
α-Borylalkyl radicals: their distinctive reactivity in modern organic synthesis
Nivesh
Kumar†
,
Reddy Rajasekhar
Reddy†
,
Nadim
Eghbarieh
and
Ahmad
Masarwa
 *
*
Institute of Chemistry, The Hebrew University of Jerusalem, Safra Campus, Givat Ram, Jerusalem 9190401, Israel. E-mail: Ahmad.Masarwa1@mail.huji.ac.il
First published on 27th November 2019
Abstract
Organoborons are extremely important for synthetic organic chemistry; they can serve as advanced intermediates for a variety of transformations. Such a well-known transformation involves the loss of the boron moiety, creating alkyl radicals. Although these originally developed protocols for alkyl radical generation remain in active use today, in recent years their α-boryl carbon-centred radicals have been joined by a new array of radical generation strategies that offer a unique reactivity to forge a wider diversity of organoborons that often operate under mild and benign conditions. Herein, we will highlight the stability and reactivity of α-borylalkyl radicals and their remarkably recent advances in order to further utilise them for C–C and C–heteroatom bond formation. Their use for this purpose has been reported over the last decade in an attempt to guide the synthetic community. Various transition-metal and metal-free methods for their generation are presented, and more advanced photoredox approaches are discussed, mainly for the period of 2009–2019.
 Nivesh Kumar | Dr Nivesh Kumar was born in New Delhi, India. He received his MSc degree from Delhi University, New Delhi in 2012 and his PhD degree from the Indian Institute of Science Education and Research (IISER)-Bhopal, India under the guidance of Prof. Alakesh Bisai in 2017. In 2018, he joined the group of Dr Ahmad Masarwa at The Hebrew University of Jerusalem, Israel. |
 Reddy Rajasekhar Reddy | Dr Reddy Rajasekhar Reddy was born in Palukudoddi, Kurnool, India. He received his MSc degree from Yogi Vemana University, Kadapa, India in 2012 and his PhD degree from the Indian Institute of Science Education and Research (IISER)-Bhopal, India under the guidance of Prof. Prasanta Ghorai in 2018. In the same year, he joined the group of Dr Ahmad Masarwa at The Hebrew University of Jerusalem, Israel. |
 Nadim Eghbarieh | Nadim Eghbarieh was born in Israel in 1995. He completed his BSc degree in 2019, in chemistry biology, at The Hebrew university of Jerusalem. Currently he is a graduate student in Masarwa's lab. He is working in the field of organoboron chemistry. |
 Ahmad Masarwa | Dr Ahmad Masarwa received his PhD degree in Chemistry from the Technion-Israel Institute of Technology in 2013 under the supervision of Professor Ilan Marek. Later he moved to the University of California, Berkeley (USA) where he completed his postdoctoral studies working in the research group of Professor Richmond Sarpong. In 2016, Ahmad began his independent research at The Hebrew University of Jerusalem, Israel. |
1. Introduction
Organoborons1 are organometallic equivalents, which are considered to be one of the most valuable reagents in modern organic synthesis, providing rapid access to a wide array of transformations including the construction of C–C and C–heteroatom bonds.1a,b These compounds have gained increasing popularity because of the steric and electronic properties of the Lewis acidic boron moiety, which can be easily tuned just by changing the substitution around it, which allows diverse reactivity, as a stoichiometric reagent and as well as a catalyst.1c,dNowadays the presence of boron functionality in natural products and synthetic drugs has appeared repeatedly. For example, the antibiotics aplasmomycin, boromycin, and tartolon-B contain boron in the form of borate complex.2 Drugs such as Tavaborole, Bortezomib and others also contain a boron functional group in them.3 To synthesise such motifs, metal and metal-free strategies are available;4 however, radical pathways could provide a unique reactivity to obtain complex structures transformed to a greater extent.
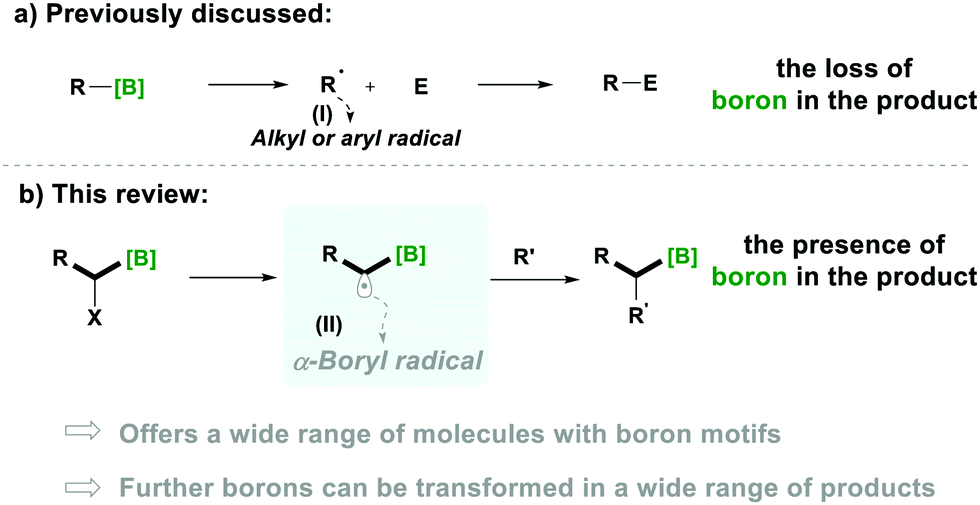
Notably, organoboron5a–i derivatives have become a convenient source for alkyl radicals and provide a green alternative to tin-based radical chemistry.5j–n The generation of alkyl radicals (Type-I) from organoboron derivatives, through the loss of the boron functionality, is a well-established chemistry, which results in further utilisation of these radicals in various transformations (Scheme 1a).5i–m
 | ||
| Scheme 1 General schematic representation of α-boryl radicals. | ||
In comparison, the generation of α-alkyl-radicals from a boron moiety (Type-II), without losing boron, is less known and their special reactivity is expected to offer a respectable range of transformations for the synthesis of organic molecules in which the boron functionality is left intact for further utilisation (Scheme 1b).
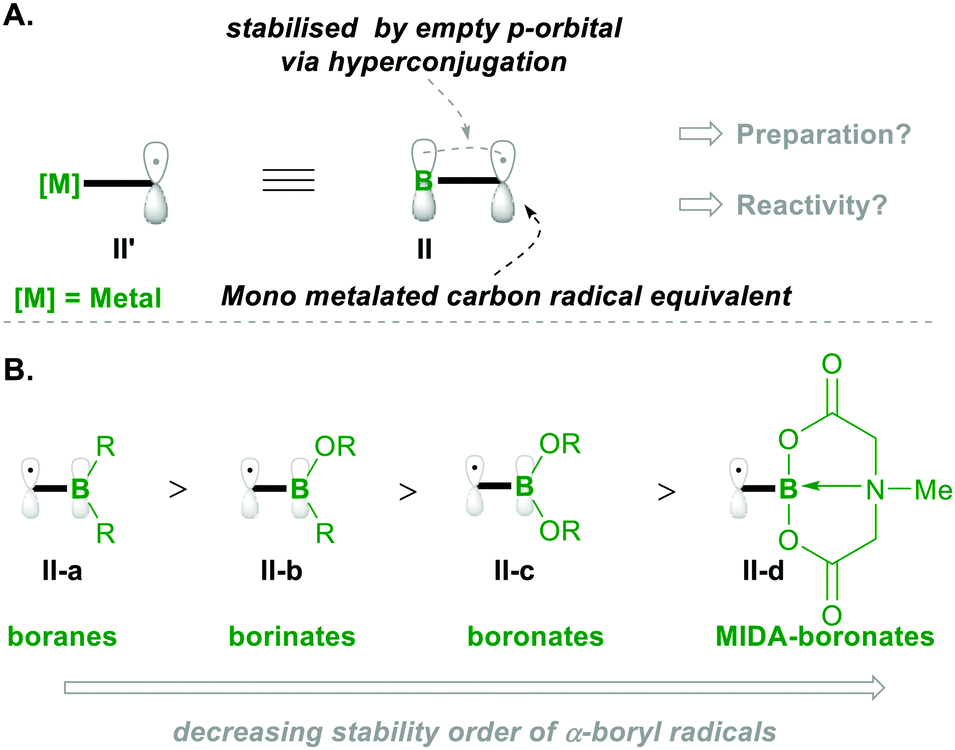
Before going into the discussion, let us first introduce the characteristics of the α-boryl radical species (which is the equivalent of the α-metalated radical; see II′, Scheme 2a) in terms of stability and electronics, based on its substitution pattern. The α-boryl radicals are more stabilised than the secondary alkyl-radicals by 14.5 kcal mol−1. Similarly, the α-boryl radical, obtained by H-abstraction, at the α-position, from Et3B was found to be more stable than the corresponding methyl radical of toluene.6 These α-boryl radicals (e.g., II) are stabilised by the empty p-orbital of boron through hyperconjugation-like structures, as depicted in Scheme 2A. Upon introduction of oxygen substituents on boron, the stabilisation of the corresponding α-boryl radicals decreases because the empty p-orbital of boron delocalises with nonbonding electrons on oxygen, resulting in decreased availability of the empty p-orbital of boron for radical stabilisation (Scheme 2B).7 Based on the number of oxygen substitutions on boron, the radical stabilisation order will be boranes II-a > borinates II-b > boronates II-c. The radical of MIDA-boronate (MIDA: N-methyliminodiaceticacid) is the least stable compared with others because the p-orbital is connected to nitrogen (in II-d).7,8
 | ||
| Scheme 2 Stabilisation of the α-boryl radical by an adjacent empty p-orbital. | ||
In this review, we would like to provide information on the reactions that are involved in the generation of α-boryl carbon-centred radical intermediates II and their utilisation for the synthesis of various important structural motifs. The key to the success of these reactions largely depends on the good matching philicity of the radical with alkene and other electrophiles. Additionally, this review will provide the various ranges of reactions of α-boryl radicals with electrophiles.
2. Synthetic methods of α-boryl radicals and their applications in synthesis
The α-boryl radical species (e.g., II) from different precursors have been generated in several ways for the synthesis of various organic frameworks with boron functionality. In general, for understanding the purpose of initiating the α-boryl alkyl radical, the generation has been classified into the following three categories, based on the reaction conditions:2.1 Using radical initiators
2.2 Visible light-mediated reactions
2.3 Miscellaneous reactions of three-component reactions
2.1 Initiation of the α-boryl radical by a radical initiator
In this category, the formation of α-boryl radicals was mainly promoted by radical initiators such as B(alkyl)3/bromine, azobisisobutyronitrile (AIBN) with stannates or with tris(trimethylsilyl)silane (TTMSS), and lauroyl peroxide (DLP).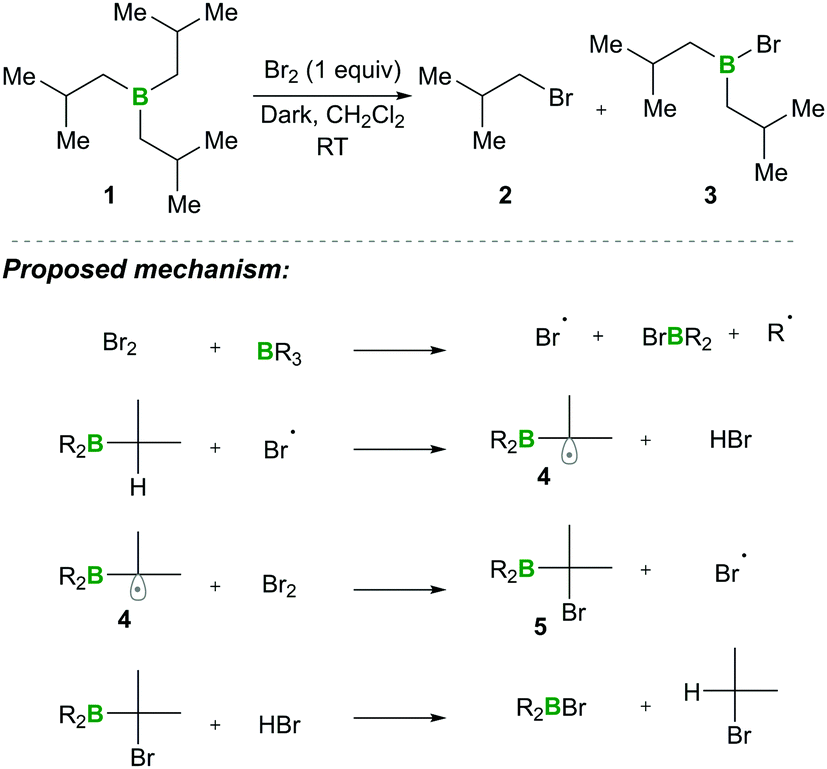 | ||
| Scheme 3 Anti-Markovnikov hydrobromination of alkenes and its mechanism. | ||
In 1973 the same group extended the above strategy for studying competitive bromination on B-isopropyl-9-borabicyclo[3.3.1]nonane 6.10 Interestingly, they obtained complete selectivity of bromination at an isopropyl group over a bicyclic ring (Scheme 4). This selectivity can result from the interaction between the odd electron and a vacant p-orbital (in 9), which is much more facile, due to the perfect degree of overlapping, than the C–H bond at the bridgehead position α to boron (highlighted in red in 6).10 Moreover, in this bromination study Lane and Brown found that the α-(C–H) position in relation to boron is 5.5 times more reactive than the tertiary hydrogen of cumene, owing to the same reason of the stabilisation of the empty p-orbital of boron.
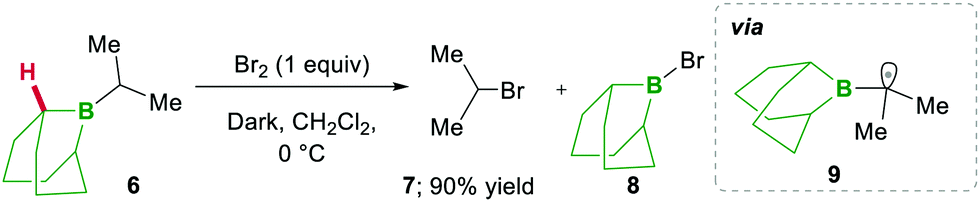 | ||
| Scheme 4 Competitive anti-Markovnikov bromination of B-isopropyl-9-borabicyclo[3.3.1]nonane. | ||
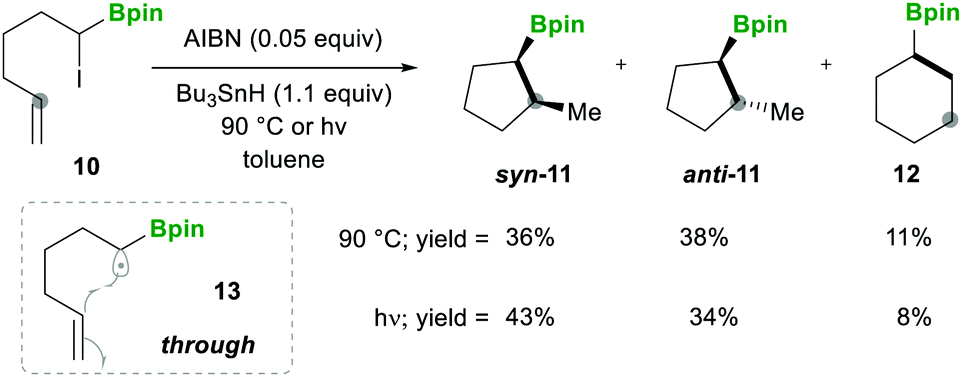 | ||
| Scheme 5 Intramolecular α-boryl carbon-centred radical cyclisation. | ||
Analogously, Batey's12 research group reported intermolecular additions of α-boryl radicals (see 18), generated from α-iodoalkylboranes 14, which are then coupled with electron-rich as well as electron-deficient alkenes. In this reaction, electron-deficient alkene 15 yields less yield because of the telomerisation of radical adducts with electron-deficient alkenes. However, electron-rich alkene 15 afforded good yields and no side reactions were observed (Scheme 6). Interestingly, when they performed a competitive reaction between electron-deficient and electron-rich alkenes, the addition occurred preferentially to the electron-deficient alkene.12
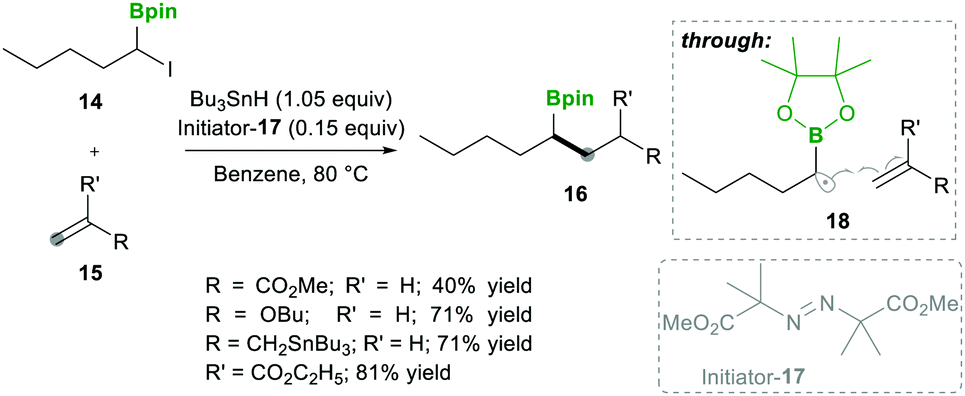 | ||
| Scheme 6 An intermolecular α-boryl carbon-centred radical coupled with alkenes. | ||
Later on, in 1999, the same group13 described the one-pot intramolecular additions of α-boryl radicals to tethered electron-rich alkenes 20 (or alkynes 21) via22a to 23a, when the organoboron products 23a were oxidised to 1,3-diols 20a. When they used the same conditions as above, they observed 5-exo cyclized product 23a with low yield, but upon the addition of tris(trimethylsilyl)silane, they obtained good yields. This reagent is known to trap the radicals with lower efficiency than tributyltin hydride does; ultimately, it reduces the degree of direct reduction (Scheme 7). Furthermore, the reaction results in good diastereoselectivity up to 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :81.13
:81.13
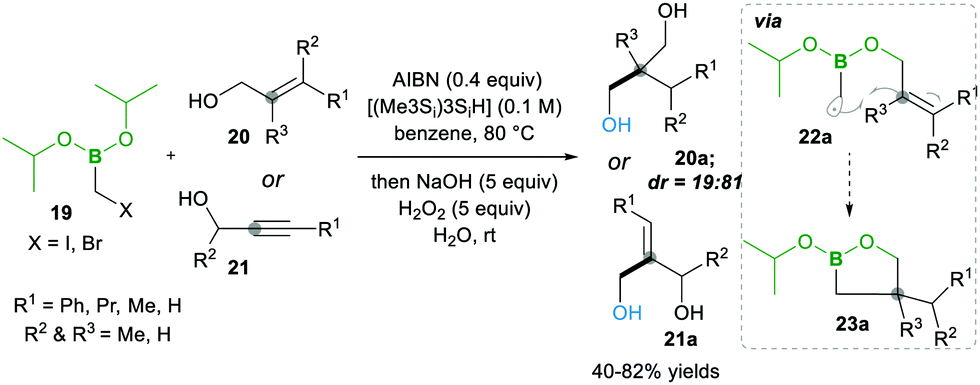 | ||
| Scheme 7 The α-boryl carbon-centred radical coupled with alkenes and alkynes. | ||
In 2019, Ouchi and Nishikawa14 carried out the polymerisation of isopropenyl boronate pinacol ester (IPBpin) 24 by taking advantage of the stabilisation of radicals adjacent to the boron through the empty p-orbital 26. This moderate stabilisation of radicals allows a reaction towards the chain growth of the polymer and suppresses the chain transfer reaction, which leads to a greater chain length of the polymer. Here IPBpin 24 was treated with AIBN and 2-cyano-2-propyl dodecyl trithiocarbonate (CPDT) as a chain-transfer agent by reversible addition–fragmentation chain-transfer polymerisation (RAFT), obtaining polymers, e.g., 25, with high molecular weights (Scheme 8).
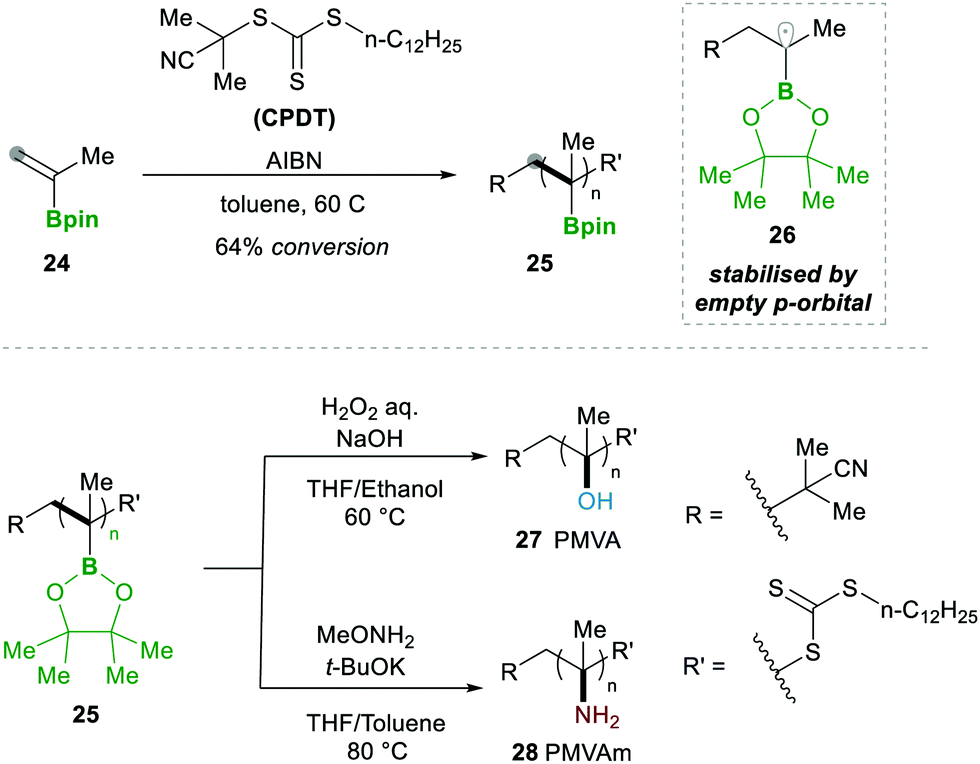 | ||
| Scheme 8 RAFT polymerisation of IPBpin 24. | ||
Later, to determine the significance of an empty p-orbital (in 26), they compared IPBpin 24 polymerisation with isopropenyl boronate carrying an N-methyliminodiacetic acid ester (IPBmida), which has no such boron p-empty orbital. Predictably, IPBmida was not polymerised, in comparison with IPBpin 24. Furthermore, they utilised the presence of boron in skeleton 25, which was transformed to alcohols 27 and amines 28 by the oxidation of pinacolatoboronate ester for the synthesis of poly(a-methyl vinyl alcohol) (PMVA) and poly(a-methyl vinyl amine) (PMVAm), respectively (Scheme 8).14
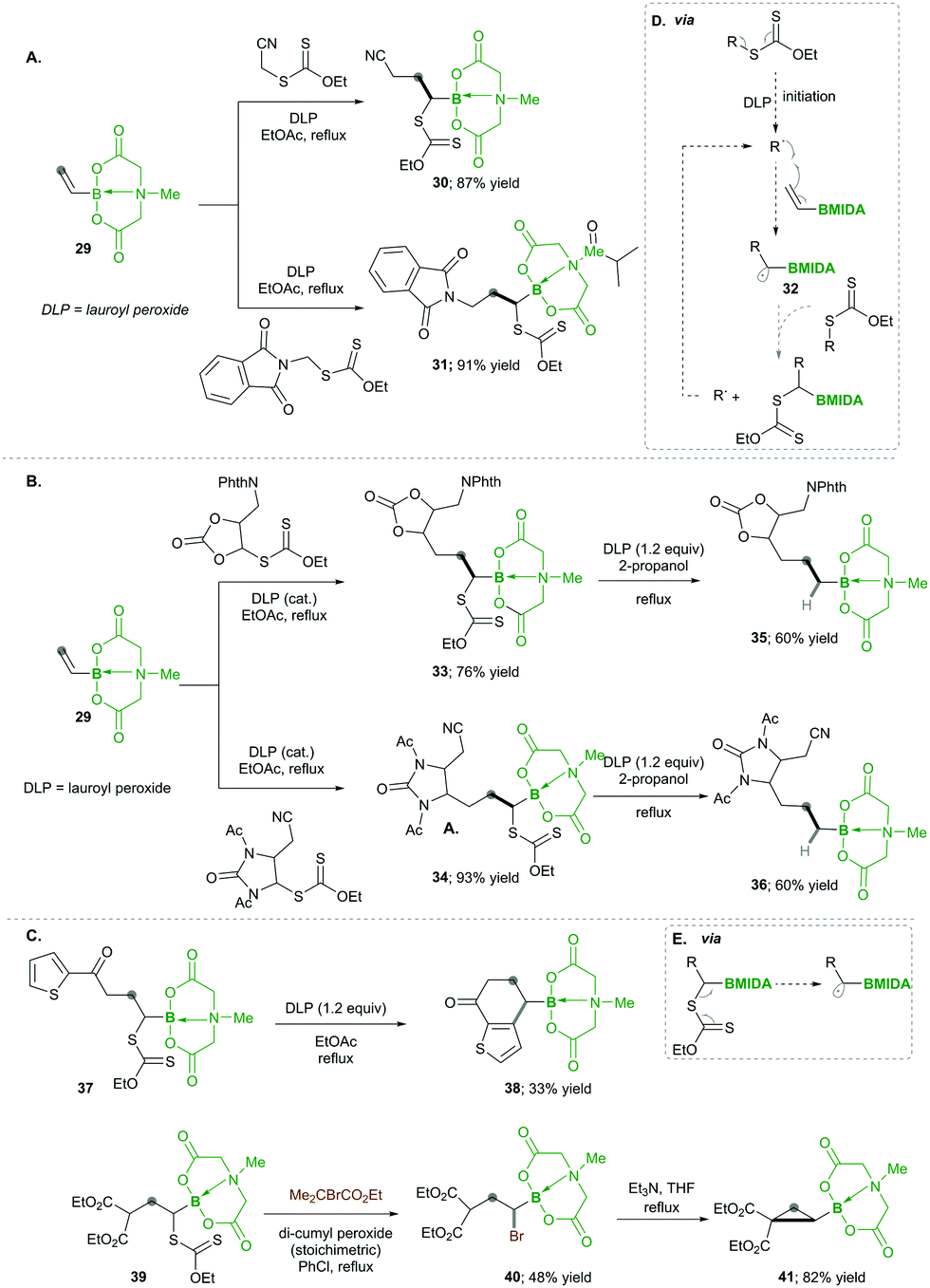 | ||
| Scheme 9 Radical chemistry of vinyl MIDA boronates. | ||
Finally, based on their high reactivity, the products, e.g., 37 and 39, were successfully utilised for different synthetic transformations (via box D) such as ring fusion with the heteroaromatic ring, which afforded the corresponding product 38 in low yields; the xanthates in 39 were replaced with bromine, following base-mediated nucleophilic substitution forms, which substituted cyclopropane 41 with good yields (Scheme 9C).
Consequently, in 2019, they utilised carbon-radical 45, which is similar to pinacolatoboronate in functionality.16 It was generated from α-boronylxanthates 42 for the synthesis of the terminal and internal boronates. The α-boronylxanthates 42 were reacted with different substituted alkenes 43 under DLP as a radical initiator and refluxed at 100 °C, yielding products 44 in moderate yields (Scheme 10).16
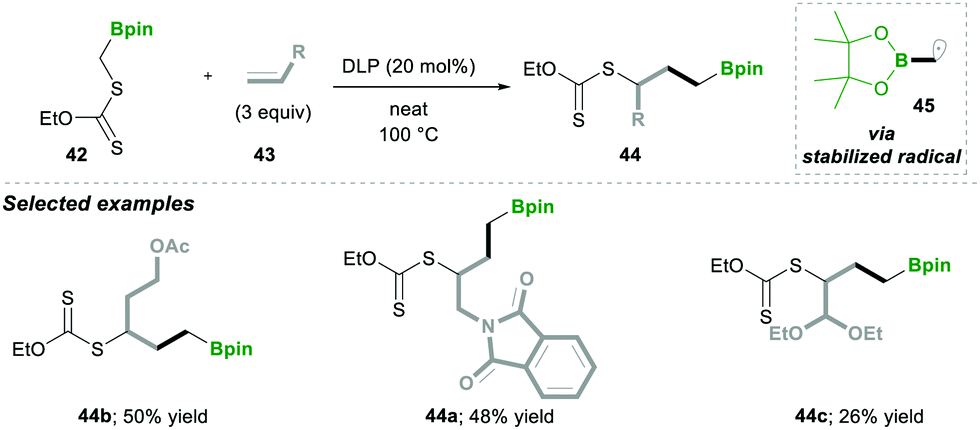 | ||
| Scheme 10 Synthesis of terminal boranes. | ||
Accordingly, they used vinyl-B(DEA) 46 when they started to diversify the organoboron products through a sequential radical reaction, as described in Scheme 11. In this case, vinyl-B(DEA) 46 forms a non-stabilised radical intermediate 51, which is similar to the previous case of the B(MIDA) radical intermediates e.g., 42, this allows the rapid formation of multi-substituted boronylxanthates 46. Later the B(DEA) functionality of the product was interconverted to pinacolatoborane esters 47, which were further treated under similar radical reactions and reacted with a wide range of alkenes 48, affording internal boronylxanthates 49. Finally, the reduction of the xanthate group in 49 with silyl hydride yielded the internal boranes 50 with good to excellent yields (Scheme 11). This strategy was also expanded for the synthesis of structurally complex internal boranes 50a–50f with very good yields.
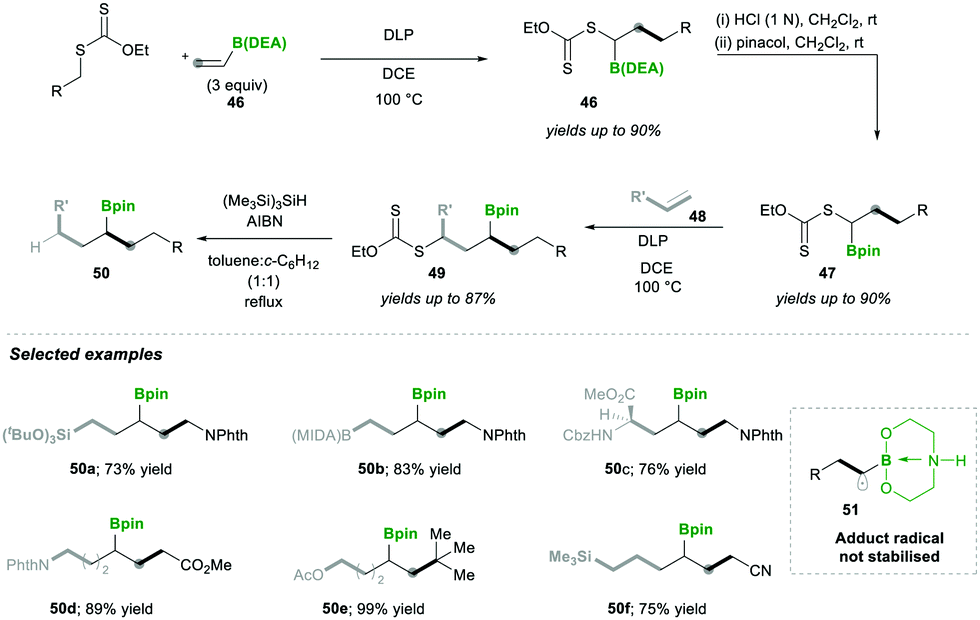 | ||
| Scheme 11 Synthesis of internal boranes. | ||
Additionally, they found that increasing the steric hindrance of the substituent in boronates, e.g., the benzopinacolato groups in 52, results in greater hydrolytic stability, which ultimately reduces the formation of side products.17 Regarding their hypothesis, they achieved better yields than with their simple pinacolate analogues 42. Further increasing the substitution at the α-position of boron with methyl, e.g., 52a, greatly increased the yield of product 54 (Scheme 12Avs.Scheme 12B). This clearly means that radical 55 is much more stabilised prior to the addition of alkenes 53 (Scheme 12).17
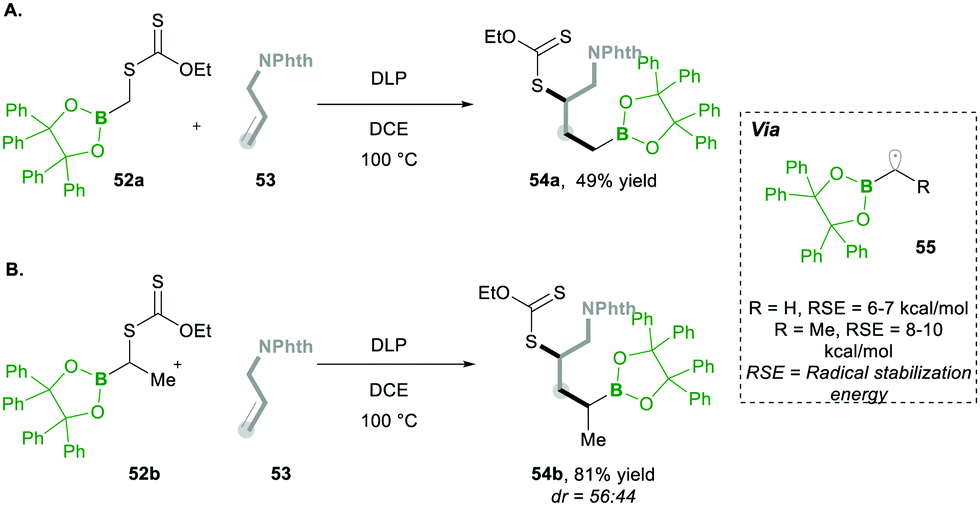 | ||
| Scheme 12 Synthesis of benzopinacolato boranes. | ||
Recently Renaud18 and co-workers utilised a similar radical initiator, i.e., DLP, as Zard's15–17 group for the atom transfer radical addition (ATRA) scenario. Here ICH2Bpin 56 and terminal alkene 57 were reacted in the presence of DLP as a radical initiator, affording 3-iodoalkyl borane 58. Furthermore, 3-iodoalkyl borane 58 was treated with a nucleophile (TBAF), leading to the cyclopropane product 59 with moderate to good yields (38–77%). In this reaction, various substituted terminal alkenes were tolerated well under these reaction conditions and the corresponding cyclopropanes 59a–59d were obtained (Scheme 13). Mechanistically, first, radical 60 is generated and then added via the ATRA step to alkene 57, which leads to 58, followed by the Lewis base-mediated intramolecular 1,3-elimination of 62 to finally afford the cyclopropanation product 59.18
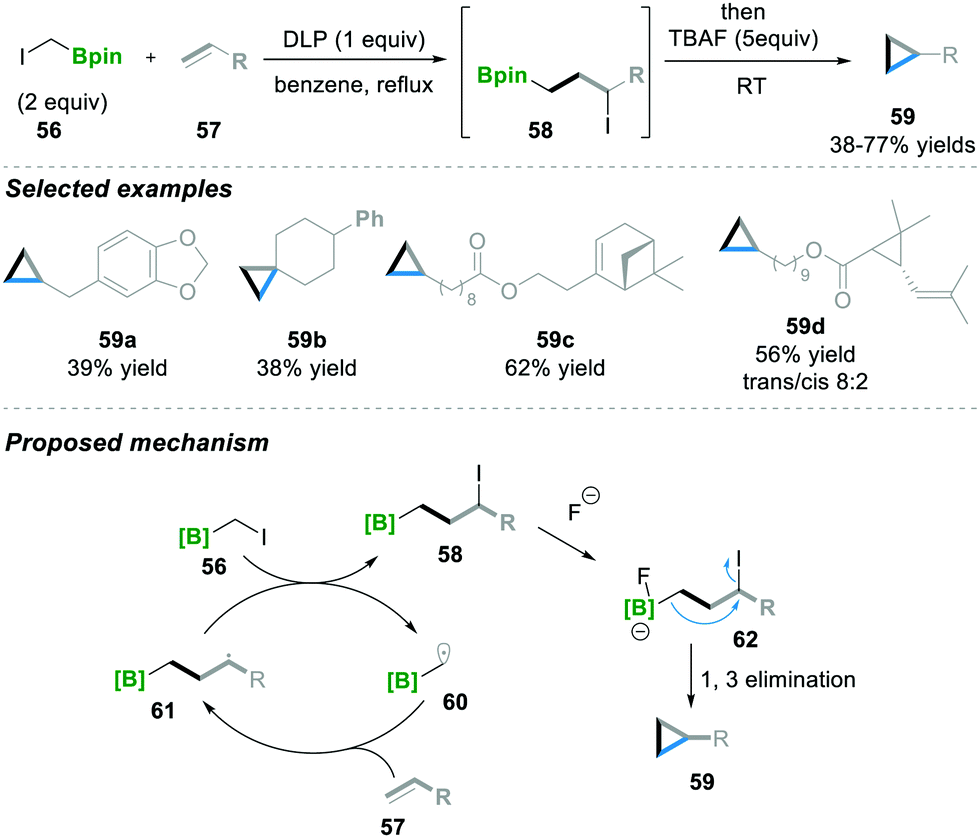 | ||
| Scheme 13 Cyclopropanation via ATRA and its mechanism. | ||
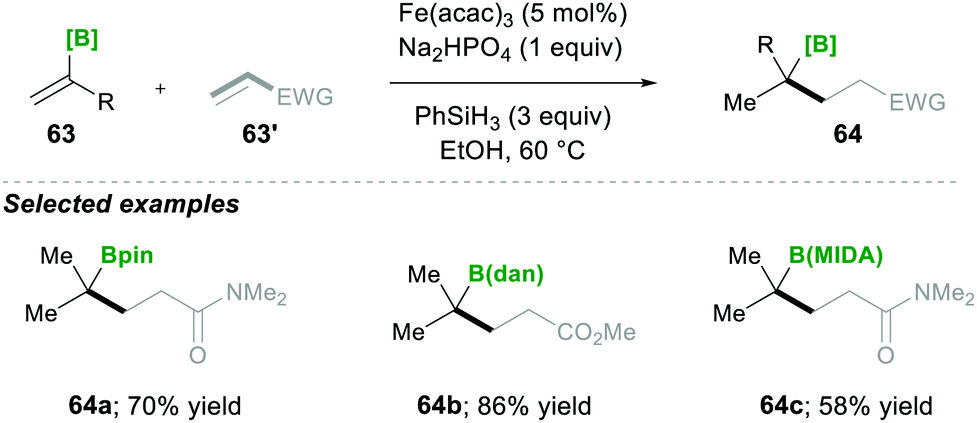 | ||
| Scheme 14 Iron catalysed radical-based Markovnikov alkene hydrofunctionalisation. | ||
Based on mechanistic investigations, they proposed the mechanism as depicted in Scheme 15. The first step of the reaction mechanism involves the formation of a bridged dimeric Fe complex 66 on the solvolysis of Fe(acac)3 with ethanol. This bridged complex reacts with PhSi(OEt)H2, which was generated from PhSiH3 upon reaction with Fe(II) species, yielding a hydride-bridged iron complex 67. This hydride-bridged complex forms an active species L2Fe(III)H. L2Fe(III)H then reacts with olefin 63, which forms tertiary radical 65via hydrofunctionalisation. Furthermore, the radical species 65 was added to the Michael acceptor, followed by reduction with Fe(II) and finally, upon protonation, yielded the desired product 64.19
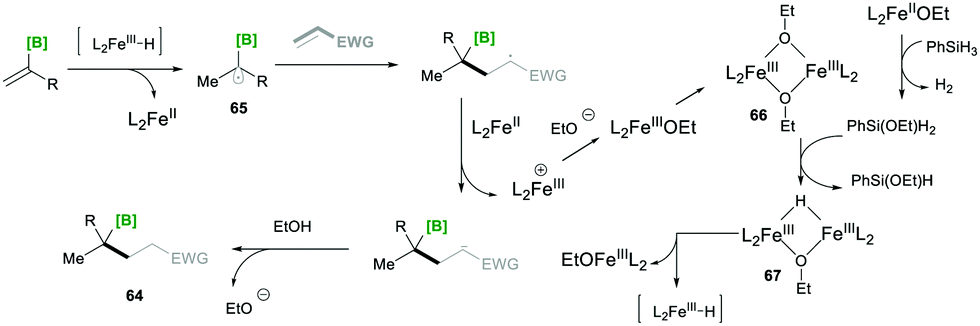 | ||
| Scheme 15 Proposed mechanism for the iron-catalysed radical-based Markovnikov alkene hydrofunctionalisation. | ||
In 2019, the Yorimitsu group20 reported the transition-metal-free diborylation of alkynes as an efficient approach for preparing 1,2-diborylated alkanes 71 or alkenes 72. The reaction was proposed to proceed via the 1,2-diboryl-1,2-dimetalloalkane intermediates 69–70. First, a single-electron reduction of the alkyne 68 generated radical anion 73, which readily attacked B2pin2, affording a borate intermediate. This borate intermediate followed an inter-metallate rearrangement (a cross-over experiment suggested that this step yielded a mixture of inter- and intra-products if there is a B2nep2 and B2pin2 mixture); an attack took place on the sterically less hindered carbon, which furnished the intermediate 69. This again takes a single electron, finally forming dianion 70, which in the presence of an electrophile afforded 1,2-diborylated alkanes 71. Alternatively, treatment with oxidant lithium iodide yielded 1,2-diborylated alkene 72 with E-isomer as the major product. Consequently, using the aforementioned method, they prepared a variety of 1,2-diborylated alkanes 71a–c in good yield and with moderate diastereoselectivity (Scheme 16).20
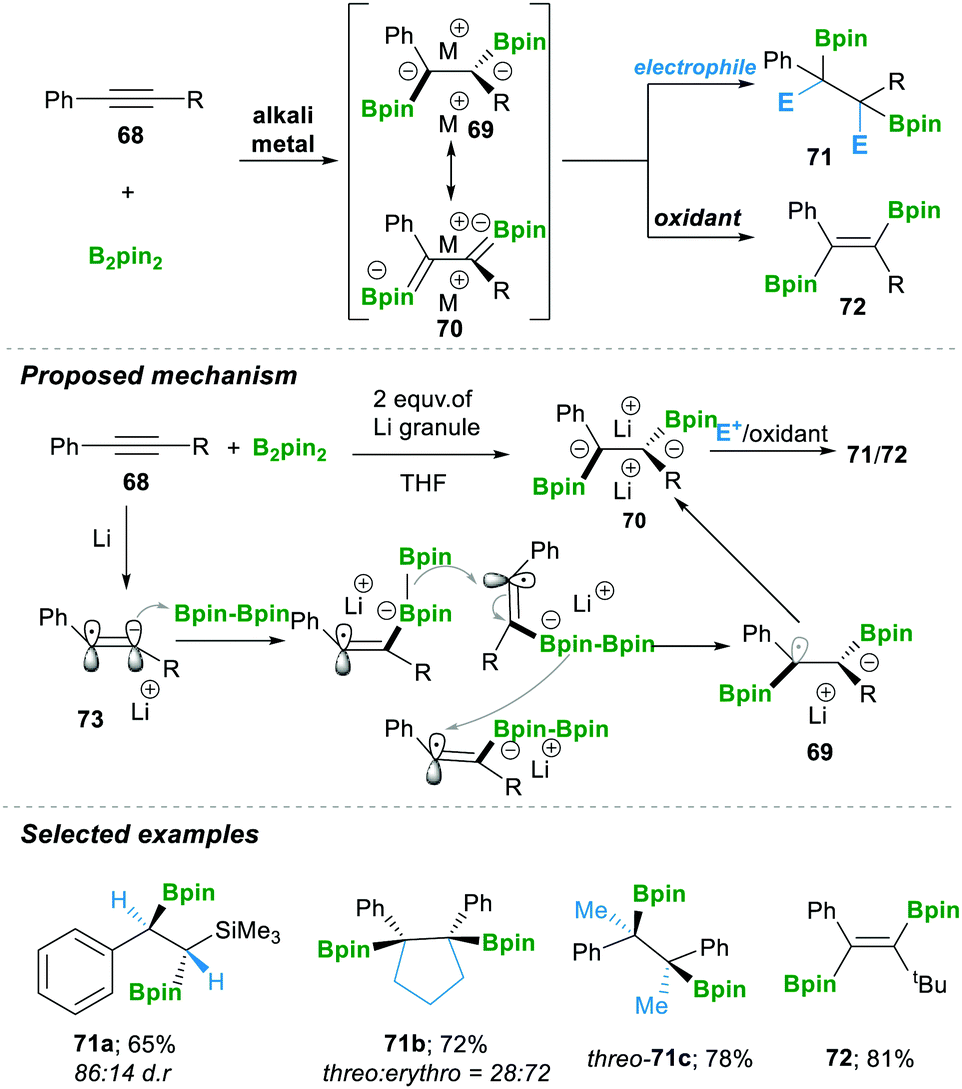 | ||
| Scheme 16 Transition-metal-free diborative reduction of alkynes’ direct access to vicinal-bisboronate esters. | ||
2.2 Visible light-mediated reactions for the formation of α-boryl radicals
These visible light-mediated reactions, in general, include two types: radical-induced 1,2-migrations and radical addition to vinyl boronic esters, vinyl boronate esters, and boronate complexes.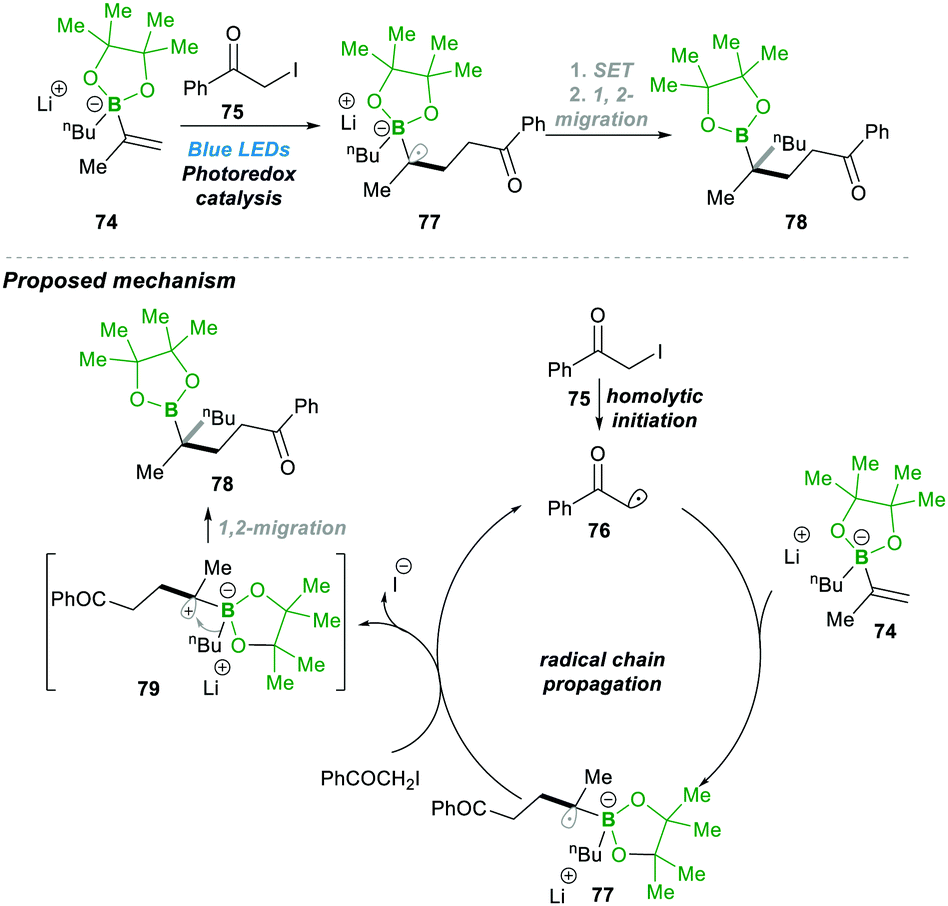 | ||
| Scheme 17 Visible light-mediated alkylation of vinyl-boronates via the 1,2-migratory rearrangement. | ||
Similarly, in 2018, the same group developed an enantiospecific three-component alkylation reaction on indoles 80 and furan 81′ following the boronate complex photoredox strategy.22 This method was found to be highly efficient; it allowed introducing a diverse class of functional groups in the final products 86–86′. The mechanism pathway is followed by the reaction described in Scheme 18; first, the photocatalyst Ru(II) undergoes irradiation with visible light to generate an excited Ru(II)-catalyst, which becomes reduced by electron-rich sacrificial boronate complex 87 (this was observed in the reaction mixture 85) via a single electron transfer (SET) step.
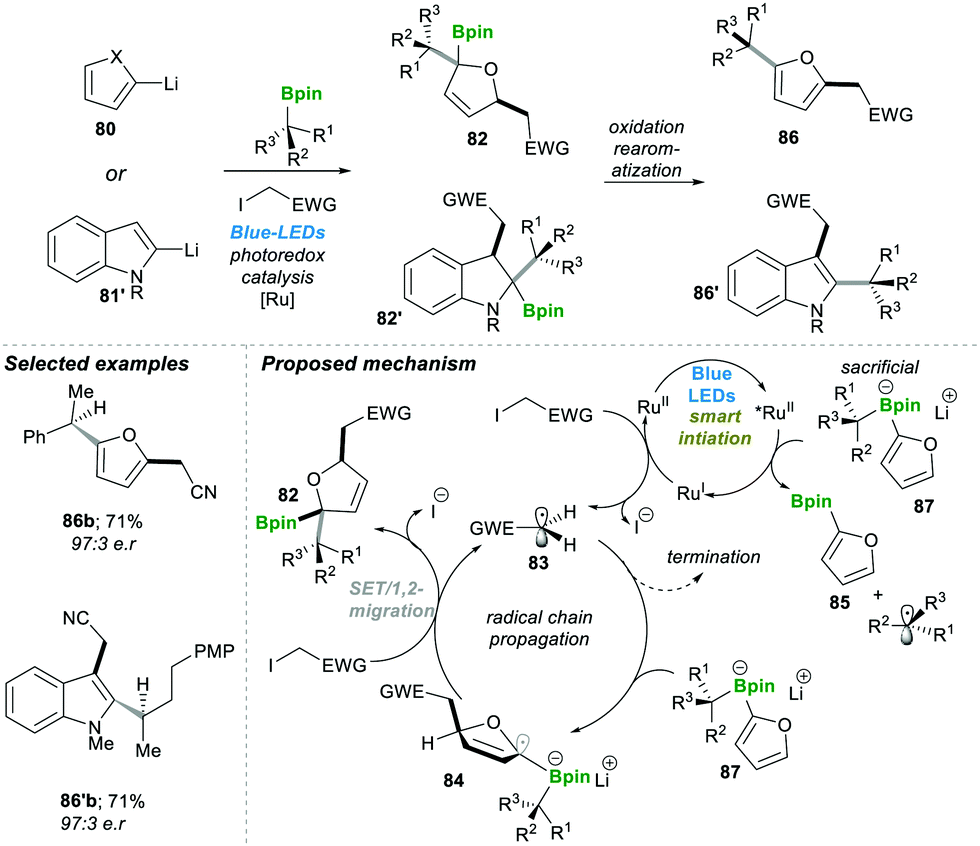 | ||
| Scheme 18 Radical-induced enantiospecific three-component alkylation reactions on indoles and furans. | ||
This electron-rich, reduced Ru(I)-catalyst breaks the C–I bond by the SET pathway, resulting in an electrophilic radical 83, and recycles the Ru(II) photocatalyst back to complete the catalytic cycle. Radical 83 attacks electron-rich furyl boronate 87, leading to an α-boronate radical 84. This α-boronate radical 84 undergoes SET reaction which yields the zwitterionic species 83, which undergoes a 1,2-metallate rearrangement to furnish a boronic ester 82. Oxidative re-aromatisation in the presence of molecular iodine and K2CO3 finally affords the desired 2,5-disubstituted furans 86 (Scheme 18). The reaction was found to be highly efficient and affords the product in good yield and with a high enantiomeric ratio (in 86b–86′b).22
Very recently, Shi23 and co-workers reported an unprecedented approach for synthesising gem-Bis(boryl)alkanes5b–f90. In this protocol they generated an alkenyl diboronate intermediate 89, starting from alkenyl Grignard reagents 88, which are added to bis(pinacolato)diboron (B2pin2), which then under visible-light photoredox catalytic conditions reacts with a wide range of alkyl halide 91, resulting in a wide range of highly substituted gem-diboryl alkanes 90 in moderate to good yield (Scheme 19).
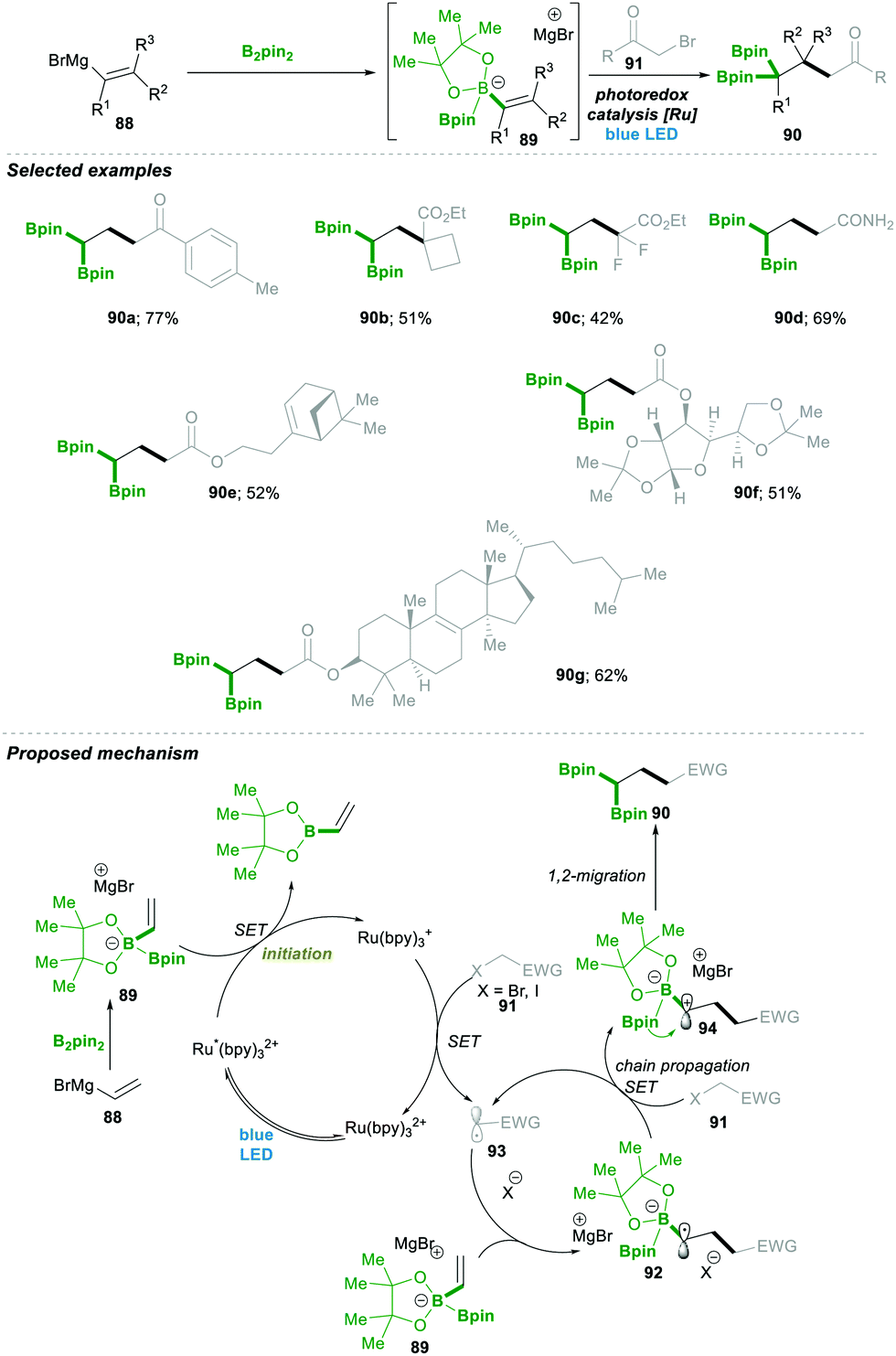 | ||
| Scheme 19 Radical-triggered 1,2-boryl-migration approach for gem-diboryl alkanes. | ||
Under the aforementioned reaction conditions, even a complex structure was attractively tolerated (90a–d, 90g); the reaction condition was found to be very mild and tolerated a wide range of functional groups (90e–f) (Scheme 19). After a detailed mechanistic study and more than one quantum yield (experimentally found to be Φ = 49.8), it was found that the reaction followed a radical chain pathway. Vinyl Grignard 88 was added in the first step to electrophilic bis(pinacolato)diboron (B2pin2), leading to intermediate 89. This electron-rich sacrificial bis-boronate intermediate 89 transferred a single-electron to excited Ru(II)-catalyst (Ru*(bpy)22+), generating a reduced Ru(I)-complex. Then the reduced Ru(I)-complex followed a single electron transfer with alkyl halide 91, generating a carbonyl stabilised electrophilic-radical 93 and regenerated the active Ru(bpy)22+ catalyst. The highly reactive radical 93 attacked the electron-rich boronate complex 89, leading to an electron-rich α-bisboryl radical 92. This α-bisboryl radical 92 afforded a single electron to the alkyl halide, leading to α-bisboryl carbocation 94, which then followed a spontaneous 1,2-boryl migration, furnishing the desired gem-bis(boryl)alkane product 90. The alkyl radical that formed in the previous catalytic step propagated a chain reaction, leading to very high quantum yield (Φ = 49.8) from the reaction (Scheme 19).23
Aggarwal's24 research group has continued to open up a new avenue by adding radical 95 to the σ-bonds of highly strained bicyclobutyl-boronate 98, leading to a highly substituted boronic ester containing cyclobutyl motif 99 in a very efficient manner (Scheme 20). In situ, compound 97, along with 102, generates the bicyclobutyl-boronate complex 98, upon addition of the electrophilic radical 95 (generated from alkyl halides 96), leading to 1,3-alkylated cyclobutyl boronic esters 99 in a very good yield and with complete stereospecificity. Since boronate esters are electron rich, nucleophilic radicals (generated from electronically rich alkyl halides), they were ineffective as expected. The mechanism includes as a first step an electrophilic radical 95 generated photochemically from alkyl iodides 96. This attacked the σ-bond of a highly strained bicyclobutane-boronate complex 98, which yielded an electron-rich cyclobutane boronate radical 100. The thus formed cyclobutane radical 100 followed SET with alkyl iodide 96, leading to cyclobutane-boronate carbocation 101. This zwitterionic intermediate prefers to undergo an energetically favoured 1,2-metalate alkyl migration, affording the desired 1,3-disubstituted cyclobutyl boronic esters 99 with excellent stereoselectivity. The reaction condition was mild, with a wide substrate and functional group range; even natural compounds with complex structures worked smoothly and afforded the desired product 99c in a moderate yield of 51% and with very high dr >20:1. The stereoselectivity is thought to come from the single-electron transferring step (100 to 101), since the cyclobutane boronate radical 100 has a rigid four-membered ring and alkyl iodide needs to approach it from the less hindered side; the hindrance is caused by the 3-position group of cyclobutane (see 100′ in the shaded portion of the box of Scheme 20). For the same reason, the following step of 1,2-metalate alkyl migration goes from the opposite lobe of the p-orbital (see 101). This 1,2-metalate rearrangement step is much faster than the C–B bond rotation in cyclobutane boronate carbocation 101; consequently, only the cis-cyclobutane product was formed during the course of the reaction (Scheme 20).24
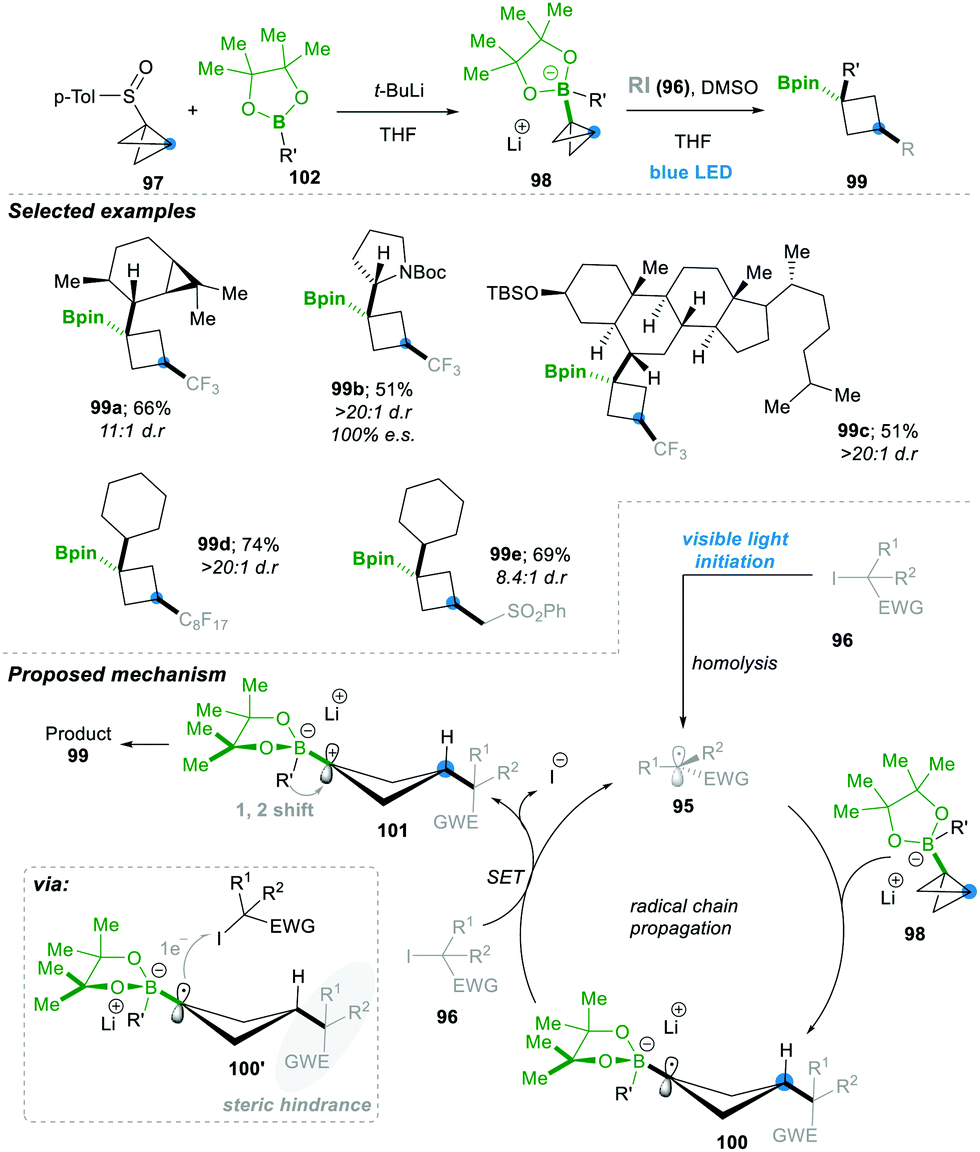 | ||
| Scheme 20 Radical-triggered stereocontrolled approach for cyclobutyl boronic ester synthesis. | ||
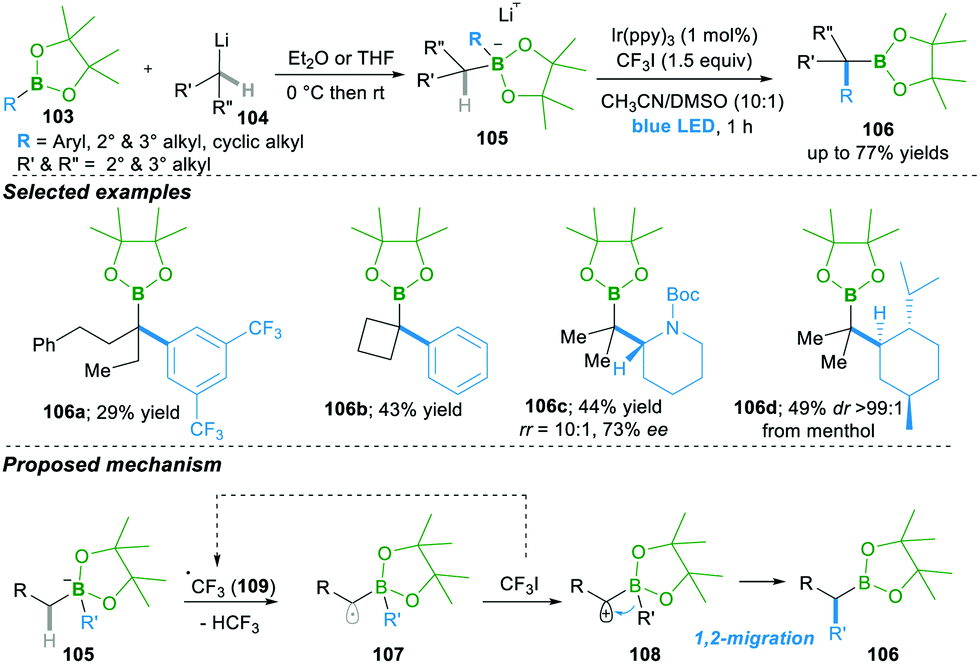
Very recently, Studer25a and co-workers reported smart initiation by a visible light mediated reaction leading to the α-C–H bond functionalisation of boronate complex 105, which is obtained from the addition of alkyl lithium 104 to boronic esters 103, followed by 1,2-migration, yielding the α-functionalised boron compounds 106 (Scheme 21).25a
 | ||
| Scheme 21 H-atom transfer induced the α-functionalisation of borates and its mechanism. | ||
In this reaction, in the presence of iridium as a photocatalyst, trifluoromethyl radical 109 forms, which then undergoes the regeoselective abstraction of an α-C–H proton of boronate complex 105, forming α-radical anion 107. This α-radical anion 107 in the presence of oxidant CF3I results in the zwitter-ion 108, followed by 1,2-migration, yielding α-aryl/alkylated functionalised boron compounds 106. This method works well with various aryl and alky boronic esters, with acceptable yields. Of note, this method forms products 106c–d with retention of enantioselectivity and high diastereoselectivity via stereospecific migration (Scheme 21).25b
 | ||
| Scheme 22 Visible light-mediated decarboxylative strategy: direct access to alkyl boronic esters. | ||
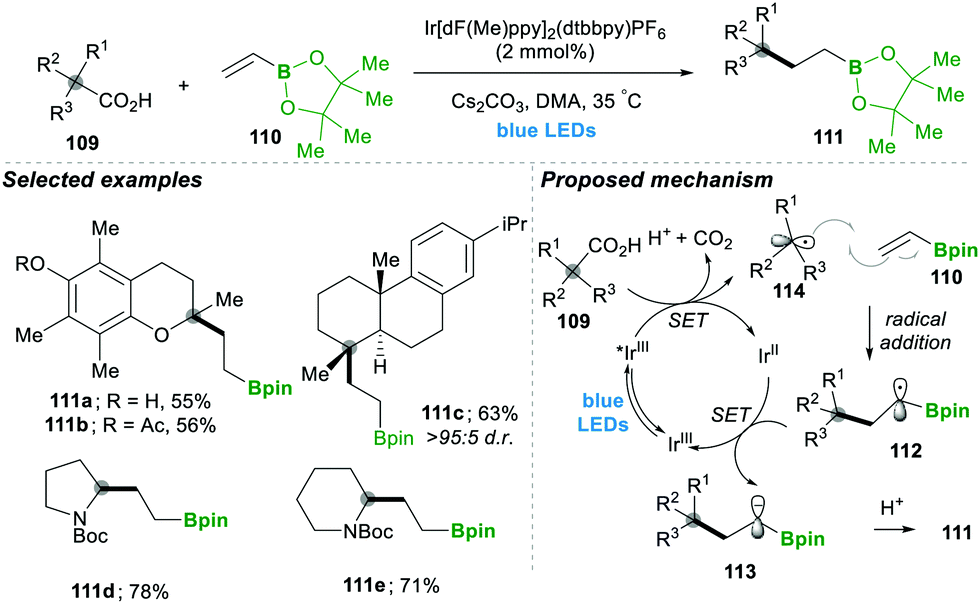
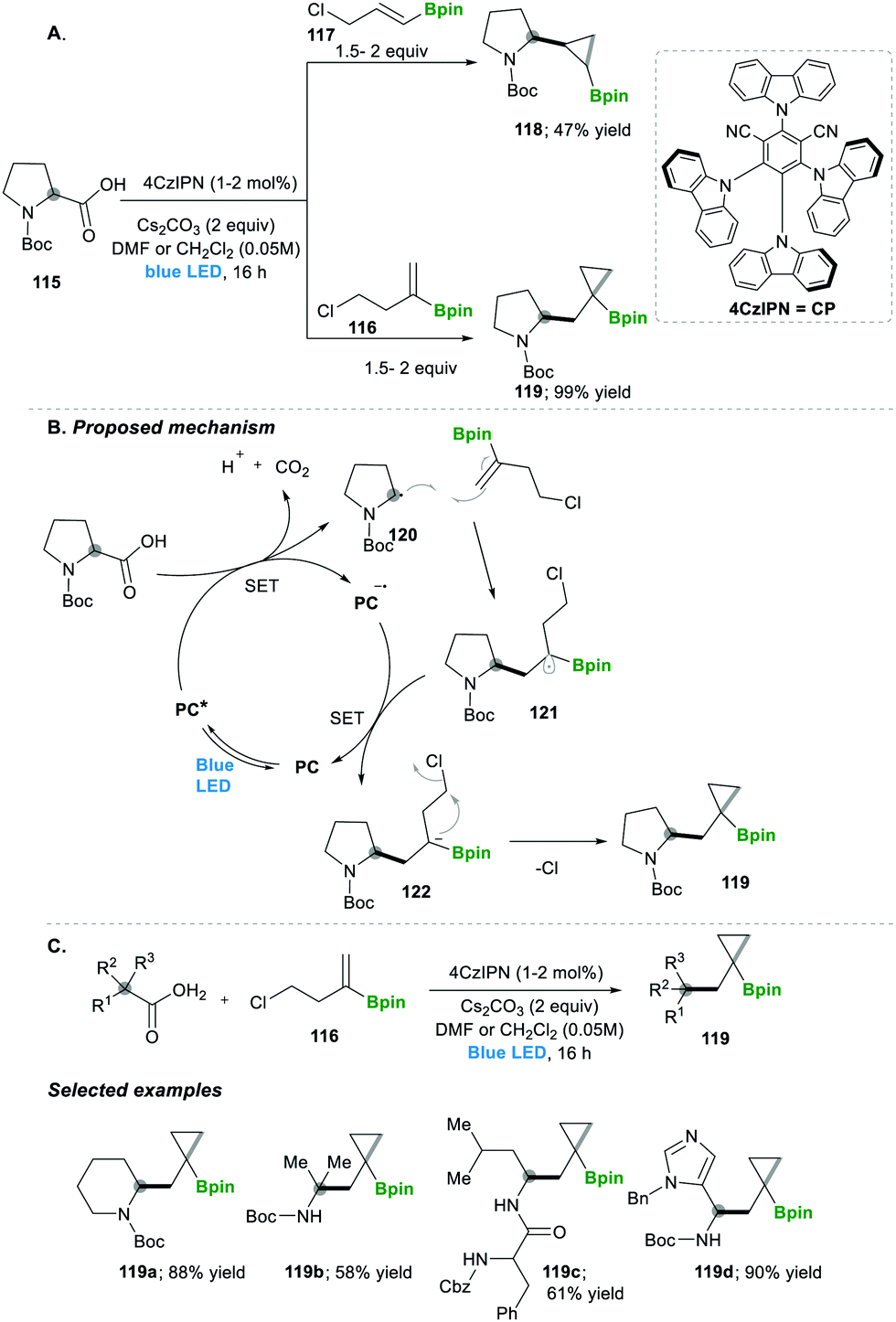
Analogously, the same research group reported on a decarboxylative radical addition and a polar cyclisation cascade for constructing a C–C bond to obtain the functionalised cyclopropanes by photoredox catalysis (Scheme 23), in which primarily used proline 115-based nucleophiles, which reacted with chloroalkylalkenes 116/117 in the presence of photoredox catalyst (4CzIPN) and caesium carbonate as a base, yielded the highly functionalised cyclopropanes 118/119 with good yields (Scheme 3A).27 Furthermore, they tested different carboxylic acids under the same reaction conditions, which provided a wide range of functionalised cyclopropanes 119a–d (Scheme 23C). Even more complex carboxylic acids also reacted well under this condition, e.g., 119c. The generality of the ring cyclisation can also be extended to five-membered rings.27 The mechanistic studies of this transformation primarily show a photocatalyst under visible light irradiation that generates the excited state catalyst, which further oxidises the carboxylic acid to a radical upon the extrusion of carbon dioxide. Later this alkyl radical 120 is added to chloroalkylalkene, which forms the boron-stabilised radical intermediate 121, which is then reduced by the oxidized photocatalyst forming stabilised carbanion 122. Finally, 3-exo-tert cyclisation provides highly functionalised cyclopropanes 119 (Scheme 23B).27
 | ||
| Scheme 23 Visible light-mediated decarboxylative strategy: radical addition and polar cyclisation cascade. | ||
2.3 Miscellaneous reactions of the three-component strategy
Reactions that did not fit into the above categories have been discussed here and are listed below.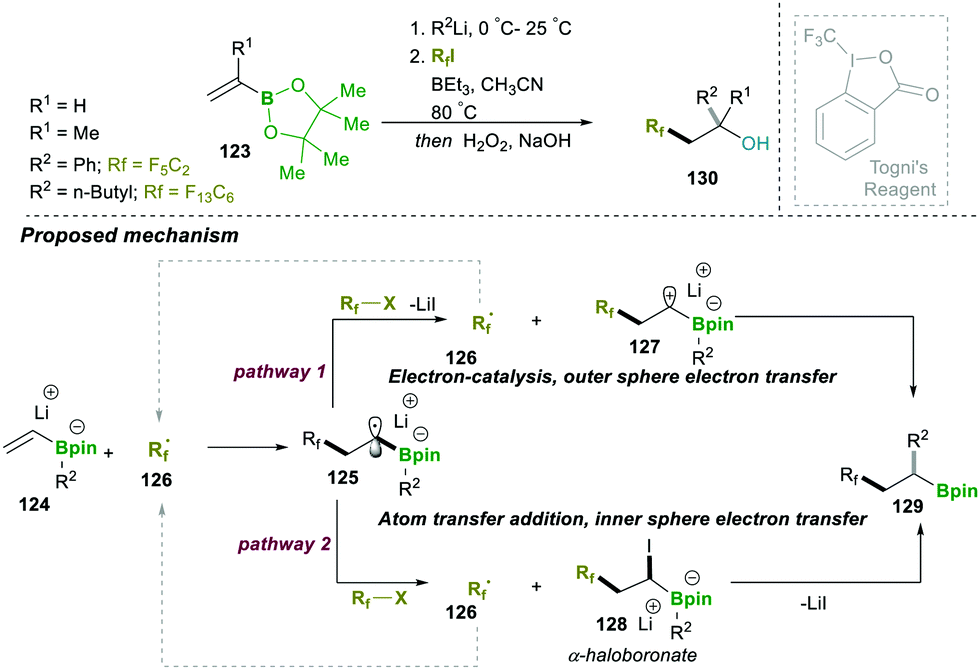 | ||
| Scheme 24 Radical-induced three-component alkylation reactions of alkenylboronates. | ||
This can take place by two possible pathways: the radical anion intermediate 125 can further undergo either outer sphere electron transfer (electron-catalysis, pathway 1) or inner sphere electron transfer (atom transfer addition, pathway 2). In the case of electron-catalysis, a single electron transfer from the radical anion intermediate 125 to an alkyl halide takes place, which yielded zwitterionic species 127 and regenerated perfluroralkyl radical 126. The previously formed zwitterionic intermediate 127 followed a spontaneous 1,2-metallate rearrangement and afforded the desired boronic esters 129. The second path, atom transfer addition, follows an iodide abstraction by a vinylboronate radical anion intermediate 125, leading to an α-haloboronate intermediate 128, which further follows a 1,2-alkyl migration (assisted by counter cation Li+) and yields the expected product 129. To understand the proposed mechanism, Togni's reagent (CF3 source, it cannot follow the iodine transfer pathway) was also tested and the reaction worked perfectly; this result supported the likelihood of pathway 1. However, an electron-poor alkyl halide afforded the product; therefore, pathway 2 is also possible. Accordingly, the reaction mechanism could depend on the kinetic ability of an alkyl radical, depending on whether reduction or iodine atom transfer is faster (Scheme 24).28
In the same year, Aggarwal29 and co-workers reported on the trifluoromethyl radical-induced multi-component approach for 2,5-disubstituted furans 134 with good yields and excellent enantiospecificity (Scheme 25). This reaction worked via generating the boronate complex 135 from furan-2-yllithium 131, which initiates single-electron transfer, leading to electrophilic trifluoromethyl-radical 138 (initiation step). This highly reactive, electrophilic CF3-radical 138 adds to an electron-rich boronate complex 135, which leads to intermediate 136via a radical addition step. Intermediate α-boryl radical 136 then undergoes oxidation with Umemoto's reagent A, affording a highly reactive oxonium intermediate 137, which then follows the 1,2-metallate rearrangement to furnish boronic ester 134. Iodine and K2CO3 facilitate oxidative re-aromatisation, finally affording the desired 2,5-disubstituted furans 134 in good yield (Scheme 25).29
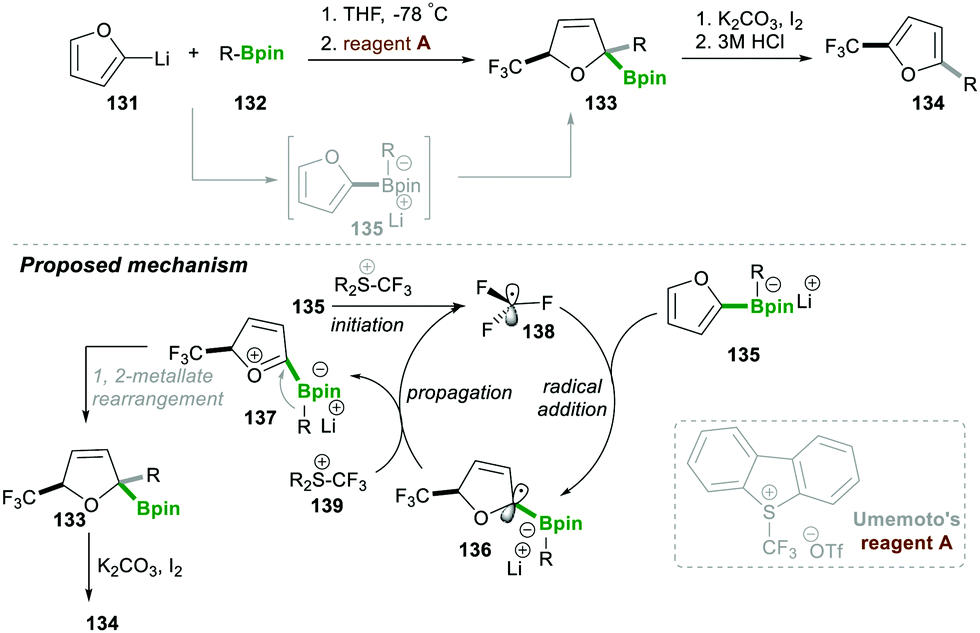 | ||
| Scheme 25 Radical-induced multi-component coupling of boronic esters and furans. | ||
Similarly to the previous case of Studer, in 2018 Renaud and co-workers showed the three-component alkylation reaction of alkenylboronic esters 140, α-halocarbonyl and organolithium (Scheme 26). The reaction was found to be highly efficient and worked on a wide range of halides and organolithium reagents.25a This operationally simple method offered two C–C bond formations, along with the generation of a quaternary-carbon centre (α- to boronic esters, 146) in a single step. Three possible competing reaction pathways were proposed: in pathway 1, radical-142 reacts with R–X via atom transfer, leading to α-haloboronate 143, which further follows a 1,2-metalate-rearrangement to forge 146. In pathway 2, single-electron-transfer by intermediate 142 provides an inverse-boron ylide 144 and a reduced halide, the former of which undergoes a fast intramolecular 1,2-migration, leading to 146. In the last possible pathway 3, 142 undergoes a radical 1,2-migration, resulting in boron-centred radical anion 145, which in fact is an initial step for a chain reaction.
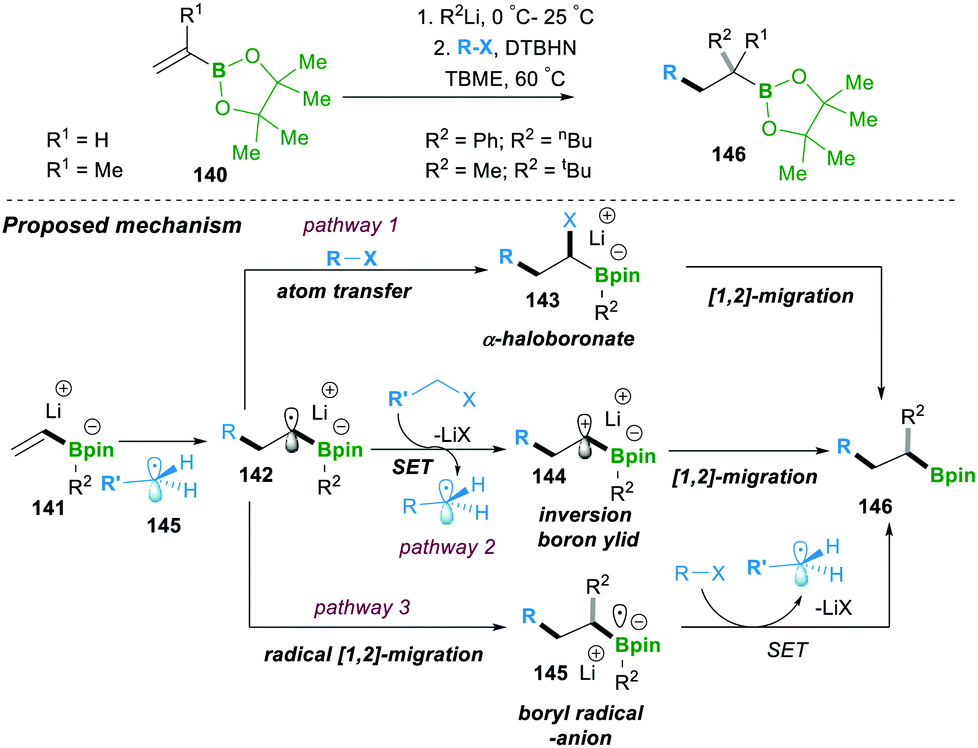 | ||
| Scheme 26 Radical-induced three-component alkylation reactions of alkenylboronates. | ||
However, investigations and a controlled study of the mechanism and additional literature supported by Studer and Aggarwal (see Schemes 20 and 24 for details) revealed that pathway 2 (the inverse ylide mechanism) was most likely to explain the three-component alkylation reactions of alkenylboronates better than the two others shown in Scheme 26.25a
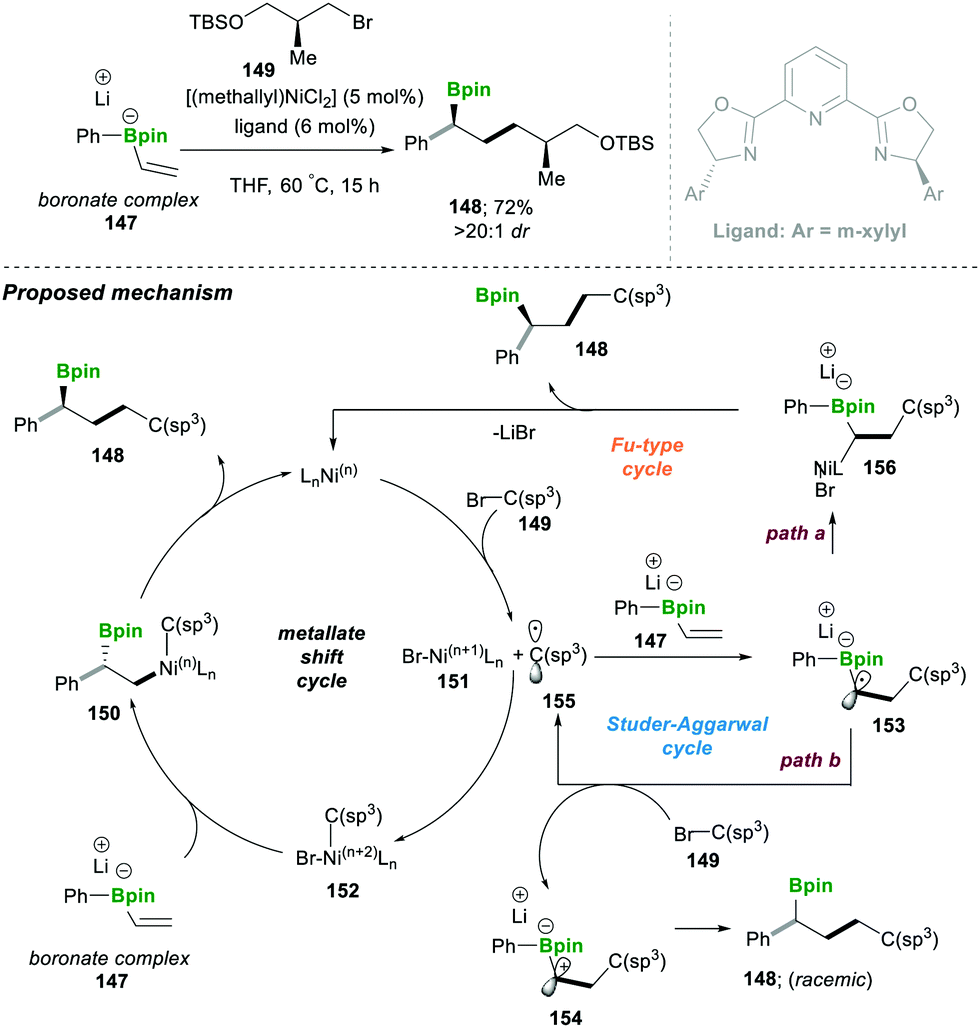 | ||
| Scheme 27 Ni-catalysed radical-induced enantioselective conjunctive cross-coupling. | ||
To better understand the exact path followed by the reaction, which depends on the nature of the radical formed during the first step, the substrate is completely controlled. The electrophilic radical followed the Studer–Aggarwal cycle. It attacks the electron-rich vinyl boronate complex in a much faster step, following a radical chain reaction. In the case of a non-stabilised carbon radical, formed in the first step, it follows the Fu-type cycle. A detailed mechanistic investigation suggested that it follows the conjunctive coupling path, affording the β-boronate Ni-complex, which follows a two-electron reductive elimination pathway to afford a stereospecific product (Scheme 27).30
In a subsequent work of 2019, a neutral boron-based enantioselective three-component approach was reported by the same research group.31 In this report, 10 mol% of Ni(II)-catalyst and 13 mol% of chiral diamine ligand (L) mixed with organozinc reagent 157, alkyl halide 158 and neutral organoboron 159 afforded the desired product 160 in moderate yield and with high enantio and diastereoselectivity. A series of mechanistic experiments suggested that a reaction following a radical pathway experiment, e.g., in the presence of a TEMPO reaction did not afford product 160. The proposed catalytic pathway includes in the first step the homolytic cleavage of the C–I bond (in 158), which takes place in the presence of the Ni(I)–R complex 163, leading to carbon-centred radical 161 and Ni(II)-complex (164). The formed carbon-centred radical 161, when added to alkenyl-pinacole boron ester 159, yields a more stable α-boryl radical 162, which when combined with Ni(II)-complex 164 by single-electron oxidation, affords intermediate Ni(III) 165. This advanced intermediate 165 undergoes a facile reductive elimination step, regenerating active Ni(I)-iodide 166 catalyst, along with the desired product 160. It worth mentioning that in the absence of vinylBpin 159 the homo and cross-coupling products A and B were also observed, following a similar mechanism. This method was found to be highly general and worked with a wide range of substrate scopes with functional group tolerance. In all the cases, desired products were obtained in good yield and with very high er and dr (Scheme 28).31
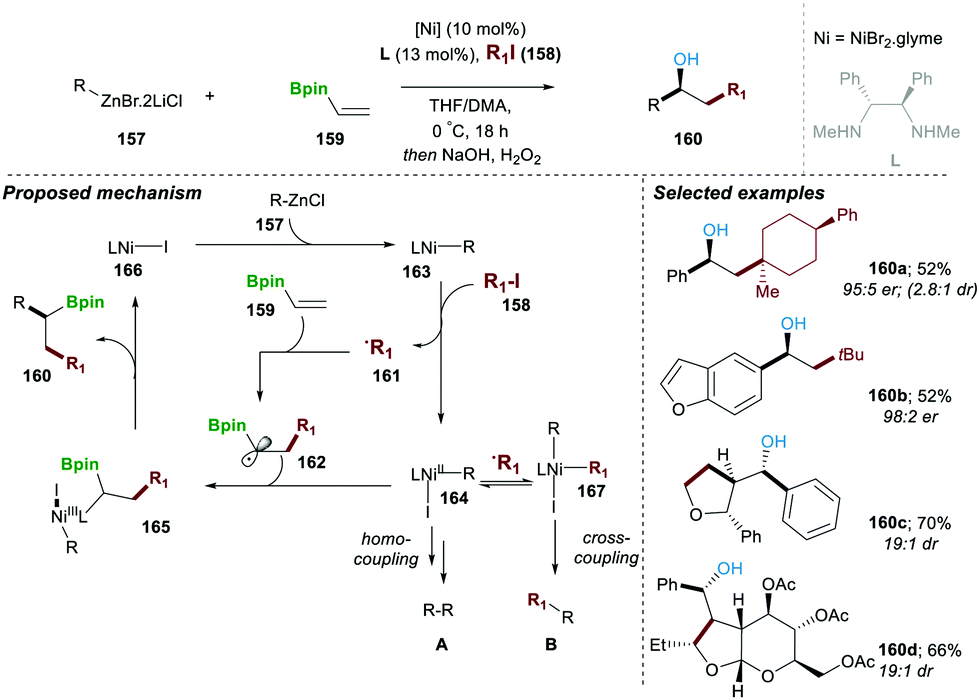 | ||
| Scheme 28 Ni-catalysed radical-induced enantioselective multi-component cross-coupling. | ||
3. Conclusions and outlook
For more than a century, organoboron chemistry has offered novel routes towards fabricating synthetically highly valuable compounds. For example, organoborons are widely used in constructing C–C/C–heteroatom bonds by activating the C–B bond. Additionally, they can be involved in radical chemistry. Their radical strategies complement the existing approaches for the formation of new bonds. This radical approach generally relies on the cleavage of the C–B bond to create alkyl/aryl radicals. Thanks to this radical approach, new bond formation can be achieved; however, the product will lose the boron moiety. By overcoming this main limitation, of losing the boron group by radical transformation, a new reactivity in the field of organoboron chemistry can be designed. In recent years, organoboron radical chemistry has elegantly opened up new pathways without losing the valuable boron moiety; this includes the growing field of α-boryl carbon-centred radical intermediates, e.g., II, which were developed by many research groups.In this review, we wish to emphasise the importance of radical II generation from organoborons, leading to (a stable and) synthetically useful α-boron carbon-centred radical intermediate II, which is further followed by forged new connections. For its generation we chiefly focused on a few main strategies: (1) using radical initiators: in this strategy, the formation of α-boryl radicals was mainly promoted by radical initiators such as the B(alkyl)3/bromine system, AIBN (with stannates or TTMSS), and DLP, (2) visible-light mediated reactions including the photoredox catalysis via SET reactions, (3) three-component coupling reactions, (4) metal and metal-free mediated reactions, and (5) radical addition to vinyl boronic esters, vinyl boronate esters, and boron ate complexes.
As highlighted in this review, II's unique stability and reactivity have been discussed in depth, many challenging transformations have been accomplished, and a good mechanistic understanding of these processes has been acquired over the last ten years.
Owing to the potential for the synthesis of α-boron carbon-centred radical species II, this field will continue to grow rapidly, and more appealing transformations are expected to appear in the years to come.
Future approaches to expand the synthetic utility of these α-boryl carbon-centred radical intermediates can be through the generation of more stabilized radical analogues, such as 1,1-diboryl-radical and the 1,1-borylsilyl-radical. Such studies are already ongoing in our research group.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. M. is grateful to the Azrieli Foundation for the receipt of an Azrieli Fellowship. N. K. and R. R. R. are thankful to HUJI for a postdoctoral fellowship.Notes and references
- (a) The Roles of Boron and Silicon, Susan E. Thomas; Oxford Chemistry Primers No. 1, 1991; (b) Organometallics, ed. C. Elschenbroich, 3rd edn, 2006, Wiley-VCH, Weinheim, ISBN: 3-527-29390-6 Search PubMed; (c) J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31 CrossRef CAS; (d) B. Rao and R. Kinjo, Chem. – Asian J., 2018, 13, 1279 CrossRef CAS PubMed.
- (a) S. Touchet, F. Carreaux, B. Carboni, A. Bouillon and J.-L. Boucher, Chem. Soc. Rev., 2011, 40, 3895 RSC; (b) V. M. Dembitsky, A. A. A. Al Quntar and M. Srebnik, Chem. Rev., 2011, 111, 209 CrossRef CAS PubMed; (c) S. J. Baker, J. W. Tomsho and S. J. Benkovic, Chem. Soc. Rev., 2011, 40, 4279 RSC; (d) R. Smoum, A. Rubinstein, V. M. Dembitsky and M. Srebnik, Chem. Rev., 2012, 112, 4156 CrossRef CAS PubMed; (e) D. B. Diaz and A. K. Yudin, Nat. Chem., 2017, 9, 731 CrossRef CAS PubMed.
- (a) J. Adams, V. J. Palombella, E. A. Sausville, J. Johnson, A. Destree, D. D. Lazarus, J. Maas, C. S. Pien, S. Prakash and P. J. Elliott, Cancer Res., 1999, 59, 2615–2622 CAS; (b) S. J. Baker, Y.-K. Zhang, T. Akama, A. Lau, H. Zhou, V. Hernandez, W. Mao, M. R. K. Alley, V. Sanders and J. J. Plattner, J. Med. Chem., 2006, 49, 4447 CrossRef CAS PubMed; (c) A. Maynard, et al. , J. Med. Chem., 2014, 57, 1902–1913 CrossRef CAS PubMed.
- (a) P. Renaud, A. Beauseigneur, A. Brecht-Forster, B. Becattini, V. Darmency, S. Kandhasamy, F. Montermini, C. Ollivier, P. Panchaud, D. Pozzi, E. M. Scanlan, A.-P. Schaffner and V. Weber, Pure Appl. Chem., 2007, 79, 223 CAS; (b) G. Duret, R. Quinlan, P. Bisseret and N. Blanchard, Chem. Sci., 2015, 6, 5366 RSC; (c) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850 CrossRef CAS PubMed.
- (a) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091 CrossRef CAS PubMed; (b) R. Nallagonda, K. Padala and A. Masarwa, Org. Biomol. Chem., 2018, 16, 1050 RSC; (c) N. Kumar, R. R. Reddy and A. Masarwa, Chem. – Eur. J., 2019, 25, 8008 CrossRef CAS PubMed; (d) N. Miralles, R. J. Maza and E. Fernandez, Adv. Synth. Catal., 2018, 360, 1306 CrossRef CAS; (e) J. Royes, A. B. Cuenca and E. Fernández, Eur. J. Org. Chem., 2018, 2728 CrossRef CAS; (f) C. Wu and J. Wang, Tetrahedron Lett., 2018, 59, 2128 CrossRef CAS; (g) D. S. Matteson and R. W. H. Mah, J. Am. Chem. Soc., 1963, 85, 2599 CrossRef CAS; (h) D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174 CrossRef CAS PubMed; (i) J. Schmidt, J. Choi, A. T. Liu, M. Slusarczyk and G. C. A. Fu, Science, 2016, 354, 1265 CrossRef CAS PubMed; (j) C. Ollivier and P. Renaud, Chem. Rev., 2001, 101, 3415 CrossRef CAS PubMed; (k) J. K. Matsui, D. N. Primer and G. A. Molander, Chem. Sci., 2017, 8, 3512 RSC; (l) N. Oehlrich, F. Rombouts and G. A. Molander, J. Org. Chem., 2013, 78, 4615 CrossRef PubMed; (m) I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194 CrossRef CAS PubMed; (n) K. Foo, E. Sella, I. Thome, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5279 CrossRef CAS PubMed.
- (a) J. Grotewold, E. A. Lissi and J. C. Scaiano, J. Organomet. Chem., 1969, 19, 431 CrossRef CAS; (b) C. F. Lane and H. C. Brown, J. Am. Chem. Soc., 1970, 92, 7212 CrossRef; (c) J. E. Grotewold, E. A. Lissi and J. C. Scaiano, J. Chem. Soc. B, 1971, 1187 RSC; (d) A. G. Davies and B. P. Roberts, in Free Radicals, ed. J. K. Kochi, Wiley, New York, 1973, ch. 10, pp. 547–587 Search PubMed.
- (a) D. J. Pasto, R. Krasnansky and C. Zercher, J. Org. Chem., 1987, 52, 3062 CrossRef CAS; (b) J. C. Walton, A. J. McCarroll, Q. Chen, B. Carboni and R. Nziengui, J. Am. Chem. Soc., 2000, 122, 5455 CrossRef CAS; (c) H. Wang, Y.-F. Zengb, W.-X. Lva and D.-H. Tana, Synlett, 2018, 1415 CrossRef CAS.
- Q. Huang and S. Z. Zard, Org. Lett., 2018, 20, 5304 CrossRef CAS PubMed.
- C. F. Lane and H. C. Brown, J. Am. Chem. Soc., 1970, 92, 7212 CrossRef.
- C. F. Lane and H. C. Brown, J. Am. Chem. Soc., 1974, 96, 311 CrossRef.
- N. Guennouni, F. Lhermitte, S. Cochard and B. Carboni, Tetrahedron, 1995, 51, 6999 CrossRef CAS.
- R. A. Batey, B. Pedram, K. Yong and G. Baquer, Tetrahedron Lett., 1996, 37, 6847 CrossRef CAS.
- R. A. Batey and D. V. Smil, Tetrahedron Lett., 1999, 40, 9183 CrossRef CAS.
- T. Nishikawa and M. Ouchi, Angew. Chem., Int. Ed., 2019, 58, 12435 CrossRef CAS PubMed.
- B. Q. Sire and S. Z. Zard, J. Am. Chem. Soc., 2015, 137, 6762 CrossRef PubMed.
- Q. Huang and S. Z. Zard, Org. Lett., 2018, 20, 5304 CrossRef CAS PubMed.
- Q. Huang, J. Michalland and S. Z. Zard, Angew. Chem., Int. Ed., 2019, 58, 16936 CrossRef CAS PubMed.
- N. D. C. Tappin, W. Michalska, S. Rohrbach and P. Renaud, Angew. Chem., Int. Ed., 2019, 58, 1–6 CrossRef PubMed.
- J. C. Lo, D. Kim, C.-M. Pan, J. T. Edwards, Y. Yabe, J. Gui, T. Qin, S. Gutiérrez, J. Giacoboni, M. W. Smith, P. L. Holland and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 2484 CrossRef CAS PubMed.
- F. Takahashi, K. Nogi, T. Sasamori and H. Yorimitsu, Org. Lett., 2019, 21, 4739 CrossRef CAS PubMed.
- M. Silvi, C. Sandford and V. K. Aggarwal, J. Am. Chem. Soc., 2017, 139, 5736 CrossRef CAS PubMed.
- M. Silvi, R. Schrof, A. Noble and V. K. Aggarwal, Chem. – Eur. J., 2018, 24, 4279–4282 CrossRef CAS PubMed.
- B. Zhao, Z. Li, Y. Wu, Y. Wang, J. Qian, Y. Yuan and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 9448–9452 CrossRef CAS PubMed.
- M. Silvi and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 9511 CrossRef CAS PubMed.
- (a) N. D. C. Tappin, M. G.-G. Lux and P. Renaud, Chem. – Eur. J., 2018, 24, 11498 CrossRef CAS PubMed; (b) D. Wang, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2019, 141(36), 14126 CrossRef CAS PubMed.
- A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155 CrossRef CAS PubMed.
- C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 15430 CrossRef CAS.
- M. Kischkewitz, K. Okamoto, C. M.-Lichtenfeld, and A. Studer Science 2017, 355, 936. Follow-up reports based on these findings are the following: (a) M. Kischkewitz, C. Gerleve and A. Studer, Org. Lett., 2018, 20, 3666 CrossRef CAS PubMed; (b) C. Gerleve, M. Kischkewitz and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 2441 CrossRef CAS PubMed; (c) M. Kischkewitz and A. Studer, Org. Synth., 2018, 95, 205–217 CrossRef CAS; (d) F. Clausen, M. Kischkewitz, K. Bergander and A. Studer, Chem. Sci., 2019, 10, 6210 RSC.
- Y. Wang, A. Noble, C. Sandford and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 1810 CrossRef CAS PubMed.
- G. J. Lovinger and J. P. Morken, J. Am. Chem. Soc., 2017, 139, 17293 CrossRef CAS PubMed.
- M. Chierchia, P. Xu, G. J. Lovinger and J. P. Morken, Angew. Chem., Int. Ed., 2019, 58, 4245 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |