
Highly efficient near IR photosensitizers based-on Ir–C bonded porphyrin-aza-BODIPY conjugates†
Jinfeng Zhoua,
Lizhi Gaib,
Zhikuan Zhoua,
John Mack*c,
Kejing Xud,
Jianzhang Zhaod,
Hailin Qiua,
Kin Shing Chan*b and
Zhen Shen*a
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210046, China. E-mail: zshen@nju.edu.cn
bDepartment of Chemistry, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong, China. E-mail: ksc@cuhk.edu.hk
cDepartment of Chemistry, Rhodes University, Grahamstown, 6140, South Africa. E-mail: j.mack@ru.ac.za
dState Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, China
First published on 21st July 2016
Abstract
Two hybrid organic–metal complexes based-on Ir–C bonded porphyrin-aza-BODIPY conjugates have been facilely synthesized through group 9 iridium porphyrin with an aza-BODIPY axial ligand. The conjugates combine the advantages of the near-IR (NIR) absorption of the aza-BODPY dye (>700 nm) with the efficient singlet oxygen production of iridium porphyrin (ΦΔ = 79–85%). The experimental results demonstrate that the axial ligand of aza-BODIPY largely affects the optical and photophysical properties of the conjugates due to the strong interaction between 5d orbitals of the Ir(III) ion and the aza-BODIPY π-system in the ground state, which differs considerably from previous reports.
Introduction
The photosensitized production of singlet oxygen is significant in a range of areas from photooxidation, DNA damage, photodynamic therapy (PDT) of cancer, to polymer science.1 Many transition metal complexes such as Pt(II), Ru(II), Ir(III), Rh(III), etc., show efficient intersystem crossing (ISC) due to heavy atom effects, and as a result, induce singlet oxygen production.2 Unfortunately, the major drawback, which has greatly hampered their wide applications, is that the absorption bands at the red end of the visible region and the neighboring portion of the NIR tend to be relatively weak, since only forbidden ligand-to-metal and metal-to-ligand charge transfer (LMCT and MLCT) transitions lie in this spectral region. Therefore, the preparation of stable strongly near IR-absorbing transition metal complexes would be highly desirable. One of the most promising strategies to obtain those materials is to link the transition metal complexes with another more strongly NIR absorbing fluorophore.BF2-chelated tetraarylazadipyrromethenes (aza-BODIPYs) are a versatile class of chromophores with high extinction coefficients and moderate fluorescence in the NIR region, but they show poor singlet oxygen quantum yield.3 Porphyrins, the most significant pigment in nature, have attracted increasing interest because of their successful utilization across a wide range of research disciplines.4 Although the porphyrin structure can be readily modified and can coordinate most elements in the periodic table,5 they absorb weakly in the NIR region. To address these challenges, one of the most promising strategies is to link the porphyrin complex with another more strongly NIR absorbing fluorophore. The porphyrin–BODIPY conjugates have been the focus of considerable interest in recent years due to their unique photophysical properties and structural functionalities. To date, the vast majority of the literature in this regard has investigated intramolecular electron and energy-transfer processes within the xy-plane of the porphyrin π-system.4a,6 There are only a very limited number of examples where the second fluorophore is linked to the metal center of the porphyrin complex as an axial ligand through an sp3 hybridized linker unit, such as an oxygen7 or nitrogen atom.8 Although these dyads may have strong absorption bands in the visible or NIR regions, the excitation energy harvested by the chromophore cannot be funneled efficiently into the coordination centre and hence to the triplet excited state manifold, so the triplet quantum yield is not enhanced evidently.
In this study, a aza-BODIPY moiety is directly bonded to the metal core of the porphyrin complex through a Ir–C(aryl) bond. This not only results in strong NIR region absorption, but also efficient singlet oxygen quantum yields. The photophysical properties have been investigated using steady-state and time-resolved absorption spectra, MCD spectroscopy and TD-DFT calculations.
Experimental section
Materials and instrumentations
Unless otherwise noted, all reagents were purchased from commercial suppliers and directly used without further purification. Hexane for chromatography was distilled from anhydrous calcium chloride. Benzene and benzene-d6 were distilled from sodium and were stored in a Teflon-capped tube under nitrogen gas prior to use. All reactions carried out in Teflon screw-capped tube under N2 with the mixture degassed for three freeze–thaw–pump cycles were wrapped with aluminum foil to prevent undesired photochemical reactions. The reaction mixtures in Teflon-screw-capped tubes were heated on heat blocks in heaters. The crude mixture was dried under high vacuum.1H and 13C NMR spectra were recorded on a Bruker Avance III 400 at 400 MHz and 700 MHz, respectively. Chemical shifts were referenced with the residual solvent protons of benzene-d6 (δ = 7.15 ppm) and CDCl3 (δ = 7.26 ppm) in 1H NMR spectra as the internal standards. Chemical shifts (δ) were reported as part per million (ppm) in (δ) scale downfield from tetramethylsilane (TMS). Coupling constants (J) were reported in Hertz (Hz). MALDI-TOF MS data were measured on Bruker Daltonics autoflexII mass spectrometer. High resolution mass spectra (HRMS) were recorded on a ThermoFinnigan MAT 95 XL mass spectrometer. Fast atom bombardment spectra were measured with 3-nitrobenzyl alcohol (NBA) as the matrix. UV-visible absorption spectra were recorded with a SHIMADZU UV-2550 spectrometer. Magnetic circular dichroism (MCD) spectra were recorded using a Jasco J-725 spectrodichrometer and equipped with a Jasco permanent magnet that produces a magnetic field of 1.5 T. A FluoroLog-3 spectrofluorometer equipped with a 450 W xenon lamp and near-IR accessories (InGaAs detector) was used to measure the 1O2 emission spectrum.
Singlet oxygen quantum yield determination
Singlet oxygen quantum yields of the photosensitizers were calculated by monitoring the photooxidation of 1,3-diphenyl isobenzofuran (DPBF) a known singlet oxygen scavenger and using MB as the standard (ΦΔ = 0.57 in dichloromethane).9 The mixture of photosensitizer and DPBF was irradiated with a 690 nm laser beam at a power of 100 mW cm−2 at 30 s intervals. The absorbance was measured after each irradiation and a decrease in the absorption band intensity for DPBF at 409 nm was observed. The following equation was used to calculate the singlet oxygen quantum yield of sensitizer:9where ΦΔ(x) is the single oxygen quantum yield of sample, the ‘x’ and ‘std’ subscripts denote the sample and MB standard, respectively, S denotes the slope of a plot of the change in absorbance for DPBF at 410 nm vs. the irradiation time, and F is the absorption correction factor, which is given by F = 1–10−OD (where OD represents the optical density of sample and MB at the irradiation wavelength).
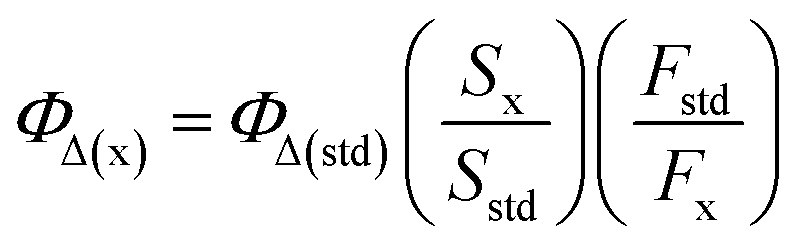
Theoretical calculations
Geometry optimization and TD-DFT calculations were carried out for IrIII(tpp)Ph, 1a and 1b using the B3LYP functional of the Gaussian09 software packages10 with 6-31G(d) basis sets both in vacuo and in the presence of dichlomethane using the polarized continuum model (PCM). In a similar manner, the B3LYP optimized geometries were then used to carry out TD-DFT calculations using the Coulomb-attenuated B3LYP functional (CAM-B3LYP) functional with 6-31G(d) basis sets. The CAM-B3LYP functional was used to calculate the electronic absorption properties based on the time-dependent (TD-DFT) method, since it includes a long-range correction of the exchange potential, which incorporates an increasing fraction of Hartree–Fock (HF) exchange as the interelectronic separation increases. The B3LYP functional has been found to overestimate the degree of configurational interaction between the B excited state of porphyrinoids and higher energy ππ* states.11Synthetic procedures
1a. IrIII(ttp)(CO)Cl (18.0 mg, 0.019 mmol), aza-BODIPY-a (13.7 mg, 0.021 mmol) and K2CO3 (53.9 mg, 0.39 mmol) were added to benzene (1.0 mL) and the mixture was degassed for three freeze–pump–thaw cycles in a Teflon screw-capped tube and filled with N2. The reaction was then heated at 150 °C for 48 h. The solvent was then removed under vacuum and the crude product was purified by column chromatography over silica eluted with CH2Cl2/hexane (1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give compound 1a (17.1 mg, 0.012 mmol, 61%). Rf = 0.68 (CH2Cl2/hexane = 1:1). 1H NMR (400 MHz, C6D6): δ 1.12 (d, 2H, J = 8.4 Hz), 2.42 (s, 12H), 5.40 (d, 2H, J = 4.3 Hz), 5.55 (s, 1H), 6.35 (s, 3H), 6.86–7.05 (m, 4H), 7.26–7.31 (m, 6H), 7.38–7.43 (m, 6H), 7.69 (d, 2H, J = 5.8 Hz), 7.91 (d, 2H, J = 7.7 Hz), 8.01 (d, 4H, J = 7.6 Hz), 8.22 (d, 4H, J = 7.6 Hz), 8.97 (s, 8H). 13C NMR (175 MHz, CDCl3): δ 21.58, 116.63, 117.39, 123.59, 123.77, 124.07, 124.30, 127.52, 127.75, 128.32, 128.36, 129.15, 129.30, 129.76, 129.99, 130.11, 130.82, 131.22, 131.58, 131.74, 131.89, 133.72, 134.13, 137.42, 138.48, 139.62, 142.87, 143.89, 145.69, 146.11, 156.24, 161.01. HRMS (FABMS): calcd for [C80H56BBrF2IrN7]+ ([M]+): m/z 1435.3467. Found: m/z 1435.3489.
:1) to give compound 1a (17.1 mg, 0.012 mmol, 61%). Rf = 0.68 (CH2Cl2/hexane = 1:1). 1H NMR (400 MHz, C6D6): δ 1.12 (d, 2H, J = 8.4 Hz), 2.42 (s, 12H), 5.40 (d, 2H, J = 4.3 Hz), 5.55 (s, 1H), 6.35 (s, 3H), 6.86–7.05 (m, 4H), 7.26–7.31 (m, 6H), 7.38–7.43 (m, 6H), 7.69 (d, 2H, J = 5.8 Hz), 7.91 (d, 2H, J = 7.7 Hz), 8.01 (d, 4H, J = 7.6 Hz), 8.22 (d, 4H, J = 7.6 Hz), 8.97 (s, 8H). 13C NMR (175 MHz, CDCl3): δ 21.58, 116.63, 117.39, 123.59, 123.77, 124.07, 124.30, 127.52, 127.75, 128.32, 128.36, 129.15, 129.30, 129.76, 129.99, 130.11, 130.82, 131.22, 131.58, 131.74, 131.89, 133.72, 134.13, 137.42, 138.48, 139.62, 142.87, 143.89, 145.69, 146.11, 156.24, 161.01. HRMS (FABMS): calcd for [C80H56BBrF2IrN7]+ ([M]+): m/z 1435.3467. Found: m/z 1435.3489.
1b. IrIII(ttp)(CO)Cl (15.8 mg, 0.017 mmol), aza-BODIPY-b (12.3 mg, 0.019 mmol) and K2CO3 (47.0 mg, 0.34 mmol) were added to benzene (1.0 mL) and the mixture was degassed for three freeze–pump–thaw cycles in a Teflon screw-capped tube and filled with N2. The reaction was then heated at 150 °C for 72 h. The solvent was then removed under vacuum and the crude product was purified by column chromatography over silica eluted with CH2Cl2/hexane (1
:1) to give compound 1b (11.4 mg, 0.008 mmol, 46%). Rf = 0.64 (CH2Cl2/hexane = 1:1). 1H NMR (400 MHz, C6D6): δ 1.27 (d, 2H, J = 8.3 Hz), 2.41 (s, 12H), 5.48 (s, 1H), 5.83 (d, 2H, J = 8.7 Hz), 6.28 (s, 1H), 6.91 (d, 3H, J = 7.9 Hz), 7.02–7.08 (m, 5H), 7.23 (d, 4H, J = 7.4 Hz), 7.35 (d, 4H, J = 7.3 Hz), 7.41–7.47 (m, 4H), 7.72 (d, 2H, J = 7.5 Hz), 7.93 (d, 4H, J = 7.4 Hz), 8.13 (d, 4H, J = 7.4 Hz), 8.80 (s, 8H). 13C NMR (175 MHz, CDCl3): δ 21.59, 116.77, 120.30, 121.80, 123.86, 124.12, 124.35, 127.49, 127.62, 128.33, 128.35, 128.81, 129.05, 129.35, 130.50, 130.87, 131.48, 131.55, 131.62, 131.69, 132.95, 133.57, 134.25, 137.44, 138.45, 139.58, 142.74, 143.28, 143.84, 146.75, 151.88, 161.94. HRMS (FABMS): Calcd for [C80H56BBrF2IrN7]+ ([M+]): m/z 1435.3467. Found: m/z 1435.3479.
Results and discussion
Synthesis
Aza-BODIPY-a,12 aza-BODIPY-b,12 IrIII(ttp)Ph (ttp = 5, 10, 15, 20-tetra-p-tolylporphyrinato dianion)13 and IrIII(ttp)COCl (ref. 13) were prepared according to literature procedures. In the IrIII(ttp)-aza-BODIPY conjugates (1a and 1b), the IrIII(ttp) ring was linked through the para-positions of phenyl substituents of the aza-BODIPY fluorophore. Conjugates 1a and 1b were obtained in satisfactory yields by a facile one-step reaction of the appropriate aza-BODIPY precursor with IrIII(ttp)COCl in the presence of K2CO3 in benzene under a N2 atmosphere (Scheme 1, for synthetic details see the Experimental section). The molecular structures were characterized by 1H and 13C NMR spectroscopy and HR-MS.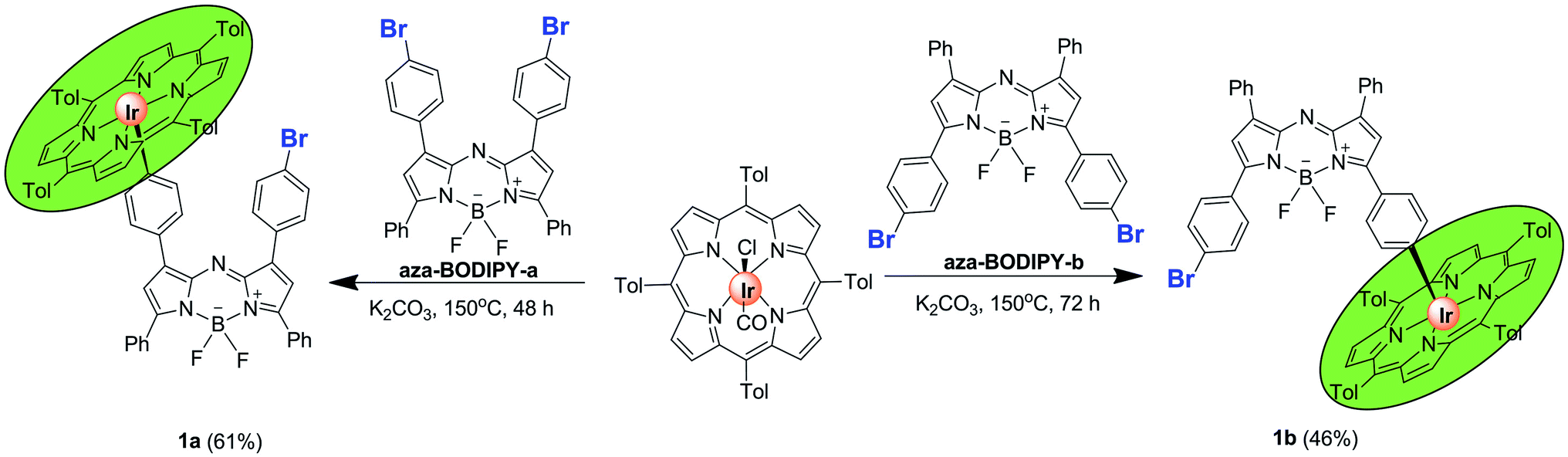 | ||
| Scheme 1 Synthetic procedures for conjugates 1a and 1b. | ||
Photophysical properties and computational studies
When the UV-visible absorption spectra of the Ir(ttp)-aza-BODIPY conjugates are compared with the IrIII(ttp)Ph and aza-BODIPY molecules in toluene (Fig. 1), it becomes clear that in addition to the presence of porphyrin B (or Soret) band at ca. 410 nm, which is almost unaffected compared with that of the IrIII(ttp)Ph component, new bands appeared in the NIR region at longer wavelength than those of the corresponding aza-BODIPY dyes. The absorption data of the conjugates and reference compounds in a wide range of solvents are listed in Table 1 and Table S1.† In the case of 1a, the main aza-BODIPY absorption band at 655 nm is replaced by two new absorption bands at 628 nm and 685 nm. In contrast, 1b has one major absorption band at 739 nm that is significantly red shifted by 76 nm compared with that of aza-BODIPY-b, suggesting a very strong ground-state interaction with the IrIII(ttp) moiety. These results are in marked contrast to what has been reported previously for porphyrin–BODIPY arrays, where the absorption spectra closely match the sum of the spectra of the component moieties of the molecule.6a,6d,6e,7a,8,14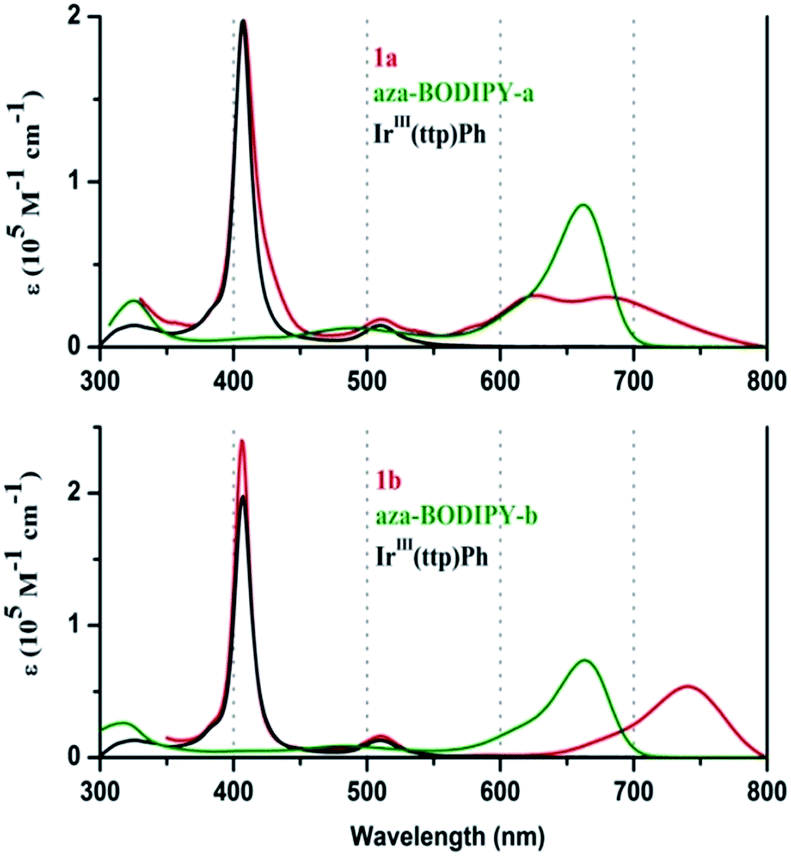 | ||
| Fig. 1 Absorption spectra of 1a, 1b and the reference compounds in toluene. | ||
| Component | λabs [nm] (logε [M−1 cm−1]) |
ΦΔa |
|---|---|---|
| a Singlet oxygen (1O2) quantum yield determined in O2 saturated dichloromethane.9b Excited by a 690 nm laser using MB as the standard.c Excited by a 671 nm laser using MB as the standard.d Excited by a 405 nm laser using Rose Bengal (RB) as the standard.17e No singlet oxygen produced upon excitation with a 690 nm laser. | ||
| 1a | 411 (4.75), 512 (3.76), 628 (4.06), 685 (4.11) | 0.85b |
| 1b | 406 (5.38), 510 (4.21), 739 (4.73) | 0.79b |
| aza-BODIPY-a | 484 (3.87), 662 (4.79) | 0.006c, 0.005d |
| aza-BODIPY-b | 483 (4.00), 663 (4.93) | 0.008c |
| IrIII(ttp)Ph | 409 (5.41), 512 (4.28) | N. De |
A detailed analysis of magnetic circular dichroism (MCD) spectral data for 1a and 1b (Fig. 2 and S5†) can be used to definitively assign the main spectral bands, since significant MCD intensity is only anticipated for the absorption bands that are associated with the MIII(ttp) macrocycle.15 Much weaker induced magnetic dipole transition moments are anticipated for the excited states associated with the main spectral bands of the aza-BODIPY moiety due to the absence of a π-conjugated macrocycle. The intense bands beyond 600 nm in the spectra of conjugates 1a and 1b can be unambiguously assigned to the aza-BODIPY moiety on this basis. TD-DFT spectra calculated in vacuo and in the presence of dichloromethane using PCM provide further support for this (Fig. 2 and S5†). As is usually the case with porphyrin and BODIPY spectra, the energies of the spectral bands are significantly overestimated when a comparison is made with the experimental data.16 The band morphologies and crossover points of the derivative-shaped pseudo-![[scr A, script letter A]](https://www.rsc.org/images/entities/char_e520.gif) 1 terms in the MCD spectra of 1a and 1b are largely unaffected when a comparison is made with the spectra of the IrIII(ttp)Ph reference compound (Fig. 2), so the bands at ca. 510 and 410 nm in the spectra of conjugates 1a and 1b can be readily assigned as the IrIII(ttp) Q and B bands, respectively. Much greater differences in wavelength are observed when a comparison is made with the spectra of the corresponding aza-BODIPYs, due to the effect that replacing the bromine para-substituent on one of the phenyl moieties with a iridium metal ion has on the energies of the HOMO and LUMO of the aza-BODIPY chromophore (Fig. 3 and S6†). TD-DFT calculations at the CAM-B3LYP/6-31G(d) of theory predict that the HOMO of 1a is located mainly on the π-system of the IrIII(ttp) moiety and the LUMO is located on both the metal center and aza-BODIPY, while those of 1b are both located primarily on the aza-BODIPY (Fig. 3). For example, there is a strong interaction between the highest occupied MO of the aza-BODIPY chromophore of 1b and one of the d MOs of the central metal ion, which demonstrates why the presence of a direct metal–carbon bond results in a stronger interaction between the aza-BODIPY and IrIII(ttp) moieties than is present in the porphyrin–BODIPY dyads that have been reported previously. Since the highest occupied MOs of the aza-BODIPY and IrIII(ttp) moieties lie very close in energy, minor changes in the linkage position of the metal ion can result in marked changes in the spectroscopic properties.
1 terms in the MCD spectra of 1a and 1b are largely unaffected when a comparison is made with the spectra of the IrIII(ttp)Ph reference compound (Fig. 2), so the bands at ca. 510 and 410 nm in the spectra of conjugates 1a and 1b can be readily assigned as the IrIII(ttp) Q and B bands, respectively. Much greater differences in wavelength are observed when a comparison is made with the spectra of the corresponding aza-BODIPYs, due to the effect that replacing the bromine para-substituent on one of the phenyl moieties with a iridium metal ion has on the energies of the HOMO and LUMO of the aza-BODIPY chromophore (Fig. 3 and S6†). TD-DFT calculations at the CAM-B3LYP/6-31G(d) of theory predict that the HOMO of 1a is located mainly on the π-system of the IrIII(ttp) moiety and the LUMO is located on both the metal center and aza-BODIPY, while those of 1b are both located primarily on the aza-BODIPY (Fig. 3). For example, there is a strong interaction between the highest occupied MO of the aza-BODIPY chromophore of 1b and one of the d MOs of the central metal ion, which demonstrates why the presence of a direct metal–carbon bond results in a stronger interaction between the aza-BODIPY and IrIII(ttp) moieties than is present in the porphyrin–BODIPY dyads that have been reported previously. Since the highest occupied MOs of the aza-BODIPY and IrIII(ttp) moieties lie very close in energy, minor changes in the linkage position of the metal ion can result in marked changes in the spectroscopic properties.
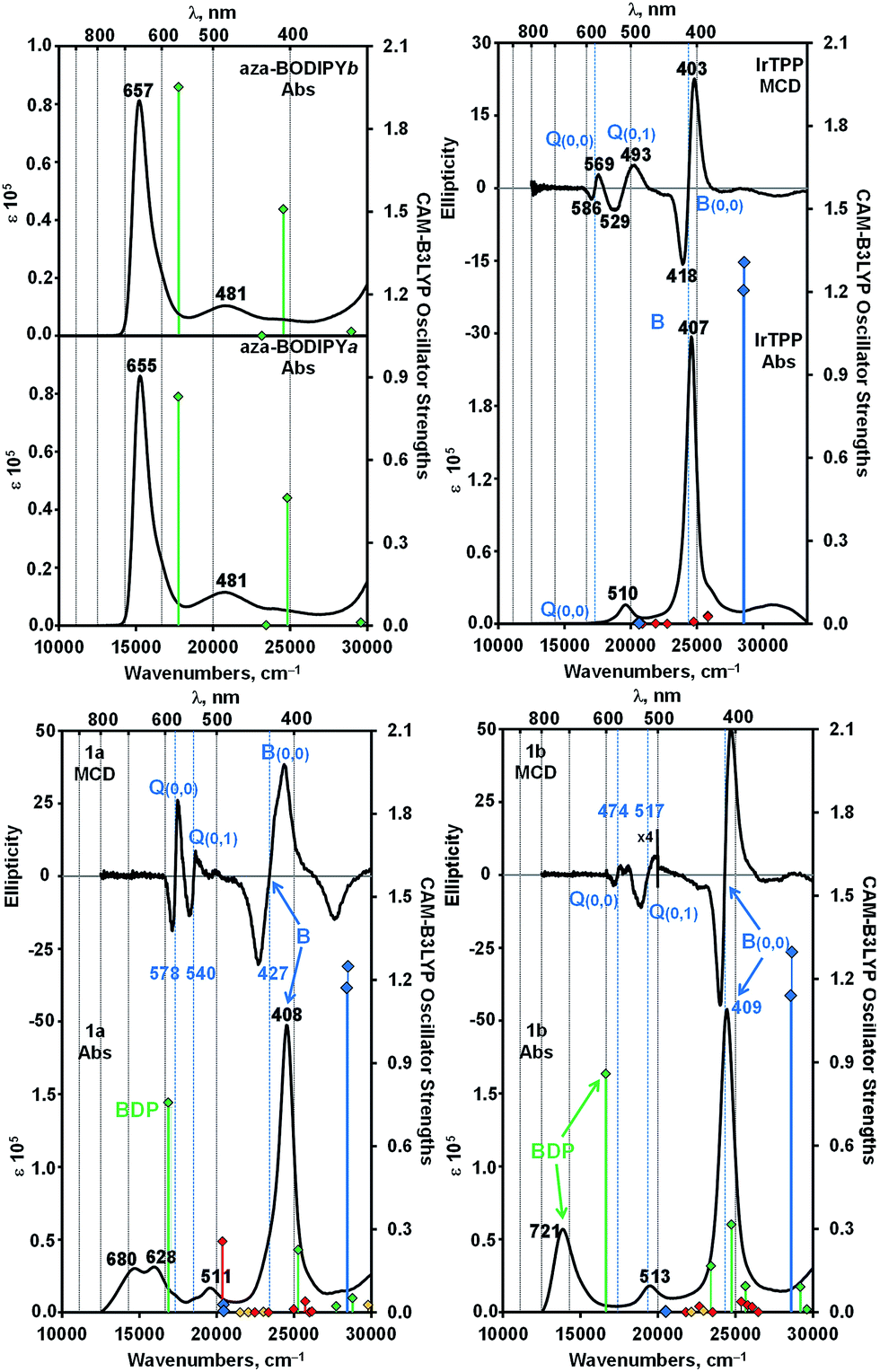 | ||
| Fig. 2 Absorption and MCD spectra of conjugates 1a, 1b, aza-BODIPY-a, aza-BODIPY-b and IrIII(ttp)Ph in dichloromethane (c = 5.0 × 10−6 M). TD-DFT spectra calculated for B3LYP geometries with the CAM-B3LYP functional and 6-31G(d) basis sets are plotted against secondary axes. Blue diamonds are used to highlight the Q and B bands of the π-system of the Ir(ttp) moiety, while green diamonds are used to highlight transition associated with the aza-BODIPY π-system. Red and yellow diamonds are used to highlight transitions involving the 5d orbitals of the Ir(III) ion and charge transfer transitions between the IrIII(ttp) and aza-BODIPY moieties, respectively. Vertical blue lines are used to highlight the crossover points of the pseudo-1 terms in the MCD spectra that are associated with the Q and B bands of Gouterman's 4-orbital model.15 | ||
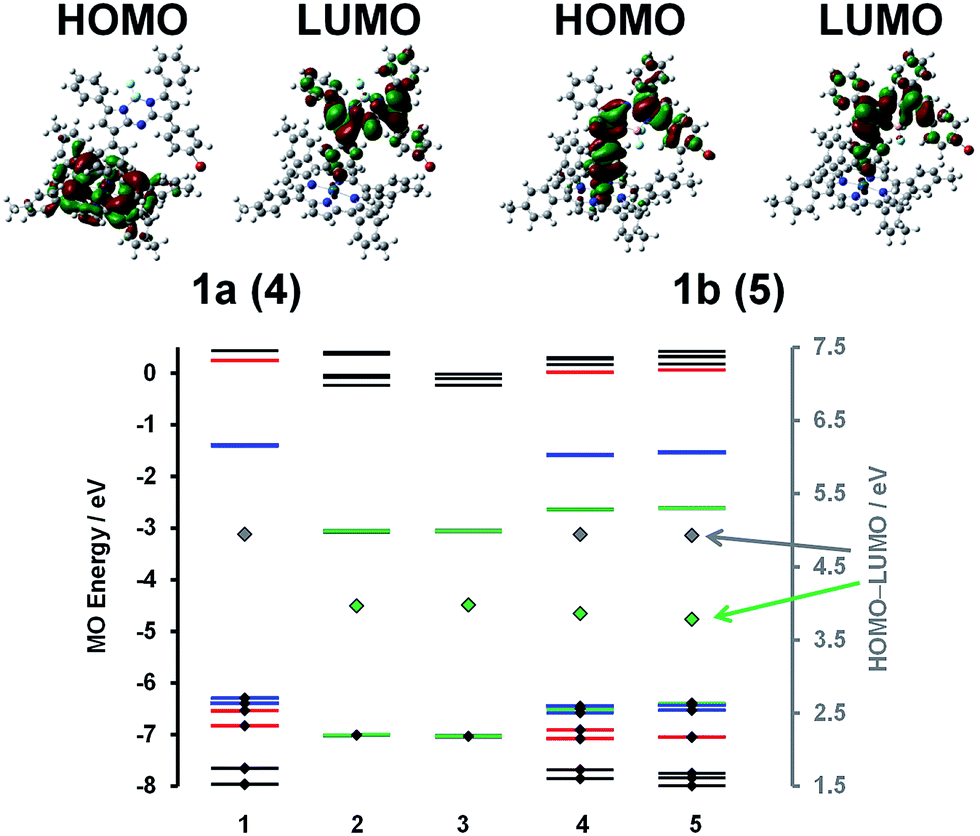 | ||
| Fig. 3 Angular nodal patterns of the HOMO and LUMO of 1a and 1b at an isosurface of 0.02 a.u (TOP). MO energies of IrIII(ttp)Ph (1), aza-BODIPY-a (2), aza-BODIPY-b (3), 1a (4) and 1b (5). The HOMO–LUMO gaps for the BODIPY and IrIII(ttp) moieties are plotted against a secondary axis and are denoted with large green and gray diamonds, respectively. The four frontier π-MOs of the π-system of the IrIII(ttp) moiety are highlighted with blue lines, while red and green lines are used to denote the 5d orbitals of the Ir(III) ion and the HOMO and LUMO of the aza-BODIPY π-system, respectively. Small black diamonds are used to highlight occupied MOs. | ||
Photosensitizing property in the NIR region
Although porphyrins and their derivatives have been widely used in PDT, their major shortcoming in this regard is their relatively weak absorption at the red end of the visible region. Aza-BODIPYs have high extinction coefficients in the NIR region, but they show poor singlet oxygen production ability. In order to investigate the photosensitizing capability of Ir(ttp)-aza-BODIPY conjugates and the reference compounds IrIII(ttp)Ph and aza-BODIPYs, the singlet oxygen quantum yield (ΦΔ) values were evaluated by using a steady-state method with 3-diphenyl isobenzofuran (DPBF) as the 1O2 scavenger and methylene blue (MB) as the standard (ΦΔ = 0.57 in dichloromethane) (Table 1).9 In air-saturated dichloromethane, conjugates 1a and 1b induced drastic changes in the DPBF absorption at 410 nm upon photo-irradiation with a 690 nm laser beam (100 mW cm−2). The values of ΦΔ = 0.85 for 1a and 0.79 for 1b were obtained, which are much larger than that of most previously reported photosensitizers.9,18 In contrast, negligible singlet oxygen quantum yields for IrIII(ttp)Ph and aza-BODIPY was observed under the same condition. Simultaneously, a more direct method for detecting singlet oxygen was also used based on monitoring the phosphorescence spectrum of 1O2 radiative decay around 1270 nm. Significant 1O2 phosphorescence intensity was observed in the presence of 1a and 1b upon excitation of a 5.0 μM solution at both 405 and 690 nm (Fig. S10†). The quantum efficiencies of 1O2 production are almost the same once the differing optical densities and laser intensities are taken into consideration (Table S2, ESI†).Photostability
It is well known that good photostability upon repetitive excitation is highly desirable in practical applications. To assess the photostability of the Ir(ttp)-aza-BODIPY conjugates, the photobleaching kinetics of target compounds were measured in dichloromethane with a 671 nm laser beam at a power of 100 mW cm−2, using MB as a reference (Fig. 4). 36% of the MB reference solution was degraded after 60 min irradiation. In contrast, 1b was almost unaffected under the same irradiation conditions and there was a 15% degradation for 1a. It is obvious, therefore, that the conjugates exhibit remarkable degree of photostability. Additionally, conjugates 1a and 1b also have high thermal stabilities and do not decompose at temperatures below 300 °C.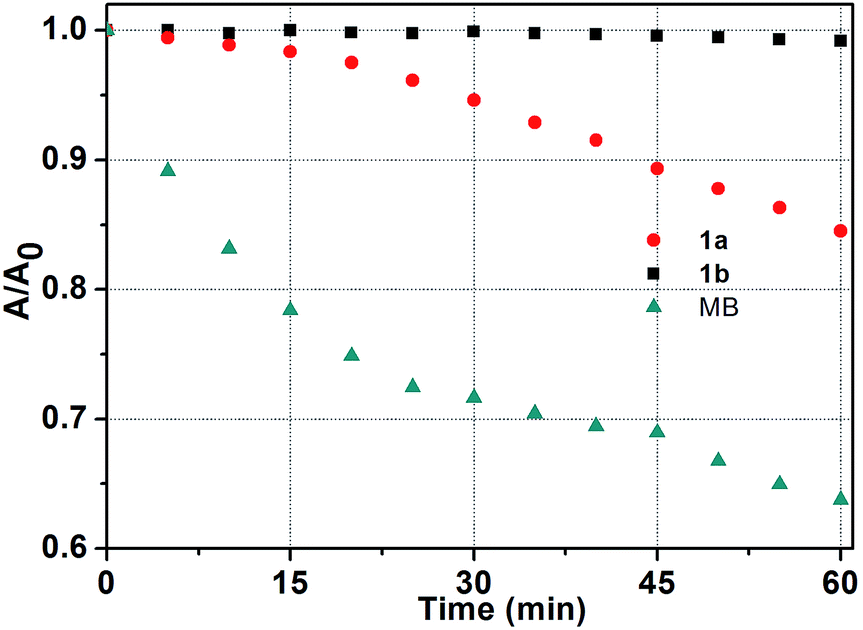 | ||
| Fig. 4 Photostability of 1a, 1b and MB in dichloromethane at 5 min intervals determined using a laser beam (671 nm, 100 mW cm−2) over an irradiation period of 60 min (c = 5.0 μM, 298 K). | ||
Conclusions
In conclusion, we have designed and synthesized two remarkably photostable and efficient singlet oxygen production NIR dyes, which are hybrid organic–metal complexes based-on Ir–C bonded porphyrin-aza-BODIPY conjugates through the facile synthesis of a group 9 iridium porphyrin with an aza-BODIPY axial ligand. The NIR region absorbing aza-BODIPY moiety can be easily modified and this has been found to have a significant impact on the observed spectral properties. The results clearly demonstrate that this novel approach holds promise as a strategy for overcoming the two main bottlenecks that have been encountered during dye research in the NIR region, firstly the relatively weak absorption of transition-metal porphyrins in this spectral region and secondly, the lack of ISC in organic fluorophores. It is now possible to exploit the advantages of both types of compound by forming an orthogonal conjugate with a direct metal–C bond. More broadly, the molecular design and building block approach we have developed should enable the development of a far wider series of stable NIR absorbing triplet photosensitizers, which will be of interest both in fundamental studies and for practical applications in issues such as photodynamic therapy, photocatalysis, photovoltaics and upconversion.Acknowledgements
This work was supported by the Major State Basic Research Development Program of China (Grant No. 2013CB922101 & 2011CB808704), the National Natural Science Foundation of China (No. 21371090) to ZS. Scientific Research Foundation of Graduate School of Nanjing University to L.G, and a NSFC/NRF China-South Africa research cooperation program (CS08-L07 and UID: 95421) to ZS and JM, and an NRF of South Africa CSUR grant (93627) to JM. The theoretical calculations were carried out at the Centre for High Performance Computing in Cape Town.Notes and references
- (a) J. W. Snyder, I. Zebger, Z. Gao, L. Poulsern, P. K. Frederiksen, E. Skovsen, S. P. Mcilroy, M. Klinger, L. K. Andersen and P. R. Ogilby, Acc. Chem. Res., 2004, 37, 894 CrossRef CAS PubMed; (b) P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181 RSC.
- J. Z. Zhao, W. H. Wu, J. F. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323 RSC.
- (a) P. A. Bouit, K. Kamada, P. Feneyrou, G. Berginc, L. Toupet, O. Maury and C. Andraud, Adv. Mater., 2009, 21, 1151 CrossRef CAS; (b) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed; (c) H. Lu, J. Mack, Y. C. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778 RSC.
- (a) T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 4, 943–969 RSC; (b) L. L. Li and E. W. Diau, Chem. Soc. Rev., 2013, 42, 291 RSC; (c) K. A. Carter, S. Shao, M. I. Hoopes, D. Luo, B. Ahsan, V. M. Grigoryants, W. Song, H. Huang, G. Zhang, R. K. Pandey, J. Geng, B. A. Pfeifer, C. P. Scholes, J. Ortega, M. Karttunen and J. F. Lovell, Nat. Commun., 2014, 5, 3546 Search PubMed; (d) M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340 RSC; (e) S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Gratzel, Nat. Chem., 2014, 6, 242 CrossRef CAS PubMed.
- (a) Y. Chang, H. C. Chen, Z. K. Zhou, Y. Zhang, C. Schütt, R. Herges and Z. Shen, Angew. Chem., Int. Ed., 2012, 51, 12801 CrossRef CAS PubMed; (b) J. Mack, K. Lobb, T. Nyokong, Z. Shen and N. Kobayashi, J. Porphyrins Phthalocyanines, 2012, 16, 833 CrossRef CAS; (c) J. W. Tian, L. Ding, H. J. Xu, Z. Shen, H. X. Ju, L. Jia, L. Bao and J. S. Yu, J. Am. Chem. Soc., 2013, 135, 18850 CrossRef CAS PubMed; (d) D. Wu, A. B. Descalzo, F. Weik, F. Emmerling, Z. Shen, X. Z. You and K. Rurack, Angew. Chem., Int. Ed., 2008, 47, 193 CrossRef CAS PubMed; (e) H. J. Xu, J. Mack, A. B. Descalzo, Z. Shen, N. Kobayashi, X. Z. You and K. Rurack, Chem.–Eur. J., 2011, 17, 8965 CrossRef CAS PubMed; (f) H. J. Xu, J. Mack, D. Wu, Z. L. Xue, A. B. Descalzo, K. Rurack, N. Kobayashi and Z. Shen, Chem.–Eur. J., 2012, 18, 16844 CrossRef CAS PubMed; (g) Z. L. Xue, Z. Shen, J. Mack, D. Kuzuhara, H. Yamada, T. Okujima, Z. Ono, X. Z. You and N. Kobayashi, J. Am. Chem. Soc., 2008, 130, 16478 CrossRef CAS PubMed; (h) Z. K. Zhou, Y. Chang, S. Shimizu, J. Mack, C. Schütt, R. Herges, Z. Shen and N. Kobayashi, Angew. Chem., Int. Ed., 2014, 53, 6563 CrossRef CAS PubMed.
- (a) M. T. Whited, S. T. Djurovich, A. C. Durrell, C. W. Schlenker, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2011, 133, 88 CrossRef CAS PubMed; (b) T. Lazarides, G. Charalambidis, A. Vuillamy, M. Reglier, E. Klontzas, G. Froudakis, S. Kuhri, D. M. Guldi and A. G. Coutsolelos, Inorg. Chem., 2011, 50, 8926 CrossRef CAS PubMed; (c) F. R. Li, S. I. Yang, Y. Z. Ciringh, J. Seth, C. H. Martin, D. L. Singh, D. Kim, R. R. Birge, D. F. Bocian, D. Holten and J. S. Lindsey, J. Am. Chem. Soc., 1998, 120, 10001 CrossRef CAS; (d) Q. Q. Hu, Y. Z. Zhu, S. C. Zhang, Y. Z. Tong and J. Y. Zheng, Dalton Trans., 2015, 44, 15523 RSC; (e) F. D'Souza, A. N. Amin, M. E. El-Khouly, N. K. Subbaiyan, M. E. Zandler and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 654 CrossRef PubMed; (f) A. Eggenspiller, A. Takai, M. E. El-Khouly, K. Ohkubo, C. P. Gros, C. Bernhard, C. Goze, F. Denat, J. M. Barbe and S. Fukuzumi, J. Phys. Chem. A, 2012, 116, 3889 CrossRef CAS PubMed.
- (a) T. Shiragami, K. Tanaka, Y. Andou, S. I. Tsunami, J. Matsumoto, H. Luo, Y. Araki, O. Ito, H. Inoue, M. Yasuda and J. Photochem, J. Photochem. Photobiol., A, 2005, 170, 287 CrossRef CAS; (b) T. Lazarides, S. Kuhri, G. Charalambidis, M. K. Panda, D. M. Guldi and A. G. Coutsolelos, Inorg. Chem., 2012, 51, 4193 CrossRef CAS PubMed; (c) M. K. Panda, T. Lazarides, G. Charalambidis, V. Nikolaou and A. G. Coutsolelos, Eur. J. Inorg. Chem., 2015, 468 CrossRef CAS.
- P. Mondal, A. Chaudhary and S. P. Rath, Dalton Trans., 2013, 42, 12381 RSC.
- (a) Y. C. Yang, Q. L. Guo, H. C. Chen, Z. K. Zhou, Z. J. Guo and Z. Shen, Chem. Commun., 2013, 49, 3940 RSC; (b) X. S. Ke, Y. Chang, J. Z. Chen, J. Tian, J. Mack, X. Cheng, Z. Shen and J. L. Zhang, J. Am. Chem. Soc., 2014, 136, 9598 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, M. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) J. Mack, Y. Asano, N. Kobayashi and M. J. Stillman, J. Am. Chem. Soc., 2005, 127, 17697 CrossRef CAS PubMed; (b) J. Mack, M. Bunya, Y. Shimizu, H. Uoyama, N. Komobuchi, T. Okujima, H. Uno, S. Ito, M. J. Stillman, N. Ono and N. Kobayashi, Chem.–Eur. J., 2008, 14, 5001 CrossRef CAS PubMed; (c) D. Kuzuhara, J. Mack, H. Yamada, T. Okujima, N. Ono and N. Kobayashi, Chem.–Eur. J., 2009, 15, 10060 CrossRef CAS PubMed; (d) H.-J. Xu, J. Mack, D. Wu, Z.-L. Xue, A. B. Descalzo, K. Rurack, N. Kobayashi, Z. Shen, Chem.–Eur. J., 2012, 18, 16844 Search PubMed; (e) M. Kon-no, J. Mack, N. Kobayashi, M. Suenaga, K. Yoza and T. Shinmyozu, Chem.–Eur. J., 2012, 18, 13361 CrossRef CAS PubMed.
- A. Gorman, J. Killoran, C. F. O'Shea, T. Kenna, W. M. Gallagher and D. F. O'Shea, J. Am. Chem. Soc., 2004, 126, 10619 CrossRef CAS PubMed.
- C. W. Cheung and K. S. Chan, Organometallics, 2011, 30, 4269 CrossRef CAS.
- (a) G. N. Lim, E. Maligaspe, M. E. Zandler and F. D'Souza, Chemistry, 2014, 20, 17089 CrossRef CAS PubMed; (b) H. J. Xu, A. Bonnot, P. L. Karsenti, A. Langlois, M. Abdelhameed, J. M. Barbe, C. P. Gros and P. D. Harvey, Dalton Trans., 2014, 43, 8219 RSC.
- (a) J. Mack, M. J. Stillman and N. Kobayashi, Coord. Chem. Rev., 2007, 251, 429 CrossRef CAS; (b) N. Kobayashi and K. Nakai, Chem. Commun., 2007, 40, 4077 RSC.
- (a) J. Mack, N. Kobayashi and Z. Shen, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2012, vol. 23, pp. 282–351 Search PubMed; (b) J. Mack, J. Stone and T. Nyokong, J. Porphyrins Phthalocyanines, 2014, 18, 630 CrossRef CAS.
- R. W. Redmond and J. N. Gamlin, Photochem. Photobiol., 1999, 70, 391 CrossRef CAS PubMed.
- (a) L. Hou, X. Zhang, T. C. Pijper, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 910 CrossRef CAS PubMed; (b) Y. C. Lai and C. C. Chang, J. Mater. Chem. B, 2014, 2, 1576 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Methods and synthesis, 1H and 13C NMR spectra, and additional experimental and calculated optical spectra. See DOI: 10.1039/c6ra10131c |
| This journal is © The Royal Society of Chemistry 2016 |