
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical desulfurative formation of C–N bonds through selective activation of inert C(sp3)–S bonds†
Shaopeng
Guo‡
a,
Yujun
Li‡
a,
Qing-Han
Li
*b and
Ke
Zheng
 *a
*a
aKey Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, 610064, P. R. China. E-mail: kzheng@scu.edu.cn
bKey Laboratory of General Chemistry of the National Ethnic Affairs Commission, College of Chemistry and Environment, Southwest Minzu University, Chengdu, PR China. E-mail: lqhchem@swun.edu.cn
First published on 1st February 2024
Abstract
In this study, we introduce an efficient, metal-free electrocatalytic desulfurative protocol for forming C–N bonds by selectively activating inert C(sp3)–S bonds of alkyl thioethers. This method offers a straightforward and environmentally friendly approach for modification of heterocyclic compounds from readily accessible thioethers. Preliminary mechanistic investigations suggest that the reaction proceeds via a carbocation intermediate. Furthermore, successful synthesis on a 10-gram scale was achieved in a continuous flow electrochemical reactor.
Organosulfur compounds are not only commonly found in natural products, pharmaceuticals, and functional materials but also serve as vital intermediates and precursors in the field of organic chemistry.1 The activation of inert C–S bonds to form C–C and C–heteroatom bonds has garnered significant attention in organic chemistry. Over the past few decades, transition metal-catalyzed coupling reactions have established themselves as a robust platform for the transformation of C–S bonds.2 This approach typically involves metal insertion into the carbon–sulfur bond to facilitate its cleavage, followed by the desired transformation.2 However, despite the effectiveness of transition metals, certain limitations persist. For instance, sulfur atoms can strongly coordinate with metals, leading to catalyst deactivation, and sometimes additional sacrificial reagents are required to achieve catalyst turnover.3 Additionally, higher reaction temperatures are often necessary to facilitate oxidative addition to transition metals.4 Furthermore, the sulfur-containing compounds frequently employed in these reactions are typically C(sp2)–S bonds or reactive high-valence sulfur species such as sulfones/sulfonium salts.2a,2b,2d In contrast, the study of widely available C(sp3)–S bonds has been notably scarce and remains relatively underexplored, particularly in the context of metal-free catalysis.5,6
Over the past decade, organic electrochemistry has garnered significant attention due to its precise electron-transfer processes and its unique utilization of electrons as redox reagents that leave no trace.7 Electrochemistry offers a potent tool for activating compounds containing sulfide groups.8 Consequently, the selective oxidation of thioethers proceeds seamlessly in electrocatalysis, as it does not rely on sulfur atom coordination to the catalyst.9 Early in 2004, Yoshida's research group introduced the “cation pool” method for generating carbocations (Scheme 1a). In this method, the electrochemical oxidation of glycosylated phenylthioethers at temperatures as low as −78 °C yields a reservoir of discrete alkoxy carbocation intermediates, which can subsequently react with nucleophilic reagents.9e,9h Lei and colleagues recently reported electrooxidatively induced C–S bond cleavage for the synthesis of disulfides from thioethers. This process initiates with the formation of a radical cation intermediate, which subsequently decomposes into a carbocation and a thiyl radical, enabling S–S bond formation (Scheme 1b, left).10 Malins, Connal, and co-workers developed an electroauxiliary-assisted oxidation of glutamine residues for the modification of peptide substrate via C(sp3)–S cleavage of thioethers.9a Most recently, Lundberg's group has also contributed to the field by reporting electroreductive desulfurative transformations utilizing thioethers as alkyl radical precursors.11 These transformations include hydrodesulfurization, carboxylation, and Giese-type cross-coupling. However, it is worth noting that the achievement of high conversion of the starting feedstock and high yields in these processes requires the use of various sacrificial anode materials (Scheme 1b, right).
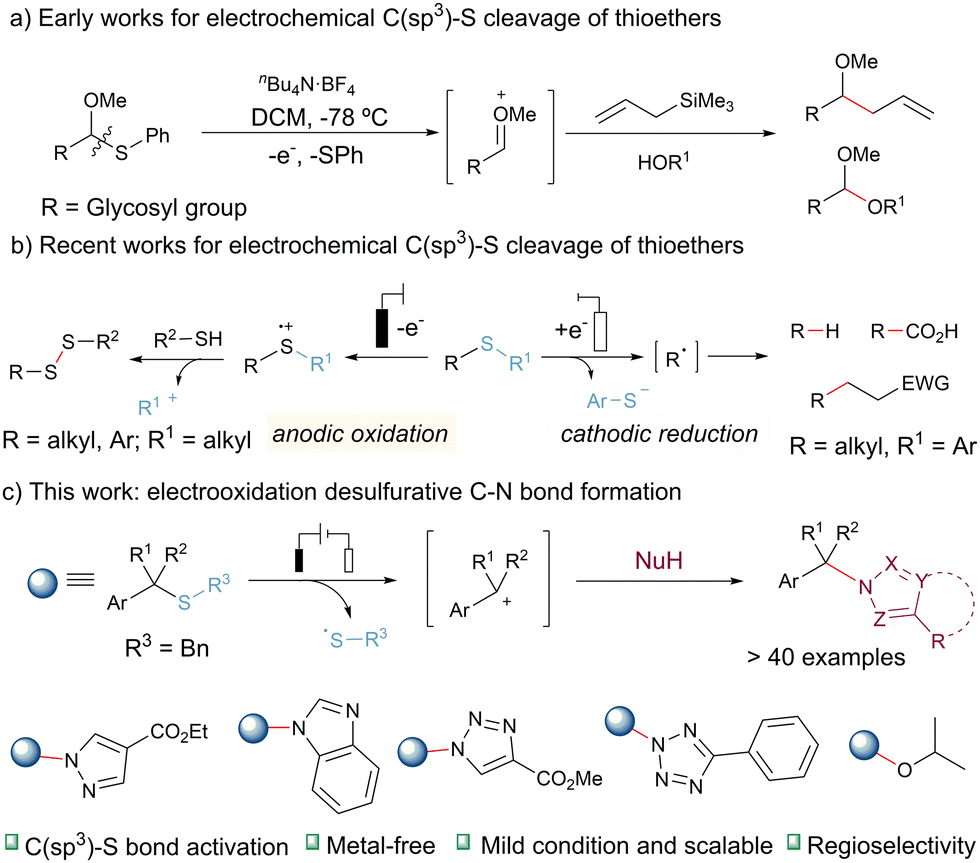 | ||
| Scheme 1 C–S bond activation strategy. | ||
In this study, we present an innovative example of electrocatalytic C(sp3)–S bond activation for the construction of C–N bonds (Scheme 1c). The process leverages the carbocation resulting from the decomposition of the radical cation intermediate as an electrophilic reagent. This electrochemical method demonstrates a broad substrate scope, encompassing over 40 examples of thioethers, heterocycles (pyrazole, triazole, and tetrazole), and alcohols. Moreover, the reaction can be readily scaled up to a 10-gram level using a continuous-flow system operating under mild conditions. This electrooxidation strategy do not need for sacrificial anodes, costly transition metal catalysts, or external chemical oxidants.
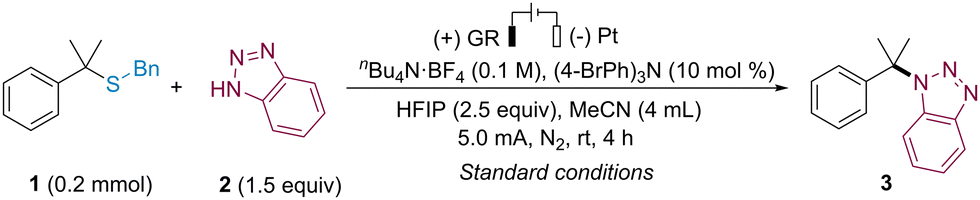
We conducted an investigation into the electrochemical desulfurative N-alkylation using alkyl thioether 1 and benzotriazole 2 as model substrates in an undivided cell (Table 1). After a series of experiments, we successfully obtained the desired N-alkylation product 3 in 80% yield under a constant current of 5 mA and in a N2 (Table 1, entry 1, more details see ESI†). When alternative organic redox mediators such as 9,10-Dicyanoanthracen (DCA) or TEMPO were used, lower yields were observed (Table 1, entries 2 and 3). In the absence of an electron transfer mediator, the desired product was obtained in 60% yield (Table 1, entry 4). The choice of solvent was critical, with CH3CN providing the best results, while no product was obtained in DMF or acetone (Table 1, entries 5–7). Lower yields were achieved without the addition of HFIP or by replacing HFIP with tBuOH (Table 1, entries 8 and 9). Explorations into other solvents, additives, and electrode materials did not yield improved results (more details see ESI†). Prolonged electrolysis to 6 hours led to a significant decrease in yield (Table 1, entry 10). The current density played a crucial role in the reaction, with an increase or decrease in electric current resulting in lower yields (see ESI†). The reaction could occur in an air atmosphere, albeit with a decreased yield of 55% (Table 1, entry 11). Control experiments confirmed the essential nature of electricity for this reaction, as no reaction occurred in the absence of an electric current (Table 1, entry 12).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yieldsb (%) |
| a Standard conditions: carbon rod (ϕ 6 mm) anode, Pt plate (15 mm × 10 mm × 0.1 mm) cathode, constant current (5 mA), thioether 1 (0.2 mmol), benzotriazole 2 (0.3 mmol), nBu4NBF4 (0.1 M), (4-BrPh)3N (10 mol%), HFIP (2.5 equiv.), CH3CN (4.0 mL), room temperature, N2, undivided cell, 4 h. b Isolated yield. | ||
| 1 | None | 80 |
| 2 | DCA instead of (4-BrPh)3N | 70 |
| 3 | TEMPO instead of (4-BrPh)3N | N.R. |
| 4 | Without (4-BrPh)3N | 60 |
| 5 | DCM instead of MeCN | 30 |
| 6 | DMF instead of MeCN | N.R. |
| 7 | Acetone instead of MeCN | N.R. |
| 8 | t BuOH instead of HFIP | 60 |
| 9 | Without HFIP | 58 |
| 10 | 6 h | 34 |
| 11 | Air | 55 |
| 12 | Without current | N.R. |
With optimized reaction conditions in hand, we proceeded to explore the scope of this electrochemical method. As illustrated in Scheme 2, various tertiary thioethers proved to be compatible with the reaction, providing the N-alkylation products 3–18 in good yields. Substrates featuring phenyl rings with electron-donating groups generally exhibited better results, yielding 78–85% compared to those with electron-withdrawing groups (4–5, 10vs. 6–8). Furthermore, the thioethers with condensed ring or multiple aryl groups were also well-tolerated, resulting in the N-alkylation products 11–15 in 60–78% yields. Thioethers bearing large steric hindrance in their alkyl substituents were efficiently converted to the corresponding N-alkylation products 16–18 in 64–91% yield. Several secondary alkyl thioethers were also smoothly converted to the corresponding products 19–21 in 52–80% yields. Diphenyl substituted thioether delivered N-alkylation product 19 in 80% good yield. Thiophene-substituted thioether was well-tolerated in this method, yielding the N-alkylation product 21 in 73% yield. Moreover, the success of gram scale synthesis through a flow electrochemical reactor (5, 1.0 g, 75% yield) demonstrates promising outcomes for potential industrial applications. Encouraged by the results presented above, we proceeded to investigate the applicability of various heterocycles in our electrochemical protocol. As depicted in Scheme 2, benzotriazoles featuring Me, F, Cl, Br, CF3, and CN substituents on the benzene ring exhibited the capacity to trap the carbocation, affording the corresponding N-alkylation products in moderate to high yields (22–28, 31–71%). Notably, diversely substituted benzotriazoles yielded N1/N3 mixtures, with the exception of the 4-Me-substituted benzotriazole, which exclusively delivered the N1-selective product. Furthermore, various azoles, such as pyrazole, triazole, and tetrazole, were found to be suitable substrates for this electrochemical protocol, providing the products with 20–84% yields (29–36). In addition to these heterocyclic substrates, a range of common alcohols, including methanol, isopropanol, tert-butanol, and ethylene glycol, were found to be compatible with the reaction, acting as nucleophilic reagents in the reaction to capture the carbocation. This resulted in the formation of the corresponding ether products with good outcomes (37–40, 65–79%). The use of deuterium oxide (D2O) led to the formation of the corresponding deuterium-substituted products 41 and 42 in yields of 78% and 77%, respectively. Additionally, this electrochemical desulfurative N-alkylation reaction proved effective in the late-stage functionalization of complex molecules, including natural products such as Celestolide, Podophyllotoxin, and GeMfibrozil-derived thioethers, with 65–92% yields for products 43–45. Remarkably, Valsartan with a carboxyl acid group was also well-tolerated in this electrocatalytic system (46, 67%). To further underscore the utility of our electrocatalytic protocol, we conducted a scaled-up reaction in a continuous flow electrochemical reactor. Encouragingly, under slightly modified conditions (more details see ESI†), the reaction proceeded smoothly at a 10.0 g scale (60 mmol), affording the product 10 in 65% isolated yield (11.15 g, Scheme 2, bottom right).
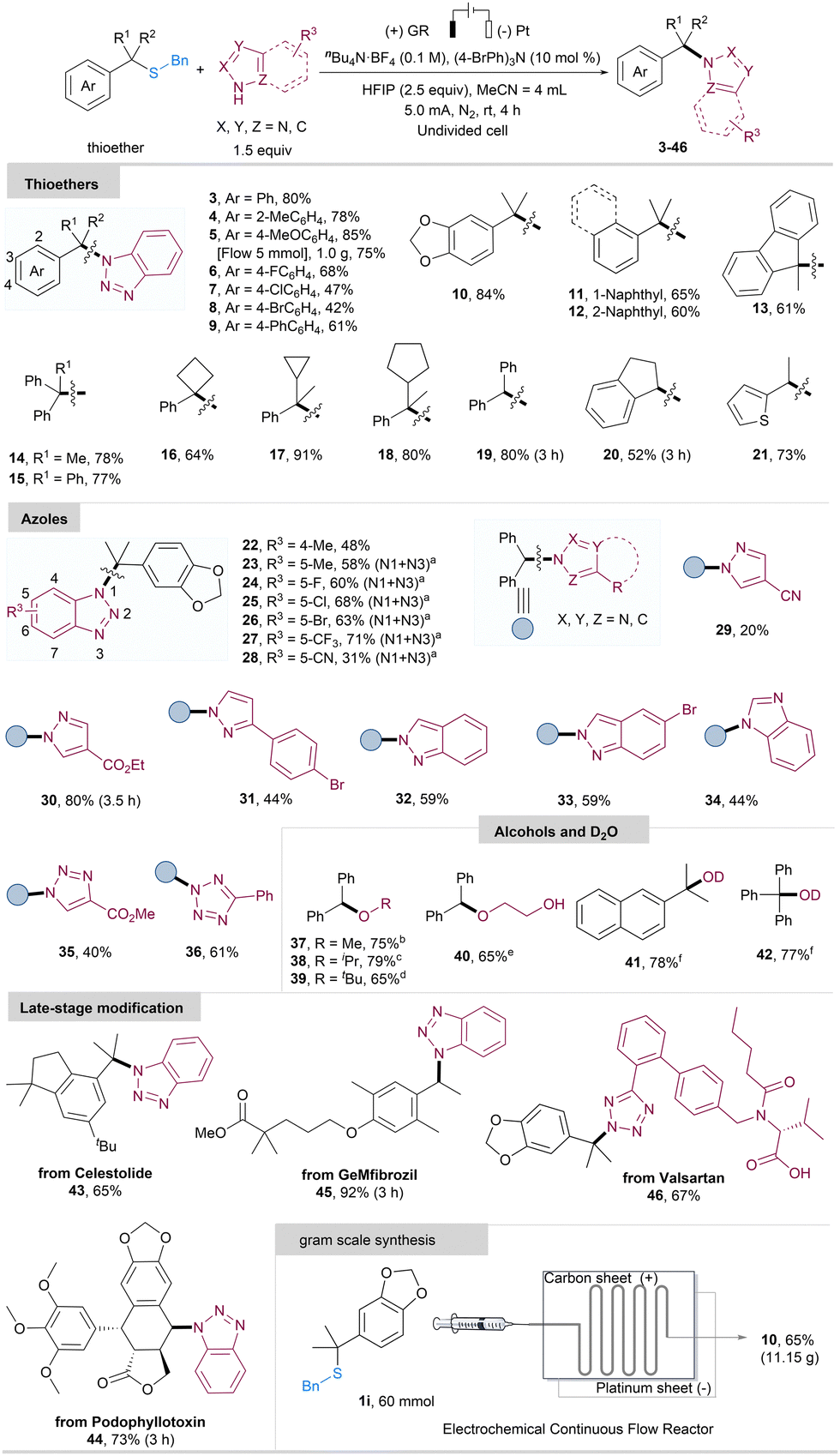 | ||
Scheme 2 Scope of desulfurization N-alkylation. Unless otherwise mentioned, reactions were performed with standard conditions. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) determined by 1H NMR. bMeCN/MeOH = 3/1 (v/v) mL. cMeCN/iPrOH = 3/1 (v/v) mL. dMeCN/tBuOH = 3/1 (v/v) mL. eMeCN/Ethylene glycol = 3/1 (v/v) mL. fMeCN/D2O = 4/0.18 (v/v) mL. Isolated yield. determined by 1H NMR. bMeCN/MeOH = 3/1 (v/v) mL. cMeCN/iPrOH = 3/1 (v/v) mL. dMeCN/tBuOH = 3/1 (v/v) mL. eMeCN/Ethylene glycol = 3/1 (v/v) mL. fMeCN/D2O = 4/0.18 (v/v) mL. Isolated yield. | ||
To shine light on the mechanism of this electrochemical desulfurative N-alkylation reaction, a series of several control experiments were conducted (more details see ESI†). As depicted in Fig. 1a, the reaction of thioether 1r with diphenylphosphine oxide under standard conditions yielded the radical coupling product 48 in 40% yield and the sulfur radical dimerization product 49 in 25% yield, respectively (Fig. 1a, top). This outcome strongly suggested the generation of sulfur radicals within the system, which subsequently engaged in cross-coupling with phosphine radicals.12 Furthermore, when the corresponding thioether 1p was used as the substrate, the N-alkylation product 17 was obtained in 91% yield, and no discernible radical ring-opening product was observed (Fig. 1a, middle). These results collectively indicated a sulfur radical cation was produced at the anode, leading to the decomposition into a carbocation and a thiyl radical. The capture of the carbocation by the nucleophilic reagent benzotriazole 2, resulting in the formation of addition product 17. Moreover, to gain insights into the role of HFIP in the reaction, the corresponding ether 1x was subjected to electrolysis with benzotriazole 2 under standard conditions, with no detection of N-alkylation product 19 (Fig. 1a, bottom). This observation suggested that the ether 1x does not serve as an intermediate in the transformation.
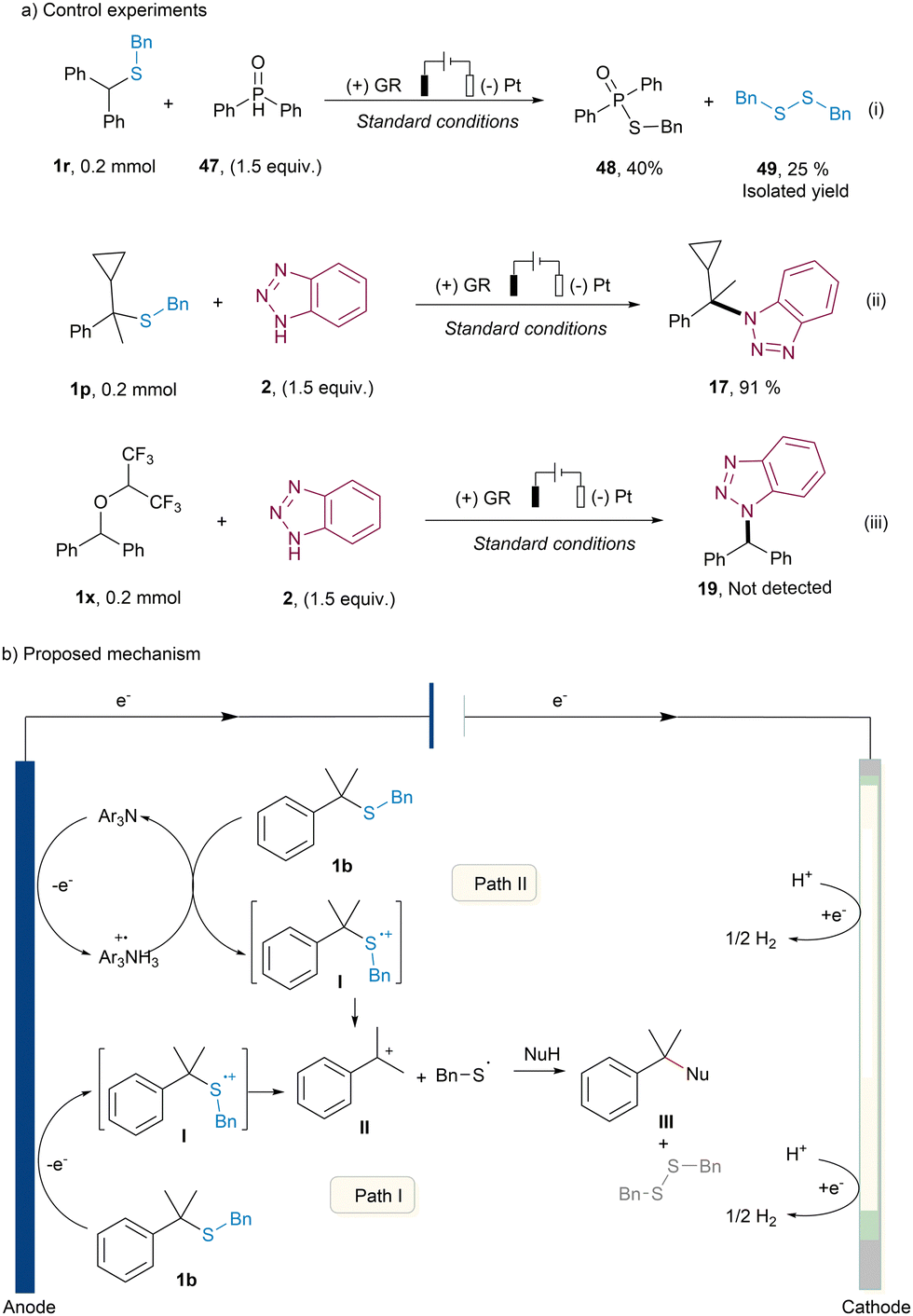 | ||
| Fig. 1 (a) Control experiments, (b) proposed mechanism. | ||
Based on the above mechanistic results and the previous report,10 two plausible mechanisms are proposed in Fig. 1b. In the primary pathway (Path I), thioether 1b undergoes direct oxidation at the anode, yielding a sulfur radical cation intermediate I. This intermediate subsequently undergoes cleavage to form a carbocation II, along with a sulfur radical. The resulting carbocation II is then engaged by nucleophilic reagents, yielding the desired product III. On the cathode side, HFIP functions as a hydrogen donor, facilitating the reduction of hydrogen ions to form H2, thus maintaining charge balance within the overall cell. Alternatively, an indirect pathway (Path II) can also give rise to the sulfur radical cation intermediate I. In this case, the catalyst triarylamine undergoes one-electron oxidation at the anode, generating a triarylamine radical cation intermediate with inherent oxidizing properties. This radical cation subsequently oxidizes the corresponding thioether 1b, leading to the formation of the pivotal intermediate I.
In summary, we introduce an innovative electrochemical protocol for activating and transforming C(sp3)–S bonds with excellent efficiency, enabling the construction of C–N bonds from thioethers under transition-metal free conditions. Furthermore, we have developed a flow electrochemical method that allows synthesis on a 10-gram scale, demonstrating its potential for practical and large-scale applications. Preliminary mechanistic experiments suggest that the reaction proceeds through a carbocation intermediate pathway. This electrocatalytic protocol offers an efficient method for activating thioethers in desulfurative transformations under mild conditions, expanding the methodological toolkit in this field.
We thank the Xiaoming Feng laboratory (SCU) for access to equipment. We acknowledge support from the National Natural Science Foundation of China (No. 22371195), Sichuan University (No. 2020SCUNL204 and No. 2023SCUH0081), and Fundamental Research Funds for the Central Universities.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) N. Wang, P. Saidhareddy and X. Jiang, Nat. Prod. Rep., 2020, 37, 246–275 RSC; (b) M. Wang and X. Jiang, ACS Sustainable Chem. Eng., 2022, 10, 671–677 CrossRef CAS.
- (a) M. Nambo, Y. Maekawa and C. M. Crudden, ACS Catal., 2022, 12, 3013–3032 CrossRef CAS; (b) M. Nambo and C. M. Crudden, Chem. Rec., 2021, 21, 3978–3989 CrossRef CAS PubMed; (c) J. Lou, Q. Wang, P. Wu, H. Wang, Y.-G. Zhou and Z. Yu, Chem. Soc. Rev., 2020, 49, 4307–4359 RSC; (d) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed; (e) L. Wang, W. He and Z. Yu, Chem. Soc. Rev., 2013, 42, 599–621 RSC.
- (a) N.-N. Ma, X.-B. Hu, Y.-S. Wu, Y.-W. Zheng, M. Ma, X.-Q. Chu, H. Xu, H. Luo and Z.-L. Shen, Org. Lett., 2023, 25, 1771–1775 CrossRef CAS PubMed; (b) Z. Li, Q. Shi, X. Ma, Y. Li, K. Wen, L. Qin, H. Chen, W. Huang, F. Zhang, Y. Lin, T. J. Marks and H. Huang, Nat. Commun., 2022, 13, 144 CrossRef CAS PubMed; (c) L. S. Liebeskind and J. Srogl, J. Am. Chem. Soc., 2000, 122, 11260–11261 CrossRef CAS.
- (a) M. Zhang, B. B. Zhang, Q. Lin, Z. Jiang, J. Zhang, Y. Li, S. Pei, X. Han, H. Xiong, X. Liang, Y. Lin, Z. Wei, F. Zhang, X. Zhang, Z. X. Wang, Q. Shi and H. Huang, Angew. Chem., Int. Ed., 2023, 62, e20230637 Search PubMed; (b) Z. Lian, B. N. Bhawal, P. Yu and B. Morandi, Science, 2017, 356, 1059–1063 CrossRef CAS PubMed.
- (a) K. Yang, Q. Li, Z. Li and X. Sun, Chem. Commun., 2023, 59, 5343–5364 RSC; (b) G. Li, Y. Nieves-Quinones, H. Zhang, Q. Liang, S. Su, Q. Liu, M. C. Kozlowski and T. Jia, Nat. Commun., 2020, 11, 2890 CrossRef CAS PubMed; (c) F. Dénès, C. H. Schiesser and P. Renaud, Chem. Soc. Rev., 2013, 42, 7900–7942 RSC.
- (a) R. Fan, S. Liu, Q. Yan, Y. Wei, J. Wang, Y. Lan and J. Tan, Chem. Sci., 2023, 14, 4278–4287 RSC; (b) Y. Wang, L. F. Deng, X. Zhang, Z. D. Mou and D. Niu, Angew. Chem., Int. Ed., 2021, 60, 2155–2159 CrossRef CAS PubMed; (c) M. O. Zubkov, M. D. Kosobokov, V. V. Levin, V. A. Kokorekin, A. A. Korlyukov, J. Hu and A. D. Dilman, Chem. Sci., 2020, 11, 737–741 RSC; (d) M. Lanzi, J. Merad, D. V. Boyarskaya, G. Maestri, C. Allain and G. Masson, Org. Lett., 2018, 20, 5247–5250 CrossRef CAS PubMed.
- (a) T. S. Chen, H. Long, Y. Gao and H. C. Xu, Angew. Chem., Int. Ed., 2023, 62, e202310138 CrossRef CAS PubMed; (b) Z. W. Hou, D. J. Liu, P. Xiong, X. L. Lai, J. Song and H. C. Xu, Angew. Chem., Int. Ed., 2021, 60, 2943–2947 CrossRef CAS PubMed; (c) T. Sheng, H.-J. Zhang, M. Shang, C. He, J. C. Vantourout and P. S. Baran, Org. Lett., 2020, 22, 7594–7598 CrossRef CAS PubMed; (d) J. Xiang, M. Shang, Y. Kawamata, H. Lundberg, S. H. Reisberg, M. Chen, P. Mykhailiuk, G. Beutner, M. R. Collins, A. Davies, M. Del Bel, G. M. Gallego, J. E. Spangler, J. Starr, S. Yang, D. G. Blackmond and P. S. Baran, Nature, 2019, 573, 398–402 CrossRef CAS PubMed; (e) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed; (f) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (g) J. Wu, Y. Zhou, Y. Zhou, C.-W. Chiang and A. Lei, ACS Catal., 2017, 7, 8320–8323 CrossRef CAS.
- (a) X. Zhou, C. Ni, L. Deng and J. Hu, Chem. Commun., 2021, 57, 8750–8753 RSC; (b) D. J. Wang, K. Targos and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 21503–21510 CrossRef CAS PubMed; (c) T. Sawamura, K. Takahashi, S. Inagi and T. Fuchigami, Angew. Chem., Int. Ed., 2012, 124, 4489–4492 CrossRef.
- (a) D. Karipal Padinjare Veedu, L. A. Connal and L. R. Malins, Angew. Chem., Int. Ed., 2022, 62, e202215470 CrossRef PubMed; (b) B. Deore, J. E. Ocando, L. D. Pham and C. A. Sanhueza, J. Org. Chem., 2022, 87, 5952–5960 CrossRef CAS PubMed; (c) S. Manmode, K. Matsumoto, T. Nokami and T. Itoh, Asian J. Org. Chem., 2018, 7, 1719–1729 CrossRef CAS; (d) N. Tanaka, F. Ohnishi, D. Uchihata, S. Torii and J. Nokami, Tetrahedron Lett., 2007, 48, 7383–7387 CrossRef CAS; (e) T. Nokami, A. Shibuya, H. Tsuyama, S. Suga, A. A. Bowers, D. Crich and J.-I. Yoshida, J. Am. Chem. Soc., 2007, 129, 10922–10928 CrossRef CAS PubMed; (f) V. Dinoiu, Rev. Roum. Chim., 2007, 52, 453–466 CAS; (g) K. Mitsudo, T. Kawaguchi, S. Miyahara, W. Matsuda, M. Kuroboshi and H. Tanaka, Org. Lett., 2005, 7, 4649–4652 CrossRef CAS PubMed; (h) S. Suzuki, K. Matsumoto, K. Kawamura, S. Suga and J.-I. Yoshida, Org. Lett., 2004, 6, 3755–3758 CrossRef CAS PubMed; (i) J. Nokami, M. Osafune, Y. Ito, F. Miyake, S. Sumida and S. Torii, Chem. Lett., 1999, 28, 1053–1054 CrossRef; (j) G. Balavoine, A. Gref, J. C. Fischer and A. Lubineau, Tetrahedron Lett., 1990, 31, 5761–5764 CrossRef CAS; (k) C. Amatore, A. Jutand, J. M. Mallet, G. Meyer and P. Sinay, J. Chem. Soc., Chem. Commun., 1990, 718–719 RSC.
- Y. Li, H. Wang, Z. Wang, H. Alhumade, Z. Huang and A. Lei, Chem. Sci., 2023, 14, 372–378 RSC.
- J. Kuzmin, J. Röckl, N. Schwarz, J. Djossou, G. Ahumada, M. Ahlquist and H. Lundberg, Angew. Chem., Int. Ed., 2023, 62, e202304272 CrossRef CAS PubMed.
- Y. Li, Q. Yang, L. Yang, N. Lei and K. Zheng, Chem. Commun., 2019, 55, 4981–4984 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc00142g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |