
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiastereoselective access to C,C-glycosyl amino acids via iron-catalyzed, auxiliary-enabled MHAT coupling†
Mylène
Lang
 ,
Damien
Tardieu
,
Benoit
Pousse
,
Philippe
Compain
* and
Nicolas
Kern
*
,
Damien
Tardieu
,
Benoit
Pousse
,
Philippe
Compain
* and
Nicolas
Kern
*
Laboratoire d’Innovation Moléculaire et Applications (LIMA), UMR 7042, Université de Strasbourg/Université de Haute-Alsace/CNRS, ECPM, 25 rue Becquerel, 67087, Strasbourg, France. E-mail: philippe.compain@unistra.fr; nicolas.kern@unistra.fr
First published on 19th February 2024
Abstract
Access to C,C-glycosyl amino acids as a novel class of glycomimetics is reported by means of radical generation, intermolecular addition and stereoselective reduction via a metal-induced hydrogen atom transfer (MHAT) sequence. The ‘matched’ coupling of exo-D-glycals with an enantiopure dehydroalanine bearing a (R)-configured benzyl oxazolidinone enables a singular case of two-fold diastereocontrol under iron catalysis. In the common exo-D-glucal series, the nature of the C-2 substituent was found to play a key role from both reactivity and stereocontrol aspects.
Glycoproteins, as ubiquitous components of mammal tissues, are involved in multiple biological processes such as cell signaling, growth regulation and immunity.1,2 In the realm of pharmaceutical research, linking an amino acid chain to sugar(s) also stands as an established way to improve stability towards proteases and increase membrane permeability.3 As the overwhelming majority of natural glycopeptides comprise enzymatically labile C–O or C–N linkages (with C-mannosyl tryptophan as the sole exception4), unnatural C-glycopeptides both furnish useful probes to facilitate the elucidation of essential processes in vivo, as well as stable drug or vaccine candidates.5 This promising potential has therefore aroused intense synthetic efforts to construct C-linked sugar amino acid (SAA) building blocks.6,7 Formal “deletion” of the linking O- or N-atom ideally calls for the direct coupling of a pyranosyl unit activated at C-18 with a functionalized two-carbon synthon bearing the (masked) AA motif. Such step-economic and convergent disconnection however adds challenges, especially regarding stereoselectivity and functional orthogonality. Notably, Ackermann and Li & Liu independently demonstrated the virtue of Pd-catalyzed C(sp2)–H and C(sp3)–H activation to assemble a persilylated iodoglycal and auxiliary-tagged enantiopure amino acid (Fig. 1a).9 While excellent yields and diastereoselectivities (β to nitrogen, i.e. at C-1′) could be achieved, efficient introduction of key O- or N-functionality at the C-2 site in such adducts can be nontrivial. Another mild approach involves the generation of glycosyl radicals10 and trapping with dehydroalanine-type acceptors (DHAs). Very recently, two photoinduced variants emanated from the groups of Di Carmine using pyranosyl bromides11 and those from Xie, Walczak & Zhu who relied on furanosyl and pyranosyl dihydropyridyl esters (Fig. 1b).12 Glycosyl radical additions to achiral and/or peptide-linked DHA derivatives typically proceed with axial selectivity but low control at the AA centre (α to nitrogen, i.e. at C-2′), emphasizing the poor level of induction imparted from the sugar and AA moieties. This trend, observed early by Kessler,13 appears as a longstanding issue. Therefore, Xie, Walczak & Zhu employed the (S)-enantiomer of the Karady–Beckwith acceptor and achieved high control in most cases through cis-selective reduction by hydrogen atom transfer (HAT), as documented.14
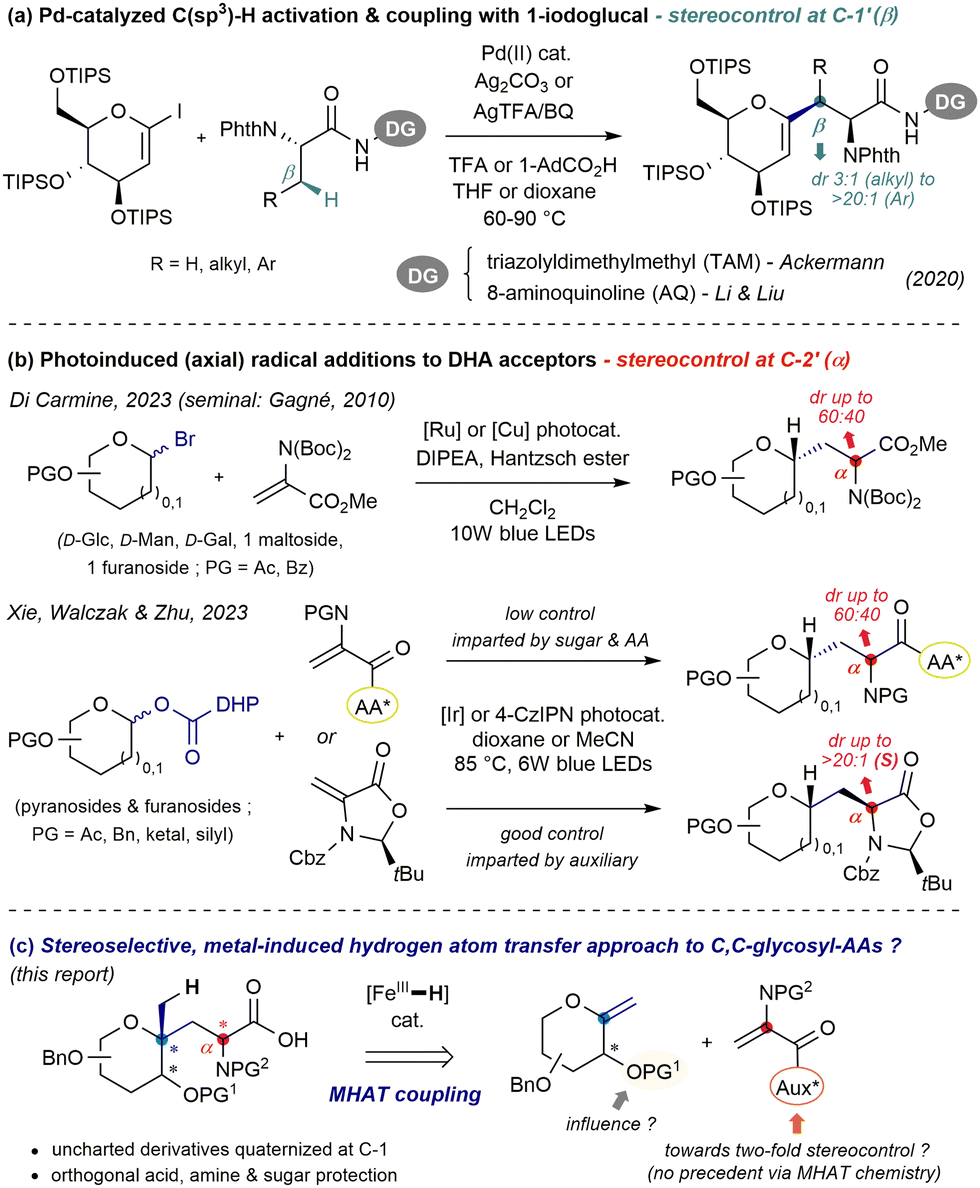 | ||
| Fig. 1 Recent convergent strategies to assemble C-glycosyl amino acids from pyranosides and (C2) amino acid synthons (a) and (b) and aims of this work (c). | ||
In connection with our search for neo-glycoconjugates and interest in novel transformations of exo-glycals,15 we questioned the viability of an MHAT (metal-induced HAT)16,17 coupling to form C,C-glycoaminoacids, unusual adducts embedding an atypical fully substituted pseudoanomeric carbon.18 Several intramolecular cyclization strategies were examined, all of which met with failure due to the low stability of DHA-acylated exo-glycals. While an intermolecular approach first appeared daunting in terms of two-fold stereocontrol (yet unreported for Fe-HAT Baran–Giese type coupling, and including an exocyclic centre), we were curious whether the inherent low stereoinduction inferred from the pyranoside (vide supra) could be enforced. We envisioned that such a task may be achievable through the identification of an appropriate “matched” nonracemic radical acceptor, along with careful choice of the protection patterns – especially for the C-2 hydroxyl moiety, in the vicinity of subsequently formed open-shell species (Fig. 1c).
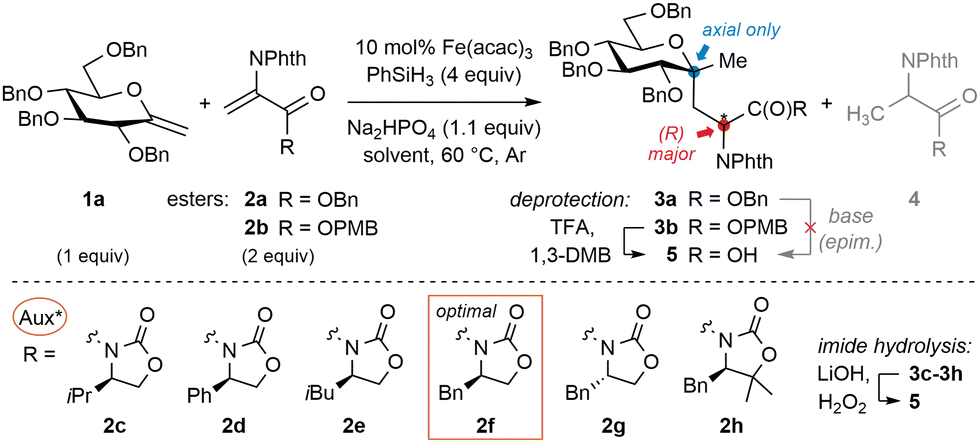
To begin to answer this question, O-benzyl-N-phthaloyl DHA-based acceptor 2a was selected (Table 1). Screening of coupling conditions (catalytic Fe(acac)3, phenylsilane and Na2HPO4 buffer in EtOH) applied to a mixture of perbenzylated exo-glycal 1a (1 equiv.) and 2a (2 equiv.) first suggested that productive radical generation from exo-glucals calls for specific dilute conditions and moderate catalyst loading (entries 1–4). Along with targeted adduct 3a, alanine imidoester 4a was identified as the main side product. Competitive reduction of C-radicophiles has been scarcely mentioned in the Fe-HAT literature,19 and may be favored here considering the captodative character of the product radical,20 partly overriding the usual cross-coupling chemoselectivity attributed to polar effects. This may also explain the use of excess electron-rich alkenes in related protocols,19,21 inconvenient with valuable exo-glycals. However, formal hydrogenation of 1a and telomerization13 were fully avoided.22 Since chromatographical separation of 3a and 4a proved tedious, base-mediated hydrolysis of the benzyl ester resulted in the epimerization of the neoformed AA stereocentre. Substitution of 2a for PMB ester 2b (entry 5, conditions [A]) allowed quantitative and epimerization-free acid-mediated dealkylation with TFA in conjunction with 1,3-dimethoxybenzene (DMB), yielding free acid 5.23 Akin to 2a, acceptor 2b appeared almost insoluble at 25 °C and sparingly at 60 °C; therefore inclusion of cosolvents was evaluated. Use of minimal EtOH in THF (16 equiv. to reach full conversion; entry 6, conditions [B]) resulted in close efficiency at the cost of reaction rate, which may be rationalized from the involvement of the alcohol in the rate-determining iron hydride generation step.15 Overall, all experiments involving achiral acceptor 2b furnished products 3 (or 5) in moderate d.r. (close to 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). We thus moved forward by evaluating DHA-tagged chiral oxazolidinones 2c–2h.21,24 Unlike esters, imide coupling adducts could be carefully hydrolyzed to 5 without epimerization using minimal LiOH and H2O2, enabling comparison of the stereochemical course with reactions of 2b. First, (R)-configured isopropyl derivative 2c underwent addition with lower diastereoselectivity (entry 7). Phenyl analog 2d only led to traces of the coupling adduct, while isobutyryl analog 2e reacted poorly with the glycosyl radical without impacting stereoinduction (entries 8 and 9). Gratifyingly, a ‘matched’ case was disclosed with benzyl surrogate 2f (i.e. Evans’ auxiliary,25 entry 10), which reacted efficiently and induced good diastereocontrol (88:12) under conditions [A]. As the stereodetermining redox termination step involves the protic solvent,26 system [B] with minimal EtOH was evaluated and induced a slight enhancement (entry 11). Since acceptor 2f displayed better solubility in THF, decreasing the reaction temperature to 25 °C allowed even higher control but with lower efficiency (entry 12). Consecutively, the (S)-enantiomer 2g was probed to be ‘mismatched’ (entry 13). The gem-dimethyl surrogate of the optimal auxiliary27 acted as a potent inductor, but the apparent lower stability of parent acceptor 2h only enabled moderate yields (entry 14). The observation that all auxiliaries and conditions afforded the same major diastereoisomer of 5 stands in line with previous reports involving pyranosides, where the degree but not the direction of stereoselectivity could be influenced.13,14 Furthermore, closer examination of related work12 reveals that stereoinduction in the termination step is usually lower for pyranosides bearing equatorial O- or N-functionality at C-2. In contrast to mannosides or 2-deoxy derivatives for example, their orientation may partly impede the population of a favorable conformer for stereoselective termination (vide infra). A series of exo-D-glucals diversely protected at C-2 were thus examined (Fig. 2a).‡ With relatively bulky silyl groups (1b & 1c) negligible conversions were observed while DHA reduction predominated. The solvolytically unstable free-hydroxyl derivative 1d only led to the corresponding adduct 6 in low yield and d.r. Pleasingly, stable and less hindered methoxymethyl (MOM) derivative 1e led to the highest yield and stereoselectivity (94:6) observed. Orthogonal removal of the auxiliary in 7 and acid-mediated lactonization to 8 were straightforward and allowed unambiguous assignment of the (R)-configuration of the AA by 1H NOESY analysis. In view of the importance of 2-amino-C-glycosides,28 coupling of the seldom (2-acetamido)exo-glycal291f could be achieved in moderate yield and stereoselectivity, granting with adduct 9 a first entry to 2-amido-C,C-glycosidic motifs. Considering these results, we speculate that (1) steric impediment of the olefin in 1 is detrimental to radical generation; (2) alongside competitive processes efficiency is also correlated to the chemical stability of the often sensitive exo-glycal; and (3) high stereoselectivities are obtained when the C-2 position of the sugar is relatively unhindered. As mentioned above, if the optimized acceptor and conditions enable enforcement of two-fold stereocontrol in the D-gluco series, extension to related important pyranosides should be feasible. This was probed with exo-D-mannal 1g and exo-D-galactal 1h, whose corresponding coupling adducts 10 and 11 were successfully isolated after direct hydrolysis (Fig. 2b). As notable, selective MOM protection at C-2 was not mandatory here to achieve high control. The (very) decent chemical yields obtained from these fragile, electron-rich terminal exo-glycals again testify of the relative mildness of the Fe-HAT conditions. Although optimized for pyranosides, the protocol also allowed the efficient conversion of furanoside 1i into adduct 12 with significant control at the AA centre (Fig. 2c). The main limitations concern trisubstituted15 and electronically deactivated difluoroolefins,30 which do not undergo MHAT at a sufficient rate with respect to DHA derivatives (Fig. 2d). On the other hand, sulfonylhydrazone acceptors used for reductive alkylation31 either seem to: (1) lack sufficient reactivity towards tertiary glycosyl radicals (alkylsulfonyl derivatives), whose reduction thus leads to formal alkene hydrogenation, or (2) undergo nonselective fragmentation (arylsulfonyl derivatives).
:3). We thus moved forward by evaluating DHA-tagged chiral oxazolidinones 2c–2h.21,24 Unlike esters, imide coupling adducts could be carefully hydrolyzed to 5 without epimerization using minimal LiOH and H2O2, enabling comparison of the stereochemical course with reactions of 2b. First, (R)-configured isopropyl derivative 2c underwent addition with lower diastereoselectivity (entry 7). Phenyl analog 2d only led to traces of the coupling adduct, while isobutyryl analog 2e reacted poorly with the glycosyl radical without impacting stereoinduction (entries 8 and 9). Gratifyingly, a ‘matched’ case was disclosed with benzyl surrogate 2f (i.e. Evans’ auxiliary,25 entry 10), which reacted efficiently and induced good diastereocontrol (88:12) under conditions [A]. As the stereodetermining redox termination step involves the protic solvent,26 system [B] with minimal EtOH was evaluated and induced a slight enhancement (entry 11). Since acceptor 2f displayed better solubility in THF, decreasing the reaction temperature to 25 °C allowed even higher control but with lower efficiency (entry 12). Consecutively, the (S)-enantiomer 2g was probed to be ‘mismatched’ (entry 13). The gem-dimethyl surrogate of the optimal auxiliary27 acted as a potent inductor, but the apparent lower stability of parent acceptor 2h only enabled moderate yields (entry 14). The observation that all auxiliaries and conditions afforded the same major diastereoisomer of 5 stands in line with previous reports involving pyranosides, where the degree but not the direction of stereoselectivity could be influenced.13,14 Furthermore, closer examination of related work12 reveals that stereoinduction in the termination step is usually lower for pyranosides bearing equatorial O- or N-functionality at C-2. In contrast to mannosides or 2-deoxy derivatives for example, their orientation may partly impede the population of a favorable conformer for stereoselective termination (vide infra). A series of exo-D-glucals diversely protected at C-2 were thus examined (Fig. 2a).‡ With relatively bulky silyl groups (1b & 1c) negligible conversions were observed while DHA reduction predominated. The solvolytically unstable free-hydroxyl derivative 1d only led to the corresponding adduct 6 in low yield and d.r. Pleasingly, stable and less hindered methoxymethyl (MOM) derivative 1e led to the highest yield and stereoselectivity (94:6) observed. Orthogonal removal of the auxiliary in 7 and acid-mediated lactonization to 8 were straightforward and allowed unambiguous assignment of the (R)-configuration of the AA by 1H NOESY analysis. In view of the importance of 2-amino-C-glycosides,28 coupling of the seldom (2-acetamido)exo-glycal291f could be achieved in moderate yield and stereoselectivity, granting with adduct 9 a first entry to 2-amido-C,C-glycosidic motifs. Considering these results, we speculate that (1) steric impediment of the olefin in 1 is detrimental to radical generation; (2) alongside competitive processes efficiency is also correlated to the chemical stability of the often sensitive exo-glycal; and (3) high stereoselectivities are obtained when the C-2 position of the sugar is relatively unhindered. As mentioned above, if the optimized acceptor and conditions enable enforcement of two-fold stereocontrol in the D-gluco series, extension to related important pyranosides should be feasible. This was probed with exo-D-mannal 1g and exo-D-galactal 1h, whose corresponding coupling adducts 10 and 11 were successfully isolated after direct hydrolysis (Fig. 2b). As notable, selective MOM protection at C-2 was not mandatory here to achieve high control. The (very) decent chemical yields obtained from these fragile, electron-rich terminal exo-glycals again testify of the relative mildness of the Fe-HAT conditions. Although optimized for pyranosides, the protocol also allowed the efficient conversion of furanoside 1i into adduct 12 with significant control at the AA centre (Fig. 2c). The main limitations concern trisubstituted15 and electronically deactivated difluoroolefins,30 which do not undergo MHAT at a sufficient rate with respect to DHA derivatives (Fig. 2d). On the other hand, sulfonylhydrazone acceptors used for reductive alkylation31 either seem to: (1) lack sufficient reactivity towards tertiary glycosyl radicals (alkylsulfonyl derivatives), whose reduction thus leads to formal alkene hydrogenation, or (2) undergo nonselective fragmentation (arylsulfonyl derivatives).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Acceptor | Solvent(s)/conc. (M) | t (h) | Yield (%)/product | d.r.b |
| a Estimated yield by 1H NMR (isolated material contaminated with 4a). b Determined by 1H NMR analysis of partly purified 3a or pure 5. c 20 mol% [Fe]. d Formation of unseparable side products. e Isolated yield after acidic hydrolysis to 5. f 16 equiv. EtOH as additive. g Confirmed by HPLC. h At 25 °C. [A] & [B]: optimal conditions. | |||||
| 1 | 2a | EtOH (0.1) | 1.5 | 60 (3a)a | 69:31 |
| 2 | 2a | EtOH (0.01) | 16 | 52 (3a)a | 71:29 |
| 3 | 2a | EtOH (0.03) | 4 | 77 (3a)a | 73:27 |
| 4c | 2a | EtOH (0.03) | 2 | 40 (3a)ad | nd |
| 5 [A] | 2b | EtOH (0.03) | 4 | 76 (5)e | 69:31 |
| 6 [B] | 2b | THF (0.03)f | 16 | 66 (5)e | 71:29 |
| 7 | 2c | EtOH (0.03) | 4 | 50 (5)e | 58:42 |
| 8 | 2d | EtOH (0.03) | 4 | Traced | nd |
| 9 | 2e | EtOH (0.03) | 4 | 21 (5)e | 70:30 |
| 10 [A] | 2f | EtOH (0.03) | 4 | 75 (5)e | 88:12g |
| 11 [B] | 2f | THF (0.03)f | 16 | 82 | 92:8g |
| 12h | 2f | THF (0.03)f | 16 | 45 | 96:4g |
| 13 | 2g | EtOH (0.03) | 4 | 67 | 74:26 |
| 14 | 2h | EtOH (0.03) | 4 | 31 | 93:7g |
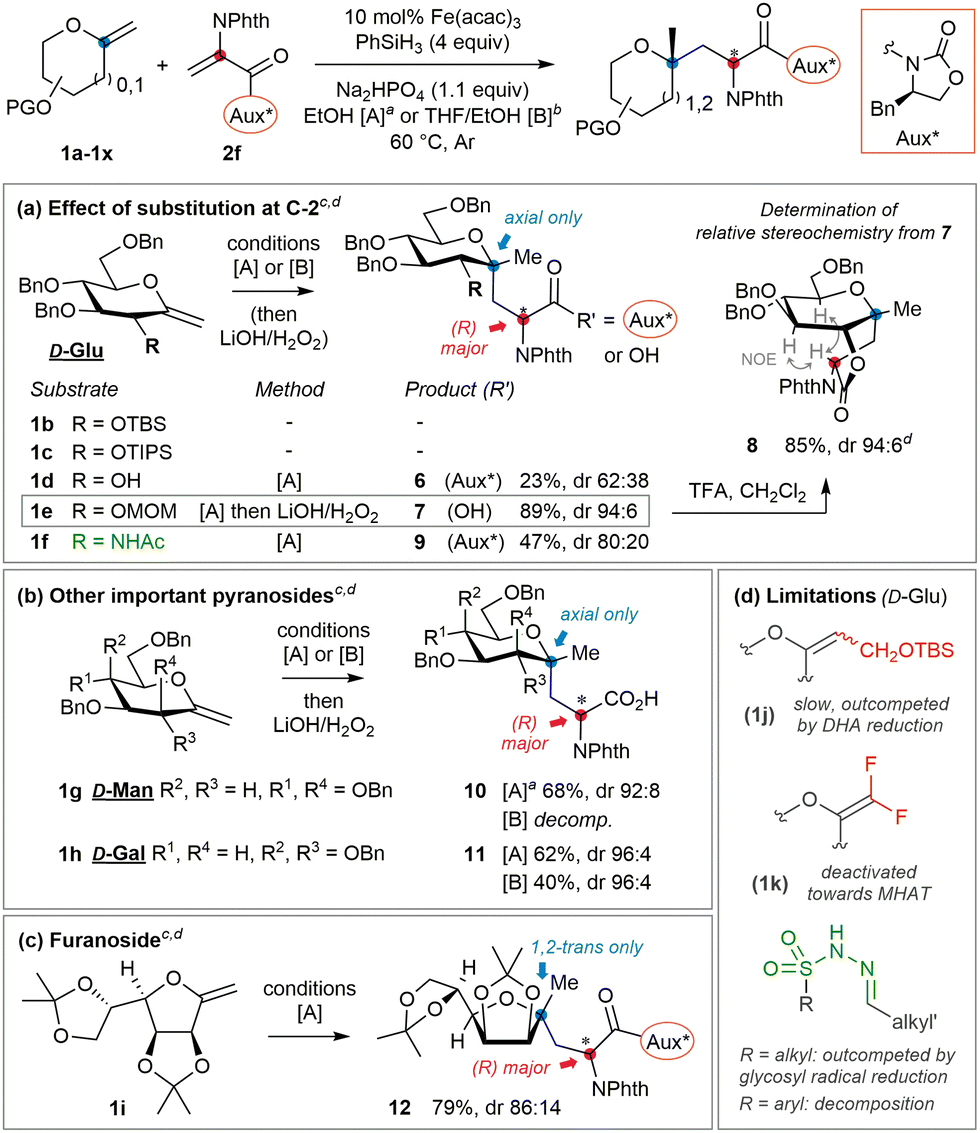 | ||
| Fig. 2 (a) Identification of the optimal exo-glucal protection pattern and derivatization. (b) Application to mannosyl and galactosyl surrogates. (c) Application to furanoside. (d) Main limitations observed through this study. a Reaction time: 3–4 h. b Reaction time: 12–16 h (full conv obs). c Isolated yields. d dr determined by 1H NMR analysis of isolated products due to overlaps. | ||
Based on our results and taking account of Poli & Holland's studies,26 a tentative model for the stereocontrolled formation of the AA centre may be envisioned from two perspectives (Fig. 3). Following generation of an iron(III) enolate via inner-sphere electron transfer from Fe(II)(acac)2 species (a), a nonchelated model – expectable considering a moderately Lewis acidic and coordinatively saturated Fe(III) complex – may explain the preference for si-face ‘concerted’ protonation from a metal-bound EtOH molecule leading to the (R)-configuration while accounting for the effect of substituents at C-2, in close vicinity of the aryl moiety presented by the auxiliary. Additional shielding of the re-face is imposed by the angular methyl group, while the flat phthalimide can partly rotate to minimize steric interactions with the former methyl and the auxiliary, also restricting the rotation of the pseudoanomeric C–C bond. Similar considerations may apply to a proton-coupled electron transfer process (b), in which a conjugated π-type radical in its ground state would adopt a similar planar geometry, poised to undergo stereoselective PCET from alcohol-bound Fe(II) species.
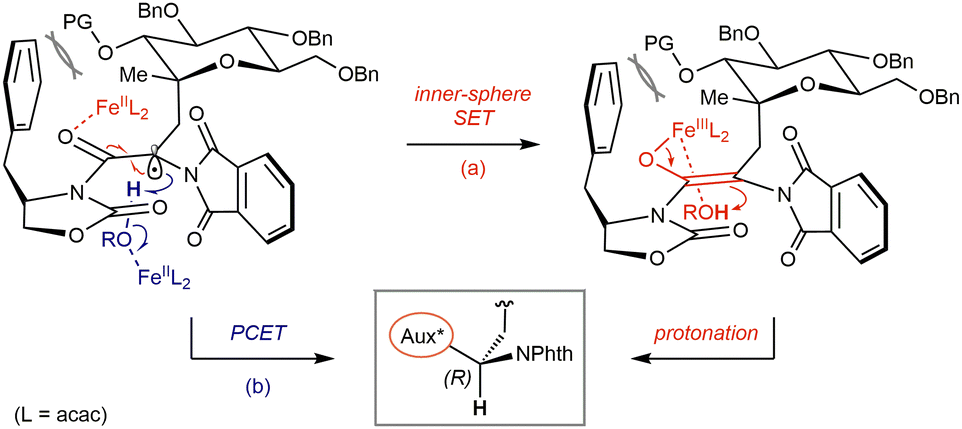 | ||
| Fig. 3 Proposed models for stereoselective termination via SET/protonation (a) or proton-coupled electron transfer (b) in the D-gluco series. | ||
In conclusion, a novel entry to rare C,C-glycosyl amino acids was established. Guided by a systematic study of substituents and auxiliary effects, high two-fold stereocontrol was achieved in the direct assembly of exo-glycals with dehydroalanine acceptors, representing a first case using the MHAT intermolecular coupling pathway. Further development of new catalytic stereocontrolled reactions of exo-glycals is underway and will be reported in due course.
NK gratefully acknowledges support from Unistra (IDEX grant W19RAT27). ML and DT thank the French Ministry of Research (MRT fellowships). Dr E. Wasielewski and M. Chessé are thanked for their support. Dr X. Bugaut is thanked for insightful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. Saxon and C. R. Bertozzi, Science, 2001, 291, 2357 CrossRef CAS PubMed; R. A. Dwek, Chem. Rev., 1996, 96, 683 CrossRef PubMed.
- K. T. Schjoldager, Y. Narimatsu, H. J. Joshi and H. Clausen, Nat. Rev. Mol. Cell Biol., 2020, 21, 729 CrossRef CAS PubMed; G. W. Hart, J. Biol. Chem., 2019, 294, 2211 CrossRef PubMed; A. Varki, Glycobiology, 2017, 27, 3 CrossRef PubMed.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309 CrossRef CAS PubMed; S. V. Moradi, W. M. Hussein, P. Varamini, P. Simerska and I. Toth, Chem. Sci., 2016, 7, 2492 RSC; C. R. Apostol, M. Hay and R. Polt, Peptides, 2020, 131, 170369 CrossRef PubMed.
- S. Minakata, S. Manabe, Y. Inai, M. Ikezaki, K. Nishitsuji, Y. Ito and Y. Ihara, Molecules, 2021, 26, 5258 CrossRef CAS PubMed.
- D. P. Gamblin, E. M. Scanlan and B. G. Davis, Chem. Rev., 2009, 109, 131 CrossRef CAS PubMed; S. A. W. Gruner, E. Locardi, E. Lohof and H. Kessler, Chem. Rev., 2002, 102, 491 CrossRef PubMed; T. P. Hogervorst, R. J. Eveline Li, L. Marino, S. C. M. Bruijns, N. J. Meeuwenoord, D. M. Filippov, H. S. Overkleeft, G. A. van der Marel, S. J. van Vliet, Y. van Kooyk and J. D. C. Codée, ACS Chem. Biol., 2020, 15, 728 CrossRef PubMed; U. Westerlind, Beilstein J. Org. Chem., 2012, 8, 804 CrossRef PubMed.
- For reviews see: Z. Yuan, C. Zhu, Y. Zhang and Y. Rao, Eur. J. Org. Chem., 2022, e202201036 CrossRef CAS; A. Dondoni and A. Marra, Chem. Rev., 2000, 100, 4395 CrossRef PubMed; H. Herzner, T. Reipen, M. Schultz and A. Kunz, Chem. Rev., 2000, 100, 4495 CrossRef PubMed.
- For selected recent as well as classical approaches see: Y.-H. Liu, Y.-N. Xia, T. Gulzar, B. Wei, H. Li, D. Zhu, Z. Hu, P. Xu and B. Yu, Nat. Commun., 2021, 12, 4924 CrossRef CAS PubMed; Y. Wei, Q. Wang and M. J. Koo, Angew. Chem., Int. Ed., 2023, 62, e202214247 CrossRef PubMed; Y.-N. Ding, N. Li, Y.-C. Huang, Y. An and Y.-M. Liang, Org. Lett., 2022, 24, 4519 CrossRef PubMed; R. Mao, S. Xi, S. Shah, M. J. Roy, A. John, J. P. Lingford, G. Gäde, N. E. Scott and E. D. Goddard-Borger, J. Am. Chem. Soc., 2021, 143, 12699 CrossRef PubMed; P. Ji, Y. Zhang, Y. Wei, H. Huang, W. Hu, P. A. Mariano and W. Wang, Org. Lett., 2019, 21, 3086 CrossRef PubMed; C. Palomo, M. Oiarbide, A. Landa, M. C. González-Rego, J. M. García, A. González, J. M. Odriozola, M. Martín-Pastor and A. Linden, J. Am. Chem. Soc., 2002, 124, 8637 CrossRef PubMed; A. Nuzzi, A. Massi and A. Dondoni, Org. Lett., 2008, 10, 4485 CrossRef PubMed; G. J. McGarvey, T. E. Benedum and F. W. Schmidtmann, Org. Lett., 2002, 21, 3591 CrossRef PubMed; D. E. Paterson, F. K. Griffin, M.-L. Alcaraz and R. J. K. Taylor, Eur. J. Org. Chem., 2002, 1323 CrossRef.
- For complementary methods of anomeric C–C bond formation in C-diglycoside synthesis see: D. C. Koester, E. Kriemen and D. B. Werz, Angew. Chem., Int. Ed., 2013, 52, 2985 CrossRef CAS PubMed; G. Hirai, T. Watanabe, K. Yamaguchi, T. Miyagi and M. Sodeoka, J. Am. Chem. Soc., 2007, 129, 15420 CrossRef PubMed; S. Palmier, B. Vauzeilles and J.-M. Beau, Org. Biomol. Chem., 2003, 1, 1097 RSC.
- J. Wu, N. Kaplaneris, S. Ni, F. Kaltenhaüser and L. Ackermann, Chem. Sci., 2020, 11, 6521 RSC; Y. Liu, Y. Wang, W. Dai, W. Huang, Y. Li and H. Liu, Angew. Chem., Int. Ed., 2020, 59, 3491 CrossRef CAS PubMed.
- C. Wang, R. Qi and Z. Xu, Chem. – Eur. J., 2023, 29, e202203689 CrossRef CAS PubMed ; for reviews on radical glycosylation see: Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281 CrossRef PubMed; L.-Y. Xu, N.-L. Fan and X.-G. Hu, Org. Biomol. Chem., 2020, 18, 5095 RSC; A. Chen, L. Xu, Z. Zhou, S. Zhao, T. Yang and F. Zhu, J. Carbohydr. Chem., 2021, 40, 361 CrossRef.
- L. Poletti, A. Massi, D. Ragno, F. Droghetti, M. Natali, C. De Risi, O. Bortolini and G. Di Carmine, Org. Lett., 2023, 25, 4862 CrossRef CAS PubMed ; for seminal work see: R. S. Andrews, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2010, 49, 7274 CrossRef PubMed.
- A. Chen, S. Zhao, Y. Han, Z. Zhou, B. Yang, L.-G. Xie, M. A. Walczak and F. Zhu, Chem. Sci., 2023, 14, 7569 RSC.
- H. Kessler, V. Wittmann, M. Köck and M. Kottenhahn, Angew. Chem., Int. Ed. Engl., 1992, 31, 902 CrossRef.
- G. A. Adamson, A. L. J. Beckwith and C. L. L. Chai, Aust. J. Chem., 2004, 57, 629 CrossRef CAS; J. R. Axon and A. L. J. Beckwith, J. Chem. Soc., Chem. Commun., 1995, 549 RSC.
- D. Tardieu, M. Desnoyers, C. Laye, D. Hazelard, N. Kern and P. Compain, Org. Lett., 2019, 21, 7262 CrossRef CAS PubMed; H. Liu, A. Laporte, D. Tardieu, D. Hazelard and P. Compain, J. Org. Chem., 2022, 87, 13178 CrossRef PubMed.
- J. C. Lo, D. Kim, C.-M. Pan, J. T. Edwards, Y. Yabe, J. Gui, T. Qin, S. Gutiérrez, J. Giacoboni, M. W. Smith, P. L. Holland and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 2484 CrossRef CAS PubMed; J. C. Lo, J. Gui, Y. Yabe, C.-M. Pan and P. S. Baran, Nature, 2014, 516, 343 CrossRef PubMed.
- J. L. M. Matos, S. A. Green and R. A. Shenvi, Org. React., 2019, 100, 383 Search PubMed; S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912 CrossRef CAS PubMed; S. A. Green, S. W. M. Crossley, J. L. M. Matos, S. Vásquez-Céspedes, S. L. Shevick and R. A. Shenvi, Acc. Chem. Res., 2018, 51, 2628 CrossRef PubMed; J. Wu and Z. Ma, Org. Chem. Front., 2021, 8, 7050 RSC.
- M. Richard, A.-S. Felten, C. Didierjean, M. Ruiz-Lopez, Y. Chapleur and N. Pellegrini-Moïse, Eur. J. Org. Chem., 2014, 7364 CrossRef CAS; F. Schweizer and O. Hindsgaul, Carbohydr. Res., 2006, 341, 1730 CrossRef PubMed; S. Bera, B. Chatterjee and D. Mondal, RSC Adv., 2016, 6, 77212 RSC.
- V. A. Larionov, N. V. Stoletova, V. I. Kovalev, A. F. Smol’yakov, T. F. Savel’yeva and V. I. Maleev, Org. Chem. Front., 2019, 6, 1094 RSC; B. Wu and R. Zhu, ACS Catal., 2020, 10, 510 CrossRef CAS.
- While additions forming captodative radicals witness increased exothermicity and lowered activation barriers, electron affinity is no more correlated to reaction enthalpy and thus polar stabilization decreases. See: R. Arnaud, N. Bugaut, V. Vetere and V. Barone, J. Am. Chem. Soc., 1998, 120, 5733 CrossRef CAS.
- H. Zhang, H. Li, H. Yang and H. Fu, Org. Lett., 2016, 18, 3362 CrossRef CAS PubMed.
- Presence of excess acceptor alongside the nontrivial stabilization conferred by the imide makes this noteworthy, see: S. Han and S. Z. Zard, Org. Lett., 2014, 20, 5386 CrossRef PubMed ; Lowering the 1a
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2 ratio to 1:1.5 led to partial conversion of 1a, while increasing it to 1:3 resulted in lower yields.
:2 ratio to 1:1.5 led to partial conversion of 1a, while increasing it to 1:3 resulted in lower yields. - M. E. Jung and P. Koch, Tetrahedron Lett., 2011, 52, 6051 CrossRef CAS.
- M. P. Sibi and J. Ji, Angew. Chem., Int. Ed. Engl., 1996, 35, 190 CrossRef CAS.
- J. R. Gage and D. A. Evans, Org. Synth., 1990, 68, 77 CrossRef CAS.
- D. Kim, S. M. W. Rahaman, B. Q. Mercado, R. Poli and P. L. Holland, J. Am. Chem. Soc., 2019, 141, 7473 CrossRef CAS PubMed.
- T. Herrmann, L. L. Smith and A. Zakarian, J. Am. Chem. Soc., 2012, 134, 6976 CrossRef PubMed; M. Pérez-Palau, N. Sanosa, P. Romea, F. Urpí, R. López, E. Gómez-Bengoa and M. Font-Bardia, Org. Lett., 2021, 23, 8852 CrossRef PubMed.
- C. Nativi, F. Papi and S. Roelens, Chem. Commun., 2019, 55, 7729 RSC; C. H. Röhrig, M. Takhi and R. R. Schmidt, Synlett, 2001, 1170 CrossRef CAS; D. Urban, T. Skrydstrup, C. Riche, A. Chiaroni and J.-M. Beau, Chem. Commun., 1996, 1883 RSC.
- Y. Leshch, A. Jacobsen, J. Thimm and J. Thiem, Org. Lett., 2013, 15, 4948 CrossRef CAS PubMed.
- T. F. Herpin, W. B. Motherwell, B. P. Roberts, S. Roland and J.-M. Weibel, Tetrahedron, 1997, 53, 15085 CrossRef CAS.
- H. T. Dao, C. Li, Q. Michaudel, B. D. Maxwell and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 8046 CrossRef CAS PubMed; M. Saladrigas, J. Bonjoch and B. Bradshaw, Org. Lett., 2020, 22, 684 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details including synthesis and characterization. See DOI: https://doi.org/10.1039/d3cc06249j |
| ‡ Due to the varying polarities of adducts, from this stage the auxiliary was cleaved only if required for an easier purification. |
| This journal is © The Royal Society of Chemistry 2024 |