
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silver(I)-catalyzed highly para-selective phosphonation of 2-aryloxazolines†
Peng-Cheng
Cui
a and
Guan-Wu
Wang
 *abc
*abc
aHefei National Research Center for Physical Sciences at the Microscale and Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: gwang@ustc.edu.cn
bKey Laboratory of Functional Molecular Solids, Ministry of Education, Anhui Laboratory of Molecule-Based Materials, and School of Chemistry and Materials Science, Anhui Normal University, Wuhu, Anhui 241002, P. R. China
cState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou, Gansu 730000, P. R. China
First published on 26th February 2024
Abstract
A silver-catalyzed phosphonation of 2-aryloxazolines has been accomplished. This protocol provides highly regioselective access to para-phosphonation products with good functional group tolerance and moderate to good yields via cross-dehydrogenation coupling. Mechanistic studies have shown that para-phosphonation products are obtained via a radical pathway. Furthermore, the directing oxazoline group in the para-phosphonation products is removable and can be converted to benzoic esters.
Benzoic acid derivatives are widely used in drugs and agrochemicals.1 Hence, their chemical modifications have potential applications. In the past few decades, transition-metal-catalyzed C–H bond activation has attracted extensive attention from chemists because of its high efficiency, atom economy and good functional group tolerance.2 In this regard, modifications of benzoic acid derivatives utilizing C–H activation have been significantly developed.3 Remote C–H activation of benzoic acid derivatives is relatively rare compared to ortho-C–H functionalization. To achieve remote C–H functionalization of benzoic acid derivatives, more complex auxiliary groups need to be utilized and tend to result in regioisomeric mixtures.4 Thus, it is urgent to develop new methodologies for the para-functionalization of benzoic acid derivatives using easily available directing groups. The conversion of benzoic acids to 2-aryloxazolines is a commonly used method.
On the other hand, phosphorus-containing compounds exhibit a wide range of applications in pharmaceuticals, materials and catalysis.5 In recent decades, transition-metal-catalyzed C–H functionalization to construct C–P bonds has been used as an important methodology for the synthesis of organophosphorus compounds.6 In 2006, Ishii and coworkers published the Mn(II)/Co(II)/O2 redox-couple catalyzed phosphorylation of arenes; although providing a simple approach to the preparation of organophosphorus compounds, the site-selectivity was relatively poor (Scheme 1a).7 For good selectivity, functional group-directed C–H activation protocols are particularly valuable. In 2013, the Yu group reported ortho-phosphorylation of arenes via heterocycle-directed ortho-palladation (Scheme 1b).8 Subsequently, the Chen and Yu groups described a process for the ortho-phosphorylation of arenes using an inexpensive copper catalyst (Scheme 1b).9 Since phosphorus reagents strongly coordinate with metals, a slow release of phosphorus reagents is required to ensure that the reaction proceeds smoothly. In 2019, the Wen, Zhang and Xu groups demonstrated a costly rhodium-catalyzed methodology to achieve ortho-phosphonation of electron-rich arylamine derivatives, and the use of oxidants was avoided because of the employed electrochemical oxidation (Scheme 1b).10 In 2013, the Zhu and Cheng groups realized the Ag(I)/K2S2O8-mediated ortho-phosphorylation of electron-deficient arenes via a radical pathway (Scheme 1c).11 In 2020, the Liang group reported the visible-light-induced para-CAr–H phosphonation reactions with electron-rich arenes by cross-dehydrogenation coupling (CDC) (Scheme 1d).12 As part of our continuing interest in remote C–H functionalization,13 herein we disclose the silver-catalyzed highly para-phosphonation of electron-deficient 2-aryloxazolines (Scheme 1e).
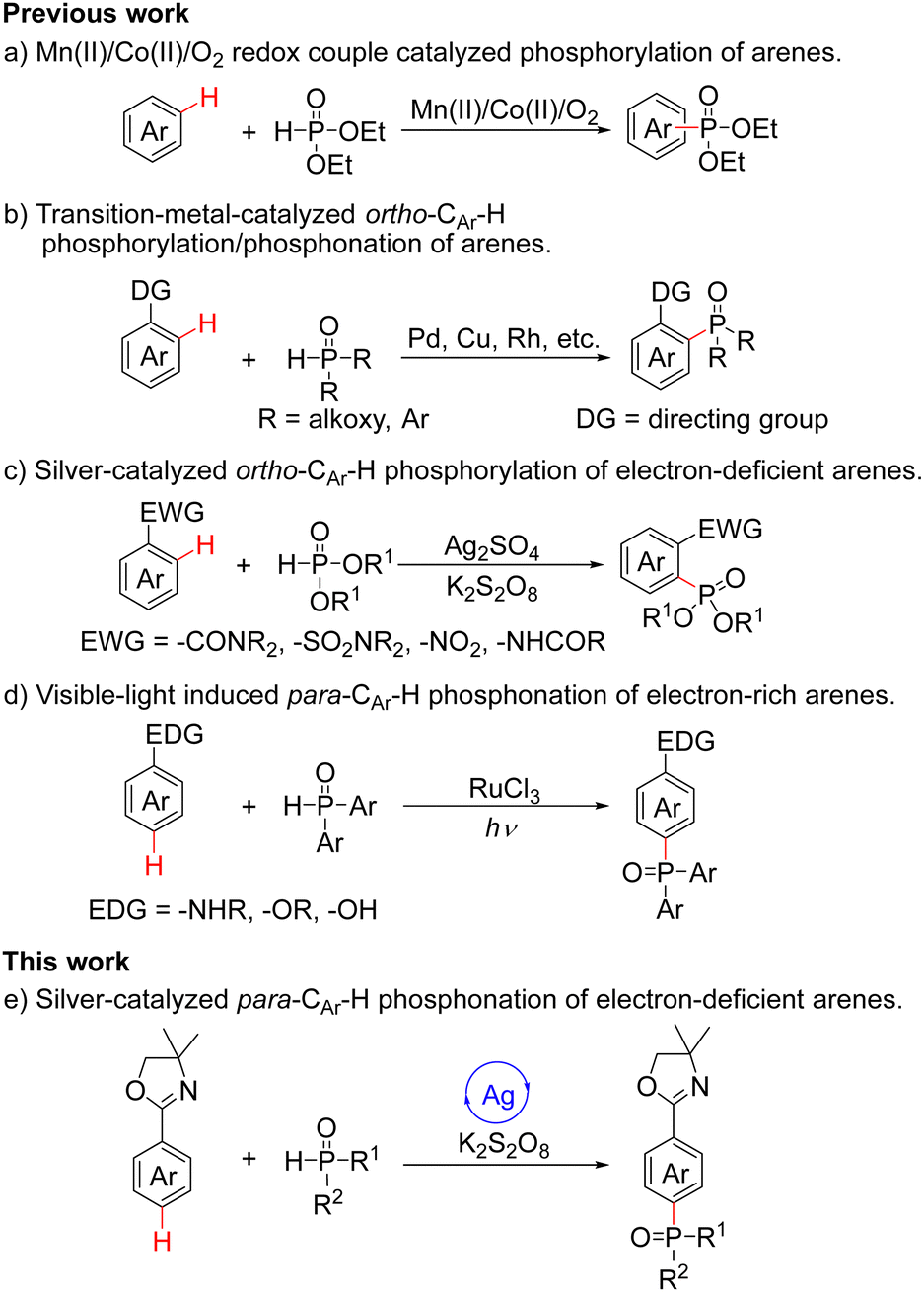 | ||
| Scheme 1 Previous (a–d) and this (e) work on transition-metal-catalyzed C–H phosphorylation/phosphonation of arenes. | ||
In our initial investigation, we chose 4,4-dimethyl-2-phenyl-4,5-dihydrooxazole (1a) and di-p-tolylphosphine oxide (2a) as model substrates to screen the reaction conditions (see Table S1 in the ESI† for details). After extensive screenings, the optimal conditions were established as follows: with 10 mol% AgNTf2 as the catalyst, 3.0 equiv. of K2S2O8 as the oxidant, 0.5 equiv. of pivalic acid (PivOH) as the additive and MeCN as the solvent, the reaction of 1a and 2a (3.0 equiv.) performed best at 120 °C for 24 h under an argon atmosphere to give 3aa in 73% yield.
With the optimal conditions in hand, we then investigated the scope and functional group tolerance of 2-aryloxazolines 1. As shown in Table 1, the reactions of 1a–i with 2a proceeded smoothly to afford para-phosphonation products 3aa–ia in moderate to good yields. First, 1a, which had an unsubstituted phenyl group, afforded 3aa in 73% yield under optimal conditions. Substrate 1b with an ortho-substituted Me group was able to provide 3ba in 61% yield. Substrates 1c–d with the ortho-substituted F and CO2Me groups could deliver 3ca and 3da in 46% and 42% yields, respectively. Substrates 1e–f with the meta-substituted OMe and F groups also proceeded smoothly to afford 3ea–fa in 41–78% yields. To our delight, when 3,5-dimethoxy-substituted substrate 1g was employed, 3ga was isolated in 53% yield. In addition, 2-(2-fluoro-5-methoxyphenyl)-4,4-dimethyl-4,5-dihydrooxazole (1h) could also react with 2a, and 3ha was obtained in 63% yield. These findings also provided a novel strategy for synthesizing multisubstituted arenes. For 4,4-dimethyl-2-(naphthalen-1-yl)-4,5-dihydrooxazole (1i), 3ia could be produced in a yield of 49%.
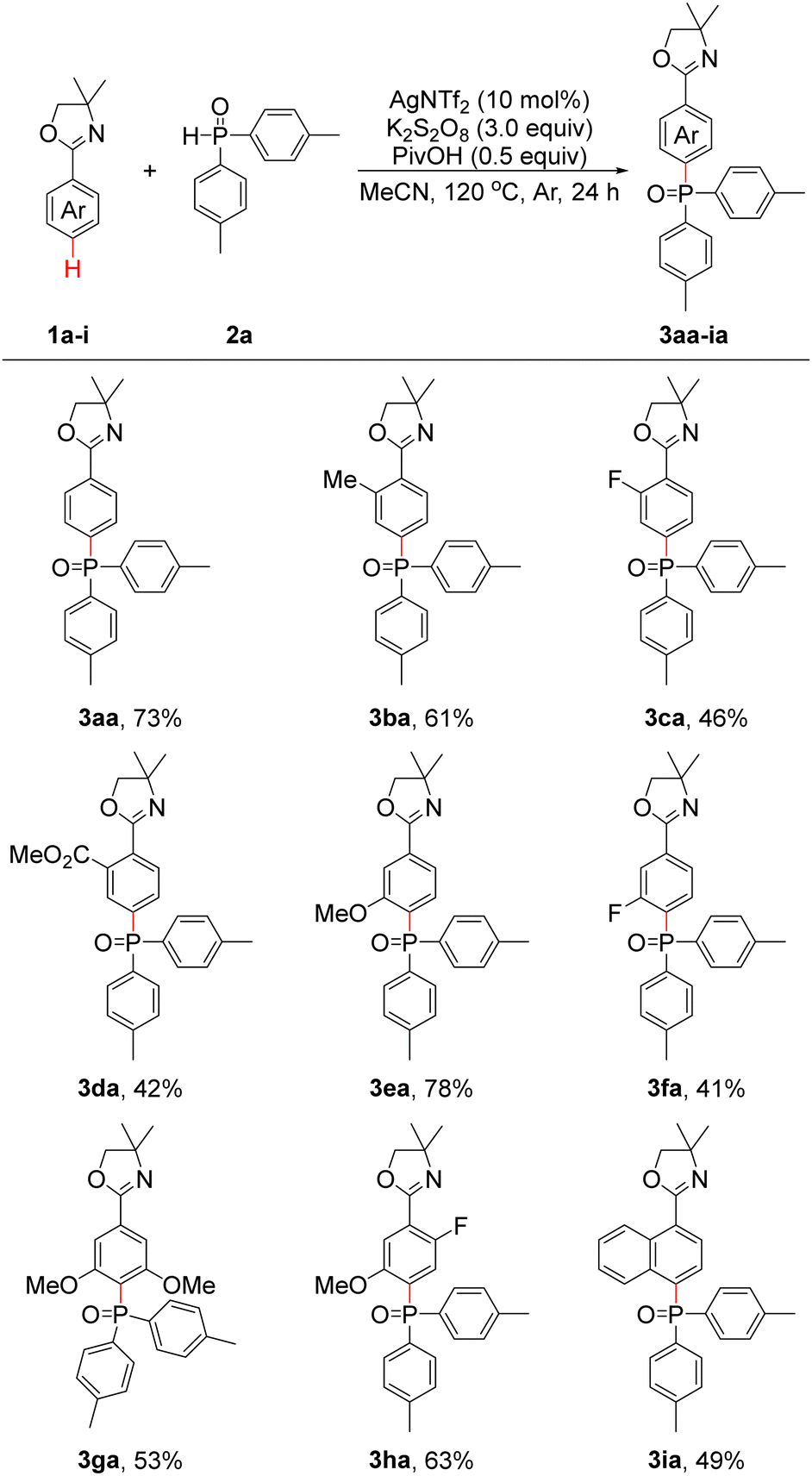
To further explore the generalizability of the substrates, the reactions of 1a with a series of diarylphosphine oxides (2b–n), ethyl phenylphosphinate (2o) and dialkylphosphine oxides (2p and 2q) were investigated under optimal conditions. As shown in Table 2, diphenylphosphine oxide (2b) was found to be suitable under our optimal conditions, and 3ab was obtained in 67% yield. When bis(m-tolyl)phosphine oxide (2c) and bis(m-methoxyphenyl)phosphine oxide (2d) were employed, 3ac and 3ad were obtained in 56% and 63% yields, respectively. If bis(p-methoxyphenyl)phosphine oxide (2e) was used, 3ae was obtained in 68% yield. Bis(p-fluorophenyl)phosphine oxide (2f) and bis(p-chlorophenyl)phosphine oxide (2g) proceeded well to produce 3af and 3ag in 62% and 47% yields, respectively. Delightedly, when bis(3,5-dimethylphenyl)phosphine oxide (2h) and bis(3,5-di-tert-butylphenyl)phosphine oxide (2i) were applied, 3ah and 3ai were generated in 61% and 58% yields, respectively. Subsequently, phenyl(tolyl)phosphine oxides (2j and 2l), phenyl(methoxyphenyl)phosphine oxides (2k and 2m) and phenyl(p-fluorophenyl)phosphine oxide (2n) were also applicable under optimal conditions, and 3aj–an could be obtained in 59–71% yields. As a result, the synthesis of phosphine oxide products with three different aryl groups is made possible. To our delight, when 2o was investigated, 3ao was generated in 37% yield. Products 3ap and 3aq could be isolated in 72% and 44% yields, respectively, when dibenzylphosphine oxide (2p) and dicyclohexylphosphine oxide (2q) were applied. Unfortunately, other substrates such as di(thiophen-2-yl)phosphine oxide and diethyl phosphonate failed to provide the desired products.
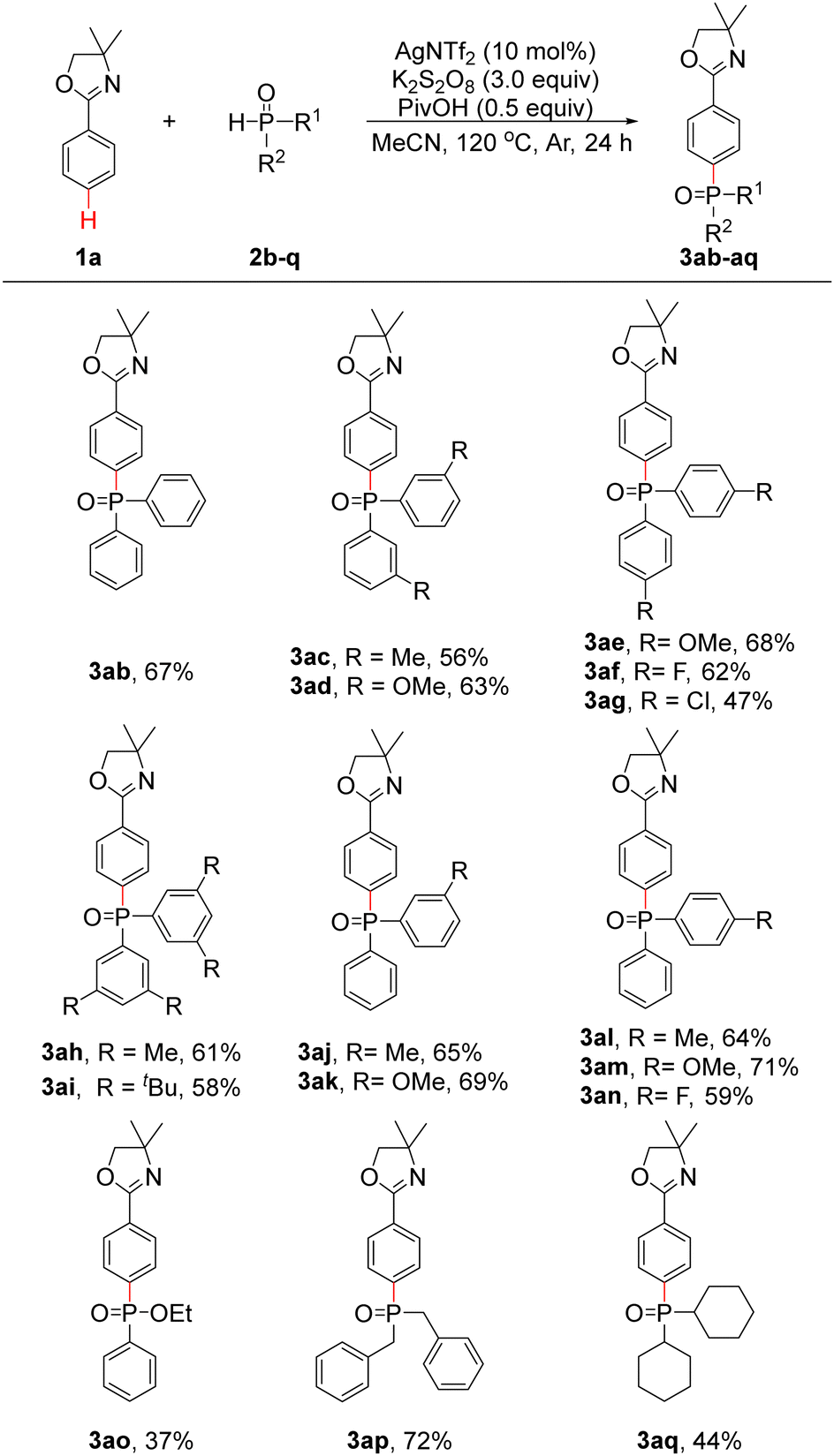
To gain insight into the reaction mechanism, additional experiments were conducted. First, 3aa could not be obtained when 3.0 equiv. of the radical scavenger 2,2,6,6-tetramethylpiperidinooxy (TEMPO) was added under optimal conditions, and the TEMPO–2a adduct could be detected by high resolution mass spectrometry (HRMS) (Scheme 2a and Fig. S1, ESI†). This result showed that a radical pathway may be involved in the reaction. Second, when 1a-D5 was employed under optimal conditions, pure 3aa-D4 (Fig. S2, ESI†) was obtained in 61% yield. In addition, pure 3aa-D4 (Fig. S3, ESI†) could also be isolated in 54% yield with an additional 1.0 equiv. of H2O. These results indicated that no D/H scrambling occurred on the phenyl ring of 3aa-D4 and implied that no ortho C–H activation process took place (Scheme 2b). Third, an intermolecular kinetic isotope effect (KIE) experiment was performed, and the kH/kD value was determined to be 5.7 (Fig. S4, ESI†); when 2a was reduced to 0.5 equiv., the kH/kD value was 4.0 (Fig. S5, ESI†), suggesting that C–H cleavage may be involved in the rate-determining step (Scheme 2c).
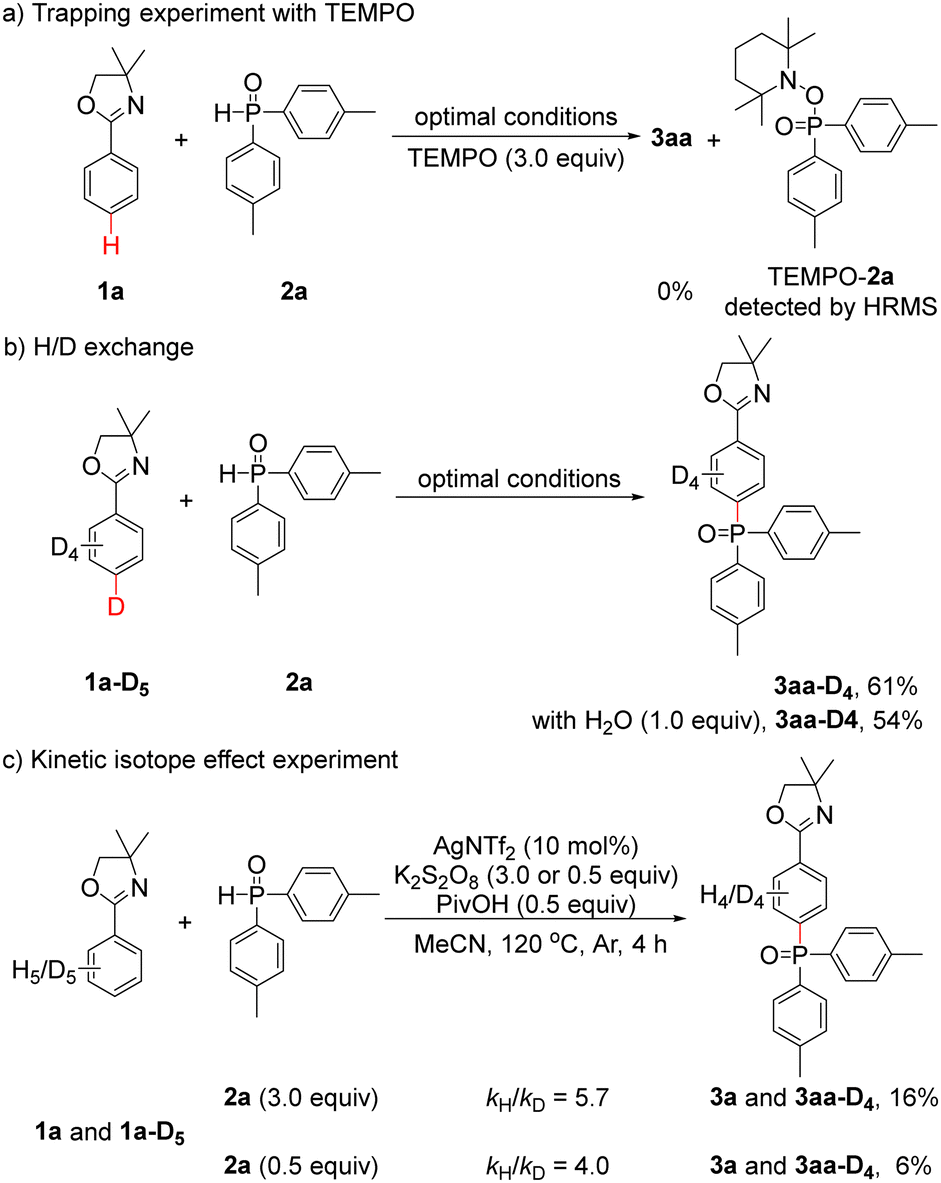 | ||
| Scheme 2 Preliminary mechanistic studies. | ||
Based on the above findings of mechanistic studies and previous reports, a plausible reaction mechanism is shown in Scheme 3.11,14–18 First, 2-aryloxazoline 1 interacts with the silver catalyst to form the intermediate A.14 Similar oxazolinium ion can also be generated in the presence of PivOH and participates in subsequent steps as the intermediate A.15 Meanwhile, phosphine oxide 2 is directly oxidized by the persulfate dianion (S2O82−) to give the key P-centered radical B together with a hydrogen sulfate anion (HSO4−) and a sulfate radical anion (SO4˙−) (Scheme 3, path a).16 Alternatively, 2 can exist in rapid equilibrium with the corresponding tautomeric isomer 2′.17 The Ag2+ salt can oxidize 2′ to afford a Ag+ species and cation radical B′, which may lead to the P-centered radical B after losing a proton; and the formed Ag+ salt can be oxidized by a S2O82− to regenerate Ag2+ together with a sulfate dianion (SO42−) and a SO4˙− (Scheme 3, path b).11,18 Radical B then regioselectively undergoes a radical addition to the para position of A to give the intermediate C, which is subsequently oxidized and deprotonated by SO4˙− to give intermediate D. Finally, the para-phosphonation product 3 is obtained by removal of the silver catalyst to complete the catalytic cycle. The oxidant K2S2O8 was mandatory and could promote the para-phosphonation alone, albeit in a low yield of 13% (Table S1, entries 22 and 23, ESI†). The beneficial effects of the silver salt (Table S1, entry 24, ESI†) and PivOH (Table S1, entry 25, ESI†) might originate from the activation of 1 by the formation of A and oxazolinium ion, respectively. Furthermore, the silver salt can also be employed as an oxidant to generate B.
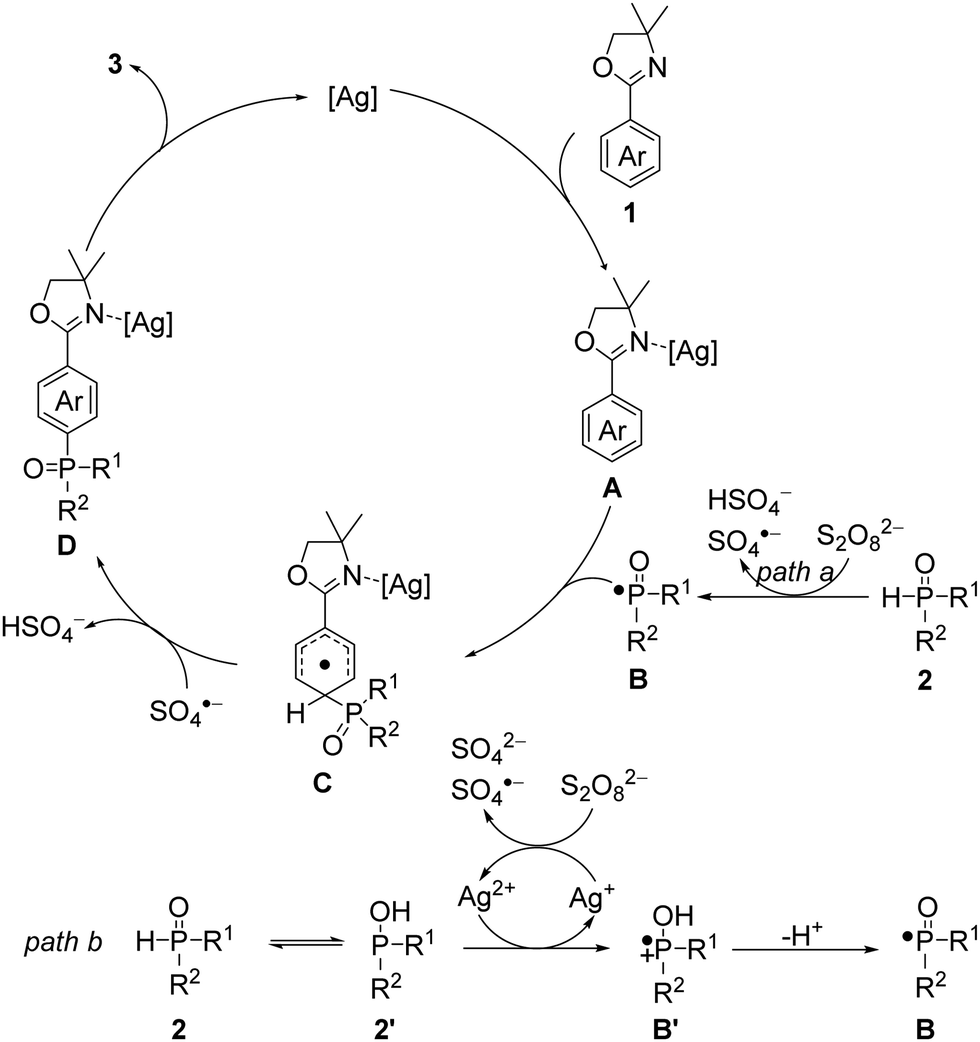 | ||
| Scheme 3 Proposed mechanism. | ||
To demonstrate the synthetic utility and importance of the present protocol, the reaction of 1a with 2b was carried out at the 5.0-mmol scale, and the para-phosphonation product 3ab was isolated in 51% yield (Scheme 4a). We chose 3ap as an example for further transformation, its oxazoline group could be easily hydrolysed under acidic conditions, providing the para-phosphonated benzoic acid derivative 4 in 74% yield (Scheme 4b).19
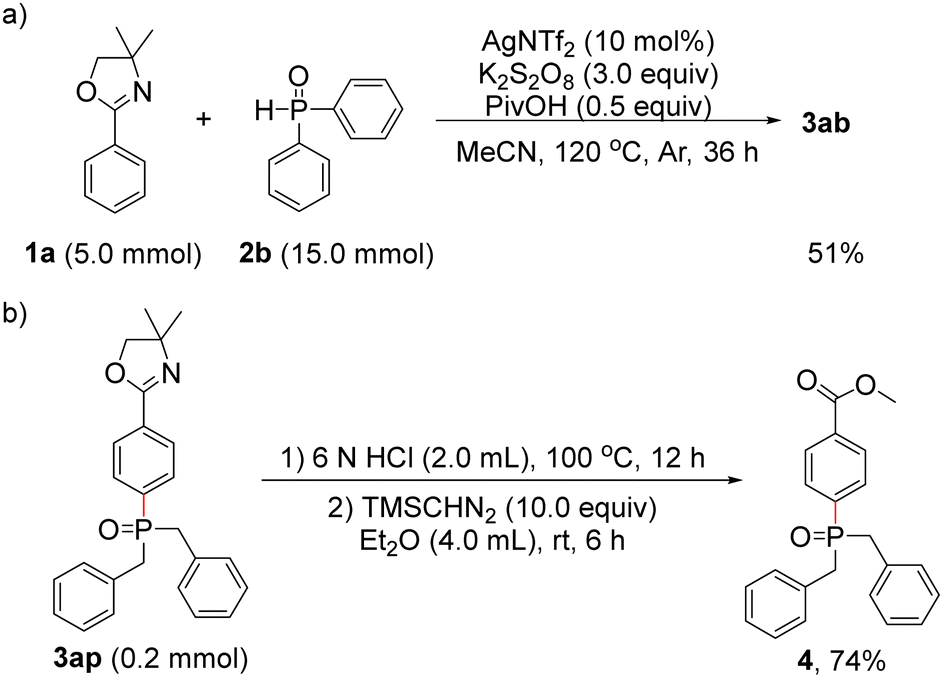 | ||
| Scheme 4 (a) Gram-scale synthesis of 3ab. (b) Further synthetic transformation of 3ap. | ||
In summary, a precise and efficient approach for the silver-catalyzed para-selective phosphonation of 2-aryloxazolines via cross-dehydrogenation coupling has been demonstrated. This protocol is able to provide highly regioselective para-phosphonation products and tolerates a wide range of functional groups. Furthermore, the directing oxazoline group in the products is removable and can be easily hydrolysed to give para-phosphonated benzoic esters. Preliminary mechanistic studies support that the para-phosphonation products are generated via a radical pathway. It is anticipated that the present protocol will also be valuable in pharmaceutical and materials science.
We are grateful for the financial support from the Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDB20000000).
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Lamberth and J. Dinges, Bioactive carboxylic compound classes: pharmaceuticals and agrochemicals, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2016 Search PubMed.
- For selected reviews, see: (a) M. P. Drapeau and L. J. Gooßen, Chem. – Eur. J., 2016, 22, 18654 CrossRef PubMed; (b) M. Shang, et al. , Synthesis, 2016, 4381 CAS; (c) Y. Wei, et al. , Chem. Rev., 2017, 117, 8864 CrossRef CAS PubMed; (d) C. Sambiagio, et al. , Chem. Soc. Rev., 2018, 47, 6603 RSC.
- For selected examples, see: (a) X.-G. Zhang, et al. , J. Am. Chem. Soc., 2012, 134, 11948 CrossRef CAS PubMed; (b) H. Li, et al. , ACS Catal., 2018, 8, 4777 CrossRef CAS; (c) A. S. Trita, et al. , Angew. Chem., Int. Ed., 2018, 57, 14580 CrossRef CAS PubMed; (d) Z. Li, et al. , Science, 2021, 372, 1452 CrossRef CAS PubMed; (e) Z. Li, et al. , J. Am. Chem. Soc., 2022, 144, 18109 CrossRef CAS PubMed.
- (a) S. Li, et al. , Nat. Commun., 2016, 7, 10443 CrossRef PubMed; (b) M. Li, et al. , Org. Lett., 2019, 21, 540 CrossRef CAS PubMed.
- For selected reviews, see: (a) M. A. Shameem and A. Orthaber, Chem. – Eur. J., 2016, 22, 10718 CrossRef CAS PubMed; (b) G. P. Horsman and D. L. Zechel, Chem. Rev., 2017, 117, 5704 CrossRef CAS PubMed; (c) H. Guo, et al. , Chem. Rev., 2018, 118, 10049 CrossRef CAS PubMed; (d) P. Finkbeiner, et al. , J. Med. Chem., 2020, 63, 7081 CrossRef CAS PubMed; (e) C. Xie, et al. , ACS Cent. Sci., 2021, 7, 536 CrossRef CAS PubMed.
- For selected reviews, see: (a) S. Strekalova, et al. , Curr. Org. Chem., 2019, 23, 1756 CrossRef CAS; (b) L. Chen, et al. , Adv. Synth. Catal., 2020, 362, 1724 CrossRef CAS; (c) F. Xu and Y. Hui, Curr. Org. Synth., 2021, 18, 377 CrossRef CAS PubMed.
- T. Kagayama, et al. , Org. Lett., 2006, 8, 407 CrossRef CAS PubMed.
- C.-G. Feng, et al. , J. Am. Chem. Soc., 2013, 135, 9322 CrossRef CAS PubMed.
- S. Wang, et al. , Chem. Commun., 2014, 50, 12718 RSC.
- Z.-J. Wu, et al. , Angew. Chem., Int. Ed., 2019, 58, 16770 CrossRef CAS PubMed.
- X. Mao, et al. , Eur. J. Org. Chem., 2013, 4245 CrossRef CAS.
- X.-Y. Gou, et al. , Chem. Commun., 2020, 56, 4704 RSC.
- (a) Z.-Y. Li, et al. , Chem. – Eur. J., 2017, 23, 3285 CrossRef CAS PubMed; (b) K. Jing, et al. , ACS Catal., 2018, 8, 11875 CrossRef CAS; (c) P.-C. Cui, et al. , Org. Lett., 2023, 25, 2663 CrossRef CAS PubMed; (d) P.-C. Cui and G.-W. Wang, Org. Lett., 2023, 26, 427 CrossRef PubMed.
- (a) P. Liu, et al. , Nat. Commun., 2018, 9, 4637 CrossRef PubMed; (b) Z. Jiao, et al. , Angew. Chem., Int. Ed., 2018, 57, 6294 CrossRef CAS PubMed; (c) Z. Tang, et al. , Angew. Chem., Int. Ed., 2022, 61, e202208089 CrossRef CAS PubMed.
- Z. Guo, et al. , J. Phys. Chem. B, 2023, 127, 4820 CrossRef CAS PubMed.
- (a) H.-Y. Zhang, et al. , Chem. Commun., 2015, 51, 4101 RSC; (b) P. Xu, et al. , Org. Lett., 2016, 18, 1143 CrossRef CAS PubMed.
- K. Sun, et al. , Chem. Commun., 2015, 51, 12111 RSC.
- (a) C.-B. Xiang, et al. , J. Org. Chem., 2012, 77, 7706 CrossRef CAS PubMed; (b) K. Sun, et al. , Asian J. Org. Chem., 2019, 8, 2042 CrossRef CAS.
- C.-C. Yuan, et al. , Org. Chem. Front., 2017, 4, 1867 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization data, 1H NMR, 13C NMR, 19F NMR, 31P NMR and HRMS of compounds 3 and 4. See DOI: https://doi.org/10.1039/d3cc06241d |
| This journal is © The Royal Society of Chemistry 2024 |