
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tri- and hexanuclear heterometallic Ni(II)–M(II) (M = Ca, Sr and Ba) bis(salamo)-type complexes: synthesis, structure and fluorescence properties†
Xiu-Yan Donga,
Xiao-Yan Lia,
Ling-Zhi Liua,
Han Zhanga,
Yu-Jie Dingb and
Wen-Kui Dong *a
*a
aSchool of Chemical and Biological Engineering, Lanzhou Jiaotong University, Lanzhou, Gansu 730070, PR China. E-mail: dongwk@126.com
bSchool of Biological & Chemical Engineering, Anhui Polytechnic University, Wuhu 241000, PR China
First published on 16th October 2017
Abstract
Three heterometallic Ni(II)–M(II) (M = Ca, Sr and Ba) complexes, two discrete heterotrinuclear complexes [Ni2(L)Ca(OAc)2(CH3OH)2]·2C2H5OH·2CHCl3 (1) and [Ni2(L)Sr(OAc)2(CH3OH)2]·2CH3OH·2CH2Cl2 (2) and a discrete heterohexanuclear dimer [Ni2(L)Ba(OAc)2(CH3OH)2(H2O)]2·2CH3OH (3), were synthesized with a naphthalenediol-based acyclic bis(salamo)-type ligand (H4L), and characterized by elemental analyses, IR, UV-vis spectra, fluorescence spectra and X-ray crystallography. The heterometallic complexes were acquired by the reaction of H4L with 2 equiv. of Ni(OAc)2·4H2O and 1 equiv. of M(OAc)2 (M = Ca, Sr and Ba). The crystal structures of complexes 1–3 have been determined by single-crystal X-ray diffractions. Owing to the different nature of the N2O2 and O6 sites of the ligand H4L, the introduction of two different metal(II) atoms to the site-selective moiety, leads to the two Ni(II) atoms occupied both the N2O2 sites, an alkaline earth metal atom occupied the O6 site of the ligand (L)4− unit, respectively. Furthermore, the fluorescence properties have been discussed.
Introduction
The condensation between an aldehyde and an amine leading to a salamo-type bisoxime was first described by Nabeshima's group.1 Structurally, salen-type ligands2 are nitrogen analogue of an aldehyde or ketone in which the carbonyl group (CO) has been replaced by an imine or azomethine group. The properties of salen-type ligands may be easily altered and as such they are able to coordinate metals in a highly versatile way and combine with O-donors to provide multidentate ligand systems.3 Salen-type N2O2 metal complexes are used as precursors to synthesize the oligometallic complexes owing to the high coordination ability of the phenoxo groups which can bridge two metal centers in a μ2-M–O–M fashion.4 Then, such μ2-phenoxo bridging is also particularly important for the d-block homo- and heterometallic complexes of salen-type ligands, some of which exhibit interesting catalysis,5 sensors,6 electrochemical,7 luminescence8 and magnetism.9 In recent years, our group has focused on the conversion of an acyclic molecule (bis(salamo)-type ligand) to the corresponding cyclic metal host, which can afford a larger C-shaped O6 site on the metalation of the N2O2 sites and effectively control the guest recognition.10 Thus, the O6 site of this type of ligands are particularly suitable for the larger radius of the alkaline earth and rare earth metals to afford 3d–2s and 3d–4f heterometallic complexes exhibiting better crystal structures and photochemical properties.Herein, as an extension of our previous studies,10 a new acyclic bis(salamo)-type ligand H4L is synthesized, in which two N2O2 salamo moieties share one naphthalenediol. As sizes of N2O2 and O6 cavities are different, it is possible to synthesize the heterometallic complexes.11,12 In this paper, heterometallic complexes [Ni2(L)Ca(OAc)2(CH3OH)2]·2C2H5OH·2CHCl3 (1) [Ni2(L)Sr(OAc)2(CH3OH)2]·2CH3OH·2CH2Cl2 (2) and [Ni2(L)Ba(OAc)2(CH3OH)2(H2O)]2·2CH3OH (3) have been synthesized and structurally characterized. In complex 2, two μ2-acetate ions bridge Ni(II) and Sr(II) atoms in a common μ2-fashion, another μ2-acetate ion chelates Sr(II) atom as a bidentate ligand. In complex 3, two μ2-acetate ions bridge the Ba1 and Ba1#2 atoms in a familiar μ2-fashion, finally forming a hetero-hexanuclear dimer. To our knowledge, this novel 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 ((L)4−:M2+) heterohexanuclear complex isn't reported in the bis(salamo)-type complexes.1,2e,11c
:6 ((L)4−:M2+) heterohexanuclear complex isn't reported in the bis(salamo)-type complexes.1,2e,11c
Experimental
Materials and general methods
2-Hydroxy-3-methoxybenzaldehyde (99%), methyl trioctyl ammonium chloride (90%), pyridiniumchlorochromate (98%) and borontribromide (99.9%) were purchased from Alfa Aesar. Hydrobromic acid 33 wt% solution in acetic acid was purchased from J&K Scientific Ltd. The other reagents and solvents were purchased from Shanghai Darui Chemical Fine Chemicals Company. Elemental analyses were performed by using a GmbH Vario EL V3.00 automatic elemental analysis instrument. Elemental analyses for metals were detected with an IRIS ER/S-WP-1 ICP atomic emission spectrometer. FT-IR spectra were recorded on a VERTEX70 FT-IR spectrophotometer, with samples prepared as KBr (400–4000 cm−1) pellets. UV-vis absorption spectra were recorded on a Hitachi U-3900H spectrometer. 1H and 13C NMR spectra were determined by German Bruker AVANCE DRX-400 spectrometer. Fluorescent spectra were taken on a LS-55 fluorescence photometer. X-ray single crystal structure determinations were carried out on a SuperNova, Dual, Cu at zero, Eos four-circle diffractometer. FTICR-MS spectra were obtained on a Bruker Daltonics APEX-II 47e spectrometer. Melting points were obtained with the use of an X4 microscopic melting point apparatus made by the Beijing Taike Instrument Limited Company and were uncorrected.Synthesis of the ligand H4L
The reaction steps involved in the synthesis of the bis(salamo)-type tetraoxime ligand (H4L) are shown in Scheme 1. 2,3-Dihydroxynaphthalene-1,4-dicarbaldehyde was prepared according to a literature procedure;13 1,2-bis(aminooxy)ethane14 and 2-[O-(1-ethyloxyamide)]oxime-6-methoxyphenol15 were synthesized according to an analogous method.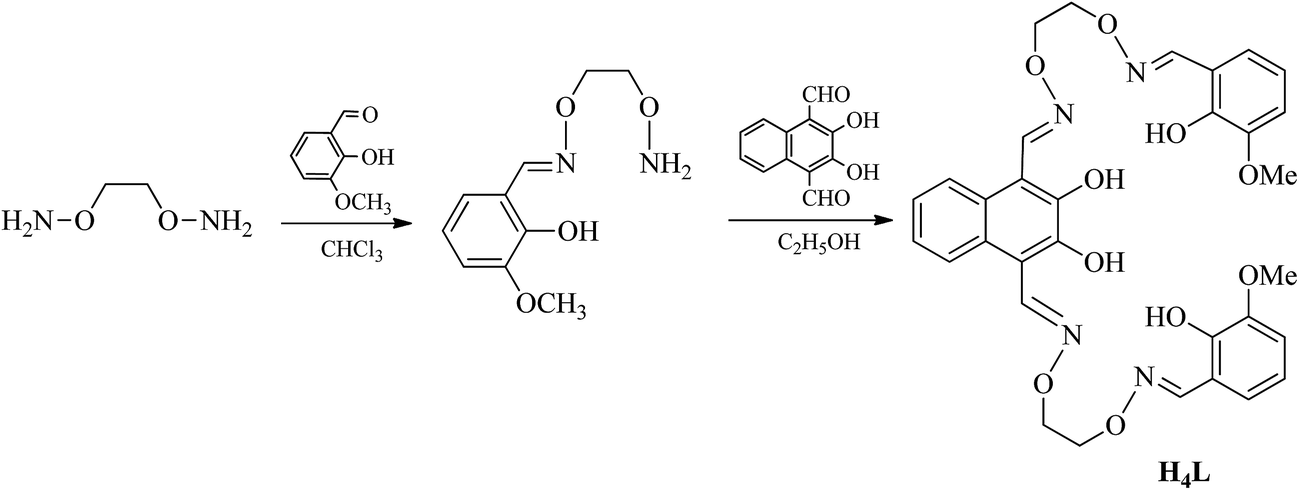 | ||
| Scheme 1 Synthetic route to the bis(salamo)-type tetraoxime ligand H4L. | ||
A solution of 2,3-dihydroxynaphthalene-1,4-dicarbaldehyde (432.08 mg, 2 mmol) in ethanol (20 mL) was added dropwise to a solution of 2-[O-(1-ethyloxyamide)]oxime-6-methoxyphenol (904.4 mg, 4 mmol) in ethanol (20 mL) under room temperature, the mixture was heated to reflux and kept refluxing for 6 h. Then cooled down to room temperature and the yellow precipitates were filtered and washed successively with ethanol and n-hexane, respectively. Several light yellow powdery solid (H4L) were obtained and collected by filtration, washed with absolute ethanol and dried under vacuum. Yield: 712.63 mg, 56.36%; mp: 172 °C; 1H NMR (CDCl3, 400 MHz) δ 11.03 (s, 2H, OH), 9.82 (s, 2H, OH), 9.14 (s, 2H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 8.29 (s, 2H, CHN), 7.97 (q, J = 3.2 Hz, 2H, ArH), 7.41 (q, J = 6.0, 2.9 Hz, 2H, ArH), 7.06–6.68 (m, 6H, ArH), 4.58 (t, 8H, CH2), 3.89 (s, 6H, CH3) (Fig. S1†). 13C NMR (DMSO, 151 MHz) δ 148.4 (s), 148.2 (s), 147.3 (s), 147.2 (s), 146.1 (s), 126.2 (s), 125.4 (s), 123.9 (s), 119.7 (s), 119.0 (s), 118.4 (s), 113.7 (s), 111.4 (s), 73.0 (s), 72.8 (s), 56.3 (s) (Fig. S2†). HRMS m/z: calc. for C32H32N4O10Na: [H4L + Na]+ 655.20, found: 655.2011 (Fig. S3†). Elemental analysis: anal. calc. for C32H32N4O10: C, 60.75; H, 5.10; N, 8.86, found (%): C, 60.38; H, 5.38; N, 8.65. IR (KBr; cm−1): 1604 [ν(CN)], 1248 [ν(Ar–O)], 3172 [ν(O–H)]. UV-vis [in methanol/dichloromethane (1:1)], λmax (nm) [2.5 × 10−5 M]: 267, 341, 356, 376.
N), 8.29 (s, 2H, CHN), 7.97 (q, J = 3.2 Hz, 2H, ArH), 7.41 (q, J = 6.0, 2.9 Hz, 2H, ArH), 7.06–6.68 (m, 6H, ArH), 4.58 (t, 8H, CH2), 3.89 (s, 6H, CH3) (Fig. S1†). 13C NMR (DMSO, 151 MHz) δ 148.4 (s), 148.2 (s), 147.3 (s), 147.2 (s), 146.1 (s), 126.2 (s), 125.4 (s), 123.9 (s), 119.7 (s), 119.0 (s), 118.4 (s), 113.7 (s), 111.4 (s), 73.0 (s), 72.8 (s), 56.3 (s) (Fig. S2†). HRMS m/z: calc. for C32H32N4O10Na: [H4L + Na]+ 655.20, found: 655.2011 (Fig. S3†). Elemental analysis: anal. calc. for C32H32N4O10: C, 60.75; H, 5.10; N, 8.86, found (%): C, 60.38; H, 5.38; N, 8.65. IR (KBr; cm−1): 1604 [ν(CN)], 1248 [ν(Ar–O)], 3172 [ν(O–H)]. UV-vis [in methanol/dichloromethane (1:1)], λmax (nm) [2.5 × 10−5 M]: 267, 341, 356, 376.
Preparation of complexes 1–3
Heterometallic complexes were synthesized by the reaction of H4L with Ni(OAc)2·4H2O and M(OAc)2 (M(II) = Ca, Sr and Ba) (Scheme 2). | ||
| Scheme 2 Synthesis of heterometallic Ni(II)–M(II) complexes 1–3. | ||
A solution of Ni(OAc)2·4H2O (9.96 mg, 0.040 mmol) in ethanol (2 mL) and Ca(OAc)2 (3.16 mg, 0.020 mmol) in water/methanol (1:3, 2 mL) were added to a solution of H4L (12.64 mg, 0.020 mmol) in chloroform (4 mL), and the resulting solution was evaporated to dryness, after which the residue was added to dichloromethane/methanol (1:1, 8 mL) and heated to dissolve it and then cooled in the refrigerator and recrystallized. The color of the mixed solution turned dark green. The mixture was filtered and the filtrate was allowed to stand at room temperature for approximately three weeks. The solvent was partially evaporated and several clear dark green prismatic single crystals of complex 1 were obtained.
A solution of Ni(OAc)2·4H2O (9.96 mg, 0.040 mmol) in methanol (2 mL) and Sr(OAc)2 (3.16 mg, 0.020 mmol) in water/methanol (1:3, 2 mL) were added to a solution of H4L (12.64 mg, 0.020 mmol) in dichloromethane (4 mL). The next steps are similar to complex 1. Complex 3 was prepared by a similar procedure as for complex 2.
Complex 1, dark green crystals, yields 12.57 mg, 48.37%. Elemental analysis: anal. calc. for C44H56Cl6CaN4O18Ni2 (%): C 40.68; H 4.34; N 4.31; Ni 9.04; Ca 3.09. Found (%): C 40.25; H 4.31; N 4.62; Ni 8.98; Ca 3.01. IR (KBr; cm−1): 1599 [ν(CN)], 1231 [ν(Ar–O)], 3413 [ν(O–H)]. UV-vis [in methanol/dichloromethane (1:1)], λmax (nm) [2.5 × 10−5 M]: 284, 370.
Complex 2, dark green crystals, yields 11.14 mg, 44.65%. Elemental analysis: anal. calc. for C42H54Cl4SrN4O18Ni2 (%): C 40.37; H 4.36; N 4.48; Ni 9.39; Sr 7.01. Found (%): C 40.25; H 4.31; N 4.62; Ni 9.26; Sr 6.92. IR (KBr; cm−1): 1597 [ν(CN)], 1233 [ν(Ar–O)], 3413 [ν(O–H)]. UV-vis [in methanol/dichloromethane (1:1)], λmax (nm) [2.5 × 10−5 M]: 284, 370.
Complex 3, dark green crystals, yields 17.26 mg, 38.69%. Elemental analysis: anal. calc. for C78H96Ba2N8O36Ni4 (%): C 41.99; H 4.34; N 5.02; Ni 10.52; Ba 12.31. Found (%): C 41.82; H 4.28; N 5.96; Ni 10.47; Ba 12.26. IR (KBr; cm−1): 1593 [ν(CN)], 1242 [ν(Ar–O)], 3402 [ν(O–H)]. UV-vis [in methanol/dichloromethane (1:1)], λmax (nm) [2.5 × 10−5 M]: 284, 370.
X-ray crystallographic analysis
The single crystals of complexes 1–3 were placed on a SuperNova, Dual, Cu at zero, Eos four-circle diffractometer. The diffraction data were collected using a graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Reflection data were corrected for Lorentz and polarization factor sand for absorption using the multi-scan method.16 The structures were solved by using the program SHELXL-2016 and Fourier difference techniques, and refined by the full-matrix least-squares method on F2. Anisotropic thermal parameters were used for the nonhydrogen atoms and isotropic parameters for the hydrogen atoms. Hydrogen atoms were added geometrically and refined using a riding model. The crystallographic data are summarized in Table 1. CCDC – 1562392 (1), 1562393 (2) and 1562394 (3)† contain the supplementary crystallographic data for this paper.| a R1 = ∑||Fo| − |Fc||/∑|Fo|.b wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2, w = [σ2(Fo2) + (0.0784P)2 + 1.3233P]−1, where P = (Fo2 + 2Fc2)/3. | |||
|---|---|---|---|
| Complex | 1 | 2 | 3 |
| Formula | C44H56Cl6CaN4O18Ni2 | C42H54Cl4SrN4O18Ni2 | C78H96Ba2N8O36Ni4 |
| Formula weight | 1299.12 | 1247.99 | 2230.14 |
| T (K) | 291(2) | 155(10) | 294(14) |
| Radiation (Å) | Mo Kα, 0.71073 | Mo Kα, 0.71073 | Mo Kα, 0.71073 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P21/n | Pbcn | P21/n |
| a (Å) | 15.4086(8) | 23.6620(5) | 15.8005(3) |
| b (Å) | 12.0628(6) | 17.8187(5) | 15.7041(3) |
| c (Å) | 29.5294(9) | 12.4666(3) | 18.2762(4) |
| α (°) | 90 | 90 | 90 |
| β (°) | 100.627(5) | 90 | 94.551(19) |
| γ (°) | 90 | 90 | 90 |
| Volume (Å3) | 5394.5(4) | 5256.2(2) | 4520.62(16) |
| Z | 4 | 4 | 4 |
| Dc (g cm−3) | 1.600 | 1.579 | 1.639 |
| Absorption coefficient (mm−1) | 1.164 | 1.996 | 1.764 |
| Θ range for data collection(°) | 1.397 to 25.997 | 3.862 to 24.112 | 3.510 to 27.218 |
| F (000) | 2680 | 2560.0 | 2264.0 |
| Index ranges | −19 ≤ h ≤ 13, | −23 ≤ h ≤ 29, | −18 ≤ h ≤ 18, |
| −14 ≤ k ≤ 13, | −21 ≤ k ≤ 21 | −14 ≤ k ≤ 18 | |
| −36 ≤ l ≤ 35, | −15 ≤ l ≤ 15, | −21 ≤ l ≤ 21, | |
| Crystal size (mm) | 0.24 × 0.22 × 0.20 | 0.21 × 0.22 × 0.24 | 0.15 × 0.17 × 0.21 |
| Reflections collected | 21343/10337 [Rint = 0.0117] |
16016/5173 [Rint = 0.050] |
17095/7925 [Rint = 0.030] |
| Independent reflection | 10337 |
5173 | 7925 |
| Data/restraints/parameters | 10337/12/642 |
5173/3/329 | 7925/45/600 |
| Final R indices [I > 2σ(I)]a | R1 = 0.0469, wR2 = 0.0959 | R1 = 0.0479, wR2 = 0.0888 | R1 = 0.0365, wR2 = 0.0760 |
| R indices (all data)b | R1 = 0.0645, wR2 = 0.0986 | R1 = 0.082, wR2 = 0.1022 | R1 = 0.0546, wR2 = 0.0874 |
Results and discussion
The ligand H4L and its corresponding metal complexes 1–3 are stable in air. The ligand H4L is remarkably soluble in DMF and DMSO, but slightly soluble in ethyl acetate, acetone, acetonitrile, methanol and ethanol. Complexes 1–3 are absolutely soluble in DMF and DMSO, but slightly soluble in chloroform, dichloromethane, methanol and ethanol at room temperature.In the 1H NMR spectrum of H4L, the peaks of methylene protons were observed ca. at 4.58 ppm, and the peaks of oxime protons were observed at 8.29 and 9.14 ppm. The OH resonances at 9.82 and 11.03 ppm strongly, respectively, showing the symmetrical structure of H4L (Fig. S1†). In the 13C NMR spectrum of H4L, the peaks of the CN carbon atoms were observed at 148.2 and 148.4 ppm, and the signals of CH3 carbon atoms were observed at 56.31 ppm (Fig. S2†).
IR spectra analyses
IR spectra of H4L and its corresponding Ni(II)–M(II) complexes 1–3 exhibit various bands in the region of 4000–400 cm−1 (Fig. 1). The O–H stretching band of the free ligand H4L has been observed at ca. 3172 cm−1 that belongs to the phenolic hydroxyl group, whereas complex 1, 2 and 3 shows a band at ca. 3413, 3413 and 3402 cm−1 that belongs to coordinated methanol molecules.17 | ||
| Fig. 1 IR spectra of the ligand H4L and its corresponding complexes 1–3. | ||
The free ligand H4L exhibits characteristic CN stretching band at ca. 1604 cm−1, which is shifted by ca. 5, 7, 11 cm−1 in complexes 1, 2 and 3, respectively, indicating that the nitrogen atoms of CN group are coordinated to the Ni(II) atoms,18 which is similar to previously reported Ni(II) complexes.19
The Ar–O stretching frequency appears at ca. 1248 cm−1 for the ligand H4L, while the Ar–O stretching frequencies in complexes 1, 2 and 3 are observed at ca. 1231, 1233 and 1242 cm−1, respectively. The Ar–O stretching frequencies are shifted to lower frequencies, indicating that the M–O bonds are formed between the metal(II) atoms and oxygen atoms of phenolic groups.20
UV-vis spectra analyses and fluorescence properties
The methanol/dichloromethane (1:1) solutions of the ligand H4L and its heterometallic Ni(II)–M(II) complexes show, as expected, almost identical UV-vis spectra (Fig. 2).
 | ||
| Fig. 2 UV-vis spectra of the ligand H4L and its corresponding complexes 1–3 in methanol/dichloromethane (1:1) (c = 2.5 × 10−5 M). | ||
The free ligand H4L shows four absorption peaks at 267, 341, 356 and 376 nm. The absorption peak at 267 nm can be assigned to the π–π* transition of the benzene rings and the other bands at 341, 356 and 376 nm can be attributed to the intra-ligand n–π* transition of the CN bonds and conjugated aromatic chromophore.21 Compared to the absorption peaks of the free ligand H4L, with the emergence of two absorption peaks at ca. 284 and 370 nm are observed in complexes 1–3, which can be assigned to π–π* type transition (MLCT). The absorption peaks of complexes 1–3 are bathochromically shifted,22 indicating coordination of (L)4− ligand unit. The coordination of metal atoms to the binding sites of N2O2 and hydroxyl oxygen of the naphthalene ring breaks the intramolecular hydrogen-bonding interactions of H4L and increases the coplanarity of the conjugated system which causes changes in the UV-vis spectra.23
The fluorescent properties of H4L was determined in DMF solution (2.5 × 10−5 mol L−1) by addition of Ni(OAc)2·4H2O, Ca(OAc)2, Sr(OAc)2 and Ba(OAc)2 in methanol/H2O (1:1) solution (1 × 10−3 mol L−1) are shown in Fig. 3.
 | ||
| Fig. 3 (a) Absorption spectra of H4L in DMF solution upon the addition of Ni2+. (b) Absorption spectra of the Ni(II) complex in DMF solution upon the addition of Ca2+. (c) Absorption spectra of the Ni(II) complex in DMF solution upon the addition of Sr2+. (d) Absorption spectra of the Ni(II) complex in DMF solution upon the addition of Ba2+. | ||
The excitation wavelengths of these complexes were measured several times by using the maximum absorption wavelengths, which is the UV-vis spectral theoretical excitation wavelength. The optimal excitation wavelength of these complexes at 378 nm with the maximum emission wavelength is 437 nm.
The fluorescence titration experiment were measured by the addition of Ni2+ is shown in Fig. 3(a), the free ligand H4L shows remarkable fluorescence quenching with maximum emission at ca. 437 nm upon the addition of Ni2+. When the added amount of the Ni2+ reached 3.0 equiv., the fluorescence emission intensity almost complete quenching and became stable. Weakened of fluorescence is possible due to the coordination of metal ion with the ligand.24 The spectroscopic titration indicated that the stoichiometric ratio between Ni2+ and ligand unit (L)4− was 3:1, which signify the Ni(II) complex was formed.6c,18b Then, Ca2+, Sr2+ and Ba2+ were added to the Ni(II) complex, respectively. As shown in Fig. 3(b)–(d), the fluorescence intensity gradually increased, when the added amount of the Ca2+ reached 1.0 equiv., the fluorescence intensity reached the maximum, which because one Ni2+ in the Ni(II) complex was replaced by one Ca2+, Sr2+ or Ba2+, respectively.6c,18b This phenomenon may be due to the difference between the radius of the Ni2+ and the alkaline earth metal ion.
Titration of Ni(II) complex with Ca2+, Sr2+ or Ba2+ were followed by fluorescence spectroscopy to determine the binding constant, respectively. As shown in Fig. S4,† the binding constant K of Ni(II) complex with Ca2+, Sr2+ and Ba2+ were estimated to be 8.14 × 103 M−1, 3.01 × 103 M−1 and 8.82 × 102 M−1 by the Benesi–Hildebrand equation (fluorescence method) way, respectively,25 which unambiguously demonstrates stronger binding ability of Ni(II) complex with Ca2+.
According to the obtained experimental data, the differences among the three heterometallic complexes are very obvious and could be utilized in host–guest systems.
Description of the crystal structures
The crystal structures of the heterometallic complexes 1–3 were determined by single-crystal X-ray diffraction. Selected bond lengths and angles of complexes 1–3 are listed in Table 2.| a Symmetry transformations used to generate equivalent atoms: #1 −x + 1, −y + 1, −z + 1 (complex 2); #2 −x + 1, y, −z + 3/2 (complex 3). | |||||
|---|---|---|---|---|---|
| Complex 1 | |||||
| Ni1–O2 | 2.020(2) | Ni1–O5 | 1.963(2) | Ni1–O14 | 2.079(2) |
| Ni1–O15 | 2.196(2) | Ni1–N1 | 2.071(3) | Ni1–N2 | 2.062(2) |
| Ni2–O6 | 1.968(2) | Ni2–O9 | 2.018(2) | Ni2–O11 | 2.082(2) |
| Ni2–O16 | 2.179(2) | Ni2–N3 | 2.066(3) | Ni2–N4 | 2.055(3) |
| Ca1–O1 | 2.609(2) | Ca1–O2 | 2.405(2) | Ca1–O5 | 2.386(2) |
| Ca1–O6 | 2.404(2) | Ca1–O9 | 2.404(2) | Ca1–O10 | 2.605(2) |
| Ca1–O12 | 2.393(2) | Ca1–O13 | 2.409(2) | ||
| O2–Ni1–O5 | 83.63(9) | O2–Ni1–O14 | 91.39(9) | O2–Ni1–O15 | 88.79(9) |
| O2–Ni1–N1 | 86.59(10) | O2–Ni1–N2 | 169.72(10) | O5–Ni1–O14 | 91.05(9) |
| O5–Ni1–O15 | 85.37(9) | O5–Ni1–N1 | 167.96(11) | O5–Ni1–N2 | 88.15(10) |
| O14–Ni1–O15 | 176.37(9) | O14–Ni1–N1 | 96.21(10) | O14–Ni1–N2 | 94.94(11) |
| O15–Ni1–N1 | 87.42(10) | O15–Ni1–N2 | 84.38(10) | N1–Ni1–N2 | 100.74(11) |
| O6–Ni2–O9 | 83.04(8) | O6–Ni2–O11 | 92.06(9) | O6–Ni2–O16 | 85.99(9) |
| O6–Ni2–N3 | 88.35(10) | O6–Ni2–N4 | 168.08(11) | O9–Ni2–O11 | 92.18(9) |
| O9–Ni2–O16 | 88.79(9) | O9–Ni2–N3 | 169.62(10) | O9–Ni2–N4 | 87.27(10) |
| O11–Ni2–O16 | 177.70(8) | O11–Ni2–N3 | 93.92(10) | O11–Ni2–N4 | 95.25(10) |
| O16–Ni2–N3 | 84.83(10) | O16–Ni2–N4 | 86.88(10) | N3–Ni2–N4 | 100.51(11) |
| O1–Ca1–O2 | 61.28(7) | O1–Ca1–O5 | 120.48(8) | O1–Ca1–O6 | 152.21(8) |
| O1–Ca1–O9 | 102.73(7) | O1–Ca1–O10 | 69.83(7) | O1–Ca1–O12 | 76.33(8) |
| O1–Ca1–O13 | 117.57(8) | O2–Ca1–O5 | 67.31(7) | O2–Ca1–O6 | 131.84(7) |
| O2–Ca1–O9 | 161.47(8) | O2–Ca1–O10 | 102.24(7) | O2–Ca1–O12 | 105.29(8) |
| O2–Ca1–O13 | 77.80(8) | O5–Ca1–O6 | 64.54(7) | O5–Ca1–O9 | 131.22(8) |
| O5–Ca1–O10 | 151.85(8) | O5–Ca1–O12 | 90.77(8) | O5–Ca1–O13 | 76.17(8) |
| O6–Ca1–O9 | 66.68(7) | O6–Ca1–O10 | 119.98(8) | O6–Ca1–O12 | 76.28(8) |
| O6–Ca1–O13 | 90.21(8) | O9–Ca1–O10 | 61.33(7) | O9–Ca1–O12 | 77.73(8) |
| O9–Ca1–O13 | 104.34(8) | O10–Ca1–O12 | 117.38(8) | O10–Ca1–O13 | 76.06(8) |
| O12–Ca1–O13 | 164.39(8) | ||||
|
|||||
| Complex 2 | |||||
| Ni1–O2 | 2.011(3) | Ni1–O5 | 1.984(2) | Ni1–O6 | 2.063(3) |
| Ni1–O8 | 2.169(3) | Ni1–N1 | 2.068(3) | Ni1–N2 | 2.042(3) |
| Sr1–O1 | 2.667(3) | Sr1–O2 | 2.517(2) | Sr1–O5 | 2.511(2) |
| Sr1–O7 | 2.525(3) | Sr1–O1#1 | 2.667(3) | Sr1–O2#1 | 2.517(2) |
| Sr1–O5#1 | 2.511(2) | Sr1–O7#1 | 2.525(3) | ||
| O2–Ni1–O5 | 86.26(10) | O2–Ni1–O6 | 91.73(10) | O2–Ni1–O8 | 87.13(10) |
| O2–Ni1–N1 | 86.72(12) | O2–Ni1–N2 | 170.41(12) | O5–Ni1–O6 | 92.21(10) |
| O5–Ni1–O8 | 86.08(11) | O5–Ni1–N1 | 170.72(12) | O5–Ni1–N2 | 86.94(12) |
| O6–Ni1–O8 | 178.00(10) | O6–Ni1–N1 | 94.07(12) | O6–Ni1–N2 | 95.27(12) |
| O8–Ni1–N1 | 87.51(12) | O8–Ni1–N2 | 85.68(12) | N1–Ni1–N2 | 99.27(14) |
| O1–Sr1–O2 | 60.20(8) | O1–Sr1–O5 | 118.79(8) | O1–Sr1–O7 | 116.48(9) |
| O1–Sr1–O1#1 | 76.99(1) | O1–Sr1–O2#1 | 108.97(8) | O1–Sr1–O5#1 | 150.00(8) |
| O1–Sr1–O7#1 | 76.83(9) | O2–Sr1–O5 | 65.81(8) | O2–Sr1–O7 | 75.92(8) |
| O2–Sr1–O1#1 | 108.97(8) | O2–Sr1–O2#1 | 167.40(8) | O2–Sr1–O5#1 | 126.78(8) |
| O2–Sr1–O7#1 | 105.89(8) | O5–Sr1–O7 | 73.26(9) | O5–Sr1–O1#1 | 150.00(8) |
| O5–Sr1–O2#1 | 126.78(8) | O5–Sr1–O5#1 | 61.16(8) | O5–Sr1–O7#1 | 92.77(9) |
| O7–Sr1–O1#1 | 76.83(9) | O7–Sr1–O2#1 | 105.89(8) | O7–Sr1–O5#1 | 92.76(9) |
| O7–Sr1–O7#1 | 163.99(10) | O1#1–Sr1–O2#1 | 60.21(8) | O1#1–Sr1–O5#1 | 118.79(8) |
| O1#1–Sr1–O7#1 | 116.49(9) | O2#1–Sr1–O5#1 | 65.81(8) | O2#1–Sr1–O7#1 | 75.93(8) |
| O5#1–Sr1–O7#1 | 73.27(9) | ||||
|
|||||
| Complex 3 | |||||
| Ni2–O5 | 2.085(3) | Ni2–O8 | 2.006(3) | Ni2–O20 | 2.017(3) |
| Ni2–O22 | 2.14(6) | Ni2–N14 | 2.077(3) | Ni2–N18 | 2.033(3) |
| Ni3–O9 | 2.005(3) | Ni3–O12 | 2.152(3) | Ni3–O13 | 2.045(3) |
| Ni3–O16 | 2.059(3) | Ni3–N25 | 2.052(3) | Ni3–N35 | 2.090(3) |
| Ba1–O8 | 2.699(3) | Ba1–O9 | 2.683(3) | Ba1–O11 | 2.788(3) |
| Ba1–O13 | 2.695(3) | Ba1–O17 | 2.897(3) | Ba1–O19 | 2.858(3) |
| Ba1–O20 | 2.696(3) | Ba1–O23 | 2.761(3) | Ba1–O15#2 | 3.128(3) |
| O5–Ni2–O8 | 88.26(11) | O5–Ni2–O20 | 91.72(12) | O5–Ni2–O22 | 177.6(15) |
| O5–Ni2–N14 | 90.56(12) | O5–Ni2–N18 | 91.04(12) | O8–Ni2–O20 | 85.98(11) |
| O8–Ni2–O22 | 90.8(10) | O8–Ni2–N14 | 173.33(13) | O8–Ni2–N18 | 86.40(12) |
| O20–Ni2–O22 | 86.0(15) | O20–Ni2–N14 | 87.50(13) | O20–Ni2–N18 | 171.80(12) |
| N14–Ni2–O22 | 90.1(9) | N18–Ni2–O22 | 91.1(14) | N14–Ni2–N18 | 100.19(14) |
| O9–Ni3–O12 | 84.24(11) | O9–Ni3–O13 | 91.41(11) | O9–Ni3–O16 | 89.72(11) |
| O9–Ni3–N25 | 174.23(13) | O9–Ni3–N35 | 83.75(12) | O12–Ni3–O13 | 87.89(12) |
| O12–Ni3–O16 | 173.76(11) | O12–Ni3– N25 | 90.16(13) | O12–Ni3–N35 | 87.00(12) |
| O13–Ni3–O16 | 90.70(12) | O13–Ni3–N25 | 86.97(13) | O13–Ni3–N35 | 173.28(11) |
| O16–Ni3–N25 | 95.83(13) | O16–Ni3–N35 | 93.92(13) | N25–Ni3–N35 | 97.39(14) |
| O8–Ba1–O9 | 57.67(7) | O8–Ba1–O11 | 107.89(8) | O8–Ba1–O13 | 122.39(8) |
| O8–Ba1–O17 | 172.94(8) | O8–Ba1–O19 | 111.07(8) | O8–Ba1–O20 | 61.13(8) |
| O8–Ba1–O23 | 90.11(9) | O9–Ba1–O11 | 145.91(8) | O9–Ba1–O13 | 65.25(8) |
| O9–Ba1–O17 | 121.39(8) | O9–Ba1–O19 | 135.21(8) | O9–Ba1–O20 | 115.47(8) |
| O9–Ba1–O23 | 65.68(9) | O11–Ba1–O13 | 115.18(9) | O11–Ba1–O17 | 68.85(8) |
| O11–Ba1–O19 | 77.65(9) | O11–Ba1–O20 | 71.30(9) | O11–Ba1–O23 | 148.41(9) |
| O13–Ba1–O17 | 56.15(8) | O13–Ba1–O19 | 113.98(9) | O13–Ba1–O20 | 168.29(8) |
| O13–Ba1–O23 | 72.47(9) | O17–Ba1–O19 | 74.75(9) | O17–Ba1–O20 | 121.90(8) |
| O17–Ba1–O23 | 95.67(9) | O19–Ba1–O20 | 56.57(8) | O19–Ba1–O23 | 71.61(9) |
| O20–Ba1–O23 | 96.91(9) | ||||
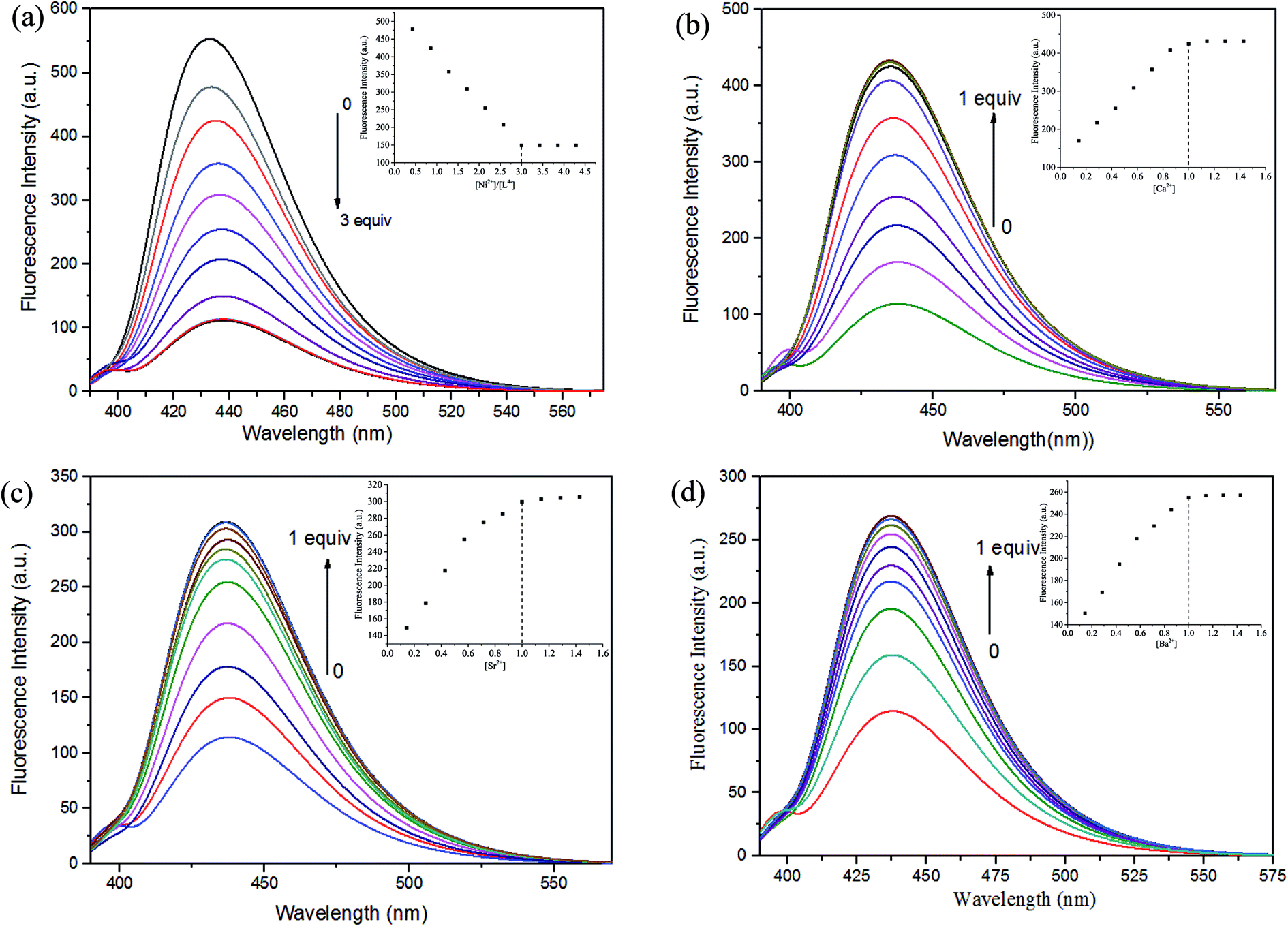
Crystal structure of complexes 1 and 2
The crystal structures and atom numberings of complexes 1 and 2 are shown in Fig. 4 and 5, respectively. X-ray crystallographic analysis of complex 1 reveals a trinuclear structure. It crystallizes in the monoclinic system, space group P21/n, and consists of two Ni(II) atoms, one Ca(II) atom, one (L)4− unit, two μ2-acetate ions, two coordinated methanol molecules and two crystallized ethanol and chloroform molecules. While complex 2 crystallizes in the orthorhombic crystal system, space group Pbcn, and consists of two Ni(II) atoms, one Sr(II) atom, one (L)4− unit, two μ2-acetate ions, two coordinated methanol molecules and two crystallized methanol and dichloromethane molecules. X-ray crystallography clearly shows the formation of complexes 1 and 2, which was isolated as dark green crystals.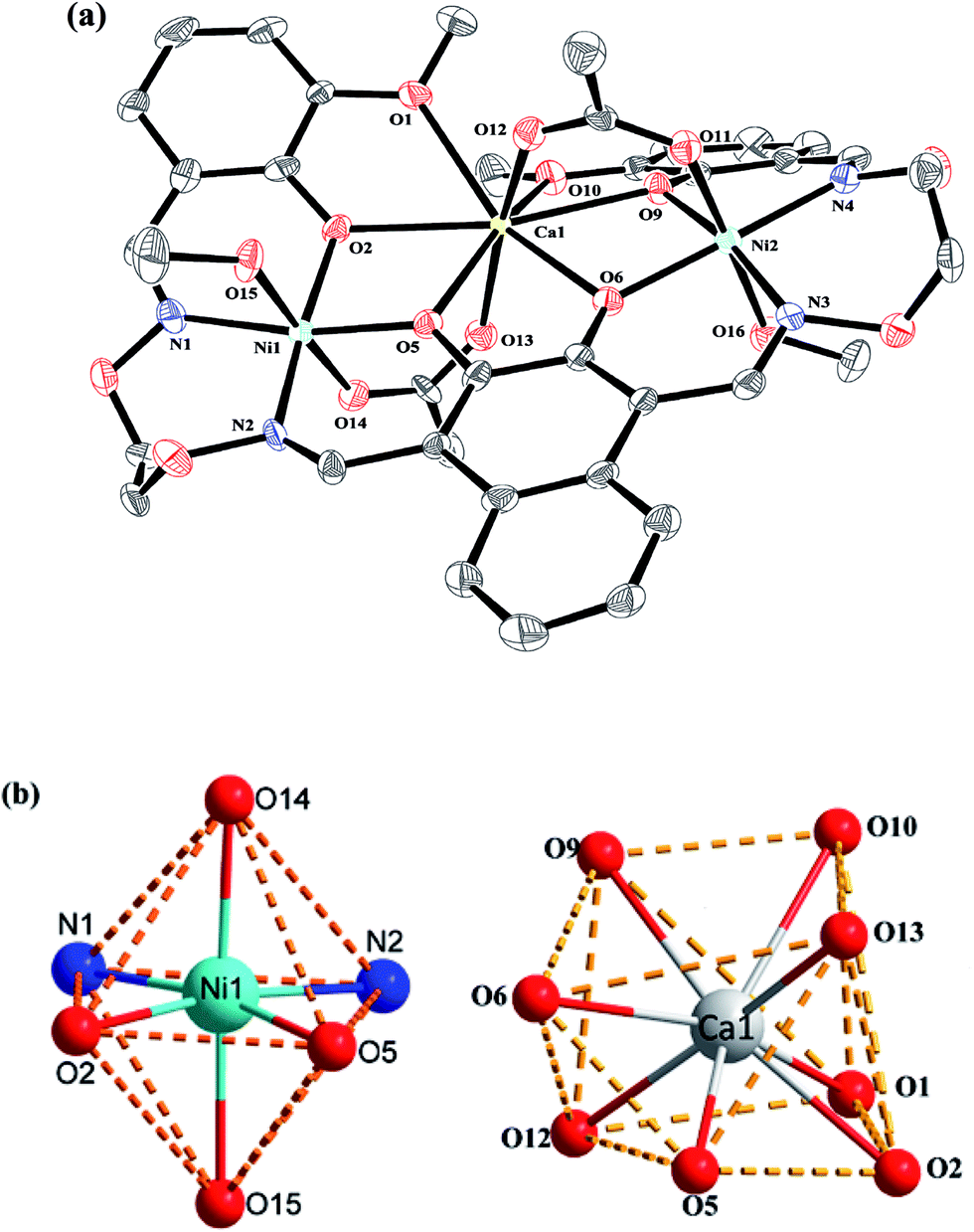 | ||
| Fig. 4 (a) View of the molecular structure of complex 1 (hydrogen atoms and solvent molecules are omitted for clarity, and thermal ellipsoids are drawn at the 30% probability level). (b) Coordination polyhedra for Ni(II) and Ca(II) atoms of complex 1. | ||
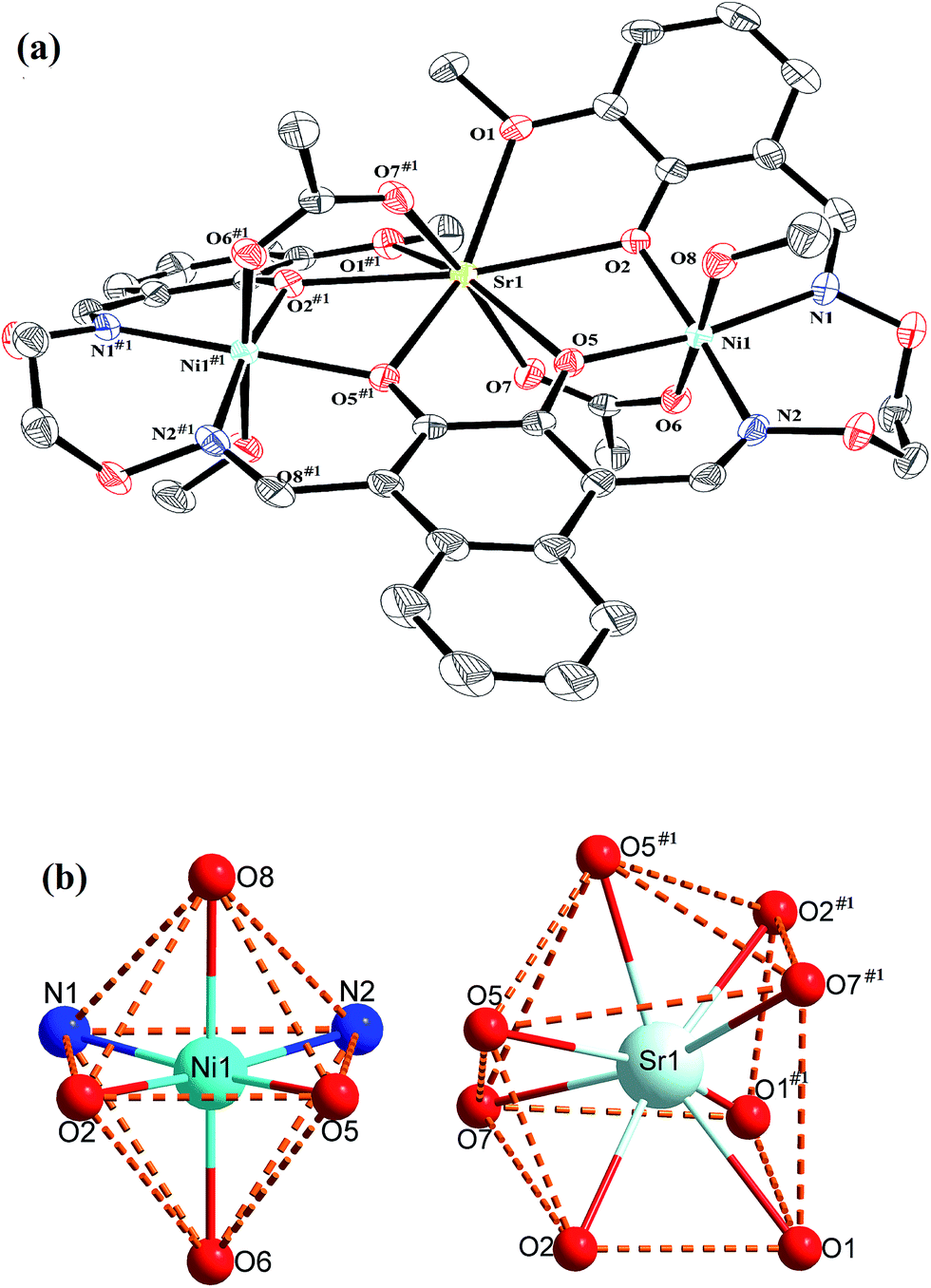 | ||
| Fig. 5 (a) View of the molecular structure of complex 2 (hydrogen atoms and solvent molecules are omitted for clarity, and thermal ellipsoids are drawn at the 30% probability level). (b) Coordination polyhedra for Ni(II) and Sr(II) atoms of complex 2. | ||
Interestingly, the formation process of complex 1 was highly cooperative. In the crystal structure of complex 1, the terminal Ni(II) atom (Ni1 or Ni2) is located in the N2O2 coordination cavity of completely deprotonated (L)4− unit, one oxygen atom (O14 or O11) from the μ2-acetate bridge and one oxygen atom (O15 or O16) from the coordinated methanol molecule. Because of Ni1 and Ni2 are symmetry related, they have identical geometries. Thus, the Ni1 and Ni2 atoms are both hexa-coordinated with slightly distorted octahedral geometries.26 While the central Ca1 atom is located in the O6 cavity, the four are phenoxy oxygen atoms (O2, O5, O6 and O9) while two others (O1 and O10) come from methoxy groups. Besides, two μ2-acetate ions bridge the two terminal Ni(II) atoms and the central Ca(II) atom in a μ2-fashion. So the Ca1 atom is octa-coordinated with a slightly distorted square antiprism geometry. The Ca1 atom of complex 1 has strong coordination with the four phenoxy oxygen atoms (O2, O5, O6 and O9) of the (L)4− units and the two oxygen atoms (O12 and O13) of the μ2-acetate ions by analyzing the distances of the eight Ca1–O bonds. The distances between the Ca1 atom and the four phenoxy oxygen atoms (O2, O5, O6 and O9) and the two μ2-acetate oxygen atoms (O12 and O13) are ranged from 2.386(2) to 2.409(2) Å, which are obviously shorter than the distances of the other two Ca1–O bonds (Ca1–O1 2.609(2) Å and Ca1–O10 2.605(2) Å). The coordination of complex 2 is similar to that of complex 1. In the crystal structure of complex 2, the distances between the Sr1 atom and the four phenoxy oxygen atoms (O2, O5, O2#1 and O5#1) and the two μ2-acetate oxygen atoms (O7 and O7#1) are ranged from 2.511(2) to 2.525(3) Å, which are obviously shorter than the distances of the other two Sr1–O bonds (Sr1–O1 2.667(3) Å and Sr1–O1#1 2.667(3) Å). Obviously, the Sr–O bond lengths in complex 2 are larger than the corresponding Ca–O bond lengths found in complex 1.
Crystal structure of complex 3
The crystal structure and atom numbering of complex 3 is shown in Fig. 6. Complex 3 crystallizes in the monoclinic crystal system, space group P21/n. In complex 3, the terminal Ni(II) atom (Ni2 or Ni3) is hexa-coordinated with a slightly distorted octahedral geometry. Where the inner N2O2 cavities of completely deprotonated (L)4− units as the basal plane, and one oxygen atom (O22) from the coordinated methanol molecule, the other oxygen atom (O5) from the coordinated H2O molecule for the Ni2 atom. Similarly, one oxygen atom (O12) from the coordinated methanol molecule, the other oxygen atom (O16) from the μ-acetate ion for the Ni3 atom. It is unexpected that the coordination of two μ2-acetate ions bridge the Ba1 and Ba1#2 atoms in a familiar μ2-fashion, finally forming a hetero-hexanuclear dimer. To our knowledge, this novel 2:6 ((L)4−:M2+) heterohexanuclear complex isn't reported in the bis(salamo)-type complexes.1,2e,11c
 | ||
| Fig. 6 (a) View of the molecular structure of complex 3 (hydrogen atoms are omitted for clarity, and thermal ellipsoids are drawn at the 30% probability level). (b) Coordination polyhedra for Ni(II) and Ba(II) atoms of complex 3. | ||
The central Ba1 atom is nona-coordinated with a slightly distorted tricapped trigonal prism geometry, which is different from the Ca1 and Sr1 atoms. The distances between the Ba1 atom and the four phenoxy oxygen atoms (O8, O9, O13 and O20) and the μ2-acetate oxygen atom (O23) are ranged from 2.683(3) to 2.761(3) Å, which are evidently shorter than the distances between the Ba1 atom and the two methoxy oxygen atoms (O17 and O19) (Ba1–O17 2.897(3) Å and Ba1–O19 2.858(3) Å) and the two μ2-acetate oxygen atoms (O11 and O15#2) (Ba1–O11 2.788(3) Å and Ba1–O15#2 3.128(3) Å).
As a result, when the two N2O2 salamo moieties are metalated with d-block transition metals, the conformation of the molecules is restricted so that the phenoxo oxygen atoms are directed inward to form an O6 cavity. Since the O6 cavity is large, the Ca(II), Sr(II) or Ba(II) atoms are suitable for this size, and will coordinate to form the C-shaped complexes.
The resulting geometries of Ni1 and Ni2 are both distorted octahedral geometries with hexa-coordinated, Ca1 and Sr1 are octa-coordinated with geometries of square antiprisms respectively. However, Ba1 has bigger size than the cavity, so the coordination of metal atoms with the methanol molecules and H2O makes the structure more stable. The resulting geometry of Ba1 is rarely tricapped trigonal prismatic geometry. As the cation radius increases, the coordination bond lengths of the central cation are distinctly becoming larger and larger. This fact suggests that the radius size of central cation is a significant factor which affects binding ability of the central O6 site. As a result, the coordinating capability in the central O6 site is in the order of Ca(II) > Sr(II) > Ba(II), which obtained the same conclusion with fluorescent titration.
Conclusion
Three heterometallic Ni(II)–M(II) (M = Ca, Sr and Ba) complexes 1–3 have been designed and synthesized. X-ray crystal structures reveal that the different nature of the N2O2 and O6 sites of the ligand H4L leads to the site-selective introduction of two different kinds of metal(II) atoms. The coordination number of Ca(II) Sr(II) and Ba(II) atoms in the O6 environment are 8, 8 and 9, respectively, and have slightly square antiprism and square tricapped trigonal prism gemtries. As a result, the coordinating capability in the central O6 site is in the order of Ca(II) > Sr(II) > Ba(II). Fluorescence titration experiments show that Ni2+ led to the fluorescence quenching of H4L. Owing to their electrostatic interaction and their size-fit principle, rare-earth(III) atoms would easier to occupy the center of the O6 position successfully, which could be used for the recognition of rare-earth(III) atoms. The related research is underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21361015 and 21761018) and the Program for Excellent Team of Scientific Research in Lanzhou Jiaotong University (201706), which are gratefully acknowledged.References
- S. Akine, T. Taniguchi and T. Nabeshima, Inorg. Chem., 2008, 47, 3255–3264 CrossRef CAS PubMed.
- (a) G. Murugavel, P. Sadhu and T. Punniyamurthy, Chem. Rec., 2016, 16, 1906–1917 CrossRef CAS PubMed; (b) Y. Xu, D. Yuan, Y. Wang and Y. Yao, Dalton Trans., 2017, 46, 5848–5855 RSC; (c) H. L. Wu, Y. C. Bai, Y. H. Zhang, G. L. Pan, J. Kong, F. R. Shi and X. L. Wang, Z. Anorg. Allg. Chem., 2014, 640, 2062–2071 CrossRef CAS; (d) X. Q. Song, P. P. Liu, Z. R. Xiao, X. Li and Y. A. Liu, Inorg. Chim. Acta, 2015, 438, 232–244 CrossRef CAS; (e) L. Chen, W. K. Dong, H. Zhang, Y. Zhang and Y. X. Sun, Cryst. Growth Des., 2017, 17, 3636–3648 CrossRef CAS; (f) X. Y. Li, L. Chen, L. Gao, Y. Zhang, S. F. Akogun and W. K. Dong, RSC Adv., 2017, 7, 35905–35916 RSC; (g) E. V. Alekseeva, I. A. Chepurnaya, V. V. Malev, A. M. Timonovb and O. V. Levina, Electrochim. Acta, 2017, 225, 378–391 CrossRef CAS; (h) R. Irie and T. Katsuki, Chem. Rec., 2004, 4, 96–109 CrossRef CAS PubMed; (i) T. Nabeshima and S. Akine, Chem. Rec., 2008, 8, 240–251 CrossRef CAS PubMed.
- (a) P. P. Liu, L. Sheng, X. Q. Song, W. Y. Xu and Y. A. Liu, Inorg. Chim. Acta, 2015, 434, 252–257 CrossRef CAS; (b) X. Q. Song, Y. Q. Peng, G. Q. Cheng, X. R. Wang, P. P. Liu and W. Y. Xu, Inorg. Chim. Acta, 2015, 427, 13–21 CrossRef CAS.
- (a) X. Y. Dong, Y. X. Sun, L. Wang and L. Li, J. Chem. Res., 2012, 36, 387–390 CrossRef CAS; (b) P. Wang and L. Zhao, Spectrochim. Acta, Part A, 2015, 135, 342–350 CrossRef CAS PubMed; (c) C. Y. Chen, J. W. Zhang, Y. H. Zhang, Z. H. Yang, H. L. Wu, G. L. Pan and Y. C. Bai, J. Coord. Chem., 2015, 68, 1054–1071 CrossRef CAS.
- (a) L. H. Li, W. K. Dong, Y. Zhang, S. F. Akogun and L. Xu, Appl. Organomet. Chem., 2017, DOI:10.1002/aoc.3818; (b) T. K. Chin, S. Endud, S. Jamil, S. Budagumpi and H. O. Lintang, Catal. Lett., 2013, 143, 282–288 CrossRef CAS.
- (a) W. K. Dong, X. L. Li, L. Wang, Y. Zhang and Y. J. Dong, Spectrochim. Acta, Part B, 2016, 229, 370–378 CAS; (b) W. K. Dong, S. F. Akogun, Y. Zhang, Y. X. Sun and X. Y. Dong, Spectrochim. Acta, Part B, 2017, 238, 723–734 CAS; (c) B. J. Wang, W. K. Dong, Y. Zhang and S. F. Akogun, Spectrochim. Acta, Part B, 2017, 247, 254–264 CAS.
- (a) W. K. Dong, J. C. Ma, L. C. Zhu, Y. Zhang and X. L. Li, Inorg. Chim. Acta, 2016, 445, 140–148 CrossRef CAS; (b) Ş. Ömer, Ö. Ö. Ümmühan, S. Nurgul, A. Burcu, S. Musa, T. Tuncay and S. Zeynel, Tetrahedron, 2016, 72, 5843–5852 CrossRef.
- (a) T. Z. Yu, K. Zhang, Y. L. Zhao, C. H. Yang, H. Zhang, L. Qian, D. W. Fan, W. K. Dong, L. L. Chen and Y. Q. Qiu, Inorg. Chim. Acta, 2008, 361, 233–240 CrossRef CAS; (b) Y. J. Dong, X. Y. Dong, W. K. Dong, Y. Zhang and L. S. Zhang, Polyhedron, 2017, 123, 305–315 CrossRef CAS; (c) W. K. Dong, J. C. Ma, Y. J. Dong, L. Zhao, L. C. Zhu, Y. X. Sun and Y. Zhang, J. Coord. Chem., 2016, 69, 3231–3241 CrossRef CAS; (d) H. L. Wu, C. P. Wang, F. Wang, H. P. Peng, H. Zhang and Y. C. Bai, J. Chin. Chem. Soc., 2015, 62, 1028–1034 CrossRef CAS; (e) W. K. Dong, J. C. Ma, L. C. Zhu, Y. X. Sun, S. F. Akogun and Y. Zhang, Cryst. Growth Des., 2016, 16, 6903–6914 CrossRef CAS; (f) L. Liu, W. Feng, X. Lu and W. K. Wong, Inorg. Chem. Commun., 2017, 75, 29–32 CrossRef CAS; (g) X. Yang and R. A. Jones, J. Am. Chem. Soc., 2005, 127, 7686–7687 CrossRef CAS PubMed; (h) F. Wang, L. Gao, Q. Zhao, Y. Zhang, W. K. Dong and Y. J. Ding, Spectrochim. Acta, Part A, 2018, 190, 111–115 CrossRef CAS PubMed.
- (a) W. K. Dong, J. C. Ma, Y. J. Dong, L. C. Zhu and Y. Zhang, Polyhedron, 2016, 115, 228–235 CrossRef CAS; (b) S. S. Zheng, W. K. Dong, Y. Zhang, L. Chen and Y. J. Ding, New J. Chem., 2017, 41, 4966–4973 RSC; (c) H. Zhang, W. K. Dong, Y. Zhang and S. F. Akogun, Polyhedron, 2017, 133, 279–293 CrossRef CAS; (d) P. Seth, S. Ghosh, A. Figuerola and A. Ghosh, Dalton Trans., 2014, 43, 990–998 RSC; (e) A. B. Canaj, M. Siczek, M. Otręba, T. Lis, G. Lorusso, M. Evangelisti and C. J. Milioset, Dalton Trans., 2016, 45, 18591–18602 RSC.
- (a) X. Y. Dong, S. F. Akogun, W. M. Zhou and W. K. Dong, J. Chin. Chem. Soc., 2017, 64, 412–419 CrossRef CAS; (b) Y. J. Dong, X. L. Li, Y. Zhang and W. K. Dong, Supramol. Chem., 2017, 29, 518–527 CrossRef CAS; (c) W. K. Dong, J. Zhang, Y. Zhang and N. Li, Inorg. Chim. Acta, 2016, 444, 95–102 CrossRef CAS; (d) W. K. Dong, F. Zhang, N. Li, L. Xu, Y. Zhang, J. Zhang and L. C. Zhu, Z. Anorg. Allg. Chem., 2016, 642, 532–538 CrossRef CAS; (e) W. K. Dong, G. Li, Z. K. Wang and D. X. Yong, Spectrochim. Acta, Part A, 2014, 133, 340–347 CrossRef CAS PubMed.
- (a) S. Akine, T. Tadokoro and T. Nabeshima, Inorg. Chem., 2012, 51, 11478–11486 CrossRef CAS PubMed; (b) S. Akine and T. Nabeshima, Dalton Trans., 2009, 47, 10395–10408 RSC; (c) J. Hao, L. L. Li, J. T. Zhang, S. F. Akogun, L. Wang and W. K. Dong, Polyhedron, 2017, 134, 1–10 CrossRef CAS.
- (a) L. Wang, J. C. Ma, W. K. Dong, L. C. Zhu and Y. Zhang, Z. Anorg. Allg. Chem., 2016, 642, 834–839 CrossRef CAS; (b) H. L. Wu, G. L. Pan, H. Wang, X. L. Wang, Y. C. Bai and Y. H. Zhang, J. Photochem. Photobiol., B, 2014, 135, 33–43 CrossRef CAS PubMed; (c) Y. A. Liu, C. Y. Wang, M. Zhang and X. Q. Song, Polyhedron, 2017, 127, 278–286 CrossRef CAS; (d) H. L. Wu, G. L. Pan, Y. C. Bai, H. Wang, J. Kong, F. Shi, Y. H. Zhang and X. L. Wang, J. Chem. Res., 2014, 38, 211–217 CrossRef CAS; (e) H. L. Wu, Y. C. Bai, Y. H. Zhang, Z. Li, M. C. Wu, C. Y. Chen and J. W. Zhang, J. Coord. Chem., 2014, 67, 3054–3066 CrossRef CAS.
- H. A. Tran, J. Collins and P. E. Georghiou, New J. Chem., 2008, 32, 1175–1182 RSC.
- D. W. Dixon and R. H. Weiss, J. Org. Chem., 1984, 49, 4487–4494 CrossRef CAS.
- S. Akine, T. Taniguchi, W. K. Dong, S. Masubuchi and T. Nabeshima, J. Org. Chem., 2005, 70, 1704–1711 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- P. P. Liu, C. Y. Wang, M. Zhang and X. Q. Song, Polyhedron, 2017, 129, 133–140 CrossRef CAS.
- (a) W. K. Dong, P. F. Lan, W. M. Zhou and Y. Zhang, J. Coord. Chem., 2016, 69, 1272–1283 CrossRef CAS; (b) W. K. Dong, S. S. Zheng, J. T. Zhang, Y. Zhang and Y. X. Sun, Spectrochim. Acta, Part A, 2017, 184, 141–150 CrossRef CAS PubMed.
- (a) X. Y. Dong, Q. P. Kang, B. X. Jin and W. K. Dong, Z. Naturforsch., 2017, 72, 415–420 CAS; (b) L. M. Pu, H. T. Long, Y. Zhang, Y. Bai and W. K. Dong, Polyhedron, 2017, 128, 57–67 CrossRef CAS.
- W. K. Dong, L. C. Zhu, J. C. Ma, Y. X. Sun and Y. Zhang, Inorg. Chim. Acta, 2016, 453, 402–408 CrossRef CAS.
- S. Akine, T. Taniguchi and T. Nabeshima, Chem. Lett., 2001, 30, 682–683 CrossRef.
- W. K. Dong, L. C. Zhu, Y. J. Dong, J. C. Ma and Y. Zhang, Polyhedron, 2016, 117, 148–154 CrossRef CAS.
- M. Tümer, H. Köksal, M. K. Sener and S. Serin, Transition Met. Chem., 1999, 24, 414–420 CrossRef.
- (a) L. Q. Chai, G. Wang, Y. X. Sun, W. K. Dong, L. Zhao and X. H. Gao, J. Coord. Chem., 2012, 65, 1621–1631 CrossRef CAS; (b) L. Q. Chai, K. Y. Zhang, L. J. Tang, J. Y. Zhang and H. S. Zhang, Polyhedron, 2017, 130, 100–107 CrossRef CAS; (c) L. Q. Chai, L. J. Tang, L. C. Chen and J. J. Huang, Polyhedron, 2017, 122, 228–240 CrossRef CAS; (d) L. Xu, L. C. Zhu, J. C. Ma, Y. Zhang, J. Zhang and W. K. Dong, Z. Anorg. Allg. Chem., 2015, 641, 2520–2524 CrossRef CAS; (e) L. Q. Chai, J. J. Huang, J. Y. Zhang and Y. X. Li, J. Coord. Chem., 2015, 68, 1224–1237 CrossRef CAS; (f) X. Q. Song, P. P. Liu, Y. A. Liu, J. J. Zhou and X. L. Wang, Dalton Trans., 2016, 45, 8154–8163 RSC.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- (a) W. K. Dong, J. C. Ma, L. C. Zhu and Y. Zhang, New J. Chem., 2016, 40, 6998–7010 RSC; (b) W. K. Dong, L. S. Zhang, Y. X. Sun, M. M. Zhao, G. Li and X. Y. Dong, Spectrochim. Acta, Part A, 2014, 121, 324–329 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1562392–1562394. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra07826a |
| This journal is © The Royal Society of Chemistry 2017 |