
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiselenolane-mediated cellular uptake†
Nicolas
Chuard
a,
Amalia I.
Poblador-Bahamonde
a,
Lili
Zong
a,
Eline
Bartolami
a,
Jana
Hildebrandt
b,
Wolfgang
Weigand
 b,
Naomi
Sakai
a and
Stefan
Matile
*a
b,
Naomi
Sakai
a and
Stefan
Matile
*a
aDepartment of Organic Chemistry, University of Geneva, Geneva, Switzerland. E-mail: stefan.matile@unige.ch; Web: http://www.unige.ch/sciences/chiorg/matile/ Tel: +41 22 379 6523
bInstitute of Inorganic and Analytical Chemistry, Friedrich-Schiller University Jena, Germany
First published on 17th January 2018
Abstract
The emerging power of thiol-mediated uptake with strained disulfides called for a move from sulfur to selenium. We report that according to results with fluorescent model substrates, cellular uptake with 1,2-diselenolanes exceeds uptake with 1,2-dithiolanes and epidithiodiketopiperazines with regard to efficiency as well as intracellular localization. The diselenide analog of lipoic acid performs best. This 1,2-diselenolane delivers fluorophores efficiently to the cytosol of HeLa Kyoto cells, without detectable endosomal capture as with 1,2-dithiolanes or dominant escape into the nucleus as with epidithiodiketopiperazines. Diselenolane-mediated cytosolic delivery is non-toxic (MTT assay), sensitive to temperature but insensitive to inhibitors of endocytosis (chlorpromazine, methyl-β-cyclodextrin, wortmannin, cytochalasin B) and conventional thiol-mediated uptake (Ellman's reagent), and to serum. Selenophilicity, the extreme CSeSeC dihedral angle of 0° and the high but different acidity of primary and secondary selenols might all contribute to uptake. Thiol-exchange affinity chromatography is introduced as operational mimic of thiol-mediated uptake that provides, in combination with rate enhancement of DTT oxidation, direct experimental evidence for existence and nature of the involved selenosulfides.
Unlike simple alkanes or peroxides, the most favorable CXXC dihedral angle θ in acyclic diselenides 1 and disulfides 2 is ∼90° (X = S, Se, Fig. 1).1 Decreasing θ in cyclic disulfides causes the increase of ring tension in 1,2-dithianes with 62° over 1,2-dithiolanes 3–4 with 27–35° to approach the maximum epidithiodiketopiperazine (ETP) 5 with ∼0°.2 Earlier, we found the dependence of the ability of cyclic disulfides as delivery vehicles of otherwise cell-impermeable cargos to the ring tension.1,2 Mechanistic studies support that the ring tension promotes dynamic covalent thiol-disulfide exchange on the cell surface1–6 to initiate uptake.1,2 Asparagusic acid (SA) derivative 4 efficiently delivers functional peptides and objects as large as intact liposomes.6
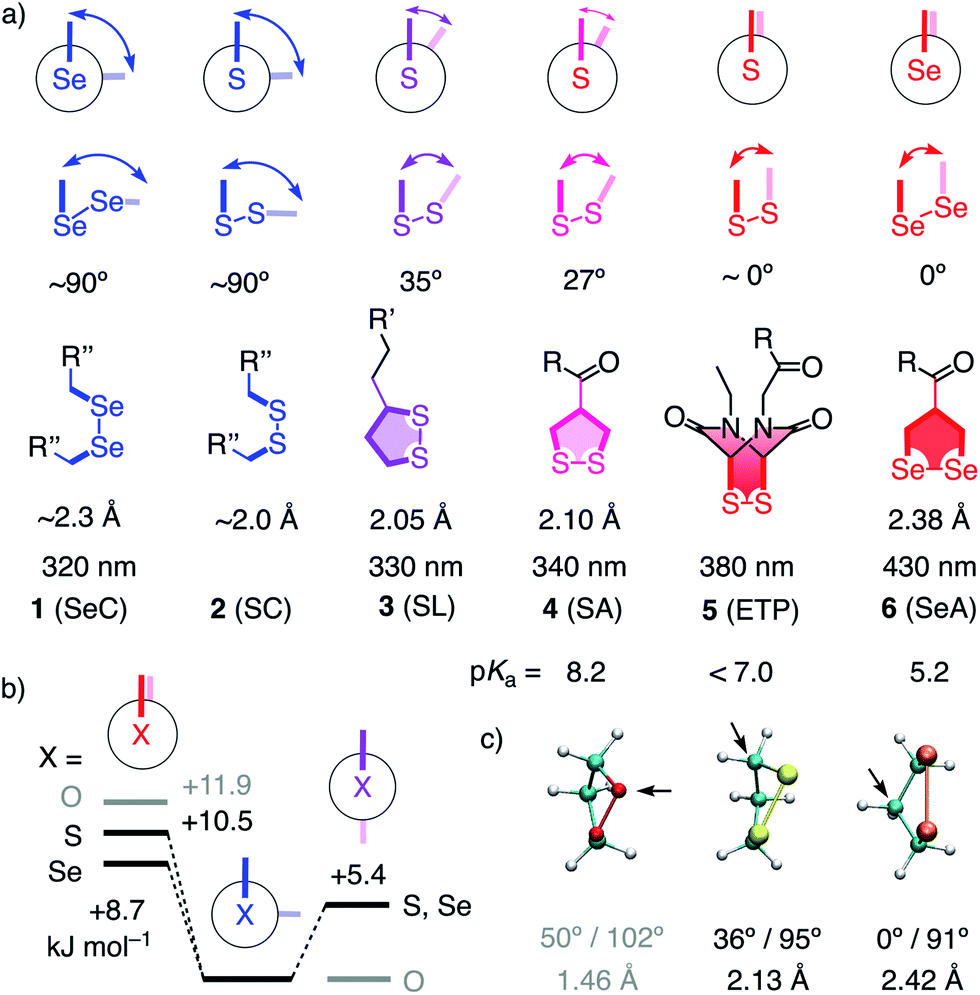 | ||
| Fig. 1 (a) Selected CXXC dihedral angles, XX bond length and absorption maxima of disulfides and diselenides, and the intrinsic pKa of one thiol or selenol in their reduced form (X = S, Se; R, R′, R′′, see Fig. 2). (b) B3LYP/6-311++G** energies of MeXXMe conformers. (c) Minimized structure of the parent 1,2-dioxo- (O = red), dithio- (S = yellow) and diselenolane (Se = orange) with angles (CXXC, CXX) and bond length (XX), arrows: endo position. | ||
This emerging power of strain-promoted thiol-mediated uptake with cyclic disulfides called for cyclic diselenides. Besides many similarities, disulfides and -selenides have also important differences. In six-membered rings, a very similar θ of ∼60° (56°) has been confirmed.7 In the crystal structure of 1,2-diselenolanes like SeA 6, the CSeSeC dihedral angle is 0.2°, contributing to an almost ideal, twist-free envelope structure of the five-membered ring.8,9 At the same time, the Se–Se bond length increases from ∼2.3 Å in relaxed diselenides 1 to 2.38 Å in 1,2-diselenolane 6, and the absorption maximum shifts 320 nm to 430 nm.8–10 The CSSC dihedrals of 1,2-dithiolanes maximize at 27° in SAs such as 4, and absorptions shift from 250 nm to maximal 340 nm.1 The crystal structures of 1,2-di-selenolanes show layers of selenium with chalcogen bonds11 of down to 3.55 Å length between the polarizable selenium atoms of different molecules.8,9 In the solid state, the strained di-selenolanes easily polymerize into a gum.9 In solution, relaxed diselenides exchange up to 7 orders of magnitude faster than disulfides.12,13 The high acidity of selenols (pKa ∼ 5.2) compared to thiols (pKa ∼ 8.2) contributes to this increased reactivity by activating both nucleophile and leaving group for selenol–diselenide exchange in neutral and also slightly acidic water.12–17 However, although exchange is possible with both thiols and selenols,12 the formation of Se–Se bonds is favored over mixed S–Se bonds.13 This selenophilicity has been used elegantly to catalyze protein folding15 and protein denaturation with di-selenides in the presence of immobilized thiols.16
Some of the characteristics of 1,2-diselenolanes are reminiscent of ETPs 5 (i.e., pKa < 7, CXXC dihedral ∼0°, Fig. 1) and could thus promise similarly outstanding uptake efficiencies with dual activation at highest tension.2 Worthwhile to note in this context is also the presence of eight out of the 25 human selenoproteins in cellular membranes, which could potentially participate in selenol–diselenide exchange reactions during the uptake of diselenides.17 Moreover, cellular uptake, particularly gene transfection, with tension-free poly(diselenide)s and poly(ditelluride)s has been reported to occur and differ sometimes favorably from uptake with poly(disulfide)s.18 In the following, we report uptake of 1,2-diselenolanes SeL 7 and SeA 6 to the cytosol without endosomal capture as with the corresponding 1,2-dithiolanes SL 3 and SA 4 or dominant escape into the nucleus as with ETP 5.
To elaborate on diselenolane-mediated uptake, green-fluorescent fluorescein isothiocyanate (FITC) conjugates were synthesized. SeL 7 was prepared from dichloride 8 (Fig. 2 and Scheme S1†). The reaction with Na2Se2 afforded the 1,2-di-selenolane scaffold as in the previously reported diseleno lipoic acid 9.19 Activation with N-hydroxysuccinimide (NHS) afforded the “SeL tag” 10, an activated ester that reacted readily with amines such as FITC-amine 11 to afford SeL 7. The green-fluorescent diselenolane SeA 6 was prepared from diseleno asparagusic acid7,8 following the same route (Scheme S2†), and the acyclic diselenide 1 was obtained by reaction of methylated diseleno cystine (SeC)20 with FITC (Scheme S3†). The control compounds SA 4 and ETP 5 were prepared following previously reported procedures.1,2
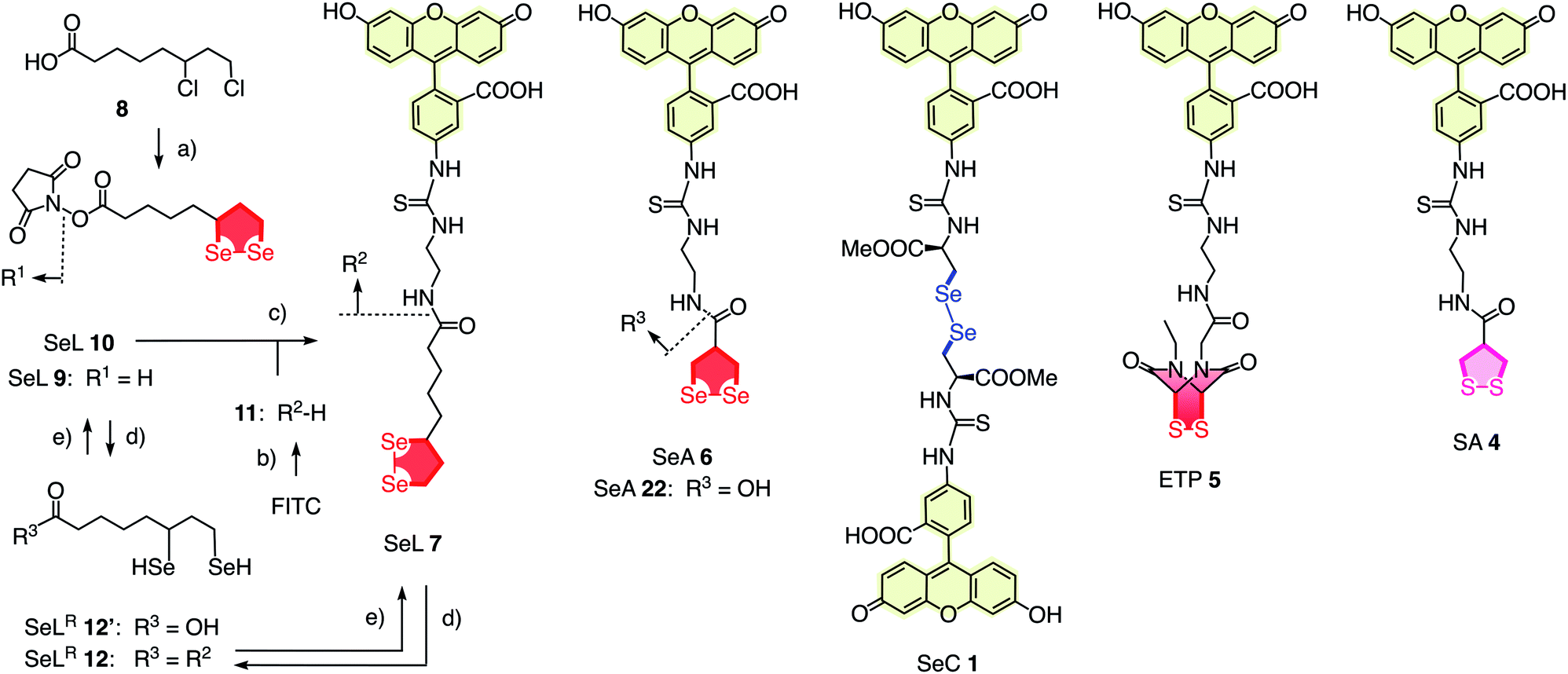 | ||
| Fig. 2 Complete structure of all transporters used (neutral form), with representative synthesis and reduction for SeL 7. (a) (1) Several steps, ref. 17, (2) NHS, DCC, THF, rt, 18 h, 53%; (b) several steps, ref. 1; (c) DMF, rt, 3 h, 14%; (d) TCEP, <5 min; (e) air. | ||
In computational studies, the minima at 90° and maxima at 0° in the conformational profiles of MeSeSeMe and MeSSMe were confirmed, whereas MeOOMe naturally minimized in anti conformation (Fig. 1b). The decreasing energy of the maxima from O to S and Se at 0° supported that ring tension in cyclic diselenides is lower than in disulfides. For unsubstituted 1,2-dioxo-, dithio- and diselenolanes, this trend resulted in envelope conformers with the chalcogen atom, the α carbon, and the β carbon in endo position and CXX angles decreasing from 102° to 95° and 91°, respectively (arrows, Fig. 1).
With tris(2-carboxyethyl)phosphine (TCEP), 1,2-diselenolane 9 was readily reduced to diselenol SeLR12′ (Fig. 2). However, neither SeA nor SeL appeared to react with various thiols 13 (Fig. 3). These results implied that either diselenides do not undergo exchange reactions with thiolates or, unlike S–S homologs 14, the selenosulfide intermediate products 15–17 ring close easily by intramolecular selenol–selenosulfide exchange. Their existence and nature was thus explored by thiol-exchange affinity chromatography (Fig. 3). Compared to a non-reactive carboxyfluorescein (CF) standard (Fig. 3a), all disulfides and diselenides showed delayed elution as expected for temporal covalent bonding with the solid phase through thiol exchange (Fig. 3b–f, solid). Although the retention time of these peaks was shortened a little (∼1 min) in the presence of DTT in the mobile phase (Fig. 3b–f, dashed), these retentions supported but did not prove transient dynamic covalent binding to the solid phase.
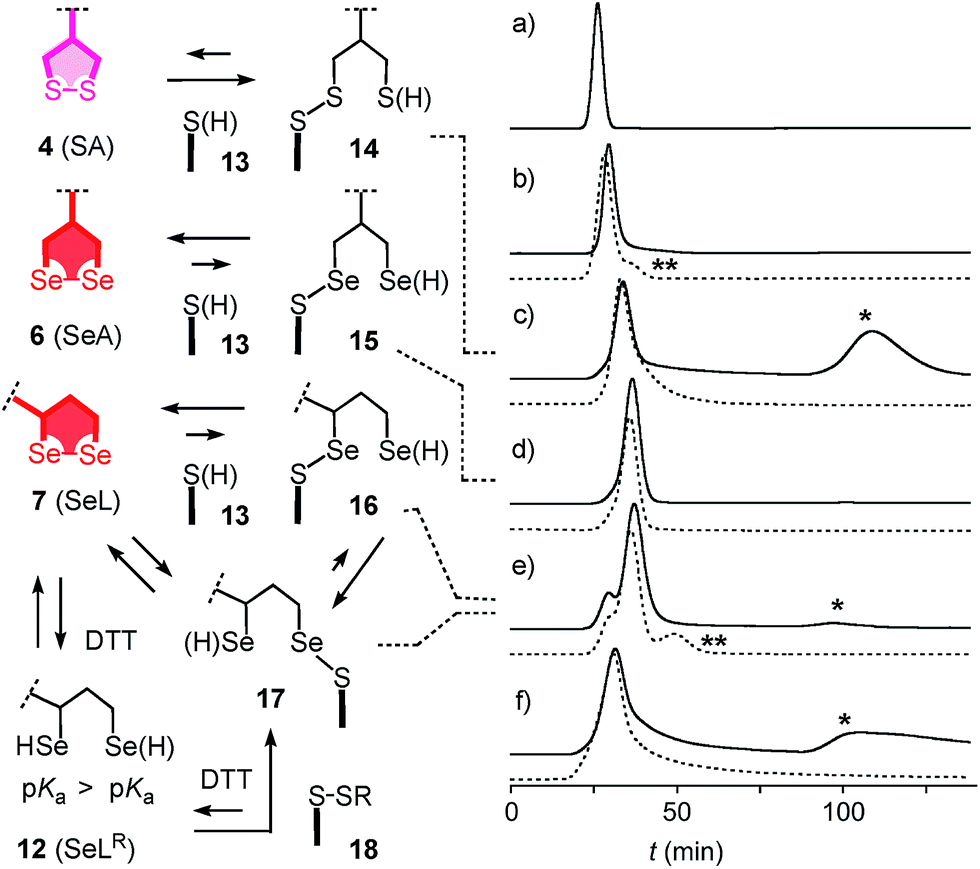 | ||
| Fig. 3 Thiol-exchange affinity chromatograms of (a) CF, (b) ETP 5, (c) SA 4, (d) SeA 6, (e) SeL 7 and (f) SeC 1 in 10 mM Tris, 0.1 M NaCl, 1 mM EDTA, pH 7.5 with a 0–50 mM DTT gradient at t = 60–70 min (solid) and constant 50 mM DTT from t = 0 (dashed). * = peaks indicative for inert binding of 1, 4 and 7 to thiols on the stationary phase by strain-releasing thiol-disulfide/diselenide exchange in the absence of DTT. ** = peaks indicative for labile binding of reduced forms (e.g. SeLR12) to disulfides on the stationary phase by thiol/selenol–disulfide exchange in the presence of DTT. Structures are shown in neutral form, acidic protons are indicated in parenthesis. | ||
With SA 4, a second broad peak eluted only upon addition of DTT to the mobile phase (Fig. 3c, *). This observation confirmed the formation and stability of the ring-opened, strain-released exchange product 14 by covalent attachment to the stationary phase. The absence of this second peak with SeA 6 indicated that ring-opened S–Se product 15, if formed on the exchange column, ring closes rapidly back to SeA 6 (Fig. 3d). This result was consistent with expectations from the reduced ring tension in 1,2-diselenolanes compared to 1,2-dithiolanes, selenophilicity and the low pKa of the selenol (Fig. 1). The importance of this low pKa was confirmed by the absence of this second DTT-dependent peak also for ETP 5 with thiols of comparable acidity (Fig. 1 and 3b).
With SeL 7, a second broad peak could be observed upon addition of DTT to the mobile phase (Fig. 3e, *). This finding provided direct experimental evidence for the occurrence of thiol-diselenide exchange with SeL 7, i.e., existence of the ring-opened S–Se product 16. This S–Se product 16 and not the constitutional isomer 17 was presumably formed first because a primary selenolate with a lower pKa is a better leaving group than the secondary selenolate with a higher pKa. However, S–Se isomer 16 was expected to exchange intramolecularly because the secondary selenol in S–Se isomer 17 with a higher pKa is a weaker nucleophile. The reduced reactivity of this S–Se isomer 17 with a secondary selenol could explain the detectability of the second, DTT-dependent peak in the affinity chromatogram of SeL 7 but not for SA 6 with primary selenols only (Fig. 3e, *). The isomerization from more reactive 16 to less reactive 17 could also account for the split of the first-eluting peak into two, a behavior that was unique for SeL 7 (Fig. 3e). An intense, very broad, DTT-dependent second peak was also observed with the acyclic diseleno cystine SeC 1 (Fig. 1 and 3f, *). The appearance of this peak was as expected because of the absence of a nearby selenol for rapid release by intramolecular exchange.
In the permanent presence of 50 mM DTT in the mobile phase, the fast eluting double peak of SeL 7 at Rt = 36.1 min was followed by smaller but significant additional peak (Fig. 3e, **). A similar peak occurred also with ETP 5 (Fig. 3b, **). A possible explanation of this observation was that DTT reduces SeL 7 to the diselenol 12 (Fig. 2), which then offers the activated primary selenolate to exchange with “hidden” disulfides 18 on the solid phase to give the more stable selenosulfide 17, which undergoes ring closure or exchanges with DTT slowly enough to appear as separate peak. The same arguments were applicable to ETP 5. This plausible explanation implied the existence of less reactive disulfides 18 on the solid phase that resist DTT but not the more reactive SeLR12 and reduced ETP 5. Although speculative, this conclusion caught our full attention because it would be consistent with unique properties of SeL 7 and ETP 5 with regard to cellular uptake (see below).
Further support of the possible thiol-diselenide exchange was obtained by measuring the oxidation kinetics of DTT 19 to DTTO20 catalyzed by diselenides.15 In the presence of one equivalent seleno lipoic acid 9, the halflife time of DTT 19 decreased from t50 ≈ 16 h (Fig. 4, ○) to t50 ≈ 6.9 h (Fig. 4, ■). This rate enhancement confirmed the occurrence of thiol/diselenide exchange, the existence of intermediate 21 with mixed S–Se bonds and fast oxidation of SeLR12′ by molecular oxygen. The catalysis of DTT oxidation by 1,2-diselenolane 9 also demonstrated that the reloading of ring tension in 9 is less favored than ring closure of DTT into the much less strained DTTO20 (Fig. 4, arrows).
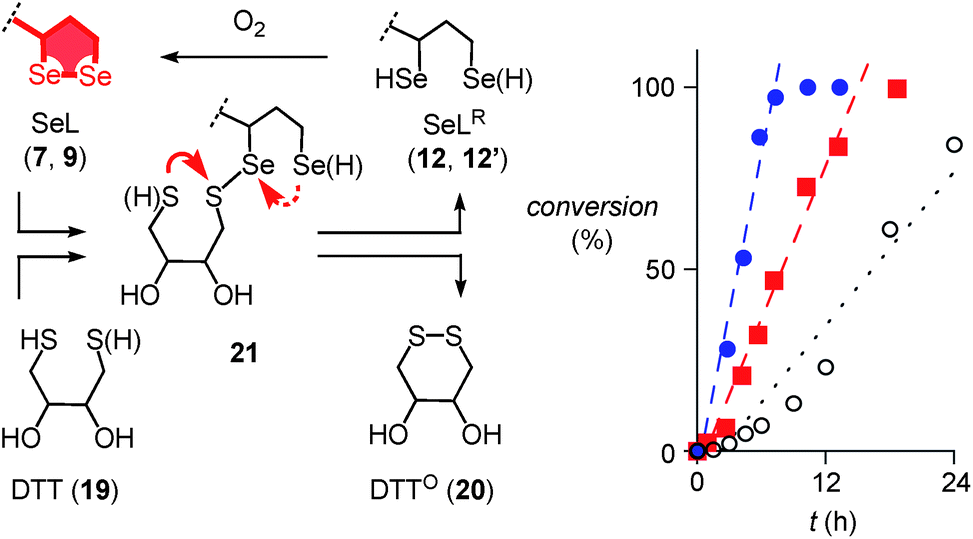 | ||
| Fig. 4 Oxidation of 500 μM 19 to 20 as a function of time in the absence (○) and the presence of 500 μM 9 (■) and 22 (●) in D2O, pD = 8.0, with linear fits to determine the t50. Structures are shown in neutral form, acidic protons are indicated in parenthesis. Competing ring closures in reactive intermediate 21 are indicated with solid (preferred) and dashed red arrows. | ||
Even more significant rate enhancements for DTT oxidation were observed with seleno asparagusic acid (SeA 22, Fig. 2 and 4, ●). Compared to 1,2-diselenolane 9, DTT lifetimes dropped further from t50 ≈ 6.9 h to t50 ≈ 3.9 h in the presence of SeA, and the final second-order rate constant increased from k ≈ 1.4 × 10−2 M−1 s−1 to k ≈ 8.9 × 10−2 M−1 s−1. These results provided the experimental evidence for thiol-diselenide exchange for SeA that could not be secured unambiguously by thiol-exchange affinity chromatography (Fig. 3d).
In summary, the passage through thiol-exchange affinity columns complemented by DTT oxidation catalysis reflected pertinent aspects of cellular uptake well (see below).1,2 The results demonstrated compatibility of 1,2-diselenolanes with strain-promoted thiol-mediated uptake. Moreover, evidence became available for dynamic covalent exchange with thiols on the cell surface, possible hopping along thiols or disulfides, facile intracellular release, possible participation of less reactive cellular disulfides and thiols, and clear differences between diselenides SeL 7, SeA 6 and SeC 1 as well as disulfides SA 4 and ETP 5.
The quantitative detection of cellular uptake was complicated by the fluorescence quenching with 1,2-diselenolanes. Compared to already strongly quenched 1,2-dithiolane SA 4, 1,2-diselenolanes SeL 7 and SeA 6 displayed even weaker emission of FITC in buffer by factors of 4.6 and 3.3, respectively (Fig. 5b, solid, blue, red vs. green). Adding the 3.4-fold quenching of free FITC by 1,2-dithiolane in 4,1 the total quenching by 1,2-diselenolane calculated to a factor of up to 15.6. Fluorescence partially recovered upon reduction with TCEP (Fig. 2, 5a and b). Compared to reduced SA 4, correction factors of 1.9 and 1.5 were obtained for SeLR12 and reduced form of SeA 6 (Fig. 5b, dashed, blue, red vs. green). For the quantification of uptake experiments, correction factors for closed cycles were applied.
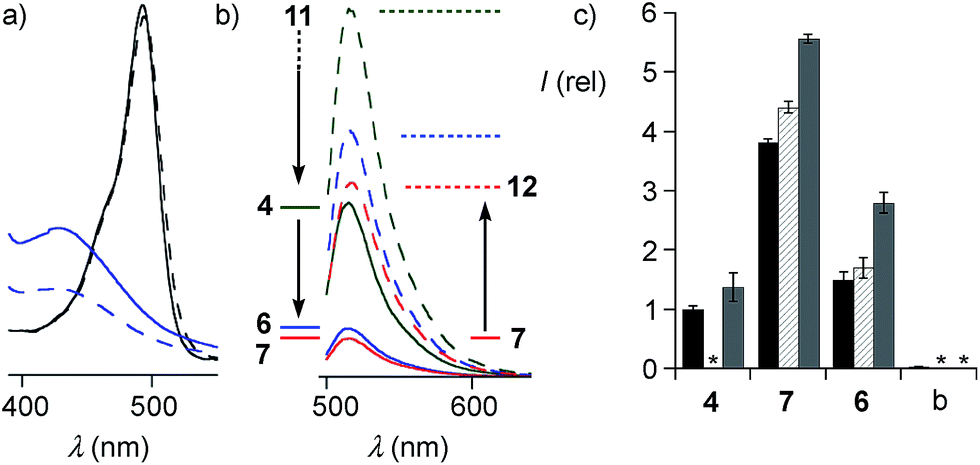 | ||
| Fig. 5 (a) Absorption spectra of the isolated motifs FITC (black) and SeA (22, blue) before (solid) and after treatment with TCEP (dashed). (b) FITC emission from conjugates SA 4 (green), SeA 6 (blue) and SeL 7 (red, 100 μM, PBS) before (solid) and after (dashed) reduction with TCEP. (c) Flow cytometry results (mean ± error of three independent measurements) for uptake of 10 μM SA 4, SeA 6 and SeL 7 and untreated (blank, b) HeLa Kyoto cells after incubation for 1 h (black), 2 h (hashed) and 4 h (grey), corrected for emission in closed form (b, solid). *Not determined. | ||
The uptake of the green-fluorescent SA 4, SeA 6 and SeL 7 into HeLa Kyoto cells was monitored by flow cytometry and confocal laser scanning microscopy (CLSM). Flow cytometry analysis with 10 μM of diselenides in Leibovitz medium at 37 °C showed a gradually increasing uptake with time, with 1,2-diselenolanes being much more active than 1,2-dithiolane SA 4, and with SeL 7 outperforming SeA 6 (Fig. 5c).
These impressions were confirmed in CLSM images, taken after one hour of incubation. Dithiolane SA 4 showed the known punctate emission characteristic for substantial capture in endosomes, together with significant homogeneous emission from the cytosol (Fig. 6a). SeA 6 showed stronger and more homogeneous emission at 50 μM but was visibly cytotoxic at 100 μM (Fig. 6b). With SeL 7, intact cells were stained brightly and homogeneously including nuclei, at 50 μM and also at 100 μM (Fig. 6c).
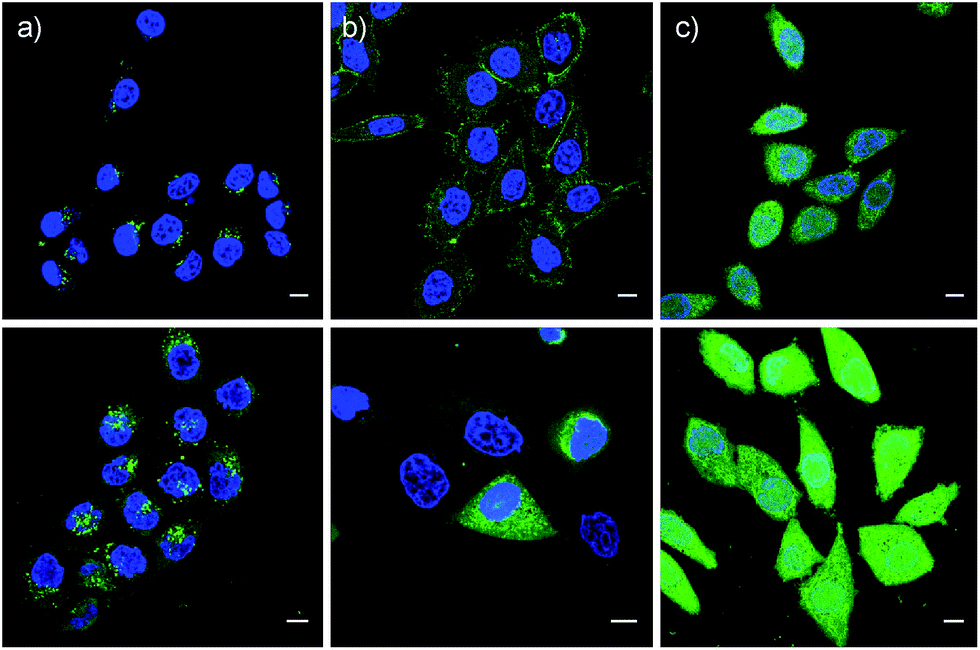 | ||
| Fig. 6 CLSM images of HeLa Kyoto cells after 1 h of incubation with 50 μM (top) and 100 μM (bottom) SA 4 (a), SeA 6 (b) and SeL 7 (c) in Leibovitz medium at 37 °C, together with Hoechst 33342 to stain the nuclei (blue). Scale bar = 10 μm. LP = 0.8% for CF and 6.5% for Hoechst. | ||
Cytotoxicity was determined with the MTT assay, an assay that reports on the metabolic activity of the cells. Like SA 4 and ETP 5, SeL 7 was not cytotoxic up to concentrations as high as 100 μM (Fig. 7a). As indicated by CLSM images (Fig. 6b), cytotoxicity started to emerge at 100 μM of SeA 6, although 10 μM CPPs remained clearly more toxic (Fig. 7a). Studies on SeA 6 were thus discontinued.
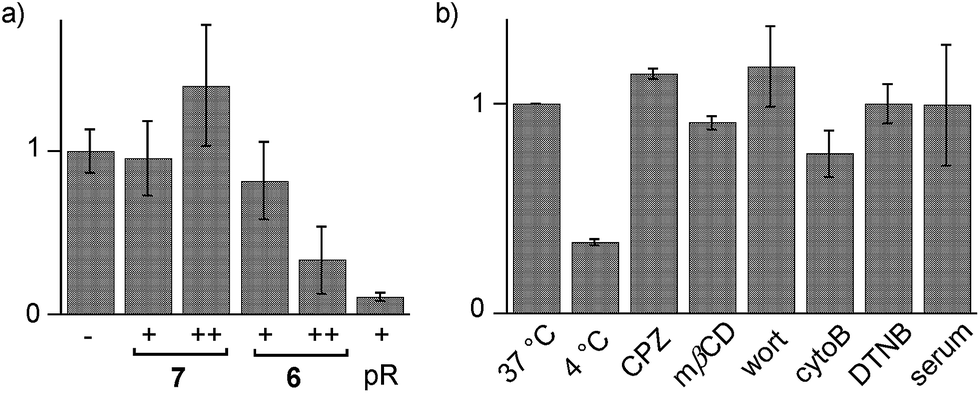 | ||
| Fig. 7 (a) Cell viability from MTT assay with 0 ((−) normalized to 1.0), 10 (+) and 100 μM (++) of SeL 7, SeA 6 or polyarginine (pR) in HeLa cells. (b) Flow cytometry data for HeLa Kyoto cells that were incubated with 10 μM SeL 7 at 37 °C (normalized to 1.0) and 4 °C, and cells that were preincubated with inhibitors of endocytosis (CPZ, mβCD, wort, cytoB) and thiol-mediated uptake (DTNB, 1.2 mM), followed by incubation with 7 at 37 °C, and incubation with 10% serum at 37 °C. Shown are average values ± error from three independent measurements. | ||
Diselenolane-mediated uptake of SeL 7 was less sensitive to temperature than ETP-mediated uptake,2 for instance (Fig. 7b). Insensitivity toward inhibitors supported that SeL does not enter cells by clathrin-mediated endocytosis (chlorpromazine; CPZ), caveolae-mediated endocytosis (methyl-β-cyclodextrin; mβCD) and macropinocytosis (wortmannin; wort, cytochalasin B; cytoB).5 The presence of 10% serum slightly decelerated uptake without reducing final efficiency significantly. As for ETP 5 but neither for SA 4 nor SL 3, uptake of SeL 7 was insensitive to DTNB, an inhibitor of conventional thiol-mediated uptake. In agreement with results from thiol-exchange affinity columns (Fig. 3), this might suggest that SeL 7 and ETP 5 can exchange also with less reactive thiols on the cell surface that are beyond reach of Ellman's reagent.
After longer incubation times, SeL 7 was efficiently delivered also at 10 μM (Fig. 8a–c). Z-stacks for SeL 7 confirmed that the homogeneous emission originates from cytosol (Fig. 8e). Corroborative support for delivery to cytosol and nucleus could be seen by the poor emission from nucleoli (Fig. 8d, arrows). Entry into the nucleus but not the nucleoli contrasted nicely to CPDs and other polycationic carriers that can accumulate in nucleoli, presumably due to interactions with polyanionic DNA,5 and to ETP 5, which targets the nucleus including nucleoli.2
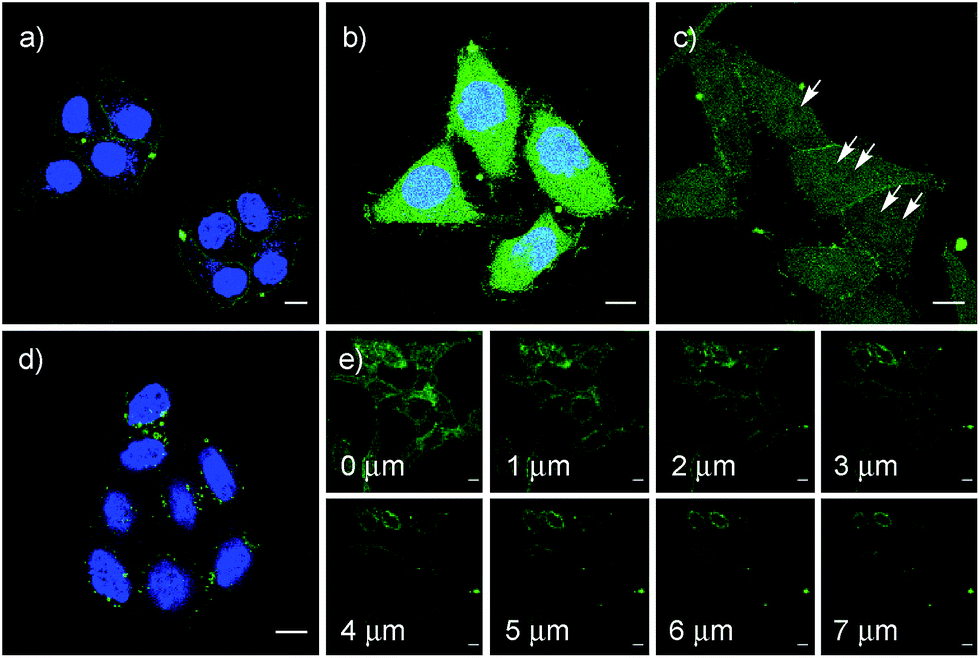 | ||
| Fig. 8 CLSM images of HeLa Kyoto cells after incubation with 10 μM SeL 7 for 1 h (a), 2 h (b) and 4 h (c) (arrows indicate selected nucleoli), z-stacks ((e) 4 h, distances in z-axis) and 10 μM SeC 1 (4 h (d)); (a), (b) and (d) together with Hoechst 33342, blue. Scale bar = 10 μm. LP = 0.8% for CF and 6.5% for Hoechst. | ||
The acyclic diseleno cystine SeC 1 showed weaker activity and mostly produced the punctate emission characteristic for endosomal capture (Fig. 8d and 9). This behavior was similar to SA 4 (Fig. 9) and in excellent agreement with mechanistic expectations from thiol exchange affinity columns (Fig. 3). More specifically, the selenosulfides obtained by thiol-diselenide exchange of SeC 1 on the cell surface are much more stable than with SeL 7 because the proximal covalent selenol for ring closure and release is missing (Fig. 3fvs.Fig. 3e). Uptake of this covalently attached selenosulfide could then occur by endocytosis and result in mostly endosomal capture, exactly as with SA 4 (Fig. 3fvs.3c and 9).6a It is understood that SeC 1 is not a perfect acyclic mimic of SeL 7 because it contains an additional fluorophore. However, this difference was expected to be irrelevant with regard to the uptake mechanism under investigation, the second fluorophore released at the cell surface should be simply washed away before imaging. The agreement between mechanistic expectations and observed results in cells and on thiol-exchange columns supported that these assumptions are correct and SeC 1 and SeL 7 properly reveal the intrinsic differences between cellular uptake of acyclic and strained cyclic diselenides. Given the demonstrated usefulness of SA 4 to deliver peptides, liposomes and polymersomes also to the cytosol along this route,6 this implied that delivery mediated by acyclic SeC 1 could be of substantial practical interest.
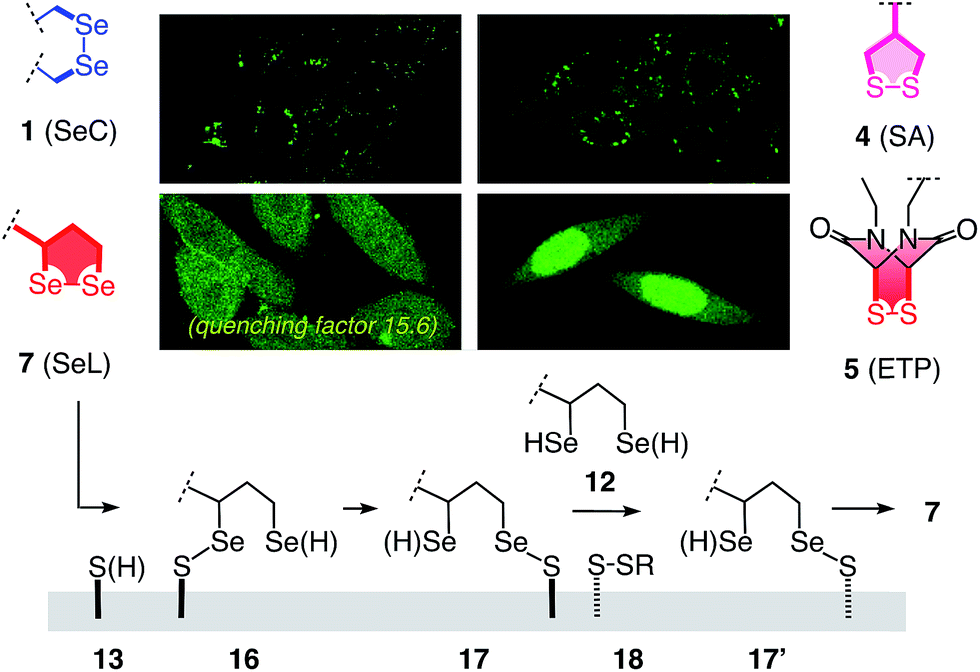 | ||
| Fig. 9 Comparative summary of intracellular location of SeC 1, SA 4, ETP 5,2 and SeL 7, and a tentative uptake mechanism for SeL 7. | ||
The clear difference of SA 4 and SeC 1 compared to ETP 5 and SeL 7 suggested that for dominant delivery to cytosol and nucleus with little endosomal capture, the release from covalent conjugates is essential (Fig. 9). The similar affinity chromatogram of the most active ETP 5 and SeL 7, particularly the unique, DTT-dependent peak (Fig. 3, **), implied the transient existence of the fully reduced form and its exchange with less reactive cellular disulfides during uptake (12, Fig. 9). For 1,2-diselenolane-mediated uptake, this added up to a putative mechanism that begins with exchange of poorly reactive thiols 13 on the cell surface with SeL 7 (insensitivity to DTNB). The obtained selenosulfide 16, covalently attached to the cell surface, rearranges to isomer 17 that is stable enough for detection by affinity chromatography (Fig. 3d*). Somewhere during uptake, thiol exchange with selenosulfide isomer 17 releases SeLR12, which exchanges with less reactive cellular disulfides 18 before it can back-oxidize to 7 (interpretation of **peak in affinity chromatogram, Fig. 3). Further exchange with other cellular thiols 13 and disulfides 18 is possible until SeL 7 or SeLR12 are released into the cytosol (Fig. 4 and 9). The result for diselenolane-mediated cytosolic delivery with SeL 7 is a quite spectacular target hopping mechanism on different oxidation levels that will be extremely difficult to clarify in detail (Fig. 9). The same conclusions apply to the similarly promising cytosolic delivery with ETP 5.
Several possible additions and alternatives to this putative uptake mechanism exist. For instance, the free selenol next to the selenosulfide in conjugates 16 or 17 could exchange with another exofacial disulfide to produce a second selenosulfide and afford a more stable conjugate, whereas further dimerization of oligomerization are unlikely due to high dilution and fast ring closure (Fig. 9). Glutathione at the cell surface could be involved and possibly initiate uptake with partially or fully reduced SeLR12, exchanging first with exofacial disulfides 18, instead of oxidized SeL 7, exchanging first with exofacial thiols 13 (Fig. 9).3p,3q Intracellular retention could contribute to uptake efficiencies and should influence intracellular location21 (uptake of underivatized CF was negligible under experimental conditions1,21). These and other uncertainties concerning a complex mode of action should not distract from the key facts disclosed in this report: (a) Cytosolic delivery by SeL 7 is outstanding (Fig. 9).22 (b) Clear differences to SeA 6 (cytotoxic, Fig. 6b and 7a; insensitive to affinity columns, Fig. 3d and e) indicate that a secondary selenol is essential (they very well reflect the distinct differences between SL and SA with regard to cellular uptake1 and polymerization on surfaces23 and in solution24). (c) Clear differences to SeC 1 (endosomal capture, Fig. 9; permanent retention on affinity columns, Fig. 3f and e) indicate that a strained cyclic diselenide is essential. (d) All trends, covering diselenides and disulfides from molecular modeling to affinity chromatography and intracellular localization, are consistent with conceptual expectations.
In summary, the move from sulfur to selenium affords the best carriers identified in this series so far. Cellular uptake mediated by 1,2-diselenolane 7, i.e., diseleno lipoic acid SeL, exceeds the performance of the best 1,2-dithiolane, i.e. SA 4 as well as the best disulfide at maximal ring tension, i.e. ETP 5, with regard to both efficiency and intracellular localization. No trace of endosomal capture as with 1,2-dithiolane 4, no trace of escape into the nucleus as with ETP 5: diselenolane 7 delivers mostly to the cytosol (Fig. 9). The fluorescence images are particularly impressive with the strong quenching exerted by 1,2-diselenolanes taken into consideration (Fig. 5). The homologous 1,2-dithiolane SL 3 is clearly less performing not only than SeL 7 but also SA 4.1
A comprehensive analysis of the nature of 1,2-diselenolanes compared to 1,2-dithiolanes reveals a combination of ring-tension, selenophilicity and the high but different acidity of primary and secondary selenols as the origin of the superb performance of 1,2-diselenolane 7. Most importantly, thiol-exchange affinity chromatography is introduced to mimic and rationalize insights on thiol-mediated cellular uptake. The similar characteristics of ETP 5 and SeL 7 recorded by this method point toward an intriguing multitarget thiol hopping mechanism along so far unexplored routes to account for the efficient cytosolic delivery of these most powerful systems.22,25
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Xavier Martin-Benlloch for contributions to synthesis, the Roux group for assistance with cell culture, the NMR, the MS 2.0 and the Bioimaging Platform for services, and the University of Geneva, the Swiss National Centre of Competence in Research (NCCR) Chemical Biology, the NCCR Molecular Systems Engineering and the Swiss NSF for financial support.Notes and references
- G. Gasparini, G. Sargsyan, E.-K. Bang, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2015, 54, 7328–7331 CrossRef CAS PubMed.
- L. Zong, E. Bartolami, D. Abegg, A. Adibekian, N. Sakai and S. Matile, ACS Cent. Sci., 2017, 3, 449–453 CrossRef CAS PubMed.
- (a) S. Aubry, F. Burlina, E. Dupont, D. Delaroche, A. Juliot, S. Laveille, G. Chassaing and S. Sagan, FASEB J., 2009, 23, 2956–2967 CrossRef CAS PubMed; (b) D. Oupický and J. Li, Macromol. Biosci., 2014, 14, 908–922 CrossRef PubMed; (c) Y. Y. Ling, J. Ren, T. Li, Y. B. Zhao and C. L. Wu, Chem. Commun., 2016, 52, 4533–4536 RSC; (d) J. Fu, C. Yu, L. Lu and S. Q. Yao, J. Am. Chem. Soc., 2015, 137, 12153–12160 CrossRef CAS PubMed; (e) P. Yuan, H. Zhang, L. Qian, X. Mao, S. Du, C. Yu, B. Peng and S. Q. Yao, Angew. Chem., Int. Ed., 2017, 56, 12481–12485 CrossRef CAS PubMed; (f) A. G. Torres and M. J. Gait, Trends Biotechnol., 2012, 30, 185–190 CrossRef CAS PubMed; (g) L. Brülisauer, N. Kathriner, M. Prenrecaj, M. A. Gauthier and J.-C. Leroux, Angew. Chem., Int. Ed., 2012, 51, 12454–12458 CrossRef PubMed; (h) A. Kichler, J. S. Remy, O. Boussif, B. Frisch, C. Boeckler, J.-P. Behr and F. Schuber, Biochem. Biophys. Res. Commun., 1995, 209, 444–450 CrossRef CAS PubMed; (i) E. P. Feener, W. C. Shen and H. J. P. Ryser, J. Biol. Chem., 1990, 265, 18780–18785 CAS; (j) P. K. Hashim, K. Okuro, S. Sasaki, Y. Hoashi and T. Aida, J. Am. Chem. Soc., 2015, 137, 15608–15611 CrossRef CAS PubMed; (k) S. A. Bode, R. Wallbrecher, R. Brock, J. C. M. van Hest and D. W. P. M. Löwik, Chem. Commun., 2014, 50, 415–417 RSC; (l) C. R. Drake, A. Aissaoui, O. Argyros, M. Thanou, J. H. G. Steinke and A. D. Miller, J. Controlled Release, 2013, 171, 81–90 CrossRef CAS PubMed; (m) S. Son, R. Namgung, J. Kim, K. Singha and J. W. Kim, Acc. Chem. Res., 2012, 45, 1100–1112 CrossRef CAS PubMed; (n) T.-I. Kim and S. W. Kim, React. Funct. Polym., 2011, 71, 344–349 CrossRef CAS PubMed; (o) M. Balakirev, G. Schoehn and J. Chroboczek, Chem. Biol., 2000, 7, 813–819 CrossRef CAS PubMed; (p) W. Gao, T. Li, J. Wang, Y. Zhao and C. Wu, Anal. Chem., 2017, 89, 937–944 CrossRef CAS PubMed; (q) T. Li, W. Gao, J. Liang, M. Zha, Y. Chen, Y. Zhao and C. Wu, Anal. Chem., 2017, 89, 8501–8508 CrossRef CAS PubMed; (r) D. Jha, R. Mishra, S. Gottschalk, K.-H. Wiesmüller, K. Ugurbil, M. E. Maier and J. Engelmann, Bioconjugate Chem., 2011, 22, 319–328 CrossRef CAS PubMed; (s) A. N. Shirazo, N. Salem El-Sayed, D. Mandal, R. K. Tiwari, K. Tavakoli, M. Etesham and K. Parang, Bioorg. Med. Chem. Lett., 2016, 26, 656–661 CrossRef PubMed.
- T. Li and S. Takeoka, Int. J. Nanomed., 2014, 9, 2849–2861 Search PubMed.
- (a) G. Gasparini, E.-K. Bang, G. Molinard, D. V. Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 CrossRef CAS PubMed; (b) P. Morelli and S. Matile, Helv. Chim. Acta, 2017, 100, e1600370 CrossRef; (c) E. Derivery, E. Bartolami, S. Matile and M. Gonzalez-Gaitan, J. Am. Chem. Soc., 2017, 139, 10172–10175 CrossRef CAS PubMed.
- (a) D. Abegg, G. Gasparini, D. G. Hoch, A. Shuster, E. Bartolami, S. Matile and A. Adibekian, J. Am. Chem. Soc., 2017, 139, 231–238 CrossRef CAS PubMed; (b) N. Chuard, G. Gasparini, D. Moreau, S. Lörcher, C. Palivan, W. Meier, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2017, 56, 2947–2950 CrossRef CAS PubMed.
- (a) T. G. Back and P. W. Codding, Can. J. Chem., 1983, 61, 2749–2752 CrossRef CAS; (b) O. Foss, K. Johnsen and T. Reistad, Acta Chem. Scand., 1964, 18, 2345–2354 CrossRef CAS.
- J. Hildebrandt, T. Niksch, R. Trautwein, N. Häfner, H. Görls, M.-C. Barth, M. Dürst, I. B. Runnebaum and W. Weigand, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 182–186 CrossRef CAS.
- T. Niksch, H. Görls, M. Friedrich, R. Oilunkaniemi, R. Laitinen and W. Weigand, Eur. J. Inorg. Chem., 2010, 74–94 CrossRef CAS.
- (a) G. Bergson, G. Claeson and L. Schotte, Acta Chem. Scand., 1962, 16, 1159–1174 CrossRef CAS; (b) G. Bergson and G. Claeson, Acta Chem. Scand., 1961, 15, 1611–1613 CrossRef CAS.
- (a) B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS PubMed; (b) S. Benz, J. López-Andarias, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2017, 56, 812–815 CrossRef CAS PubMed.
- (a) B. Rasmussen, A. Sorensen, H. Gotfredsen and M. Pittelkow, Chem. Commun., 2014, 50, 3716–3718 RSC; (b) R. J. Hondal, S. M. Marino and V. N. Gladyshev, Antioxid. Redox Signaling, 2013, 18, 1675–1689 CrossRef CAS PubMed; (c) J. Sayers, R. J. Payne and N. Winssinger, Chem. Sci., 2018 10.1039/c7sc02736b.
- D. Steinmann, T. Nauser and W. H. Koppenol, J. Org. Chem., 2010, 75, 6696–6699 CrossRef CAS PubMed.
- W. H. H. Guenther, J. Org. Chem., 1967, 32, 3931–3933 CrossRef CAS.
- (a) J. Beld, K. J. Woycechowsky and D. Hilvert, Biochemistry, 2007, 46, 5382–5390 CrossRef CAS PubMed; (b) J. Beld, K. J. Woycechowsky and D. Hilvert, Biochemistry, 2008, 47, 6985–6987 CrossRef CAS PubMed.
- J. C. Lukesh, B. VanVeller and R. T. Raines, Angew. Chem., Int. Ed., 2013, 52, 12901–12904 CrossRef CAS PubMed.
- J. Liu and S. Rozovsky, Antioxid. Redox Signaling, 2015, 23, 795–813 CrossRef CAS PubMed.
- (a) H. Xu, W. Cao and X. Zhang, Acc. Chem. Res., 2013, 46, 1647–1658 CrossRef CAS PubMed; (b) D. Yue, G. Cheng, Y. He, Y. Nie, Q. Jiang, X. Cai and Z. Gu, J. Mater. Chem. B, 2014, 2, 7210–7221 RSC; (c) Y. Wang, L. Zhu, Y. Wang, L. Li, Y. Lu, L. Shen and L. W. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 35106–35113 CrossRef CAS PubMed.
- F. Xu, Z.-Z. Yang and S.-J. Zhang, Phosphorus, Sulfur Silicon Relat. Elem., 2013, 188, 1312–1319 CrossRef CAS.
- M. Haratake, S. Matsumoto, M. Ono and M. Nakayama, Bioconjugate Chem., 2008, 19, 1831–1839 CrossRef CAS PubMed.
- S. Izumi, Y. Urano, K. Hanaoka, T. Terai and T. Nagano, J. Am. Chem. Soc., 2009, 131, 10189–10200 CrossRef CAS PubMed.
- Preliminary results support that diselenolane-mediated uptake is compatible with the cytosolic delivery of proteins. Studies toward practical applications to deliver substrates of free choice, from DNA and RNA22a to proteins5c and fluorescent probes,22b are ongoing. (a) C. Gehin, J. Montenegro, E.-K. Bang, S. Takayama, H. Hirose, S. Futaki, A. Cajaraville, S. Matile and H. Riezman, J. Am. Chem. Soc., 2013, 135, 9295–9298 CrossRef CAS PubMed; (b) Q. Verolet, M. Dal Molin, A. Colom, A. Roux, L. Guénée, N. Sakai and S. Matile, Helv. Chim. Acta, 2017, 100, e1600328 CrossRef.
- A. Carmine, Y. Domoto, N. Sakai and S. Matile, Chem.–Eur. J., 2013, 19, 11558–11563 CrossRef CAS PubMed.
- E.-K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 2088–2091 CrossRef CAS PubMed.
- A pioneering study on the delivery of acyclic selenosulfides appeared during the evaluation process of this manuscript: C. C. Tjin, K. D. Otley, T. D. Baguley, P. Kurup, J. Xu, A. C. Nairn, P. J. Lombroso and J. A. Ellman, ACS Cent. Sci., 2017, 3, 1322–1328 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures and results for all reported experiments. See DOI: 10.1039/c7sc05151d |
| This journal is © The Royal Society of Chemistry 2018 |