
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bridged [2.2.1] bicyclic phosphine oxide facilitates catalytic γ-umpolung addition–Wittig olefination†
Kui
Zhang‡
,
Lingchao
Cai‡
,
Zhongyue
Yang
,
K. N.
Houk
 and
Ohyun
Kwon
*
and
Ohyun
Kwon
*
Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095-1569, USA. E-mail: ohyun@chem.ucla.edu
First published on 18th January 2018
Abstract
A novel bridged [2.2.1] bicyclic phosphine oxide, devised to circumvent the waste generation and burdens of purification that are typical of reactions driven by the generation of phosphine oxides, has been prepared in three steps from commercially available cyclopent-3-ene-1-carboxylic acid. The performance of this novel phosphine oxide was superior to those of current best-in-class counterparts, as verified experimentally through kinetic analysis of its silane-mediated reduction. It has been applied successfully in halide-/base-free catalytic γ-umpolung addition–Wittig olefinations of allenoates and 2-amidobenzaldehydes to produce 1,2-dihydroquinolines with good efficiency. One of the 1,2-dihydroquinoline products was converted to known antitubercular furanoquinolines.
Introduction
The classic Wittig reaction has been adopted widely for the construction of carbon–carbon double bonds since its discovery in 1953.1 The reaction is, however, far from ideal in terms of its environmental impact, requiring stoichiometric amounts of organic halide, base, and tertiary phosphine and producing stoichiometric amounts of base–halide salt and phosphine oxide wastes.2 Typically, the latter is not water-soluble, co-elutes with the olefin product during chromatographic separation, and (for a solid product) readily precipitates with the product, impeding its purification, particularly in large-scale processes (e.g., those conducted on industrial scale).3 Traditionally, phosphorus ylides are prepared from corresponding phosphonium halides using stoichiometric amounts of base. Although less prevalent, ylides can also be prepared by simply mixing activated alkenes, acetylenes, and allenes with tertiary phosphines.4 In fact, halogen- and base-free Wittig olefinations between activated alkenes and aldehydes predate the venerable Morita–Baylis–Hillman reaction by several years.4a More recently, Werner, Lin, and Voituriez each reported halogen- and base-free catalytic Wittig olefinations.5 We also envisioned a halide- and base-free γ-umpolung–Wittig process.6 In our reaction, phosphine adds to the allenoate 1a to form the zwitterionic intermediate A (Scheme 1). Deprotonation of the sulfonamide 2a and γ-addition produces the ylide B, which, through a Wittig reaction, generates the 1,2-dihydroquinoline 3a and produces the phosphine oxide (condition 1; steps I → II → III). To minimize the generation of the phosphine oxide byproduct, we turned our attention to the recently developed catalytic Wittig conditions.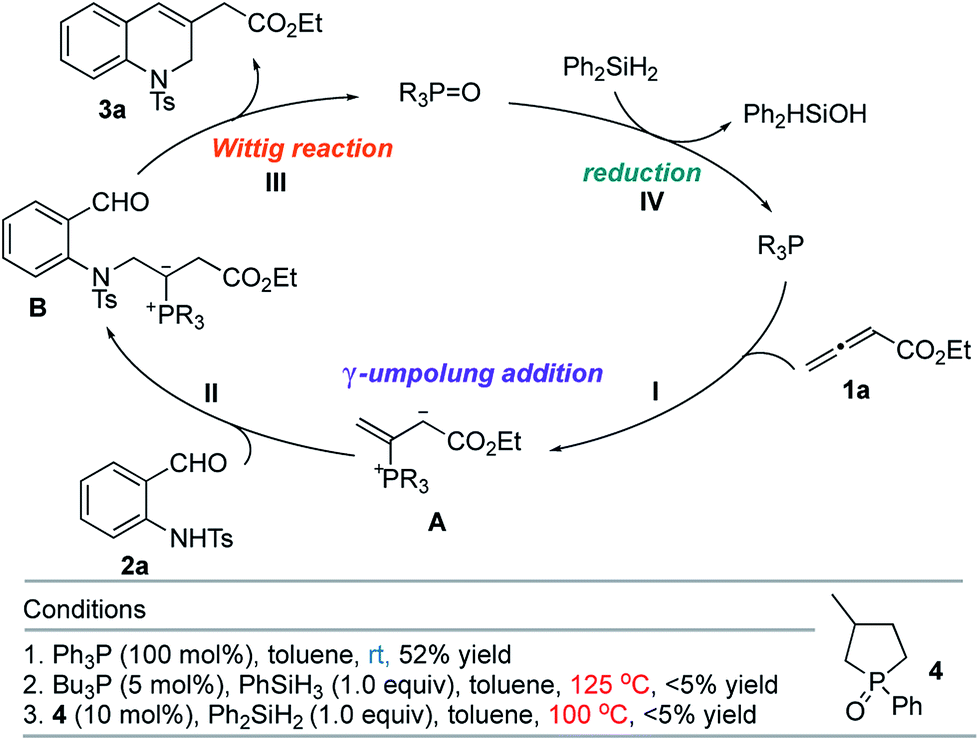 | ||
| Scheme 1 γ-Umpolung–Wittig reaction. | ||
In 2009, O'Brien and coworkers reported the first phosphine oxide-catalyzed Wittig reaction for the synthesis of alkenes, with the phosphine oxide byproduct reduced by a silane in situ back to the phosphine.7a This strategy has since been employed to promote Wittig, Appel, Staudinger, and Mitsunobu reactions, as well as other reactions that are thermodynamically driven by virtue of the formation of phosphine oxide.7–9 When we applied catalytic Bu3P5b or phospholane oxide 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7a under the reported reaction conditions, the desired dihydroquinoline was obtained in less than 5% yield (conditions 2 and 3; steps I → II → III → IV). Presumably, the allenoate 1a oligomerized under the high temperatures. Consequently, we directed our study to the development of new phosphine oxides that can be reduced under milder conditions to realize the γ-umpolung–Wittig reaction in a catalytic mode.
7a under the reported reaction conditions, the desired dihydroquinoline was obtained in less than 5% yield (conditions 2 and 3; steps I → II → III → IV). Presumably, the allenoate 1a oligomerized under the high temperatures. Consequently, we directed our study to the development of new phosphine oxides that can be reduced under milder conditions to realize the γ-umpolung–Wittig reaction in a catalytic mode.
Results and discussion
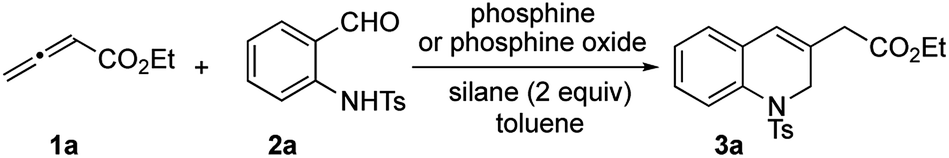
Recently reported catalytic Wittig reactions have been mainly promoted by phospholane oxide 4 and dibenzophosphole oxide 5 (Fig. 1), which are reduced faster than larger-ring phosphacycloalkane oxides or acyclic counterparts.7,8 The currently accepted mechanism for silane-mediated reduction of phosphine oxides to phosphines is based on the studies of Horner and Mislow (Scheme 2).10 Krenske demonstrated computationally that the rate-determining step (RDS) of the reduction is an intramolecular hydride transfer from silicon to phosphorus after coordination of the phosphine oxide to the silane.11 The transition state (TS) for the hydride transfer features a four-membered P–O–Si–H ring, in which both the P and Si atoms are centers of two trigonal bipyramids bridged by O and H atoms. Consequently, we surmised that a phosphine oxide with an a–P–b angle close to 90° may undergo facile silane-mediated reduction, due to minimal structural deformation upon proceeding to the TS.12 Previously, we reported the invention of the [2.2.1] bicyclic phosphine oxide 6, whose C–P–C angle in the X-ray structure was 93.3° (Fig. 1).13 Relative to the phospholane oxide 4 and the dibenzophosphole oxide 5, compound 6 is reduced faster by a silane (vide infra). Accordingly, we designed the [2.2.1] bicyclic phosphine oxide 7, which possesses a scaffold similar to that of 6.14 Computational modeling predicted a C–P–C angle of 92.6°—much closer to 90° than the corresponding value (95.4°) of its non-bridged counterpart 4.15,16 Despite its smaller endocyclic C–P–C angle (91.5°), the triarylphosphine oxide 5 is reduced at a rate similar to that of the dialkylarylphosphine oxide 4, because more electron rich alkyl-substituted phosphine oxides are reduced faster than their aryl-substituted counterparts.5b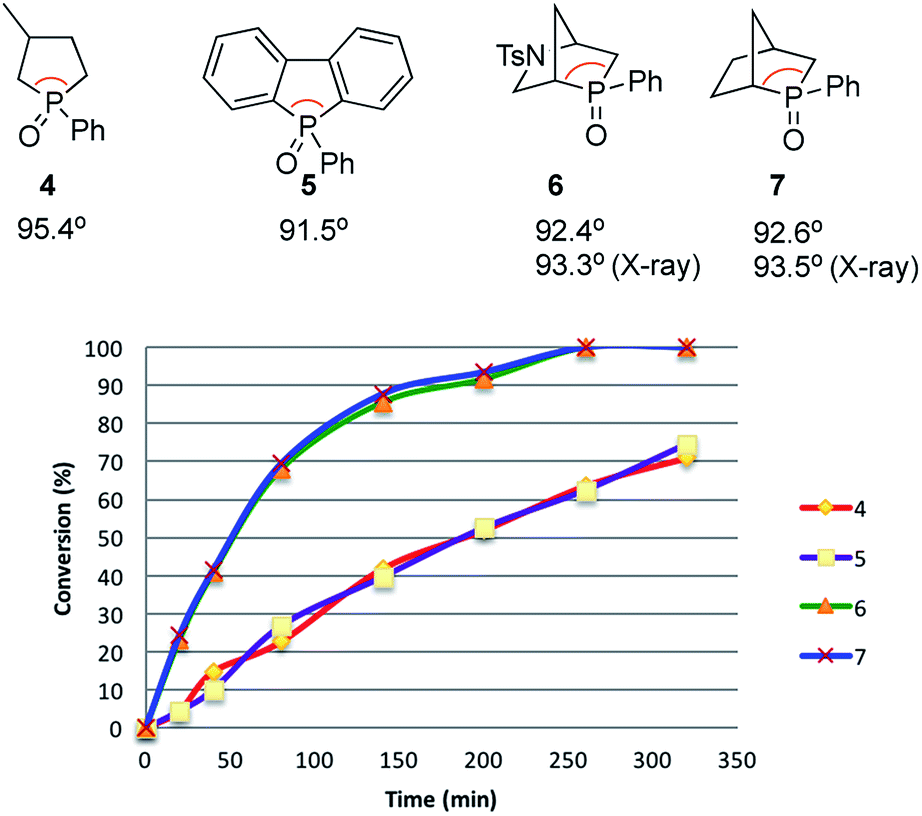 | ||
| Fig. 1 Diphenylsilane-mediated reductions of the phosphine oxides 4–7 and their computed C–P–C angles. | ||
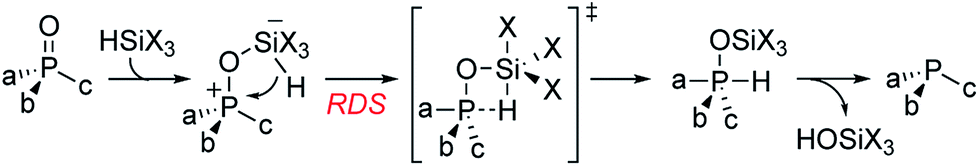 | ||
| Scheme 2 Silane-mediated reduction of a phosphine oxide. | ||
To test our hypothesis, we synthesized the phosphine oxide 7. As depicted in Scheme 3, reduction, hydroboration, and oxidation of commercially available cyclopent-3-ene-1-carboxylic acid, followed by bismesylation, produced the dimesylate 8 with 2.8:1 trans-to-cis selectivity, in 70% yield over two steps. A 2.4:1 mixture of the exo- and endo-P-phenylphosphine oxides 7 (δP = 55.8) and 7′ (δP = 57.4) was obtained in 68% isolated yield after bisalkylation with dilithium phenylphosphide and subsequent oxidative workup.17
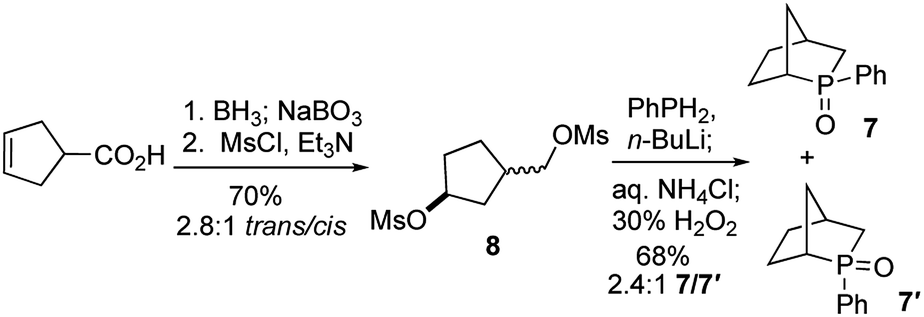 | ||
| Scheme 3 Synthesis of the phosphine oxide 7. | ||
With the phosphine oxide 7 in hand, we compared the efficiencies of the diphenylsilane-mediated reductions of the phosphine oxides 4–7. The conversions of the phosphine oxides to the phosphines (diphenylsilane, 20 equiv.; CD3CN; 80 °C; 0.2 mol L−1) were monitored using 31P NMR spectroscopy (Fig. 1).18 At 20 min, the conversions of 4–7 were 4.5, 4.5, 23.2, and 24.3%, respectively. The reductions progressed steadily, with the phosphine oxides 6 and 7 reaching full conversion after 280 min; in contrast, the phosphine oxides 4 and 5 were not fully converted into their phosphines even after 6 h. The kinetic profiles of 6 and 7 were similar, as were those of 4 and 5. The initial rates for 4 and 7 were 0.37 and 1.42 min−1, respectively.15 The ratio of these two initial rates (r4:r7 = 1:4) corresponds to a 0.8 kcal mol−1 difference in activation free energies, based on the Boltzmann distribution. Computational studies were conducted to rationalize the reactivity difference between 4 and 7. The TSs for the reductions of 4 and 7 by diphenylsilane were located using the B3LYP method in Gaussian 09, with single point energy computed under B3LYP method with Grimme D3 correction. The calculated activation free energies for substrates 4 and 7 were 30.1 and 29.3 kcal mol−1, respectively—a difference of 0.8 kcal mol−1, consistent with the experimental observation.15
Having confirmed the ready reducibility of the phosphine oxide 7 experimentally, our attention turned to applying this bridged bicyclic phosphine oxide to halide-/base-free catalytic γ-umpolung–Wittig olefination. Initially, we employed ethyl allenoate (1a) and o-(p-toluenesulfonamido)benzaldehyde (2a) as model reactants to identify suitable reaction conditions for the catalytic γ-umpolung–Wittig olefination (Table 1). We used diphenylsilane as the reducing reagent in the reactions catalyzed by the various phosphines or phosphine oxides at a reaction temperature of 80 °C (entries 1–6). Little to no catalytic turnover occurred when applying triphenylphosphine or tributylphosphine, presumably due to the lack of ring strain.19 The phosphine oxides 4–7 produced the dihydroquinoline product in yields of 62, 23, 76, and 81%, respectively. Notably, the reaction employing 7 took only 4 h to reach completion, while that of 4 was sluggish (24 h). Much starting material remained after 48 h for the reaction performed using the phosphine oxide 5. Diphenylsilane was more efficient (81%) as a reducing agent than were phenylsilane and triphenylsilane (74 and 0%, respectively) under otherwise identical reaction conditions (entries 7 and 8). A survey of reaction temperatures revealed that the yield did not improve when run at either higher (oligomerization of allenoate) or lower temperature (entries 9 and 10). The reaction concentration also had a discernible effect on the reaction efficiency, with 0.03 M being optimal (entries 11 and 12). Finally, with decreased loading of the phosphine oxide 7 to 20 and 15 mol%, the reactions were prolonged to 12 and 29 h, respectively, with diminished product yields (entries 13 and 14). To our delight, the reaction time shortened to 4 h and the yield improved to 88% when 15 mol% of the phosphine oxide 7 was pre-reduced by diphenylsilane prior to addition of the reactants (entry 15). In addition, here we added the allenoate 1a using a syringe pump to minimize its oligomerization. The yield declined to 75% when 10 mol% of the phosphine oxide 7 was employed in the same manner (entry 16). Thus, the standard reaction conditions were established: 1a (0.6 mmol, 3 equiv.), 2a (0.2 mmol), Ph2SiH2 (0.4 mmol, 2 equiv.) as the reducing agent, and the phosphine oxide 7 (0.03 mmol, 0.15 equiv.) as the catalyst at 80 °C.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Phosphine (oxide) | C (mol L−1) | Silane | Temp. (°C) | Time (h) | Yieldb (%) |
| a Performed in a vial containing 1a (0.6 mmol), 2a (0.2 mmol), a silane (0.4 mmol), and a phosphine or phosphine oxide in toluene. b Isolated yield. c Much starting material (2a) remained after 2 days. d 7 was reduced by Ph2SiH2 in toluene before adding 1a and 2a. e 1a was added using a syringe pump to prohibit oligomerization. | ||||||
| 1 | PBu3 (30%) | 0.05 | Ph2SiH2 | 80 | 24 | — |
| 2 | PPh3 (30%) | 0.05 | Ph2SiH2 | 80 | 24 | 39 |
| 3 | 4 (30%) | 0.05 | Ph2SiH2 | 80 | 24 | 62 |
| 4c | 5 (30%) | 0.05 | Ph2SiH2 | 80 | 48 | 23 |
| 5 | 6 (30%) | 0.05 | Ph2SiH2 | 80 | 12 | 76 |
| 6 | 7 (30%) | 0.05 | Ph2SiH2 | 80 | 4 | 81 |
| 7 | 7 (30%) | 0.05 | PhSiH3 | 80 | 4 | 74 |
| 8 | 7 (30%) | 0.05 | Ph3SiH | 80 | 24 | — |
| 9 | 7 (30%) | 0.05 | Ph2SiH2 | 100 | 3 | 42 |
| 10 | 7 (30%) | 0.05 | Ph2SiH2 | 70 | 24 | 38 |
| 11 | 7 (30%) | 0.1 | Ph2SiH2 | 80 | 2 | 47 |
| 12 | 7 (30%) | 0.03 | Ph2SiH2 | 80 | 6 | 83 |
| 13 | 7 (20%) | 0.03 | Ph2SiH2 | 80 | 12 | 82 |
| 14 | 7 (15%) | 0.03 | Ph2SiH2 | 80 | 29 | 75 |
| 15d,e | 7 (15%) | 0.03 | Ph2SiH2 | 80 | 4 | 88 |
| 16d,e | 7 (10%) | 0.03 | Ph2SiH2 | 80 | 13 | 75 |
To track the evolution of the phosphine oxide catalyst 7 during the catalytic cycle, the reaction mixture was examined spectroscopically over the course of the entire process under the optimized conditions. 31P NMR spectroscopy revealed that the resting state of catalyst was the phosphine oxide.15 This observation is in agreement with our premise that the RDS of the whole catalytic cycle is the reduction of the phosphine oxide to the phosphine, and that the overall reaction efficiency reflects that of the phosphine oxide's reducibility.
The generality of this catalytic γ-umpolung–Wittig olefination was probed using various allenoates and o-sulfonamidobenzaldehydes (Scheme 4).20 Electron-donating substituents on the benzaldehyde ring led to the 1,2-dihydroquinoline derivatives 3a–3f all being generated in high yields when coupling with the allenoate 1a. When using 2-(4-toluenesulfonamido)-3-methylbenzaldehyde, a prolonged reaction time was required, presumably because of steric effects; the reaction was, however, clean, furnishing the desired product 3f in quantitative yield. When 5-bromo-2-(4-toluenesulfonamido)benzaldehyde, bearing an electron-withdrawing bromine substituent, was used, the yield was 55% (3g).21 The compatibility of an aryl bromide with the current reaction is especially notable in light of its suitability for further functionalization of the dihydroquinoline products.
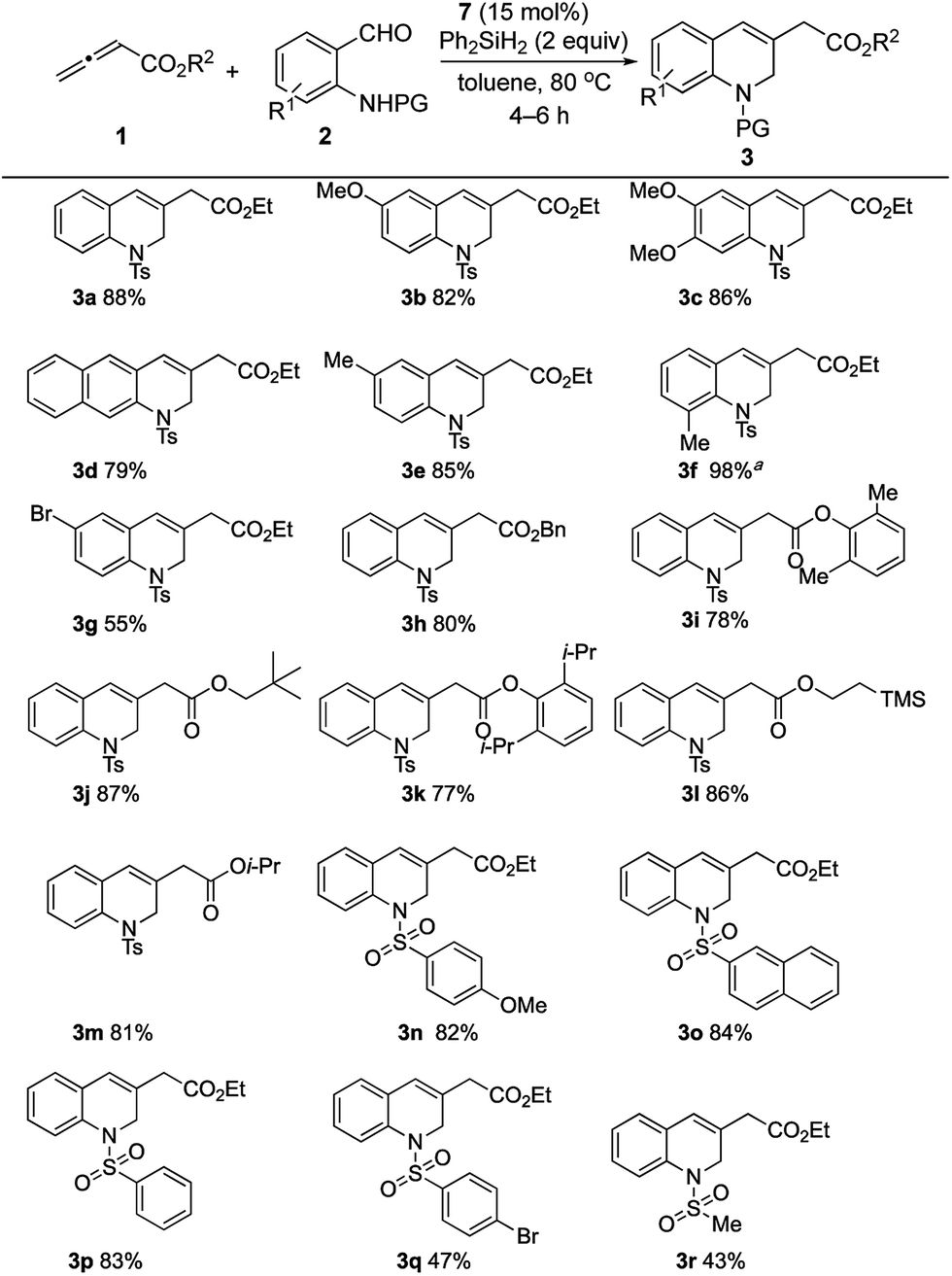 | ||
| Scheme 4 Synthesis of 1,2-dihydroquinolines. General conditions: 7 (30 μmol) and Ph2SiH2 (0.4 mmol) were stirred in toluene (0.5 mL) at 80 °C for 1 h, followed by addition of neat 2 (0.2 mmol), then 1 (0.6 mmol, 0.11 M in toluene), using a syringe pump, over 2 h. The isolated yields are shown. aReaction was complete within 20 h. | ||
We then studied the allenoate substrate scope by treating 2a with a series of allenoates, which all afforded their corresponding 1,2-dihydroquinolines (3h–3m) in high yields. γ-Phenylallenoate and α-methylallenoate did not produce the desired products, presumably due to increased steric hindrance.21 Subsequently, various protecting groups of the amide were probed. N-(2-Formylphenyl)-4-methoxybenzenesulfonamide, N-(2-formylphenyl)naphthalene-2-sulfonamide, and N-(2-formylphenyl)benzenesulfonamide produced their desired products 3n, 3o, and 3p, respectively, in good yields (82–84%). A sulfonamide with an electron-withdrawing 4-bromo group gave its expected product 3q in moderate yield, presumably because of the diminished nucleophilicities of its sulfonamide unit during the γ-umpolung addition.22 The reaction of N-(2-formylphenyl)methanesulfonamide and ethyl allenoate gave 3r in 43% yield; we suspect that the acidic methyl protons of the methanesulfonamide might have had a deleterious effect on the overall reaction efficiency.
The utility of this γ-umpolung/Wittig reaction was further explored in the synthesis of the furanoquinolines 13, which have shown antitubercular activity (Scheme 5).23 For this synthesis, we reduced the ester 3a with lithium aluminum hydride (LAH), followed by iodoetherification with N-iodosuccinimide (NIS), to obtain the furan 9 in 82% yield over two steps. The core structure 10 was obtained after deiodination with RANEY® Ni. Compound 10 was treated with Mg in MeOH under sonication to reductively cleave the tosyl group. Subsequent iodosobenzene-mediated oxidation yielded the imine 12 in 80% yield. The synthetic target 13 was obtained in 63% yield as a 1:1 mixture of diastereoisomers after nucleophilic addition of diphenylzinc.
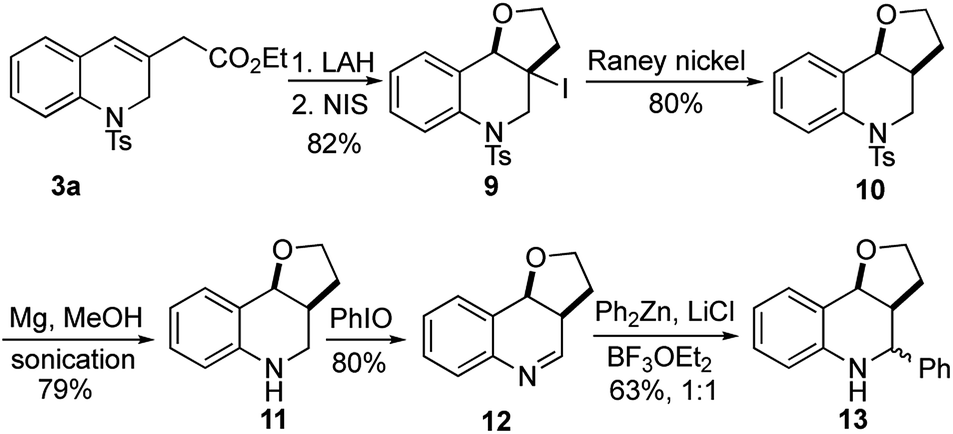 | ||
| Scheme 5 Synthesis of antitubercular furanoquinolines 13. | ||
Conclusions
We have developed a new bridged [2.2.1] bicyclic phosphine oxide 7 that displays exceptional reduction efficiency in the presence of diphenylsilane. Although the catalytic Wittig reaction has been developing rapidly for several years, there remains room for the development of phosphine oxide structures to improve the efficiency of their silane-mediated reductions. The utility of the bridged bicyclic phosphine oxide 7 was exemplified in catalytic halide- and base-free γ-umpolung–Wittig reactions of allenoates and o-sulfonamidebenzaldehydes, providing 1,2-dihydroquinolines. One of the γ-umpolung–Wittig reaction products, 3a, was elaborated in the synthesis of the antitubercular furanoquinolines 13. Further refinement of the phosphine oxide scaffold and studies of the applications of these phosphine oxides in other catalytic processes are underway in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NIH (GM071779 to O. K.) and the NSF (CHE-1361104 to K. N. H.) for financial support. We also thank Yi Chiao Fan and Andrew Smaligo for their assistance with CombiFlash and HPLC. Computational resources were provided by the UCLA Institute for Digital Research and Education (IDRE) and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (OCI-1053575).Notes and references
- (a) G. Wittig and G. Geissler, Liebigs Ann. Chem., 1953, 580, 44 CrossRef CAS; (b) B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863 CrossRef CAS; (c) P. J. Murphy and S. E. Lee, J. Chem. Soc., Perkin Trans. 1, 1999, 3049 RSC.
- (a) M. G. Russell and S. Warren, Tetrahedron Lett., 1998, 39, 7995 CrossRef CAS; (b) A. Galante, P. Lhoste and D. Sinou, Tetrahedron Lett., 2001, 42, 5425 CrossRef CAS; (c) J. Westman, Org. Lett., 2001, 3, 3745 CrossRef CAS PubMed.
- (a) C. Mercier and P. Chabardes, Pure Appl. Chem., 1994, 66, 1509 CrossRef CAS; (b) H. Ernst, Pure Appl. Chem., 2002, 74, 2213 CrossRef CAS.
- For examples of Wittig reactions of ylides generated in situ from olefins, allenes, and acetylenes, see: (a) R. Oda, T. Kawabata and S. Tanimoto, Tetrahedron Lett., 1964, 5, 1653 CrossRef; (b) A. Ramazani and A. Bodaghi, Tetrahedron Lett., 2000, 41, 567 CrossRef CAS; (c) C. K. Jung, J. C. Wang and M. J. Krische, J. Am. Chem. Soc., 2004, 126, 4118 CrossRef CAS PubMed; (d) N. T. McDougal and S. E. Schaus, Angew. Chem., Int. Ed., 2006, 45, 3117 CrossRef CAS PubMed; (e) D. Virieux, A. F. Guillouzic and H. J. Cristau, Tetrahedron, 2006, 62, 3710 CrossRef CAS; (f) Z. He, X. Tang and Z. He, Phosphorus Sulfur Relat. Elem., 2008, 183, 1518 CrossRef CAS; (g) S. Xu, S. Zeng, R. Ma, Z. Wang and Z. He, Org. Lett., 2009, 11, 3498 CrossRef CAS PubMed; (h) I. L. Odinets, Organophosphorus Chem., 2010, 39, 106 Search PubMed; (i) S. Xu, W. Zou, G. Wu, H. Song and Z. He, Org. Lett., 2010, 12, 3556 CrossRef CAS PubMed; (j) S. N. Khong, Y. S. Tran and O. Kwon, Tetrahedron, 2010, 66, 4760 CrossRef CAS PubMed; (k) M. J. Jacobsen, E. D. Funder, J. R. Cramer and K. V. Gothelf, Org. Lett., 2011, 13, 3418 CrossRef CAS PubMed; (l) S. Xu, R. Chen and Z. He, J. Org. Chem., 2011, 76, 7528 CrossRef CAS PubMed; (m) J. C. Deng and S. C. Chuang, Org. Lett., 2011, 13, 2248 CrossRef CAS PubMed; (n) S. C. Chuang, J. C. Deng, F. W. Chan, S. Y. Chen, W. J. Huang, L. H. Lai and V. Rajeshkumar, Eur. J. Org. Chem., 2012, 2606 CrossRef CAS; (o) M. Masoudi and M. Anary-Abbasinejad, Tetrahedron Lett., 2016, 57, 103 CrossRef CAS; (p) Q. Zhang, Y. Zhu, H. Jin and Y. Huang, Chem. Commun., 2017, 53, 3974 RSC; (q) C. Zhu, Y. Sun, Y. Wei and M. Shi, Adv. Synth. Catal., 2017, 359, 1263 CrossRef CAS; (r) Z. Huang, Q. Chen, X. Yang, Y. Liu, L. Zhang, T. Lu and Q. Zhou, Org. Chem. Front., 2017, 4, 967 RSC; (s) Z. Ren, M. Sun, Z. Guan and M. Ding, Synlett, 2018, 29, 106 CrossRef CAS; for a review, see: (t) A. Ramazani and A. R. Kazemizadeh, Curr. Org. Chem., 2008, 12, 59 CrossRef CAS.
- (a) M. Schirmer, S. Adomeit and T. Werner, Org. Lett., 2015, 17, 3078 CrossRef CAS PubMed; (b) Y.-L. Tsai and W. Lin, Asian J. Org. Chem., 2015, 4, 1040 CrossRef CAS; (c) M.-L. Schirmer, S. Adomeit, A. Spanneberg and T. Werner, Chem.–Eur. J., 2016, 22, 2458 CrossRef CAS PubMed; (d) N. Saleh and A. Voituriez, J. Org. Chem., 2016, 81, 4371 CrossRef CAS PubMed; (e) C. Lee, T. Chang, J. Yu, G. M. Reddy, M. Hsiao and W. Lin, Org. Lett., 2016, 18, 3758 CrossRef CAS PubMed; (f) N. Saleh, F. Blanchard and A. Voituriez, Adv. Synth. Catal., 2017, 359, 2304 CrossRef CAS.
- For examples of γ-umpolung reactions, see: (a) H.-J. Cristau, J. Viala and H. Christol, Tetrahedron Lett., 1982, 23, 1569 CrossRef CAS; (b) B. M. Trost and C. J. Li, J. Am. Chem. Soc., 1994, 116, 3167 CrossRef CAS; (c) C. Zhang and X. Lu, Synlett, 1995, 645 CrossRef CAS; (d) Z. Chen, G. Zhu, Q. Jiang, D. Xiao, P. Cao and X. Zhang, J. Org. Chem., 1998, 63, 5631 CrossRef CAS; (e) Z. Pakulski, O. M. Demchuk, J. Frelek, R. Luboradzki and K. M. Pietrusiewicz, Eur. J. Org. Chem., 2004, 3913 CrossRef CAS; (f) D. Virieux, A. F. Guillouzic and H. J. Cristau, Tetrahedron, 2006, 62, 3710 CrossRef CAS; (g) S. W. Smith and G. C. Fu, J. Am. Chem. Soc., 2009, 131, 14231 CrossRef CAS PubMed; (h) Y. K. Chung and G. C. Fu, Angew. Chem., Int. Ed., 2009, 48, 2225 CrossRef CAS PubMed; (i) J. Sun and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 4568 CrossRef CAS PubMed; (j) R. Sinisi, J. Sun and G. C. Fu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20652 CrossRef CAS PubMed; (k) Y. Fujiwara, J. Sun and G. C. Fu, Chem. Sci., 2011, 2, 2196 RSC; (l) R. J. Lundgren, A. Wilsily, N. Marion, C. Ma, Y. K. Chung and G. C. Fu, Angew. Chem., Int. Ed., 2013, 52, 2525 CrossRef CAS PubMed; (m) Q. F. Zhou, K. Zhang and O. Kwon, Tetrahedron Lett., 2015, 56, 3273 CrossRef CAS PubMed; (n) K. Zhang, L. Cai, X. Jiang, M. A. Garcia-Garibay and O. Kwon, J. Am. Chem. Soc., 2015, 137, 11258 CrossRef CAS PubMed.
- For examples of catalytic Wittig reactions, see: (a) C. J. O'Brien, J. L. Tellez, Z. S. Nixon, L. J. Kang, A. L. Carter, S. R. Kunkel, K. C. Przeworski and G. A. Chass, Angew. Chem., Int. Ed., 2009, 48, 6836 CrossRef PubMed; (b) C. J. O'Brien, F. Lavigne, E. E. Coyle, A. J. Holohan and B. J. Doonan, Chem.–Eur. J., 2013, 19, 5854 CrossRef PubMed; (c) C. J. O'Brien, Z. S. Nixon, A. J. Holohan, S. R. Kunkel, J. L. Tellez, B. J. Doonan, F. L. Coyle, F. Lavigne, L. J. Kang and K. C. Przeworski, Chem.–Eur. J., 2013, 19, 15281 CrossRef PubMed; (d) E. E. Coyle, B. J. Doonan, A. J. Holohan, K. A. Walsh, F. Lavigne, E. H. Krenske and C. J. O'Brien, Angew. Chem., Int. Ed., 2014, 53, 12907 CrossRef CAS PubMed; (e) T. Werner, M. Hoffmann and S. Deshmukh, Eur. J. Org. Chem., 2014, 6873 CrossRef CAS; (f) T. Werner, M. Hoffmann and S. Deshmukh, Eur. J. Org. Chem., 2014, 6630 CrossRef CAS; (g) S. Rommel, C. Belger, J. Begouin and B. Plietker, ChemCatChem, 2015, 7, 1292 CrossRef CAS; (h) T. Werner, M. Hoffmann and S. Deshmukh, Eur. J. Org. Chem., 2015, 3286 CrossRef CAS; (i) M. Hoffmann, S. Deshmukh and T. Werner, Eur. J. Org. Chem., 2015, 4532 CrossRef CAS; (j) L. Wang, M. Sun and M. Ding, Eur. J. Org. Chem., 2017, 2568 CrossRef CAS; for commentaries, see: (k) I. J. S. Fairlamb, ChemSusChem, 2009, 2, 1021 CrossRef CAS PubMed; (l) S. P. Marsden, Nat. Chem., 2009, 1, 685 CrossRef CAS PubMed.
- For other reactions involving in situ reduction of phosphine oxides, see: (a) J. R. Harris, M. T. Haynes II, A. M. Thomas and K. A. Woerpel, J. Org. Chem., 2010, 75, 5083 CrossRef CAS PubMed; (b) H. A. van Kalkeren, S. H. A. M. Leenders, C. R. A. Hommersom, F. P. J. T. Rutjes and F. L. van Delft, Chem.–Eur. J., 2011, 17, 11290 CrossRef CAS PubMed; (c) A. D. Kosal, E. E. Wilson and B. L. Ashfeld, Angew. Chem., Int. Ed., 2012, 51, 12036 CrossRef CAS PubMed; (d) H. A. van Kalkeren, J. J. Bruins, F. P. J. T. Rutjes and F. L. van Delft, Adv. Synth. Catal., 2012, 354, 1417 CrossRef CAS; (e) H. A. van Kalkeren, F. L. van Delft and F. P. J. T. Rutjes, Pure Appl. Chem., 2013, 85, 817 CAS; (f) H. A. van Kalkerente, C. Grotenhuis, F. S. Haasjes, C. A. Hommersom, F. P. J. T. Rutjes and F. L. van Delft, Eur. J. Org. Chem., 2013, 7059 CrossRef; (g) L. Wang, Y. Wang, M. Chen and M.-W. Ding, Adv. Synth. Catal., 2014, 356, 1098 CrossRef CAS; (h) D. C. Lenstra, P. J. T. Rutjes and J. Mecinovic, Chem. Commun., 2014, 50, 5763 RSC; (i) H. Bel Abed, O. Mammoliti, O. Bande, G. V. Lommen and P. Herdewijin, Org. Biomol. Chem., 2014, 12, 7159 RSC; (j) J. A. Buonomo and C. C. Aldrich, Angew. Chem., Int. Ed., 2015, 54, 13233 CrossRef; (k) W. Zhao, P. K. Yan and A. T. Radosevich, J. Am. Chem. Soc., 2015, 137, 616 CrossRef CAS PubMed; (l) K. Fourmy and A. Voituriez, Org. Lett., 2015, 17, 1537 CrossRef CAS PubMed; (m) D. Hirose, M. Gazvoda, J. Košmrlj and T. Taniguchi, Org. Lett., 2016, 18, 40 CrossRef PubMed; (n) T. V. Nykaza, T. S. Harrison, A. Ghosh, R. A. Putnik and A. T. Radosevich, J. Am. Chem. Soc., 2017, 139, 6839 CrossRef CAS PubMed; for reviews, see: (o) S. P. Marsden, in Sustainable Catalysis, ed. P. J. Dunn, K. K. Hii, M. J. Krische and M. T. Williams, John Wiley & Sons, Hoboken, New Jersey, 2013, ch. 15, pp. 339–361 Search PubMed; (p) H. A. van Kalkeren, F. L. van Delft and F. P. J. T. Rutjes, ChemSusChem, 2013, 6, 1615 CrossRef CAS PubMed; (q) H. A. van Kalkeren, A. L. Bloom, F. P. J. T. Rutjes and M. A. J. Huijbregts, Green Chem., 2013, 15, 1255 RSC; (r) A. Voituriez and N. Saleh, Tetrahedron Lett., 2016, 57, 4443 CrossRef CAS; (s) Z. Lao and P. H. Toy, Beilstein J. Org. Chem., 2016, 12, 2577 CrossRef CAS PubMed.
- For examples of redox-neutral phosphine oxide catalysis processes, see: (a) T. W. Campbell, J. J. Monagle and V. S. Foldi, J. Am. Chem. Soc., 1962, 84, 3673 CrossRef CAS; (b) S. P. Marsden, A. E. McGonagle and B. McKeever-Abbas, Org. Lett., 2008, 10, 2589 CrossRef CAS PubMed; (c) R. M. Denton, J. An and B. Adeniran, Chem. Commun., 2010, 46, 3025 RSC; (d) R. M. Denton, X. Tang and A. Przeslak, Org. Lett., 2010, 12, 4678 CrossRef CAS PubMed; (e) R. M. Denton, J. An, B. Adeniran, A. J. Blake, W. Lewis and A. M. Poulton, J. Org. Chem., 2011, 76, 6749 CrossRef CAS PubMed; (f) R. M. Denton, J. An, P. Lindovska and W. Lewis, Tetrahedron, 2012, 68, 2899 CrossRef CAS; (g) J. An, X. Tang, J. Moore, W. Lewis and R. M. Denton, Tetrahedron, 2013, 69, 8769 CrossRef CAS; (h) J. An, R. M. Denton, T. H. Lambert and E. D. Nacsa, Org. Biomol. Chem., 2014, 12, 2993 RSC.
- (a) L. Horner and W. D. Balzer, Tetrahedron Lett., 1965, 6, 1157 CrossRef; (b) K. Naumann, G. Zou and K. Mislow, J. Am. Chem. Soc., 1969, 91, 2788 CrossRef CAS; (c) P. D. Henson, K. Naumann and K. Mislow, J. Am. Chem. Soc., 1969, 91, 5645 CrossRef CAS; (d) K. Naumann, G. Zon and K. Mislow, J. Am. Chem. Soc., 1969, 91, 7012 CrossRef CAS. For a review, see: (e) D. Herault, D. H. Nguyen, D. Nuel and G. Buono, Chem. Soc. Rev., 2015, 44, 2508 RSC.
- (a) E. H. Krenske, J. Org. Chem., 2012, 77, 1 CrossRef CAS PubMed; (b) E. H. Krenske, J. Org. Chem., 2012, 77, 3969 CrossRef CAS PubMed.
- (a) N. Dunn, M. Ha and A. T. Radosevich, J. Am. Chem. Soc., 2012, 134, 11330 CrossRef CAS PubMed; (b) S. M. McCarthy, Y. Lin, D. Devarajan, J. W. Chang, H. P. Yennawar, R. M. Rioux, D. H. Ess and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 4640 CrossRef CAS PubMed.
- (a) I. P. Andrews and O. Kwon, Chem. Sci., 2012, 3, 2510 RSC; (b) C. E. Henry, Q. Xu, Y. C. Fan, T. J. Martin, L. Belding, T. Dudding and O. Kwon, J. Am. Chem. Soc., 2014, 136, 11890 CrossRef CAS PubMed; (c) L. Cai, K. Zhang and O. Kwon, J. Am. Chem. Soc., 2016, 138, 3298 CrossRef CAS PubMed.
- The [2.2.1] bicyclic phosphine oxide S1 has been reported previously; see: Y. Kashman and A. Rudi, Tetrahedron Lett., 1976, 17, 2819 CrossRef . In the ground state, the phenyl ring of the phosphine oxide S1 is aligned coplanar with the P
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond to avoid repulsion with the bridgehead methyl group; in contrast, the phenyl ring of the phosphine oxide 7 is aligned perpendicular to the PO bond. The increased conjugation between the phenyl group and PO in S1 increases the electron density at the phosphorus center and shortens the ring P–C bonds, thereby increasing the C–P–C angle by 0.3°. The calculated activation barrier for the diphenylsilane-mediated reduction of S1 is 30.4 kcal mol−1; experimentally, its reduction was also slower than that of 7, presumably because of the steric congestion around the phosphorus center.15
O bond to avoid repulsion with the bridgehead methyl group; in contrast, the phenyl ring of the phosphine oxide 7 is aligned perpendicular to the PO bond. The increased conjugation between the phenyl group and PO in S1 increases the electron density at the phosphorus center and shortens the ring P–C bonds, thereby increasing the C–P–C angle by 0.3°. The calculated activation barrier for the diphenylsilane-mediated reduction of S1 is 30.4 kcal mol−1; experimentally, its reduction was also slower than that of 7, presumably because of the steric congestion around the phosphorus center.15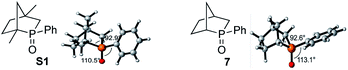 .
. - See the ESI for more details.†.
- We also considered the phosphetane oxide, anticipating that it may possess an angle closer to 90°. Nevertheless, computation predicted an angle of 80.5° for 2,2,3,4,4-pentamethyl-1-phenylphosphetane oxide (S2) and a known X-ray structure revealed an endocyclic C–P–C angle of 77° (D. C. R. Hockless, Y. B. Kang, M. A. McDonald, M. Pabel, A. C. Willis and S. B. Wild, Organometallics, 1996, 15, 1301). We also performed a kinetic study for the diphenylsilane-mediated reduction of S2; it behaved better than the phosphine oxides 4 and 5, but slightly worse than the phosphine oxides 6 and 7. When we ran the catalytic γ-umpolung–Wittig olefination employing S2, we observed no reaction at all, probably because of its low nucleophilicity.15.
- CCDC 1489646 contains the supplementary crystallographic data for 7.†.
- Based on our previous study13b of the phosphine oxide 6, we assumed that 7 would be configurationally stable during the reduction cycle, as was indeed the case. In contrast, the phosphine oxide 7′ produced a 10:1 mixture of endo-/exo-P-phenylphosphines when reduced using diphenylsilane or trichlorosilane. Compounds 7 and 7′ displayed almost identical kinetic profiles during the reduction cycle.15 We chose 7 to conduct the subsequent catalysis reactions, because it was the major isomer during the synthesis.
- (a) G. Keglevich, M. Fekete, T. Chuluunbaatar, A. Dobó, V. Harmat and L. Töke, J. Chem. Soc., Perkin Trans. 1, 2000, 4451 RSC; (b) E. Vedejs, O. Daugulis, L. A. Harper, J. A. MacKay and D. R. Powell, J. Org. Chem., 2003, 68, 5020 CrossRef CAS PubMed.
- Use of the trifluoromethyl ketone substrate 4-methyl-N-[2-(2,2,2-trifluoroacetyl)phenyl]benzenesulfonamide in place of the aldehyde substrate did not provide the corresponding dihydroquinoline product.
- γ-Methylallenoates are known to react with salicylaldehyde (derivatives)s to provide tetrahydrobenzopyrans through the phosphine-catalyzed γ-umpolung addition followed by δ-addition: (a) X. Meng, Y. Huang, H. Zhao, P. Xie, J. Ma and R. Chen, Org. Lett., 2009, 11, 991 CrossRef CAS PubMed; (b) R. Ma, S. Xu, X. Tang, G. Wu and Z. He, Tetrahedron, 2011, 67, 1053 CrossRef CAS.
- Dehalogenation of arylhalides in the presence of phenylsilane at 150 °C in dioxane has been reported previously.7i The authors of that paper noted, however, that phenylsilane-mediated dehalogenation usually requires radical initiators: D. O. Jang, Synth. Commun., 1997, 27, 1023 CrossRef CAS.
- A. Kumar, S. Srivastava, G. Gupta, V. Chaturvedi, S. Sinha and R. Srivastava, ACS Comb. Sci., 2011, 13, 65 CrossRef CAS PubMed.
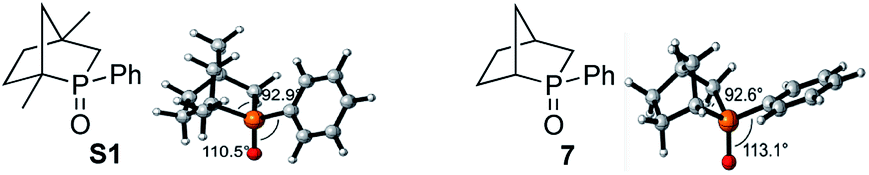
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1489646. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc04381c |
| ‡ K. Z. and L. C. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |