiao, H. Wang, Y. Zheng, G. A. Kamat, M. S. Kim, S. F. Bent, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2022, 7, 94–106 CrossRef CAS.
- Q. Liu, C. W. Hsu, T. L. Dzwiniel, K. Z. Pupek and Z. Zhang, Chem. Commun., 2020, 56, 7317–7320 RSC.
- Q. Liu, W. Jiang, J. Xu, Y. Xu, Z. Yang, D. J. Yoo, K. Z. Pupek, C. Wang, C. Liu, K. Xu and Z. Zhang, Nat. Commun., 2023, 14, 3678 CrossRef CAS PubMed.
- E. Lebegue, J. Agullo and D. Belanger, ChemSusChem, 2018, 11, 219–228 CrossRef CAS PubMed.
- A. J. Lucio and S. K. Shaw, J. Phys. Chem. C, 2015, 119, 12523–12530 CrossRef CAS.
- P. Jankowski, W. Wieczorek and P. Johansson, Energy Storage Mater., 2019, 20, 108–117 CrossRef.
- D. Aurbach, M. Daroux, P. Faguy and E. Yeager, J. Electroanal. Chem. Interfacial Electrochem., 1991, 297, 225–244 CrossRef CAS.
- B. D. Adams, J. Zheng, X. Ren, W. Xu and J. G. Zhang, Adv. Energy Mater., 2017, 8, 1702097 CrossRef.
- Z. Yu, P. E. Rudnicki, Z. Zhang, Z. Huang, H. Celik, S. T. Oyakhire, Y. Chen, X. Kong, S. C. Kim, X. Xiao, H. Wang, Y. Zheng, G. A. Kamat, M. S. Kim, S. F. Bent, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2022, 7, 94–106 CrossRef CAS.
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef CAS PubMed.
- J. Zheng, X. Fan, G. Ji, H. Wang, S. Hou, K. C. DeMella, S. R. Raghavan, J. Wang, K. Xu and C. Wang, Nano Energy, 2018, 50, 431–440 CrossRef CAS.
- S. Angarita-Gomez and P. B. Balbuena, Phys. Chem. Chem. Phys., 2020, 22, 21369–21382 RSC.
- J. Z. Lee, T. A. Wynn, M. A. Schroeder, J. Alvarado, X. Wang, K. Xu and Y. S. Meng, ACS Energy Lett., 2019, 4, 489–493 CrossRef CAS.
- N. Shpigel, M. D. Levi and D. Aurbach, Energy Storage Mater., 2019, 21, 399–413 CrossRef.
- V. Dargel, N. Shpigel, S. Sigalov, P. Nayak, M. D. Levi, L. Daikhin and D. Aurbach, Nat. Commun., 2017, 8, 1389 CrossRef PubMed.
- P. G. Kitz, M. J. Lacey, P. Novak and E. J. Berg, Anal. Chem., 2019, 91, 2296–2303 CrossRef CAS PubMed.
- Y. Zhou, M. Su, X. Yu, Y. Zhang, J. G. Wang, X. Ren, R. Cao, W. Xu, D. R. Baer, Y. Du, O. Borodin, Y. Wang, X. L. Wang, K. Xu, Z. Xu, C. Wang and Z. Zhu, Nat. Nanotechnol., 2020, 15, 224–230 CrossRef CAS PubMed.
- C. Wan, M. Y. Hu, O. Borodin, J. Qian, Z. Qin, J.-G. Zhang and J. Z. Hu, J. Power Sources, 2016, 307, 231–243 CrossRef CAS.
- H. Kim, F. Wu, J. T. Lee, N. Nitta, H.-T. Lin, M. Oschatz, W. I. Cho, S. Kaskel, O. Borodin and G. Yushin, Adv. Energy Mater., 2015, 5 Search PubMed.
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S. C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715–722 CrossRef CAS PubMed.
- C. V. Amanchukwu, Z. Yu, X. Kong, J. Qin, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2020, 142, 7393–7403 CrossRef CAS PubMed.
- Z. Zhang, J. Yang, W. Huang, H. Wang, W. Zhou, Y. Li, Y. Li, J. Xu, W. Huang, W. Chiu and Y. Cui, Matter, 2021, 4, 302–312 CrossRef CAS.
- C. Busà, M. Belekoukia and M. J. Loveridge, Electrochim. Acta, 2021, 366, 137358 CrossRef.
- X. Ren, L. Zou, S. Jiao, D. Mei, M. H. Engelhard, Q. Li, H. Lee, C. Niu, B. D. Adams, C. Wang, J. Liu, J.-G. Zhang and W. Xu, ACS Energy Lett., 2019, 4, 896–902 CrossRef CAS.
- M. Carboni, A. G. Marrani, R. Spezia and S. Brutti, Chemistry, 2016, 22, 17188–17203 CrossRef CAS PubMed.
- J. Alvarado, M. A. Schroeder, T. P. Pollard, X. Wang, J. Z. Lee, M. Zhang, T. Wynn, M. Ding, O. Borodin, Y. S. Meng and K. Xu, Energy Environ. Sci., 2019, 12, 780–794 RSC.
- P. Zhang, H. Jin, T. Wang and M. Wang, Electrochim. Acta, 2018, 277, 116–126 CrossRef CAS.
- A. A. Leitch, X. Yu, C. M. Robertson, R. A. Secco, J. S. Tse and R. T. Oakley, Inorg. Chem., 2009, 48, 9874–9882 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. R. Jow, K. Xu, O. Borodin and M. Ue, Electrolytes for Lithium and Lithium-Ion Batteries, Springer, New York, 2014, vol. 58, pp. 371–401 Search PubMed.
- J. Xiao, Q. Li, Y. Bi, M. Cai, B. Dunn, T. Glossmann, J. Liu, T. Osaka, R. Sugiura, B. Wu, J. Yang, J.-G. Zhang and M. S. Whittingham, Nat. Energy, 2020, 5, 561–568 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee00296b |
| This journal is © The Royal Society of Chemistry 2024 |