
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light activated coumarin photocages: an interplay between radical and organobase generation to govern thiol–ene polymerizations†
Meghan T.
Kiker‡
 ,
Ain
Uddin‡
,
Lynn M.
Stevens
,
Kun-You
Chung
,
Pengtao
Lu
and
Zachariah A.
Page
*
,
Ain
Uddin‡
,
Lynn M.
Stevens
,
Kun-You
Chung
,
Pengtao
Lu
and
Zachariah A.
Page
*
Department of Chemistry, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: zpage@utexas.edu
First published on 2nd August 2023
Abstract
Photobase generators (PBGs) are an attractive tool for the latent and spatially governed formation of polymer networks with uniform topologies due to the anionic step-growth mechanistic pathway they unlock. Despite the significant advances made in PBG frameworks, utility in rapid, visible light driven polymer formation and associated structure–reactivity relationships remain scarce. Herein, five coumarinylmethyl PBGs bearing caged tetramethylguanidine (TMG) were synthesized and systematically examined for inducing thiol–ene polymerizations, while benchmarking against the classic ortho-nitrobenzyl (oNB) PBG framework. Quantification of photopolymerization kinetics and bond scission quantum yields with real-time Fourier transform infrared and steady-state UV-vis absorption spectroscopies revealed an increase in the disparity between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and S–H conversion for derivatives halogenated at the 3-position. Alternatively, incorporation of a π-extended styryl moiety at the same position decreased the conversion gap and enabled uncaging with a blue LED (470 nm). This gap was attributed to the concurrent activation of radical chain-growth and base-catalyzed step-growth mechanisms, which was tempered through the (sub-)stoichiometric addition of tetramethylpiperidinyloxy (TEMPO). These findings paint a detailed picture of PBGs for thiol–ene polymerizations that will inform the selection and optimization of future light-activated catalysts and enable advanced manufacturing of tailored soft materials.
C and S–H conversion for derivatives halogenated at the 3-position. Alternatively, incorporation of a π-extended styryl moiety at the same position decreased the conversion gap and enabled uncaging with a blue LED (470 nm). This gap was attributed to the concurrent activation of radical chain-growth and base-catalyzed step-growth mechanisms, which was tempered through the (sub-)stoichiometric addition of tetramethylpiperidinyloxy (TEMPO). These findings paint a detailed picture of PBGs for thiol–ene polymerizations that will inform the selection and optimization of future light-activated catalysts and enable advanced manufacturing of tailored soft materials.
Introduction
Light-triggered polymerizations offer spatiotemporal control that has enabled transformative manufacturing technologies, including (stereo)lithography for microelectronics and dentistry.1,2 Mechanistically, photoinduced homolytic bond scission to generate radicals that initiate a chain-growth polymerization of acrylic resins has dominated the industry. This arises in-part from the efficient photolysis and correspondingly rapid curing (i.e., conversion from liquid monomeric resin to solid polymeric network in ∼seconds to minutes). However, the uncontrolled free radical chain-growth process results in a non-ideal, heterogenous polymer network that is often more brittle and stiff relative to analogous ideal uniform architectures achieved by anionic step-growth polymerizations.3,4 Photobase generators (PBGs) have emerged as a means to address this challenge, whereby light-triggered uncaging (i.e., release) of a base catalyzes a step-growth polymerization to form ideal networks (Fig. 1A).5–7 To this end, PBGs with carbamate linked aliphatic amines and tetramethylguanidine (TMG) have been successfully applied to polymer formation via base-catalyzed thiol-Michael8–12 and alcohol-13 or thiol-isocyanate14 addition. Still, challenges remain in both the speed of polymer formation and contemporary reliance on high energy ultraviolet (UV) light to trigger base formation, necessitating a systematic examination of PBGs for polymerization.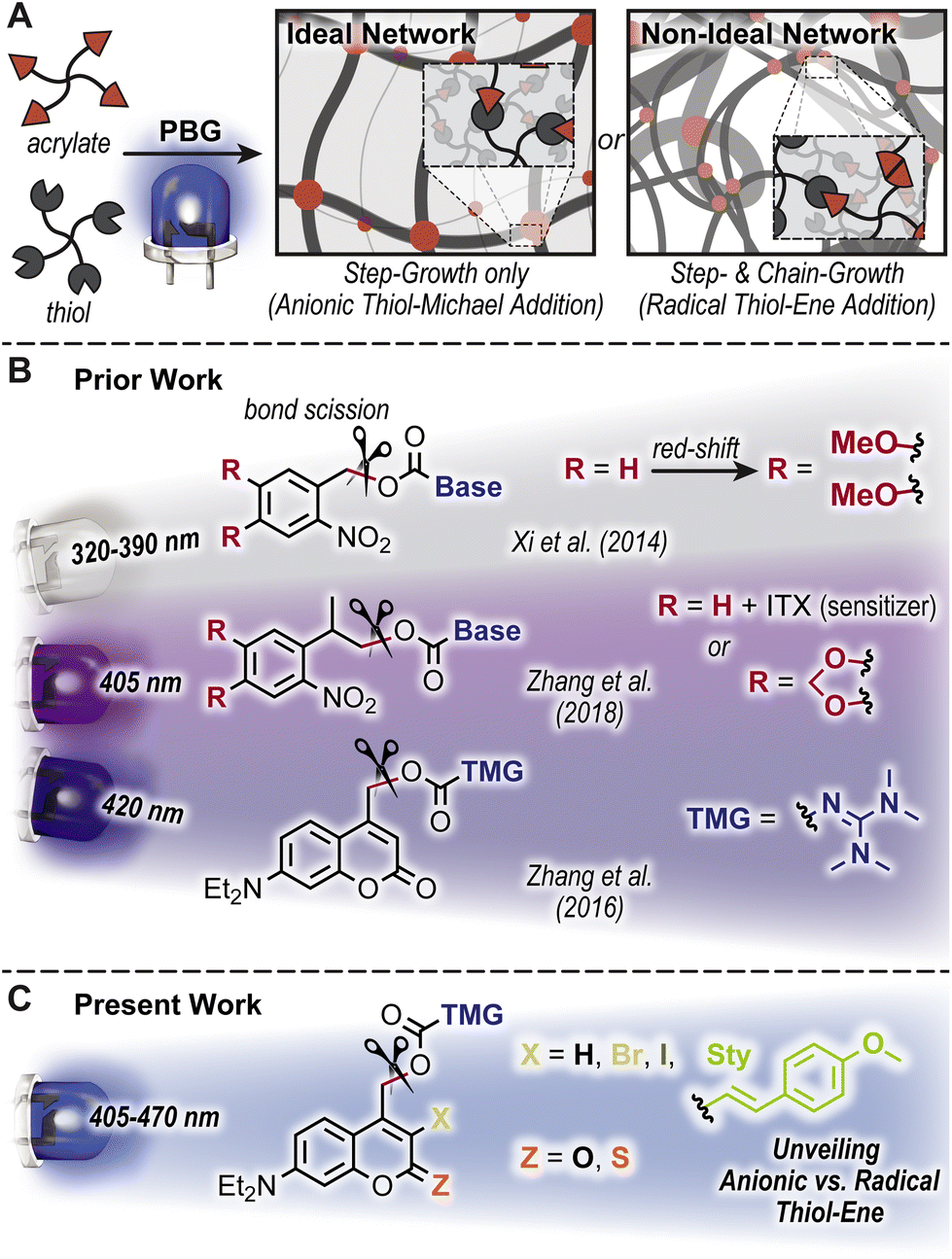 | ||
| Fig. 1 Overview of thiol-acrylate reactions activated by photobase generators (PBGs). (A) Formation of ideal (i.e., uniform) and non-ideal network topologies via anionic step-growth and radical chain-growth pathways, respectively. (B) Prior PBGs used to form thiol-acrylate networks, from UV-to-violet light activation of ortho-nitrobenzyl and coumarinylmethyl scaffolds. (C) Present systematic study on tetramethylguanidine (TMG) derivatized coumarin PBGs and their influence on the mechanism of polymer network formation. | ||
The desirable features offered by visible-light activated (wavelength, λ > 400 nm) PBGs include reduced scattering,15 increased penetration depth,16 greater functional group tolerance, biocompatibility,15,17,18 and potential for wavelength-selective multimaterial fabrication.19–25 These features have inspired the design and utility of novel PBGs in polymer synthesis. The two predominant classes of visible-light activated PBGs used in polymer formation include ortho-nitrobenzyl (oNB) and coumarinylmethyl scaffolds. General strategies to red-shift absorption include the introduction of electron donating substituents and halogens, as well as extending π-conjugation. For example, Xi et al. demonstrated that 3,4-dimethoxy substitution of an oNB PBG increased molar absorptivity in the long-wave UV region, which in-turn raised uncaging yields useful for thiol-Michael polymerization (λmax = 368 nm) (Fig. 1B).9 Alternatively, Zhang et al. demonstrated that uncaging with a violet LED (λmax = 405 nm) was possible via sensitization of an analogous oNB PBG using isopropylthioxanthone (ITX) or directly with an oNB PBG containing a cyclic 3,4-methylenedioxy substituent (Fig. 1B).11 Furthermore, [7-(diethylamino)coumarin-4-yl]methyl (DEACM) was recently developed as a visible light reactive photocage with applications in photo-polymerization and -crosslinking, drug release monitoring, and tissue scaffolds.26–31 For example, Zhang et al. introduced DEACM as a scaffold for caging TMG capable of catalyzing thiol-Michael polymerizations upon exposure to violet LEDs (λmax = 420 nm) (Fig. 1B).12 The proposed uncaging mechanism of DEACM consists of heterolytic bond scission at the carbamate, followed by decarboxylation and release of TMG.12 However, recent reports demonstrate that heterolysis can be accompanied by radical formation,32–36 which results in a combined step- and chain-growth polymerization, and mitigates the formation of an ideal network architecture. As this had not been carefully examined in these PBG-catalyzed polymerizations, it warranted a closer look.
Herein, five coumarin-TMG derivatives are synthesized and systematically characterized with relation to polymer formation (Fig. 1C). Polymerization kinetics and photolysis quantum yields were quantitatively characterized using a combination of real-time Fourier transform infrared (RT-FTIR) and UV-vis absorption spectroscopies, which unveiled radical contributions to the thiol–ene reactions. Finally, 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) was employed as a radical mediator to bias polymerizations towards base-catalyzed thiol-Michael addition, which provided greater insight into the contributions of step- vs. chain-growth mechanisms that result from using different PBGs.
Results and discussion
Coumarinylmethyl was selected as the photocage core for systematic examination owing to its strong absorption in the visible region (λ ≈ 400–470 nm) and tunable photophysical properties. Five TMG-containing photocage derivatives were synthesized (Fig. 2A and ESI† for details), which were inspired by recent examples where halogenation31,37,38 and π-extension with styryl moieties39 at the 3-position and 2-thiocarbonyl substitution20,39 resulted in red-shifted absorption and increased photolysis yields. For clarity, the following coumarin-TMG (CTMG) photocage naming system is used throughout: Z–X-CTMG, where Z (2-position) = O or S, and X (3-position) = H, Br, I, or Sty (styryl). Notably, O–H-CTMG (= DEACM)12 and oNB-TMG served as benchmarks for the remaining four novel CTMG derivatives. Four monomers, 1,6-hexanediol diacrylate (HDDA) and tetra(ethylene glycol) diacrylate (TEGDA) and 1,6-hexanedithiol and 3,6-dioxa-1,8-octanedithiol (DODT) served as readily available diene's and dithiols, respectively (Fig. 2A, right).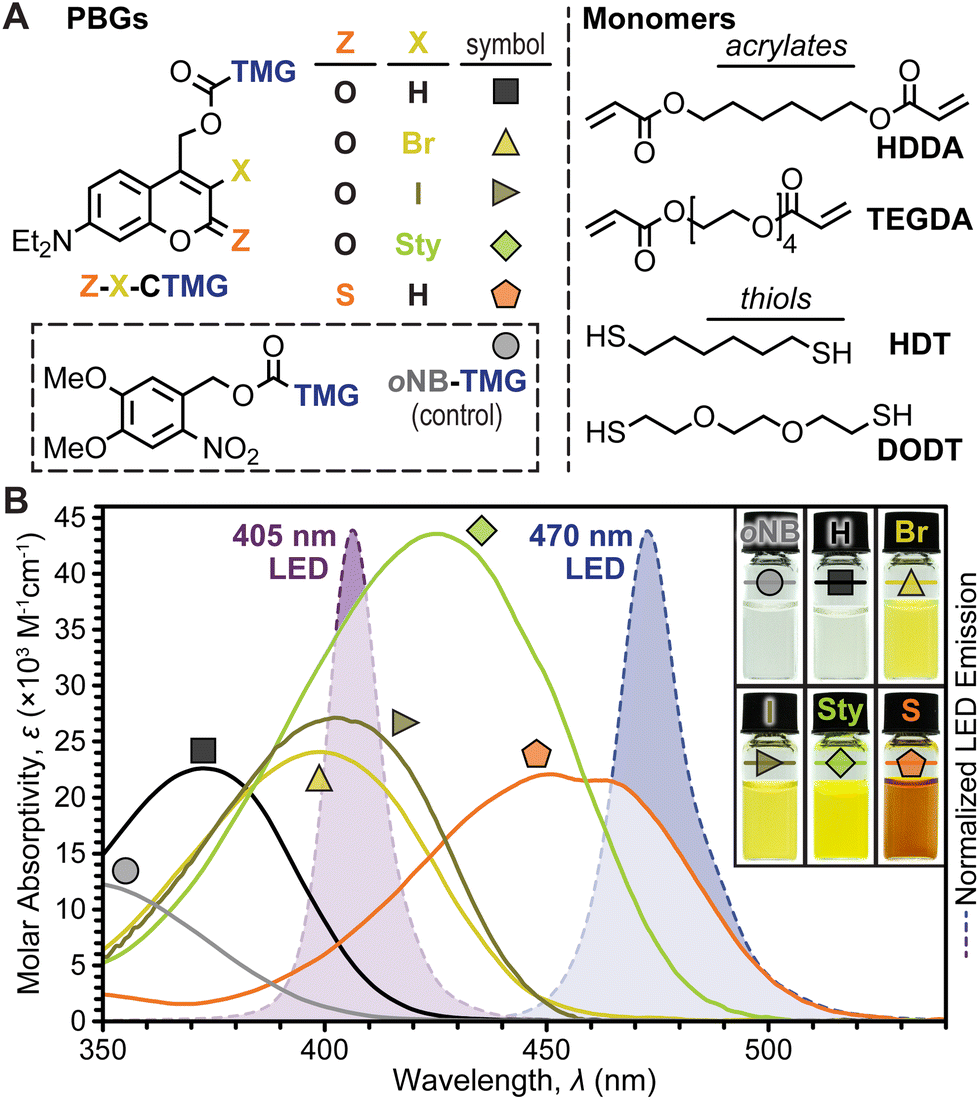 | ||
| Fig. 2 Resin composition and absorption profiles. (A) Chemical structures for PBGs (left) and monomers (right). (B) Molar absorptivity of PBGs overlaid onto LED emission profiles. Inset: photographs of PBGs in solution (4 mM in CH3CN). | ||
To quantify the uncaging efficiency, it was first necessary to characterize molar absorptivity (ε) for each photocage derivative. Using UV-vis absorption spectroscopy, samples were measured in dilute solution (10−5 M in CH3CN), providing peak wavelengths (λmax) of 349, 372, 499, 402, 425, and 454 nm for oNB-TMG and O–H-, O–Br-, O–I-, O-Sty-, and S–H-CTMG, respectively, along with corresponding εmax values of 2.8 ± 0.2, 22 ± 2, 24 ± 2, 27 ± 2, 43 ± 3, and 22 ± 2 × 103 M−1 cm−1 (Fig. 2B and Table S1†). Fluorescence spectra and fluorescence quantum yields (Φf) were additionally characterized to provide insight into the influence of chemical variations on excited state properties for the different derivatives (Table S1, Fig. S1, S2, and eqn (SE1)†). Emission maxima were 463, 494, and 516 nm for O–H-, O–Br-, and O-Sty-CTMG, respectively, along with corresponding Φf values of 0.30, 0.23, and 0.41. The O–I- and S–H-CTMG derivatives were non-emissive, indicating the presence of alternative non-radiative decay pathways, such as intersystem crossing to long-lived spin-triplet excited states, which can potentially increase photouncaging quantum yield (Φun), as described later.
To showcase relative LED absorption for each photocage derivative, spectral profiles were overlaid with the violet (λmax = 405 nm) and blue (λmax = 470 nm) LED emission profiles used herein (Fig. 2B). The relative number of photons absorbed by each photocage was quantified by integrating the overlap between their absorption and LED emission profiles (intensity, I = 100 mW cm−2), providing the following values normalized to the strongest absorber (O-Sty-CTMG, 405 nm LED): 0.01, 0.17, 0.66, 0.75, 1.00, and 0.33 for the 405 nm LED for oNB-TMG and O–H-, O–Br-, O–I-, O-Sty-, and S–H-CTMG, respectively, along with corresponding values of <0.01, <0.01, 0.02, 0.04, 0.38, and 0.86 for the 470 nm LED (Fig. 2B and Table S2†). Therefore, if Φun values were equivalent, the fastest TMG release, and thus highest polymerization rate would occur from using O-Sty-CTMG at 405 nm and S–H-CTMG at 470 nm, given their largest degree of absorption overlap with the respective LED emission profile.
Photopolymerization rates were next characterized under ambient conditions (i.e., room temperature and in air) using real-time Fourier transform infrared (RT-FTIR) spectroscopy during LED exposure (Fig. 3A). Photopolymerizable resins were prepared using equimolar quantities of thiol and ene monomers, along with each photocage at a concentration of 0.4 mol% (14.5 mM) relative to total monomer (photocage concentration optimization provided in Fig. S3†). Notably, due to differences in purity between commercial dithiol and diacrylate monomers, a slight mismatch in initial SH-to-ene ratios was anticipated, with an excess of SH being <10 mol% (see ESI† for detail). Resins with O–H-CTMG were loaded between glass slides separated by 100 μm shims and irradiated with a violet or blue LED at a fixed intensity of 100 mW cm−2 for all experiments, unless otherwise noted. Using RT-FTIR, conversion (ρ) of monomer to polymer was elucidated by tracking the disappearance of the CC vinylic overtone at ∼6150 cm−1 and S–H stretch at ∼2580 cm−1.40,41
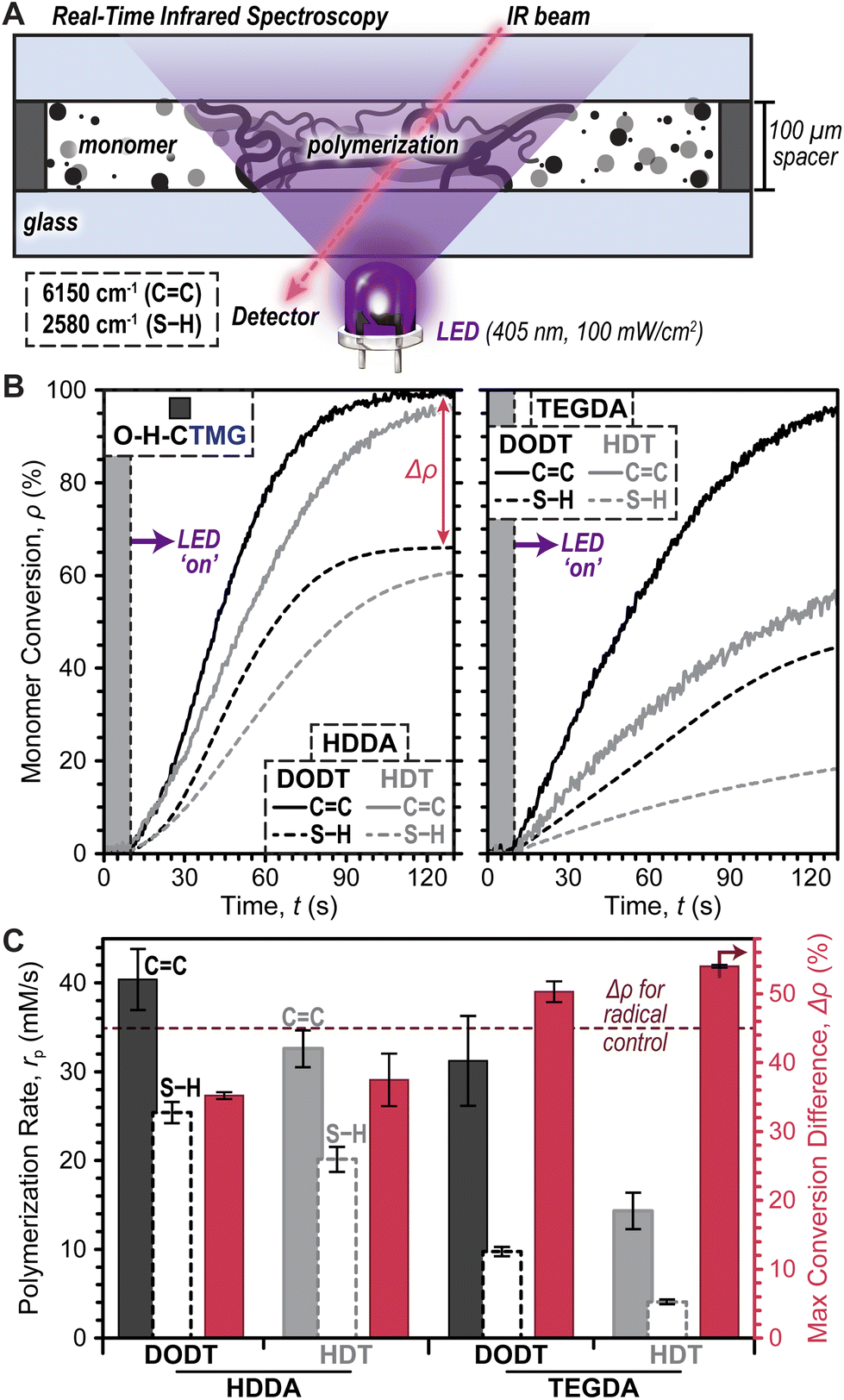 | ||
| Fig. 3 Photopolymerization resin optimization. (A) Illustration of the transmission real-time Fourier transform infrared (RT-FTIR) spectroscopy setup used to monitor monomer to polymer conversion (ρ) during LED irradiation. (B) Photopolymerization kinetics as a function of diacrylate and dithiol composition. (C) Maximum polymerization rate (rp) and difference in maximum CC and S–H conversion (Δρ) for different resin compositions. Dashed line represents Δρ for the HDDA–DODT resin cured with bisacylphosphine oxide as a radical photoinitiator. Averages were calculated from a minimum of three samples and error bars represent ±1 standard deviation from the mean. | ||
To identify an optimal resin for further studies, the effect of diacrylate and dithiol composition on polymerization rate (rp) and difference in maximum CC and S–H conversion (Δρ) (SE2) was characterized using combinations of HDDA or TEGDA (diacrylates) with HDT or DODT (dithiols) and O–H-CTMG as the photocage (Fig. 3B). Data was collected for 10 seconds prior to turning the violet LED ‘on’, followed by 7 minutes (420 seconds) of irradiation to reach maximum CC and S–H conversion. Results from the first 2 minutes (120 seconds) of irradiation are shown in Fig. 3B for clarity (Fig. S4 and Table S3† for full data). In all cases excellent temporal control was demonstrated, with no observable conversion in the dark, followed by immediate conversion (i.e., no inhibition) upon turning the violet LED ‘on’. It was empirically observed that HDDA resulted in faster polymerizations relative to TEGDA (Fig. 3B, left), while DODT outperformed HDT (Fig. 3B, right).
Quantification of photopolymerization kinetics for resins comprising HDDA and DODT revealed an rp of 40 ± 3 mM s−1 (based on CC conversion), which was ∼1.2–2.8× larger than the other combinations. Additionally, this resin resulted in the highest maximum CC (∼100%) and S–H (∼65%) conversion, in-turn providing the smallest Δρ equal to 35.2 ± 0.5% (Fig. 3C and Table S3†). For the case of stoichiometrically balanced CC and S–H functionality, a purely step-growth mechanism would result in a Δρ = 0%. Thus, the large Δρ observed here suggested that ene–ene chain-growth mechanisms were occurring in these photopolymerizations. As an additional control expected to provide a maximum amount of ene–ene addition, the same HDDA–DODT resin was prepared using a classic radical photoinitiator, phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide (BAPO, 0.4mol%), in place of O–H-CTMG. Irradiation of this resin with the same 405 nm LED (0.2 mW cm−2) resulted in a Δρ of 44.8 ± 1.2% (Fig. S5 and Table S4†). The lower Δρ value for resins containing O–H-CTMG relative to those with BAPO suggests that anionic thiol-Michael addition (step-growth) competes with the radical ene–ene (chain-growth) polymerization. To maximize polymerization rate and bias the reactions towards step-growth addition, future experiments utilized HDDA and DODT as the diacrylate and dithiol, respectively.
Comparisons between all six photocages was accomplished next under fixed conditions; HDDA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DODT (1:1), 0.4 mol% photocage, and 100 mW cm−2 405 nm LED exposure (Fig. 4A). Both rp based on CC and S–H conversion along with Δρ were quantified for each photosystem. In general, O–H-, O–Br-, and O–I-CTMG (Fig. 4A, left) were considerably faster than the longer wavelength absorbing O-Sty- and S–H-CTMG, along with the oNB-TMG control (Fig. 4A, right). Furthermore, the rp based on CC conversion followed the same general trends as that observed for S–H conversion (i.e., as one increased so did the other). Quantification of rp gave values (based on CC conversion) of 40 ± 3, 162 ± 16 and 77 ± 8 mM s−1 in the halogenated series of increasing atomic number from O–H- to O–Br- to O–I-CTMG (Fig. 4B). However, Δρ also increased correspondingly from 35.2 ± 0.5% (O–H-) to 50.3 ± 1.6% (O–Br-) and 45.0 ± 2.7% (O–I-), respectively, which suggests additional ene–ene addition (i.e., chain-growth) reactions occur with halogenated photocages. In contrast to these three derivatives, the O-Sty- and S–H-CTMG, along with the oNB-TMG control, had considerably smaller rp values (based on CC conversion) of 28 ± 1, 4 ± 2 and 17 ± 1 mM s−1, respectively (Table S5†). However, Δρ values were also notably smaller, at ∼12% for both O-Sty-CTMG and oNB-TMG, which suggests that these derivatives have a larger extent of stoichiometric thiol–ene step-growth addition relative to the previous three photocages. This is hypothesized to arise from the intramolecular uncaging mechanism for both the O-Sty-CTMG39 and oNB-TMG PBGs, which may foster heterolytic bond scission via stabilization of the (partially) charged transition states.
:DODT (1:1), 0.4 mol% photocage, and 100 mW cm−2 405 nm LED exposure (Fig. 4A). Both rp based on CC and S–H conversion along with Δρ were quantified for each photosystem. In general, O–H-, O–Br-, and O–I-CTMG (Fig. 4A, left) were considerably faster than the longer wavelength absorbing O-Sty- and S–H-CTMG, along with the oNB-TMG control (Fig. 4A, right). Furthermore, the rp based on CC conversion followed the same general trends as that observed for S–H conversion (i.e., as one increased so did the other). Quantification of rp gave values (based on CC conversion) of 40 ± 3, 162 ± 16 and 77 ± 8 mM s−1 in the halogenated series of increasing atomic number from O–H- to O–Br- to O–I-CTMG (Fig. 4B). However, Δρ also increased correspondingly from 35.2 ± 0.5% (O–H-) to 50.3 ± 1.6% (O–Br-) and 45.0 ± 2.7% (O–I-), respectively, which suggests additional ene–ene addition (i.e., chain-growth) reactions occur with halogenated photocages. In contrast to these three derivatives, the O-Sty- and S–H-CTMG, along with the oNB-TMG control, had considerably smaller rp values (based on CC conversion) of 28 ± 1, 4 ± 2 and 17 ± 1 mM s−1, respectively (Table S5†). However, Δρ values were also notably smaller, at ∼12% for both O-Sty-CTMG and oNB-TMG, which suggests that these derivatives have a larger extent of stoichiometric thiol–ene step-growth addition relative to the previous three photocages. This is hypothesized to arise from the intramolecular uncaging mechanism for both the O-Sty-CTMG39 and oNB-TMG PBGs, which may foster heterolytic bond scission via stabilization of the (partially) charged transition states.
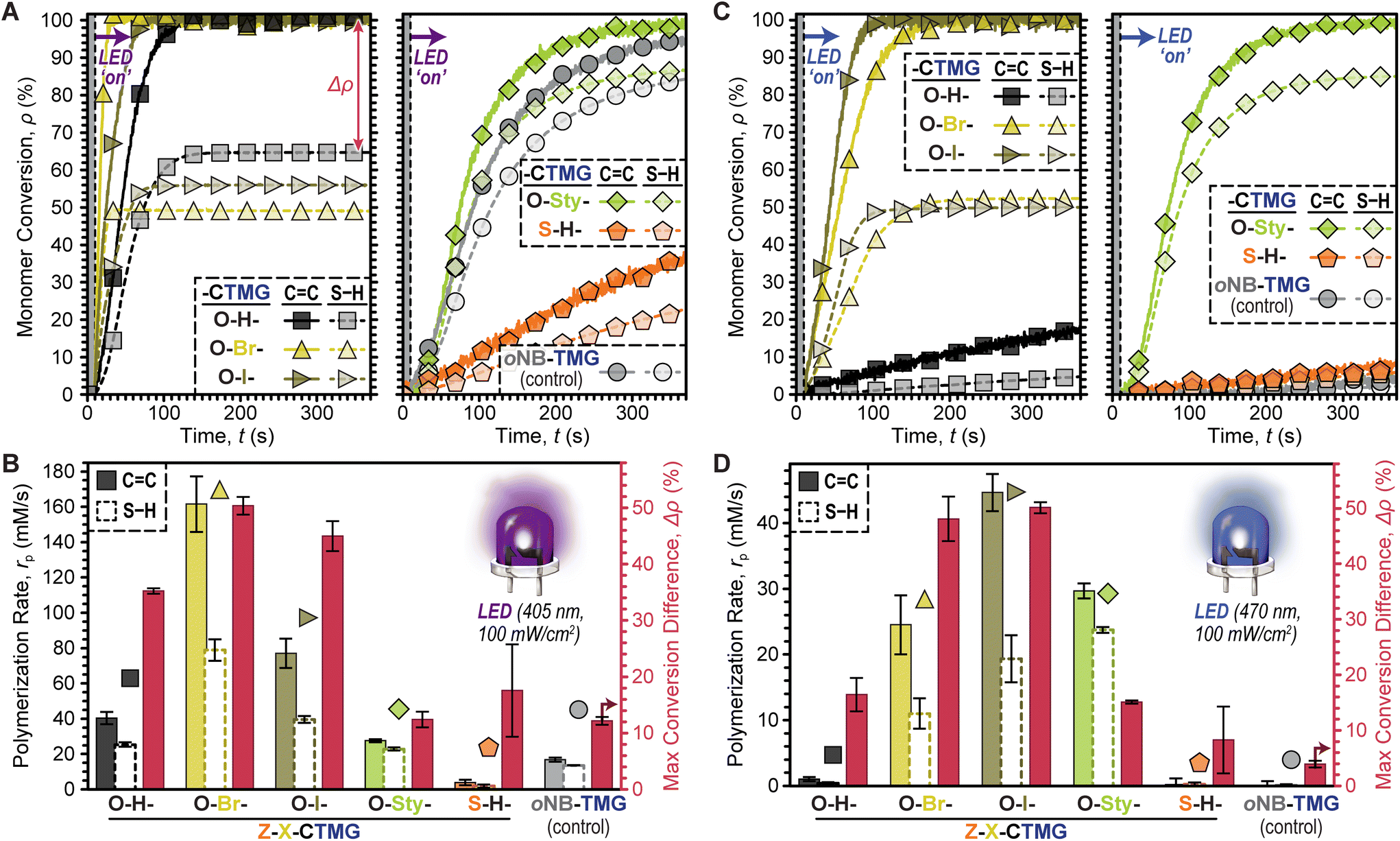 | ||
| Fig. 4 Survey of photopolymerization kinetics and relative conversions for HDDA + DODT resins with six different photocages. (A/B) Results from exposure to a 405 nm (violet) LED, 100 mW cm−2. (C/D) Results from exposure to a 470 nm (blue) LED, 100 mW cm−2. LEDs were turned ‘on’ 10 seconds into each measurement, showing excellent temporal control given little to no inhibition period. Averages were calculated from a minimum of three samples and error bars represent ±1 standard deviation from the mean. | ||
To further probe the composition–reactivity relationships the same six resins were subjected to blue LED exposure (470 nm, 100 mW cm−2) under otherwise identical conditions (Fig. 4C & D). The most notable result was that all samples polymerized more slowly under blue light as compared to violet light, except for those containing O-Sty-CTMG. Additionally, samples with O–Br- and O–I-CTMG showed rapid polymerizations with both violet and blue LED irradiation (max conversion in a few minutes). Specifically, rp values (based on CC conversion) were 27 ± 5, 43 ± 3, and 28 ± 1 for samples containing O–Br-, O–I-, and O-Sty-CTMG (Table S6†). However, samples with O-Sty-CTMG resulted in a low Δρ value of 15.1 ± 0.2% as compared to the ∼50% values observed for samples containing either O–Br- or O–I-CTMG control. Furthermore, despite its relatively strong absorption of blue light, samples with S–H-CTMG were slow to polymerize. Overall, these results suggest that O-Sty-CTMG is a good candidate to bias thiol-acrylate resins towards step-growth polymerizations, while also enabling the use of longer wavelength (i.e., 470 nm) LEDs relative to the traditional CTMG and oNB-TMG derivatives.
To better understand the driving forces behind the measured polymerization kinetics, uncaging quantum yields (Φun), or the number of “uncaging” events per photon absorbed, were estimated using RT-FTIR in an attenuated total reflectance (ATR) mode (Fig. 5A, Fig. S6–S12, Table S7, and eqn (SE3) and (SE4)†). This was accomplished by dissolving each photocage in DODT (0.5 mol%) and monitoring the carbamate decomposition (∼1710 cm−1)42 during controlled LED irradiation. Notably, this allows for quantification of C–O bond scission, but does not directly track release of TMG, nor does it indicate whether scission occurs in a homolytic or heterolytic fashion. Thus, values will be referred to as scission quantum yields (Φs) for clarity. Overall, the Φs values were small (<0.5%), but in general they tracked with rp (i.e., higher Φs corresponded to a higher rp) (Fig. 5B). Specific Φs values were 0.06 ± 0.01%, 0.29 ± 0.03%, 0.35 ± 0.05%, 0.04 ± 0.01%, and 0.04 ± 0.01% for O–H-, O–Br-, O–I-, O-Sty-, and S–H-CTMG, respectively. Notably, Φs for the CTMG derivatives were ∼1–2 orders of magnitude smaller than the oNB-TMG control (Φs = 5.7 ± 0.3%), which was determined by monitoring the C–N stretch of the carbamate (∼1550 cm−1),43 along with directly measuring TMG release by HPLC (Fig. S13 and S14†), with the latter made possible by the relatively large TMG yield. Despite the disparity in Φs, similar rp values under violet LED exposure were measured for samples containing O-Sty-CTMG relative to oNB-TMG (Fig. 4A & B), which was attributed to the ∼100-fold increase in violet LED photons absorbed by the former (Fig. 2B and Table S7†). Thus, by leveraging increased photon absorption and tailoring composition to enhance Φun, opportunities exist to improve the efficiency of visible light photouncaging of organobases for rapid light-induced polymerizations towards ideal (uniform) networks.
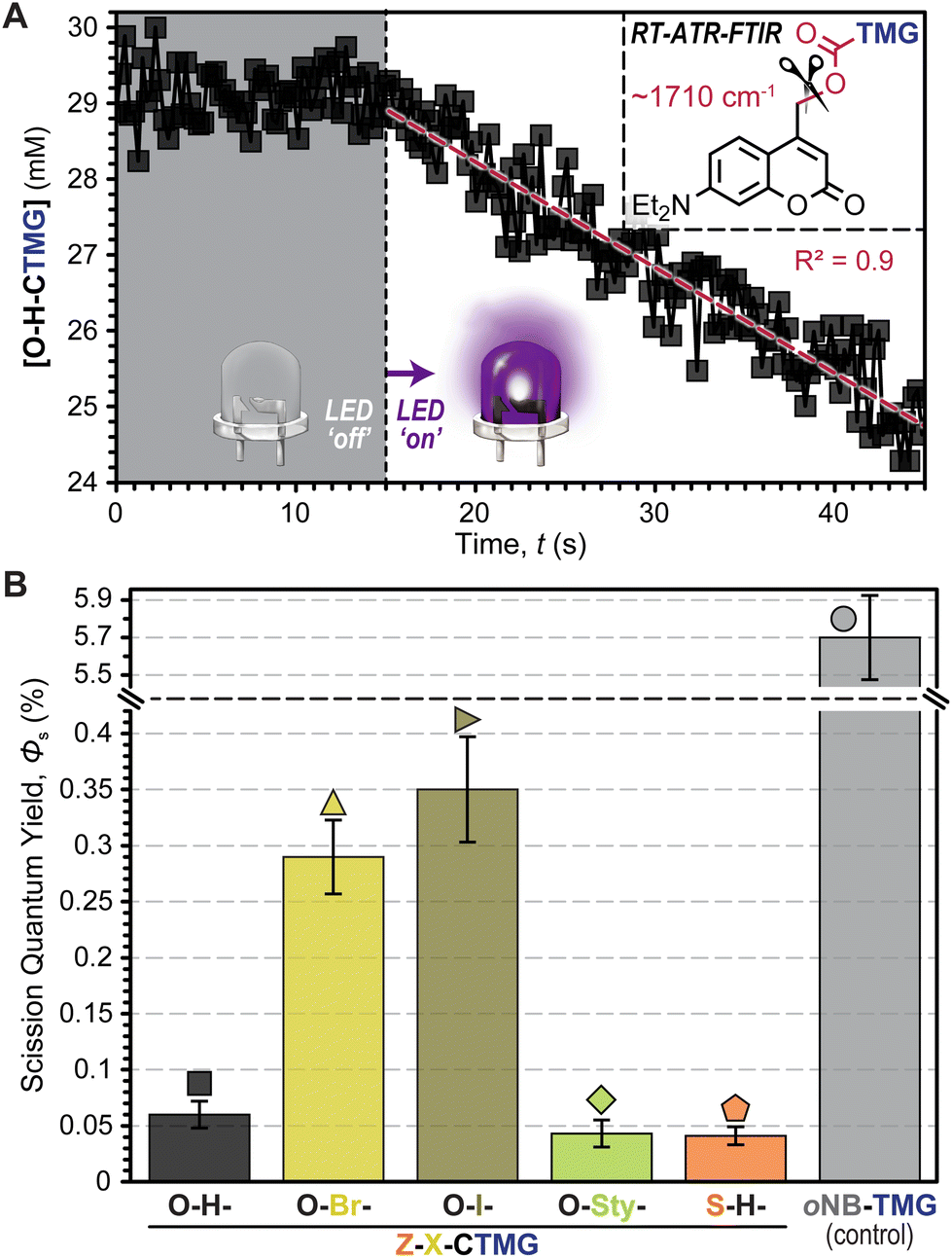 | ||
| Fig. 5 Estimating photouncaging quantum yields. (A) Carbamate bond scission was tracked and quantified using RT-FTIR in an attenuated total reflectance (ATR) configuration during violet LED irradiation, using 0.5 mol% photocages in DODT. (B) Bar graph showing bond scission quantum yields (Φs) for each derivative. Dashed line represents break in data for clarity. Averages were calculated from a minimum of three samples and error bars represent ±1 standard deviation from the mean. | ||
At this stage it was hypothesized that the CTMG photocages were undergoing a mixture of heterolytic and homolytic bond scission at the 4-methyl carbamate position, resulting in the formation of ionic and radical species, respectively (Fig. 6A and Scheme S7†). In-turn, this would lead to a combination of anionic (thiol-Michael) step-growth polymerization and radical thiol–ene and ene–ene step- and chain-growth polymerization, respectively. Previous reports suggest that radical formation in O-H-coumarinylmethyl photocages occurs via intersystem crossing (ISC) post-bond homolysis.32–36 However, given the relatively slow kinetics of bond scission, it was also postulated that homolysis may occur directly, particularly in the event that ISC to long lived spin-triplet excited states is competitively fast. To test this conjecture, excited state dynamics were probed using time-correlated single photon counting (TCSPC) fluorescence spectroscopy (Fig. S15–S17 and Table S8†) and nanosecond transient absorption spectroscopy (Fig. S18 and S19†). Overall, singlet excited state lifetimes in degassed CH3CN ranged from 2.3–4.0 ns for O–Br- and O–H-CTMG, respectively. Low fluorescence signal for O–I-CTMG and S–H-CTMG precluded TCSPC measurements, however these derivatives showed the presence of a long-lived (∼2–3 μs) triplet–triplet absorption band at 450 nm (ref. 44) (Figs S18 and S19†), indicating that ISC prior to bond scission occurs and could lead to direct radical formation via bond homolysis.
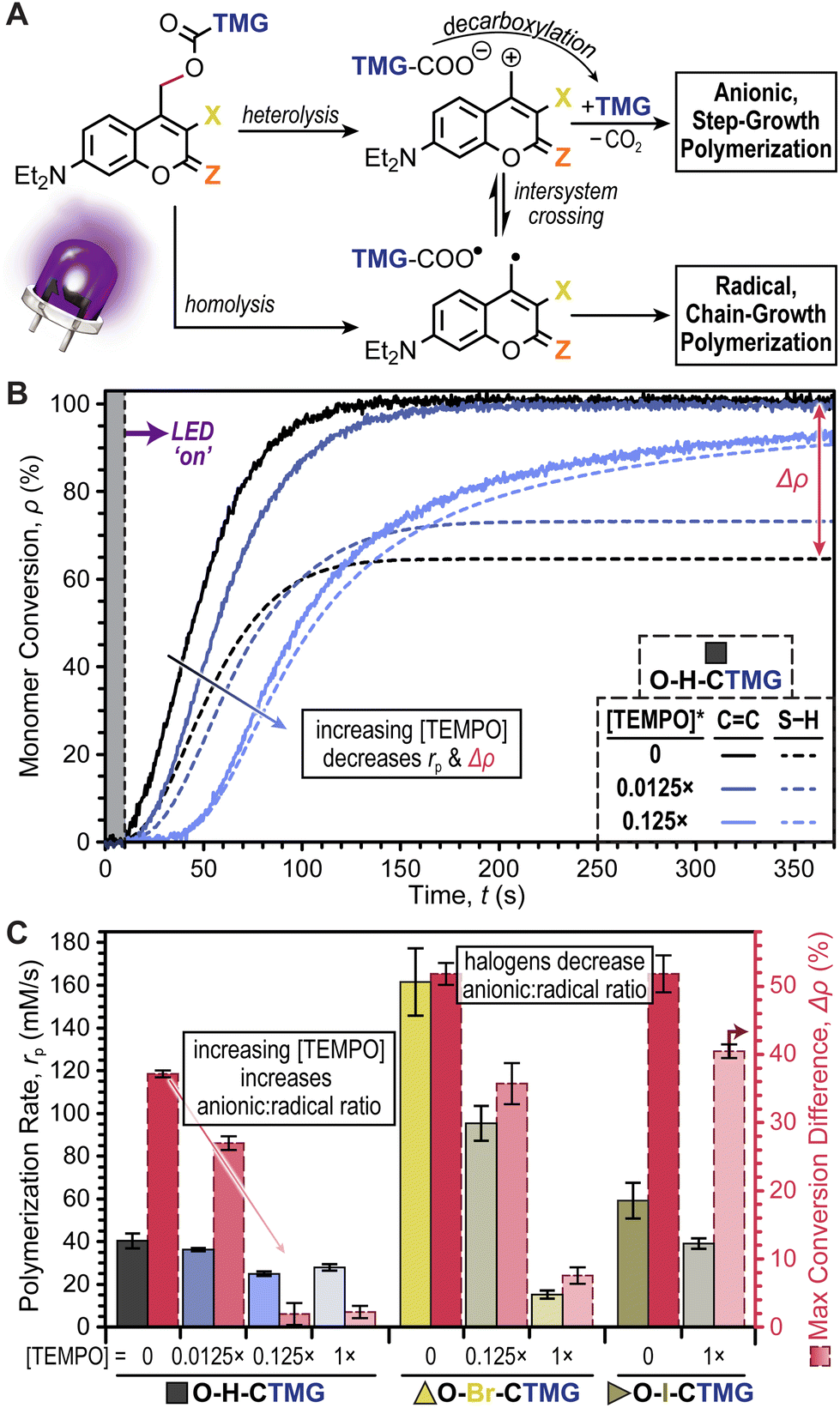 | ||
| Fig. 6 Understanding radical effects. (A) Proposed simplified mechanistic pathways leading to anionic step- vs. radical chain-growth polymerizations. (B) Representative photopolymerization kinetics for HDDA + DODT resin comprising O–H-CTMG and varying amounts of TEMPO, where 1× represents equimolar quantities of photocage and TEMPO. (C) Bar graph showing the effect of TEMPO on rp and Δρ for O–H-, O–Br-, and O–I-CTMG. Averages were calculated from a minimum of three samples and error bars represent ±1 standard deviation from the mean. | ||
In response to the potential formation of radical species that could induce a chain-growth polymerization, we decided to systematically examine the effect of TEMPO as a radical scavenger. The halogenated series, O–H-, O–Br-, and O–I-CTMG, was selected for this study given their relatively large Δρ values, ranging from ∼35–50% in the absence of TEMPO (Fig. 6B & C). For the case of O–H-CTMG, sub-stoichiometric addition of TEMPO resulted in stark decreases in Δρ, while also slightly decreasing rp (Fig. 6B). Specifically, adding 0.0125× and 0.125× TEMPO relative to O–H-CTMG resulted in Δρ values of 29.2 ± 1.0 and 3.5 ± 1.6%, respectively (Fig. 6C and Table S9†). However, rp (based on CC conversion) were only moderately impacted, decreasing from 40 ± 3 (no TEMPO) to 36 ± 1 and 25 ± 1 mM s−1 for 0.0125× and 0.125×, respectively. Increasing the amount of TEMPO to 1× (equimolar with O–H-CTMG) resulted in no significant change in Δρ or rp relative to the 0.125× TEMPO case. These results suggest that the addition of TEMPO is a simple strategy to preclude chain-growth polymerizations and foster the formation of more uniform polymer network topologies.
In contrast to O–H-CTMG, the halogenated analogues required larger quantities of TEMPO to cause a decrease in Δρ. For O–Br-CTMG, 0.125× TEMPO only decreased Δρ to 38.5 ± 3.0%, while 1× TEMPO lowered it to 9.3 ± 1.2%, but at the cost of rp decreasing by ∼10-fold, from 162 ± 16 mM s−1 (no TEMPO) to 15 ± 2 mM s−1 (Fig. 6C, Fig. S20, and Table S10†). Furthermore, the O–I-CTMG derivative maintained a high Δρ of 43.2 ± 0.6% with 1× TEMPO (Fig. S21 and Table S11†). These results suggest that the presence of halogens on the coumarinylmethyl photocage scaffold biases the system to form radicals over releasing base to catalyze thiol-Michael, or related addition reactions useful in step-growth polymerizations. Thus, relative to the halogenated analogues O–H-CTMG is the better suited photobase generator if the objective is to prepare polymers via a purely step-growth mechanism.
Conclusions
In summary, five coumarinylmethyl PBGs were synthesized and systematically characterized in-terms of their utility to catalyze rapid (∼minutes to max monomer conversion) thiol–ene polymerizations upon exposure to violet (405 nm) and blue (470 nm) LEDs. Photopolymerization kinetics and bond scission quantum yields were determined using real-time Fourier transform infrared (RT-FTIR) spectroscopy, revealing key structure–reactivity relationships. Notably, congruent free radical chain-growth and anionic step-growth polymerization mechanisms were identified and correlated to PBG composition. Halogenation at the 3-position (e.g., going from O–H- to O–Br- and O–I-CTMG) resulted in an increase in polymerization rate, but also an increase in the disparity between maximum CC and S–H conversion (i.e., larger Δρ), indicative of greater radical contributions to the polymerization mechanism. In contrast, the π-extended styryl derivative (O-Sty-CTMG) appeared to have reduced radical character (i.e., more anionic, step-growth character), which was reminiscent of the oNB-TMG PBG control. Addition of TEMPO to the halogenated CTMG series further affirmed the presence of radicals as Δρ was reduced with sub-stoichiometric quantities of TEMPO, which highlighted a simple method (i.e., TEMPO addition) to bias photopolymerizations towards a step-growth mechanism. Finally, quantification of bond scission quantum yield (Φs) provided low values for CTMG derivatives (<0.5%) relative to the oNB-TMG control (nearly 6%), despite comparable photopolymerization rates under equivalent conditions. This was attributed to improved visible light absorption, which was ∼100× larger for the CTMG derivatives compared to oNB-TMG considering the violet, 405 nm LED. These results also demonstrate the opportunities that exist to improve photobase-promoted polymerizations through further catalyst-design and optimization. It is envisioned that these findings will both inform the selection of PBGs for emergent light-driven materials syntheses (e.g., additive manufacturing) towards the formation of uniform (ideal) polymer networks, while guiding new catalyst designs with control over radical vs. anionic mechanisms to drive efficient photopolymerizations.
Author contributions
Conceptualization (M. T. K., A. U., Z. A. P.); methodology (M. T. K., A. U., L. M. S., K. Y., P. L., Z. A. P.); investigation (M. T. K., A. U., L. M. S., K. Y., P. L.); visualization (M. T. K., L. M. S., Z. A. P.); funding acquisition (Z. A. P.); project administration (Z. A. P.); supervision (Z. A. P.); writing – original draft (M. T. K., Z. A. P.); writing – review & editing (M. T. K., A. U., L. M. S., K. Y., P. L., Z. A. P.).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge primary support by the National Science Foundation under grant no. MSN-2107877 (MTK, AU, KC, PL, ZAP; synthesis and characterization, supervision, and writing). Partial support was provided by the Robert A. Welch Foundation under grant no. F-2007 (ZAP; partial materials and supplies support) and Research Corporation for Science Advancement under award #28184 (LMS, ZAP; partial materials and supplies support). We acknowledge Prof. Sean Roberts and his research group for the use of a Magnitude Instruments enVISion spectrometer for nanosecond transient absorption measurements.References
- J. Huang, Q. Qin and J. Wang, Processes, 2020, 8, 1138 CrossRef CAS.
- F. A. Rueggeberg, Dent. Mater., 2011, 27, 39–52 CrossRef CAS PubMed.
- X. Zhao, X. Chen, H. Yuk, S. Lin, X. Liu and G. Parada, Chem. Rev., 2021, 121, 4309–4372 CrossRef CAS PubMed.
- Y. Gu, J. Zhao and J. A. Johnson, Angew. Chem., 2020, 132, 5054–5085 CrossRef.
- N. Zivic, P. K. Kuroishi, F. Dumur, D. Gigmes, A. P. Dove and H. Sardon, Angew. Chem., 2019, 131, 10518–10531 CrossRef.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119–191 CrossRef PubMed.
- B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. E. Kirkpatrick, K. S. Anseth and C. N. Bowman, Chem. Rev., 2021, 121, 6915–6990 CrossRef CAS PubMed.
- S. Chatani, T. Gong, B. A. Earle, M. Podgorski and C. N. Bowman, ACS Macro Lett., 2014, 3, 315–318 CrossRef CAS PubMed.
- W. Xi, H. Peng, A. Aguirre-Soto, C. J. Kloxin, J. W. Stansbury and C. N. Bowman, Macromolecules, 2014, 47, 6159–6165 CrossRef CAS PubMed.
- X. Zhang, W. Xi, S. Huang, K. Long and C. N. Bowman, Macromolecules, 2017, 50, 5652–5660 CrossRef CAS PubMed.
- X. Zhang, W. Xi, G. Gao, X. Wang, J. W. Stansbury and C. N. Bowman, ACS Macro Lett., 2018, 7, 852–857 CrossRef CAS PubMed.
- X. Zhang, W. Xi, C. Wang, M. Podgórski and C. N. Bowman, ACS Macro Lett., 2016, 5, 229–233 CrossRef CAS PubMed.
- N. Zivic, N. Sadaba, N. Almandoz, F. Ruipérez, D. Mecerreyes and H. Sardon, Macromolecules, 2020, 53, 2069–2076 CrossRef CAS.
- H. Matsushima, J. Shin, C. N. Bowman and C. E. Hoyle, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3255–3264 CrossRef CAS.
- S. Zakeri, M. Vippola and E. Levänen, Addit. Manuf., 2020, 35, 101177 CAS.
- A. Bagheri and J. Jin, ACS Appl. Polym. Mater., 2019, 1, 593–611 CrossRef CAS.
- F. Dumur, Catalysts, 2019, 9, 736 CrossRef CAS.
- P. Xiao, J. Zhang, F. Dumur, M. A. Tehfe, F. Morlet-Savary, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Prog. Polym. Sci., 2015, 41, 32–66 CrossRef CAS.
- P. Lu, D. Ahn, R. Yunis, L. Delafresnaye, N. Corrigan, C. Boyer, C. Barner-Kowollik and Z. A. Page, Matter, 2021, 4, 2172–2229 CrossRef CAS.
- I. Elamri, C. Abdellaoui, J. Kaur Bains, K. Felicitas Hohmann, S. Lakshmi Gande, E. Stirnal, J. Wachtveitl and H. Schwalbe, J. Am. Chem. Soc., 2021, 143, 10596–10603 CrossRef CAS PubMed.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS PubMed.
- K. Kalayci, H. Frisch, C. Barner-Kowollik and V. X. Truong, Adv. Funct. Mater., 2020, 30, 1908171 CrossRef CAS.
- J. L. Pelloth, P. A. Tran, A. Walther, A. S. Goldmann, H. Frisch, V. X. Truong and C. Barner-Kowollik, Adv. Mater., 2021, 33, 2102184 CrossRef CAS PubMed.
- V. X. Truong, J. Bachmann, A. N. Unterreiner, J. P. Blinco and C. Barner-Kowollik, Angew. Chem.,– Int. Ed., 2022, 61, e202113076 CrossRef CAS PubMed.
- T. Rapp and C. DeForest, ChemRxiv, 2022. This content is a preprint and has not been peer-reviewed. https://doi.org/10.26434/chemrxiv-2022-hlbk6.
- A. M. Schulte, G. Alachouzos, W. Szymański and B. L. Feringa, J. Am. Chem. Soc., 2022, 144, 12421–12430 CrossRef CAS PubMed.
- R. Klimek, M. Asido, V. Hermanns, S. Junek, J. Wachtveitl and A. Heckel, Chem. – Eur. J., 2022, 28, e202200647 CrossRef CAS PubMed.
- P. Liu, B. Li, C. Zhan, F. Zeng and S. Wu, J. Mater. Chem. B, 2017, 5, 7538–7546 RSC.
- S. R. Trenor, A. R. Shultz, B. J. Love and T. E. Long, Chem. Rev., 2004, 104, 3059–3078 CrossRef CAS PubMed.
- T. Ohba, K. Suyama and M. Shirai, React. Funct. Polym., 2006, 66, 1189–1197 CrossRef CAS.
- M. Bojtár, A. Kormos, K. Kis-Petik, M. Kellermayer and P. Kele, Org. Lett., 2019, 21, 9410–9414 CrossRef PubMed.
- C. Hamerla, C. Neumann, K. Falahati, J. Von Cosel, L. J. G. W. Van Wilderen, M. S. Niraghatam, D. Kern-Michler, N. Mielke, M. Reinfelds, A. Rodrigues-Correia, A. Heckel, J. Bredenbeck and I. Burghardt, Phys. Chem. Chem. Phys., 2020, 22, 13418–13430 RSC.
- J. D. Rule and J. S. Moore, Macromolecules, 2005, 38, 7266–7273 CrossRef CAS.
- N. Kamatham, J. P. Da Silva, R. S. Givens and V. Ramamurthy, Org. Lett., 2017, 19, 3588–3591 CrossRef CAS PubMed.
- M. A. Takano and M. Abe, Org. Lett., 2022, 24, 2804–2808 CrossRef CAS PubMed.
- T. R. Albright and A. H. Winter, J. Am. Chem. Soc., 2015, 137, 3402–3410 CrossRef CAS PubMed.
- K. Kalayci, H. Frisch, C. Barner-Kowollik and V. X. Truong, Chem. Commun., 2022, 58, 6397–6400 RSC.
- X. J. Tang, Y. Wu, R. Zhao, X. Kou, Z. Dong, W. Zhou, Z. Zhang, W. Tan and X. Fang, Angew. Chem., Int. Ed., 2020, 59, 18386–18389 CrossRef CAS PubMed.
- Q. Lin, L. Yang, Z. Wang, Y. Hua, D. Zhang, B. Bao, C. Bao, X. Gong and L. Zhu, Angew. Chem., Int. Ed., 2018, 57, 3722–3726 CrossRef CAS PubMed.
- H. Y. Park, C. J. Kloxin, M. F. Fordney and C. N. Bowman, Dent. Mater., 2012, 28, 888–893 CrossRef CAS PubMed.
- E. Pretsch, P. Bühlmann and M. Badertscher, Structure determination of organic compounds: Tables of spectral data, 2009 Search PubMed.
- C. Hamerla, C. Neumann, K. Falahati, J. Von Cosel, L. J. G. W. Van Wilderen, M. S. Niraghatam, D. Kern-Michler, N. Mielke, M. Reinfelds, A. Rodrigues-Correia, A. Heckel, J. Bredenbeck and I. Burghardt, Phys. Chem. Chem. Phys., 2020, 22, 13418–13430 RSC.
- F. Mallamace, C. Corsaro, D. Mallamace, S. Vasi, C. Vasi and G. Dugo, Comput. Struct. Biotechnol. J., 2015, 13, 33–37 CrossRef CAS PubMed.
- S. Nad and H. Pal, J. Phys. Chem. A, 2000, 104, 673–680 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of coumarin derivatives and oNB control, NMR, HRMS, UV-Vis spectroscopy, fluorimetry, ns-TA, RT-FTIR spectroscopy, emission and uncaging quantum yield calculations. See DOI: https://doi.org/10.1039/d3py00771e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |