
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRing-opening polymerization of β-thiobutyrolactone catalyzed by phosphazenes†
Hui
Li
,
Sophie M.
Guillaume
 * and
Jean-François
Carpentier
*
* and
Jean-François
Carpentier
*
Univ Rennes, CNRS, ISCR-UMR 6226, 35000 Rennes, France. E-mail: sophie.guillaume@univ-rennes.fr; jean-francois.carpentier@univ-rennes.fr
First published on 26th July 2023
Abstract
Thiolactones that give access to polythioesters (PTEs) upon ring-opening polymerization (ROP) have recently attracted increasing attention especially due to their biocompatibility and improved degradability among other properties. We report herein on the ROP of the simplest four-membered ring thiolactone, namely racemic β-thiobutyrolactone (rac-TBL), mediated by commercially available phosphazene bases, affording the corresponding poly(3-thiobutyrolactone) (P3TB). The reactions proceeded at room temperature, in toluene or in THF, with the apparent reactivity increasing monotonously with the basicity and steric hindrance of the phosphazene (BEMP ∼ tBu-P1 < tBu-P2 < tBu-P4). This trend suggests that initiation of the ROP proceeds, at least to some extent, by abstraction of one acidic methylene hydrogen from the monomer, to generate eventually a phosphazenium–thiocarboxylate ion-pair species that next acts as the initiating/chain-propagating species. ROP performed in THF was kinetically controlled affording P3TBs with defined molar mass and fair dispersity, some features of a living ROP. Detailed 1H, 13C, 31P and DOSY NMR spectroscopic, MALDI-ToF/ESI mass spectrometric and TGA studies conducted on the polymers produced using tBu-P4 supported the concomitant formation of {[tBu-P4]H}+ and essentially α-thiocrotonate,ω-[thiocarboxylate]− end-capped linear P3TBs; yet, the presence of cyclic P3TBs could not be excluded. The molar mass values, as determined by NMR analysis from this thiocrotonate end-group (Mn,NMR), increased linearly with the monomer conversion, yet with a nonzero intercept, suggesting slow initiation vs. propagation. The molar mass of the recovered P3TBs was moderately controlled (Mn,SEC = 7.5–18.5 kg mol−1, Mn,NMR = 5.2–22.4 kg mol−1), displaying relatively narrow dispersities (ĐM = 1.39–1.52 in THF). This work provides the second example of chemical synthesis of P3TB.
Introduction
The replacement of metal-based catalysts with small organic molecules has emerged as an appealing polymerization approach, in particular in the context of polymer materials designed for microelectronics, or pharmaceutical and biomedical applications.1 Organocatalysts are of interest due to their high chemical stability, relatively low cost, ease of handling and, for some of them, relative non-toxicity towards living organisms. A wide range of commercially available simple organic molecules is available for the ring-opening polymerization (ROP) of heterocyclic monomers, such as sulfonic and phosphoric acids, N-heterocyclic carbenes (NHCs), amines, amidines, guanidines, phosphines, and phosphazene bases.2–8A variety of organocatalysts have been investigated in the ROP of four-membered β-lactones, a class of challenging monomers because of their reluctance to ring open, despite their high ring strain. Mechanisms at play were demonstrated to depend drastically on the nature of the catalyst/initiator system as dictated by its basic, nucleophilic or dual behavior, and also quite importantly, on the chemical nature of the monomer itself and on the reaction conditions. Early attempts using the simple β-butyrolactone (BL) were conducted by the groups of Hedrick and Waymouth with guanidines and amidines, yet at a too low temperature (50 °C) to enable polymerization.9 Our group later demonstrated that 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP) can act as a nucleophilic initiator in the ROP of BL10 or benzyl β-malolactonate (MLABn),11 at 60 °C under bulk (neat) conditions. These reactions gave the corresponding α-phosphazene,ω-crotonate telechelic polyesters (Scheme 1), as supported by NMR and MALDI-ToF MS analyses, and were proposed to proceed via the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 organobase-N-acylcrotonate intermediate,10 followed by propagation through monomer O-acyl bond cleavage. On the other hand, in the ROP of 4-alkoxymethylene-β-propiolactones (BPLOR, R = Me, Bn, SiMe2tBu) initiated by BEMP, the latter organocatalyst acts as a basic pre-initiator. It was proposed that BEMP abstracts one of the methylene hydrogen at the α-position of the carbonyl of the rac-BPLOR monomer, thereby leading to the formation of an α,β-unsaturated carboxylate species as the “real” initiator, which next propagates via monomer O-alkyl bond cleavage, forming ion-pair active {α-crotonate,ω-COO}−{[BEMP]H}+ PBLOR chains that eventually return α-crotonate,ω-COOH telechelic PBLOR upon acidic workup (Scheme 1).12
:1 organobase-N-acylcrotonate intermediate,10 followed by propagation through monomer O-acyl bond cleavage. On the other hand, in the ROP of 4-alkoxymethylene-β-propiolactones (BPLOR, R = Me, Bn, SiMe2tBu) initiated by BEMP, the latter organocatalyst acts as a basic pre-initiator. It was proposed that BEMP abstracts one of the methylene hydrogen at the α-position of the carbonyl of the rac-BPLOR monomer, thereby leading to the formation of an α,β-unsaturated carboxylate species as the “real” initiator, which next propagates via monomer O-alkyl bond cleavage, forming ion-pair active {α-crotonate,ω-COO}−{[BEMP]H}+ PBLOR chains that eventually return α-crotonate,ω-COOH telechelic PBLOR upon acidic workup (Scheme 1).12
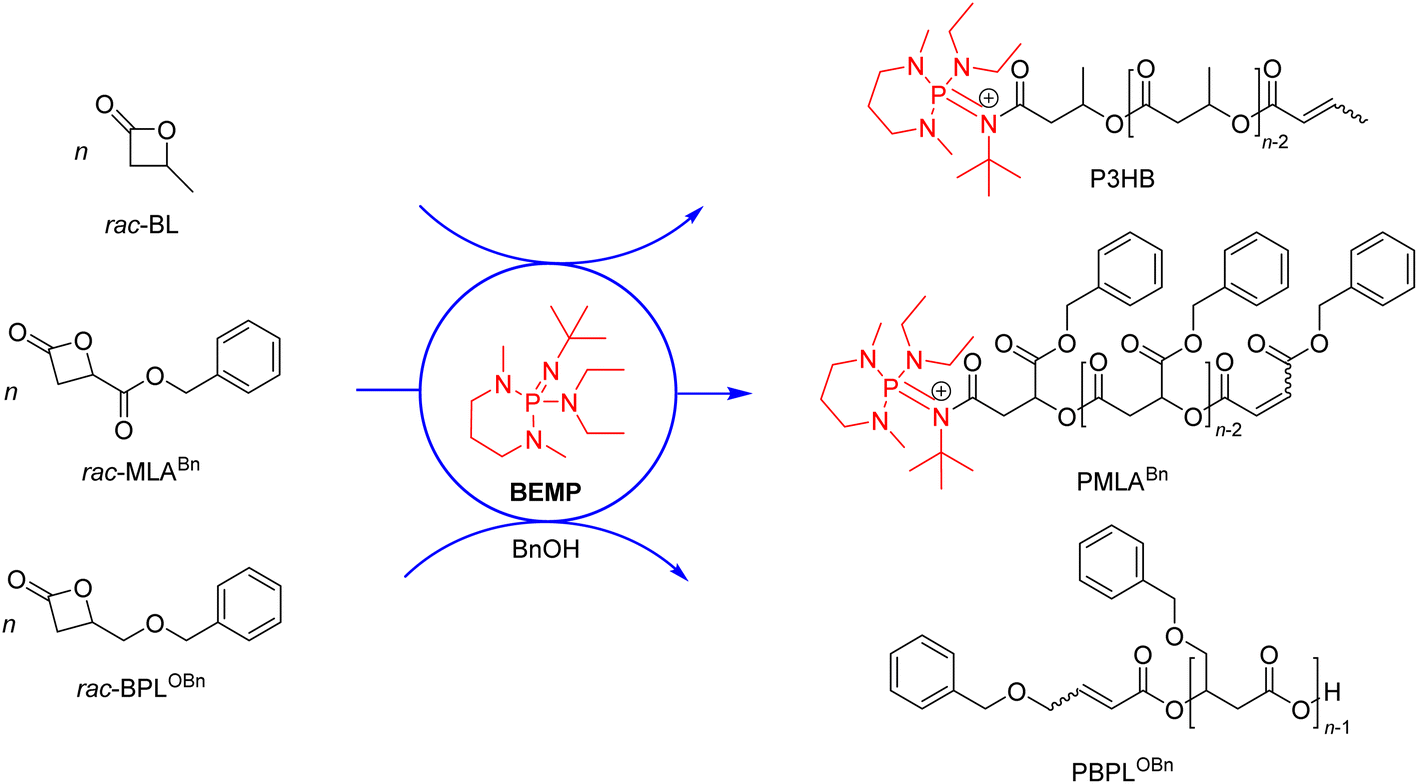 | ||
| Scheme 1 Ring-opening polymerization of β-butyrolactone (BL) and functional β-propiolactone derivatives mediated by BEMP in bulk.10–12 | ||
We herein report on our preliminary studies related to the extension of this organocatalyzed chemistry to the ROP of β-thiobutyrolactone (TBL), the thio analogue of BL (Fig. 2). The literature showcases that poly(3-thiobutyrate) (P3TB), the resulting polythioester (PTE), was first isolated by a fermentation approach using E. coli cultured with thioalkanoic acids, thereby generating pure isotactic P3TB (presumably linear) exhibiting a high molar mass (Mn = ca. 175000 g mol−1, ĐM = 1.9).13 Recently, we described the first chemical synthesis of P3TB by ROP of rac-TBL using yttrium-based catalysts, which provides access to cyclic P3TB with either high isoselectivity or high syndiotactic bias, depending on the nature of the achiral ancillary ligand used.14 A few examples of the ROP of β-thiolactones towards PTEs have been reported.15 The ROP of (S)-α-p-toluenesulfonamido-β-propiothiolactone, initiated by water or benzyl thioalcohol, or even thermally without any initiator, was briefly reported in the 1960s.16 The ROP of a cysteine-derived β-thiolactone proceeded in the presence of a thiol as the initiator which attacks the carbonyl to open the monomer ring by C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–S bond cleavage.17 In both the latter examples, the resulting P3TEs have relatively low molar mass and broad dispersity, most likely as the result of extensive transthioesterification side-reactions. More recently, the ROP of a gem-dimethyl-substituted β-thiolactone catalyzed by DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) in the presence of benzyl mercaptan (BnSH) as the initiator returned the corresponding P3TE with a high molar mass (Mn = 162600 g mol−1, ĐM = 1.53), while also demonstrating the propensity to degrade back to the original monomer.18 Also, more generally, PTEs prepared by ROP of cyclic thiolactones are currently attracting renewed attention given their inherent valuable properties in homopolymers as well as in copolymers, especially in relation to their degradation ability.15,19–22
O)–S bond cleavage.17 In both the latter examples, the resulting P3TEs have relatively low molar mass and broad dispersity, most likely as the result of extensive transthioesterification side-reactions. More recently, the ROP of a gem-dimethyl-substituted β-thiolactone catalyzed by DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) in the presence of benzyl mercaptan (BnSH) as the initiator returned the corresponding P3TE with a high molar mass (Mn = 162600 g mol−1, ĐM = 1.53), while also demonstrating the propensity to degrade back to the original monomer.18 Also, more generally, PTEs prepared by ROP of cyclic thiolactones are currently attracting renewed attention given their inherent valuable properties in homopolymers as well as in copolymers, especially in relation to their degradation ability.15,19–22
In the present work, we focused our attention on the ROP of rac-TBL mediated by several ubiquitous phosphazene organocatalysts, namely BEMP, tBu-P1, tBu-P2, and tBu-P4 (Scheme 2). Due to their high basicity, non-nucleophilic nature and good solubility in various organic solvents, phosphazenes have been widely investigated for the ROP and ring-opening copolymerization of different types of monomers, including lactones but also epoxides, and cyclic carbonates.23,24 The performance of phosphazenes highly depends on their basicity: higher basicity usually improves the polymerization rate, yet often at the expense of side-reactions, such as chain transfer, especially inter- and intra-molecular transesterification reactions, which in turn broaden the dispersity of the resulting polymers.23,24 Also, similarly to many other basic organocatalysts, phosphazenes often convert exogenous protic reagents/co-initiators, such as alcohols and thiols, into nucleophilic initiators through deprotonation. Hence, judicious choice of a phosphazene with a suitable basicity matching the target monomer, possibly combined with a co-initiator, is critical for the achievement of a controlled polymerization.
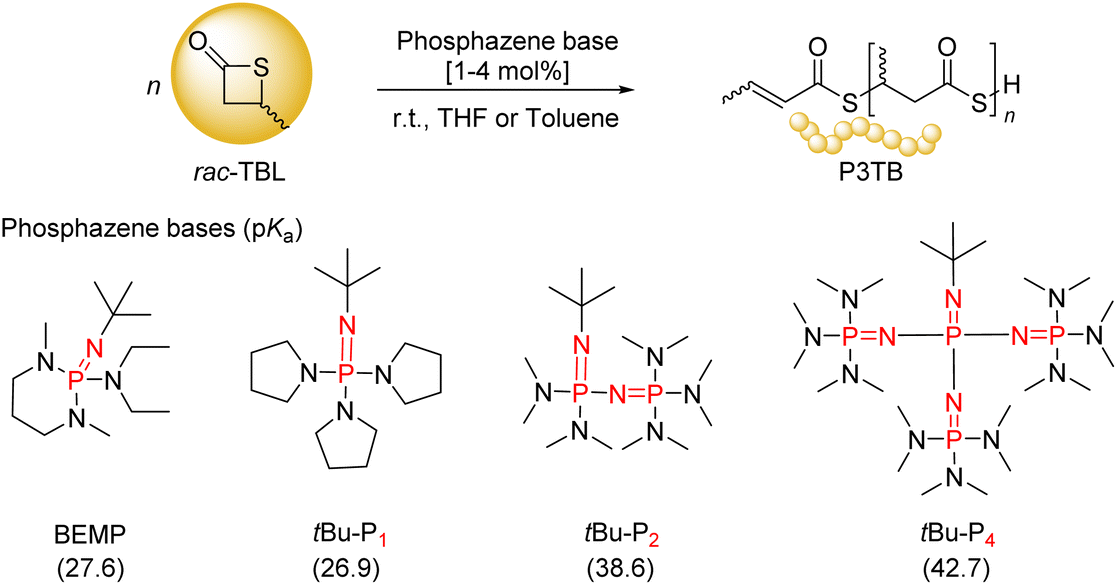 | ||
| Scheme 2 Common phosphazene bases (and their pKa values measured in MeCN)23,24 used in this study in the ROP of rac-TBL. | ||
Experimental section
General considerations
All polymerizations were carried out in Schlenk-type glassware on a vacuum dual-manifold line or in a glove box operating under argon (<5 ppm H2O, O2). rac-Thiobutyrolactone (rac-TBL) was synthesized according to the previously reported procedure.14 HPLC-grade organic solvents were first purged extensively with nitrogen during filling 20 L solvent reservoirs, and then, for toluene, dried by passage through Q-5 supported copper catalyst stainless steel columns. Tetrahydrofuran and toluene were degassed and dried over sodium/benzophenone followed by distillation under argon. Benzyl alcohol (BnOH) and benzyl mercaptan (BnSH) were purified by distillation from CaH2. 2-tert-Butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP, 1.0 M in hexanes), tert-butylimino-tri(pyrrolidino)phosphorane (tBu-P1), 1-tert-butyl-2,2,4,4,4-pentakis(dimethylamino)-2λ5,4λ5-catenadi(phosphazene) (tBu-P2, 2.0 M in THF) and 1-tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis-[tris(dimethylamino)phosphoranylidenamino]-2λ5,4λ5-catenadi(phosphazene) (tBu-P4, 0.8 M in hexanes) were purchased from Aldrich and the solvents were removed under vacuum prior to use.1H (500 MHz), 13C{1H} (125 MHz), 31P{1H} (202 MHz) and 2D DOSY (500 MHz) NMR spectra were recorded on Bruker Avance III HD 500, Av-III 400 or Av-III 300 spectrometers at 25 °C, fitted with a direct broadband 5 mm probe head (BBO). 13C NMR spectra were typically acquired using the Bruker standard zgpg30 sequence, with an acquisition time of 1.05 s and a relaxation delay of 5.00 s.
Monomer conversions were calculated from 1H NMR spectra of the crude polymer samples in CDCl3 using the integration ratio of the methine hydrogens of P3TB (δ 3.90 ppm) and the methine hydrogen of the TBL monomer (δ 3.73 ppm). The molar mass of P3TB samples was also determined by 1H NMR analysis in CDCl3 from the relative intensities of the signals of the P3TB repeating unit methine hydrogen (δ 3.90 ppm) and the signals of the thiocrotonate chain-end (δ (ppm): 1.84 (CH3); 6.03 (CO–CHCH(CH3)); 6.86 (CO–CHCH(CH3))).
Number- and weight-average molar mass (Mn,SEC and Mw,SEC) and dispersity (ĐM = Mw/Mn) values of the polymers were determined by size-exclusion chromatography (SEC) in THF at 30 °C (flow rate = 1.0 mL min−1) on Polymer Laboratories PL50 apparatus equipped with a refractive index detector and a set of two ResiPore PLgel 3 μm MIXED-D 300 × 7.5 mm columns. The polymer samples were dissolved in THF (2 mg mL−1). All elution curves were calibrated with polystyrene standards (Mn range = 580–126000 g mol−1); the Mn,SEC values of the P3TB reported in all tables are uncorrected for the possible difference in the hydrodynamic radius vs. that of polystyrene.
The onset decomposition temperatures (Td, defined as the temperature for 5% weight loss) of the polymers were measured by Thermogravimetric Analysis (TGA) on Mettler-Toledo TGA-DSC-1 apparatus under dry nitrogen flow. Polymer samples were heated from ambient temperature to 500 °C at a rate of 10 °C min−1.
Recording of high-resolution Matrix Assisted Laser Desorption Ionization-Time of Flight (MALDI-ToF) mass spectrometry analyses was attempted using an ULTRAFLEX III TOF/TOF spectrometer (Bruker Daltonik Gmbh, Bremen, Germany) in positive ion mode at CRMPO (ScanMAT UAR 2025 CNRS, Université de Rennes). Spectra were recorded using reflectron mode and an accelerating voltage of 25 kV or higher. A mixture of a freshly prepared solution of the polymer in CH2Cl2 (HPLC grade) and DCTB (trans-2-(3-(4-tert-butylphenyl)-2-methyl-2-propenylidene)-malononitrile) in CH2Cl2 (HPLC grade, 10 mg mL−1), and a MeOH or CH2Cl2/CH3CN (50:50 v/v) solution of the cationizing agent (NaI, 10 mg mL−1) were prepared. The solutions were combined in a 1:1:1 (v/v/v) ratio of matrix-to-sample-to-cationizing agent. The resulting solution (0.25–0.5 µL) was deposited onto the sample target (Prespotted AnchorChip PAC II 384/96 HCCA) and air or vacuum dried. High-resolution ElectroSpray-Ionization (ESI) mass spectra were recorded in positive ion mode in methanol suspensions (ca. 10 μg mL−1) with a MeOH solution of the cationizing agent (NaI, 10 mg mL−1), with direct infusion (10 μL min−1), using a maXis 4G instrument (Bruker Daltonik Gmbh, Bremen, Germany) at CRMPO (ScanMAT UAR 2025 CNRS, Université de Rennes).
General polymerization procedures
Polymerizations were performed either in a glass vial (5 mL) inside the glovebox for room temperature runs, or in a Schlenk flask (100 mL) interfaced to the dual-manifold Schlenk line for runs using a heating bath. The vial/flask was charged in the glovebox with a predetermined amount of phosphazene base, with or without an added co-initiator, and solvent. For experiments performed at room temperature, polymerization was initiated by rapid addition, in the glovebox, to the reaction vessel, of a pre-weighted amount of TBL using a syringe. For runs using an external heating bath, the flask containing the phosphazene and the solvent was closed, taken out of the glovebox, connected to the dual-manifold Schlenk line and then immersed in the bath at the desired temperature. After equilibration at the targeted reaction temperature, the polymerization was in this case initiated by the rapid addition of a predetermined amount of the monomer using a gastight syringe. After the desired period, the polymerization was quenched by addition of ‘wet/undried’ commercial-grade hexanes (0.5 mL). An aliquot (0.1 mL) was taken from the reaction mixture and used to determine the monomer conversion by 1H NMR analysis. The crude mixture was next added dropwise into “wet” commercial-grade hexanes (100 mL, used as received) (for some reactions, it was pre-quenched by adding a few drops of a 5wt% benzoic acid solution in CHCl3) and the viscous polymer precipitated. Following the removal of the supernatant solution, the thus recovered polymer was dissolved in CH2Cl2 and precipitated once more to ensure the removal of all of the unreacted monomer, and finally dried in a vacuum oven at room temperature until a constant weight was achieved.Results and discussion
Our first exploratory experiments on the ROP of TBL were performed using BEMP (Scheme 2) with a [rac-TBL]0/[BEMP]0 feed ratio of 50:1 (mol mol−1) at 60 °C under bulk conditions (neat monomer), based on our previous work on the ROP of β-lactones (Scheme 1).10–12 Under these conditions, the conversion of TBL reached 99% in less than 3 h (Table 1, entry 1); this is an apparent reactivity similar to that observed with rac-BL, but higher than with rac-BPLORs (at 60 °C, bulk) with TOF values (mol(monomer) mol(BEMP)−1 h−1) of TBL = 87, BL = 79,10 BPLOAll = 13,11 and BPLOBn = 1511. Conducting the reaction at room temperature at a loading ratio of 50:1 in toluene enabled achieving 52% monomer conversion after 7 h (entry 2); this is remarkable as the ROP of BL and BPLORs does not proceed under these much milder conditions. In both bulk or solution polymerization, the molar masses of the P3TBs measured by SEC (uncorrected values) were much higher than the ones calculated from the (converted) monomer-to-BEMP ratio, and the dispersity was relatively broad (ĐM = 1.51 and 1.79, respectively); this possibly denotes distinct hydrodynamic ratios of P3TBs vs. polystyrene used as SEC standards, but the Mn values determined by NMR based on thiocrotonate end-groups (Mn,NMR; vide infra) were also significantly higher than the calculated ones. Hence, this discrepancy is rather indicative of poor, slow initiation efficiency of BEMP, and/or undesirable side-reactions.
| Entry | Organic base (P)b | ROHc | [TBL]0/[P]0/[ROH]0 | Solvent | Reaction time (h) | TBL Conv.d (%) | M n,calc (kg mol−1) | M n,NMR (kg mol−1) | M n,SEC (kg mol−1) | Đ M |
|---|---|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions unless otherwise stated: [rac-TBL]0 = 1.0 M, room temperature. b P = phosphazene base. c ROH = alcohol added to catalyst P. d Conversion of TBL as determined by 1H NMR analysis of the crude polymerization mixture (refer to the experimental section). e Molar mass calculated according to Mn,calc = [TBL]0/[P]0 × Conv.TBL × MTBL, with MTBL = 204 g mol−1. f Molar mass determined from 1H NMR based on the thiocrotonate end-group signal (refer to the experimental section). g Number-average molar mass (Mn,SEC) and dispersity (ĐM) as determined by SEC analysis in THF at 30 °C using polystyrene standards (uncorrected values). h Polymerization performed at 60 °C. i All P3TB chains may not be linear and/or end-capped by a thiocrotonate moiety; refer to the discussion. j Molar mass calculated considering that added BnOH or BnSH would act as an initiator/transfer agent. | ||||||||||
| 1h | BEMP | — | 50:1:0 |
Bulk | 0.5 | 87 | 4.4 | 13.5i | 16.4 | 1.54 |
| 2 | BEMP | — | 50:1:0 |
Toluene | 7 | 52 | 2.6 | 18.0i | 8.7 | 1.79 |
| 3 | tBu-P1 | — | 50:1:0 |
Toluene | 7 | 59 | 3.0 | 9.0 | 9.2 | 1.68 |
| 4 | tBu-P2 | — | 50:1:0 |
Toluene | 7 | 66 | 3.4 | 6.8 | 7.5 | 1.88 |
| 5 | tBu-P4 | — | 50:1:0 |
Toluene | 7 | 90 | 4.6 | 9.5 | 8.2 | 1.60 |
| 6 | tBu-P4 | — | 100:1:0 |
Toluene | 17.5 | 94 | 9.6 | 22.4 | 12.4 | 1.64 |
| 7 | tBu-P4 | — | 25:1:0 |
THF | 4 | 93 | 2.4 | 5.2 | 11.7 | 1.52 |
| 8 | tBu-P4 | — | 50:1:0 |
THF | 8 | 81 | 4.1 | 10.7 | 15.5 | 1.39 |
| 9 | tBu-P4 | — | 100:1:0 |
THF | 17 | 94 | 9.6 | 16.4 | 17.2 | 1.47 |
| 10 | tBu-P4 | — | 200:1:0 |
THF | 42 | 82 | 16.7 | 17.7 | 18.5 | 1.42 |
| 11 | tBu-P4 | BnOH | 100:1:1 |
THF | 19 | 87 | 8.9 | 13.8 | 15.4 | 1.43 |
| 12 | tBu-P4 | BnOH | 100:1:2 |
THF | 19 | 86 | 4.4j | 13.2 | 14.6 | 1.47 |
| 13 | tBu-P4 | BnOH | 100:1:5 |
THF | 19 | 88 | 1.8j | 15.5 | 15.6 | 1.45 |
| 14 | tBuP4 | BnSH | 50:1:1 |
Toluene | 7 | 73 | 3.8 | 5.0 | 8.2 | 1.58 |
| 15 | tBuP4 | BnSH | 100:1:1 |
Toluene | 17.5 | 73 | 7.5 | 6.7 | 13.2 | 1.31 |
| 16 | tBuP4 | BnSH | 50:1:1 |
THF | 7.6 | 83 | 4.3 | 23.2 | 10.7 | 1.38 |
The ROP of TBL was next investigated under the same conditions (i.e., 50 equiv. of TBL, 1.0 M in toluene, room temperature) with tBu-P1, and the superbases tBu-P2 and tBu-P4. After 7 h of reaction, the conversion of TBL reached 59%, 66% and 90%, respectively (Table 1, entries 3–5). Hence, the apparent reactivity increased monotonously with the basicity of the phosphazene in the order of BEMP ∼ tBu-P1 < tBu-P2 < tBu-P4. At the same time, the steric hindrance of phosphazenes should also be taken into account: the sterically bulkier the phosphazene (BEMP < tBu-P1 ∼ tBu-P2 < tBu-P4), the looser the ion-pair active species formed, which should favorably enhance the chain propagation rate. This observed reactivity trend suggests that initiation of the ROP may proceed, at least to some extent, by abstraction of one of the relatively acidic α-carbonyl methylene hydrogens from the monomer, to generate eventually a phosphazenium–thiocarboxylate ion-pair active species, the latter acting as the initiating/chain-propagating species (vide infra). While the molar mass values as determined by SEC were similar to those calculated from 1H NMR analysis (vide infra), and increased proportionally to the monomer loading (compare entries 5 vs. 6), they were still higher than the values calculated from the monomer-to-phosphazene ratio while all featuring a rather broad dispersity (ĐM = 1.68–1.88). The latter dispersity values are broader than those of polyesters prepared with BEMP from rac-BL and rac-BPLORs (ĐM = 1.05–1.23 and 1.09–1.53, respectively).10,11 This is likely due, at least to some extent, to transthioesterification reactions that occur more easily with PTEs as compared to their PHA oxo-analogues, since thioester bonds are more reactive than similar oxoester bonds.15
The ROP of rac-TBL mediated by tBu-P4, next investigated in THF, a more polar solvent (ε = 7.39 vs. 2.38 for toluene), returned similar results (Table 1, entries 7–10). The reactions proceeded apparently at about the same rate (compare e.g. entries 5 vs. 8 and 6 vs. 9). The molar mass of the resulting P3TBs, as determined by both NMR and SEC analyses, increased monotonously with the [TBL]0/[tBu-P4]0 feed ratio in the range 25–100, but apparently saturated at a higher monomer loading of 200 (Mn,NMR = 17700 g mol−1, Mn,SEC (uncorrected) = 18500 g mol−1; entry 10). Again, these experimental molar masses were higher than those calculated from the monomer-to-phosphazene ratio. The dispersity values were somehow lower than those of the P3TBs prepared in toluene (DM = 1.42–1.52 vs. 1.54–1.88, respectively), yet in the same range as the cyclic P3TBs we recently reported (DM = 1.21–1.68).14 This possibly arises from the more polar solvent which better stabilizes the ion-pair active species (vide infra). On the other hand, the presence of cyclic P3TB within the polymer samples may also rationalize that NMR molar mass values are larger than expected and that the dispersity is broadened, due to easy transthioesterification reactions, as aforementioned.
Addition of 1–5 equiv. of benzyl alcohol (BnOH vs. tBu-P4) did not change the polymerization rate or the characteristics of the recovered P3TBs (compare entries 11–13 vs. 10). Also, the ROP reactions conducted in the presence of one equiv. of BnSH proceeded slightly more slowly in toluene or with similar rates to that in THF, as compared to those conducted in the presence of tBu-P4 without a co-initiator, as judged by monomer conversions (compare entries 5, 6 vs. 14, 15 and entries 8 vs. 16, respectively). The experimental Mn,SEC values of the corresponding P3TBs determined by SEC were also very similar, no matter BnSH was added or not (compare also entries 5, 6 vs. 14, 15 and entries 8 vs. 16, respectively). The Mn,NMR values determined by NMR analysis from the thiocrotonate end-groups are quite close to the theoretical values for the P3TBs prepared in the presence of BnSH in toluene (entries 14 and 15), but revealed much higher for the one prepared in THF (entry 16). In all cases, no signals assignable to a BnS end-group/initiating group could be identified in the NMR spectra of the P3TBs (Fig. S9–S12†). This suggests that such protic compounds, BnOH and BnSH, do not act as chain-transfer agents, and that they are not involved in the initiation process of these phosphazene-mediated ROPs of TBL. We previously made similar observations in the ROP of BL mediated by BEMP, with no apparent effect of added ROH.11 This contrasts with the ROP of the gem-dimethyl-substituted β-thiolactone catalyzed by DBU where the presence of benzyl mercaptan as an initiator is mandatory.18
For these different reasons, the polymerizations were not further investigated in the presence of BnSH (or BnOH) as a possible co-initiator and we focused on ROP experiments conducted solely with tBu-P4. NMR monitoring of the kinetics of the ROP of TBL promoted by tBu-P4 in THF-d8 indicated a first-order reaction (Fig. 1) with the formation of thiocrotonate groups (vide infra) at the very early stage of the polymerization. This is consistent with the aforementioned initiation process proceeding via the abstraction of one acidic methylene hydrogen by the phosphazene base. The plot of the molar mass values calculated by NMR (Mn,NMR) against the monomer conversion is almost perfectly linear, with the slope at the theoretical value (i.e., MW of TBL) of 204 g mol−1, yet with a non-zero intercept (Fig. 2). This first highlighted some features of a “living” ROP. These observations are reminiscent of those described by Waymouth and coworkers in the zwitterionic ROP of lactide mediated by N-heterocyclic carbene organocatalysts.25 In this earlier work, linear plots of Mn values against monomer conversion with a nonzero intercept, combined with narrow dispersities (ĐM < 1.3), were interpreted, as supported by modeling of the kinetics, in terms of a living polymerization with a slower initiation rate (ki) than the propagation rate (kp); the initiation period, corresponding to the formation of zwitterions, hence resulted, at low conversions, in higher molar mass than those predicted simply on the basis of the monomer-to-initiator ratio and conversion.
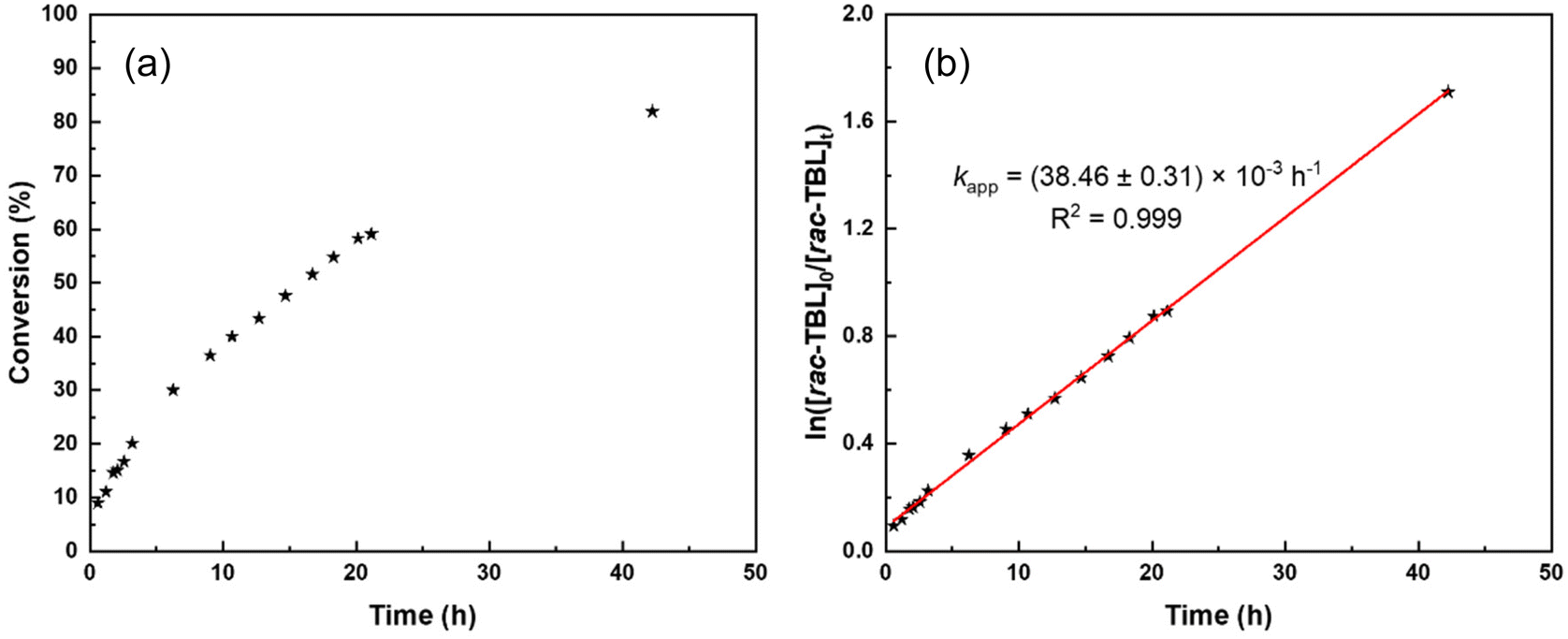 | ||
| Fig. 1 Detailed kinetics of the ROP of rac-TBL by tBu-P4 (without co-initiator) in THF-d8 at a feed ratio of 200:1 at room temperature (Table 1, entry 10); (a) (left): conversion of rac-TBL as a function of reaction time; (b) (right): semi-logarithmic first-order plot as a function of reaction time. | ||
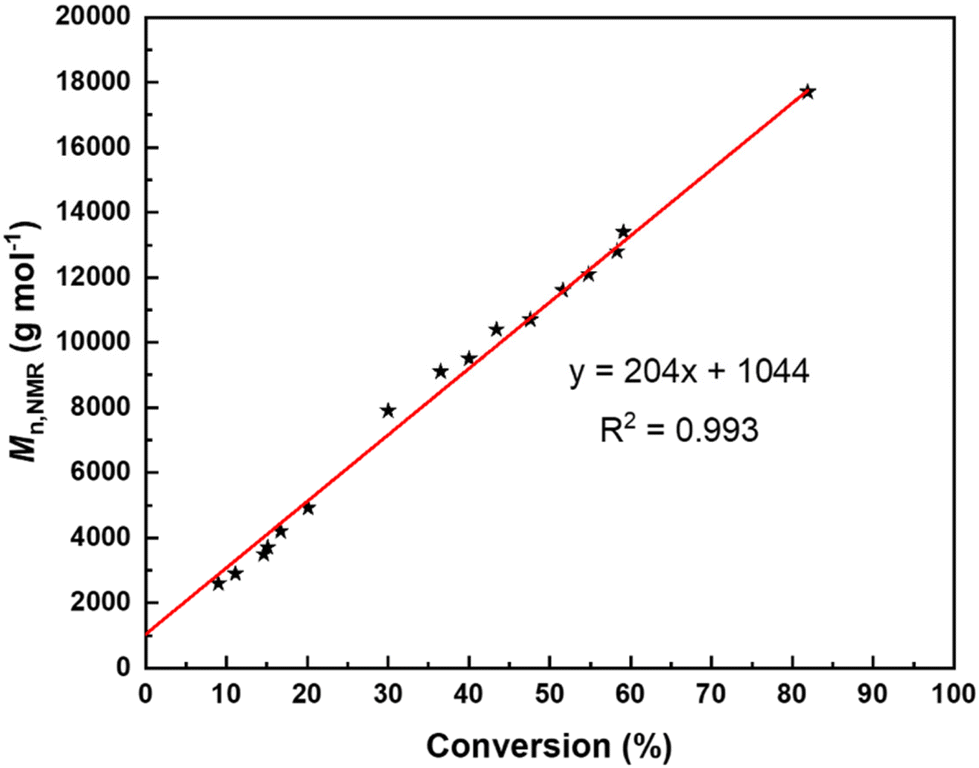 | ||
| Fig. 2 Molar mass values as calculated by NMR based on the thiocrotonate end-group obtained from the ROP of rac-TBL mediated by tBu-P4 (without a co-initiator) in THF-d8 at a feed ratio of 200:1 at room temperature (Table 1, entry 10) as a function of rac-TBL conversion. | ||
The microstructure of the P3TBs was then investigated by NMR spectroscopy. The 1H and 13C{1H} NMR spectra of all polymers, regardless of the solvent used (Fig. 3 and 4; see also the ESI†) clearly displayed the signals characteristic of the TBL repeating units, consistent with those of atactic P3TBs, as prepared in our previous work.14 In addition, a terminal thiocrotonate moiety was unambiguously observed in all cases: e.g. the 1H NMR spectra all featured the characteristic resonances at δ 6.86 ppm (CH3CHCHC(O)S), 6.04 ppm (CH3CHCHC(O)S) and 1.85 ppm (CH3CHCHC(O)S). A DOSY NMR analysis (Fig. 5) conducted on a P3TB prepared with tBu-P4 in toluene revealed that the TBL repeating units and the thiocrotonate group correspond to the same diffusion coefficient, hence supporting that the thiocrotonate moiety is an end-group associated with the P3TB chains. Also, the DOSY spectrum confirmed the presence of {[tBu-P4]H}+ that distinctively showcased a different and greater diffusion coefficient. Altogether, these observations suggest that ROP of TBL proceeds through deprotonation of the acidic hydrogen of TBL by tBu-P4, and more generally by phosphazene organocatalysts, to generate eventually a phosphazenium-thiocarboxylate initiating/chain-propagating ion-pair active species (vide infra). Alternatively, elimination of small molecules from the main-chain of polyhydroxyalkanoates such as P3HB involving the acidic hydrogen α to the carbonyl group is a quite common phenomenon that generates polymer chains with crotonate end-groups; a similar process may also be considered in P3TB and would eventually lead to thiocrotonate end-groups. Although the latter hypothesis seems less likely than the initial deprotonation of TBL by the phosphazene, as hinted by the formation of thiocrotonate groups observed from the very early stage of the ROP (vide supra, Fig. 1 and 2), it cannot be ruled out.
 | ||
| Fig. 3 1H NMR spectrum (500 MHz, CDCl3, 25 °C) of a P3TB prepared with tBu-P4 in toluene (Table 1, entry 6). | ||
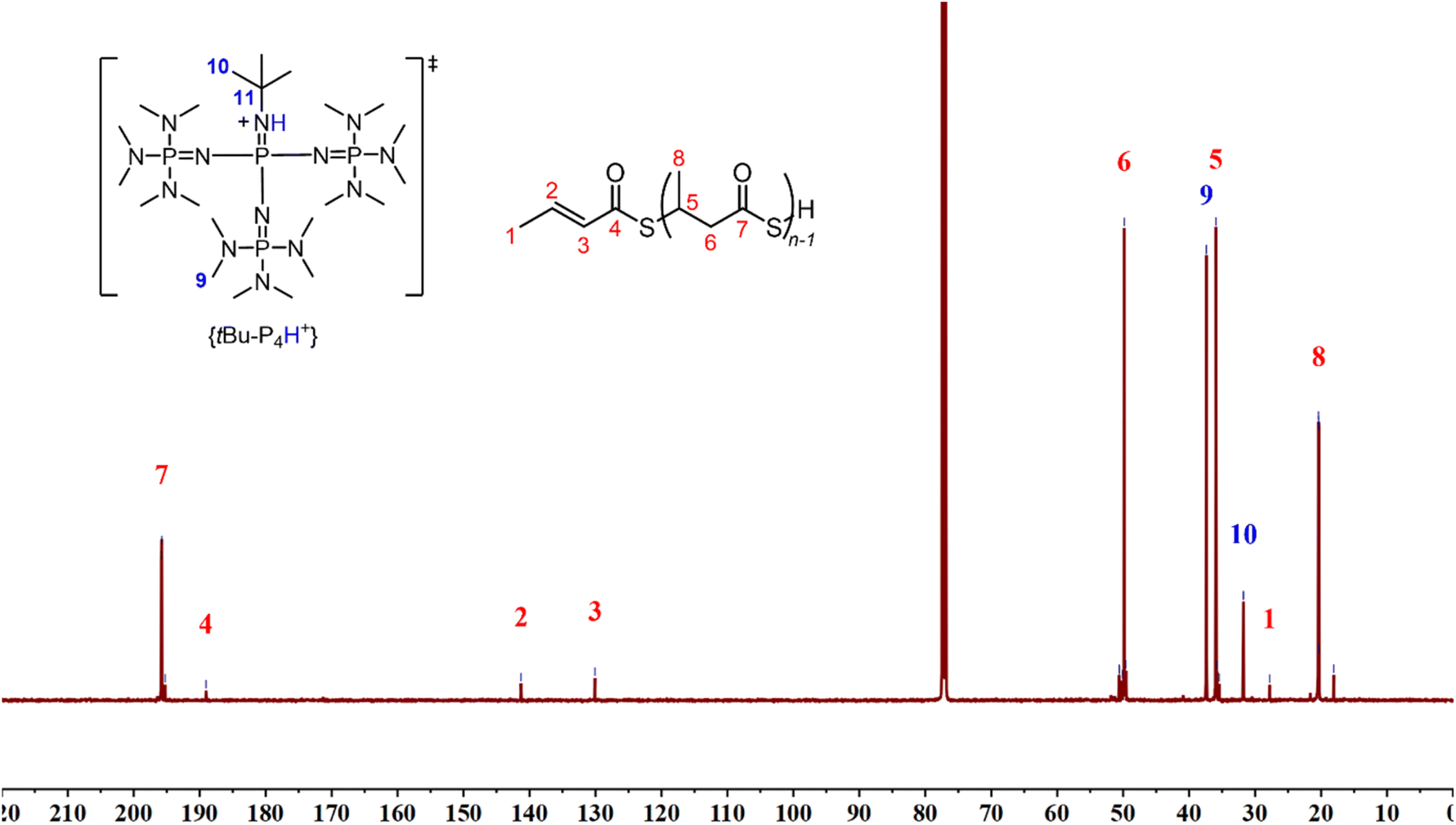 | ||
| Fig. 4 13C{1H} NMR spectrum (125 MHz, CDCl3, 25 °C) of a P3TB prepared with tBu-P4 in toluene (Table 1, entry 7) (tertiary C11 carbon signal of {[tBu-P4]H}+ possibly appears at δ ca. 51 ppm). | ||
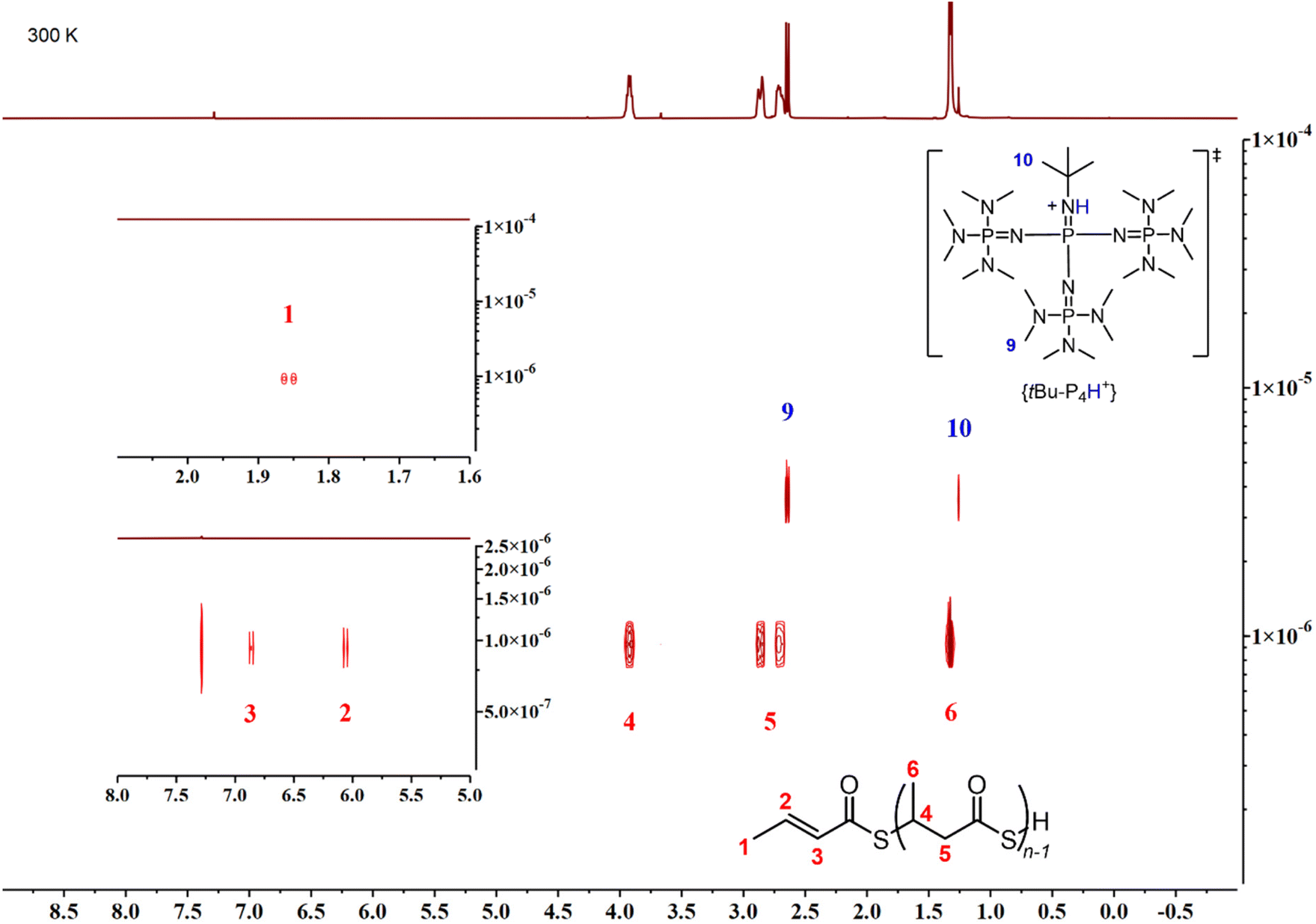 | ||
| Fig. 5 DOSY NMR spectrum (500 MHz, CDCl3, 25 °C) of a P3TB prepared with tBu-P4 in toluene (Table 1, entry 6); the bottom-left and upper-left insers correspond to zoomed regions of the thiocrotonate hydrogens. | ||
To gain further insights, we monitored the stoichiometric reaction between tBu-P4 and TBL in C6D6 at room temperature by 31P NMR spectroscopy (Fig. S28†). This analysis revealed the near-quantitative formation of the phosphazenium cation {[tBu-P4]H}+ (δ(31P) 13.20 and −23.52 ppm; δ(1H) 2.64 ppm (Me2N–P), 1.25 ppm (tBu–NP)) within a few minutes. This observation is in line with the reported findings on the ROP of γ-butyrolactone promoted by tBu-P4 in the absence of a co-initiator, who also evidenced the quantitative formation of the phosphazenium cation {[tBu-P4]H}+.26 The corresponding 1H NMR spectrum is not very informative (Fig. S29†): it features broadened signals with the most intense ones at δ values of ca. 1.56, 2.55 and 2.95 ppm assignable to the {[tBu-P4]H}+ cation; a weak, broad signal is observed at δ ca. 7.5 ppm which may correspond to the NH+ in this cation. In addition, a very broad signal is observed at δ 3–4 ppm but no distinct signals assignable to a putative thiocrotonate species of the type CH3CHCHC(O)S− could be observed (in particular no signals were observed in the region δ 6–7 ppm). This contrasts with the aforementioned work which reported, in addition to the signals for the {[tBu-P4]H}+ cation, a set of sharp signals in the region δ ca. 3.0–4.25 ppm that were assigned to an anionic γ-butyrolactone dimer which can exist in either the ring-retention or ring-opening form, the ratio of which can be varied with the reaction conditions (substrate ratio and reaction time).26 Hence, the NMR analysis did not provide any hint on the nature of the anionic species that pairs with the {[tBu-P4]H}+ cation resulting from the reaction of tBu-P4 and TBL.
As evidenced by DOSY NMR analysis, the ROP of rac-TBL promoted by tBu-P4 in toluene at room temperature gives a single population of macromolecules, that is α-thiocrotonate,ω-[thiocarboxylate-phosphazenium] end-capped linear P3TB. Based on the relative intensity of the thiocrotonate and TBL repeating unit signals, the molar mass of the resulting P3TBs can thus be, in principle, easily determined by 1H NMR. For example, the ROP of rac-TBL promoted by tBu-P4 at a [TBL]0/[tBu-P4]0 feed ratio of 50:1 in toluene gave the corresponding P3TB with Mn,NMR = 9500 g mol−1. While this value is, to some extent, close to the (uncorrected) value measured by SEC (Mn,SEC = 8200 g mol−1), it is twice the expected value based on a fast, fully efficient initiation (Mn,calc = 4600 g mol−1; Table 1, entry 5) (vide infra). Note that, however, as shown in Table 1 and as aforementioned, Mn,NMR values do not necessarily match those determined by SEC. These discrepancies between the expected and experimental molar mass values may suggest the possible presence of cyclic P3TB along with linear macromolecular chains. The formation of some cyclic PTEs, which cannot be fully assessed by NMR spectroscopic analyses, would be further consistent with a ROP of the thiolactone mediated by ion-pair active species. Indeed, organocatalytic zwitterionic polymerization of cyclic esters is known to promote the formation of cyclic polymers.27,28
The prepared P3TBs were also analyzed by mass spectrometry (MS). MALDI-ToF- MS using trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]-malononitrile (DCTB), which revealed an effective matrix for such MS analyses of P3TBs produced by yttrium catalysts,14 proved ineffective in the present work: for all P3TB samples prepared from organocatalysts tBu-P4, tBu-P2 and BEMP, prepared either in toluene or in THF, no signal but the one attributed to the protonated phosphazene base was observed, even upon increasing the laser power to very high levels. Only in the case of a P3TB sample prepared with tBu-P1, the MALDI-ToF mass spectrum recorded at very high laser power returned signals for several populations of macromolecules, among which cyclic P3TB or linear α-thiocrotonate,ω-[thiocarboxylic] end-capped P3TB was unambiguously identified (Table 1, entry 3 and Fig. S30, S31†); note, indeed, that both cyclic P3TB and α-thiocrotonate,ω-[thiocarboxylic] end-capped P3TB have the same molecular formula and hence cannot be differentiated by MS. In a control experiment, a cyclic P3TB sample prepared by yttrium catalysis,14 was mixed with a small amount (ca. 1 wt%) of BEMP, and the resulting sample was subjected to MALDI-ToF MS analysis. Again, the MS signal of PTE macromolecules completely vanished. Unfortunately, phosphazene bases obviously, even if present in a small amount, for a yet obscure reason, are highly detrimental to obtaining MALDI-ToF mass spectra of such P3TB samples.
Informative results could be further gained by performing ESI-MS analysis (in the positive mode) of a relatively low molar mass P3TB prepared with tBu-P4 in THF (Table 1, entry 7) (Fig. 6). In addition to the very intense signal for the {[tBu-P4]H}+ cation (m/z = 634.4352), several series of macromolecular ions could be observed, three of which were unambiguously assigned to (i) (as the most intense one) α-thiocrotonate,ω-thiocarboxylic end-capped linear P3TB or cyclic P3TB (i.e., both have the same molecular weight), (ii) α-thiocrotonate,ω-carboxylic end-capped linear P3TB and (iii) α-thiocrotonate,ω-methoxycarbonyl end-capped linear P3TB (see Fig. S32–S34† for details of the ESI-MS spectra). Note that the former two populations are also observed in the MALDI-ToF mass spectrum. The observation of the latter two populations in the ESI mass spectrum can be accounted for, by hydrolysis and esterification by methanol (used as an ESI matrix) of the former population, respectively, although it cannot be deciphered whether it is linear or cyclic.
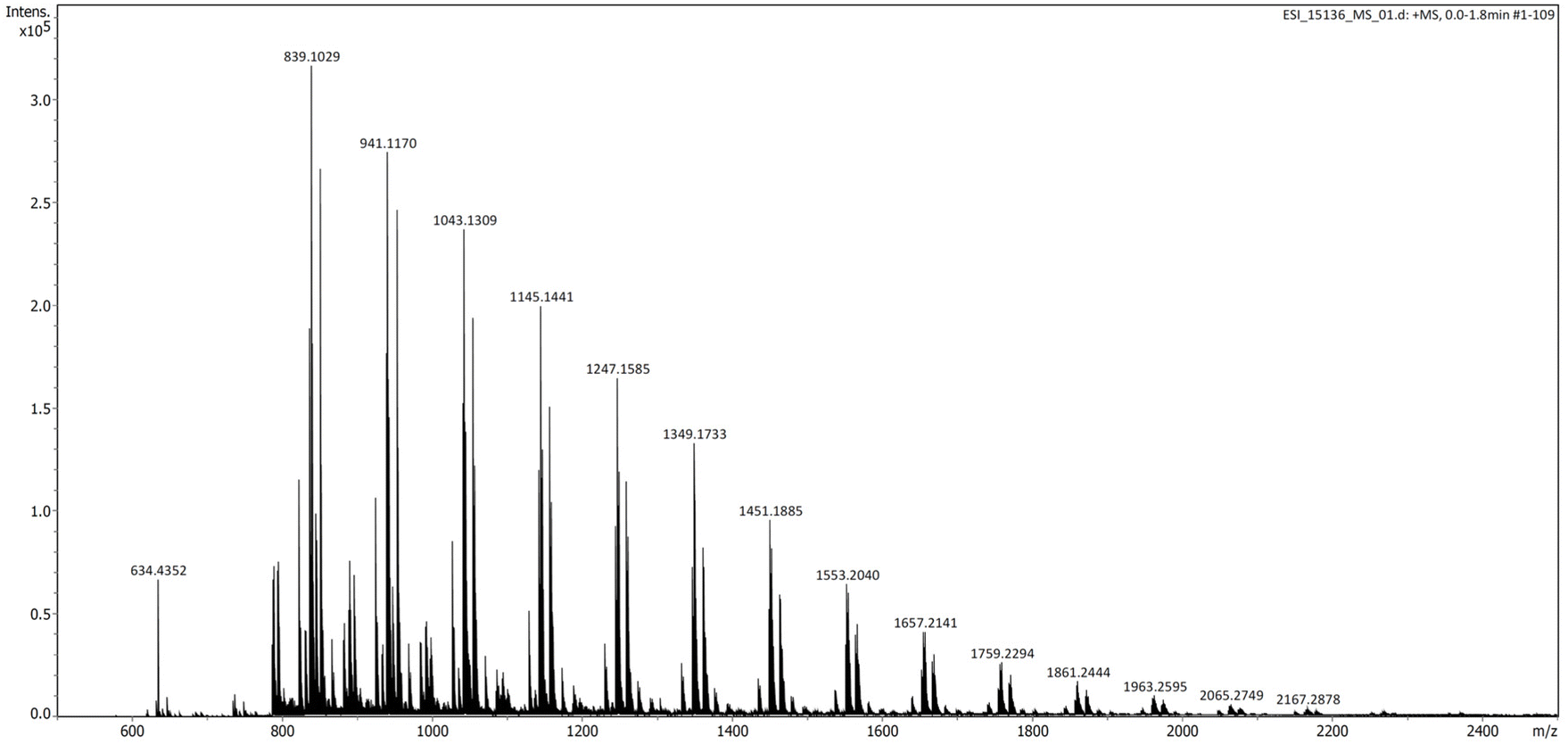 | ||
| Fig. 6 ESI mass spectrum (positive mode, MeOH) of a P3TB prepared with tBu-P4 in THF (Table 1, entry 7) (intensity of the signals above m/z = 880 has been enhanced). The most intense series (with peak picking values) corresponds to linear {CH3CH=CHC(O)S[C4H6OS]n−1H + Na}+ or cyclic {[C4H6OS]n + Na}+ with, for n = 8, m/z exp = 839.1029 vs. m/z theo = 839.1012; for n = 9, m/z exp = 941.1170 vs. m/z theo = 941.1152; for n = 10, m/z exp = 1043.1309 vs. m/z theo = 1043.1291; for n = 11, m/z exp = 1145.1441 vs. m/z theo = 1145.1431; for n = 12, m/z exp = 1247.1585 vs. m/z theo = 1247.1570. | ||
To possibly gain further insight into the macromolecular topology and possibly evaluate the presence of linear and/or cyclic P3TB in the isolated samples, TGA experiments were conducted on four different, representative P3TBs (Table 1, entries 2, 3, 9 and 10; Fig. S35–S38†). The polymers prepared with BEMP and tBu-P1 (Mn,SEC = 8700 and 9200 g mol−1) featured decomposition temperatures (Td, defined as the temperature for 5% weight loss) of 192 and 196 °C, respectively; these values are somewhat lower than that of a ‘pure’ cyclic P3TB with similar molar mass prepared by yttrium catalysis (Td = 205 °C, Mn,SEC = 7800 g mol−1).14 On the other hand, these values are significantly higher than those of the polymers prepared using tBu-P4 (Td = 168–170 °C), even though these polymers have a higher apparent molar mass (Mn,SEC = 17200–18500 g mol−1). The latter Td values are much lower than any of those observed for P3TBs prepared by yttrium catalysis or with NaOMe (Td = 186–230 °C).14 Cyclic P3TBs are anticipated to feature higher thermal stability as compared to their linear counterparts, as reported for other polyesters.29 Hence, overall, the above observations suggest that the P3TBs prepared using BEMP and tBu-P1 may be mixtures of cyclic and linear macromolecules, while those prepared using tBu-P4 could be enriched in linear topology.
Conclusion
The results of our preliminary studies on the ROP of TBL mediated by different phosphazenes, namely BEMP, tBu-P1, tBu-P2, and tBu-P4, are documented herein, emphasizing mechanistic insights. NMR spectroscopic analyses conducted on P3TBs produced using tBu-P4 do support the formation of α-thiocrotonate,ω-[thiocarboxylate-phosphazenium] end-capped linear P3TBs through a ROP mediated by ion-pair active species along with S-alkyl bond cleavage. This suggests that such bulky superbases behave essentially as basic, non-nucleophilic initiators, although the exact nature of the putative anionic initiating species generated in the very early-stage remains unclear (Scheme 3). On the other hand, regarding the P3TBs produced with less basic and less sterically hindered phosphazenes such as BEMP and tBu-P1, we suggest that the latter ones may trigger the ROP of this thiolactone also through nucleophilic attack at the carbonyl group of rac-TBL with S-acyl bond cleavage, ultimately affording {α-[BEMP/tBu-P1]+,ω-SH}P3TB chains (Scheme 3). Both mechanisms may likely compete and take place at the same time, accounting in part for the uncontrolled molar mass of the resulting polymers. ROP of TBL performed in THF proceeds, to some extent, with some “living” features affording P3TB with a molar mass of up to Mn,NMR = 17.7 kg mol−1 and ĐM = 1.42. | ||
| Scheme 3 Putative initiating steps at play in the ROP of TBL promoted by different phosphazenes. | ||
Author contributions
J.-F. C and S. M. G. conceived the project and directed the research. H.L. designed and conducted experiments and analyzed the results with the help of colleagues acknowledged above. All authors contributed to the initial draft and revised subsequent versions by reviewing the entire manuscript.Conflicts of interest
The authors declare no conflict-of-interest.Acknowledgements
We are grateful for the financial support provided by the China Scholarship Council (CSC Ph.D. grant No. 201804910783 to H.L.). We thank Jérôme Ollivier, Philippe Jehan, Fabian Lambert and Elsa Caytan for SEC, MS and NMR analyses. Part of this work has been developed using NMR spectrometers from CRMPO (UAR 2025 ScanMAT, CNRS-Université de Rennes) facility.References
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Catalysis as an Enabling Science for Sustainable Polymers, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- W. N. Ottou, H. Sardon, D. Mecerreyes, J. Vignolle and D. Taton, Update and Challenges in Organo-Mediated Polymerization Reactions, Prog. Polym. Sci., 2016, 56, 64–115 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. Lohmeijer and J. L. Hedrick, Organocatalytic Ring-Opening Polymerization, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Organocatalysis: Opportunities and Challenges for Polymer Synthesis, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- M. Fevre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, N-Heterocyclic, Carbenes (NHCs) as Organocatalysts and Structural Components in Metal-Free Polymer Synthesis, Chem. Soc. Rev., 2013, 42, 2142–2172 RSC.
- A. P. Dove, Organic Catalysis for Ring-Opening Polymerization, ACS Macro Lett., 2012, 1(12), 409–1412 Search PubMed.
- A. Khalil, S. Cammas-Marion and O. Coulembier, Organocatalysis Applied to the Ring-Opening Polymerization of β-Lactones: A Brief Overview, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 657–672 CrossRef CAS.
- J. Xu, X. Wang, J. Liu, X. Feng, Y. Gnanou and N. Hadjichristidis, Ionic H-bonding Organocatalysts for the Ring-Opening Polymerization of Cyclic Esters and Cyclic Carbonates, Prog. Polym. Sci., 2022, 125, 101484 CrossRef CAS.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Guanidine and Amidine Organocatalysts for Ring-Opening Polymerization of Cyclic Esters, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- C. G. Jaffredo, J. F. Carpentier and S. M. Guillaume, Controlled ROP of β-Butyrolactone Simply Mediated by Amidine, Guanidine, and Phosphazene Organocatalysts, Macromol. Rapid Commun., 2012, 33, 1938–1944 CrossRef CAS PubMed.
- C. G. Jaffredo, J.-F. Carpentier and S. M. Guillaume, Organocatalyzed Controlled ROP of β-Lactones towards Poly(hydroxyalkanoate)s: From β-Butyrolactone to Benzyl β-Malolactone Polymers, Polym. Chem., 2013, 4, 3837–3850 RSC.
- R. M. Shakaroun, P. Jéhan, A. Alaaeddine, J.-F. Carpentier and S. M. Guillaume, Organocatalyzed Ring-Opening Polymerization (ROP) of Functional β-Lactones: New Insights into the ROP Mechanism and Poly(hydroxyalkanoate)s (PHAs) Macromolecular Structure, Polym. Chem., 2020, 11, 2640–2652 RSC.
- T. Lutke-Eversloh, A. Fischer, U. Remminghorst, J. Kawada, R. H. Marchessault, A. Bogershausen, M. Kalwei, H. Eckert, R. Reichelt, S. J. Liu and A. Steinbuchel, Biosynthesis of Novel Thermoplastic Polythioesters by Engineered Escherichia coli, Nat. Mater., 2002, 1, 236–240 CrossRef PubMed.
- H. Li, J. Ollivier, S. M. Guillaume and J.-F. Carpentier, Tacticity Control of Cyclic Poly(3-Thiobutyrate) Prepared by Ring-Opening Polymerization of Racemic β-Thiobutyrolactone, Angew. Chem., Int. Ed., 2022, 61, e202202386 CAS.
- For a recent account, see: H. Li, S. M. Guillaume and J.-F. Carpentier, Polythioesters Prepared by Ring-Opening Polymerization of Cyclic Thioesters and Related Monomers, Chem. – Asian J., 2022, e202200641 CAS.
- D. Fleš and V. Tomašić, Preparation of Poly-(S)-(−)-[α-(p-toluenesulfonamido)-β-propiothiolactone], J. Polym. Sci., Part B: Polym. Lett., 1968, 6, 809–813 CrossRef.
- M. Suzuki, K. Makimura and S. Matsuoka, Thiol-Mediated Controlled Ring-Opening Polymerization of Cysteine-Derived β-Thiolactone and Unique Features of Product Polythioester, Biomacromolecules, 2016, 17, 1135–1141 CrossRef CAS PubMed.
- W. Xiong, W. Chang, D. Shi, L. Yang, Z. Tian, H. Wang, Z. Zhang, X. Zhou, E.-Q. Chen and H. Lu, Geminal Dimethyl Substitution Enables Controlled Polymerization of Penicillamine-Derived β-Thiolactones and Reversed Depolymerization, Chem, 2020, 6, 1831–1843 CAS.
- T. J. Bannin and M. K. Kiesewetter, Poly(thioester) by Organocatalytic Ring-Opening Polymerization, Macromolecules, 2015, 48, 5481–5486 CrossRef CAS PubMed.
- V. Puchelle, Y. Latreyte, M. Girardot, L. Garnotel, L. Levesque, O. Coutelier, M. Destarac, P. Guégan and N. Illy, Functional Poly(ester-alt-sulfide)s Synthesized by Organo-Catalyzed Anionic Ring-Opening Alternating Copolymerization of Oxiranes and γ-Thiobutyrolactones, Macromolecules, 2020, 53, 5188–5198 CrossRef CAS.
- N. Illy and E. Mongkhoun, Thiolactone Chemistry, a Versatile Platform for Macromolecular Engineering, Polym. Chem., 2022, 13, 4592–4614 RSC.
- O. Ivanchenko, S. Mazières, S. Harrisson and M. Destarac, On-Demand Degradation of Thioester/Thioketal Functions in Vinyl Pivalate-Derived Copolymers with Thionolactones, Macromolecules, 2023, 56, 4163–4171 CrossRef CAS.
- L. Zhang, F. Nederberg, R. C. Pratt, R. M. Waymouth, J. L. Hedrick and C. G. Wade, Phosphazene Bases: A New Category of Organocatalysts for the Living Ring-Opening Polymerization of Cyclic Esters, Macromolecules, 2007, 40, 4154–4158 CrossRef CAS.
- S. Liu, C. Ren, N. Zhao, Y. Shen and Z. Li, Phosphazene Bases as Organocatalysts for Ring-Opening Polymerization of Cyclic Esters, Macromol. Rapid Commun., 2018, 39, e1800485 CrossRef PubMed.
- D. A. Culkin, W. Jeong, S. Csihony, E. D. Gomez, N. P. Balsara, J. L. Hedrick and R. M. Waymouth, Zwitterionic Polymerization of Lactide to Cyclic Poly(Lactide) by Using N–Heterocyclic Carbene Organocatalysts, Angew. Chem., 2007, 119, 2681–2684 CrossRef.
- M. Hong and E. Y.-X. Chen, Towards Truly Sustainable Polymers: A Metal-Free Recyclable Polyester from Biorenewable Non-Strained γ-Butyrolactone, Angew. Chem., Int. Ed., 2016, 55, 4188–4193 CrossRef CAS PubMed.
- Y. A. Chang and R. M. Waymouth, Recent Progress on the Synthesis of Cyclic Polymers via Ring-Expansion Strategies, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2892–2902 CrossRef CAS.
- H. A. Brown and R. M. Waymouth, Zwitterionic Ring-Opening Polymerization for the Synthesis of High Molecular Weight Cyclic Polymers, Acc. Chem. Res., 2013, 46, 2585–2596 CrossRef CAS PubMed.
- M. Hong and E. X.-Y. Chen, Completely recyclable biopolymers with linear and cyclic topologies via ring-opening polymerization of γ-butyrolactone, Nat. Chem., 2016, 8, 42–49 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00707c |
| This journal is © The Royal Society of Chemistry 2023 |