
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical sensor based on lead ion-imprinted polymer particles for ultra-trace determination of lead ions in different real samples†
Xuan Luoa,
Weihong Huanga,
Qingye Shib,
Wanzhen Xu*a,
Yu Luanc,
Yanfei Yangc,
Huajie Wangb and
Wenming Yang *b
*b
aSchool of the Environment and Safety Engineering, Jiangsu University, Zhenjiang 212013, China. E-mail: xwz09@ujs.edu.cn
bSchool of Materials Science and Engineering, Jiangsu University, Zhenjiang 212013, China. E-mail: ywm@ujs.edu.cn
cZhenjiang Institute for Drug Control of Jiangsu Province, Zhenjiang 212013, China
First published on 13th March 2017
Abstract
In this paper, a self-manufactured lead(II)-selective electrode, which was based on the use of lead(II) ion imprinted polymer particles (IIPs) to detect lead ions, was studied using differential pulse voltammetry. The synthesis and preparation of the IIPs was by precipitation polymerization with methacrylic acid (function monomer and lead-binding ligand), ethylene glycol dimethacrylate (the cross-linker) 2,2-azobisisobutyronitrile (the initiator) and lead ions (the template ion) in an acetonitrile solution. The IIPs were prepared to establish the electrochemical sensor by leaching out the lead(II) from the imprinted polymer particles, and a carbon paste electrode was modified with lead(II) IIPs. There is a distinct difference in the response between the electrodes modified with IIPs and the electrodes modified with NIPs, which included the open circuit sorption of Pb2+ on the electrode and its reduction to its metallic form. The influence factors on the response behavior of the electrodes were investigated and optimized. The results show the introduced sensor was linear from 1.0 × 10−9 to 7.5 × 10−7 mol L−1, and the limit of detection (LOD) was 1.3 × 10−11 mol L−1 (S/N = 3). Finally, the sensor was successfully applied to the trace determination of lead ions in various real samples.
1. Introduction
Heavy metal pollution refers to the environmental pollution caused by heavy metals or their compounds. With the rapid development of city expansion and industrial production, heavy metal exposure directly or indirectly into the surroundings because of non-standard discharge, such as mining, metallurgy, electroplating, chemical fertilizer production, papermaking, pesticides and other manufacturing and improper handling, batteries, and electronic waste, has increased. When heavy metals intermix in the surroundings, they will continue to occur in the environment but they do not decompose, pollute the soil, air and water, accumulate in livestock, crops and hydrobios and enter the human body through food chain enrichment. Lead(II), a heavy metal, leads to plumbism for the vital organs of animals and humans, such as anemia, hepatopathy, serious brain damage, kidney malfunctioning, hematological damage, etc.1 Thus, lead contamination has become a focus. Therefore, a detection method should be found to test lead(II) in order to prevent the damage from poisoning.For this purpose, several methods have been proposed, especially by means of a wide range of spectrometric methods, such as electrothermal atomization atomic absorption spectrometry (ET-AAS),2,3 sequential multi-element flame atomic absorption spectrometry,4 graphite furnace atomic absorption spectrometry (GFAAS),5 electrothermal atomic absorption spectrometry,6 inductively coupled plasma-mass spectrometry (ICP-MS),7 hydride generation atomic absorption spectrometry8 and flame atomic absorption spectrometry (FAAS).9 These methods have the advantages of high sensitivity, selectivity and accuracy, but still have weakness, such as the expensive cost, professional operating and time consumption. Thus, different kinds of sensors, including optical,10 potentiometric11 and voltammetric12,13 sensors, have been widely used for lead determination. These highly adopted techniques for the determination of metal ions have the advantages of high sensitivity, low cost, easy operation and etc. In order to enhance the sensitivity and selectivity of the electrochemical determination of lead(II), chemically modified materials on the electrode have been attractive to researchers in the past decades. The modifiers used include bismuth,14,15 organic chelating groups,16,17 PAN-incorporated Nafion,18 zeolite,19 SiO2–Al2O3 mixed-oxide,20 clay nano particles21 and silica.22 Moreover, a suitable selectivity is not provided by these materials toward lead(II) in some situations with interfering ions like Cu(II), Ag(I), etc. Therefore, there is still a pressing need to propose a high selective modifier for testing lead(II). Molecular imprinting technology is a potential method for lead(II) detection.
Molecular imprinting technology has unique advantages such as superior specificity and predetermined selectivity. Molecularly imprinted polymers (MIPs) demonstrate good chemical and thermal stability against environmental interference.23 To extract target molecules from complex samples accurately, this method has become one of the most widely used techniques in the field of separation and enrichment materials in recent years.
Ion-imprinted polymers (IIPs) are similar to MIPs with different recognition for target ions after preparing IIPs, which preserve the virtues of MIPs. The prepared polymers show selective binding of a specific cation and can be equipped with complexing agents through the coordination bond to form stable intermolecular forces matching the charge, coordination geometry and size of the target cation.24
In this paper, a self-manufactured carbon paste electrode sensor was prepared for the detection of lead ions, and they use an ion-imprinted polymer technique combined with an electrochemical detection method. Ion-imprinted polymer techniques have high selectivity, and electrochemistry detection methods have excellent sensitivity. This new ion-imprinted polymer modified carbon paste sensor is high selective for lead ions, easy to operate and provides cheap, sensitive detection of low levels of lead ions in aqueous solution.
2. Experimental
2.1 Materials
Paraffin oil, activated charcoal, pure graphite powder, methanol (Augmented Reality), PbCO3, acetic acid (HOAc), anhydrous sodium acetate, hydrogen peroxide (H2O2) and thiourea were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). The acetate buffer solutions (0.2 mol L−1) were prepared with CH3COONa and CH3COOH. Methacrylic acid, ethylene glycol dimethacrylate (EGDMA) and 2,2-azobisisobutyronitrile (AIBN) were obtained from Aladdin Chemistry (Shanghai, China). Acetonitrile was purchased from Lingfeng Chemical Reagent Co., Ltd (Shanghai, China). Hydrochloric acid (HCl) was from Yongfeng Chemical Reagent Co., Ltd. (Jiangsu, China). Nitric acid (HNO3) was purchased from Yangzhou Hubao Chemical reagents Co., Ltd. (Yangzhou, China).2.2 Instrument
All the electrochemical measurements were obtained with a three-electrode system using a CHI760E electrochemical workstation (Chenhua, Shanghai, China) with a conventional three electrode configuration. A self-manufactured lead(II) ion-imprinted polymer modified carbon paste electrode (IIP-CPE) was employed as the working electrode with a potassium chloride (KCl) saturated calomel electrode (SCE) and a Pt wire electrode as the reference and auxiliary electrodes, respectively. A personal computer was used to save all the electrochemical data and perform the processing. Fourier Transform Infrared (FT-IR) spectra (4000–400 cm−1) in KBr were recorded using a Nicolet NEXUS-470FT-IR spectrometer (USA). The data of the single point surface area at P/Po was obtained with Micromeritics TriStar II3020 (America). The real sample pretreatment was used with a MSP-6600 microwave digestion system (Rayleigh, China). The pH measurement was performed with a Sartorius model PB-21 pH/mV meter (Germany). Scanning electron microscopy (SEM) was performed using a Zeiss Merlin Compact instrument (Germany).2.3 Synthesis of lead(II) ion imprinted polymer (IIP) and non-imprinted polymer particles (NIP)
In order to prepare the Pb(II) imprinted polymer, 18 mmol of methacrylic acid and 6 mmol of PbCO3 were added to 25 mL acetonitrile and stirred for 24 h at room temperature in a 100 mL glass flask. The mixture was then centrifuged and the supernatant liquid was separated for the synthesis of the polymer. EGDMA (25 mmol) was used as the cross-linker and 0.03 g AIBN the initiator, and they were dissolved in 8 mL acetonitrile and added to the supernatant solution. At the same time, dissolved oxygen in the resulting mixture was removed by purging with high purity nitrogen for 20 min. The polymerization was carried out in an oil bath at 65 °C for 24 h. The prepared polymer particles were washed with ethanol to remove the unreacted materials. Then, a 1 M HCl solution and 0.1 M thiourea solution were used to remove the target ions from the polymer particles. Finally, the prepared particles were washed with plenty of distilled water and dried under vacuum at 60 °C in a desiccator until a powder formed (Fig. 1). The non-imprinted polymer was also synthesized in the same way without the template lead ions.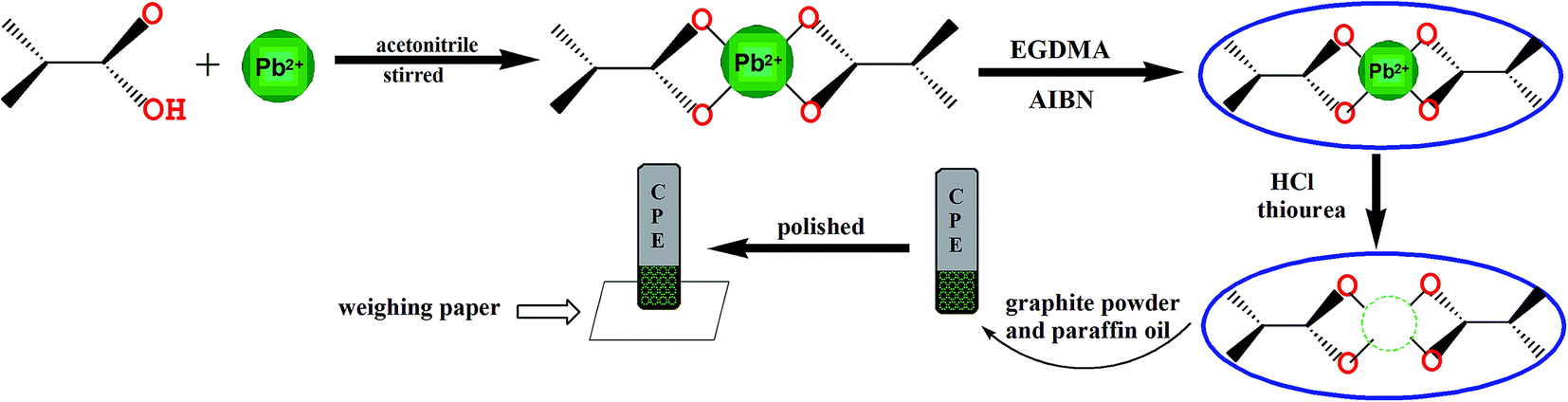 | ||
| Fig. 1 The fabrication process for the preparation of lead-imprinted polymer particles and IIP-CPE. | ||
2.4 Preparation of IIP-CPE
The ion-imprinted polymer modified carbon paste electrode (IIP-CPE) was prepared according to the optimized conditions. A mixture of leached IIP (20% w/w), graphite powder and paraffin oil (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30% w/w) were milled in a mortar with a pestle to construct the mixed paste of the leached IIP-CPE. The prepared paste was used to fill a hole (3 mm in diameter) at the end of a carbon paste working electrode and compacted. Then, the working electrode was polished on the weighing paper to make sure the surface was smooth. To extrude a thin layer of the electrode surface, the electrode can be reused after each experiment by renewing the electrode surface on the weighing paper (Fig. 1).
:30% w/w) were milled in a mortar with a pestle to construct the mixed paste of the leached IIP-CPE. The prepared paste was used to fill a hole (3 mm in diameter) at the end of a carbon paste working electrode and compacted. Then, the working electrode was polished on the weighing paper to make sure the surface was smooth. To extrude a thin layer of the electrode surface, the electrode can be reused after each experiment by renewing the electrode surface on the weighing paper (Fig. 1).
2.5 Real sample pretreatment
The microwave digestion system used for digesting flour and rice samples. These two samples were weighed, ground to powder using a mortar and pestle and sieving, then weighed again before digestion. In this paper, powdered flour and rice samples (0.5 g of flour powder, 0.5 g of rice powder) were precisely added into two microwave digestion tanks followed by the addition of 10 mL concentrated HNO3 and 1 mL concentrated H2O2. After 2 h of reaction at ambient temperature, the mixtures were digested in the microwave digestion system. After digesting, the mixtures were heated and gently boiled until about 2 mL of digestion solution were left. Subsequently, 5 mL of deionized water were added, and the resulting solution was boiled again to 1 mL. After cooling, the solutions were diluted to 50 mL with acetate buffer, and the pH was adjusted to 5 for the detection. For the detection of the water sample, the polyethylene bottles filled with samples were washed with detergent, water, diluted nitric acid and water in sequence. The samples (50 mL), which were taken from Yudai River (Jiangsu University) and tap water, were filtered through a cellulose filter membrane (pore size 0.22 μm) immediately. Then, the water samples were adjusted to a pH of 5.0 with acetate buffer according to optimized experimental conditions.2.6 Determination procedure
Electrochemical measurements were performed by differential pulse voltammetry (DPV). A 3-step procedure was followed for the voltammetric detection of lead(II) using the prepared electrodes. The electrodes were firstly incubated in Pb2+ solutions at pH 5.0 for 80 s. Then, they were rinsed and placed in the electrochemical cell containing 10 mL of 0.12 M HCl. Before the DPV scan, a negative pre-potential of −1.0 V was applied to the electrode for 40 s to reduce the recognized target ions. Finally, the differential pulse voltammetry scan was conducted from −0.8 V to 0.0 V.3. Results and discussion
3.1 Characterization of IIP
Scanning electron microscopy (SEM), Fourier Transformed Infrared Spectroscopy (FTIR) and surface area were used to represent the prepared nanosized imprinted polymer.The morphology of the lead-IIP was evaluated by scanning electron microscopy and is shown in Fig. 2(a). Fig. 2(a) showed the lead-IIP is 1 μm and 100 nm. Different polymeric particle sizes ranged from 60 nm to 280 nm. As a consequence, the lead-IIP can be used as the selective solid phase for the detection of trace lead ions.
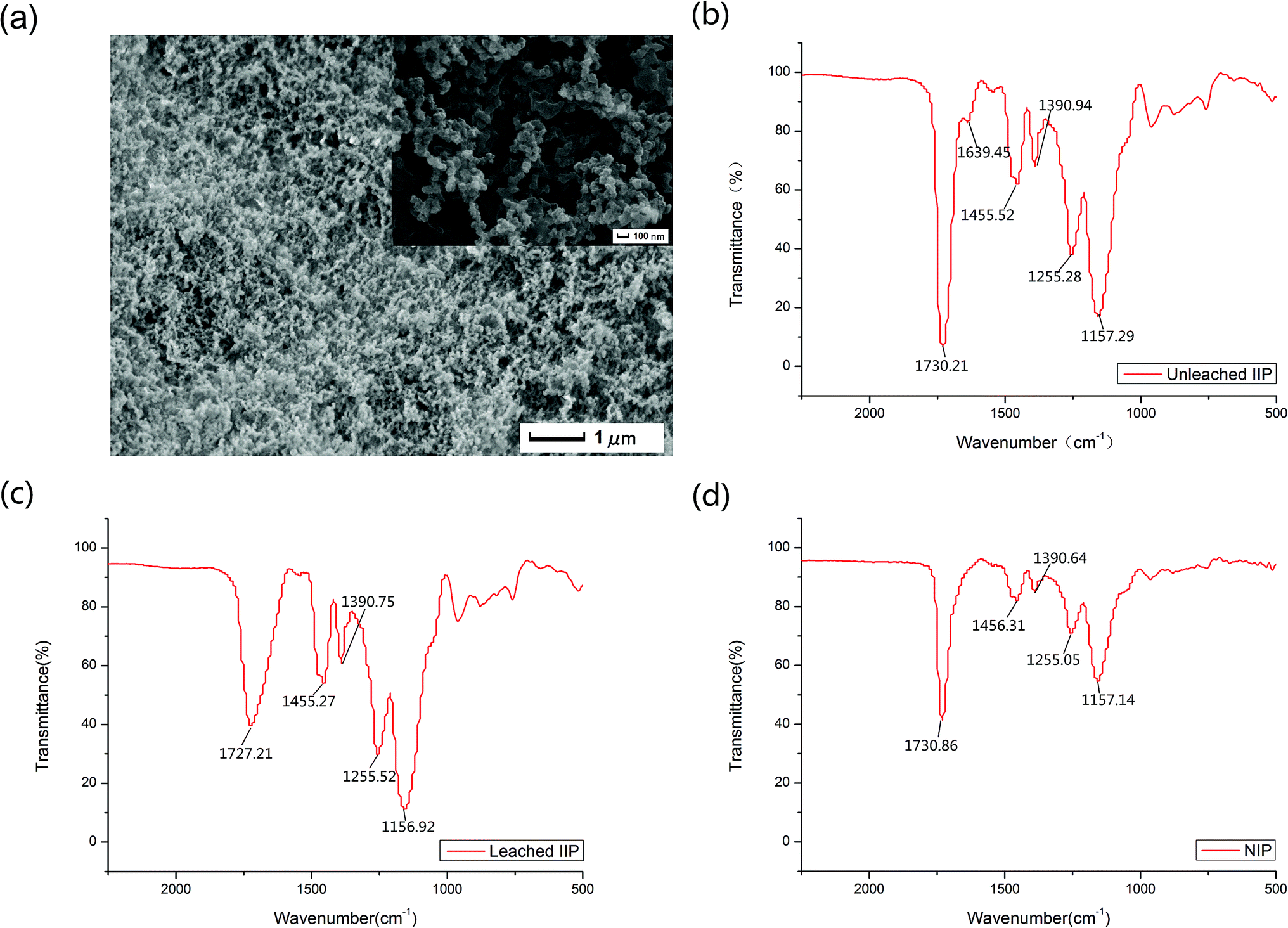 | ||
| Fig. 2 (a) SEM image of lead(II) imprinted polymer particles. (b) FT-IR spectra of unleached lead(II) imprinted polymer particles. (c) FT-IR spectra of leached lead(II) imprinted polymer particles. (d) FT-IR spectra of non-imprinted polymer particles. | ||
In all recorded regions, except the region of ∼1500–1700 cm−1, there was no considerable difference among the FT-IR spectra of the unleached IIP, leached IIP and NIP. The FT-IR spectra are shown in Fig. 2(b)–(d), respectively. In Fig. 2(b), the peak at 1730.21 cm−1 is ascribed to the νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the carboxylic acid group of methacrylic acid, and the strong stretching vibration bands at 1727.21 cm−1 and 1730.86 cm−1 in Fig. 3(c) and (d) were situated mainly at the surface of the polymeric particles. An evident band for the unleached IIP can be observed at 1639.45 cm−1, ascribed to the –CO, and linked to Pb2+ coordination bonding. This peak is less strong than that located at 1730.21 cm−1 because these types of –CO groups are mainly situated in the interior sections of the IIP particles. After washing the unleached IIP, by reason of the removal of the Pb2+ ions to a considerable decrease at the height of the vibration band of the –CO coordinated to Pb2+. The above analysis can be evidence for the existence and proper interaction of the selective recognition sites in the IIP particles, which are created during the imprinting process.
O of the carboxylic acid group of methacrylic acid, and the strong stretching vibration bands at 1727.21 cm−1 and 1730.86 cm−1 in Fig. 3(c) and (d) were situated mainly at the surface of the polymeric particles. An evident band for the unleached IIP can be observed at 1639.45 cm−1, ascribed to the –CO, and linked to Pb2+ coordination bonding. This peak is less strong than that located at 1730.21 cm−1 because these types of –CO groups are mainly situated in the interior sections of the IIP particles. After washing the unleached IIP, by reason of the removal of the Pb2+ ions to a considerable decrease at the height of the vibration band of the –CO coordinated to Pb2+. The above analysis can be evidence for the existence and proper interaction of the selective recognition sites in the IIP particles, which are created during the imprinting process.
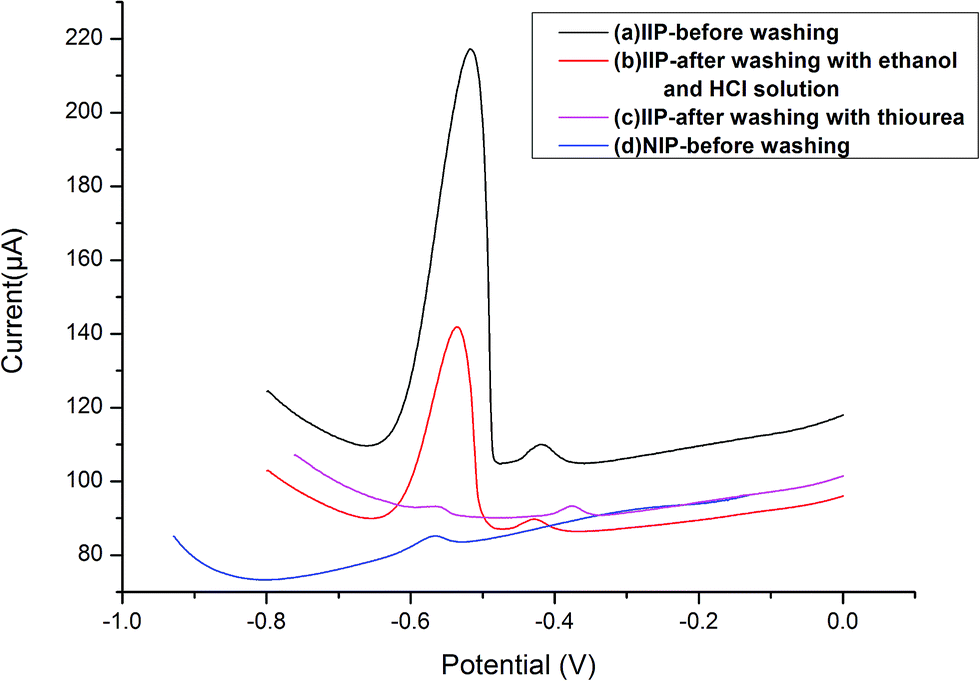 | ||
| Fig. 3 Comparison of differential pulse voltammetry responses of the IIP-CP electrode and NIP-CP electrode with and without the washing step; [Pb2+] = 5.0 × 10−7 mol L−1, extraction time 80 s, stripping voltammetry conditions: E-conditioning = −1.0 V, conditioning time = 40 s, scan rate = 0.1 V s−1. | ||
As evidence by the N2 sorption test, the imprinted polymer particles showed a specific surface area of 134.89 m2 g−1, whereas the non-imprinted polymer particles only had a specific surface area of 79.42 m2 g−1. These indicated that the surface area of the imprinted polymer particles was larger than the non-imprinted particles. More binding sites for the template ions formed on the surface of the imprinted particles.
3.2 Optimisation of lead(II) extraction conditions on the electrode response
:7. That means the amount of paraffin oil was at 30%, and in the 70% of the solid phase, the amount of IIP/graphite changed. Under the same experimental conditions for the extraction and determination procedures, the voltammetric signals were tested at a concentration of 5.0 × 10−7 mol L−1 lead. For good conductivity, carbon black, activated charcoal and graphite powder were chosen to mix with paraffin oil at the confirmed composition percentage, and tested using cyclic voltammetry in an acetic acid buffer solution (pH = 5) without added ion-imprinted polymers. As shown in Fig. S1,† the graphite powder mixed with paraffin had the best current response. Carbon black, activated charcoal and graphite powder are allotropes of carbon with different structures, and when they mixed with paraffion oil in the same proportion, the influence of the different spatial structures leads to various conductivities. The IIP-CPE with graphite powder mixed with paraffin was chosen for the following studies.Not only the experimental materials influence the current, but also the amount of IIP in the modified CPE played a key role in the response characteristics of the IIP-CPEs. As shown in Fig. S2,† the voltammetric signal was increased with the increasing IIP content, and the chosen composition was 5%, 10%, 15%, 20% and 25%. Up to 20%, with the increasing percentage of the IIP, the IIP-CPE voltammetric signal decreased considerably because of the conductivity of the electrode. Therefore, the selected percentage for a graphite powder/IIP/paraffin oil electrode was 50%/20%/30% as the optimal condition for the preparation of IIP-CPE in following studies.
3.3 Comparison of the IIP-CP electrode with the NIP-CP electrode for absorption properties
The carbon paste electrode modified with IIP or NIP was incubated in the Pb2+ solution (5.0 × 10−7 mol L−1). Then, the electrode was inserted into the electrochemical cell, and a negative potential of −1.0 V was applied to the electrode followed by a different pulse voltammetry technique for the test of lead ions. As shown in Fig. 3, the obtained signal for IIP-CPE before washing (voltammogram, a) is noticeably higher than that of NIP-CPE (voltammogram, d), indicating the selective functional cavities in the IIP-CPE were formed, and the selectivity of IIP-CPE is superior to NIP-CPE because of the functional cavities. After washing the electrode modified with IIP to remove the lead ions (voltammogram a, b and c), the electrode signals decreased in voltammograms a, b and c, which means the selective functional cavities in the IIP were created in the polymerization procedure. When the IIP was washed with ethanol, HCl solution and thiourea solutions, unreacted substances and weak cohesion were washed from the surface sites of IIP.3.4 Selectivity of IIP-CPE
The selectivity of IIP in the IIP-CPE was investigated by incubation in different solutions, including a mixture of Pb2+ (5.0 × 10−7 mol L−1) and some potential interfering cations at various concentrations, and subsequently analyzed by different pulse voltammetry. Sample Pb2+ voltammograms obtained are in the first column of Fig. 4(a)–(d) and S6,† as a comparison with other columns of different cations at various concentrations. It can be seen that the presence of Cd2+, Co2+, Cu2+ and Fe3+ in the Pb2+ solution, at identical concentrations, possessed negligible effects on the voltammetric signal of lead ions. In Fig. 4(a) and S6,† the presence of Cd2+ at a Cd2+/Pb2+ ratio of 50 and Co2+ at a Co2+/Pb2+ ratio of 100 did not affect significantly the Pb2+ signal. However, in Fig. 4(b) the presence of Cu2+ at a Cu2+/Pb2+ ratio of 10 possessed an effect on the Pb2+ signal. By increasing the Cu2+/Pb2+ ratio to 50, the voltammetric signal of Pb2+ further decreased, indicating that for occupying the selective sites in the IIP, these two different kinds of ions have a competitive relationship. The same behavior can be seen in the case of Fe3+ in Fig. 4(c). The addition of CN− bonded with Cu2+ decreases the influence on the detection of Pb2+ using the self-manufactured electrode. In Fig. 4(d), the presence of Mn2+ at a Mn2+/Pb2+ ratio of 75 cannot influence the current signal of Pb2+, but with increasing the Mn2+/Pb2+ ratio to 100 the signal of Pb2+ slightly decreased. These observations indicate that a sophisticated arrangement of carboxylic acid groups in the pre-designed cavity of an IIP led to an interesting selectivity for the target ion. The interference levels of several ions, including Cd2+, Co2+, Zn2+, Cr3+, Hg2+, Ag+, Mn2+, Cu2+, Fe3+ and alkali, which are usually present with lead ions in complex matrices, are summarized in Table S1.† The tolerance limit was established as the maximum concentration of foreign species that caused a relative error of 5% in the analytical signal. This table shows that the presence of alkaline cations as well as other interfering cations such as Cd2+, Co2+, Zn2+, Hg2+, Cr3+, Ag+ do not significantly influence the electrode signal. A 100-fold excess of Mn2+, 15-fold excess of Fe3+ and 10-fold excess of Cu2+ influence the detection of Pb2+ significantly. By adding CN− (0.005 mol L−1) in the Pb2+ solution, the Pb2+ signal of the prepared sensor reached a 25-fold and 100-fold excess for Fe3+ and Cu2+, decreasing the interference effect of Cu2+ and Fe3+. The interference of Mn2+ slightly influenced Pb2+.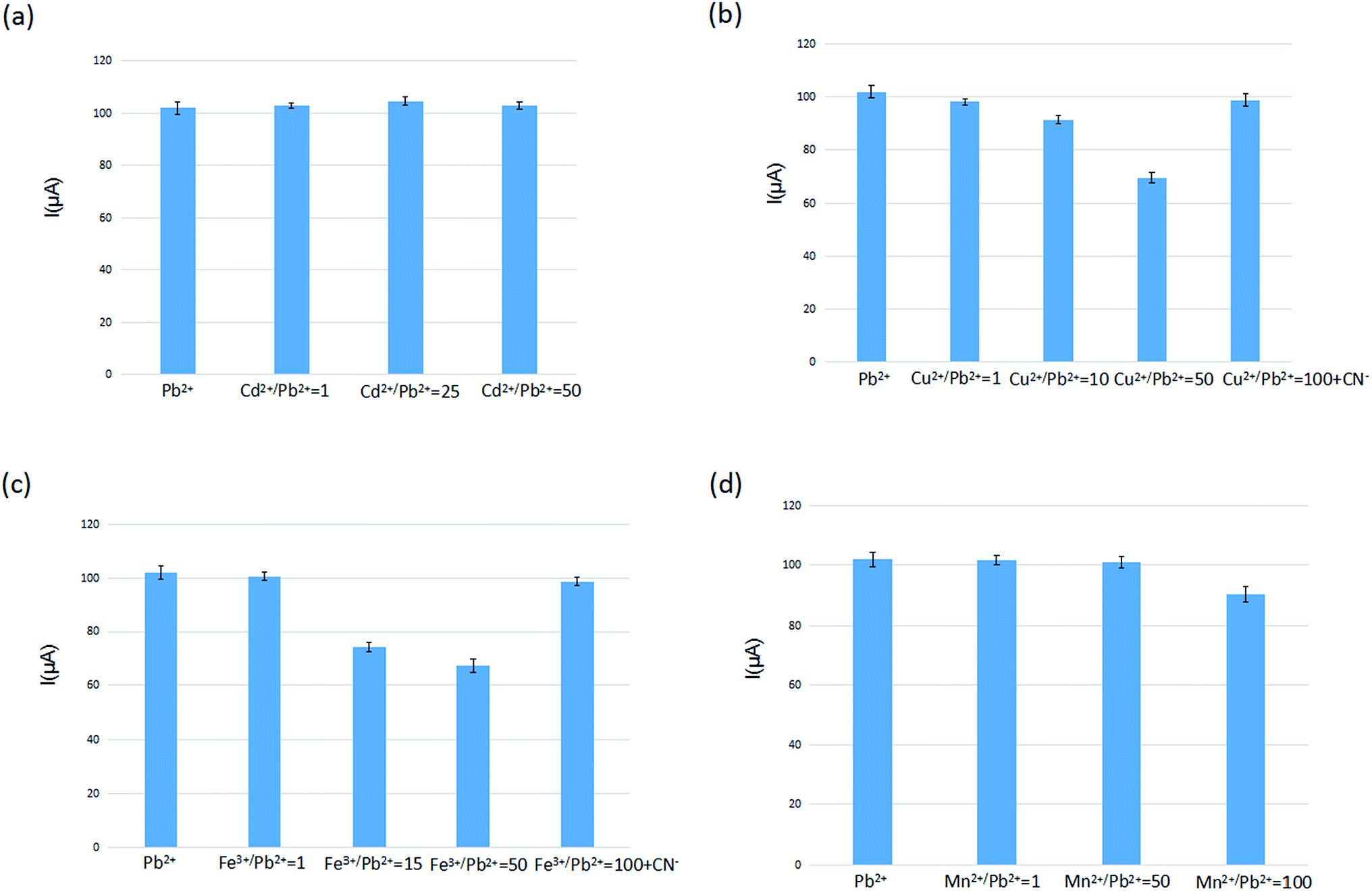 | ||
| Fig. 4 Comparison of the electrode selectivity of Pb2+ ion in the presence of difference interfere ions ((a) Cd2+ (b) Cu2+ (c) Fe3+ (d) Mn2+) at various concentrations: [Pb2+] = 5.0 × 10−7 mol L−1, extraction time 80 s, stripping voltammetry conditions: E-conditioning = −1.0 V, conditioning time = 40 s, scan rate = 0.1 V s−1 (n = 5). | ||
3.5 Analytical characterization of IIP-CPE
In order to evaluate the reproducibility of the IIP-CP electrode, 5.0 × 10−7 mol L−1 Pb2+ solution was determined by five separately produced electrodes. Experimental results were used to calculate an RSD% equal to 2% for the described determination. Moreover, five separate determinations of Pb2+ by the same electrode resulted in an RSD% = 4.76% (n = 5). Thus, the repeatability of the method can be referenced. After refreshing the electrode surface by polishing the electrode surface on weighing paper, the linear range of the prepared sensor was detected in the Pb2+ solution of different concentrations. In Fig. 5, the calibration graph of the produced sensor showed a linear relationship over the Pb2+ concentration in the range of 1.0 × 10−9 to 7.5 × 10−7 mol L−1 with a detection limit of 1.3 × 10−11 mol L−1 (S/N = 3).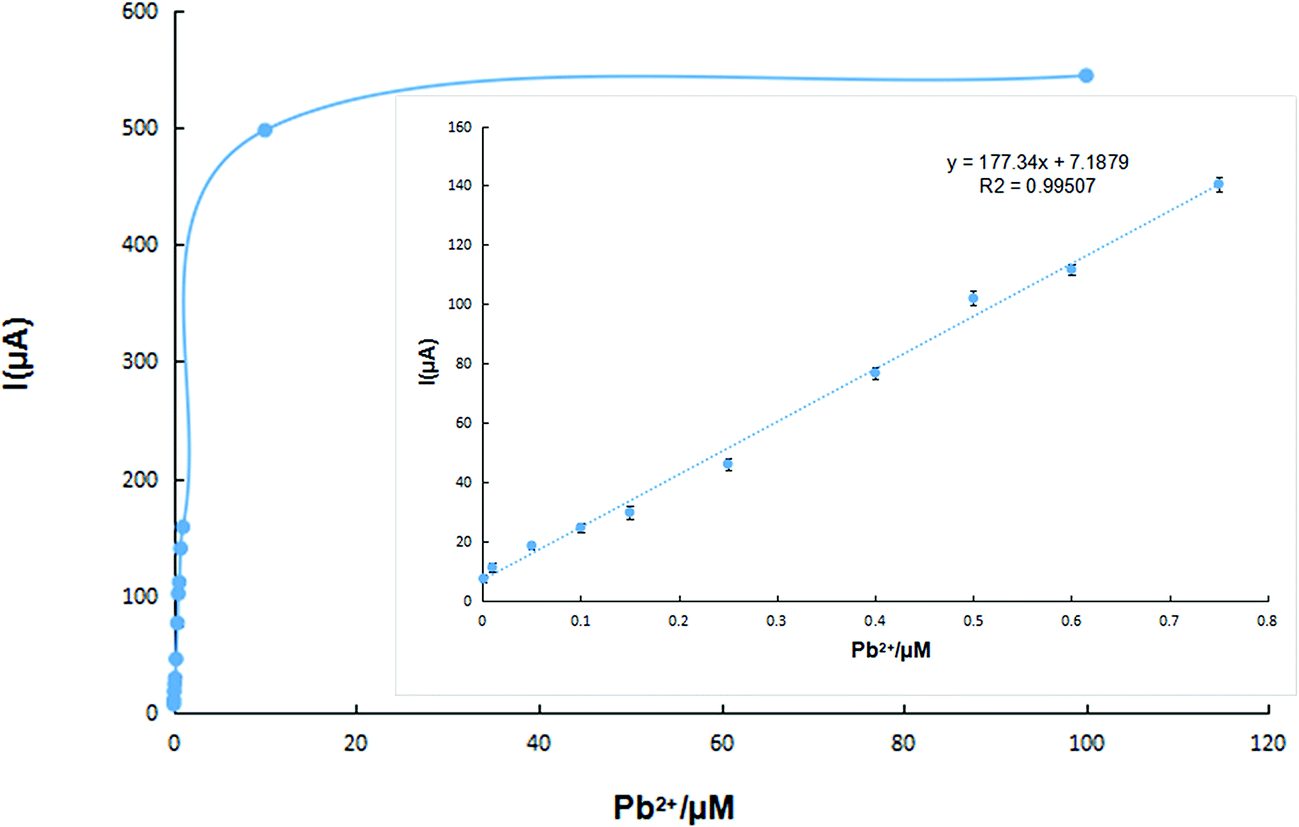 | ||
| Fig. 5 Calibration curve of the lead(II) ion sensor in the optimized conditions; inset: the linear range of the calibration curve (n = 5). | ||
3.6 Real sample detection
To evaluate the feasibility of the developed electrochemical sensor for routine analysis, the IIP-CP electrode was applied to detect lead(II) in four kinds of samples, which are flour, rice, tap water and Yudai River water. Thus, 50 mL of those samples were spiked with different concentrations of lead(II). Then, the determination method was executed step by step, according to the proposed procedure. The results of the above studies are summarized in Table 1 and clearly certify the applicability of this method for determination of Pb2+ in various real samples.3.7 Comparison of the figures of the prepared sensor with those of the previous method
In Table 2, the manufactured IIP-CP sensor is compared with other previous voltammetric sensors with a number of analytical characteristics including the linear dynamic range (LDR), limit of detection (LOD) and interference levels of Hg2+, Cd2+, Cu2+ and Fe3+ in the literature. As can be seen, the IIP-CP electrode shows very good analytical characteristics compared to the majority of the depicted sensors. The IIP-CP electrode has the lowest limit of detection among the list voltammetric sensors.19,25–30| Electrode | LDR (1.0 × 10−9 M) | LOD (1.0 × 10−9 M) | Interference level | Ref. | |||
|---|---|---|---|---|---|---|---|
| Hg2+ | Cu2+ | Cd2+ | Fe3+ | ||||
| IIP nanoparticles-CPE | 0.1–10 | 0.03 | >100 | >100 | >100 | >150 | 25 |
| 10–1000 | |||||||
| Carbon paste modified with 1,4-bis(prop-2-enyloxy)-9,10-anthraquinone | 2–10000 |
1 | >100 | <1 | >30 | >100 | 26 |
| Carbon electrode modified with montmorillonite-bismuth | 4.8–115 | 0.96 | <1 | <1 | — | — | 27 |
| Carbon paste modified with zeolite | 25–100 | 17.3 | <1 | <1 | <1 | <1 | 19 |
| Three-dimensional nanoporous gold leaf modified gold electrode | 4.83–965.3 | 0.48 | No | No | — | — | 28 |
| L-Cysteine/graphene modified glassy carbon electrode | 5.01–299.7 | 2.17 | — | >100 | — | — | 29 |
| IIP-CPE | 1–750 | 0.013 | No | >10 | No | >15 | This work |
| Exfoliated nano-zirconium phosphate | 50–2600 | 0.019 | — | No | — | — | 30 |
4. Conclusion
In this paper, the self-manufactured carbon paste electrode sensor based on the IIP was made for the determination of lead ions. The imprinted electrochemical sensor was linear from 1.0 × 10−9 to 7.5 × 10−7 mol L−1, and the limit of detection (LOD) was 1.3 × 10−11 mol L−1 (S/N = 3). The results show the outstanding selectivity and sensitivity for the electrochemical determination of lead ions, and it resists the common potential interfering agents for good application in real water and food samples. The selective electrochemical sensor will be widely used because of its ease of preparation and low-cost.Acknowledgements
This work was sponsored by the National Natural Science Foundation of China (No. 21677064), Jiangsu Natural Science Foundation of China (No. BK20151323, BK20151337), Postdoctoral Science Foundation of China (No. 2015T80515), the Scientific Research Foundation of Jiangsu University (No. Y14A105, Y14A125).References
- M. Behbahani, H. S. Abandansari, M. Salarian, M. Babapour, A. Bagheri and M. R. Nabid, Microchim. Acta, 2013, 181, 129–137 CrossRef.
- M. A. Belarra, C. Crespo, M. Resano and J. R. Castillo, Spectrochim. Acta, Part B, 2000, 55, 865–874 CrossRef.
- H. F. Maltez, D. L. Borges, E. Carasek, B. Welz and A. J. Curtius, Talanta, 2008, 74, 800–805 CrossRef CAS PubMed.
- L. A. Portugal, H. S. Ferreira, W. N. L. dos Santos and S. L. C. Ferreira, Microchem. J., 2007, 87, 77–80 CrossRef CAS.
- M. G. A. Korn, G. L. dos Santos, S. M. Rosa, L. S. G. Teixeira and P. V. de Oliveira, Microchem. J., 2010, 96, 12–16 CrossRef CAS.
- J. Biasino, J. R. Dominguez and J. Alvarado, Talanta, 2007, 73, 962–964 CrossRef CAS PubMed.
- E. R. Yourd, J. F. Tyson and R. D. Koons, Spectrochim. Acta, Part B, 2001, 56, 1731–1745 CrossRef.
- D. K. Korkmaz, N. Ertaş and O. Y. Ataman, Spectrochim. Acta, Part B, 2002, 57, 571–580 CrossRef.
- M. Shamsipur, F. Raoufi and H. Sharghi, Talanta, 2000, 52, 637–643 CrossRef CAS PubMed.
- W. H. Chan, R. H. Yang, T. Mo and K. M. Wang, Anal. Chim. Acta, 2002, 460, 123–132 CrossRef CAS.
- M. R. Ganjali, N. Motakef-Kazami, F. Faridbod, S. Khoee and P. Norouzi, J. Hazard. Mater., 2010, 173, 415–419 CrossRef CAS PubMed.
- E. Kirowa-Eisner, M. Brand and D. Tzur, Anal. Chim. Acta, 1999, 385, 325–335 CrossRef CAS.
- Y. Wu, N. B. Li and H. Q. Luo, Sens. Actuators, B, 2008, 133, 677–681 CrossRef CAS.
- S. Lee, S. Bong, J. Ha, M. Kwak, S.-K. Park and Y. Piao, Sens. Actuators, B, 2015, 215, 62–69 CrossRef CAS.
- K. Pokpas, N. Jahed, O. Tovide, P. G. Baker and E. I. Iwuoha, Int. J. Electrochem. Sci., 2014, 9, 5092–5115 CAS.
- L. Fan, J. Chen, S. Zhu, M. Wang and G. Xu, Electrochem. Commun., 2009, 11, 1823–1825 CrossRef CAS.
- F. Zavarise, D. Merli and A. Profumo, Anal. Chim. Acta, 2010, 668, 177–181 CrossRef CAS PubMed.
- Z. Hu, C. J. Seliskar and W. R. Heineman, Anal. Chim. Acta, 1998, 369, 93–101 CrossRef CAS.
- S. Senthilkumar and R. Saraswathi, Sens. Actuators, B, 2009, 141, 65–75 CrossRef CAS.
- M. Ghiaci, B. Rezaei and R. J. Kalbasi, Talanta, 2007, 73, 37–45 CrossRef CAS PubMed.
- S. Yuan, W. Chen and S. Hu, Talanta, 2004, 64, 922–928 CrossRef CAS PubMed.
- I. Cesarino, J. Braz. Chem. Soc., 2007, 18, 810–817 CrossRef CAS.
- G. Vlatakis, L. I. Andersson, R. Müller and K. Mosbach, Nature, 1993, 361, 645–647 CrossRef CAS PubMed.
- T. P. Rao, R. Kala and S. Daniel, Anal. Chim. Acta, 2006, 578, 105–116 CrossRef CAS PubMed.
- M. K. Bojdi, M. H. Mashhadizadeh, M. Behbahani, A. Farahani, S. S. H. Davarani and A. Bagheri, Electrochim. Acta, 2014, 136, 59–65 CrossRef CAS.
- M. F. Mousavi, A. Rahmani, S. M. Golabi, M. Shamsipur and H. Sharghi, Talanta, 2001, 55, 305–312 CrossRef CAS PubMed.
- L. Luo, X. Wang, Y. Ding, Q. Li, J. Jia and D. Deng, Appl. Clay Sci., 2010, 50, 154–157 CrossRef CAS.
- Y. Li, J. Liu, F. Yu, H. Tang, F. Zhao, B.-C. Ye, W. Chen and X. Lv, J. Electroanal. Chem., 2015, 758, 78–84 CrossRef CAS.
- W. Zhou, C. Li, C. Sun and X. Yang, Food Chem., 2016, 192, 351–357 CrossRef CAS PubMed.
- L. Wang, W. H. Xu, R. Yang, T. Zhou, D. Hou, X. Zheng, J. H. Liu and X. J. Huang, Anal. Chem., 2013, 85, 3984–3990 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25791g |
| This journal is © The Royal Society of Chemistry 2017 |