Structural diversity and chemical synthesis of peroxide and peroxide-derived polyketide metabolites from marine sponges
Matthew D.
Norris
and
Michael V.
Perkins
*
School of Chemical and Physical Sciences, Flinders University, GPO Box 2100, Adelaide, SA 5001, Australia. E-mail: mike.perkins@flinders.edu.au
Received
23rd November 2015
First published on 10th May 2016
Abstract
Covering: up to early 2016
Marine sponges are widely known as a rich source of natural products, especially of polyketide origin, with a wealth of chemical diversity. Within this vast collection, peroxide and peroxide-derived secondary metabolites have attracted significant interest in the fields of natural product isolation and chemical synthesis for their structural distinction and promising in vitro antimicrobial and anticancer properties. In this review, peroxide and peroxide-derived polyketide metabolites isolated from marine sponges in the past 35 years are summarised. Efforts toward their synthesis are detailed with a focus on methods that utilise or attempt to elucidate the complex biosynthetic interrelationships of these compounds beyond enzymatic polyketide synthesis. Recent isolations, advances in synthetic methodology and theories of biogenesis are highlighted and critically evaluated.
Matthew D. Norris
Matthew Norris received his B.Sc. with first-class honours in Chemistry in 2011. He is currently completing his Ph.D. at the School of Chemical and Physical Sciences, Flinders University under the supervision of Michael Perkins. His research has focused on the synthesis of peroxide-derived secondary metabolites from marine sponges with a strong emphasis on developing a better understanding of their biosynthetic origin. He received the Australian Fulbright (WG Walker) Scholarship to work with Erik Sorensen at the Department of Chemistry, Princeton University in 2014.
Michael V. Perkins
Michael Perkins gained a Ph.D. from the University of Queensland in 1990 under the guidance of W. Kitching. He carried out post doctoral work on natural product synthesis with Ian Paterson at the University of Cambridge and then with Lew Mander at the Australian National University. He joined the Faculty of Flinders University in 1995. His main research interest is in the area of synthesis of natural products of polyketide origin.
1. Introduction
Organic extracts of marine sponges from all corners of the globe often exhibit promising in vitro antimicrobial and anticancer properties, prompting further exploration to identify and characterise the constituent metabolites. In 1978, Faulkner examined the crude ethanol extracts of Plakortis halichondrioides and attributed the sample's notable growth inhibition of Staphylococcus aureus and Escherichia coli to the major product isolated, plakortin.1 Structural elucidation by spectroscopic methods and chemical degradation revealed 1,2-dioxane 1 as the identity of plakortin.2 Near the same time, a novel peroxyketal metabolite, chondrillin (2), was isolated from a marine sponge of the genus Chondrilla.3 Compound 2 and a number of natural analogues were shown to be cytotoxic against mouse leukaemia cells and were activators of sarcoplasmic reticulum Ca2+ ATPase.4,5
With unique peroxyheterocyclic core structures and compelling therapeutic potential, compounds 1 and 2 have inspired numerous research programs in the fields of natural product isolation, chemical synthesis and medicinal chemistry since the late 1970s. The class has now expanded to include many new peroxide and peroxide-derived heterocycles and carbocycles with biological significance; and novel structural elements that have been targeted in the development of new synthetic methodology. A number of review articles on this topic have appeared in recent years. Most have focused on structural determination and biological analyses of marine endoperoxides;6–13 and one, from 2004 focused on methods developed for their chemical synthesis.14 However, none have detailed the complex biosynthetic interrelationships of these compounds beyond enzymatic polyketide synthesis. Now, we present a critical review discussing the structural elucidation, chemical synthesis and therapeutic potential of this important compound class, with special focus on their intriguing structural similarities and differences from a biogenetic perspective.
This review summarises the peroxide and peroxide-derived metabolites of polyketide origin reported since the discovery of 1 and 2. Theories of biogenesis, both generally accepted and speculative; and examples of the use of chemical synthesis to demonstrate or utilise this understanding, are highlighted. Notably, it appears that the occurrence of many structurally novel polyketides in this family can be attributed to non-enzymatic modification or rearrangement of peroxide progenitors, which themselves are formed in enzymatic condensation and hydroperoxidation processes. This article is therefore presented in four parts: the first section summarises peroxide metabolites; the second section discusses related compounds most likely derived from base-mediated rearrangement; the third section details natural products that appear to arise from endoperoxide reduction; and the fourth section comments on the therapeutic potential of this compound class. Additionally, oxidative adducts are discussed where necessary.
2. Peroxide metabolites from marine sponges
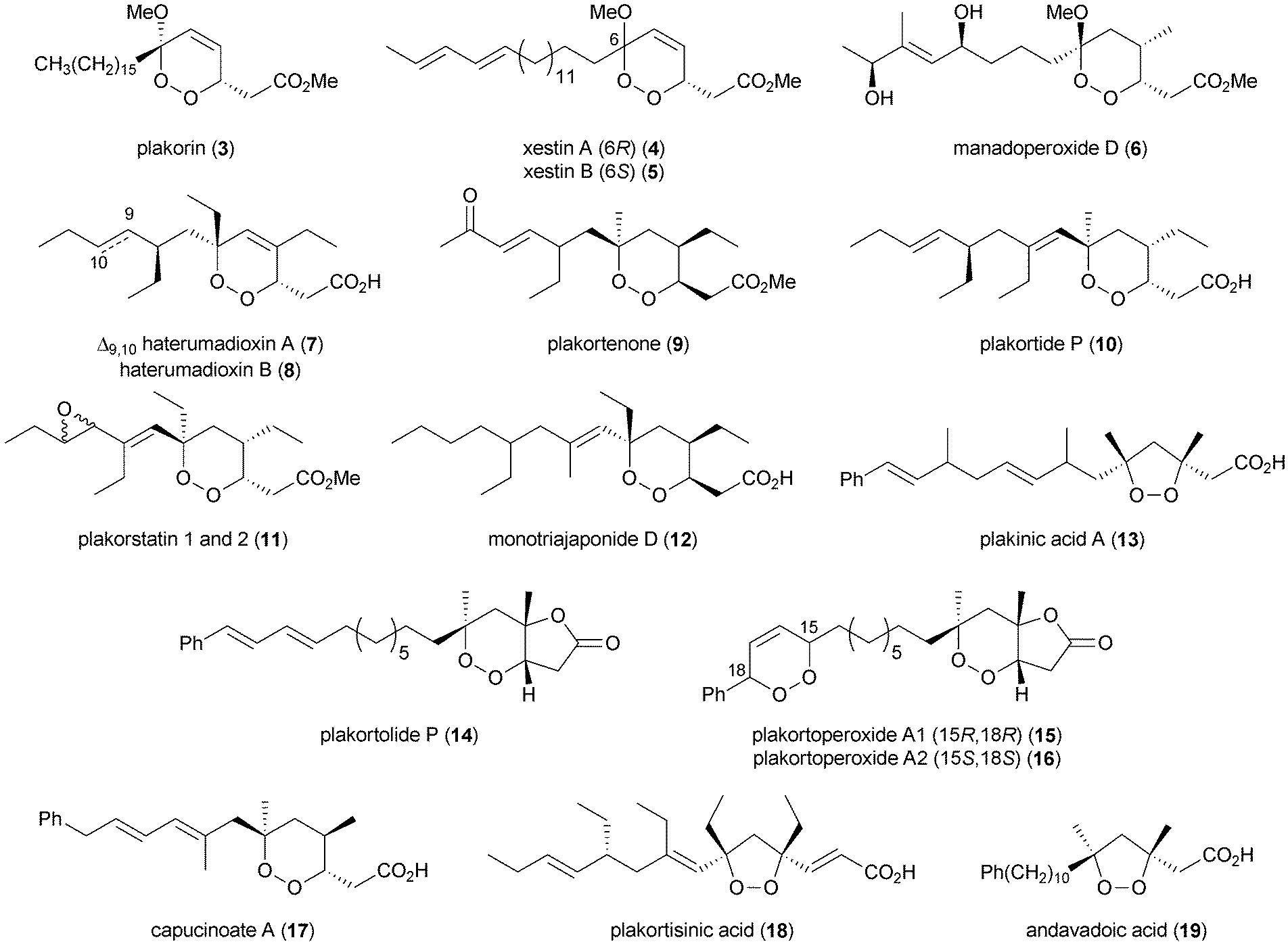
Since the discovery of 1 and 2, a suite of related metabolites have been isolated from marine sponges. These include plakorin (3),4 xestins A (4), B (5),5 manadoperoxides A–C, D (6), E–K,15–17 manadodioxans A–E,18 haterumadioxins A (7), B (8),19 plakortenone (9),20 plakortides E–O, P (10), Q–U, Z, AA,20–32 plakorstatin 1 and 2 (11),33 monotriajaponides B, C, D (12),34 plakinic acids A (13), B–P,32,35–43 plakortolides A–O, P (14), Q–W,40–48 plakortoperoxides A1 (15), A2 (16), B–D,48 capucinoate A (17),49 plakortisinic acid (18),50 andavadoic acid (19),46 and related compounds (Fig. 1).6,7,13,51–62 Following global interest in antimalarial peroxide natural products such as artemisinin,63,64 compounds in this class are routinely screened for in vitro activity against strains of Plasmodia (Section 5.1).11,12,65,66 Testing for cytotoxic action against cancer cell types is also commonplace (Section 5.2).8–10 Despite many encouraging results, the availability of marine endoperoxides is usually very poor and methods for their chemical synthesis are still in development.
Fig. 1 Selected peroxide metabolites of polyketide origin isolated from marine sponges (absolute stereochemistry not implied).
2.1 Peroxide biosynthesis
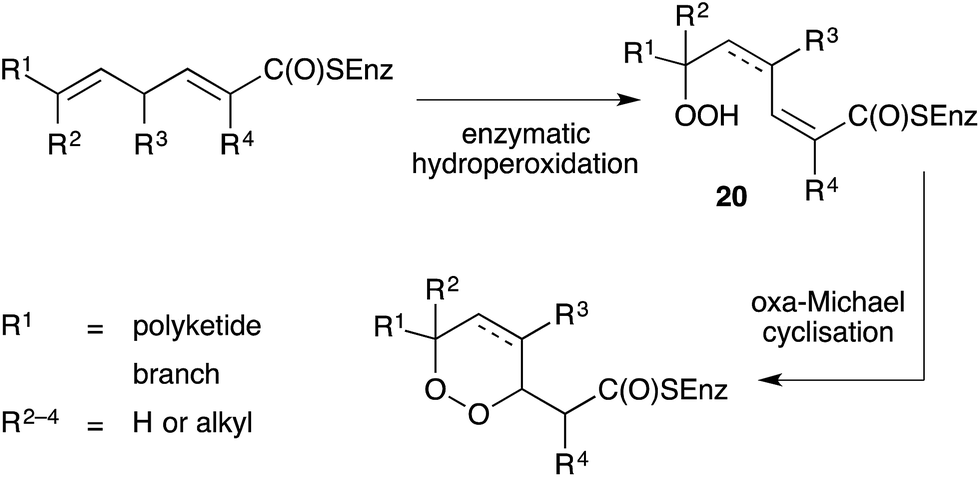
Importantly, all the above-mentioned endoperoxides are isolated as optically active compounds and are thus believed to arise from the enzymatic inclusion of molecular oxygen to an unsaturated polyketide scaffold. Capon first proposed a model for enzymatic hydroperoxidation of 2,5-dienoic acids to yield straight-chain peroxides (20), which then undergo oxa-Michael cyclisation to generate enantiomerically enriched 1,2-dioxanes (Scheme 1).67 The model was initially developed to illustrate biosynthesis of the norterpene peroxide sigmosceptrellin D,67,68 but has since been extended to rationalise the formation of other polyketide endoperoxides, including those related to plakortin.47 Recently, metagenomic analyses of several marine sponges have provided insight to the complexity of the metabolic processes of these organisms and their symbiotic microbiome.69,70
Scheme 1 Biosynthesis of polyketide endoperoxides based on Capon's model.
2.2 Chemical synthesis of peroxide metabolites
Natural products of this family, characterised by highly substituted peroxyheterocyclic core structures with specific absolute configuration, present unique challenges as targets in the field of chemical synthesis. A variety of approaches have been explored, most involving molecular oxygen or hydrogen peroxide as key reagents to construct the endoperoxide system.
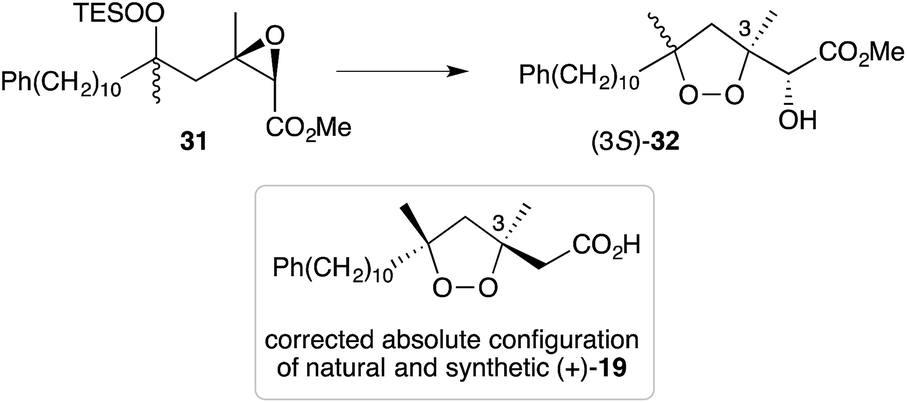
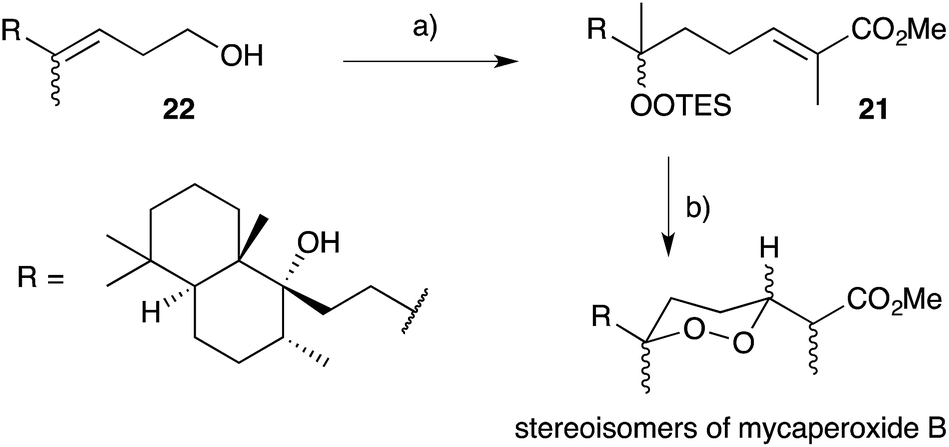
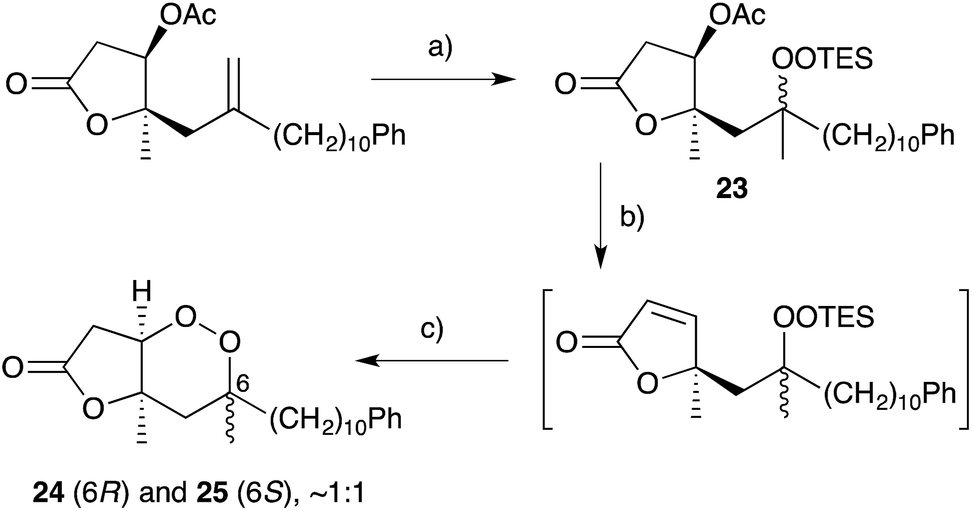
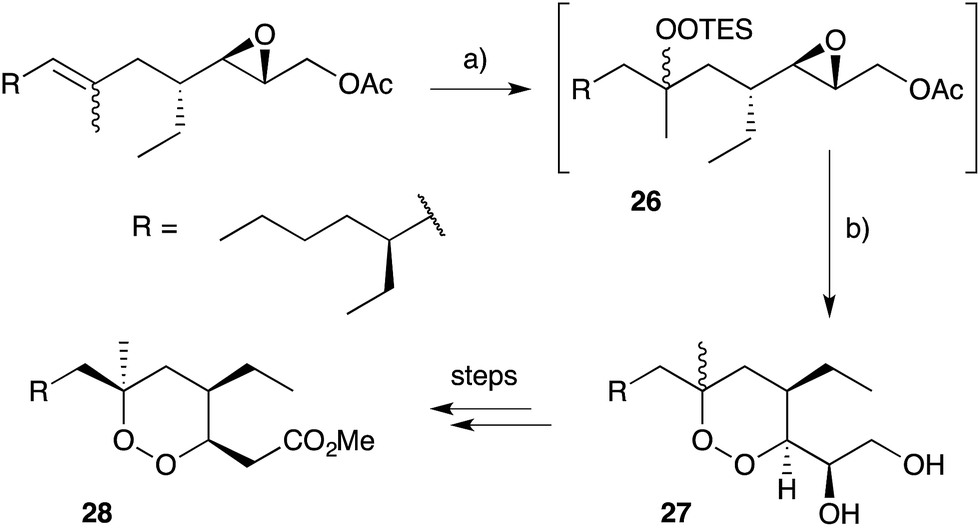
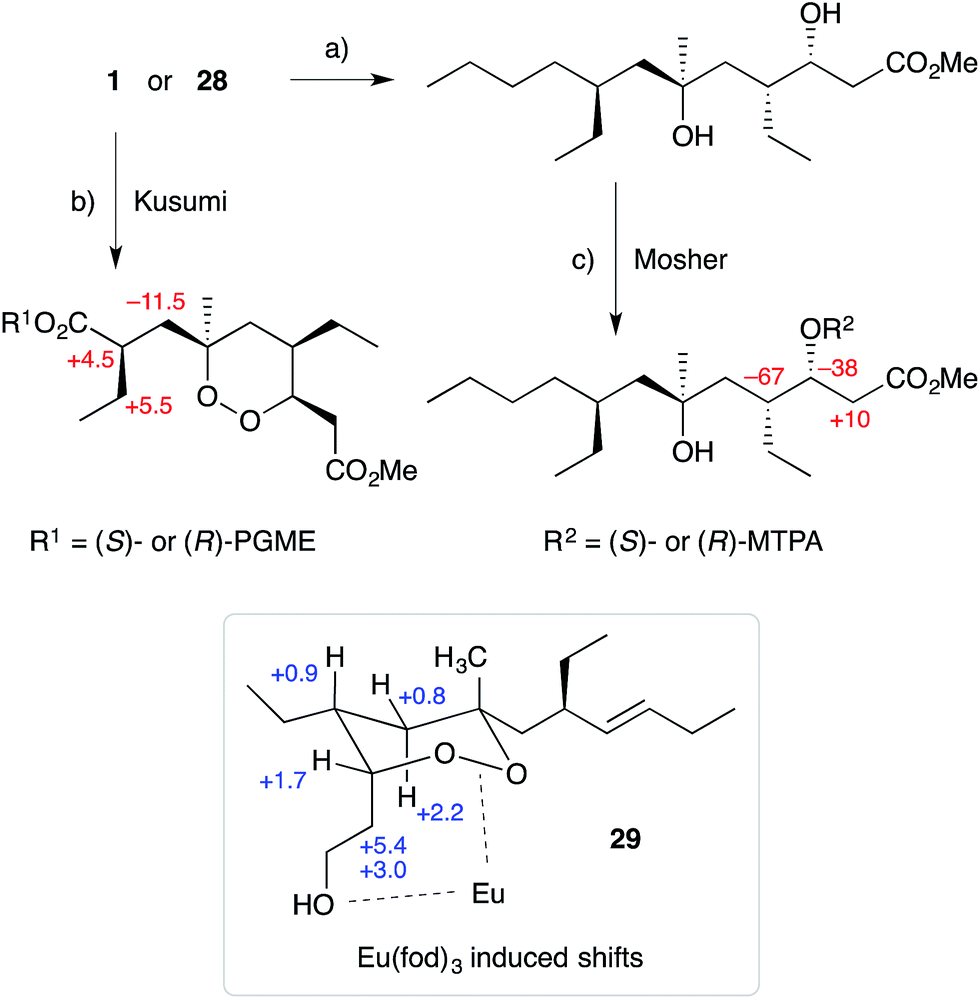
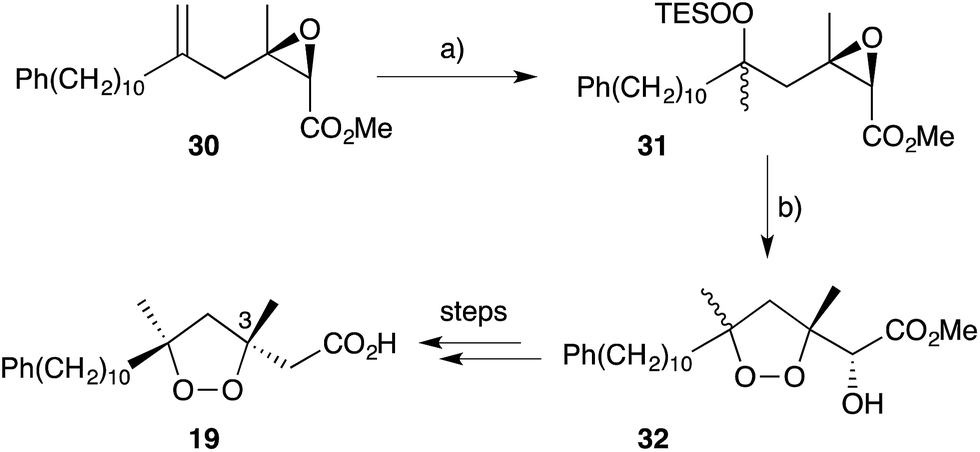
Cobalt(II)-catalysed peroxysilylation developed by Mukaiyama and Isayama71,72 is used extensively to generate open hydroperoxide substrates, which are poised for cyclisation to 5- and 6-membered rings. Although the oxidation itself proceeds with little facial selectivity, stereocontrol in the subsequent cyclisation event has proven effective. Inspired by Capon's model (Section 2.1), Harwood accessed compound 21 by peroxysilylation of olefin 22 and effected oxa-Michael cyclisation to the pendant α,β-unsaturated ester yielding a number of stereoisomers related to mycaperoxide B (Scheme 2).73,74 Similarly, Mukaiyama's method allowed the synthesis of lactone 23, which underwent conjugate-base elimination and oxa-Michael cyclisation to construct both plakortolide E (24) and ent-plakortolide I (25) in 2012 (Scheme 3).75 The asymmetric total synthesis of 24 and 25 prompted clarification of the relative and absolute configuration of the natural products.47,75,76 Campiani and co-workers were able to obtain peroxysilane 26 by the same methodology, before initiating 6-exo-tet hydroperoxide cyclisation to dioxane 27 (Scheme 4).77–79 In a number of subsequent steps, the first total synthesis of 9,10-dihydroplakortin (28) was accomplished and its absolute configuration was confirmed.77–79 Notably, the relative and absolute configuration of 28 and its dehydro analogue 1 were established from the natural material through application of Kusumi's and Mosher's methods to selected products of chemical degradation (Scheme 5).2 Lanthanide-induced shift experiments of alcohol 29 with Eu(fod)3 were also used to assign the relative stereochemistry of 1.1 Finally, Vatéle employed a similar hydroperoxide cyclisation approach toward the synthesis of andavadoic acid (19), effecting oxidation of olefin 30 to peroxysilyl ether 31, which was followed by 5-exo-tet cyclisation to dioxolane intermediate 32 (Scheme 6).80–82
Scheme 2 Synthesis of mycaperoxide B diastereomers by Mukaiyama–Isayama peroxidation and cyclisation. (a) O2, Co(modp)2, Et3SiH, DCE, r.t.; then DMP, CH2Cl2, r.t., 2 h; then (CH3)C(PPh3)CO2Me, CH2Cl2, r.t., overnight, 21% over 3 steps; (b) PPTS, EtOH, r.t., 6 h; then Et3N, MeOH, r.t., 2 d, 8% over 2 steps.
Scheme 3 Synthesis of plakortolide E (24) and ent-plakortolide I (25) by Mukaiyama–Isayama peroxidation and cyclisation. (a) O2, Co(thd)2, Et3SiH, DCE, r.t., 2 h, 94%; (b) DBU, THF, 0 °C, 3 h; then (c) TFE, TBAF, 0 °C, 3 h, 75% over 2 steps.
Scheme 4 Synthesis of 9,10-dihydroplakortin (28) by Mukaiyama–Isayama peroxidation and 6-exo-tet cyclisation. (a) O2, Co(thd)2, Et3SiH, DCE, r.t., 5 h; then (b) Amberlyst, CH2Cl2, r.t., 18 h; then K2CO3, MeOH, 0 °C, 3 h, 78% over 3 steps.
Scheme 5 Application of Kusumi's and Mosher's methods to determine the absolute configuration of plakortin (1) and dihydroplakortin (28); and Eu(fod)3 induced shift data to determine relative configuration of 1. Change in chemical shift due to Kusumi's and Mosher's method expressed as shift of (R)-PGME or (R)-MTPA compound subtracted from shift of (S)-PGME or (S)-MTPA compound (in Hz) and shown as red numbers. Change in shift due to incorporation of Eu(fod)3 (in ppm) shown as blue numbers. (a) H2 (1 atm), 10% Pd/C, EtOH, r.t., 5 h; (b) KMnO4, NaIO4, Na2CO3, tBuOH, H2O, 37 °C, 20 h; then (S)- or (R)-PGME hydrochloride, PyBoP, HOBT, N-methylmorpholine, DMF, r.t., 3 h, 76% over 2 steps (for both (S)- and (R)-PGME amide); (c) (S)- or (R)-MTPA chloride, pyridine, r.t., overnight, 88% (for both (S)- and (R)-MTPA ester). PGME = phenylglycine methyl ester; PyBoP = benzotriazolyloxytri(pyrrolidinyl) phosphonium hexafluorophosphate; HOBT = 1-hydroxybenzotriazole; MTPA = methoxy(trifluoromethyl)phenylactate.
Scheme 6 Synthesis of andavadoic acid (19) by Mukaiyama–Isayama peroxidation and 5-exo-tet cyclisation. (a) O2, Co(thd)2, Et3SiH, DCE, r.t., 3–4 h, 86%; (b) K2CO3, MeOH, 0 °C, 3 h, 64%.
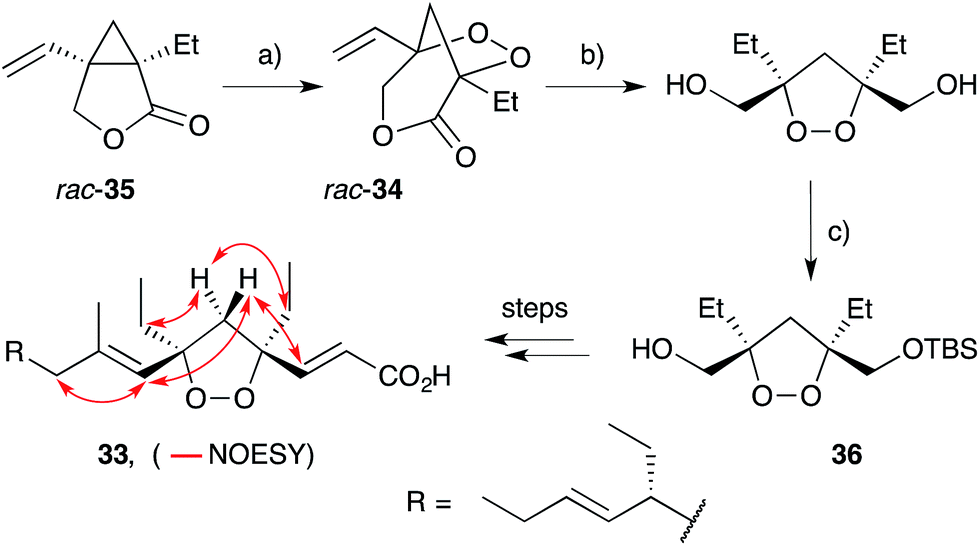
Wong and co-workers took a different approach to the synthesis of 1,2-dioxolanes en route to plakortide E (33), constructing a pivotal [3.2.1]-peroxybicycle intermediate (34) using Feldman's83–85 method for vinyl cyclopropane oxygenation (Scheme 7).86 Thus, irradiation of lactone rac-35 with a 300 W sunlamp in the presence of catalytic Ph2Se2 and AIBN under an atmosphere of oxygen effected near quantitative oxidation to rac-34. Selective reduction of the lactone ester and lipase-mediated kinetic resolution gave TBS ether 36 in >99% ee, before completing the synthesis over several steps. Using this approach, four stereoisomers of plakortide E were synthesised, establishing the relative and absolute configuration of the natural product as (4S,6R,10R)-33 (selected NOESY correlations of the natural material shown in Scheme 7).22 A similar method was later used to establish the first asymmetric total synthesis of epiplakinic acid F.87
Scheme 7 Synthesis of plakortide E (33) by Feldman vinyl cyclopropane oxygenation. Selected NOESY correlations of natural 33 shown as red arrows. (a) O2, Ph2Se2, AIBN, MeCN, 300 W lamp, r.t., 99%; (b) LiBH4, THF, 0 °C; then KO2CN = NCO2K, AcOH, CH2Cl2, 0 °C (three cycles), 70% over 2 steps; (c) TBSCl, imidazole, DMAP, DMF, 0 °C to r.t.; then lipase, vinyl acetate, hexane, 29 h, 32% over 2 steps.
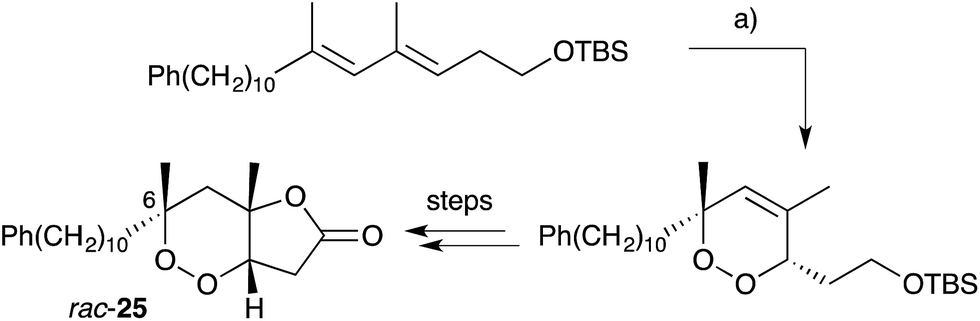
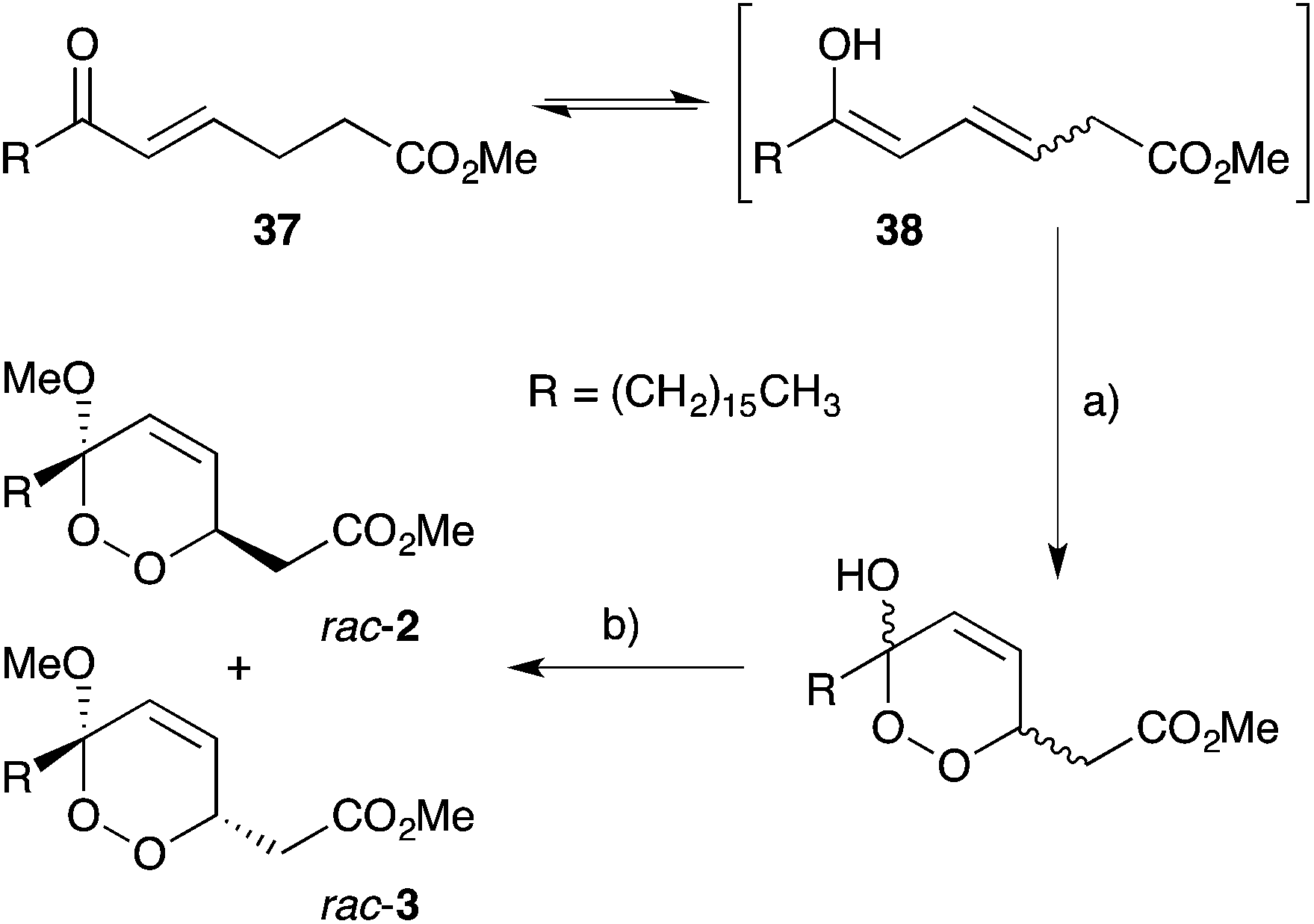
Endoperoxide substrates have also been successfully constructed from suitable straight-chain dienes by [4 + 2] cycloaddition with singlet oxygen.88 Jung89 and Taylor90 have each prepared peroxylactone analogues from diene precursors and a racemic synthesis of 6-epi-plakortolide E (rac-25) was completed in 2002 (Scheme 8).89 Steliou used a similar approach for the synthesis of two cytotoxic endoperoxides isolated from Plakortis angulospiculatus in 1990.91,92 Garson effected photooxygenation of natural plakortolide P (14), yielding synthetic plakortoperoxide A1 (15) and A2 (16) as a 1:1 mixture (Scheme 9).48 Snider constructed the peroxyketal diastereomers 2 and 3 by photooxygenation of ketone 37, presumably via the enolic tautomer 38 (Scheme 10).93–95 The first asymmetric synthesis of 2 and 3 was later achieved by photooxygenation of enantio-enriched allyl alcohols followed by hydroperoxide rearrangement.96–98 Peroxyketal structures related to 1–3 were also synthesised by Fattorusso and co-workers using manganese(III) acetate catalysed addition of molecular oxygen to β-ketoester and olefin substrates.99,100
Scheme 8 Synthesis of 6-epi-plakortolide E (rac-25) by [4 + 2] cycloaddition of singlet oxygen. (a) O2, rose bengal, CH2Cl2, MeOH, 500 W lamp, 0 °C, 6 h.
Scheme 9 Conversion of natural plakortolide P (14) to plakortoperoxides A1 (15) and A2 (16) by [4 + 2] cycloaddition of singlet oxygen. (a) O2, rose bengal bis(triethylammonium) salt, CH2Cl2, 500 W lamp, 5 °C, 3 h, 62%.
Scheme 10 Snider's racemic synthesis of chondrillin (2) and plakorin (3) by [4 + 2] oxygenation. (a) O2, rose bengal lactone, 275 W lamp, CH2Cl2, MeOH, r.t., 12 h, 73%; (b) TsOH·H2O, MeOH, r.t., 80 h, 98%.
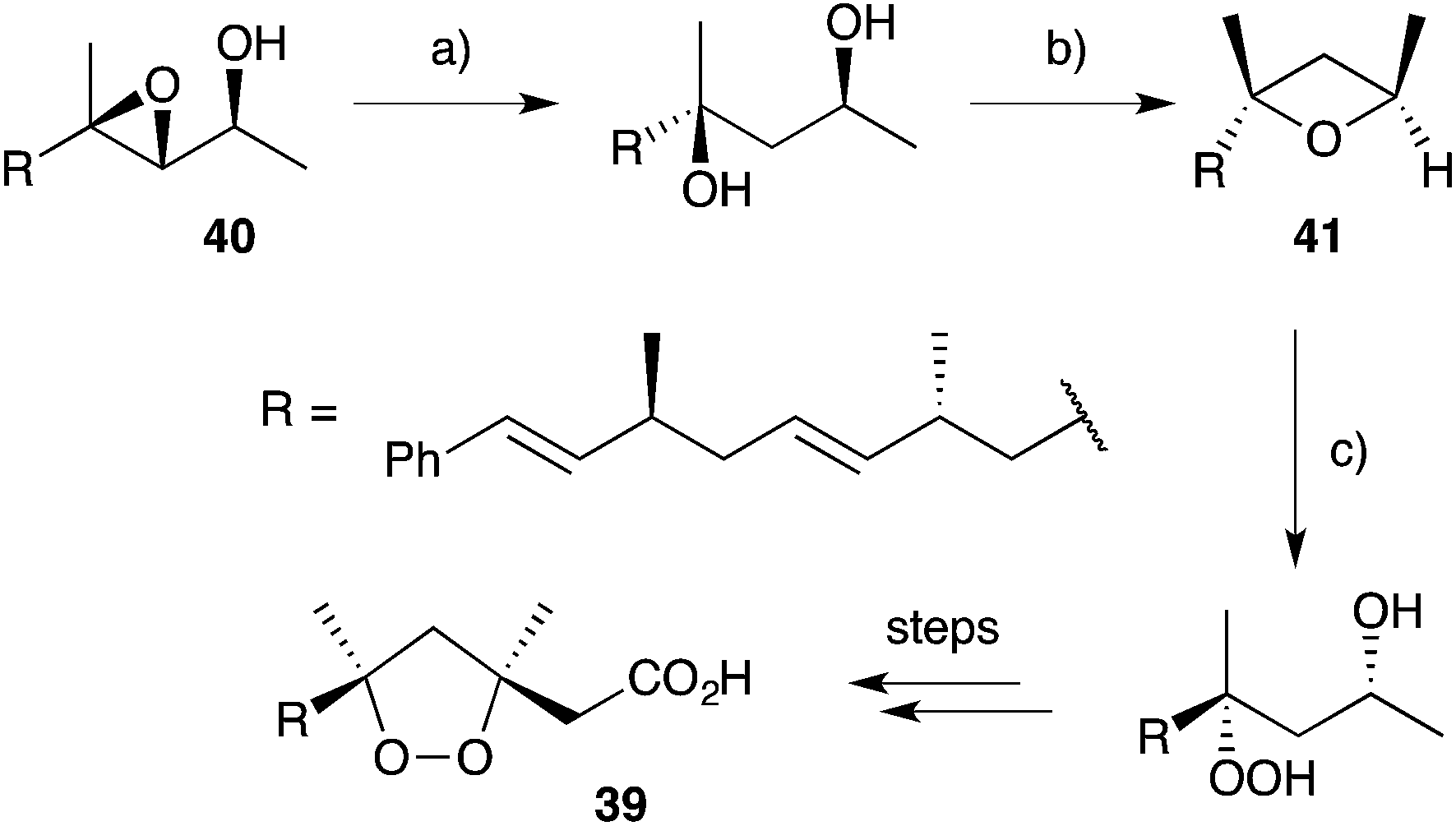
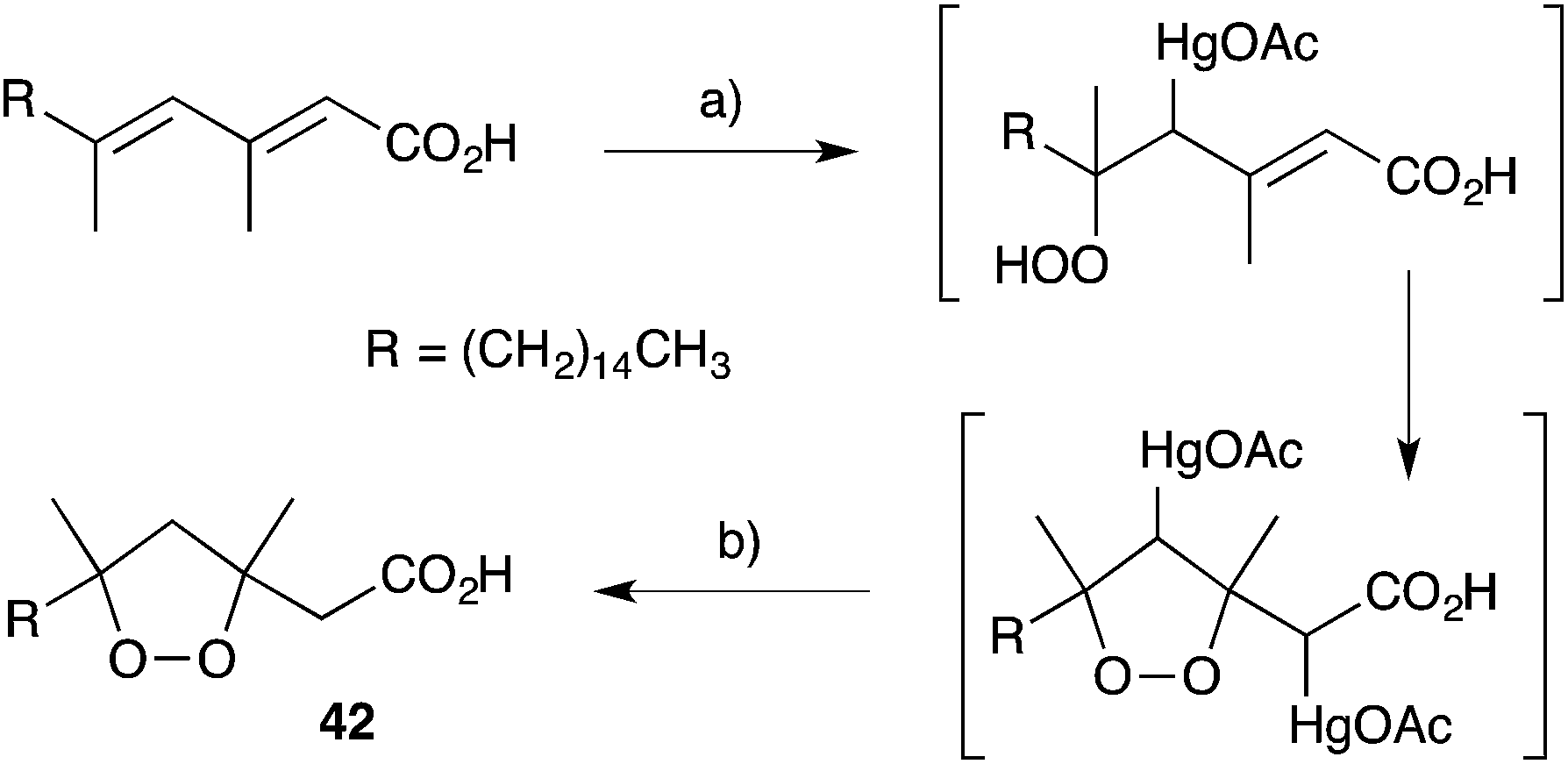
Other methods developed for the synthesis of peroxide scaffolds have involved the use of hydrogen peroxide rather than molecular oxygen.101–103 Dussault completed the synthesis of a series of plakinic acid A (13) stereoisomers (including 39) by advancing enantio-enriched α-hydroxyepoxides such as 40 (accessed by Sharpless asymmetric epoxidation of the corresponding allyl alcohol) to oxetane 41, before stereoselective ring opening with hydrogen peroxide (Scheme 11).104,105 Ring closure and acetate homologation were then achieved over a number of steps. Earlier, Bloodworth developed an effective double peroxymercuration–demercuration strategy for the synthesis of 1,2-dioxolanes, including a natural saturated analogue (42) of the plakinic acids (Scheme 12).106 Most recently, Deng and co-workers developed an enantioselective and organocatalytic method for the peroxidation of unsaturated ketones and aldehydes using cinchona alkaloid derivatives.107–109
Scheme 11 Synthesis of an isomer (39) of plakinic acid A (13). (a) Red-Al, THF, 0 °C, overnight, 97%; (b) tBuOK, TsCl, THF, r.t., 3 h, then NaH, 0 °C, overnight, 72%; (c) H2O2, TMSOTf, Et2O, THF, −78 °C, 45 min.
Scheme 12 Synthesis of a plakinic acid analogue (42) by peroxymercuration–demercuration. (a) H2O2, H2O, Hg(OAc)2, then (b) NaBH4, NaOH.
3. Metabolites derived from peroxide rearrangement
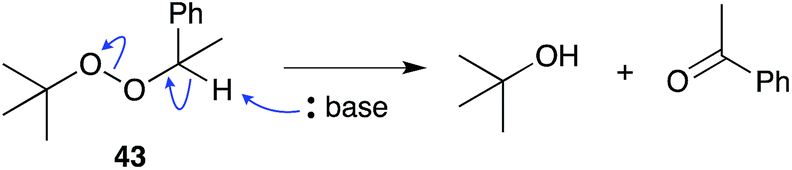
The base-catalysed rearrangement of dialkyl peroxides was first reported in the early 1950s when Kornblum and DeLaMare discovered that 1-phenylethyl tert-butylperoxide (43) decomposes in the presence of catalytic KOH, KOEt or piperidine to yield acetophenone and tert-butanol.110,111 The reaction was rationalised by abstraction of hydrogen at the less substituted α-peroxy carbon, followed either by stepwise or concerted elimination across the C–O bond. The instability of peroxide scaffolds, including 1,2-dioxanes, to base-mediated rearrangement has lead to the occurrence and isolation of many rearranged endoperoxide metabolites related to 1–3.
3.1 Untenone A and plakortic acid
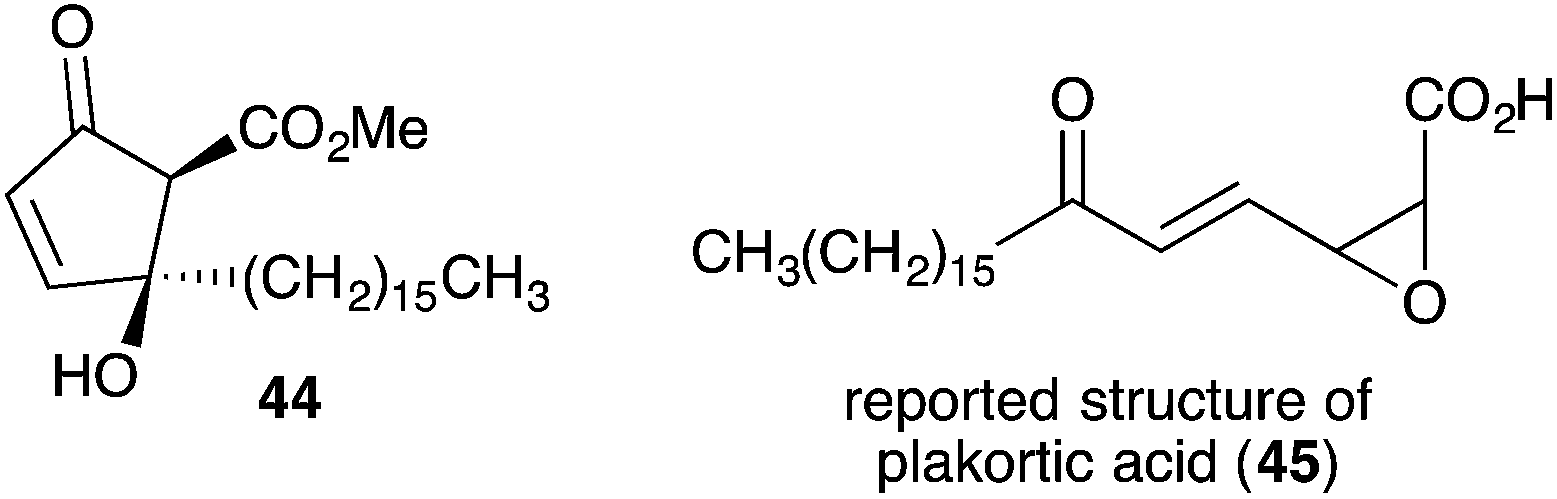
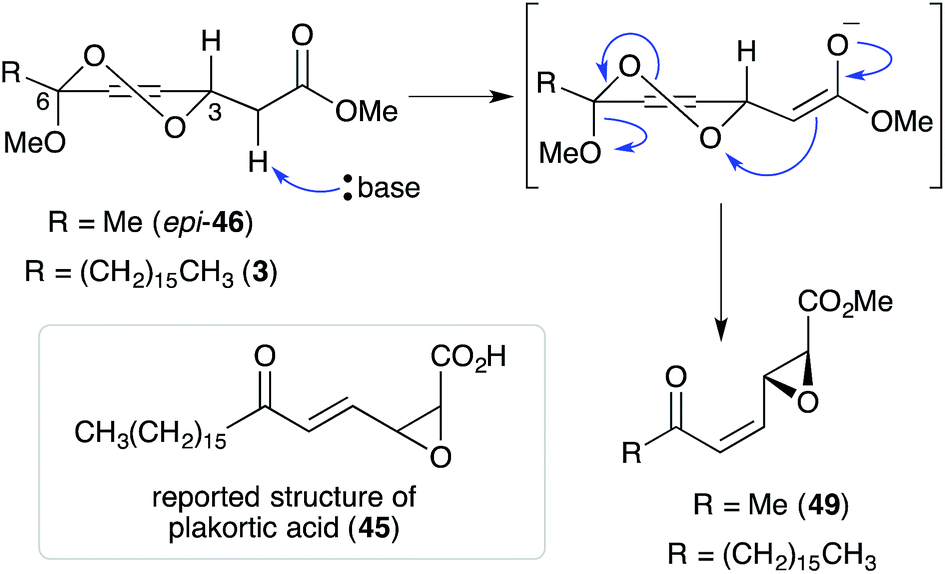
Untenone A (44)112 and plakortic acid (45)113 were each isolated from Plakortis marine sponges in 1993 and 2001, respectively. Although 44 and 45 appear to be quite different, both are closely related to chondrillin (2) and plakorin (3), sharing the same elemental composition and oxidation pattern of the central carbon chain. After completing a racemic total synthesis of 2 and 3 in the early 1990s (Section 2.2), Snider studied the base-catalysed structural rearrangements of the two diastereomers and found that they decompose via distinct pathways.95 The findings of Snider's study have, in retrospect, provided insight to the biosynthesis of 44 and 45.114
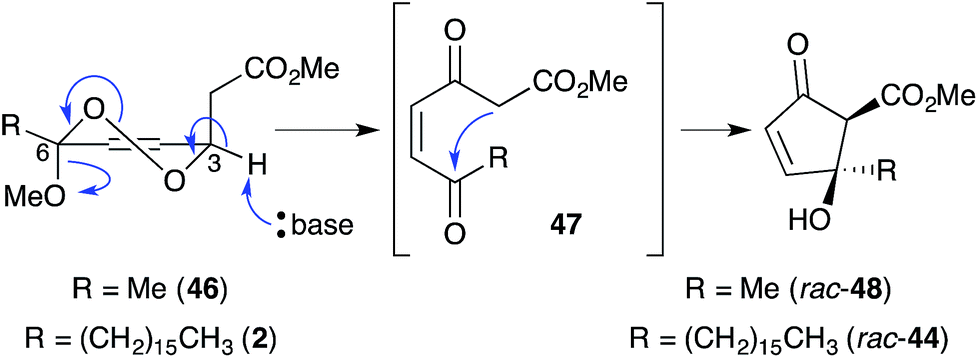
In a model system, Snider found that the rearrangement of peroxyketal heterocycles 46 and epi-46 (analogues of 2 and 3) is critically dependent on their relative stereochemistry. As shown in Scheme 13, the conformation in which the C6 alkyl substituent of 46 (a methyl analogue of 2) is pseudoequatorial places the C3 dioxane hydrogen antiperiplanar to the adjacent 1,2-dioxane bond. This facilitates Kornblum–DeLaMare rearrangement to yield methyl ester 47 (R = Me) before rapid Dieckmann condensation to give cyclopentenone rac-48, a methyl analogue of untenone A (44). In contrast, the conformation in which the C6 alkyl substituent of epi-46 (a methyl analogue of 3) is pseudoequatorial places the C3 acetate group antiperiplanar to the O–O bond (Scheme 14). This allows nucleophilic attack of the α-keto acetate carbon to the peroxide yielding oxirane 49, a structural analogue of plakortic acid (45).115 It is therefore apparent that natural 44 and 45 are likely to be derived from the rearrangement of 2 and 3, respectively. It is also important to note that 44 was isolated as a racemate112,116,117 and 45 in enantio-enriched form,113 which is consistent with the presented theory of biogenesis.
Scheme 13 Kornblum–DeLaMare rearrangement and Dieckmann condensation of chondrillin (2) analogue 46.
Scheme 14 Base-mediated decomposition of plakorin (3) analogue epi-46.
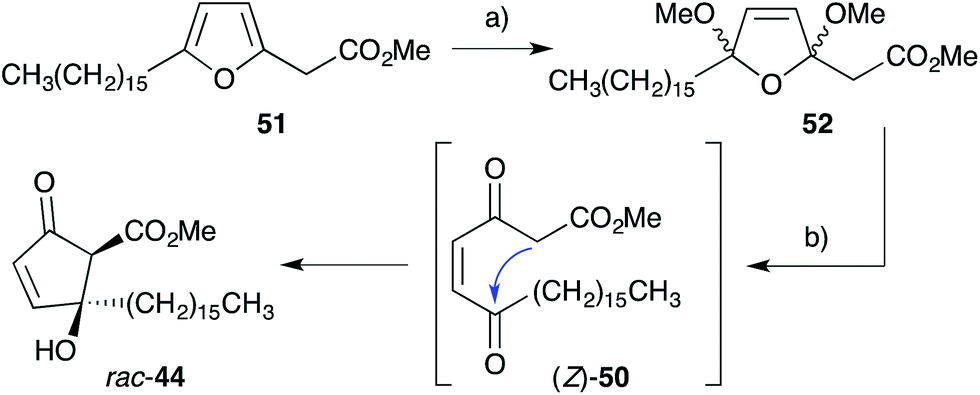
Inspired by Snider's work, Whitehead developed a synthesis of untenone A (44) by in situ generation of methyl ester (Z)-50, the postulated biosynthetic intermediate linking 2 and 44 (Scheme 15).118–120 Thus, oxidation of synthetic plakorsin A (51, Section 4.4)113 gave bismethyl acetal 52, which on hydrolysis underwent Dieckmann condensation yielding rac-44, directly. After developing an efficient total synthesis, Whitehead's study into the reactivity of rac-44 was pivotal in elucidating biosynthesis of the manzamenone family of oxylipin dimers (Section 3.2). Both racemic and asymmetric total syntheses of untenone A (44) were also accomplished by Takeda,121 Asami116 and Yamada;117 with each approach targeting modification of 5-membered carbocycles introduced in the early stages of synthesis.
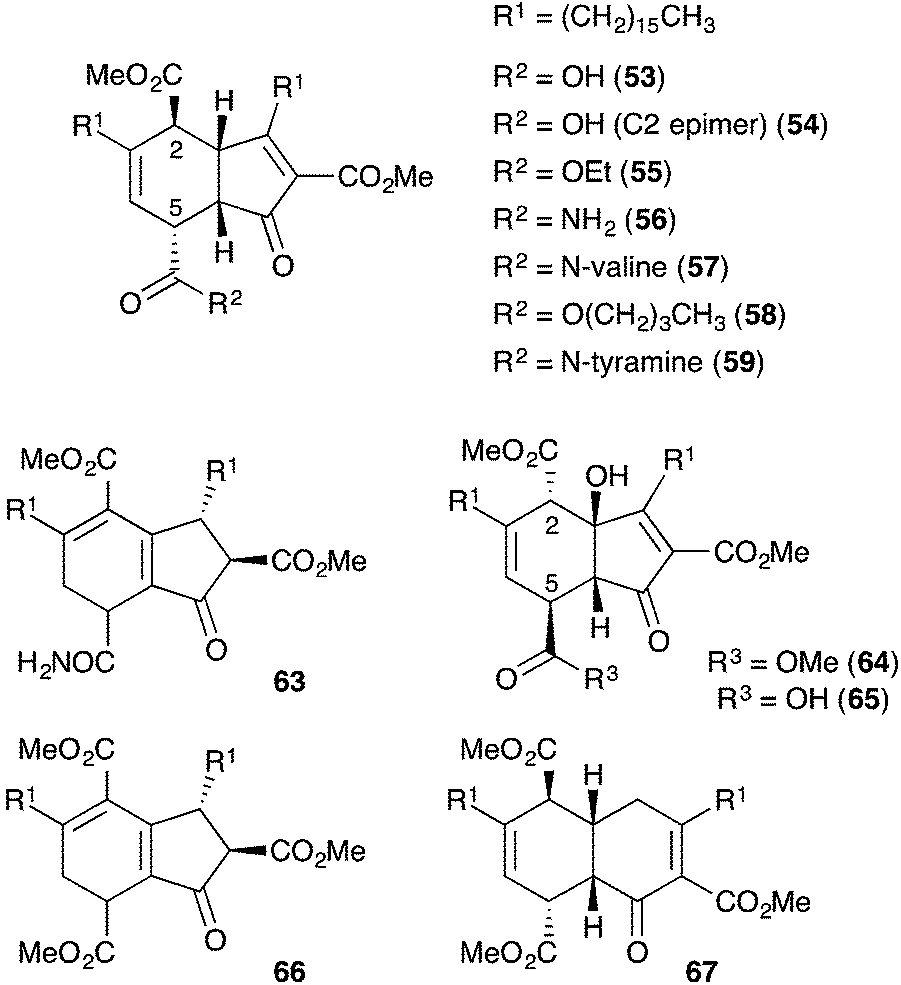
Manzamenones A–F (53–58)122 and H (59)123 were isolated by Kobayashi and co-workers from Plakortis sponges collected near Okinawa, Japan in 1992 and 1993 (Fig. 2). They share a common [4.3.0]-carbobicyclic structure differing only by the carboxy substituent at C5. Manzamenone A (53) was found to be a potent inhibitor of DNA polymerase β124,125 and manzamenones B (54) and E (57) are inhibitors of T-cell protein tyrosine phosphatase.126
Fig. 2 Reported structures of manzamenone A–F (53–58), G (67), H (59), J (63) and L–N (64–66).
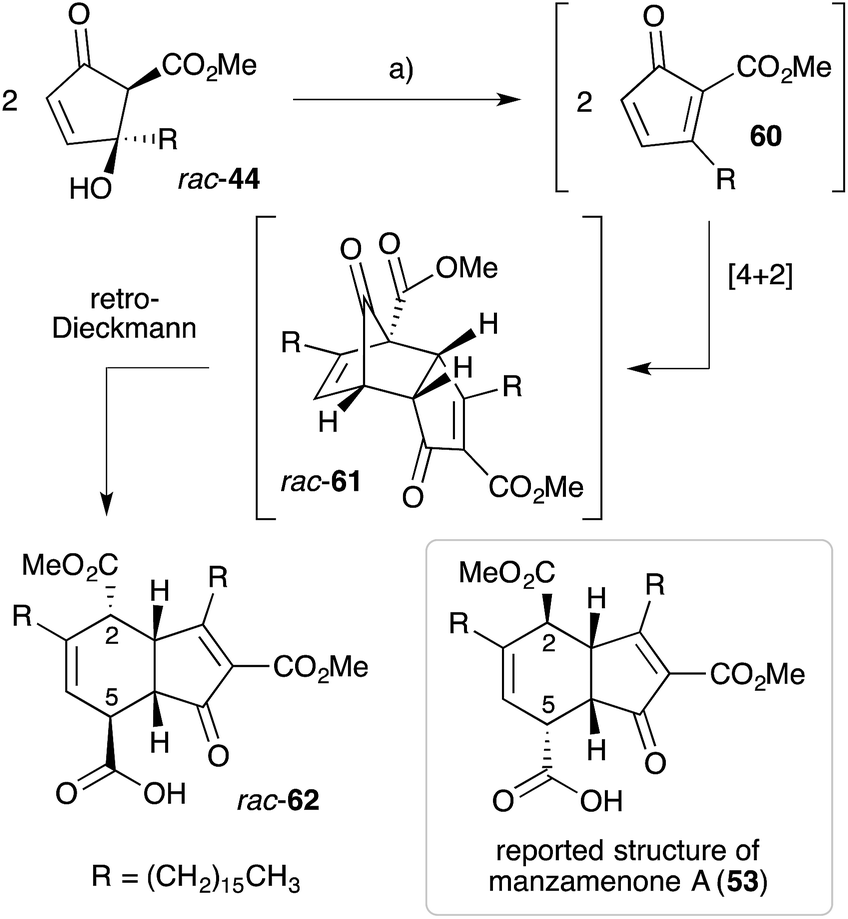
Initial speculation on the biosynthesis of 53–58 considered that the cyclohexene ring system may be constructed in a hetero-bimolecular [4 + 2] cycloaddition of untenone A (44) and a suitable diene counterpart.122,123 However, Whitehead postulated that 53–58 are in fact homodimers of untenone A (44).118–120,127 Heating a neat sample of synthetic rac-44 (Section 3.1) to it's melting point (72 °C) caused dehydration to the anti-aromatic cyclopentadiene 60 followed by rapid [4 + 2] homo-dimerisation to give endo-adduct rac-61in situ (Scheme 16). Retro-Dieckmann fragmentation of the bridgehead carbonyl with uptake of water as an incident nucleophile yielded rac-62, whose relative stereochemistry was determined by X-ray crystallography and detailed NMR analysis.120 The spectral data obtained for 62 matched that of natural manzamenone A, thus prompting stereochemical reassignment of the original structure (53) at C2 and C5. Whitehead also completed the synthesis of manzamenones C and F from rac-62 resulting in a similar revision of the original structures 55 and 58.128 The apparent pre-disposition of 44 to undergo dehydrative dimerisation yielding 62 demonstrates beyond doubt that this process is responsible for the occurrence of 53–58 in the extracts of marine sponges.129
Scheme 16 Whitehead's biomimetic synthesis and structural elucidation of manzamenone A (62). (a) Neat, 72 °C, 24 h, 48%.
Manzamenones J (63)130 and L–N (64–66),131 also isolated from Plakortis sponges, were determined to have the same carbocyclic structure as 53–58 with a different oxidation pattern of the central core and thus, are likely to be directly related (Fig. 2). However, manzamenone G (67)123 was reported as an unusual structural analogue with one carbon more than that expected for a dimer related to chondrillin (2). The structure of manzamenone G (67)123 was only tentatively assigned and its biosynthetic origin remains unclear.
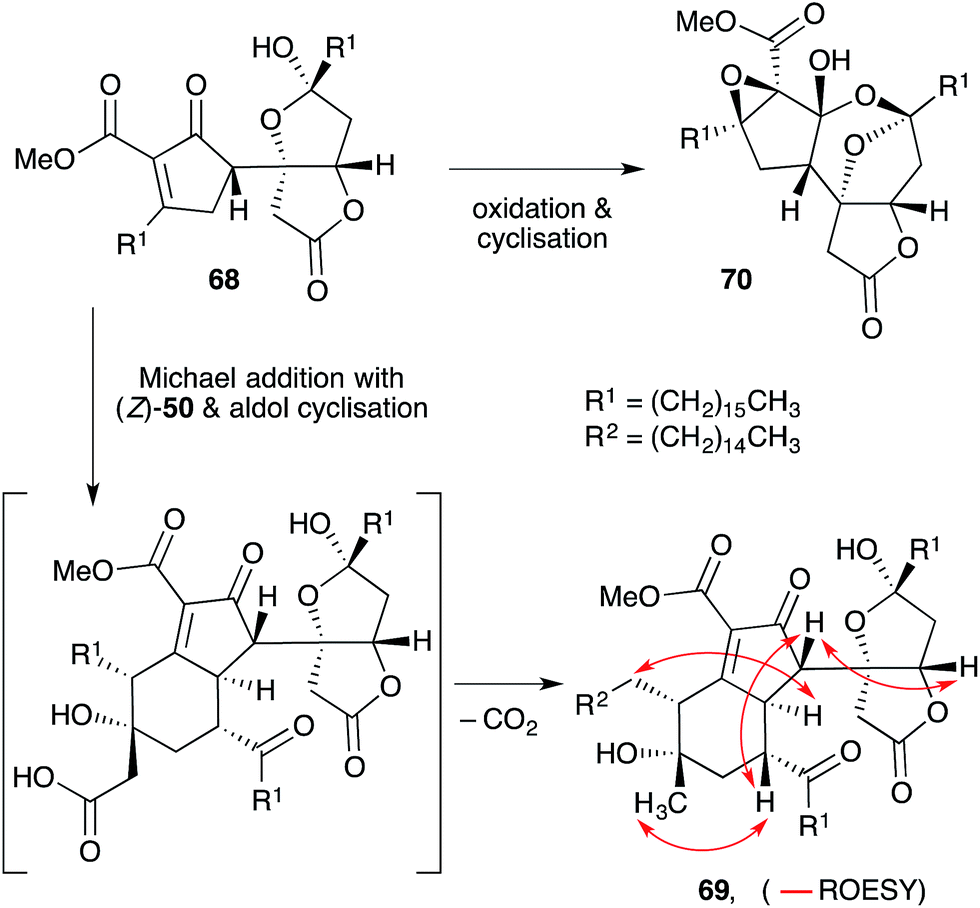
Manzamenone K (68),130,132 O (69)133 and untenolide A (70)134 also appear to be related to 2 and 3, yet are structurally distinct from 53–58 and 63–67 (Scheme 17). The central structure common to each natural product (68–70) was verified by X-ray crystallography134 and ROE spectroscopy (selected ROESY correlations of compound 69 as shown in Scheme 17);133 and found to consist of two structural elements that appear to be derived from 2 or 3 linked by a single C–C bond (most clearly seen in 68). The compounds were isolated as racemic or presumably pseudo-racemic, suggesting that they may arise from non-enzymatic dimerisation of an achiral intermediate similar to manzamenones A–F (53–58) and H (59). However, if derived from 2 and 3 it appears that a single reduction event has occurred at some stage during construction of the central scaffold. Kobayashi reported that 70 is likely to be related to 68 by oxidation and cyclisation; and 69 is likely to be related to 68via condensation with intermediate (Z)-50 (derived from 2).133–135 Despite the complexity of these unique oxylipin natural products, no further studies have commented on the biosynthesis or attempted chemical synthesis of 68–70.
Scheme 17 Structures and postulated conversion of manzamenone K (67) to manzamenone O (69) and untenolide A (70). Selected ROESY correlations of compound 69 shown as red arrows.
3.3 Plakoridines A–C
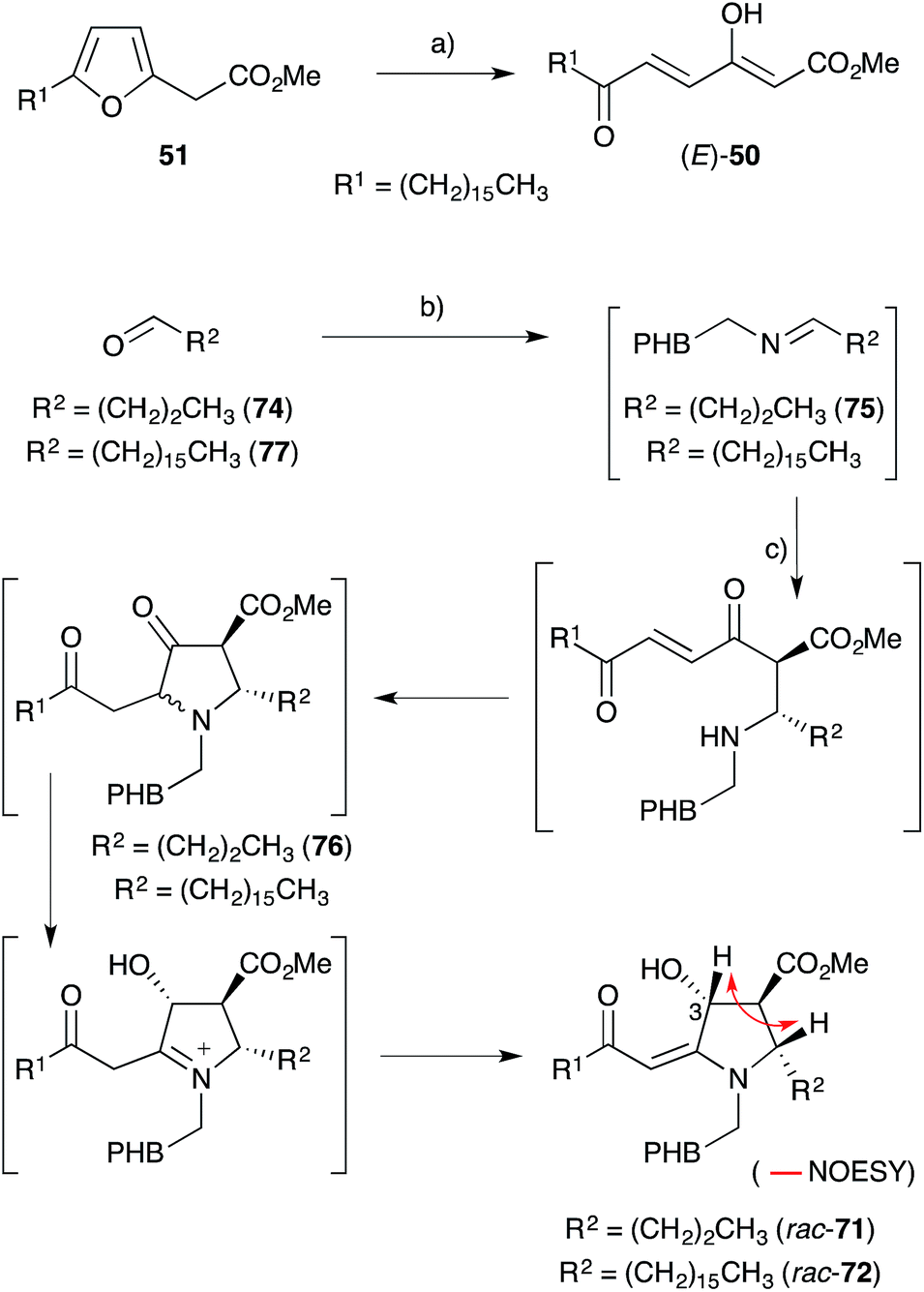
Kobayashi and co-workers also isolated three fatty acid containing alkaloids from Plakortis marine sponges collected near Okinawa. Plakoridine A (71),136 found to be an inhibitor of DNA polymerase,124 and the less active compounds plakoridines B (72)130 and C (73),137 each contain a central carbon chain with a C16 aliphatic tail common to all peroxyketal-derived natural products related to chondrillin (2, Scheme 18).
Scheme 18 Whitehead's biomimetic synthesis of plakoridine A (71) and B (72) from plakorsin A (51). Key NOESY correlation of compound rac-71 shown as a red arrow. (a) Br2, Me2CO, H2O, −20 °C to −10 °C, 6 h, 63%; (b) tyramine, CDCl3, r.t., 3 h, then MgSO4, r.t., 30 min, then (c) (E)-50, r.t., 11 d, 43% (beginning with 74) and 36% (beginning with 77). PHB = p-hydroxybenzyl.
Whitehead postulated that 71 and 72 are biosynthetically related to (E)-50,138 an intermediate derived from plakorsin A (51).139 In a single experiment, tyramine and aldehyde 74 condensed to generate imine 75in situ before addition of (E)-50 (itself prepared by bromine-mediated oxidation of synthetic 51) to give rac-71 as a 3:1 mixture of C3 epimers (Scheme 18).127,138,140 Over 11 days at room temperature, 75 and (E)-50 were presumed to undergo an intermolecular Mannich reaction followed by Michael ring closure to afford heterocycle 76. An internal redox process, resulting in reduction of the cyclopentanone carbonyl and oxidation of the neighbouring α-amino methine, then followed to yield rac-71. Relative configuration of the pyrrolidine ring was established through the analysis of vicinal coupling constants and NOE spectroscopy (key NOESY correlation of compound rac-71 shown in Scheme 18). Hexadecyl aldehyde (77) was also advanced to rac-72 using this method, demonstrating that natural 71 and 72 are most likely related to 51. Furthermore, Ma's asymmetric synthesis of (2S,3S,4R)-71 in 2000 showed that natural plakoridine A (71) was isolated as a racemate,141 which is consistent with Whitehead's theory of biogenesis. Stafford also reported a racemic synthesis of a plakoridine A lactam in 1995.142
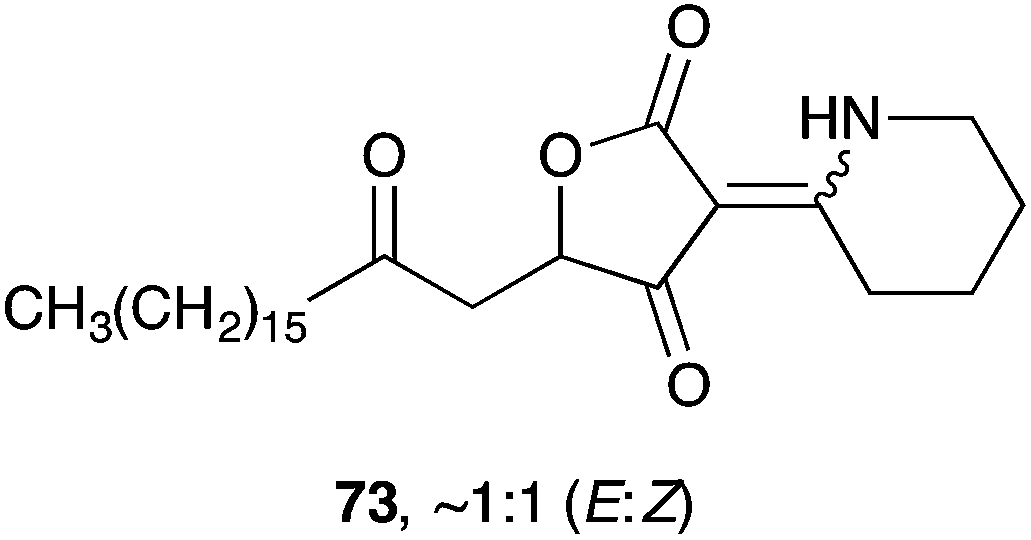
Plakoridine C (73) is structurally distinct from 71 and 72; and appears to be derived from an intermediate related to 2 or 3 by addition of piperideine or δ-lactam.137 To date, there is no published work on the synthesis of 73.
3.4 Gracilioethers B–D, plakilactones A–H and related compounds
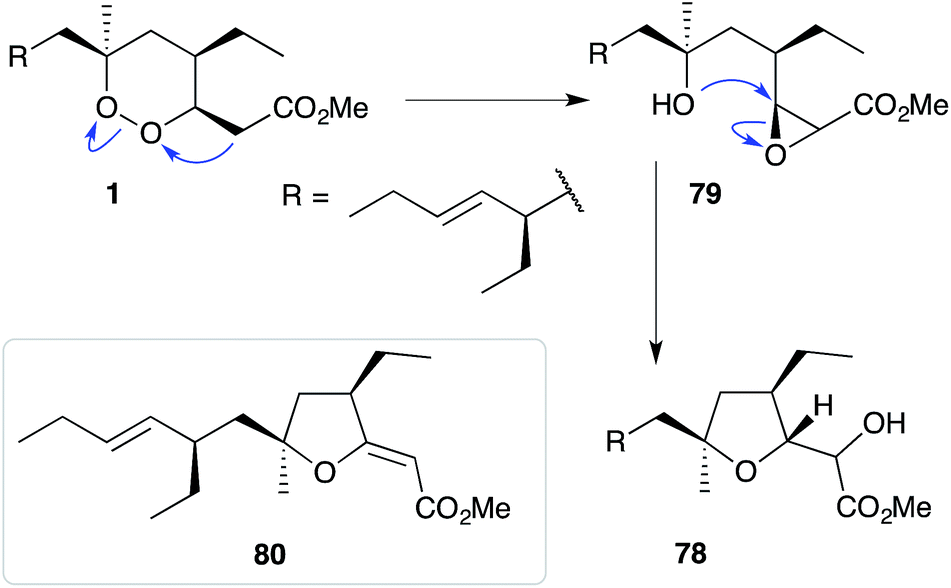
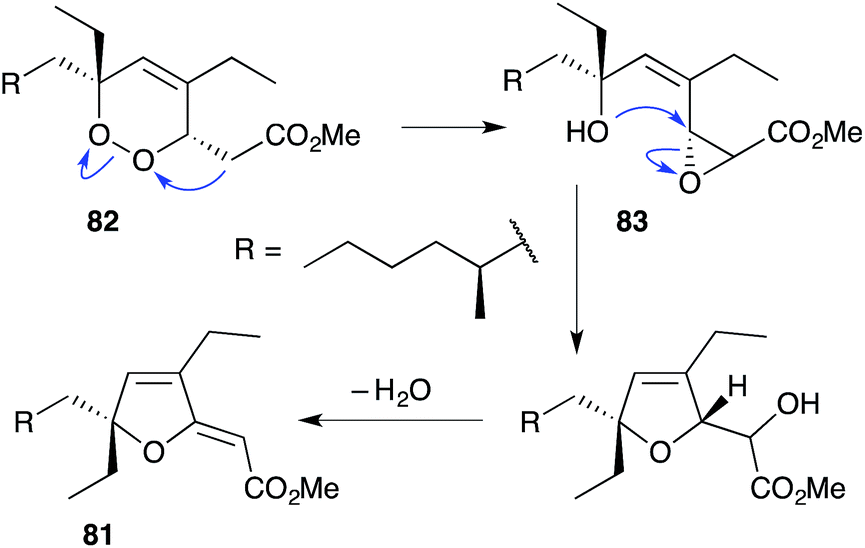
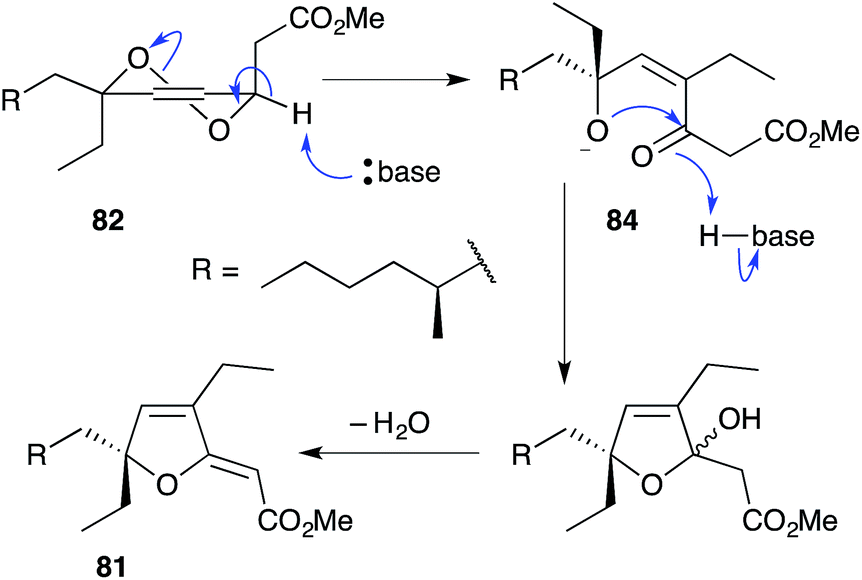
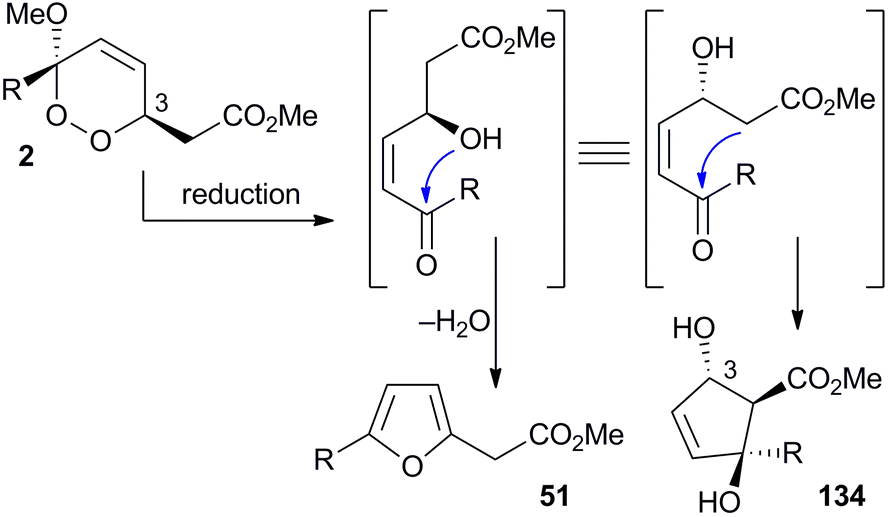
The base-catalysed rearrangements of dialkyl peroxides related to and including plakortin (1), which are structurally distinct from 2 and 3, yield furan-containing polyketide metabolites by ring contraction of the 1,2-dioxane motif. Faulkner discovered that treating 1 with NaOMe in MeOH induced rearrangement to tetrahydrofuran 78, presumably by ring closure of epoxide intermediate 79 (Scheme 19).1 This general reaction of unsaturated dialkyl peroxides has been used in the synthesis of polyether tetrahydrofuran compounds;143 and 78 has itself been linked to the sponge metabolite 80, as a likely biogenetic precursor.2 By extension, Faulkner rationalised biosynthesis of the [2(5H)-furanylidene]ethanoate (furanylidene) metabolite 81 from peroxide 82 through formation of epoxide 83 (Scheme 20).144 However, Snider's work on the stereochemical dependence of base-catalysed rearrangements of 4,5-unsaturated-1,2-dioxane peroxyketals (Section 3.1) suggests that the conversion of 82 to 81 is likely to proceed by Kornblum–DeLaMare rearrangement via hydroxy-β-ketoester 84 (Scheme 21).114 We believe a similar mechanistic course is most likely in the biosynthesis of gracilioethers B–D,145,146 spongosoritin A,147,148 spiroplakortone,149 glanvillic acids A, B49 and related compounds.51,150–152
Scheme 19 An observed ring contraction of plakortin (1).
Scheme 20 Faulkner's hypothesis for ring contraction of peroxide 82 to furanylidene 81.
Scheme 21 Postulated Kornblum–DeLaMare rearrangement of peroxide 82 to furanylidene 81.
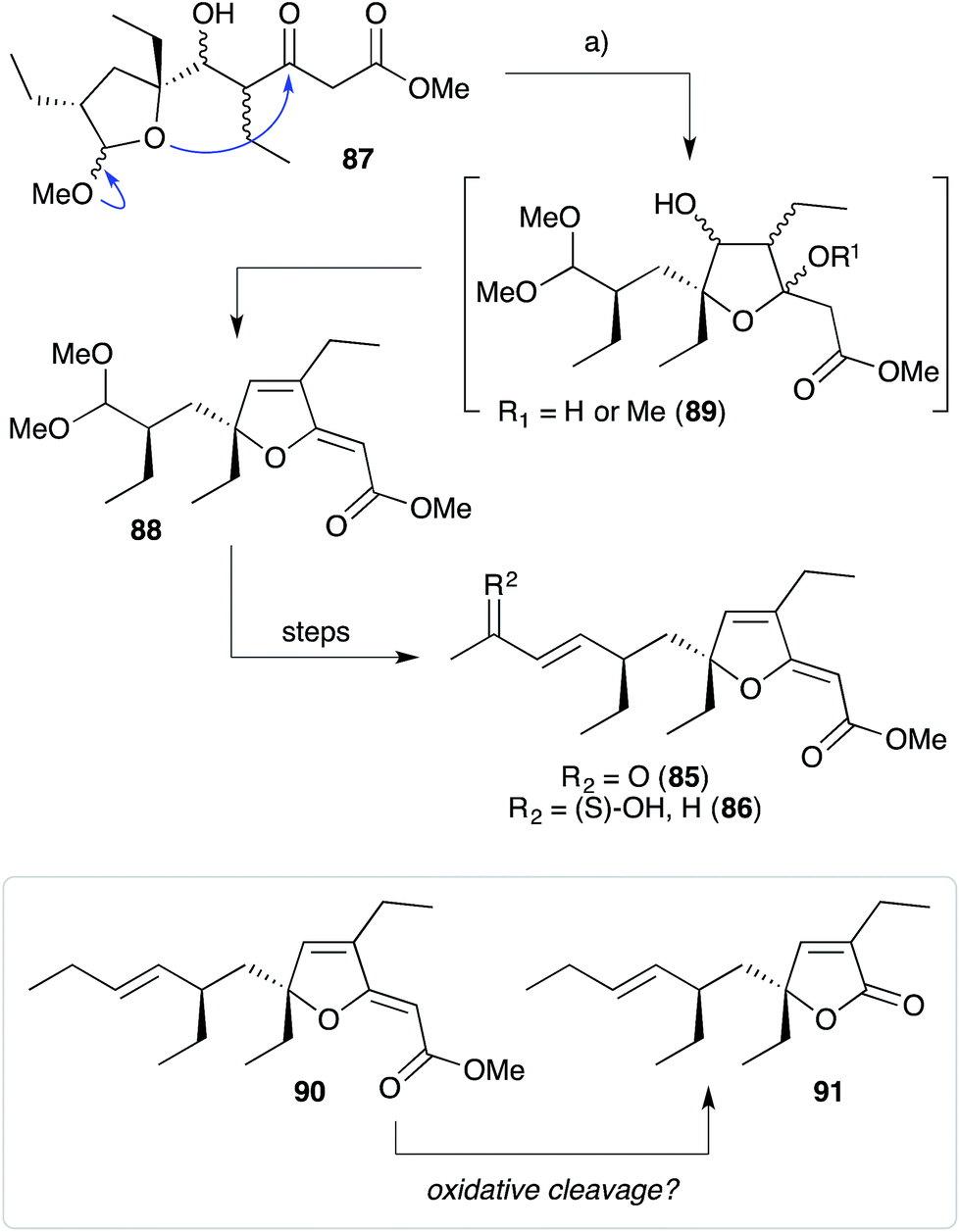
Perkins has recently demonstrated the utility of targeting such hydroxy-β-ketoester intermediates in the synthesis of furanylidene compounds and has reported the total synthesis of gracilioethers B (85) and C (86) using this approach (Scheme 22).153 Methyl acetal 87 (a synthetic analogue of 84) was converted to furanylidene 88 by transacetalisation to intermediate 89 and dehydration to the conjugated system. 85 and 86 are known agonists of peroxisome proliferator-activated receptor γ (PPARγ);146 and the related metabolite des-hydroxygracilioether C (90) was found to be cytotoxic against HCT-116 cells (human colon carcinoma).31 Ohira and co-workers completed a racemic total synthesis of 90 in 2005 using reactions of alkylidenecarbenes.154
Scheme 22 Biomimetic total synthesis of gracilioethers B (85) and C (86); and a possible biogenetic link to the plakilactone metabolites via oxidative cleavage of the furanylidene heterocycle. (a) AcCl, MeOH, CH(OMe)3, 20 °C, 15 h, 74% over 2 steps (first step not shown).
Plakilactones A–H146 and des-hydroxyplakilactone B (91) were isolated from Plakinastrella mamillaris in 2012 along with a number of furanylidene natural products, including gracilioethers B (85), C (86) and des-hydroxygracilioether C (90). The structural similarity of the plakilactone butenolides and gracilioether furanylidenes would appear to suggest that they are related by oxidative cleavage of the furanylidene enol ether, although this possible relationship is yet to be demonstrated (Scheme 22).
3.5 Gracilioethers A, E–K and hippolachnin A
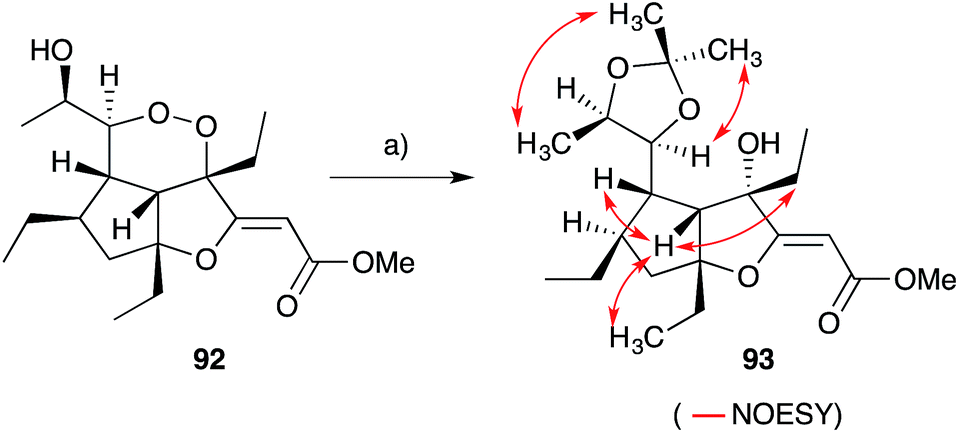
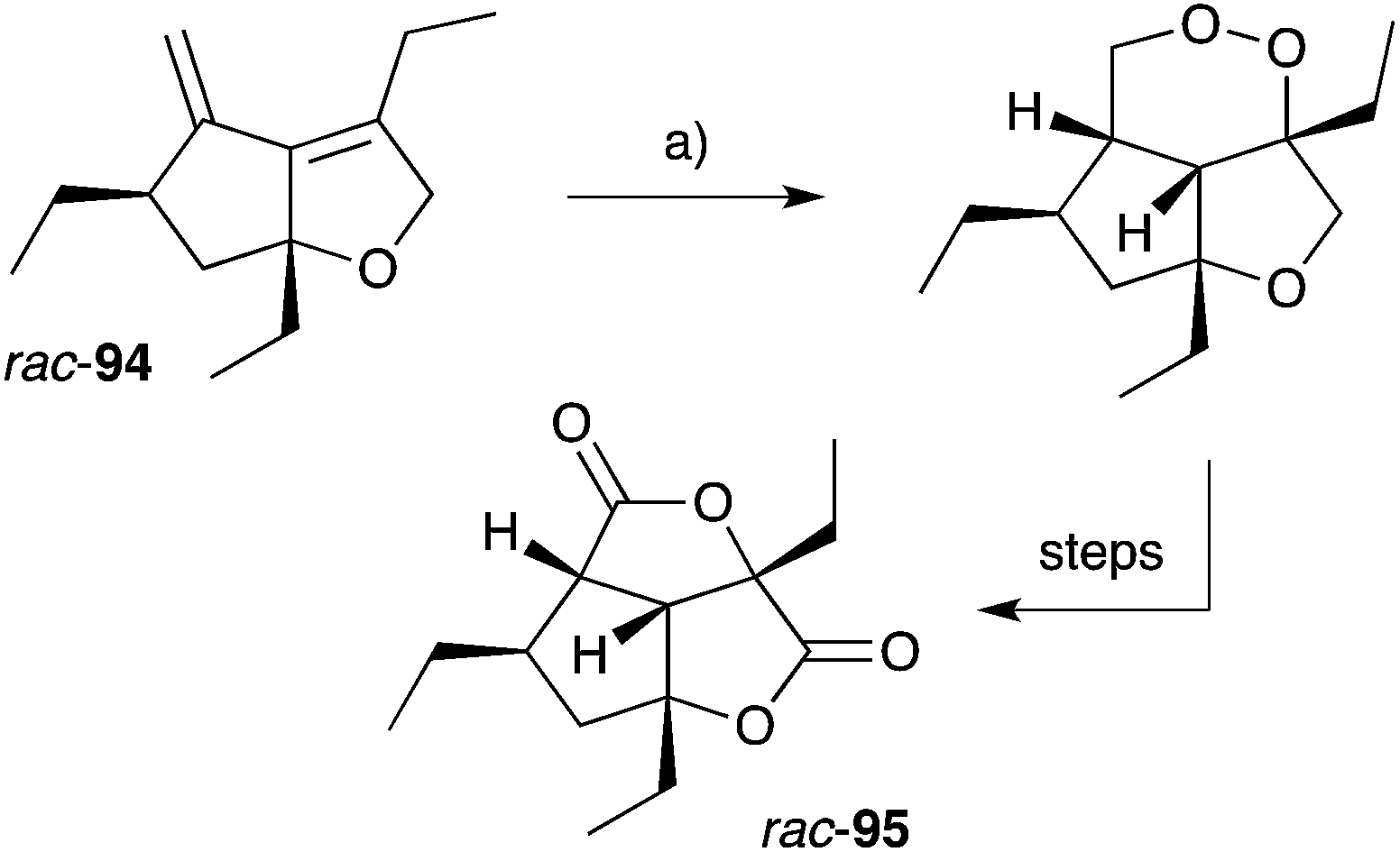
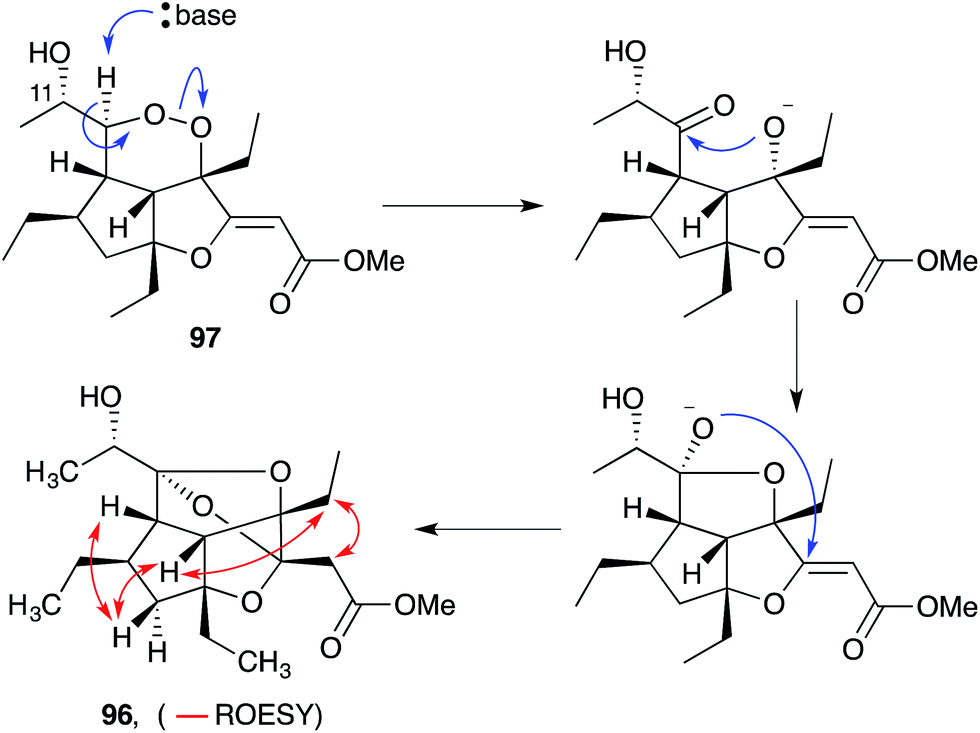
Gracilioethers A145 and E–K155,156 are complex polycyclic metabolites that also appear to be oxidative adducts of the furanylidene heterocycles, although the biosynthetic origins of these compounds remains unclear. The structure and absolute configuration of gracilioether A (92) was determined by chemical derivatisation and detailed NMR analysis of the resulting acetonide 93 (Scheme 23); and the structures of related natural products have been assigned accordingly. Wong recently demonstrated [4 + 2] addition of singlet oxygen to a reactive bicyclic diene (rac-94), forging the peroxy-tricyclic core of 92, before completing total synthesis of gracilioether F (rac-95, Scheme 24).157 Brown also completed a total synthesis of 95 in 2014 using a ketene–alkene [2 + 2] cycloaddition reaction and Baeyer–Villiger oxidation; and a carboxylic acid directed C–H oxidation to install each of the lactone rings.158 The related metabolite gracilioether K (96), whose structure was determined through detailed NMR analysis (selected ROESY correlations of compound 96 shown in Scheme 25) and by application of Mosher's method, is believed to arise from Kornblum–DeLaMare ring contraction and oxa-Michael cyclisation of (11S)-gracilioether A (97, Scheme 25),156 which itself may be constructed from gracilioether C (86) by inclusion of molecular oxygen.
Scheme 23 Structural and stereochemical elucidation of gracilioether A (92) through chemical derivatisation and detailed NMR analysis. Selected NOESY correlations of compound 93 shown as red arrows. (a) Zn, Et2O, AcOH, r.t., overnight; then (CH3)2C(OCH3)2, PPTS, CH2Cl2, r.t., overnight.
Scheme 24 Wong's synthesis of racemic gracilioether F (rac-95) by [4 + 2] oxygenation of diene rac-94. (a) O2 (bubbled), methylene blue, CH2Cl2, sunlamp (200–300 W), 0–5 °C, 2 h.
Scheme 25 Postulated base-catalysed rearrangement of (11S)-gracilioether A (97) to gracilioether K (96). Selected ROESY correlations of compound 96 shown as red arrows.
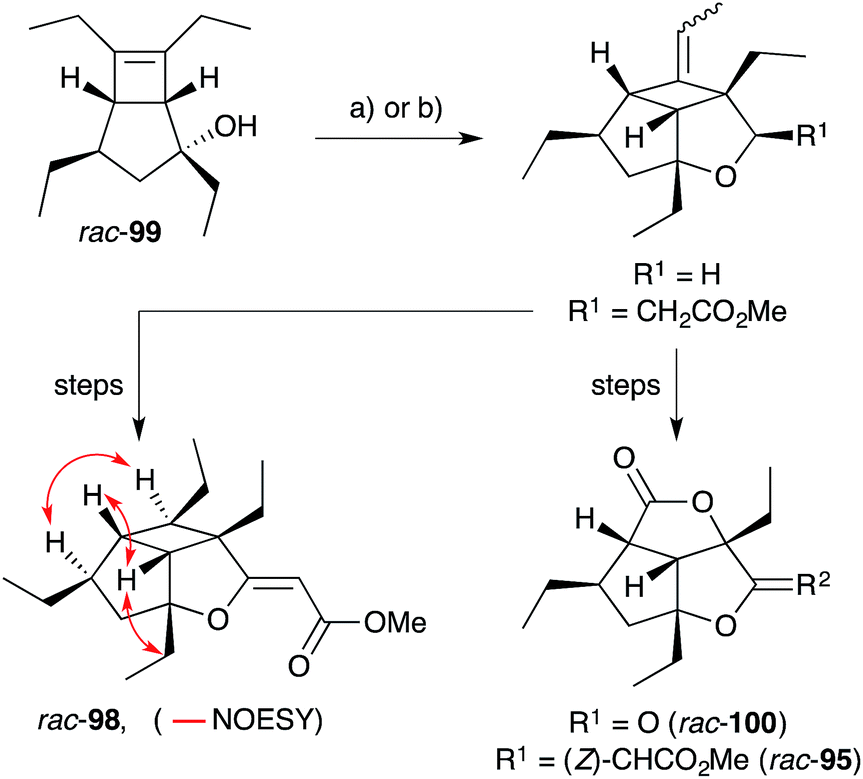
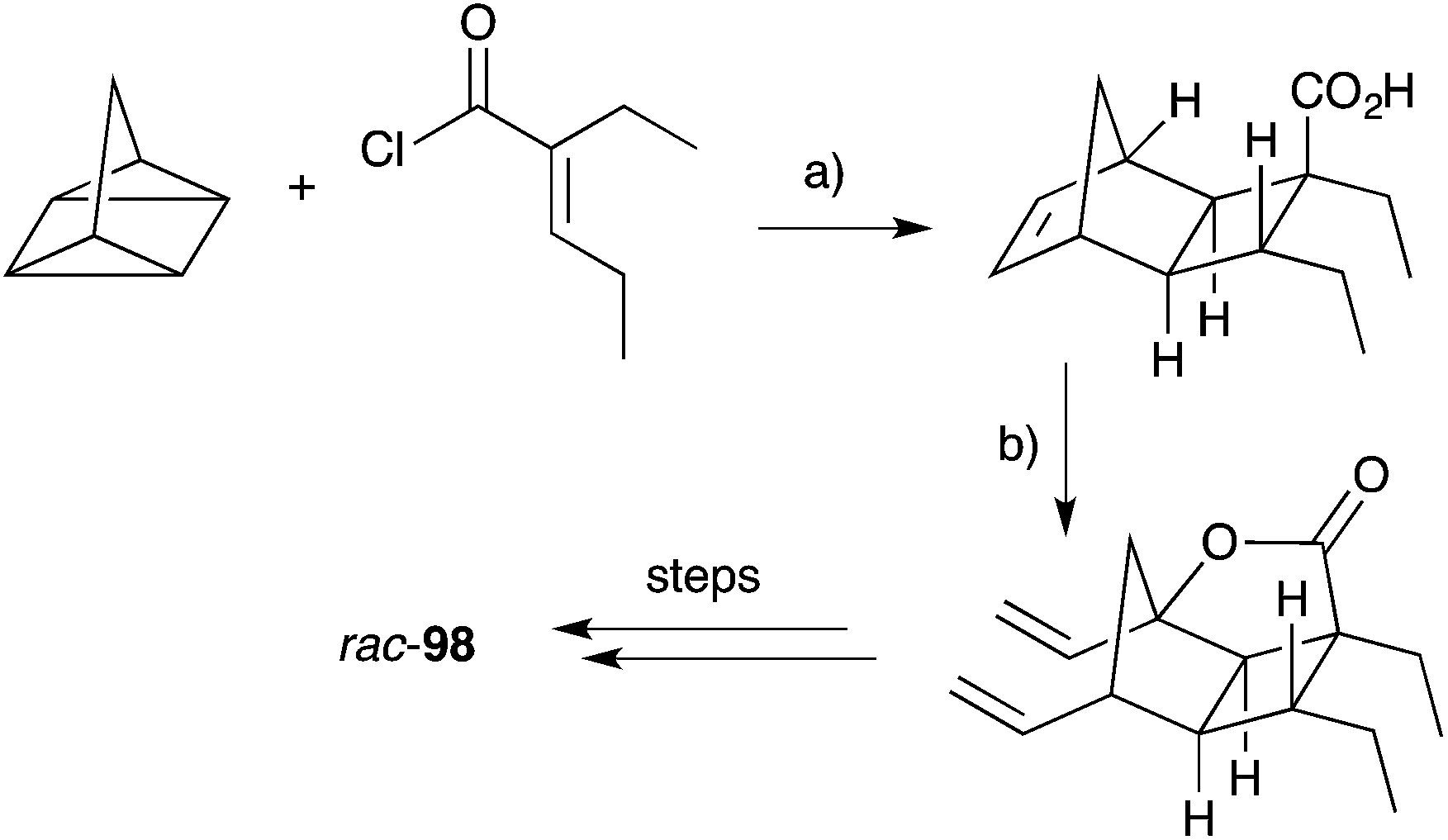
Hippolachnin A (98), isolated from Hippospongia lachne in 2013, also bears a unique tricyclic structure with a highly substituted cyclobutane ring (Scheme 26).159 It appears that 98 arises biosynthetically from an intramolecular [2 + 2] cycloaddition of 90, which itself was isolated from the same sponge extract.159 In turn, 90 is expected to arise from the dehydrative ring contraction of haterumadioxin A (7) methyl ester.160 Carreira completed the first total synthesis of rac-98 in 2015 constructing a pivotal cyclobutene intermediate 99 (Scheme 26)161en route to the natural product (selected NOESY correlations of natural 98 shown in Scheme 26);159 and later extended the approach to achieve a racemic total synthesis of gracilioethers E (rac-100) and F (rac-95).162 Brown and Wood recently published a collaborative project, which detailed the total synthesis of rac-98 in only six steps through an enabling quadricyclane cycloaddition and late-stage carboxylic acid directed C–H oxidation, similar to that developed in Brown's earlier synthesis of rac-95 (Scheme 27).163 Ghosh has also been successful in accessing the central tricyclic core, effecting a [2 + 2] enone–alkene cycloaddition of a synthetic butenolide related to 90.164
Scheme 26 Carreira's racemic synthesis of hippolachnin A (rac-98), gracilioether E (rac-100) and F (rac-95) from cyclobutene intermediate 99. Selected NOESY correlations of natural 98 shown as red arrows. (a) (CH2O)n, Sc(OTf)3, CHCl3, −78 °C to r.t., 28 h, 61% brsm (for R1 = H); (b) (E)-methyl-3-methoxyacrylate, PPTS, 80 °C, 4.5 days; then BF3·2AcOH, CH2Cl2, THF, r.t., 20 h, 62% over 2 steps (for R1 = CH2CO2Me).
Scheme 27 Brown and Wood's synthesis of racemic hippolachnin A (rac-98) via quadricyclane cycloaddition and carboxylic acid directed C–H oxidation. (a) 140 °C (microwave irradiation), 4 h, then NaOH, r.t., 24 h, 50%; (b) ethylene (1 atm), Grubbs I, CH2Cl2, r.t., 7 h; then PhSO(CH2)2SOPh·Pd(OAc)2, Cr(salen)Cl, p-benzoquinone, dioxane, H2O, 60 °C, 24 h, 64% over 2 steps.
4. Metabolites derived from peroxide reduction
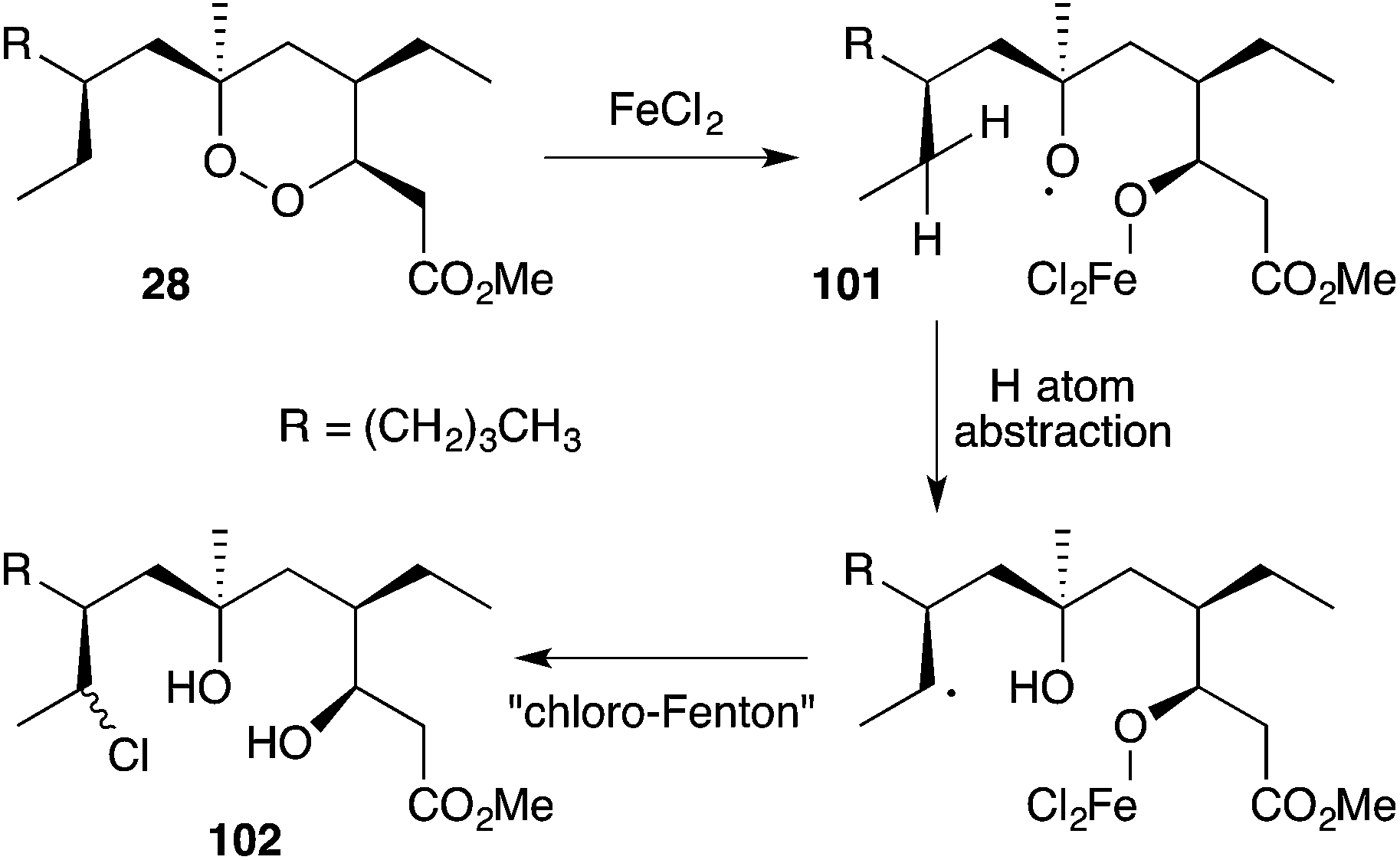
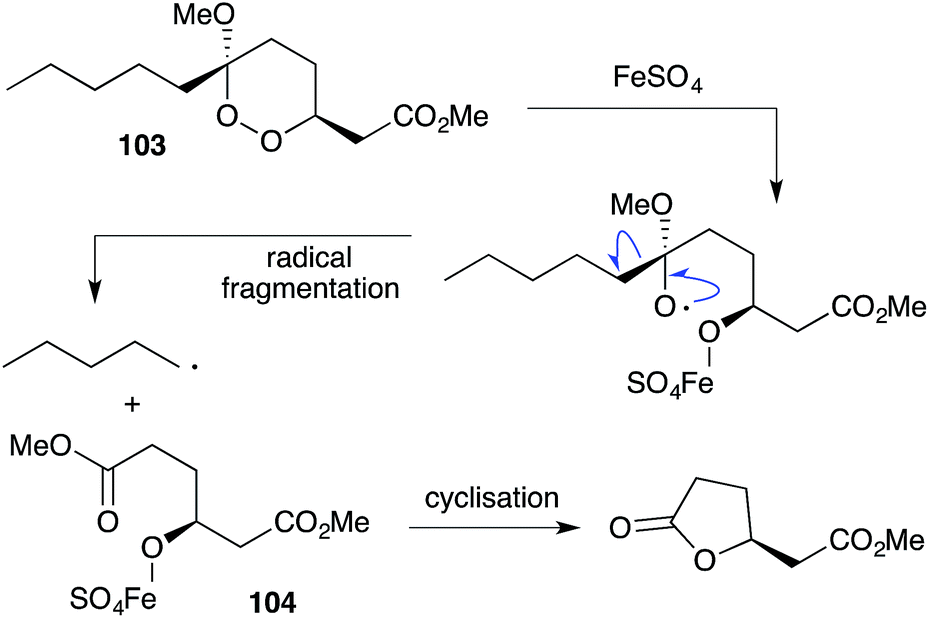
Iron(II)-mediated reduction has been identified as a likely biological mode of action of peroxide metabolites related to 1 and 2. On treatment with FeCl2, 9,10-dihydroplakortin (28) undergoes reductive cleavage of the 1,2-dioxane to give oxyradical 101, followed by H atom abstraction of the branched alkyl chain and “chloro-Fenton”37,165 reaction yielding the isolated chlorides 102 (Scheme 28).166 Peroxyketals such as 103 also undergo reduction with iron(II) followed by radical fragmentation with C–C bond scission to generate γ-hydroxyesters (by quenching of intermediate 104) or the corresponding lactones (Scheme 29),166 which themselves are isolated from marine sponge extracts.5,113,167 In each case, it is the resultant carbon-centered radical that is believed to cause cell damage and the cytotoxic effect of these metabolites.99,103,168
Scheme 28 Reaction of 9,10-dihydroplakortin (28) with iron(II) chloride.
Scheme 29 Reaction of peroxyketal 103 with iron(II) sulfate and cyclisation.
4.1 Plakortethers A–G and simplakidine A
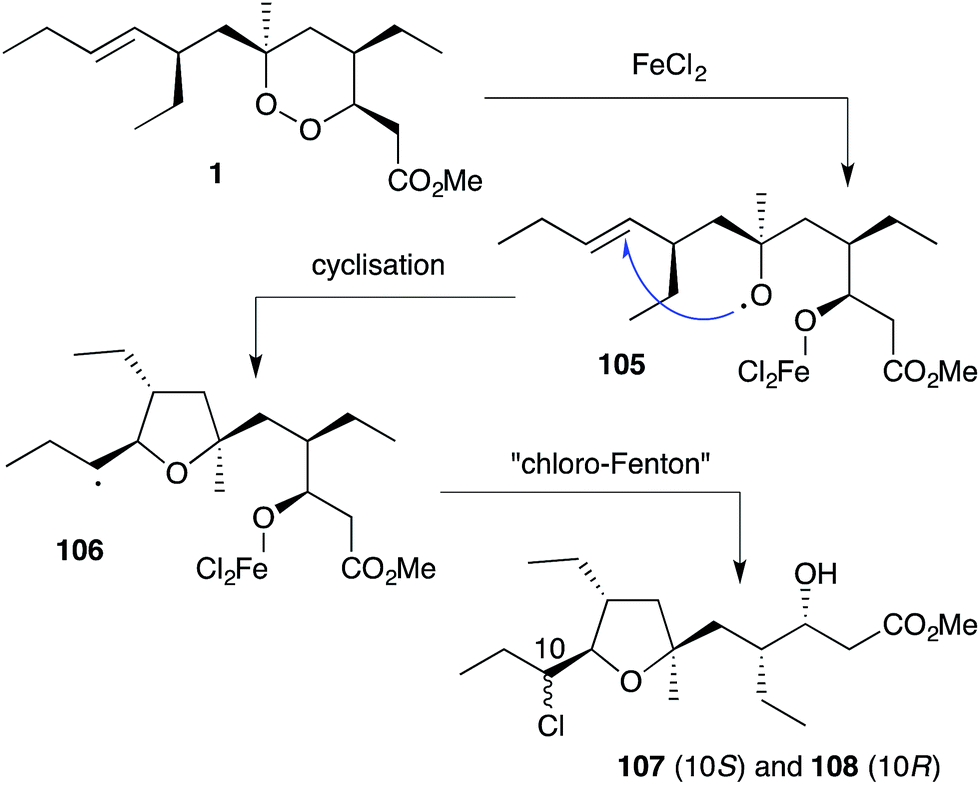
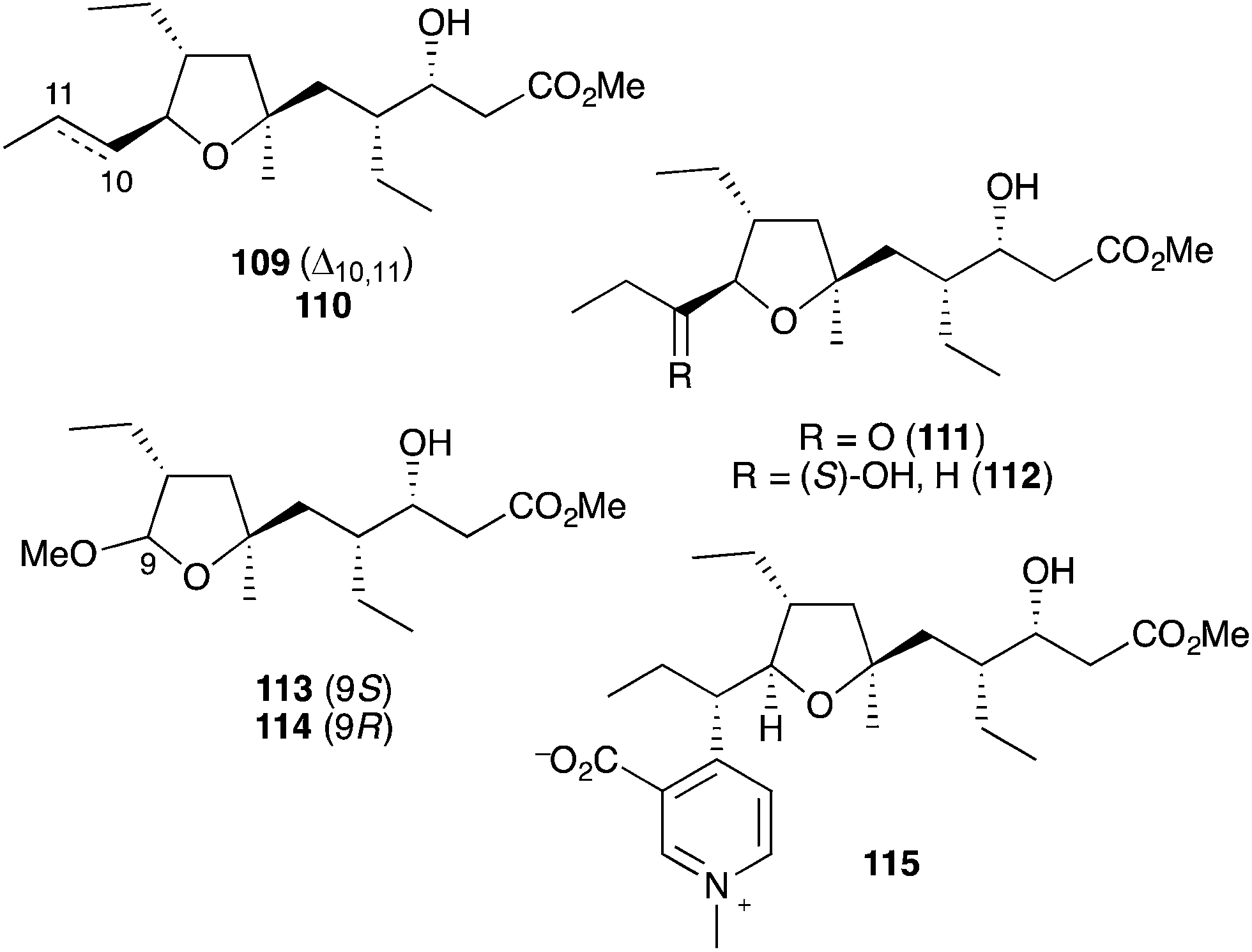
Plakortin (1) is known to undergo iron(II)-mediated reduction like dihydro-analogue 28, with concomitant cyclisation of oxyradical 105 to the pendant olefin (Scheme 30).166 When treated with FeCl2, 106 is then presumed to undergo “chloro-Fenton” reaction yielding the isolated tetrahydrofuran adducts 107 and 108, the latter isolated in 2002 from extracts of Plakortis simplex and named plakortether C (108).169 A series of co-isolates, plakortethers A (109), B (110), D–G (111–114)169 and simplakidine A (115)170 are all reported to share a similar carbon framework, indicating that each analogue most likely arises from the reductive ring opening of 1, with the termination of radical 106 taking a different mechanistic course in each case (Fig. 3). Plakortethers A (109), B (110), D (111) and E (112) are cytotoxic against the RAW 264-7 (murine macrophage) cell line in the range 7–12 μg mL−1;169 and an asymmetric total synthesis of 113 and 114 was completed by Novikov and co-workers in 2009, exploiting symmetrical aspects of the central carbon frame.171
Scheme 30 Reaction of plakortin (1) with iron(II) chloride yielding plakortether C (108).
Fig. 3 Plakortethers A (109), B (110), D–G (111–114) and simplakidine A (115) from Plakortis simplex.
4.2 Simplextones A and B
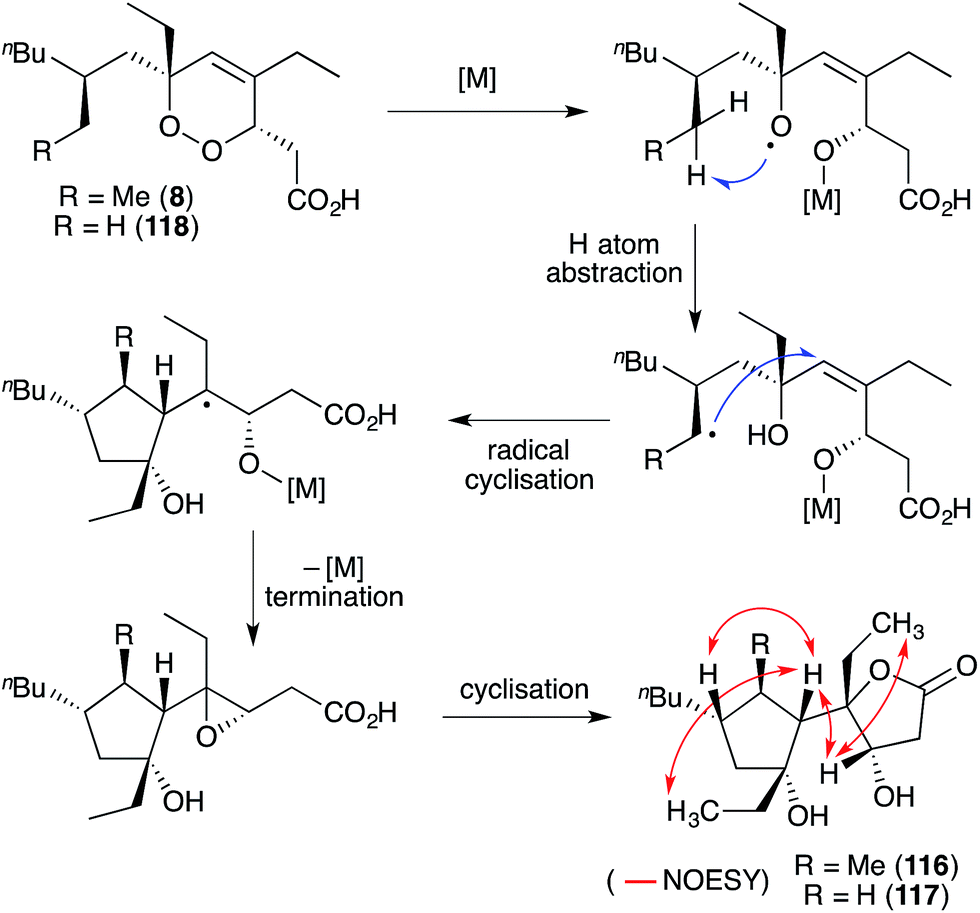
Simplextones A (116) and B (117),172 recently isolated from Plakortis simplex, have an unprecedented skeletal arrangement with a substituted cyclopentane carbocycle connected to a γ-lactone motif by a single C–C bond (Scheme 31). Their structures and absolute configuration were determined by X-ray crystallography, application of Mosher's method, detailed NMR analysis (selected NOESY correlations of compound 116 shown in Scheme 31) and quantum calculation of the CD spectra.172 The continuous carbon backbone of 116 and 117 is remarkably similar to that of 1 and hence, the plakortethers. We speculate that simplextones A (116) and B (117) may in fact result from reductive ring opening, radical cyclisation and lactonisation of the known endoperoxides haterumadioxin B (8)19 and 118,143 respectively (Scheme 31).
Scheme 31 Postulated biosynthesis of simplextones A (116) and B (117) from haterumadioxin B (8) and endoperoxide 118, respectively. Selected NOESY correlations of compound 116 shown as red arrows. M = metal.
4.3 Plakortones A–F, L, N, P and simplexolides A–E
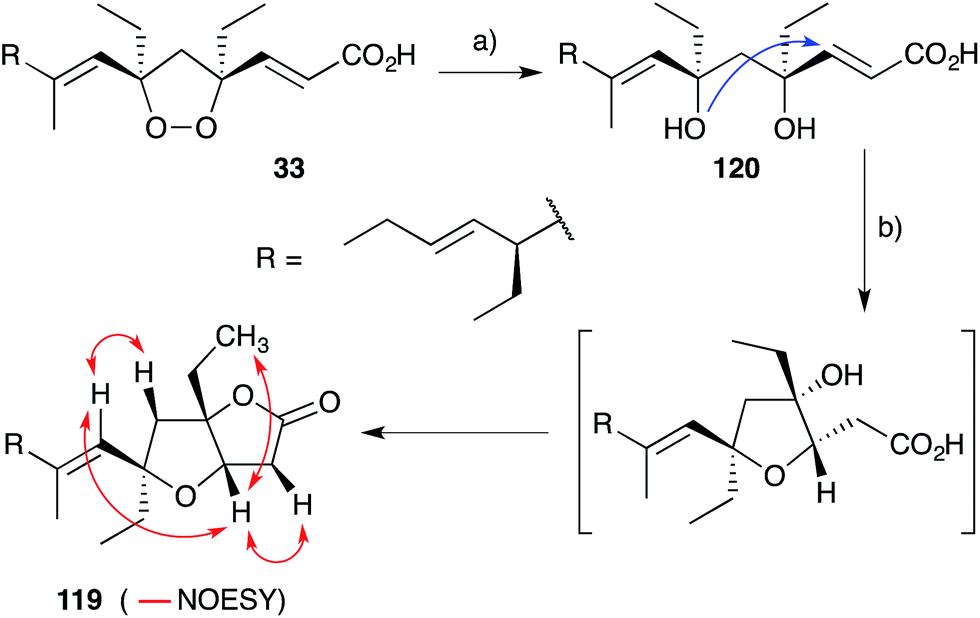
Plakortones A–F, L, N and P22,27,47,173 were isolated from marine sponges of the genera Plakortis and Plakinastrella. Each natural product is characterised by a common 2,6-dioxabicyclo[3.3.0]octan-3-one (furanolactone) motif, yet two distinct pathways of biogenesis have been identified. Following their synthesis of plakortide E (33, Section 2.2), Wong and co-workers86 demonstrated the facile conversion of 33 to plakortone B (119) by reduction to diol 120, itself related to a natural product isolated in 1993,174 oxa-Michael cyclisation and lactonisation (Scheme 32). Impressively, this conversion was achieved with full stereocontrol for the desired bicyclic framework (NOESY correlations of natural 119 shown in Scheme 32)22 providing strong evidence that natural 119 is derived from the reduction of 33. In contrast, through application of Mosher's method to determine the absolute configuration of plakortolide L (121), Garson found that seco-anologue 122 undergoes dehydration and oxa-Michael cyclisation to yield plakortone L (123), directly (Scheme 33).47 The relative structures and absolute configurations of plakortolide L and plakortone L were thus determined as (3S,4S,6S)-121 and (3S,4S,6S)-123, respectively; and the biogenesis of 123 from 121 was implicated. Plakortones A–D are activators of cardiac sarcoplasmic reticulum Ca2+ ATPase at micromolar concentrations22 and plakortones B–F exhibit in vitro cytotoxicity against WEHI 164 murine fibrosarcoma cells.173
Scheme 32 Synthesis of plakortone B (119) from plakortide E (33). Selected NOESY correlations of natural 119 shown as red arrows. (a) Zn, AcOH, CH2Cl2, 0 °C to r.t., 2 h, 99%; (b) DBU, toluene, reflux, overnight, 90%.
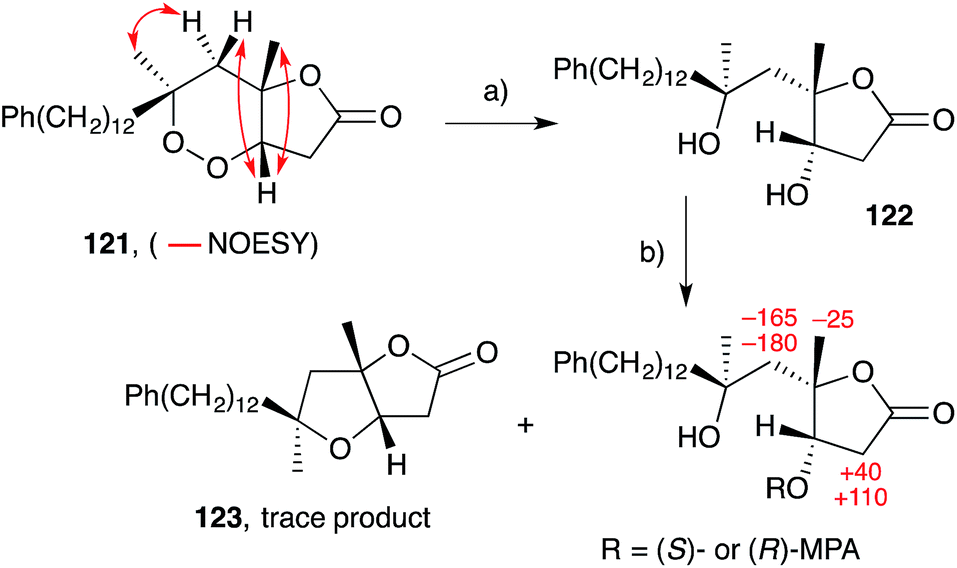
Scheme 33 Conversion of natural plakortolide L (121) to plakortone L (123). Change in chemical shift, expressed as shift of (R)-MPA compound subtracted from shift of (S)-MPA compound (in Hz) and shown as red numbers. (a) Zn, AcOH, Et2O, r.t., 16 h, 83%; (b) (S)- or (R)-MPA, DCC, DMAP, CH2Cl2, r.t., overnight. MPA = methoxyphenylacetate.
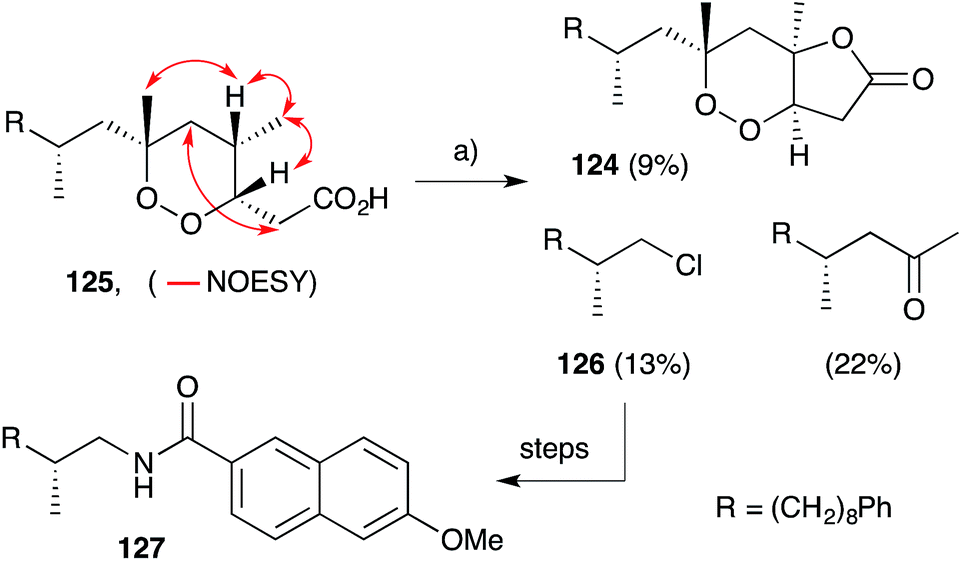
It is also interesting to note that plakortolide B (124), a structural homologue of 121, has been accessed directly from plakinic acid I (125) on treatment with FeCl2, presumably through a complex “chloro-Fenton” reaction (Scheme 34).37 The conditions also gave rise to the expected chloride 126, which was advanced to napthyl amide 127 and used to determine absolute configuration of the remaining stereocenter with a novel analytical technique, liposomal circular dichroism.37
Scheme 34 Assigning the remote stereocenter of plakinic acid I (125) by chemical derivatisation and liposomal circular dichroism. Also, biomimetic conversion of plakinic acid I (125) to plakortolide B (124) through a complex “chloro-Fenton” reaction. Key NOESY correlations of compound 125 shown as red arrows. (a) FeCl2, CH3CN, H2O, r.t., 45 min.
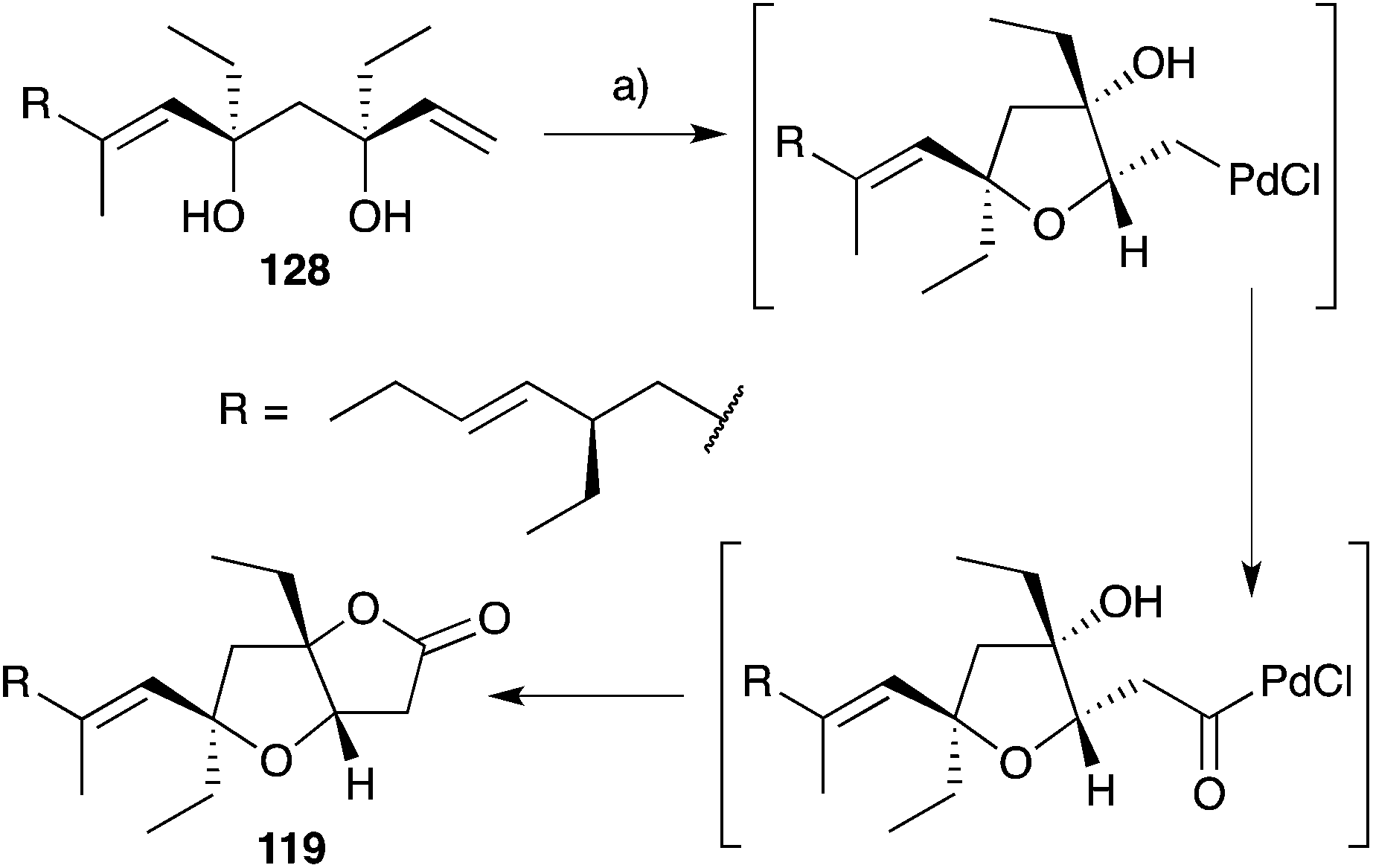
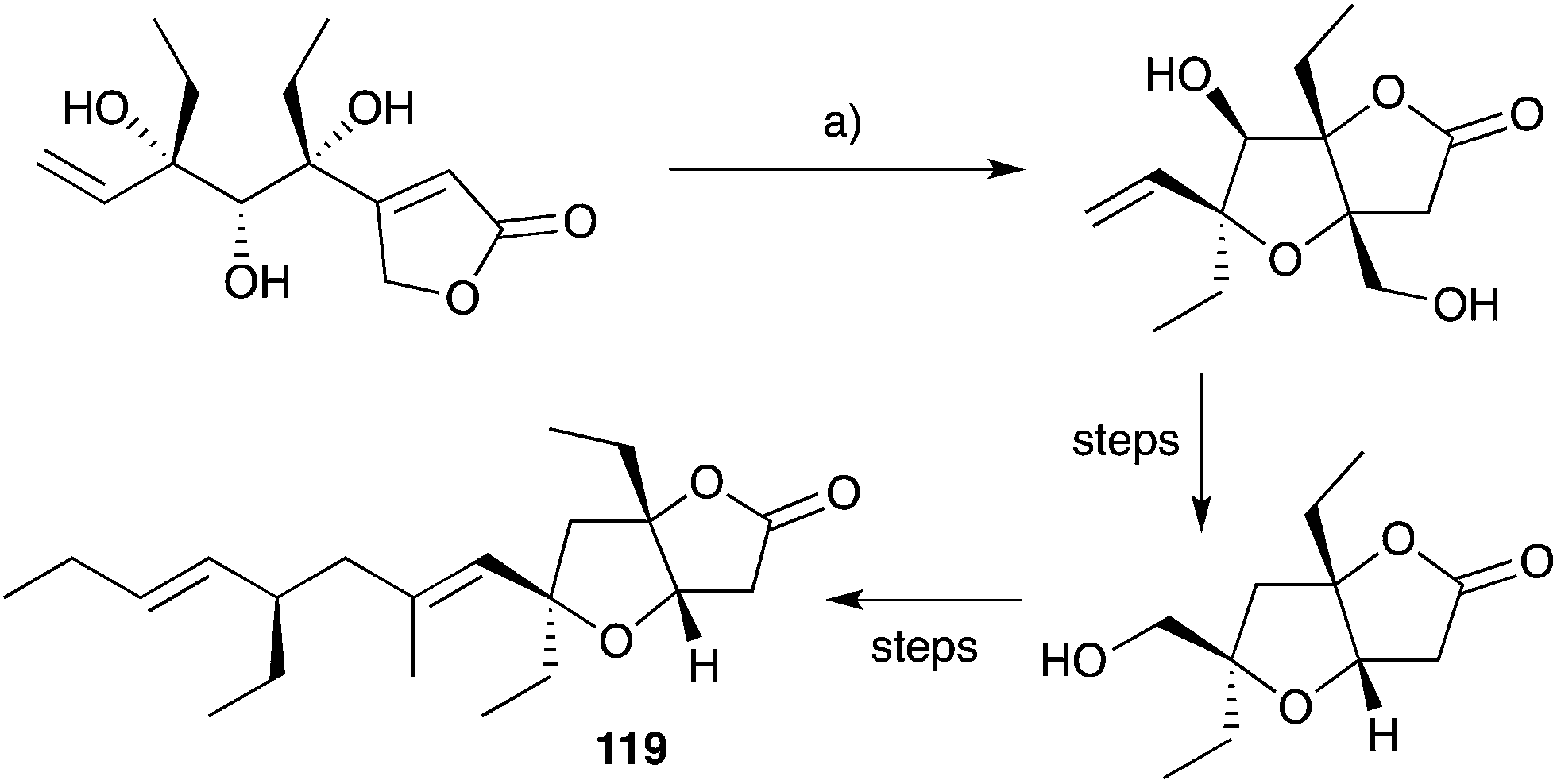
Semmelhack175,176 and Kitching177–180 have independently developed palladium-mediated carbonylation reactions of diols such as 128 (which can be considered a structural analogue of 120, the putative biosynthetic intermediate relating 33 and 119) in the synthesis of plakortone natural products (Scheme 35).176 Wong also completed the total synthesis of 119 independent to that of 33, constructing the furanolactone system by oxa-Michael cyclisation and transesterification of a suitable butenolide (once again, drawing upon the intermediacy of a diol similar to 120, Scheme 36).181,182 In other works, Mehta constructed the core bicycle from simple Morita–Baylis–Hillman adducts;183 Thornhill used an intramolecular Wittig reaction to build the furanolactone core;184 Ohira successfully effected iodolactonisation of a dihydrofuran to construct plakortone E;185 and most recently, Sugimura developed a [3 + 2] annulation strategy for the total synthesis of 123.186
Scheme 35 Synthesis of plakortone B (119) by palladium(II)-mediated carbonylation. (a) PdCl2, CuCl2, NaOAc, AcOH, CO (1 atm), 23 °C, 24 h, 75%.
Scheme 36 Synthesis of plakortone B (119) by oxa-Michael cyclisation and transesterification. (a) DBU, toluene, reflux, 72 h, 90%.
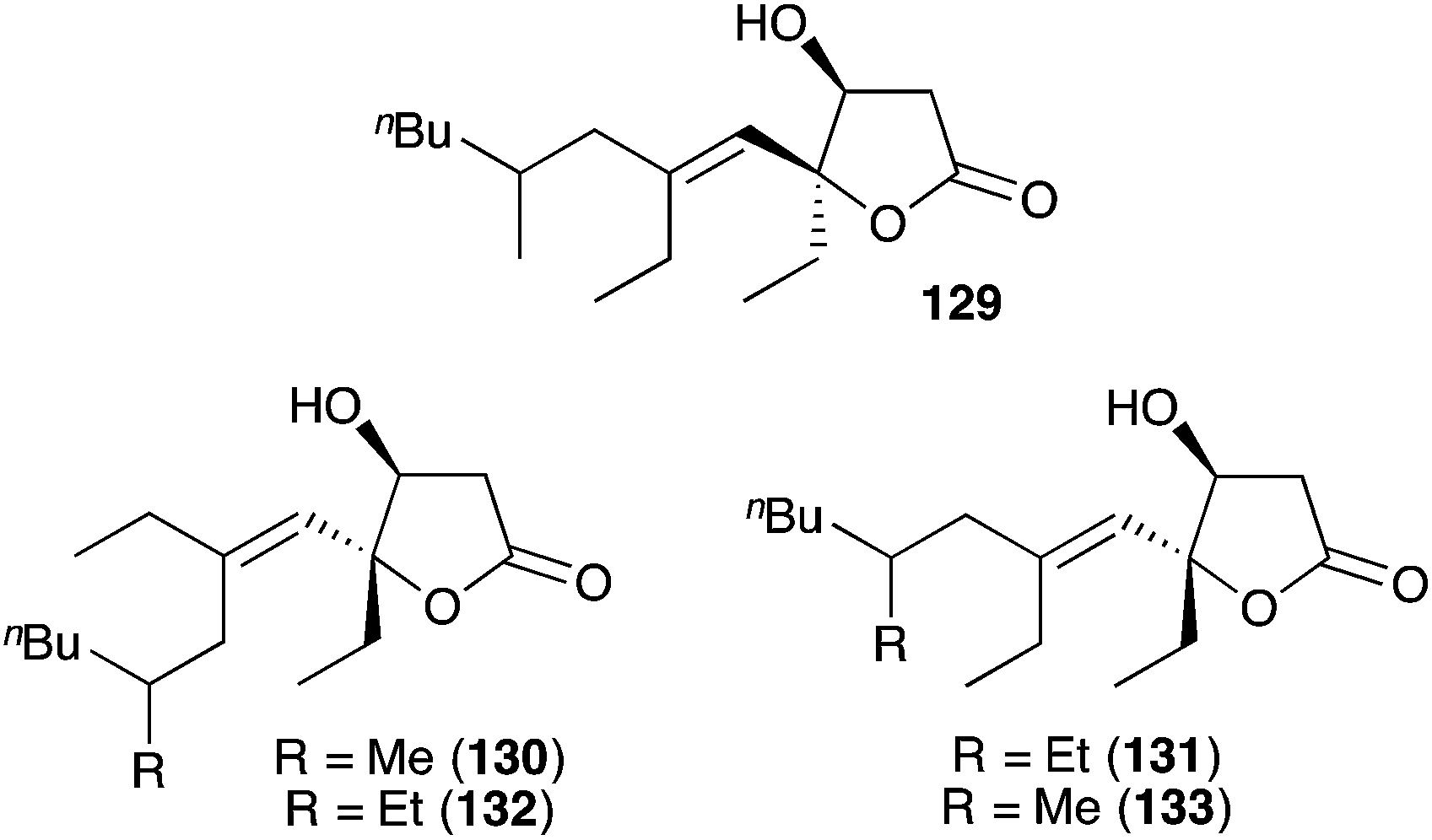
Simplexolides A–E (129–133, Fig. 4)152 represent interesting structural analogues of the plakortone natural products, which appear to arise from the tertiary alcohol dehydration of reduced peroxylactones analogous to compound 122 (Scheme 33).
Fig. 4 Simplexolides A–E (129–133) from Plakortis simplex.
4.4 Plakorsins A, B and plakevulin A
Plakorsins A (51), B113 and plakevulin A (134)187–190 were each isolated from Plakortis sponges and share a carbon framework that appears to be related to chondrillin (2, Scheme 37). While it is clear that plakorsin A (51) is likely to arise from the reduction of 2 itself, the biosynthetic origin of plakevulin A (134) is less certain. Due to their structural similarity, it has been suggested that 134 may arise by enzymatic reduction of untenone A (44).187 However, we speculate that 134 may be generated from the reduction and Dieckmann condensation of 2 (Scheme 37), in a process analogous to the accepted biosynthesis of 44 (Section 3.1). Furthermore, the absolute configuration of plakevulin A (134) at C3, as determined by asymmetric synthesis,189,190 matches that of chondrillin (2).
Scheme 37 Postulated reduction and cyclisation reactions of chondrillin (2) to give plakorsin A (51) and plakevulin A (134).
5. Therapeutic potential and biological activity
Continued interest in the isolation, structural elucidation and chemical synthesis of peroxide and peroxide-derived sponge metabolites is fuelled by their potential for development as therapeutic agents in the treatment of infectious diseases, cancers and physiological disorders. Many of the compounds presented in this review have promising activity for one or more therapeutic targets, and some exhibit selectivity for these targets against other microorganisms and non-tumour cells. However, the cytotoxicity of polyketide endoperoxides is often a factor that must be considered in the hope of producing desirable lead compounds. While a number of research groups have recently begun to address this challenge, managing the cytotoxicity of peroxide and peroxide-derived sponge metabolites remains an ongoing difficulty.
Potency has widely been attributed to the presence of 1,2-dioxane heterocycles, normally appended with a terminal acetate group, through their action as an oxidant. The rearranged congeners, where the peroxide functional group is lost, are typically less active for the desired target and also tend to have lower levels of cytotoxicity.24,44,51,144 Endoperoxides appended with a free acid are normally more potent than their corresponding esters.25,29,32,35,62 In some instances, the esterification of peroxyacids and their rearrangement to non-peroxide containing homologues has been attributed to prolonged storage in alcoholic solvents during specimen extraction.29,51
5.1 Infectious diseases
Since the discovery of peroxide metabolites as effective antimalarial agents, plakortin (1) and its related compounds have been routinely screened for antiplasmodial activity, especially against chloroquine-resistant strains. Although their mechanisms of action are still unclear, a number of studies have focused on understanding the structural requirements and limitations of plakortin-related 1,2-dioxanes as novel targets for treating malaria.
Plakortin (1) and its dihydro analogue 28 show good activity against the chloroquine sensitive D10 strain (IC50 1.12–1.26 μM) and chloroquine resistant W2 strain (IC50 0.74–0.76 μM) of Plasmodium falciparum.65 Greater potency for the resistant strain follows the same trend as artemisinin and further demonstrates that polyketide endoperoxides do not share the same mechanism of resistance as chloroquine.65 Recently, detailed studies on the structure–activity relationships (SARs) of plakortin (1) and plakortin-related scaffolds have shown that the formation of discrete carbon centered radicals plays a key role in their effect.166,191 Other SAR studies on the dioxane scaffold have generated novel synthetic compounds, which maintain antiprotozoal activity and significantly reduce cytotoxicity.88,99–101 Schwarzer recently discovered that plakortin (1) can induce lipid-peroxidation and a marked increase of the lipoperoxide breakdown product 4-hydroxynonenal, which conjugates to P. falciparum proteins critically involved in its cellular function.66
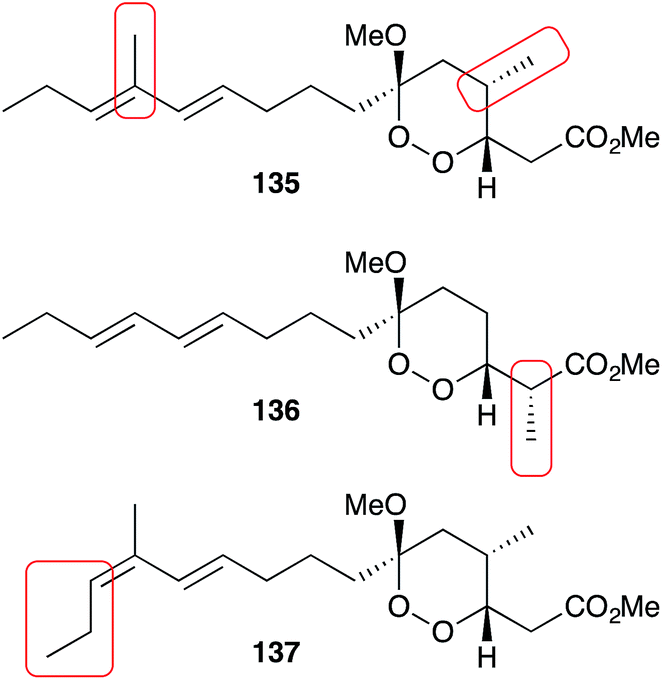
Manadoperoxides A–K were evaluated extensively for antitrypansomal activity. Most showed excellent activity against Trypanosoma brucei rhodesiense including manadoperoxide I (IC50 0.062 μg mL−1) and K (IC50 0.087 μg mL−1) with low levels of cytotoxicity against HMEC-1 (IC50 > 10 μg mL−1).15–17 Interestingly, Taglialatella–Scafati found that manadoperoxide B (135) had greater potency against T. b. rhodesience (IC50 0.003 μg mL−1) compared to P. falciparum (IC50 2.30 μg mL−1), whereas peroxyplakoric ester B3 (136) had greater potency against P. falciparum (IC50 0.040 μg mL−1) compared to T. b. rhodesience (IC50 3.61 μg mL−1); and that notably, they only differ by the placement of methyl groups on an otherwise identical core structure (Fig. 5).16 Furthermore, the isomeric compound 137 maintained activity against T. b. rhodesience (IC50 0.011 μg mL−1) but had increased cytotoxicity against L6 cells (IC50 3.80 μg mL−1).17 Further SAR-based studies revealed the importance of the peroxyketal heterocycle and length of the lipophilic tether for antiprotozoal activity.15–17
Fig. 5 The minor structural differences of manadoperoxide B (135), peroxyplakoric ester B3 (136) and 12-isomanadoperoxide B (137), which greatly effect their antiprotozoal activity and cytotoxicity.
Plakortide endoperoxides have similarly attracted interest as potential therapeutic agents for tropical diseases. Plakortide F was active against P. falciparum (IC50 0.39–0.48 μg mL−1) and cytotoxic against a number of cancer cell lines in vitro, but failed to prolong life expectancy when treating Plasmodium infected mice.27 In the same study, plakortone G was found to be highly cytotoxic with no apparent selectivity. Plakortide P (10) had antiparasitic activity against Leishmania chagasi (IC50 0.5–1.9 μg mL−1) with low toxicity against human macrophages (IC50 16.6 μg mL−1);20 and plakortide I, possessing an unsaturated ketone, was effective when tested against P. falciparum (IC50 0.57 μg mL−1) but inactive against a panel of other pathogenic bacteria and fungi.26 Plakortide E (24) was also found to be a non-competitive, slow-binding and reversible inhibitor of rhodesain from T. B. rhodesience (IC50 5 μM) without cytotoxic effects against J774.1 macrophages at 100 μM.30
Recently, the novel endoperoxide metabolite gracilioether H was reported to be active against the chloroquine resistant FC29 strain (IC50 3.3 μM) of P. falciparum with low cytotoxicity for vero cells (DT50 324 μM).160
Other metabolites in this class have also been tested for activity against pathogenic fungi and bacteria with promising results. Plakinic acid M was active toward a number of Cryptococcus fungi (MIC90 2.4–13 μM);32 manzamenone O (69) was effective against Micrococcus luteus (MIC 4 μg mL−1), Apergillis niger (IC50 8 μg mL−1) and Trichophyton mentagrophytes (IC50 8 μg mL−1);133 and hippolachnin A (98) was shown to have potent antifungal activity against Cryptococcus neoformans, Trichophyton rubrum and Microsporum gypseum (MIC 0.4 μM).155
5.2 Anticancer activity
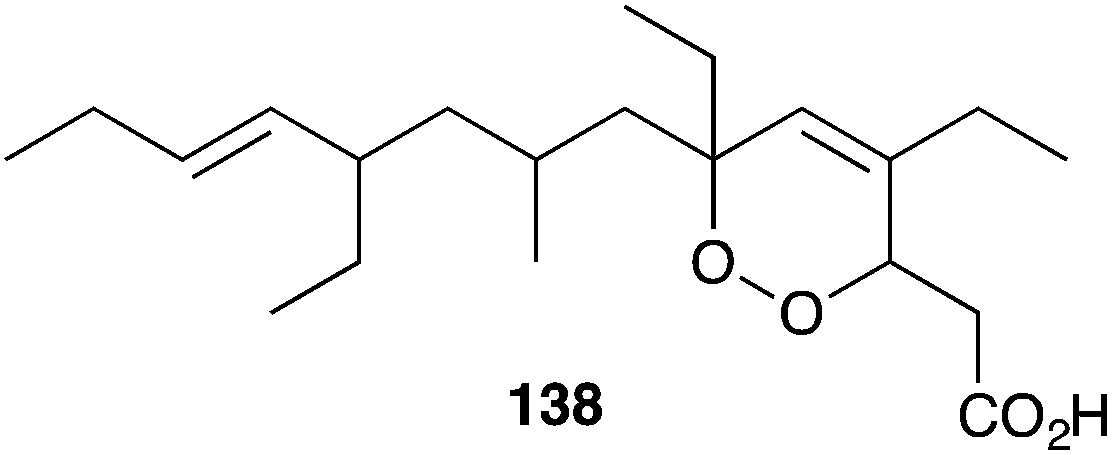
The cytotoxic effects of many peroxide and peroxide-derived sponge metabolites have prompted numerous studies into their suitability as anticancer leads. Costa-Lotufo found that plakortide P (14) and a number of structural analogues were cytotoxic against HCT-116 cells, causing arrest at the G2/M stage.31 However, the co-isolated furanylidenes des-hydroxygracilioether C (90) and spongosoritin A induced arrest at the G0/G1 stage, indicating that the two structural subclasses have distinct modes of antimitotic action. Furthermore, while the peroxides showed low selectivity for tumour cells compared to non-tumour cells, compound 90 was more selective with IC50 8.1 μM for HCT-116 and IC50 > 163 μM for MRC-5 cells.31 Plakinic acids A–D and a number of structural analogues were found to be remarkably cytotoxic against L1210 murine leukemia (IC50 0.003–0.052 μg mL−1);39 and endoperoxide 138 showed significant in vitro cytotoxicity against P388 cells (IC50 0.055 μg mL−1), but failed to induce a response in vivo.49,91
Other work has specifically targeted the inhibition of DNA polymerase (pol) enzymes. Manzamenone A (53) showed inhibition of polα (IC50 1.9 μM), β (IC50 3.2 μM) and human terminal deoxynucleotidyltransferase (IC50 2.5 μM).124,125 Untenone A (44) demonstrated greater selectivity for polα (IC50 4.3 μM);125 plakevulin A (134) was a moderate inhibitor of polγ (IC50 7.5 μg mL−1);187 and a number of synthetic analogues of plakoridine A (71) and B (72) showed greater inhibition of polα and β than the natural products.127
5.3 Physiological disorders
Peroxide-derived sponge metabolites have also been reported as useful compounds for the investigation of a range of physiological disorders, including type II diabetes, heart failure and inflammation. Gracilioether B (85) and plakilactone C were recently shown to be selective covalent agonists of peroxisome proliferator-activated receptor γ (PPARγ, EC50 2–5 μM), a known pharmacological target for the treatment of type II diabetes, undergoing thio-Michael addition of the lower binding domain cysteine residue to the unsaturated ketone of the natural products.146 Both compounds were found to regulate the expression of PPARγ-dependent genes in the liver and inhibit the generation of inflammatory mediators.146 In contrast, gracilioether C (86) was found to be a non-covalent agonist (EC50 10 μM) and its des-hydroxy homologue (90) a non-covalent antagonist.146 In a separate study, manzamenones B (54) and E (57) showed inhibition of T-cell protein tyrosine phosphatase (IC50 2.5–3.2 μM) and protein tyrosine phosphatase-1B (IC50 10.8–13.5 μM), also implicated in the treatment of type II diabetes.126
Plakortone D activated sarcoplasmic reticulum Ca2+ ATPase (EC50 2.8 μM), a protein associated with cardiac muscle relaxation abnormalities, improving calcium uptake at a similar level to that reported for gingerol.22 3-epi-Plakortin was also found to be an activator, while the longer chain co-isolates plakortides F–H were less active.23 Interestingly, the ability of polyketide endoperoxides to stimulate calcium uptake is also suggested to play a role in their antimitotic and antifungal activity.31,192
Finally, gracilioethers E (100), G and I–K were found to be agonists of pregnane-X-receptor, a novel pharmacological target for the treatment of various inflammatory and metabolic disorders, when administered in combination with rifaximin.162
6. Concluding remarks
Since their discovery in the late 1970s, plakortin (1) and chondrillin (2) have been the centerpiece of a global research effort to uncover, elucidate and synthesise peroxide and peroxide-derived metabolites from marine sponges. Literature in the chemical sciences is now laced with novel polyketide substances related to 1 and 2 and synthetic methods to effectively construct and arrange their unique and often complex structural components. These efforts, especially those targeting the chemical synthesis of peroxide-derived natural products, have increasingly become inspired by a desire to understand the fascinating decomposition pathways and inherent structural rearrangements of these peroxide substrates. Their potential as therapeutic agents for a wide range of infectious diseases, cancers and physiological disorders adds further motivation for the investigation of this class of compound. Incredibly, investigations in the field to date places the sponge metabolites chondrillin (2) and plakorin (3) as sole non-enzymatic progenitors to the natural products untenone A (44), plakortic acid (45), plakorsin A (51), manzamenone A–F (53–58) and H (59) with many others expected. Furthermore, plakortin (1) and 4,5-unsaturated-1,2-dioxane analogues such as haterumadioxin A (7) and B (8), are thought to be directly responsible for the occurrence of plakortethers A–G (108–114), simplakidine A (115), gracilioethers B (85), C (86), D, hippolachnin A (98) and spiroplakortone plus many more.
However, efforts to elucidate the biosynthesis of peroxide-related metabolites have only begun. Understanding the assembly of complex polycyclic structures such as gracilioethers A (92), E (100), F (95), G–J, K (96), manzamenones K (68), O (69), untenolide A (70), simplextones A (116) and B (117) remains a challenge of significant novelty and interest. Feasible preparation of such intricate substrates on the scale required in a pharmaceutical context will undoubtedly be aided by consideration of how they are assembled in nature.
7. Acknowledgements
We wish to thank Flinders University for funding, support, and facilities. M. D. Norris acknowledges the MF & MH Joyner PhD Scholarship in Science.
8. Notes and references
M. D. Higgs and D. J. Faulkner, J. Org. Chem., 1978, 43, 3454 CrossRefCAS.
F. Cafieri, E. Fattorusso, O. Taglialetella-Scafati and A. Ianaro, Tetrahedron, 1999, 55, 7045 CrossRefCAS.
R. J. Wells, Tetrahedron Lett., 1976, 30, 2637 CrossRef.
T. Murayama, Y. Ohizumi, H. Nakamura, T. Sasaki and J. Kobayashi, Experientia, 1989, 45, 898 CrossRefCAS.
E. Quinoa, E. Kho, L. V. Manes, P. Crews and G. J. Bakus, J. Org. Chem., 1986, 51, 4260 CrossRefCAS.
M. Jung, H. Kim, K. Lee and M. Park, Mini-Rev. Med. Chem., 2003, 3, 159 CrossRefCASPubMed.
V. M. Dembitsky, T. A. Gloriozova and V. V. Poroikov, Mini-Rev. Med. Chem., 2007, 7, 571 CrossRefCASPubMed.
V. M. Dembitsky, Eur. J. Med. Chem., 2008, 43, 223 CrossRefCASPubMed.
E. Fattorusso and O. Taglialatela-Scafati, Mar. Drugs, 2009, 7, 130 CrossRefCASPubMed.
E. Fattorusso and O. Taglialatela-Scafati, Phytochem. Rev., 2010, 9, 515 CrossRefCAS.
D.-Z. Liu and J.-K. Liu, Nat. Prod. Bioprospect., 2013, 3, 161 CrossRefCAS.
F. Rahm, P. Y. Hayes and W. Kitching, Heterocycles, 2004, 64, 523 CrossRefCAS.
C. Fattorusso, M. Persico, B. Calcinai, C. Cerrano, S. Parapini, D. Taramelli, E. Novellino, A. Romano, F. Scala, E. Fattorusso and O. Taglialatela-Scafati, J. Nat. Prod., 2010, 73, 1138 CrossRefCASPubMed.
G. Chianese, E. Fattorusso, F. Scala, R. Teta, B. Calcinai, G. Bavestrello, H. A. Dien, M. Kaiser, D. Tasdemir and O. Taglialatela-Scafati, Org. Biol. Chem., 2012, 10, 7197 RSC.
G. Chianese, F. Scala, B. Calcinai, C. Cerrano, H. A. Dien, M. Kaiser, D. Tasdemir and O. Taglialatela-Scafati, Mar. Drugs, 2013, 11, 3297 CrossRefPubMed.
M. Gushiken, I. Kagiyama, H. Kato, T. Kuwana, F. Losung, R. E. P. Mangindaan, N. J. de Voogd and S. Tsukamoto, J. Nat. Med., 2015, 69, 595 CrossRefCASPubMed.
N. Takada, M. Watanabe, A. Yamada, K. Suenaga, K. Yamada, K. Ueda and D. Uemura, J. Nat. Prod., 2001, 64, 356 CrossRefCAS.
M. H. Kossuga, A. M. Nascimento, J. Q. Reimao, A. G. Tempone, N. N. Taniwaki, K. Veloso, A. G. Ferreira, B. C. Cavalcanti, C. Pessoa, M. O. Moraes, A. M. S. Mayer, E. Hajdu and R. G. S. Berlinck, J. Nat. Prod., 2008, 71, 334 CrossRefCASPubMed.
C. Festa, S. De Marino, M. V. D'Auria, O. Taglialatela-Scafati, E. Deharo, S. Petek and A. Zampella, Tetrahedron, 2013, 69, 3706 CrossRefCAS.
A. D. Patil, A. J. Freyer, M. F. Bean, B. K. Carte, J. W. Westley, R. K. Johnson and P. Lahouratate, Tetrahedron, 1996, 52, 377 CrossRefCAS.
A. D. Patil, A. J. Freyer, B. Carte, R. K. Johnson and P. Lahouratate, J. Nat. Prod., 1996, 59, 219 CrossRefCASPubMed.
B. Harrison and P. Crews, J. Nat. Prod., 1998, 61, 1033 CrossRefCASPubMed.
E. Fattorusso, O. Taglialatela-Scafati, M. Di Rosa and A. Ianaro, Tetrahedron, 2000, 56, 7959 CrossRefCAS.
J.-F. Hu, H.-F. Gao, M. Kelly and M. T. Hamann, Tetrahedron, 2001, 57, 9379 CrossRefCAS.
D. J. Gochfeld and M. T. Hamann, J. Nat. Prod., 2001, 64, 1477 CrossRefCAS.
M. del Sol Jimenez, S. P. Garzon and A. D. Rodriguez, J. Nat. Prod, 2003, 66, 655 CrossRefCASPubMed.
F. Berrue, O. P. Thomas, C. F.-L. Bon, F. Reyes and P. Amade, Tetrahedron, 2005, 61, 11843 CrossRefCAS.
S. Oli, U. R. Abdelmohsen, U. Hentschel and T. Schirmeister, Mar. Drugs, 2014, 12, 2614 CrossRefCASPubMed.
E. A. Santos, A. L. Quintela, E. G. Ferreira, T. S. Sousa, F. d. C. L. Pinto, E. Hajdu, M. S. Carvalho, S. Salani, D. D. Rocha, D. V. Wilke, M. d. C. M. Torres, P. C. Jimenez, E. R. Silveira, J. J. La Clair, O. D. L. Pessoa and L. V. Costa-Lotufo, J. Nat. Prod., 2015, 78, 996 CrossRefCASPubMed.
M. T. Jamison, D. S. Dalisay and T. F. Molinski, J. Nat. Prod., 2016, 79, 555 CrossRefCASPubMed.
G. R. Pettit, T. Nogawa, J. C. Knight, D. L. Doubek and J. N. A. Hooper, J. Nat. Prod., 2004, 67, 1611 CrossRefCASPubMed.
M. Yanai, S. Ohta, E. Ohta, T. Hirata and S. Ikegami, Bioorg. Med. Chem., 2003, 11, 1715 CrossRefCASPubMed.
D. W. Phillipson and K. L. Rinehart, J. Am. Chem. Soc., 1983, 105, 7735 CrossRefCAS.
J. S. Sandler, P. L. Colin, J. N. A. Hooper and D. J. Faulkner, J. Nat. Prod., 2002, 65, 1258 CrossRefCAS.
D. S. Dalisay, T. Quach, G. N. Nicholas and T. F. Molinski, Angew. Chem., Int. Ed., 2009, 48, 4367 CrossRefCASPubMed.
D. S. Dalisay, T. Quach and T. F. Molinski, Org. Lett., 2010, 12, 1524 CrossRefCASPubMed.
B. S. Davidson, J. Org. Chem., 1991, 56, 6722 CrossRefCAS.
B. S. Davidson, Tetrahedron Lett., 1991, 32, 7167 CrossRefCAS.
P. A. Horton, R. E. Longley, M. Kelly-Borges, O. J. McConnell and L. M. Ballas, J. Nat. Prod., 1994, 57, 1374 CrossRefCAS.
Y. Chen, K. B. Killday, P. J. McCarthy, R. Schimoler, K. Chilson, C. Selitrennikoff, S. A. Pomponi and A. E. Wright, J. Nat. Prod., 2001, 64, 262 CrossRefCAS.
C. Jimenez-Romero, I. Ortiz, J. Vicente, B. Vera, A. D. Rodriguez, S. Nam and R. Jove, J. Nat. Prod., 2010, 73, 1694 CrossRefCASPubMed.
M. Varoglu, B. M. Peters and P. Crews, J. Nat. Prod., 1995, 58, 27 CrossRefCAS.
T. L. Perry, A. Dickerson, A. A. Khan, R. K. Kondru, D. N. Beratan, P. Wipf, M. Kelly and M. T. Hamann, Tetrahedron, 2001, 57, 1483 CrossRefCAS.
A. Rudi, R. Afanii, L. G. Gravalos, M. Aknin, E. Gaydou, J. Vacelet and Y. Kashman, J. Nat. Prod., 2003, 66, 682 CrossRefCASPubMed.
K. W. L. Yong, J. J. De Voss, J. N. A. Hooper and M. J. Garson, J. Nat. Prod., 2011, 74, 194 CrossRefCASPubMed.
K. W. L. Yong, L. K. Lambert, P. Y. Hayes, J. J. De Voss and M. J. Garson, J. Nat. Prod., 2012, 75, 351 CrossRefCASPubMed.
D. E. Williams, T. M. Allen, R. van Soest, H. W. Behrisch and R. J. Andersen, J. Nat. Prod., 2001, 64, 281 CrossRefCAS.
R. Mohammed, J. Peng, M. Kelly, M. Yousaf, E. Winn, S. Odde, Z. Bie, A. Xie, R. J. Doerksen and M. T. Hamann, Aust. J. Chem., 2010, 63, 877 CrossRefCAS.
D. B. Stierle and D. J. Faulkner, J. Org. Chem., 1979, 44, 964 CrossRefCAS.
M. Kobayashi, K. Kondo and I. Kitagawa, Chem. Pharm. Bull., 1993, 41, 1324 CrossRefCASPubMed.
A. Rudi, R. Talpir, Y. Kashman, Y. Benayahu and M. Schleyer, J. Nat. Prod., 1993, 56, 2178 CrossRefCAS.
S. I. Toth and F. J. Schmitz, J. Nat. Prod., 1994, 57, 123 CrossRefCAS.
J. C. Braekman, D. Daloze, S. De Groote, J. B. Fernandes and R. W. M. Van Soest, J. Nat. Prod., 1998, 61, 1038 CrossRefCASPubMed.
A. Fontana, M. Ishibashi and J. Kobayashi, Tetrahedron, 1998, 54, 2041 CrossRefCAS.
A. Fontana, A. Ishibashi, H. Shigemori and J. Kobayashi, J. Nat. Prod., 1998, 61, 1427 CrossRefCASPubMed.
E. Manzo, M. L. Ciavatta, D. Melck, P. Schupp, N. de Voogd and M. Gavagnin, J. Nat. Prod., 2009, 72, 1547 CrossRefCASPubMed.
Y. Feng, R. A. Davis, M. Sykes, V. M. Avery, D. Camp and R. J. Quinn, J. Nat. Prod., 2010, 73, 716 CrossRefCASPubMed.
J. S. Oh, B. S. Hwang, O.-H. Kang, D.-Y. Kwon and J.-R. Rho, Mar. Drugs, 2013, 11, 4407 CrossRefPubMed.
B. S. Hwang and J.-R. Rho, Journal of the Korean Magnetic Resonance Society, 2013, 17, 47 CrossRef.
T. R. Hoye, W. M. Alarif, S. S. Basaif, M. Abo-Elkarm, M. T. Hamann, A. E. Wahba and S.-E. N. Ayyad, ARKIVOC, 2015, 164 CAS.
J. H. McKerrow, Nat. Prod. Rep., 2015, 32, 1610 RSC.
L. Y. Kong and R. X. Tan, Nat. Prod. Rep., 2015, 32, 1617 RSC.
E. Fattorusso, S. Parapini, C. Campagnuolo, N. Basilico, O. Taglialatela-Scafati and D. Taramelli, J. Antimicrob. Chemother., 2002, 50, 883 CrossRefCASPubMed.
O. A. Skorokhod, D. Davalos-Schafler, V. Gallo, E. Valente, D. Ulliers, A. Notarpietro, G. Mandili, F. Novelli, M. Persico, O. Taglialatela-Scafati, P. Arese and E. Schwarzer, Free Radical Biol. Med., 2015, 89, 624 CrossRefCASPubMed.
S. P. D. Ovenden and R. J. Capon, J. Nat. Prod., 1999, 62, 214 CrossRefCASPubMed.
R. J. Capon, Eur. J. Org. Chem., 2001, 633 CrossRefCAS.
G. Della Sala, T. Hochmuth, V. Costantino, R. Teta, W. Gerwick, L. Gerwick, J. Piel and A. Mangoni, Environ. Microbiol. Rep., 2013, 5, 809 CrossRefCASPubMed.
G. Della Sala, T. Hochmuth, R. Teta, V. Costantino and A. Mangoni, Mar. Drugs, 2014, 12, 5425 CrossRefCASPubMed.
S. Isayama and T. Mukaiyama, Chem. Lett., 1989, 573 CrossRefCAS.
S. Isayama, Bull. Chem. Soc. Jpn., 1990, 63, 1305 CrossRefCAS.
E. M. P. Silva, R. J. Pye, C. Cardin and L. M. Harwood, Synlett., 2010, 509 CAS.
E. M. P. Silva, R. J. Pye, G. D. Brown and L. M. Harwood, Eur. J. Org. Chem., 2012, 1209 CrossRefCAS.
B. Barnych and J.-M. Vatéle, Org. Lett., 2012, 14, 564 CrossRefCASPubMed.
K. W. L. Yong, B. Barnych, J. J. De Voss, J.-M. Vatéle and M. J. Garson, J. Nat. Prod., 2012, 75, 1792 CrossRefCASPubMed.
S. Gemma, F. Marti, E. Gabellieri, G. Campiani, E. Novellino and S. Butini, Tetrahedron Lett., 2009, 50, 5719 CrossRefCAS.
S. Gemma, E. Gabellieri, S. S. Coccone, F. Marti, O. Taglialatela-Scafati, E. Novellino, G. Campiani and S. Butini, J. Org. Chem., 2010, 75, 2333 CrossRefCASPubMed.
S. Gemma, S. Kunjir, S. S. Coccone, M. Brindisi, V. Moretti, S. Brogi, E. Novellino, N. Basilico, S. Parapini, D. Taramelli, G. Campiani and S. Butini, J. Med. Chem., 2011, 54, 5949 CrossRefCASPubMed.
B. Barnych and J.-M. Vatéle, Synlett, 2011, 13, 1912 Search PubMed.
B. Barnych, B. Fenet and J.-M. Vatéle, Tetrahedron, 2013, 69, 334 CrossRefCAS.
As reproduced in Scheme 6, 5-exo-tet cyclisation of 31 was drawn in ref. 81 to give 32 with retention of configuration at C3. However, in a personal communication with the authors of this work (B. Barnych), it has been verified that this reaction should proceed with inversion at C3 and that compound 32 should be drawn with (3S) configuration, as suggested below. Consequently, the absolute configuration of natural andavadoic acid (19) is in fact (3S,5R)
.
K. S. Feldman and M. Pravez, J. Am. Chem. Soc., 1986, 108, 1328 CrossRefCAS.
K. S. Feldman and R. E. Simpson, J. Am. Chem. Soc., 1989, 111, 4878 CrossRefCAS.
K. S. Feldman and C. M. Kraebel, J. Org. Chem., 1992, 57, 4574 CrossRefCAS.
X.-Y. Sun, X.-Y. Tian, Z.-W. Li, X.-S. Peng and H. N. C. Wong, Chem.–Eur. J., 2011, 17, 5874 CrossRefCASPubMed.
X.-Y. Tian, J.-W. Han, Q. Zhao and H. N. C. Wong, Org. Biol. Chem., 2014, 12, 3686 RSC.
C. Fattorusso, M. Persico, N. Basilico, D. Taramelli, E. Fattorusso, F. Scala and O. Taglialetela-Scafati, Bioorg. Med. Chem., 2011, 19, 312 CrossRefCASPubMed.
M. Jung, J. Ham and J. Song, Org. Lett., 2002, 4, 2763 CrossRefCASPubMed.
O. Zvarec, T. D. Avery, D. K. Taylor and E. R. T. Tiekink, Tetrahedron, 2010, 66, 1007 CrossRefCAS.
S. P. Gunasekera, M. Gunasekera, G. P. Gunawardana, P. McCarthy and N. Burres, J. Nat. Prod., 1990, 53, 669 CrossRefCAS.
B. B. Snider and Z. Shi, J. Org. Chem., 1990, 55, 5669 CrossRefCAS.
B. B. Snider and Z. Shi, J. Am. Chem. Soc., 1992, 114, 1790 CrossRefCAS.
B. B. Snider, Z. Shi, S. V. O'Neil, K. D. Kreutter and T. L. Arakaki, J. Org. Chem., 1994, 59, 1726 CrossRefCAS.
P. Dussault, A. Sahli and T. Westermeyer, J. Org. Chem., 1993, 58, 5469 CrossRefCAS.
P. H. Dussault and K. R. Woller, J. Am. Chem. Soc., 1997, 119, 3824 CrossRefCAS.
P. H. Dussault, C. T. Eary and K. R. Woller, J. Org. Chem., 1999, 64, 1789 CrossRefCASPubMed.
M. Persico, A. Quintavalla, F. Rondinelli, C. Trombini, M. Lombardo, C. Fattorusso, V. Azzarito, D. Taramelli, S. Parapini, Y. Corbett, G. Chianese, E. Fattorusso and O. Taglialatela-Scafati, J. Med. Chem., 2011, 54, 8526 CrossRefPubMed.
M. Persico, S. Parapini, G. Chianese, C. Fattorusso, M. Lombardo, L. Petrizza, A. Quintavalla, F. Rondinelli, N. Basilico, D. Taramelli, C. Trombini, E. Fattorusso and O. Taglialatela-Scafati, Eur. J. Med. Chem., 2013, 70, 875 CrossRefCASPubMed.
N. Murakami, M. Kawanishi, S. Itagaki, T. Horii and M. Kobayashi, Bioorg. Med. Chem. Lett., 2002, 12, 69 CrossRefCASPubMed.
N. Murakami, M. Kawanishi, H. M. Mostaqul, J. Li, S. Itagaki, T. Horii and M. Kobayashi, Bioorg. Med. Chem. Lett., 2003, 13, 4081 CrossRefCASPubMed.
M. Kawanishi, N. Kotoku, S. Itagaki, T. Horii and M. Kobayashi, Bioorg. Med. Chem., 2004, 12, 5297 CrossRefCASPubMed.
P. H. Dussault, T. K. Trullinger and F. Noor-e-Ain, Org. Lett., 2002, 4, 4591 CrossRefCASPubMed.
P. Dai, T. K. Trullinger, X. Liu and P. H. Dussault, J. Org. Chem., 2006, 71, 2283 CrossRefCASPubMed.
A. J. Bloodworth, B. D. Bothwell, A. N. Collins and N. L. Maidwell, Tetrahedron Lett., 1996, 37, 1885 CrossRefCAS.
X. Lu, Y. Liu, B. Sun, B. Cindric and L. Deng, J. Am. Chem. Soc., 2008, 130, 8134 CrossRefCASPubMed.
L. Hu, X. Lu and L. Deng, J. Am. Chem. Soc., 2015, 137, 8400 CrossRefCASPubMed.
For a recent review on asymmetric peroxidation, see G. D. Sala and A. Lattanzi, ACS Catal., 2014, 4, 1234 CrossRef.
N. Kornblum and H. E. DeLaMare, J. Am. Chem. Soc., 1951, 73, 880 CrossRefCAS.
N. Kornblum and H. E. DeLaMare, J. Am. Chem. Soc., 1952, 74, 3079 CrossRefCAS.
M. Ishibashi, S. Takeuchi and J. Kobayashi, Tetrahedron Lett., 1993, 34, 3749 CrossRefCAS.
Y.-C. Shen, C. V. S. Prakash and Y.-H. Kuo, J. Nat. Prod., 2001, 64, 324 CrossRefCAS.
M. D. Norris and M. V. Perkins, Tetrahedron, 2013, 69, 9813 CrossRefCAS.
Epoxides 45 and 49 are reported to have a syn-oxirane, E-olefin and anti-oxirane, Z-olefin arrangement, respectively; although the data presented in literature is not directly comparable (see ref. 95 and 113). Since natural plakortic acid was originally co-isolated with 3 and base-mediated rearrangement of 3 appears to be likely, we speculate that the correct structure of natural plakortic acid may in fact be the acid of oxirane 49, where R = (CH2)15CH3 (Scheme 14 in text).
M. Asami, T. Ishizaki and S. Inoue, Tetrahedron Lett., 1995, 36, 1893 CrossRefCAS.
H. Miyaoka, T. Watanuki, Y. Saka and Y. Yamada, Tetrahedron, 1995, 51, 8749 CrossRefCAS.
S. Al-Busafi, M. G. B. Drew, T. Sanders and R. C. Whitehead, Tetrahedron Lett., 1998, 39, 1647 CrossRefCAS.
S. Al-Busafi and R. C. Whitehead, Tetrahedron Lett., 2000, 41, 3467 CrossRefCAS.
S. Al-Busafi, J. R. Doncaster, M. G. B. Drew, A. C. Regan and R. C. Whitehead, J. Chem. Soc., Perkin Trans. 1, 2002, 476 RSC.
K. Takeda, I. Nakayama and E. Yoshii, Synlett, 1994, 3, 178 CrossRef.
S. Tsukamoto, S. Takeuchi, M. Ishibashi and J. Kobayashi, J. Org. Chem., 1992, 57, 5255 CrossRefCAS.
J. Kobayashi, S. Tsukamoto, S. Takeuchi and M. Ishibashi, Tetrahedron, 1993, 49, 5955 CrossRefCAS.
M. Perpelescu, M. Tsuda, M. Suzuki, S. Yoshida and J. Kobayashi, Nat. Med., 2004, 58, 86 CAS.
F. Saito, R. Takeuchi, T. Kamino, K. Kuramochi, F. Sugawara, K. Sakaguchi and S. Kobayashi, Bioorg. Med. Chem. Lett., 2004, 14, 1975 CrossRefCASPubMed.
Y. Wakuda, T. Kubota, H. Shima, T. Okada, S. Mitsuhashi, N. Aoki, K. Kikuchi and J. Kobayashi, Mar. Drugs, 2006, 4, 9 CrossRef.
J. R. Doncaster, L. L. Etchells, N. M. Kershaw, R. Nakamura, H. Ryan, R. Takeuchi, K. Sakaguchi, A. Sardarian and R. C. Whitehead, Bioorg. Med. Chem. Lett., 2006, 16, 2877 CrossRefCASPubMed.
We expect that a similar structural reassignment is likely for compounds 56, 57 and 59.
Compounds 53–56, 58 and 59 (excluding 57, which has a valine residue attached) were reported with low magnitudes of optical rotation indicating that they may in fact have been isolated as pseudo-racemic. This is consistent with Whitehead's theory of biogenesis.
S. Takeuchi, T. Kikuchi, S. Tsukamoto, M. Ishibashi and J. Kobayashi, Tetrahedron, 1995, 51, 5979 CrossRefCAS.
T. Kubota, Y. Ishiguro, A. Takahashi-Nakaguchi, J. Fromont, T. Gonoi and J. Kobayashi, Bioorg. Med. Chem. Lett., 2013, 23, 244 CrossRefCASPubMed.
N. Tanaka, M. Asai, T. Kusama, J. Fromont and J. Kobayashi, Tetrahedron Lett., 2015, 56, 1388 CrossRefCAS.
N. Tanaka, M. Asai, A. Takahashi-Nakaguchi, T. Gonoi, J. Fromont and J. Kobayashi, Org. Lett., 2013, 15, 2518 CrossRefCASPubMed.
Y. Ishiguro, T. Kubota, J. Fromont, M. Shiro and J. Kobayashi, Tetrahedron Lett., 2010, 51, 4023 CrossRefCAS.
The acid of (Z)-50 (or (E)-50, Section 3.3) may decarboxylate prior to condensation with 68.
S. Takeuchi, M. Ishibashi and J. Kobayashi, J. Org. Chem., 1994, 59, 3712 CrossRefCAS.
Y. Ishiguro, T. Kubota, K. Ishiuchi, J. Fromont and J. Kobayashi, Tetrahedron Lett., 2009, 50, 3202 CrossRefCAS.
L. L. Etchells, A. Sardarian and R. C. Whitehead, Tetrahedron Lett., 2005, 46, 2803 CrossRefCAS.
In turn, plakorsin A (51) is expected to arise from the reduction of 2 or 3 (Section 4.4).
L. L. Etchells, M. Helliwell, N. M. Kershaw, A. Sardarian and R. C. Whitehead, Tetrahedron, 2006, 62, 10914 CrossRefCAS.
D. Ma and H. Sun, Tetrahedron Lett., 2000, 41, 1947 CrossRefCAS.
J. A. Stafford, Tetrahedron Lett., 1995, 36, 681 CrossRefCAS.
P. A. Bartlett and C. Chapuis, J. Org. Chem., 1986, 51, 2799 CrossRefCAS.
R. S. Compagnone, I. C. Pina, H. R. Rangel, F. Dagger, A. I. Suarez, M. V. R. Reddy and D. J. Faulkner, Tetrahedron, 1998, 54, 3057 CrossRefCAS.
R. Ueoka, Y. Nakao, S. Kawatsu, J. Yaegashi, Y. Matsumoto, S. Matsunaga, K. Furihata, R. W. M. van Soest and N. Fusetani, J. Org. Chem., 2009, 74, 4203 CrossRefCASPubMed.
C. Festa, G. Lauro, S. De Marino, M. V. D'Auria, M. C. Monti, A. Casapullo, C. D'Amore, B. Renga, A. Mencarelli, S. Petek, G. Bifulco, S. Fiorucci and A. Zampella, J. Med. Chem., 2012, 55, 8303 CrossRefCASPubMed.
R. J. Capon, S. Singh, S. Ali and S. Sotheeswaran, Aust. J. Chem., 2005, 58, 18 CrossRefCAS.
R. A. Epifanio, L. S. Pinheiro and N. C. Alves, J. Braz. Chem. Soc., 2005, 16, 1367 CrossRefCAS.
G. Chianese, B.-B. Gu, F. Yang, W.-H. Jiao, Y.-W. Guo, H.-W. Lin and O. Taglialetella-Scafati, RSC Adv., 2015, 5, 63372 RSC.
D. B. Stierle and D. J. Faulkner, J. Org. Chem., 1980, 45, 3396 CrossRefCAS.
Faulkner also isolated hemiacetal adducts related to 80 (ref. 51), which indicates that Kornblum–DeLaMare rearrangement may also be a prevalent decomposition pathway for saturated 1,2-dioxanes.
X.-F. Liu, Y. Shen, F. Yang, M. T. Hamann, W.-H. Jiao, H.-J. Zhang, W.-S. Chen and H.-W. Lin, Tetrahedron, 2012, 68, 4635 CrossRefCAS.
M. D. Norris, M. V. Perkins and E. J. Sorensen, Org. Lett., 2015, 17, 668 CrossRefCASPubMed.
M. Akiyama, Y. Isoda, M. Nishimoto, A. Kobayashi, D. Togawa, N. Hirao, A. Kuboki and S. Ohira, Tetrahedron Lett., 2005, 46, 7483 CrossRefCAS.
C. Festa, S. De Marino, M. V. D'Auria, E. Deharo, G. Gonzalez, C. Deyssard, S. Petek, G. Bifulco and A. Zampella, Tetrahedron, 2012, 68, 10157 CrossRefCAS.
C. Festa, C. D'Amore, B. Renga, G. Lauro, S. De Marino, M. V. D'Auria, G. Bifulco, A. Zampella and S. Fiorucci, Mar. Drugs, 2013, 11, 2314 CrossRefPubMed.
X.-Y. Shen, X.-S. Peng and H. N. C. Wong, Org. Lett., 2016, 18, 1032 CrossRefCASPubMed.
C. M. Rasik and M. K. Brown, Angew. Chem., Int. Ed., 2014, 53, 14522 CrossRefCASPubMed.
S.-J. Piao, Y.-L. Song, W.-H. Jiao, F. Yang, X.-F. Liu, W.-S. Chen, B.-N. Han and H.-W. Lin, Org. Lett., 2013, 15, 3526 CrossRefCASPubMed.
Interestingly, monotriajaponide A, isolated in 2003 (ref. 34) appears to represent a des-hydroperoxy precursor related to haterumadioxin A (7).
S. A. Ruider, T. Sandmeier and E. M. Carreira, Angew. Chem., Int. Ed., 2015, 54, 2378 CrossRefCASPubMed.
S. A. Ruider and E. M. Carreira, Org. Lett., 2016, 18, 220 CrossRefCASPubMed.
M. E. McCallum, C. M. Rasik, J. L. Wood and M. K. Brown, J. Am. Chem. Soc., 2016, 138, 2437 CrossRefCASPubMed.
R. Datta, R. J. Dixon and S. Ghosh, Tetrahedron Lett., 2016, 57, 29 CrossRefCAS.
D. T. Sawyer, J. P. Hage and A. Sobkowiak, J. Am. Chem. Soc., 1995, 117, 106 CrossRefCAS.
O. Taglialetela-Scafati, E. Fattorusso, A. Romano, F. Scala, V. Barone, P. Cimino, E. Stendardo, B. Catalanotti, M. Persico and C. Fattorusso, Org. Biomol. Chem., 2010, 8, 846 Search PubMed.
F. S. De Guzman and F. J. Schmitz, J. Nat. Prod., 1990, 53, 926 CrossRefCAS.
C. Fattorusso, G. Campiani, B. Catalanotti, M. Persico, N. Basilico, S. Parapini, D. Taramelli, C. Campagnuolo, E. Fattorusso, A. Romano and O. Taglialetela-Scafati, J. Med. Chem., 2006, 49, 7088 CrossRefCASPubMed.
C. Campagnuolo, E. Fattorusso, O. Taglialatela-Scafati, A. Ianaro and B. Pasino, Eur. J. Org. Chem., 2002, 61 CrossRefCAS.
C. Campagnuolo, C. Fattorusso, E. Fattorusso, A. Ianaro, B. Pisano and O. Taglialatela-Scafati, Org. Lett., 2003, 5, 673 CrossRefCASPubMed.
J. P. John, J. Jost and A. V. Novikov, J. Org. Chem., 2009, 74, 6083 CrossRefCASPubMed.
F. Cafieri, E. Fattorusso, O. Taglialatela-Scafati, M. Di Rosa and A. Ianaro, Tetrahedron, 1999, 55, 13831 CrossRefCAS.
A. Rudi and Y. Kashman, J. Nat. Prod., 1993, 56, 1827 CrossRefCAS.
M. F. Semmelhack and P. Shanmugam, Tetrahedron Lett., 2000, 41, 3567 CrossRefCAS.
M. F. Semmelhack, R. J. Hooley and C. M. Kraml, Org. Lett., 2006, 8, 5203 CrossRefCASPubMed.
G. C. Paddon-Jones, N. L. Hungerford, P. Hayes and W. Kitching, Org. Lett., 1999, 1, 1905 CrossRefCAS.
P. Y. Hayes and W. Kitching, J. Am. Chem. Soc., 2002, 124, 9718 CrossRefCASPubMed.
P. Y. Hayes and W. Kitching, Heterocycles, 2004, 62, 173 CrossRefCAS.
P. Y. Hayes, S. Chow, F. Rahm, P. V. Bernhardt, J. J. De Voss and W. Kitching, J. Org. Chem., 2010, 75, 6489 CrossRefCASPubMed.
H.-K. Lee and H. N. C. Wong, Chem. Commun., 2002, 2114 RSC.
X.-G. Xie, X.-W. Wu, H.-K. Lee, X.-S. Peng and H. N. C. Wong, Chem.–Eur. J., 2010, 16, 6933 CrossRefCASPubMed.
G. Mehta, B. A. Bhat and T. H. S. Kumara, Tetrahedron Lett., 2009, 50, 6597 CrossRefCAS.
C. Bittner, A. Burgo, P. J. Murphy, C. H. Sung and A. J. Thornhill, Tetrahedron Lett., 1999, 40, 3455 CrossRefCAS.
M. Akiyama, Y. Isoda, M. Nishimoto, M. Narazaki, H. Oka, A. Kuboki and S. Ohira, Tetrahedron Lett., 2006, 47, 2287 CrossRefCAS.
H. Sugimura, S. Sato, K. Tokudome and T. Yamada, Org. Lett., 2014, 16, 3384 CrossRefCASPubMed.
M. Tsuda, T. Endo, M. Perpelescu, S. Yoshida, K. Watanabe, J. Fromont, Y. Mikami and J. Kobayashi, Tetrahedron, 2003, 59, 1137 CrossRefCAS.
F. Saito, R. Takeuchi, T. Kamino, K. Kuramochi, F. Sugawara, K. Sakaguchi, S. Kobayashi, M. Tsudad and J. Kobayashi, Tetrahedron Lett., 2004, 45, 8069 CrossRefCAS.
K. Kuramochi, F. Saito, R. Takeuchi, T. Era, M. Takemura, J. Kobayashi, K. Sakaguchi, S. Kobayashi and F. Sugawara, Tetrahedron, 2006, 62, 8006 CrossRefCAS.
H. Mizutani, M. Watanabe and T. Honda, Synlett, 2005, 793 CAS.
G. Chianese, M. Persico, F. Yang, H.-W. Lin, Y.-W. Guo, N. Basilico, S. Parapini, D. Taramelli, O. Taglialatela-Scafati and C. Fattorusso, Bioorg. Med. Chem., 2014, 22, 4572 CrossRefCASPubMed.
T. Xu, Q. Feng, M. R. Jacob, B. Avula, M. M. Mask, S. R. Baerson, S. K. Tripathi, R. Mohammed, M. T. Hamann, I. A. Khan, L. A. Walker, A. M. Clark and A. K. Agarwal, Antimicrob. Agents Chemother., 2011, 55, 1611 CrossRefCASPubMed.

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: