
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-adiabatic excited state molecular dynamics of phenylene ethynylene dendrimer using a multiconfigurational Ehrenfest approach
Sebastian
Fernandez-Alberti
a,
Dmitry V.
Makhov
*b,
Sergei
Tretiak
c and
Dmitrii V.
Shalashilin
b
aUniversidad Nacional de Quilmes, Roque Saenz Peña 352, B1876BXD Bernal, Argentina
bSchool of Chemistry, University of Leeds, Leeds LS2 9JT, UK. E-mail: d.makhov@leeds.ac.uk
cCenter for Nonlinear Studies (CNLS), and Center for Integrated Nanotechnologies (CINT), Los Alamos National Laboratory, Los Alamos, NM 87545, USA
First published on 10th March 2016
Abstract
Photoinduced dynamics of electronic and vibrational unidirectional energy transfer between meta-linked building blocks in a phenylene ethynylene dendrimer is simulated using a multiconfigurational Ehrenfest in time-dependent diabatic basis (MCE-TDDB) method, a new variant of the MCE approach developed by us for dynamics involving multiple electronic states with numerous abrupt crossings. Excited-state energies, gradients and non-adiabatic coupling terms needed for dynamics simulation are calculated on-the-fly using the Collective Electron Oscillator (CEO) approach. A comparative analysis of our results obtained using MCE-TDDB, the conventional Ehrenfest method and the surface-hopping approach with and without decoherence corrections is presented.
I. Introduction
Dendrimers are highly branched conjugated macromolecules that possess well-defined regular structures1 with numerous peripheral groups, branched repeat units, and a core. Each of these components acts as an individual chromophore unit absorbing light in different characteristic ranges of the spectrum.2 Their unique geometric and energetic properties3 have made them the subject of a wide variety of technological1,4 and academic studies.5 Dendrimers have many useful properties, such as well-controlled synthesis,6 exceptional light-harvesting capabilities over a broad region of the solar spectrum, and very efficient energy funneling7 from the peripheral groups to the core. The efficient and controllable unidirectional energy transfer is associated with the π-conjugation of the regular arrays of coupled chromophore units in the dendritic macromolecule.5bThe time evolution of electronic excitation in organic conjugated materials, such as dendrimers, is determined by non-adiabatic dynamics involving multiple coupled electronic excited states. Following photoexcitation, the multiple photoinduced pathways to electronic and vibrational relaxation involve energy and/or charge transfer, internal conversion, and transition density localization/delocalization. Different theoretical approaches can deal with such photophysical processes.
A number of models, such as Pariser–Parr–Pople,8 Frenkel exciton,9 and Frenkel–Holstein,10 have been used to describe quasi one-dimensional conjugated systems. The real-time Density Matrix Renormalization Group (DMRG)11 technique is a powerful numerical method for studying these model systems. In particular, PPE dendritic models can be described using Frenkel exciton Hamiltonians12 with Coulombic coupling between excitations localized on the linear units.13
In atomistic molecular dynamics simulations, surface hopping has been proved to be an efficient method to accurately deal with non-adiabatic processes in a large variety of systems. Nevertheless, it presents certain shortcomings. First, electronic decoherence effects are not accounted for in the standard surface hopping algorithm resulting in a number of computational inconsistencies that have been addressed by implementing a large variety of alternative decoherence corrections.14 The inadequate treatment of decoherence can, in many cases, influence the accuracy of the simulated results. Moreover, there is no clear and direct strategy to improve surface hopping methods in order to introduce nuclear quantum effects such as delocalization, interferences and tunneling.
Among the most accurate fully quantum methods is time-dependent Hartree (MCTDH),15 which has been recently applied to simulate exciton dynamics in extended conjugated polymers.16 However, this approach, in its original version, requires the parameterization of potential energy surfaces. A useful alternative is represented by a variety of methods relying on trajectory-guided Gaussian basis functions (TBF). The main advantage of these methods is that they can deal with direct or ab initio molecular dynamics17 where excited state energies, gradients, and non-adiabatic coupling terms are evaluated on the fly, simultaneously with the nuclear propagation. In a variational multi-configuration Gaussian (vMCG)18 approach, the trajectories for all TBFs are calculated together from the variational principle. vMCG is similar in spirit to MCTDH and it is among the most elegant methods of direct dynamics. However, because of its complexity, on the fly vMCG so far has been implemented only for a small number of basis Gaussians.
Another group of methods17d,19 applies TBFs moving along classical trajectories. Despite that these methods are still fully quantum because these trajectories are employed only for propagating the basis, while the evolution of their amplitudes and, thus, of the total wave-function is determined by the time-dependent Schrödinger equation. The trajectories here are calculated independently allowing for a large and well-sampled basis. The methods involving branching, such as multiple spawning17d,19a or multiple cloning19g algorithms can hardly be used for dendrimers because the large number of electronic states and intersections between them would lead to the uncontrolled growth of the number of trajectories. But, on the other hand, the same makes dendrimers and other large conjugated molecules a nearly ideal object for Ehrenfest based approaches,19b–f,20 where nuclear motion is defined by the average field, and amplitudes for all electronic states are propagated together with nuclear coordinates. In such systems, a wave packet undergoes frequent transition between many coupled electronic states, and as a result its motions can be frequently well described by the Ehrenfest trajectory even on long time scales.
In recent years we have been developing a group of methods based on multicofigurational Ehrenfest ansatz for the total wave function.19d–g The central idea is to use Gaussian wavepackets guided by Ehrenfest average of the electronic states. Several variations of this idea have been presented. Our original version of ab initio MCE was using adiabatic electronic states generated by the electronic structure. Here we suggest a modification of the approach, which now has new features: each Gaussian trajectory carries its own electronic basis, which is adiabatic only in the center of the Gaussian. This is similar to the approach described in the appendix in ref. 19g, except that now we accurately take into account the overlaps between electronic states belonging to different trajectories, instead of assuming them to be Kronecker's delta as in ref. 19g. This modification makes the method applicable to large conjugated molecules where electronic states can change significantly on the length-scale of the Gaussian width. In particular, the wave function can change instantly at trivial unavoided crossings between two electronic states localized at remote parts of a large molecule.21 To distinguish the new approach from the previous work we term it as multiconfigurational Ehrenfest in time-dependent diabatic basis (MCE-TDDB). We would like to emphasize that “time-dependent diabatic basis” used here should not be confused with “diabatic basis” commonly used in many studies: the trajectories here are still calculated in adiabatic basis, and the new representation affects only matrix elements between different TBFs.
We use the proposed MCE-TDDB method to simulate the excited state dynamics of the system composed of two- and three-ring linear polyphenylene ethynylene (PPE) chromophore units linked through meta-substitution as shown in Fig. 1(a). These chromophore units correspond to the building blocks of more complex phenylethynylperylene-terminated dendrimers, such as the nanostar.5b,22 The meta-branching localizes the electronic excited states within each linear fragment, and the non-adiabatic coupling between linear fragments are responsible for quantum transitions resulting in the two-ring → three ring unidirectional electronic energy flow. Thus, this system is a good model for analyzing intramolecular electronic and vibrational energy transfer between chromophore units subsequent to the photoexcitation of dendrimer macromolecules.
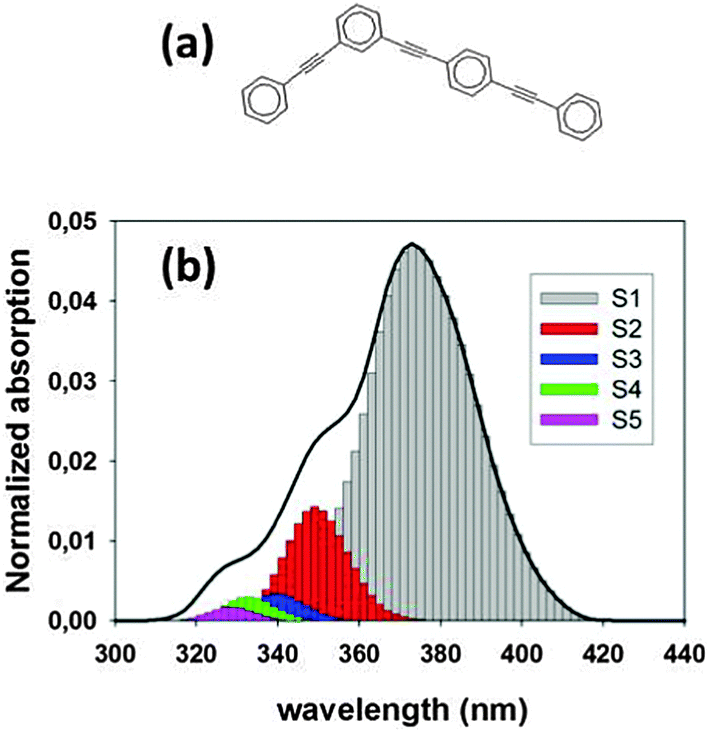 | ||
| Fig. 1 (a) Model dendritic molecule studied in this work. It involves two- and three-ring linear poly(phenylene ethynylene) units linked by meta-substitution. (b) Simulated absorption spectrum (solid line) showing the contributions of different excited states. | ||
We run the MCE-TDDB calculations in the basis of a swarm of interacting coherent state trains,19g,23 which is the most advanced basis for a given number of calculated trajectories. The excited state energies,13b,24 gradients25 and non-adiabatic coupling terms25a,26 needed to run the dynamics are calculated on the fly using the Collective Electron Oscillator (CEO) method.5a,27 The results are compared with those given by the canonical Ehrenfest approach (EHR) and by the NA-ESMD (non-adiabatic excited-state molecular dynamics)25a,28 method. The latter technique utilizes surface hopping guided by Tully's fewest-switches (FSSH) algorithm29 and was specifically developed to describe photoinduced dynamics in large organic conjugated molecules. In particular, the NA-ESMD framework has been extensively applied before to study the intramolecular energy transfer in PPE dendrimers.30
The paper is organized as follows. In Sections II and III we describe the MCE-TDDB method and computational details of simulations performed on our model system. In Section IV we present and discuss our results. Conclusions are given in Section V.
II. Theory
1. Working equations
Similar to the standard MCE approach, we represent the wave-function on a trajectory-guided basis |ψn(t)〉: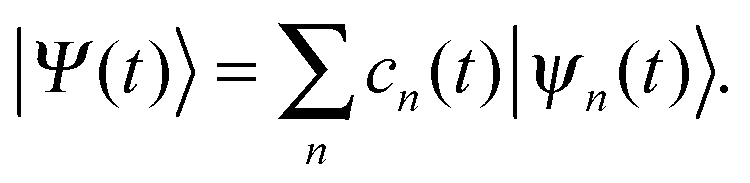 | (1) |
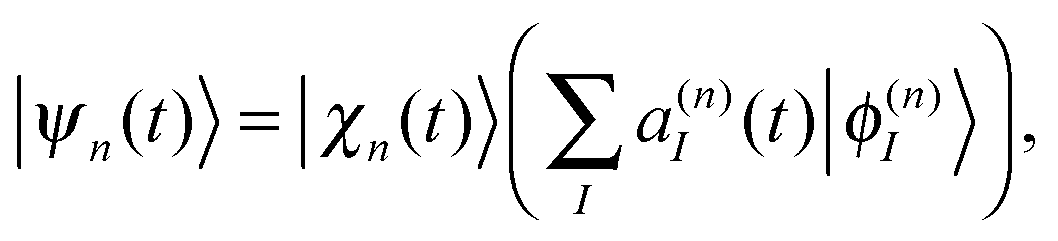 | (2) |
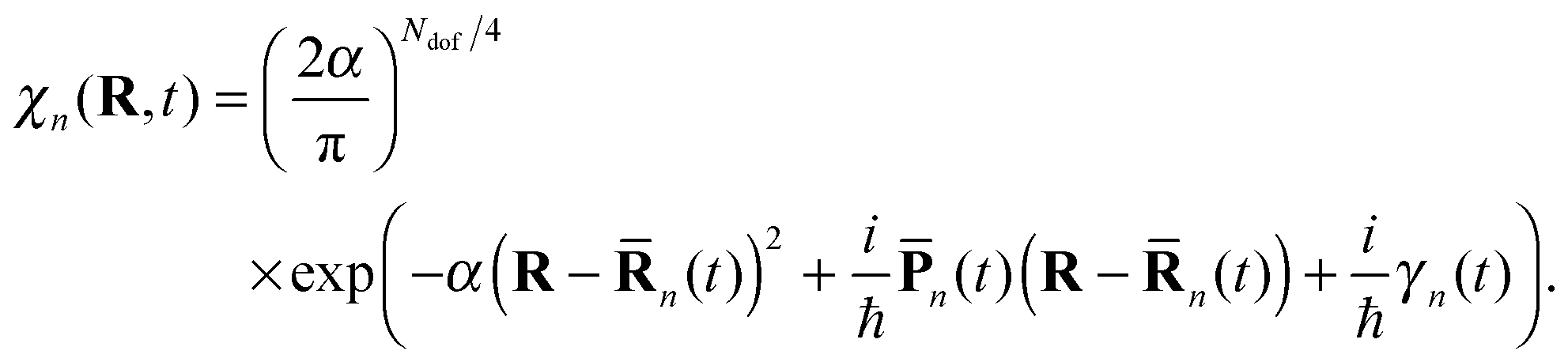 | (3) |
![[R with combining macron]](https://www.rsc.org/images/entities/b_char_0052_0304.gif) n(t) and
n(t) and ![[P with combining macron]](https://www.rsc.org/images/entities/b_char_0050_0304.gif) n(t) are the coordinate and momentum vectors of the n-th basis function center, and γn is a phase.
n(t) are the coordinate and momentum vectors of the n-th basis function center, and γn is a phase.
The basis |ϕ(n)I〉for the electronic part of wave-function can be specified in different ways. In the ab initio multiple spawning method and in the original version of the ab initio MCE approach, the electronic part is represented in the basis of adiabatic eigenstates:
| |ϕ(n)I〉 = |ϕI(r;R)〉. | (4) |
| |ϕ(n)I〉 = |ϕI(r;n(t))〉. | (5) |
However, this is not a case for the system under consideration, and a more rigorous approach must be developed. We will use electronic wave functions (5) and will take into account that the overlaps between the electronic eigenstates belonging to different Gaussians 〈ϕ(n)I|ϕ(m)J〉 can be very far from Kronecker's δIJ, even when these Gaussians are sufficiently close to each other and nuclear parts have a significant overlap.
The electronic overlaps 〈ϕ(n)I|ϕ(m)J〉 can be either calculated directly or propagated together with the basis. The latter approach is more convenient in this case, and the following equation for the time-dependence of the overlap integrals is used:
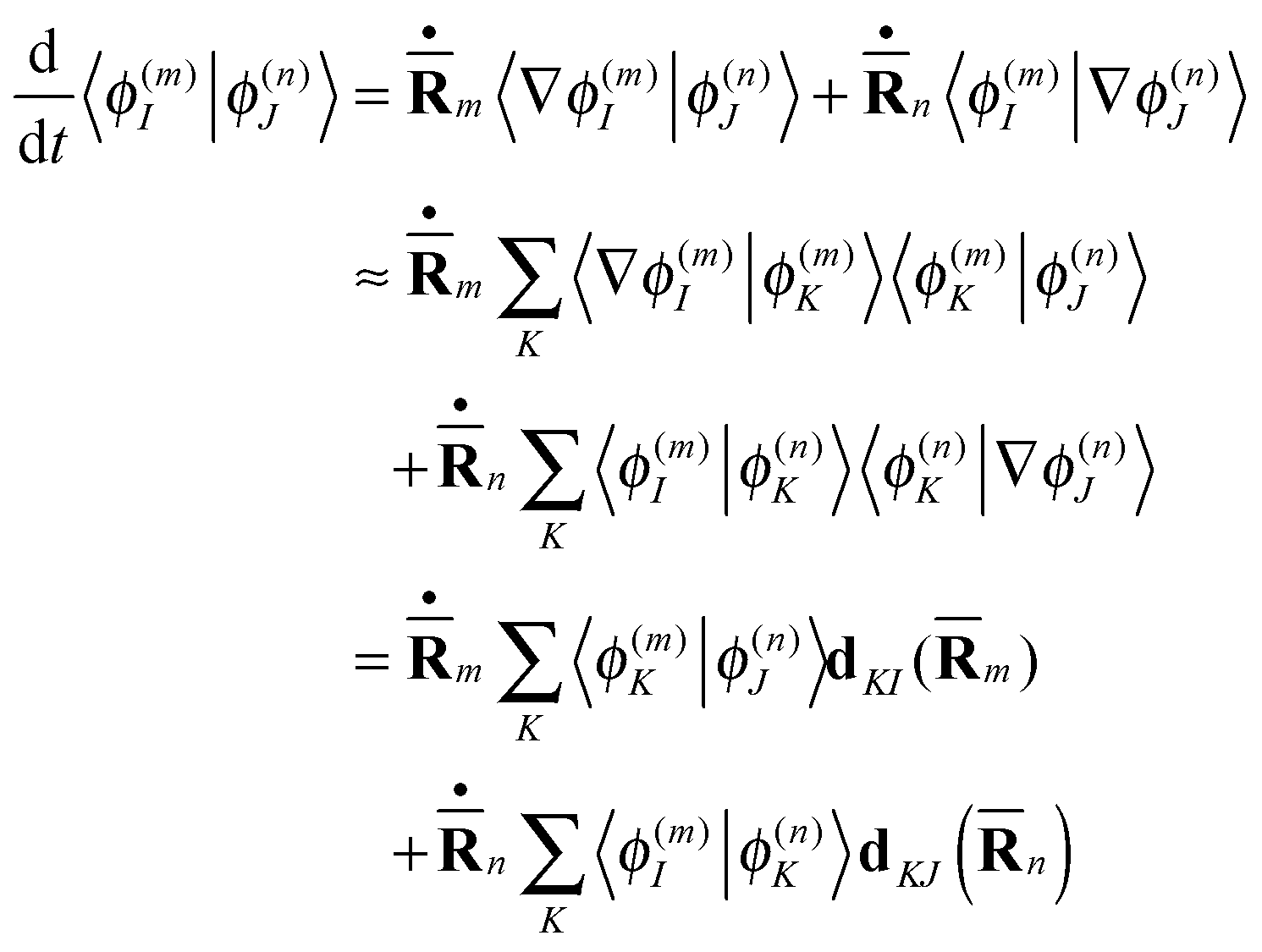 | (6) |
As before, the motion of the centers of Gaussians is determined by the usual set of Hamilton's equations
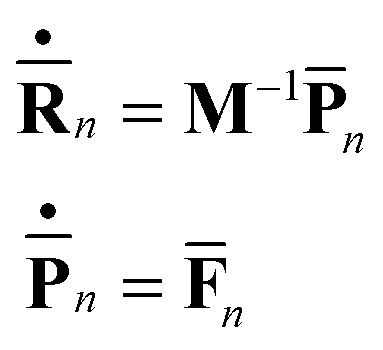 | (7) |
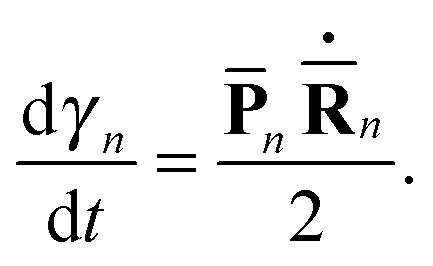 | (8) |
![[F with combining macron]](https://www.rsc.org/images/entities/b_char_0046_0304.gif) n here is an Ehrenfest force that includes both usual gradient terms and additional terms related to non-adiabatic derivative coupling:
n here is an Ehrenfest force that includes both usual gradient terms and additional terms related to non-adiabatic derivative coupling: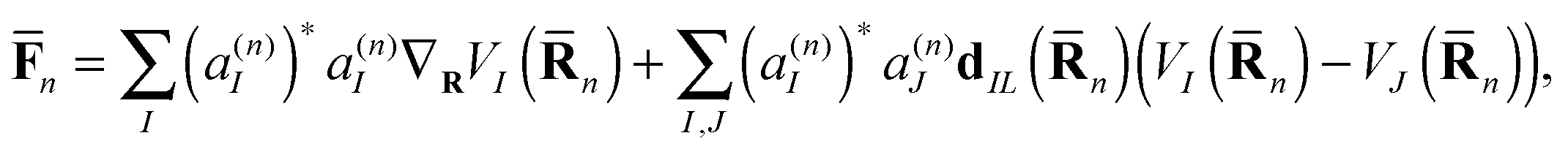 | (9) |
n) is the Ith potential energy surface and dIJ(n) = 〈ϕI|∇R|ϕJ〉 is the non-adiabatic coupling matrix element (NACME).
Also similar to our previous studies,19f,g the evolution of the Ehrenfest amplitudes a(n)I is determined by the equation
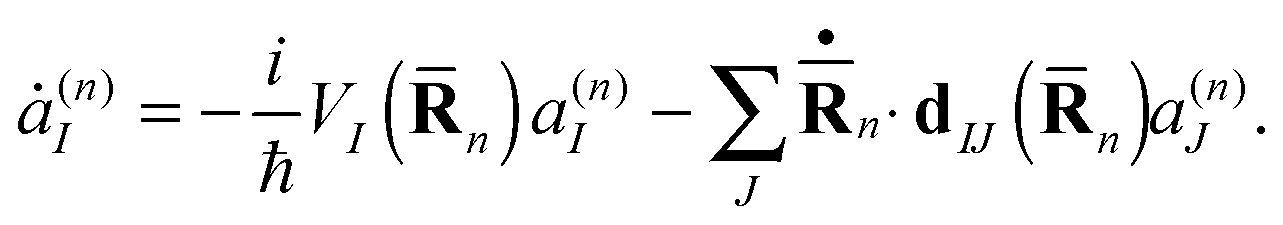 | (10) |
The above eqn (7)–(10) describe the evolution of the basis functions (2). Let us now consider the evolution of the whole wave-function (1) in the above-defined basis (2). Substituting eqn (1) into the time-dependent Schrodinger equation and taking into account non-orthogonality of the basis, we get
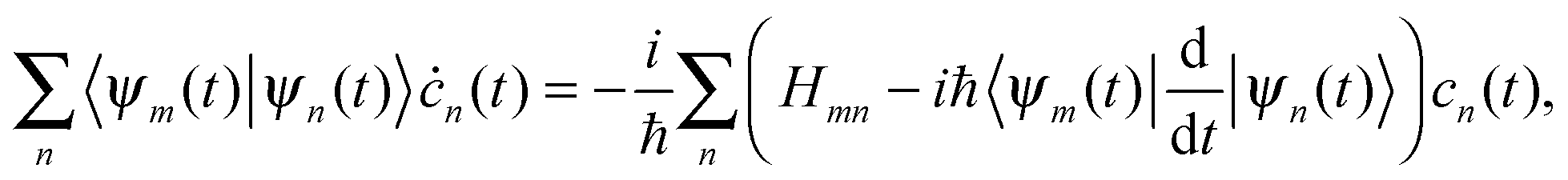 | (11) |
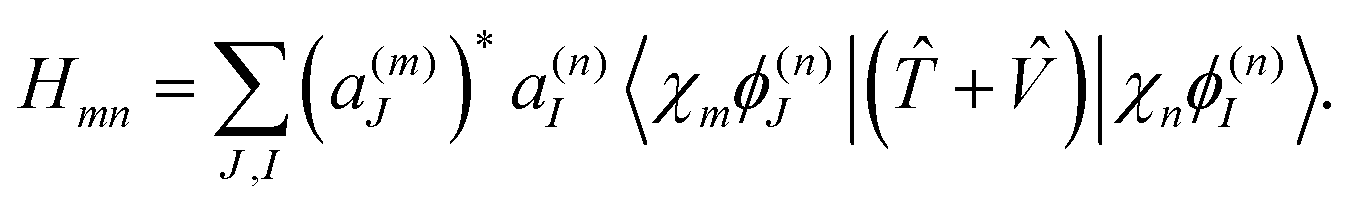 | (12) |
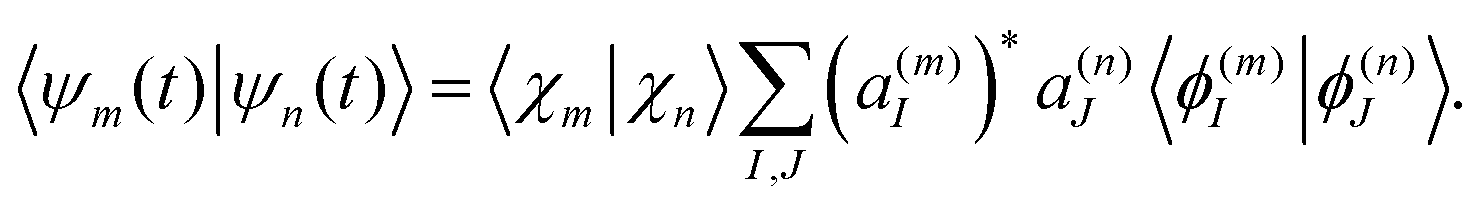 | (13) |
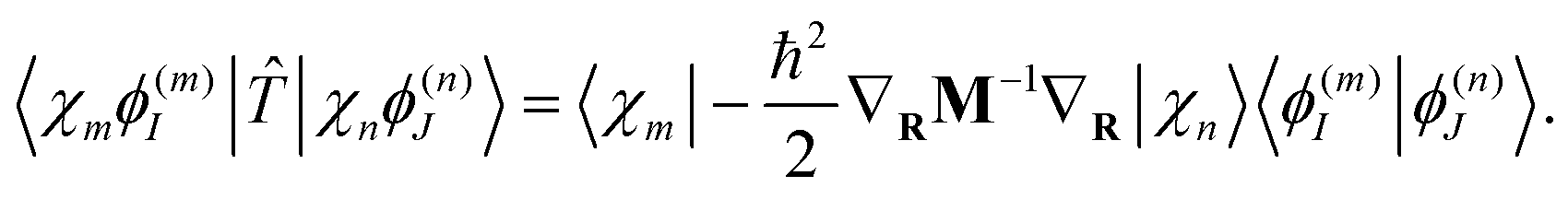 | (14) |
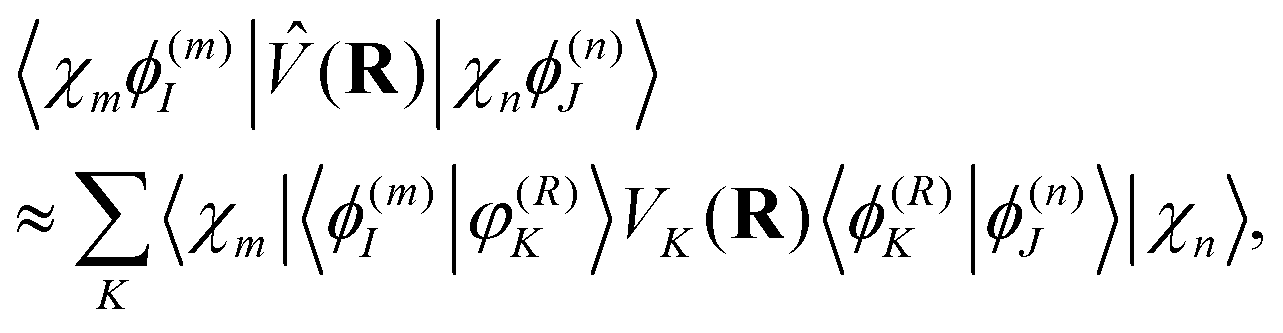 | (15) |
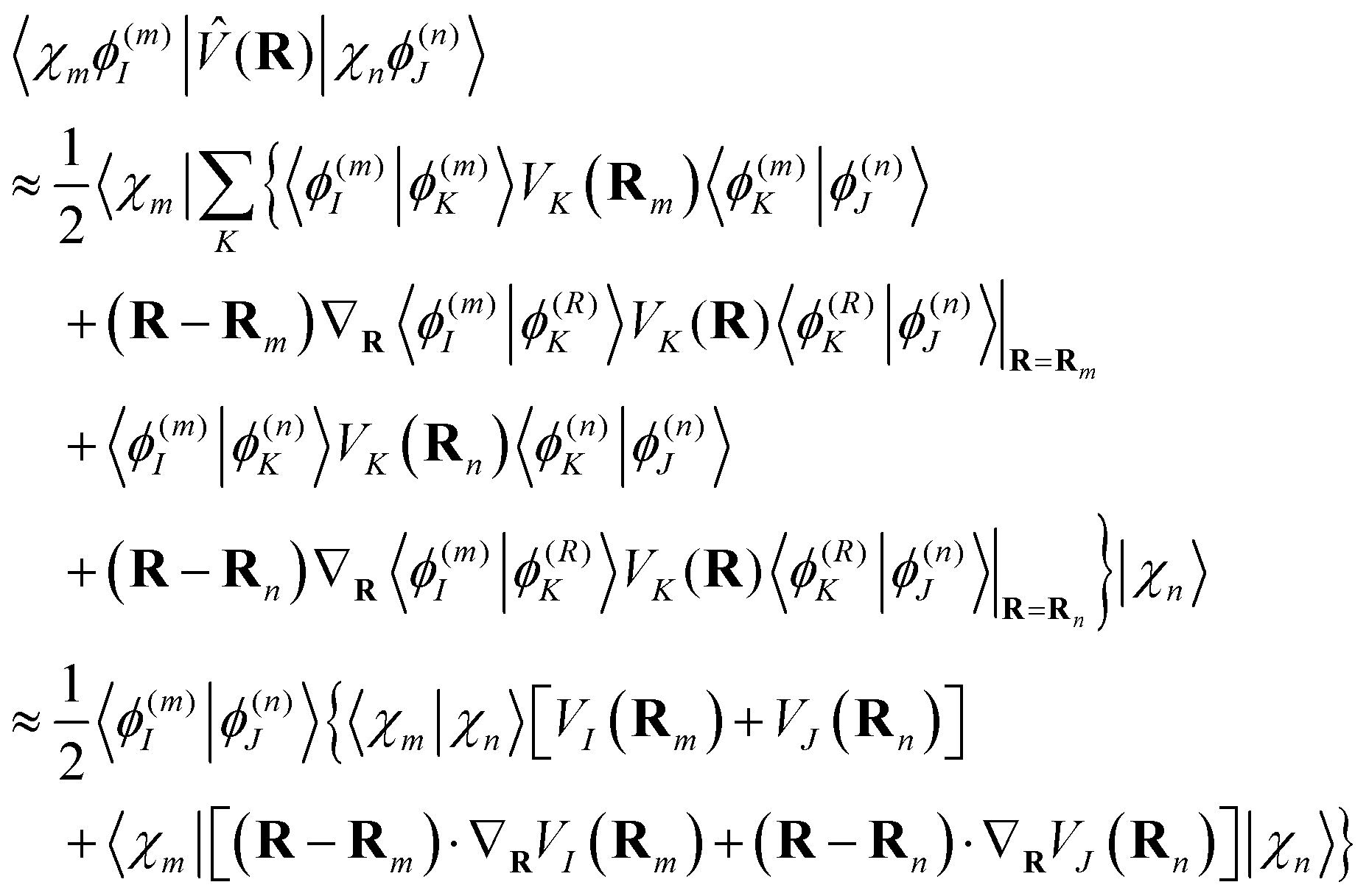 | (16) |
The term 〈Ψm(t)|d/dt|Ψn(t)〉 in eqn (11), which originates from the time dependence of the basis, can be written as
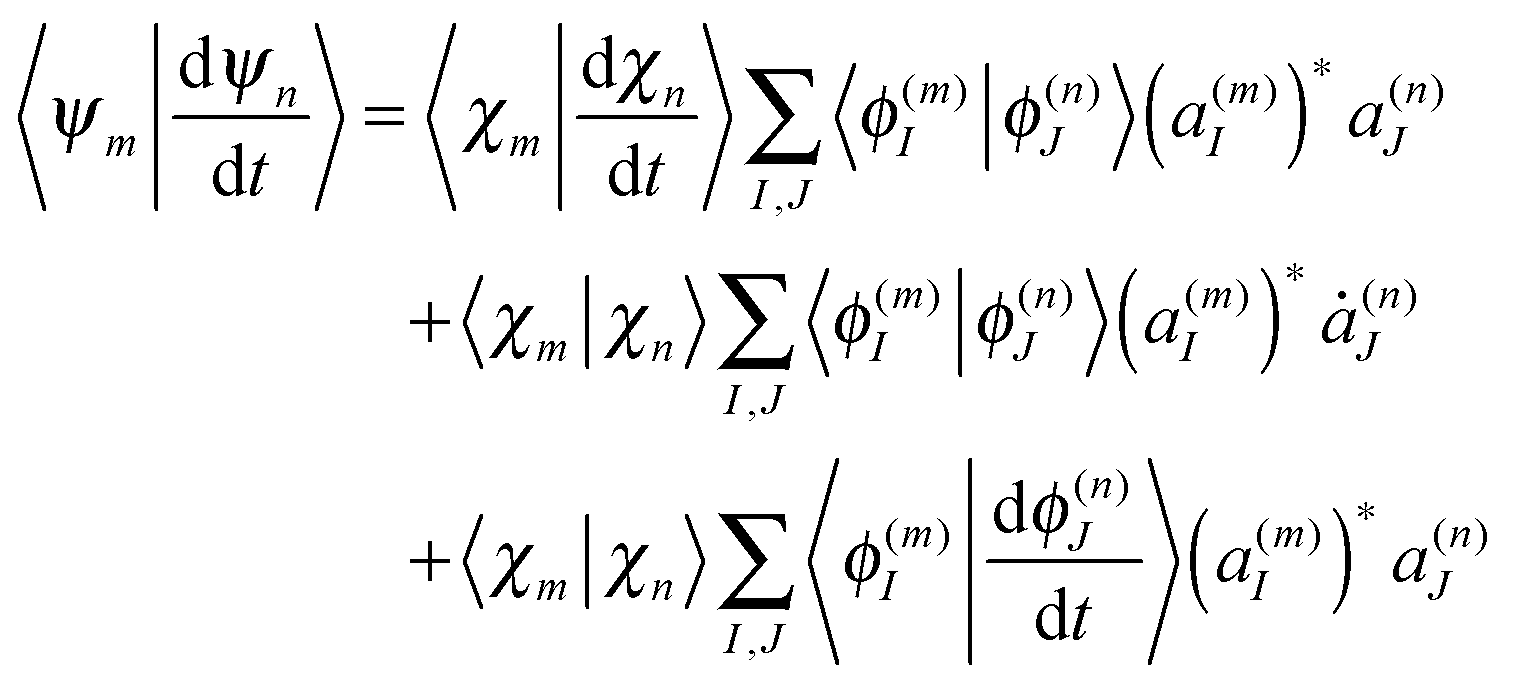 | (17) |
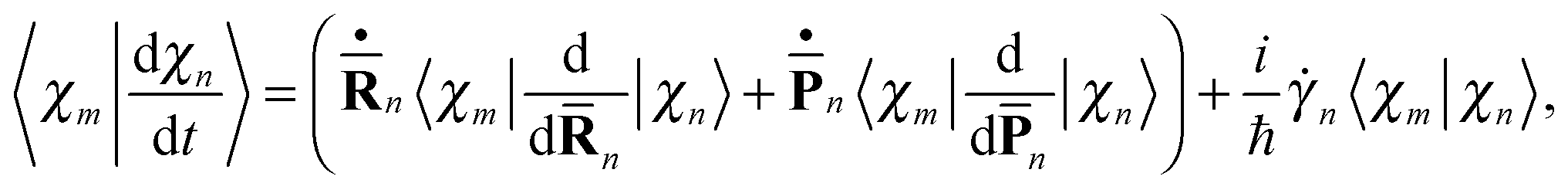 | (18) |
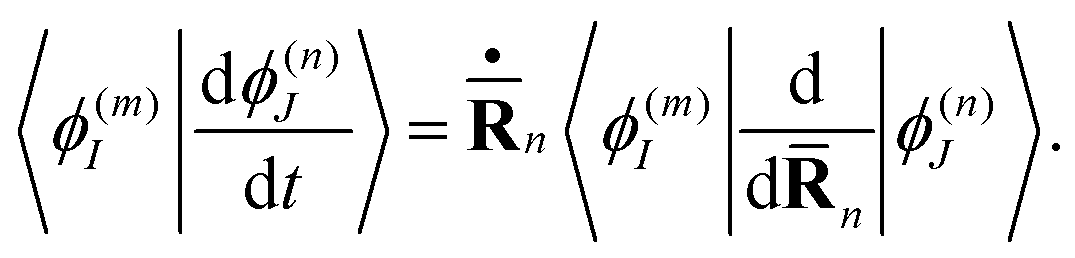 | (19) |
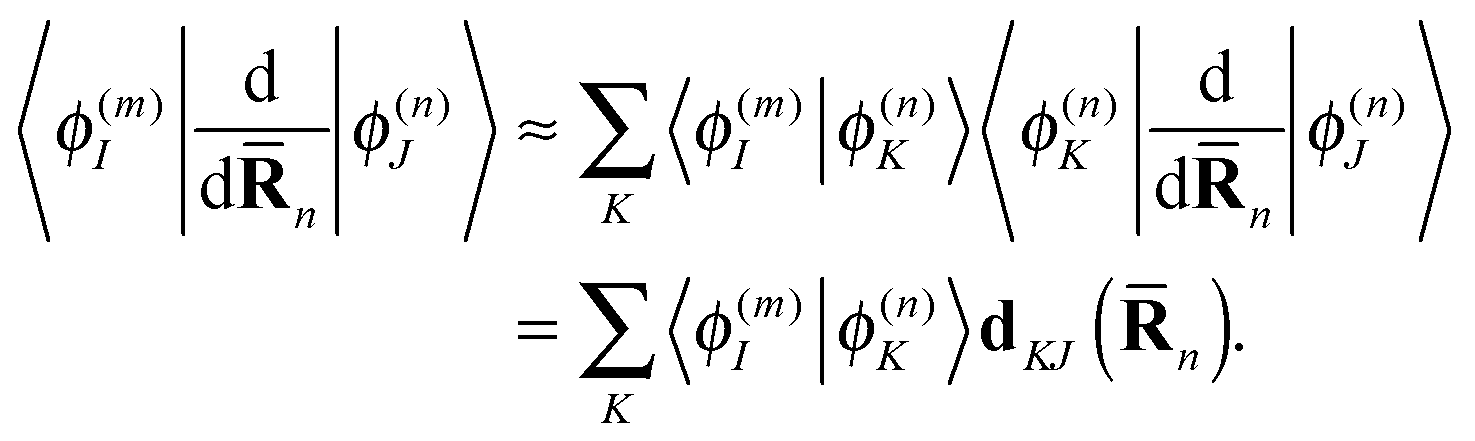 | (20) |
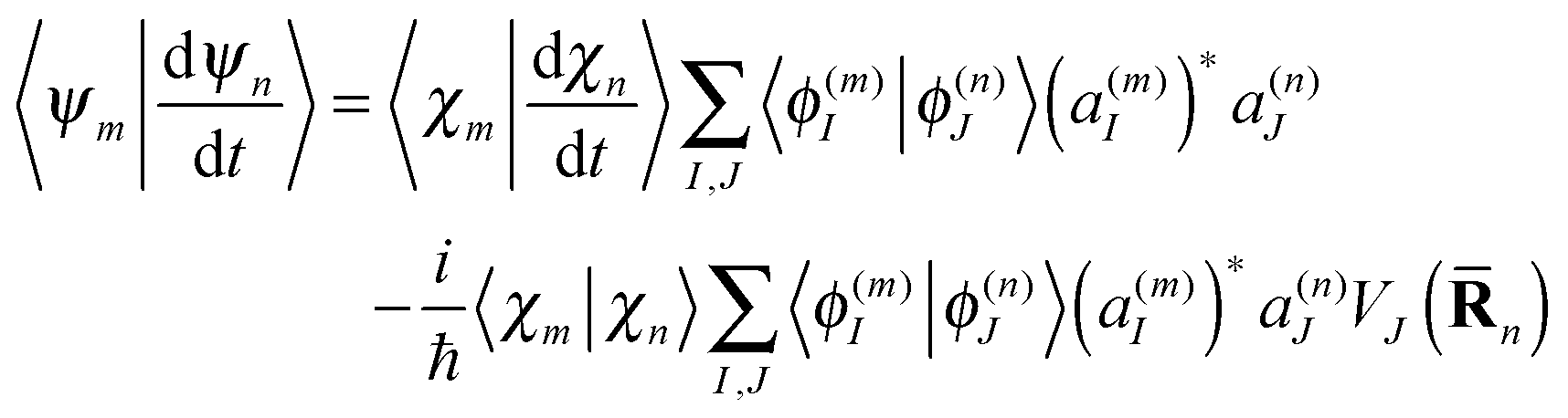 | (21) |
2. Population analysis
Population analysis is not trivial for the MCE-TDDB calculations when electronic states change very quickly because the order of electronic states can be different for different basis functions. Let us introduce the adiabatic population operator![[P with combining circumflex]](https://www.rsc.org/images/entities/i_char_0050_0302.gif) K(R) = |ϕ(R)K〉〈ϕ(R)K|, K(R) = |ϕ(R)K〉〈ϕ(R)K|, | (22) |
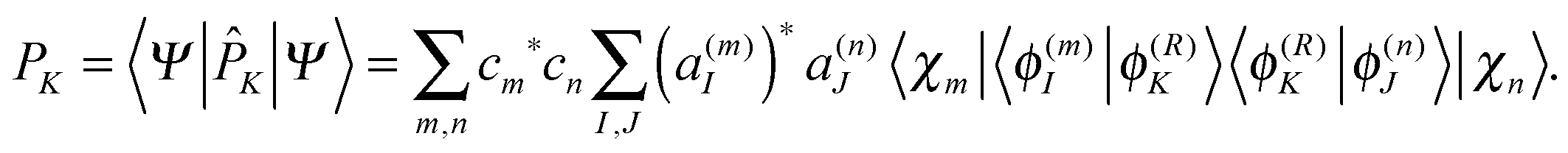 | (23) |
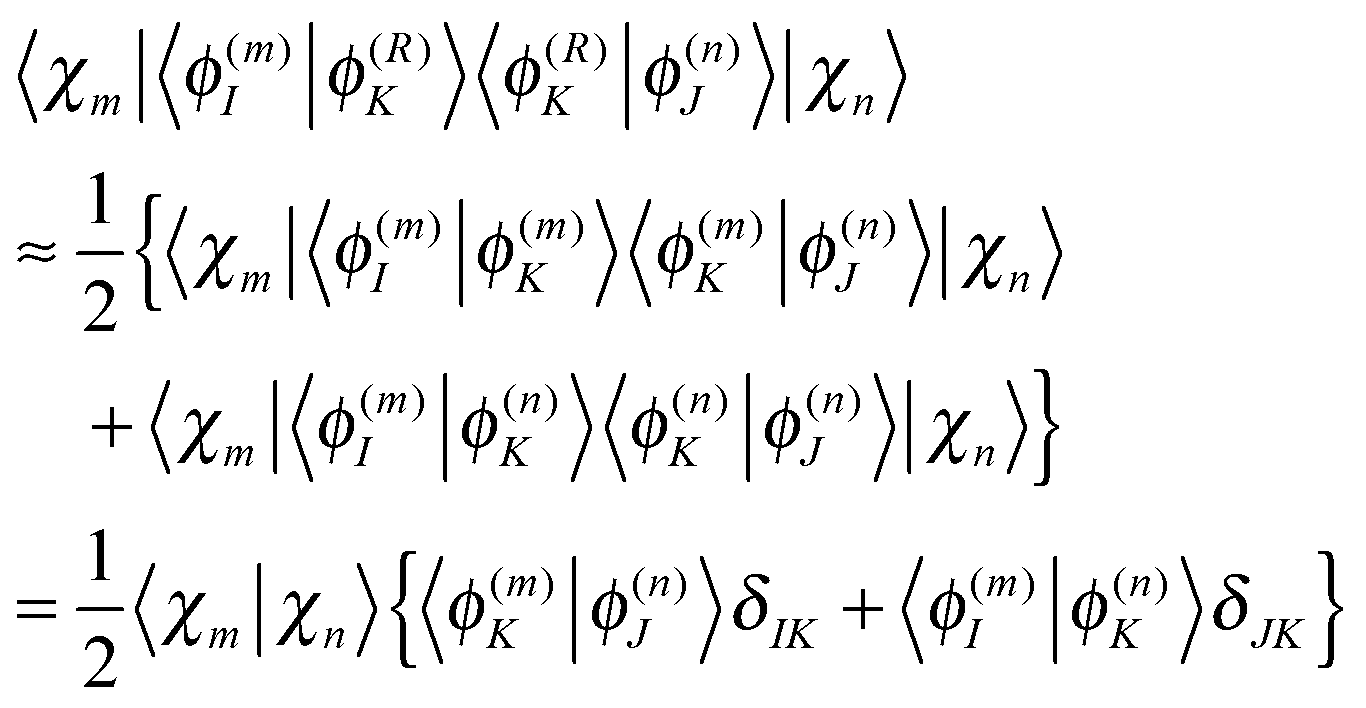 | (24) |
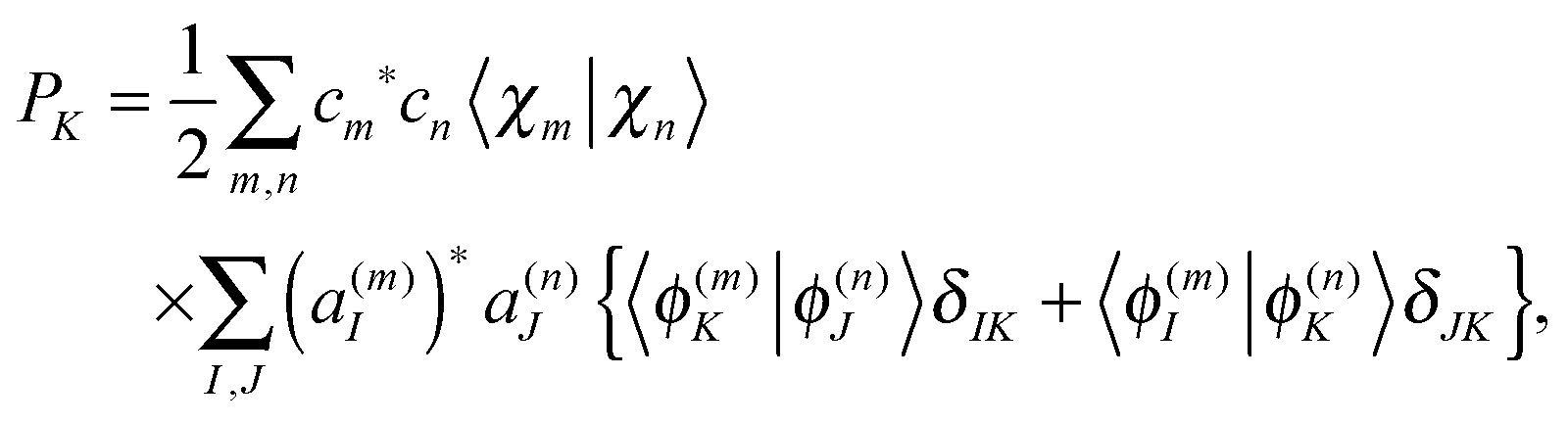 | (25) |
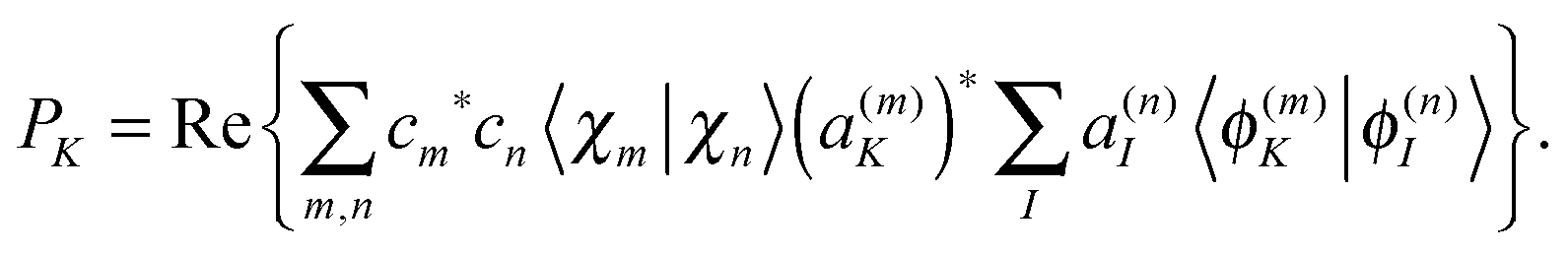 | (26) |
A similar approach can be applied for calculations of any other electronic properties, such as electronic transition density matrices reflecting spatial extent of the respective many-body electronic wave-functions. If the quantity is described by a quantum operator ![[N with combining circumflex]](https://www.rsc.org/images/entities/i_char_004e_0302.gif) , then
, then
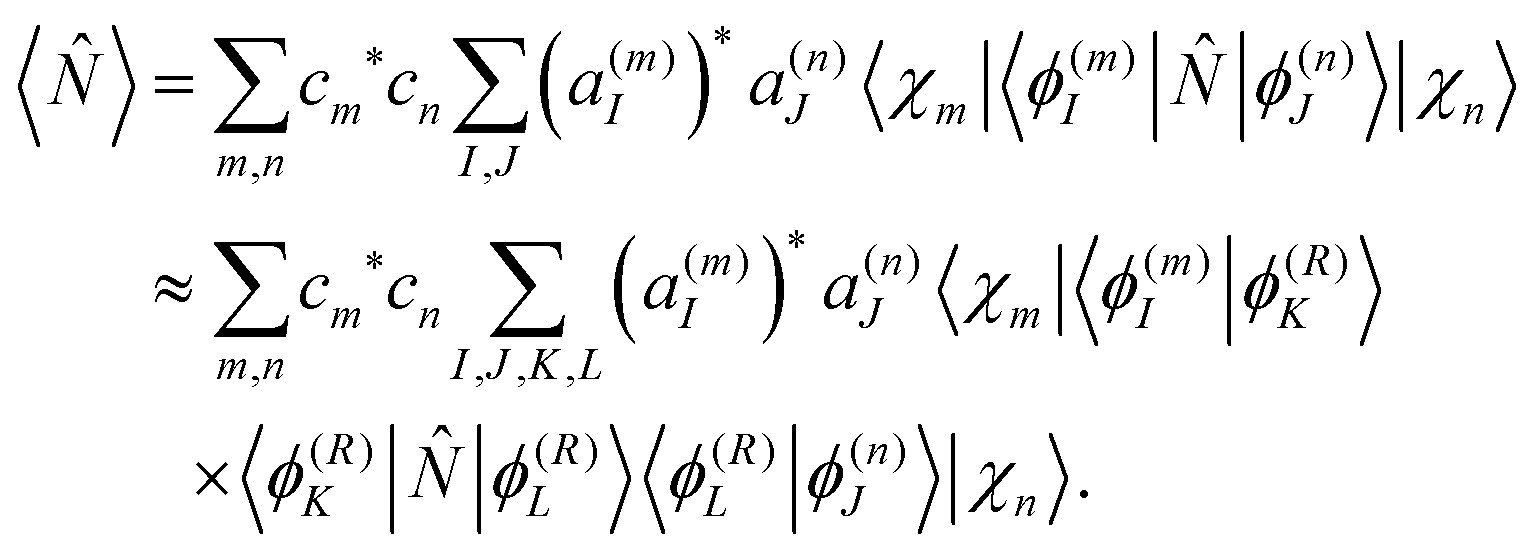 | (27) |
is real and depends only on the electronic degrees of freedom, we obtain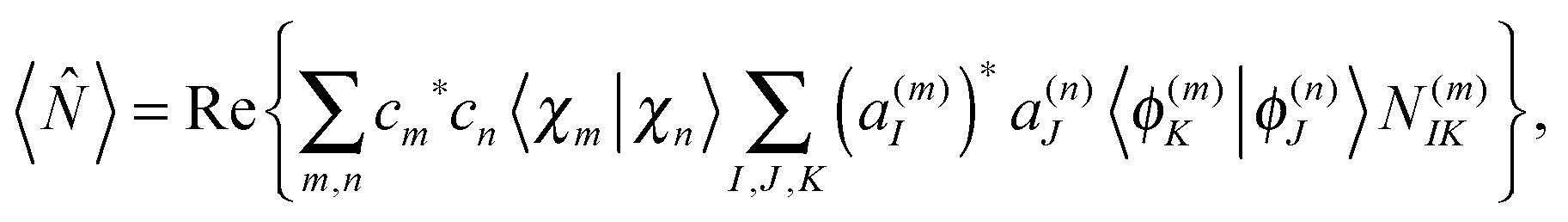 | (28) |
| N(m)IK = 〈ϕ(m)I||ϕ(m)K〉 | (29) |
between eigenstates for the center of mth Gaussian and, thus, are readily available from the electronic structure data for the trajectories.
3. Basis improvement – coherent state trains
The finite size of the basis set is the most serious limitation of the MCE and related methods, such as MS, vMCG, etc. In order to increase the size of the basis, we apply coherent state train basis sets (TBS),19g,23 where several Gaussian basis functions are moving along the same Ehrenfest trajectory but with a time-shift Δt. The most expensive part of ab initio molecular dynamics is the electronic structure calculations (energies, gradients, and NACMEs), and the computational cost can be greatly economized by using TBS because all basis functions in the train follow the same trajectory and, thus, repeatedly use the same electronic structure data. This trick allows us to increase the size of the basis by an order of magnitude practically without any additional computational cost. Fig. 2 summarizes the basis sampling ideas which were used in the current work.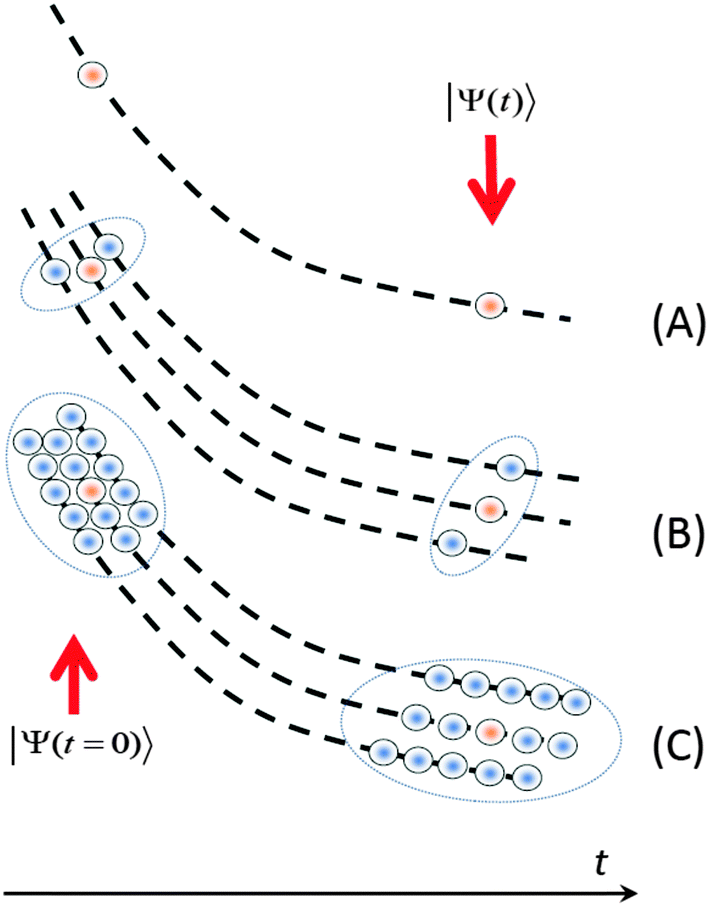 | ||
| Fig. 2 The basis sampling. (A) – a single Ehrenfest configuration; (B) – a swarm of trajectories; (C) a swarm of coherent state trains. The principle basis function is shown in orange. | ||
4. NA-ESMD framework
In this work we compare the results of our approach with those of the NA-ESMD25a,28 method. The NA-ESMD framework is based on the fewest switches surface hopping (FSSH) algorithm,29 which utilizes the same mixed electronic states as in the Ehrenfest basis functions (2), and where amplitudes a(n)I are also propagated according to the same eqn (10). The difference is that the nuclear motion in the FSSH approach is guided not by the average force but by the gradient of one “current” potential energy surface at any given time. The hops from one “current” state to another are based on the non-adiabatic coupling strengths and a stochastic switching routine. The probability of a switch at a particular time-step is related to the transition flux between the “current” and other electronic states (see eqn (10)):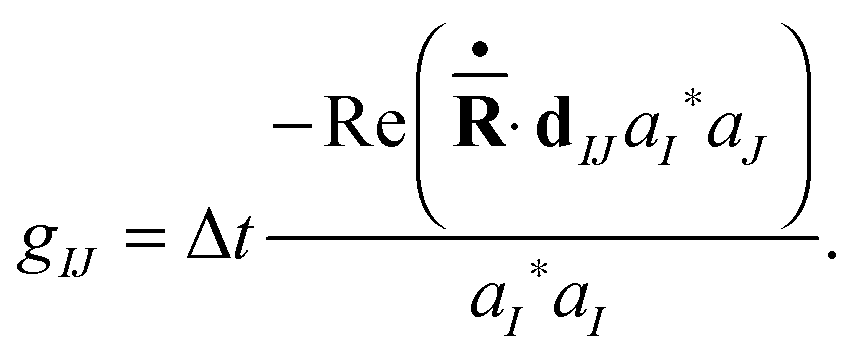 | (30) |
The FSSH algorithm implemented in NA-ESMD simulations is designed to ensure the internal consistency between the so-called classical populations and the quantum populations. The classical populations are defined as a fraction of trajectories evolving on each potential energy surface:
| PClI(t) = NI(t)/Ntraj, | (31) |
| PQI(t) = 〈|aI(t)|2〉Ntraj, | (32) |
Empirical corrections can be introduced to account for electronic decoherence.14 Following extensive numerical tests,14a we adopt the instantaneous decoherence (ID) approach, performed by resetting the quantum amplitude of the current state to unity after every attempted hop (regardless of whether hops are allowed or forbidden), thus removing the coherence of the quantum coefficients. This approach is based on the assumption that wavepackets traveling on different surfaces should immediately separate in the phase space and evolve independently. It has been previously shown14a that ID improves FSSH internal consistency between classical and quantum systems while at the same time provides results that do not depend on external parameters and maintains physical relevance.
III. Computational details
We have simulated the photoexcitation and intramolecular energy transfer between m-branched PPE units on the model molecule depicted in Fig. 1(a). Excited state energies, gradients and non-adiabatic couplings are calculated on the fly using the CEO approach.5a,27 For all simulations presented here, we use the Austin Model 1 (AM1)31 semiempirical level in combination with the configuration interaction singles (CIS) formalism to describe correlated excited states. This approach has worked well in our previous studies of similar systems.30The accuracy of excited-state energies in the AM1/CIS approximation has been benchmarked for a large variety of conjugated molecules,32 such as PPV33 and polyfluorenes.34 In the particular case of our model PPE dendritic molecule (shown at the Fig. 1(a)), vertical excitation energies for the two lowest excited states calculated at the AM1/CIS level for an AM1-optimized structure have been compared with the results obtained using TDDFT (B3LYP/6-31G* model) as well as with the experimental results,30,35 and a reasonable agreement has been achieved. More details about the benchmark calculations by SCS-AD(2), TDHF, TDDFT methods can be found in ref. 36, where the mechanism of intramolecular energy transfer suggested by NA-ESMD simulations at the AM1/CIS level30 has been confirmed.
The evaluation of the accuracy of NACME calculations is a more difficult task for any electronic structure method, because direct experimental measurements of NACME are absent. NACME can only be evaluated indirectly from electron–phonon coupling elements and Frank–Condon overlaps. Such an evaluation suggests reasonable accuracy of NACME calculated by AM1/CIS for excited state potential energy surfaces in conjugated hydrocarbons.33 Moreover, semi-quantitative direct comparisons of non-radiative relaxation rates calculated using our NA-ESMD approach and experimental time-resolved transient spectroscopy data37 supports the conclusions of ref. 33. Also, both AM1/CIS calculations38 and TDDFT36 show that the major contribution to d12(n) comes from the ethynylene stretches, which represent the reactive coordinates of the energy transfer process.
Six singlet electronic states (S1–S6) and their corresponding non-adiabatic couplings have been included in the simulations. The excited-state trajectories of 150 fs duration are propagated at constant energy in order to obtain a reasonable statistics. Time steps of 0.5 fs and 0.02 fs have been used in the ground and excited state dynamics, respectively. The Verlet finite difference algorithm39 is used to integrate the nuclear equations of motion, while the electronic coefficients are propagated using the Runge–Kutta–Verner fifth- and sixth-order method based on the code designed by Hull, Enright, and Jackson.40
In order to generate the initial conditions for excited-state dynamics, 1 ns of ground state molecular dynamics at 300 K was first performed using a Langevin friction coefficient γ of 2.0 ps−1. Snapshots of nuclei positions and momenta (conformational phase space) have been collected and used as initial conditions for subsequent photoexcitation dynamics modeling. The excited-state trajectories have been started from these initial configurations by instantaneously promoting the system to the state I selected according to a Frank–Condon window:
gI(R) = fI![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[−T2(Ωlaser − ΩI(R))2], exp[−T2(Ωlaser − ΩI(R))2], | (33) |
In order to make the comparison consistent, the same set of initial positions, momenta and excited-state populations have been used both for principle trajectories in Ehrenfest dynamics and for NA-ESMD surface hopping. Equations of motions were integrated with the same time step, and at the same level of electronic structure theory.
We performed the MCE-TDDB simulations for 100 trajectory swarms. Each swarm consists of 10 trajectories: the principle one and 9 satellites. This procedure is similar to the one used previously in model calculation,19d where first the initial wave packet created in the excited state after absorbing a photon was split into “bits” and then each “bit” was propagated separately on using a swarm of Ehrenfest configurations.
The initial conditions for the principle trajectories were generated as described above. The initial conditions for the satellites were taken23 very close to those for the principle trajectory with a random shifts ΔR and ΔP generated according to the Gaussian distribution
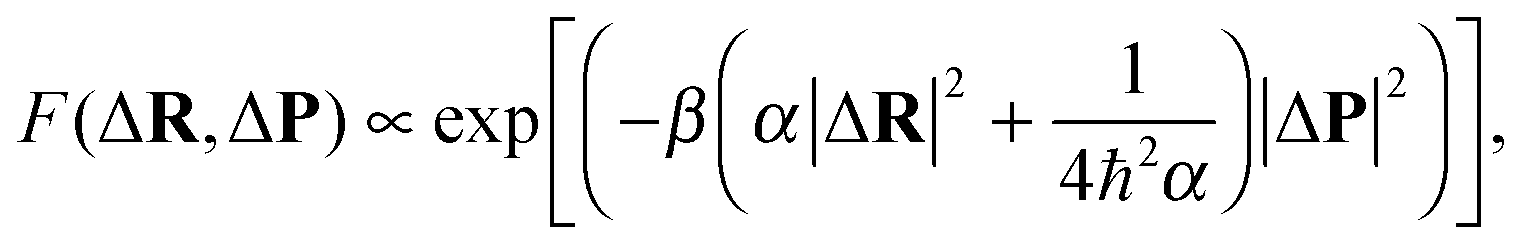 | (34) |
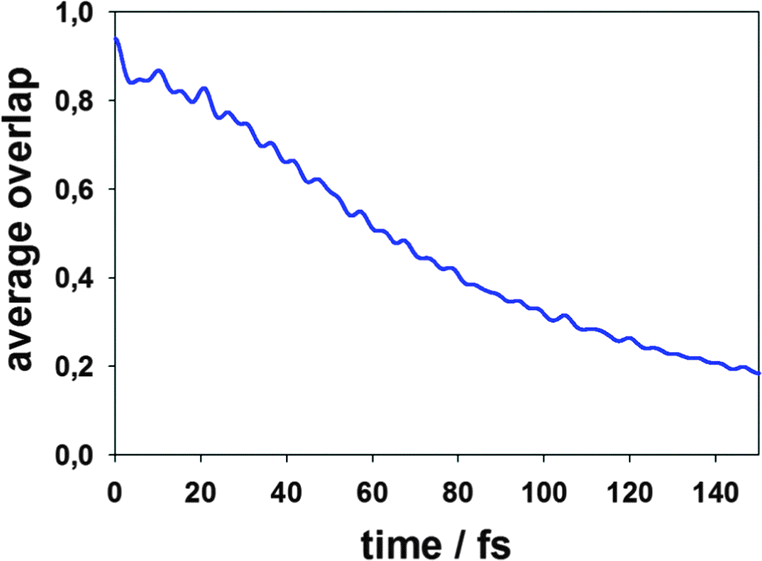 | ||
| Fig. 3 The decay of the absolute value of the average overlap between the principle and satellite trajectories in a swarm for MCE-TDDB calculations. | ||
For each trajectory in a swarm, we use a coherent state train basis of 11 basis functions. Thus each “bit” was propagated on the basis of 10 × 11 = 110 TBFs in total. A half of additional TBFs in the train are placed before and a half – after the main TBF with a time-shift of 0.6 fs, which provide the overlap of ∼0.7 between the nearest basis functions in the train. In order to avoid the necessity of backward propagation, the train basis was applied only after 3 fs of dynamics, when there was enough trajectory length to accommodate the tail half of the train; for first 3 fs, calculations were run in the basis of just 10 TBFs.
All the trajectories were calculated independently. Subsequently, the amplitudes cn(t) for all configurations in the basis were propagated using eqn (11) in a “post-processing” procedure.19g This makes the numerical procedure particularly efficient because a very large number of trajectories can be calculated in parallel. Although the total CPU time can reach years, most of it is spent on calculating trajectories using our ab initio on the fly procedure described by eqn (7)–(10) above. Quantum equations for the amplitudes use approximations (16) and (20), which allow to calculate coupling matrix elements in eqn (11) on the basis of the information already available from the calculation of the Ehrenfest trajectories. This “post-processing” technique is the central advantage of the group of methods based on our MCE methodology developed by us. The sampling techniques used in this work were tested on model systems, and it was demonstrated that the MCE can reproduce accurately fully quantum benchmark calculations.42 Thus, MCE-TDDB and related methods19f,g represent a very efficient ab initio direct dynamics methodology.
The results of ab initio MCE-TDDB calculations were compared with those given by the standard Ehrenfest (EHR) and the NA-ESMD surface hopping methods. The EHR results were obtained from the same trajectories as in the MCE-TDDB, but only the principle trajectory in each swarm was taken into account, and the populations and transition densities for these principle trajectories were simply averaged over the ensemble. The NA-ESMD surface hopping calculations28,29b,c were performed for a set of 100 excited-state trajectories with the same initial conditions as for principle trajectories in MCE-TDDB calculations. Simulations were also performed using the instantaneous decoherence corrections. More details about these kinds of simulations can be found elsewhere.14a
IV. Results
We have simulated the ultrafast intramolecular energy transfer that takes place after photoexcitation in the model branched dendritic molecule depicted in Fig. 1(a). This figure also shows the simulated absorption spectrum, in which the two largest contributions with the maxima at 378 nm (3.28 eV) and 348 nm (3.56 eV) come from S1 and S2 states corresponding to the excitation localized on two- and three-rings units respectively. There is no direct measurement of the absorption spectrum for this particular molecule. However, our calculated energies for S1 and S2 are in reasonable agreement with the values of 3.52 eV and 4.0 eV observed35 in a larger molecule, the so-called nanostar, for the corresponding electronic states, which are also localized at the same two- and three-ring units.The initial excited states are populated using a Frank–Condon window defined by a Gaussian shaped laser centered at 348 nm, which corresponds to the maximum of the contribution of the S2 state. Fig. 4 presents the average populations of the lowest four electronic states as a function of time calculated using the NA-ESMD (a, b), MCE-TDDB (c), EHR (d) methods. For NA-ESMD, both classical PClI(t) and quantum PQI(t) populations are presented. One can see that there is a significant difference between these two populations, which indicates that the so-called internal consistency requirement is violated here.
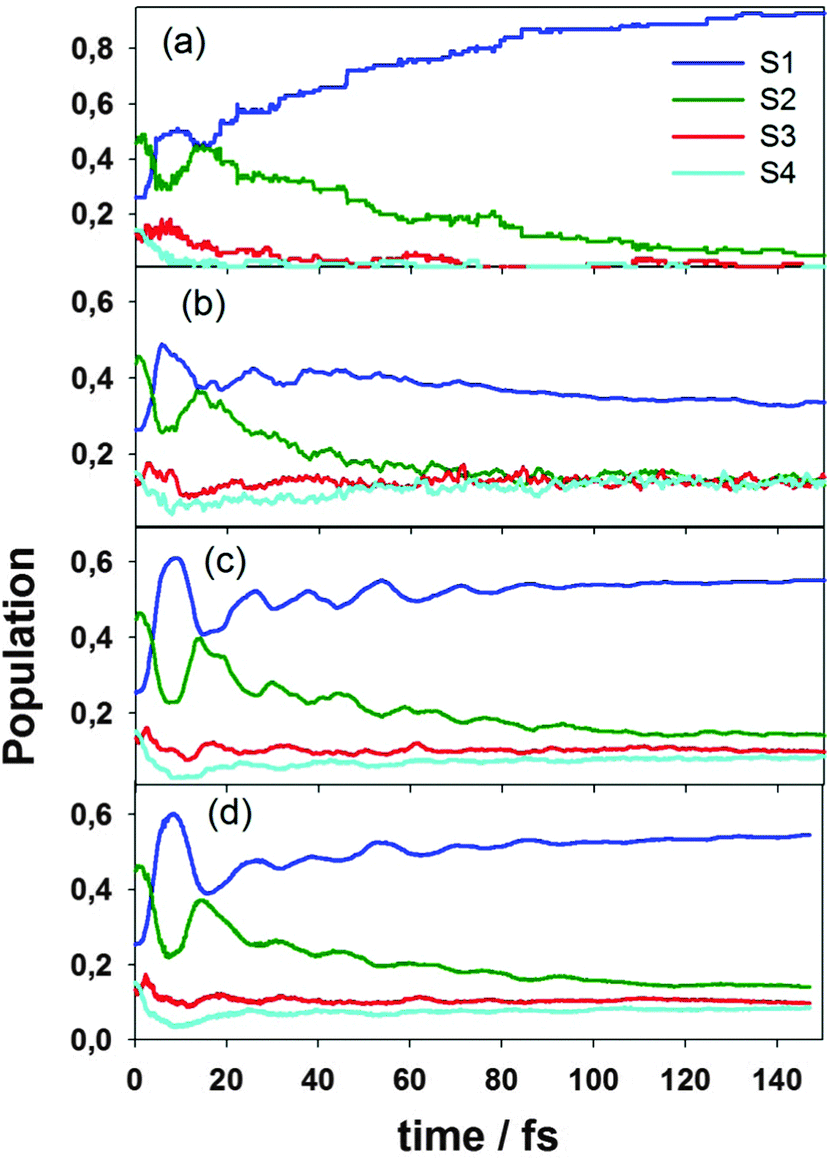 | ||
| Fig. 4 Average populations of the different electronic states as a function of time obtained from (a) NA-ESMD (classical populations), (b) NA-ESMD (quantum populations), (c) EHR, and (d) MCE-TDDB simulations. | ||
The dependencies for MCE-TDDB and EHR are very close to each other, except that MCE-TDDB, which relies on swarms and trains basis sets, produces slightly smother curves. On the one hand, this may be an indication that the Ehrenfest approach works really well for large complex systems with a singular photoproduct, such as PPE dendrimers. On the other hand, we know from our previous experience with model systems that using swarm/train basis in combination with trajectory branching (e.g., via cloning procedure19g) can further improve the convergence of the results and provide even quantitative agreement with multidimensional benchmark calculations. Cloning could introduce additional decoherence allowing basis functions for different electronic states to run away from each other. Such approaches, however, are very expensive for on the fly use, especially in the case of large molecules with many coupled electronic states, as a rapidly growing number of trajectories would require a huge amount of electronic structure calculations. We do not expect branching to change the result dramatically, but working on implementing even larger basis sets to prove that. Presented here MCE-TDDB technique provides a promising starting point for future developments of methods incorporating fully-quantum vibronic dynamics.
All population dependencies presented in Fig. 4 are similar qualitatively. In particular, one can see a noticeable oscillatory interchange of electronic populations between S1 and S2 states during the first 20 fs of dynamics after photoexcitation. The amplitude of the interchange between these states is revealed to be larger for Ehrenfest approaches with respect to surface hopping simulations. In all cases the ultrafast non-radiative decay of the electronic population on S2 takes place, and by the time of 150 fs the S1 population becomes significantly higher compared to its initial value. One can see that at the end of our excited-state dynamics, the S1 population given by the MCE-TDDB and EHR methods lies in between the classical and quantum populations PClI and PQI given by the NA-ESMD.
In order to gain insight into the origin of the main differences between surface hopping and Ehrenfest approaches, we have performed the NA-ESMD simulations using decoherence corrections. The importance of incorporating decoherence schemes in the NA-ESMD simulations applied to large extended conjugated molecules have been previously demonstrated.14a Decoherence has a large effect on the accuracy of the simulated dynamics both in terms of time scales for nonradiative relaxation and in promoting internal consistency of the FSSH algorithm. Fig. 5 shows the time-dependence of the average quantum populations of different electronic states for these NA-ESMD simulations using decoherence corrections. We observe that the oscillatory interchange of electronic populations between S1 and S2 (see Fig. 4) is damped in these simulations. This is expected because periodic collapses of the electronic wave-function to a single state eliminate the interference between these two states. After that, the population of the S1 state increases faster compared to quantum populations given by other approaches.
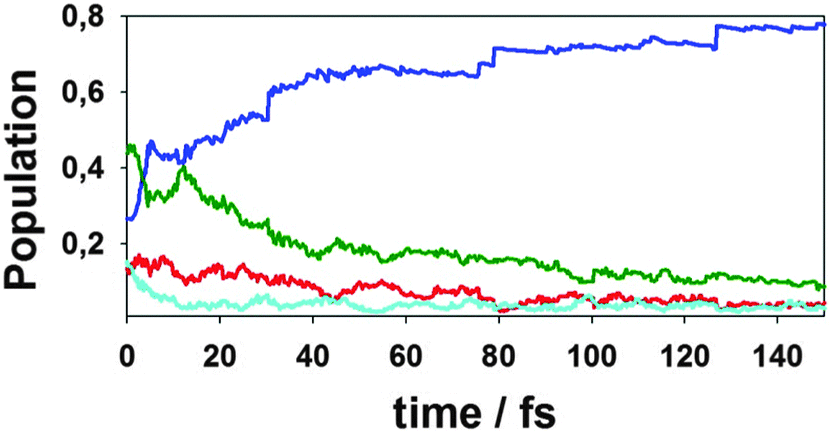 | ||
| Fig. 5 Average populations of different electronic states as a function of time obtained from NA-ESMD (quantum populations) using decoherence corrections. The colours refer to the same states as in Fig. 4. | ||
On the one hand, decoherence is not accounted for in the standard FSSH simulations, leading to internal inconsistencies of the method.14a,c,n On the other hand, instantaneous decoherence assumes that the divergent wavepackets are immediately decoupled. According to the results shown in Fig. 4 and 5, the Ehrenfest approach data fall in between quantum populations given by the NA-ESMD simulations with and without decoherence corrections. The possible explanation is that the Ehrenfest force depends on the amplitudes of all electronic states taken into account, including sufficiently low populated higher-energy states. Then, continuous population changes due to numerous intersections between a fairly large number of electronic states lead to weak quasi-random fluctuations of Ehrenfest force, which provide natural decoherence. This does not happen in the surface hopping approach where the force is usually determined by a single occupied adiabatic state, and the hops are not frequent and mostly reflect only significant population changes.
The CEO approach43 calculates the many-body eigenstates by diagonalizing the respective tetradic CIS operator in the molecular orbital (MO) basis. These CIS eigenstates are represented in the form of Nocc × Nvirt matrices (where occ and virt refer to occupied and virtual MOs), which can be transferred to the original atomic orbital (AO) basis. The resulting quantities are frequently denoted as transition density matrices (or electronic normal modes) and can be formally written as (ρI)mn ≡ 〈ϕg|cm+cn|ϕI〉, where |ϕg〉 and |ϕI〉 are the many-electron wave-functions for the ground and Ith excited states, and cm+ and cn are the electron creation and annihilation operators with indices n and m referring to the AO basis functions. Notably, owing to a unitary transformation between AO and MO basis and an orthogonality of all AO in the AM1/CIS approach, the normalization condition 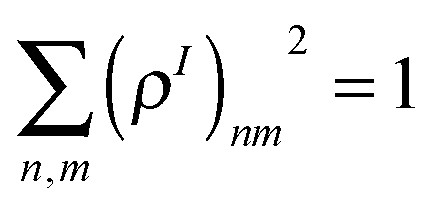 24b,25a is maintained.44 Matrix elements of the transition density matrices are subject to a straightforward interpretation: the diagonal elements of (ρI)nn reflect45 light-induced changes in an orbital n due to promotion of the molecule to the Ith excited state defining its transition dipole moment (the expectation value of the dipole operator on the transition density matrix) and, subsequently, an oscillator strength fI. In contrast, the off-diagonal elements of (ρI)nm reflect probability amplitudes of electron transfer between orbitals n and m, describing the optically induced electronic coherences and charge-transfer phenomena. Overall, the transition density matrices directly and compactly reflect the properties of many-body wave-functions even beyond the CIS framework. Subsequently, these are convenient quantities to analyze photoinduced spatial electronic/excitonic delocalization, charge-transfer, etc.13b
24b,25a is maintained.44 Matrix elements of the transition density matrices are subject to a straightforward interpretation: the diagonal elements of (ρI)nn reflect45 light-induced changes in an orbital n due to promotion of the molecule to the Ith excited state defining its transition dipole moment (the expectation value of the dipole operator on the transition density matrix) and, subsequently, an oscillator strength fI. In contrast, the off-diagonal elements of (ρI)nm reflect probability amplitudes of electron transfer between orbitals n and m, describing the optically induced electronic coherences and charge-transfer phenomena. Overall, the transition density matrices directly and compactly reflect the properties of many-body wave-functions even beyond the CIS framework. Subsequently, these are convenient quantities to analyze photoinduced spatial electronic/excitonic delocalization, charge-transfer, etc.13b
In order to obtain the fraction of the transition density localized either on the two-ring or the three-ring linear PPE units, we sum up the atomic contributions belonging to each fragment as
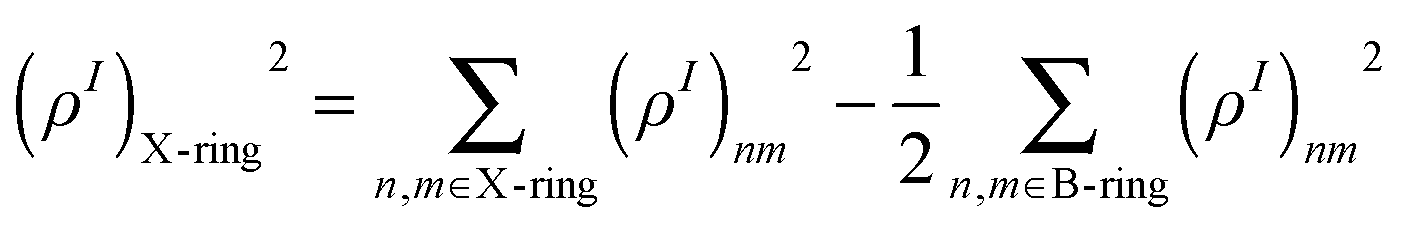 | (35) |
Fig. 6 shows localization of the electronic transition density of the four lowest electronic states at the minimum of the ground state potential energy surface. One can see that S1 and S2 states are mainly localized on the three-ring and two-ring units, respectively. This means that the dendritic PPE molecule can be represented as an ensemble of weakly coupled linear chromophore units,30c and the absorption spectra shown in Fig. 1(b) are approximately a sum of the absorption of three-ring and two-ring units:2,27a,b,46 the peak at 376 nm corresponds to the absorption of three-ring unit (S1 state), and the shoulder around 348 nm is due to the additional contribution of the two-ring unit (S2 state). Therefore, the excitation at our laser wavelength of 348 nm corresponds to an initial localization of the electronic transition density almost equally distributed between the two-ring and three-ring units. The electronic transition density of higher energy excited-states (S3 and S4) is qualitatively different from that of S1 and S2 states: it is delocalized over both two- and three-ring fragments for S3, and strongly localized for the S4 state.
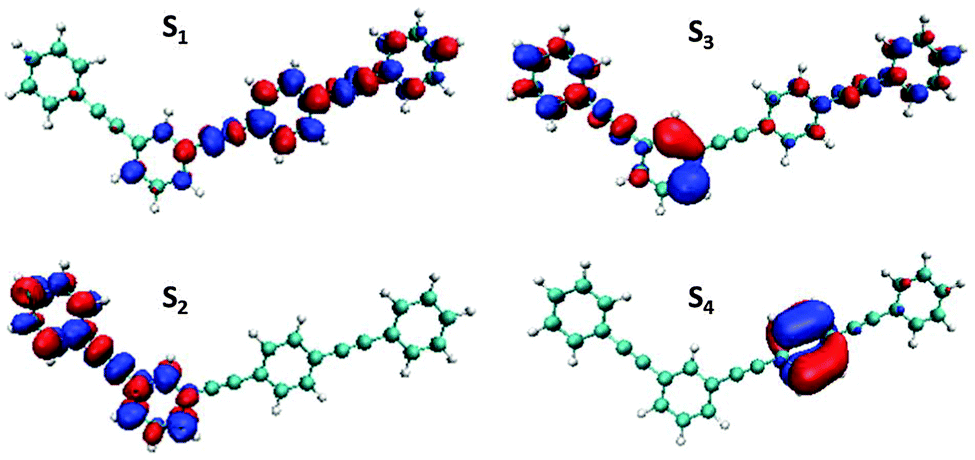 | ||
| Fig. 6 Initial localization of the electronic transition densities for the four lowest excited states. | ||
Nuclear motion mixes electronic states providing ultrafast energy transfer between the photoactive units. Fig. 7 compares the time-dependencies of the average transition density fractions localized on the two- and three-ring linear PPE units ((ρI)X-ring2) given by MCE-TDDB and NA-ESMD approaches. Note that the NA-ESMD density is calculated by averaging those of the “current” electronic state for all trajectories in the ensemble, similar to eqn (31) for classical electronic populations. The time-dependencies of transition densities are consistent with our results for the electronic populations (see Fig. 4(a) and (c)): the NA-ESMD approach shows more efficient energy migration toward the three-ring unit, as well as more efficient relaxation from S2 to the lowest state S1 associated with this transfer.
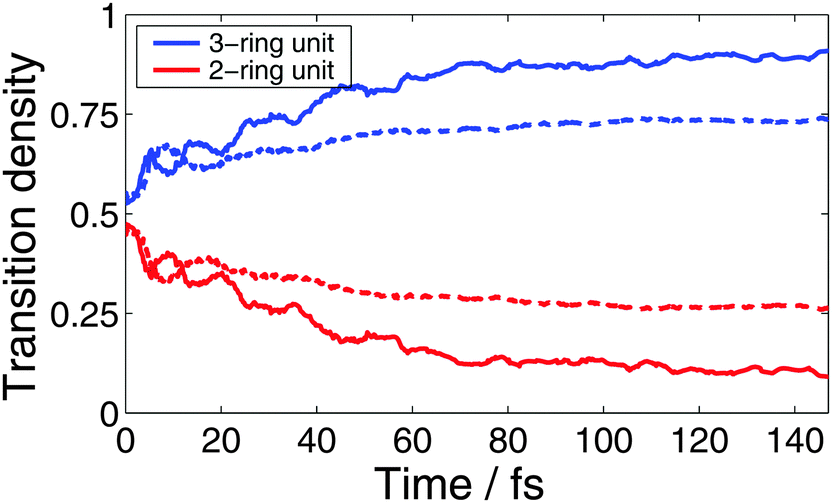 | ||
| Fig. 7 Fraction of the transition density localized in the two- and three-ring linear PPE units as a function of time obtained from NA-ESMD (solid) and MCE-TDDB (dashed) simulations. | ||
It was shown previously that the electronic energy transfer is concomitant to the intramolecular vibrational energy redistribution, and the nuclear motions in the direction of the ethynylene bonds is playing a critical role in the process.30bFig. 8 compares the distributions of the two-ring ethynylene bond length in the ground state and at S2–S1 crossing, defined as hop points for surface-hopping trajectories and as points with high NACMEs for Ehrenfest trajectories. Both types of calculations show that, in agreement with previous results,30b S2–S1 crossing is associated with ethynylene bond stretching in the two-ring part of the molecule.
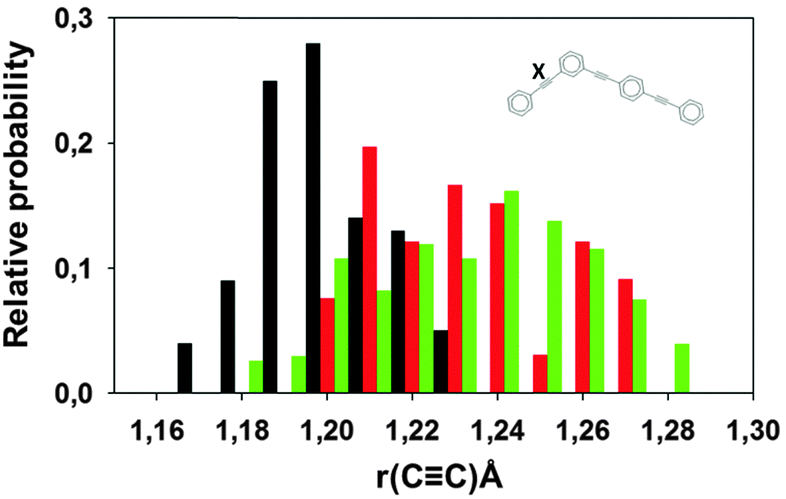 | ||
| Fig. 8 The probability distribution for the length of the ethynylene bond of the two-ring unit (labeled X in the inset) in the ground state (black), and at S2–S1 crossing: the moments of effective hops in surface hopping dynamics (red), and moments of strong non-adiabatic coupling (greater than 0.02 Hartree) in Ehrenfest dynamics (green). | ||
In Fig. 9, we plot the ethynylene bond lengths for both two- and three-ring units as a function of time. Accompanying the initial excitation of S2, this bond of the two-ring unit is initially excited. After that, its excitation relaxes together with the ultrafast decay of the S2 electronic population (Fig. 4). In contrast, the ethynylene bond of the three-ring unit becomes excited increasing its average amplitude of motion.
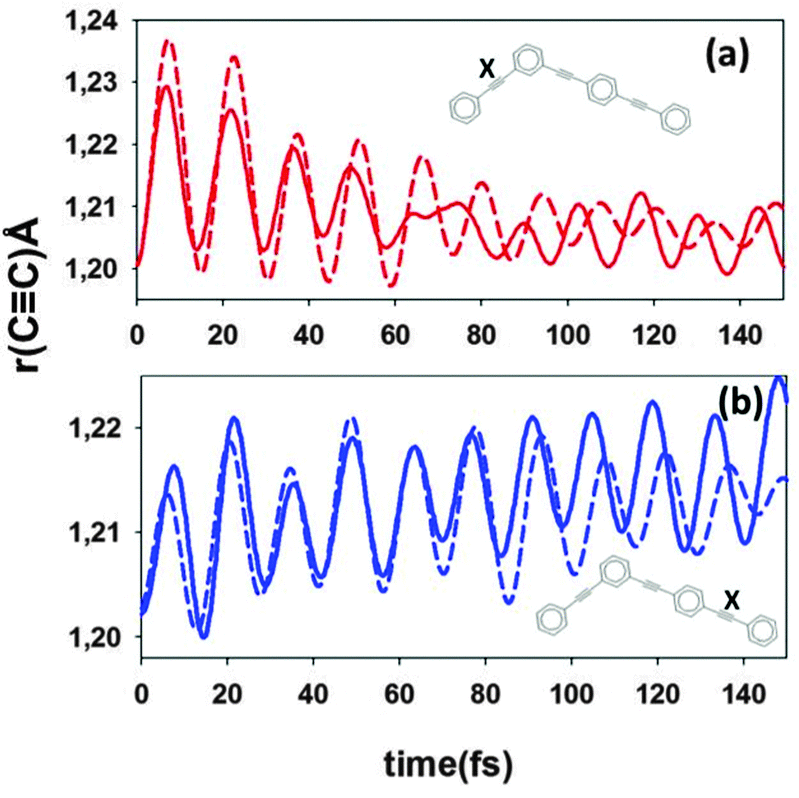 | ||
| Fig. 9 Length of particular ethynylene bonds (labeled X in the insets) as a function of time obtained from averaged NA-ESMD (solid) and Ehrenfest (dashed) simulations. | ||
The difference in the amplitude of motion for the ethynylene bonds given by different approaches is consistent with our other results. At early times of the dynamics (∼20 fs), the amplitude of motion of the ethynylene bond of the two-ring unit is larger for Ehrenfest than that for surface hopping simulation. This time corresponds to the time at which the oscillatory interchange of electronic populations between S1 and S2 states takes place (see Fig. 4). At longer times, we observe the reduction of the average vibrational amplitudes. This decay is faster for the Ehrenfest trajectories where the nuclear motion is coupled with amplitudes a(n)I for all electronic states providing better vibrational energy redistribution through continuous partial electronic transitions between many states.
Fig. 8a indicates that the ethynylene bond of the two ring unit behaves differently for NA-ESMD and the Ehrenfest approach. The non-adiabatic contribution to the Ehrenfest force n (eqn (9)), not present in NA-ESMD simulations, introduces additional vibrational coherence at earlier times of simulations in the regions with large d12(n) values. At later times, the Ehrenfest force dependence on the continuous low population changes among higher-energy states seems to result in quasi-random fluctuations that lead to vibrational decoherence. The same additional vibrational coherence is responsible for higher amplitude of the oscillation of the electronic population, which both Ehrenfest and MCE-TDDB reveal as shown in Fig. 4. This is consistent with the results of ref. 30, where a strong coupling between S1 and S2 states was shown to exist only during first 15–20 fs after photoexcitation.
V. Conclusion
In this study we report further development and a new implementation of the ab initio MCE approach suggested previously, to model excited state dynamics of realistic molecular systems beyond the Born–Oppenheimer approximation. The method treats nuclear wave packet dynamics at a fully quantum level, while the electronic structure can be described by a variety of high accuracy ab initio methods. The approach is formally exact and physical observables can be calculated unambiguously without additional assumptions. Previously, the method has been validated only on the model and more simple systems reproducing benchmark results accurately.The proposed ab initio MCE-TDDB approach is very efficient for large conjugated molecules, such as dendrimers, where the dynamics involving many continuously interacting electronic states, is well described by mean-field Ehrenfest trajectories. Although even larger basis sets with branching trajectories can be introduced, such calculations would be extremely expensive and unlikely to produce significantly different results.
As an application, here we simulate the exited state dynamics for a dendrimer molecule composed of two- and three-ring PPE chromophore units following laser pulse excitation at 348 nm. Our ab initio MCE-TDDB results are compared with those of surface hoping. Our calculations show that for dynamics involving a large number of electronic states, the approaches based on Ehrenfest trajectories naturally account for decoherence phenomena. Thus, our MCE-TDDB calculations validate decoherence corrections, previously introduced to the surface hoping technique in an ad hoc fashion.
Altogether, this makes Ehrenfest-based techniques, such as presented here MCE-TDDB, a method of choice for on the fly simulations of the excitation dynamics in large photoactive molecules.
Acknowledgements
SF-A is supported by CONICET, UNQ, ANPCyT (PICT-2014-2662). DM, DS and SF-A acknowledge the support from EPSRC through grants EP/J001481/1 and EP/N007549/1 (DM and DS). This work was performed in part at the Center for Integrated Nanotechnologies, a U.S. Department of Energy, Office of Science user facility at Los Alamos National Laboratory (LANL). LANL is operated by Los Alamos National Security, LLC, for the National Nuclear Security Administration of the U.S. Department of Energy under contract DEAC52-06NA25396.References
- A. M. Naylor, W. A. Goddard, G. E. Kiefer and D. A. Tomalia, Starburst Dendrimers. 5. Molecular Shape Control, J. Am. Chem. Soc., 1989, 111(6), 2339–2341 CrossRef CAS.
- J. L. Palma, E. Atas, L. Hardison, T. B. Marder, J. C. Collings, A. Beeby, J. S. Melinger, J. L. Krause, V. D. Kleiman and A. E. Roitberg, Electronic Spectra of the Nanostar Dendrimer: Theory and Experiment, J. Phys. Chem. C, 2010, 114(48), 20702–20712 CAS.
- D. A. Tomalia and J. M. J. Fréchet, Dendrimers and Other Dendritic Polymers, John Wiley & Sons Ltd, West Sussex, 2001 Search PubMed.
- (a) B. Balzani, M. Venturi and A. Credi, Molecular Devices and Machines: A Journey into the Nanoworld, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2003 CrossRef; (b) S. H. Bai, C. Thomas, A. Rawat and F. Ahsan, Recent Progress in Dendrimer-Based Nanocarriers, Crit. Rev. Ther. Drug Carrier Syst., 2006, 23(6), 437–495 CrossRef CAS PubMed; (c) M. Kawa and J. M. J. Fréchet, Self-Assembled Lanthanide-Cored Dendrimer Complexes: Enhancement of the Luminescence Properties of Lanthanide Ions through Site-Isolation and Antenna Effects, Chem. Mater., 1998, 10(1), 286–296 CrossRef CAS.
- (a) S. Mukamel, Photochemistry: Trees to trap photons, 1997, 388, 425–427 CAS; (b) R. Kopelman, M. Shortreed, Z. Y. Shi, W. Tan, Z. Xu, J. S. Moore, A. Bar-Haim and J. Klafter, Spectroscopic Evidence for Excitonic Localization in Fractal Antenna Supermolecules, Phys. Rev. Lett., 1997, 78, 1239 CrossRef CAS; (c) S. F. Swallen, R. Kopelman, J. S. Moore and C. Devadoss, Dendrimer photoantenna supermolecules: energetic funnels, exciton hopping and correlated excimer formation, J. Mol. Struct., 1999, 485, 585–597 CrossRef; (d) J. M. J. Fréchet, Functional polymers and dendrimers: reactivity, molecular architecture, and interfacial energy, Science, 1994, 263(51), 1710–1715 Search PubMed; (e) D. Rana and G. Gangopadhyay, Chem. Phys. Lett., 2001, 334, 314 CrossRef CAS; (f) S. F. Swallen, Z. Zhu, J. S. Moore and R. Kopelman, J. Phys. Chem. B, 2000, 104, 3988 CrossRef CAS.
- K. Inoue, Prog. Polym. Sci., 2000, 25, 453 CrossRef CAS.
- (a) J. L. Bredas, J. E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res., 2009, 42, 1691 CrossRef CAS PubMed; (b) D. G. Kuroda, C. P. Singh, Z. Peng and V. D. Kleiman, Science, 2009, 326, 263 CrossRef CAS PubMed.
- T. Dutta and S. Ramasesha, Double time window targeting technique: real-time DMRG dynamics in Pariser–Parr–Pople model, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 035115 CrossRef.
- O. R. Tozer and W. Barford, Intrachain exciton dynamics in conjugated polymer chains in solution, J. Chem. Phys., 2015, 143, 084102 CrossRef PubMed.
- O. R. Tozer and W. Barford, Exciton dynamics in disordered poly(para-phenylene vinylene) 1: ultra-fast interconversion and dynamical localization, J. Phys. Chem. A, 2012, 116, 10310 CrossRef CAS PubMed.
- (a) S. R. White, Phys. Rev. Lett., 1992, 69, 2863 CrossRef PubMed; (b) S. R. White, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 10345 CrossRef CAS; (c) Density Matrix Renormalization, ed. I. Peschel, X. Wang, K. Hallberg and M. Kaulke, Springer, New York, 1999 Search PubMed; (d) C. Raghu, Y. A. Pati and S. Ramasesha, A density matrix renormalization group study of low-lying excitations of polyacene within a Pariser–Parr–Pople model, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 035116 CrossRef; (e) M. Kumar and S. Ramasesha, A DMRG Study of the Low-Lying States of Transverse Substituted Trans-polyacetylene and Trans-polyacetylene, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 035115 CrossRef; (f) M. A. Cazalilla and J. B. Marston, Phys. Rev. Lett., 2002, 88, 256403 CrossRef CAS PubMed; (g) M. A. Cazalilla and J. B. Marston, Phys. Rev. Lett., 2003, 91, 049702 CrossRef; (h) E. Jekelmann, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 045114 CrossRef; (i) Theoretical Methods for Strongly Correlated Electrons, CRM Series in Mathematical Physics, ed. D. Sénéchal, A.-M. Tremblay and C. Bourbonnais, Springer-Verlag, New York, 2004 Search PubMed.
- (a) A. S. Davydov, Theory of Molecular Excitons, Plenum, New York, 1971 Search PubMed; (b) Excitons, ed. E. I. Rashba and M. D. Sturge, North Holland, Amsterdam, 1982 Search PubMed; (c) V. B. Broude, E. I. Rashba and E. F. Sheka, Spectroscopy of Molecular Excitons, Springer, Berlin, 1985 Search PubMed.
- (a) E. Y. Poliakov, V. Chernyak, S. Tretiak and S. Mukamel, J. Chem. Phys., 1999, 110, 8161 CrossRef CAS; (b) S. Tretiak and S. Mukamel, Chem. Rev., 2002, 102, 3171 CrossRef CAS PubMed; (c) S. Tretiak, V. Chernyak and S. Mukamel, J. Phys. Chem. B, 1998, 102, 3310 CrossRef CAS.
- (a) T. Nelson, S. Fernandez-Alberti, A. E. Roitberg and S. Tretiak, Nonadiabatic Excited-State Molecular Dynamics: Treatment of Electronic Decoherence, J. Chem. Phys., 2013, 138, 224111 CrossRef PubMed; (b) E. Neria and A. Nitzan, J. Chem. Phys., 1993, 99, 1109 CrossRef CAS; (c) J.-Y. Fang and S. Hammes-Schiffer, J. Phys. Chem. A, 1999, 103, 9399 CrossRef CAS; (d) K. Drukker, J. Comput. Phys., 1999, 153, 225 CrossRef CAS; (e) E. R. Bittner and P. J. Rossky, J. Chem. Phys., 1995, 103, 8130 CrossRef CAS; (f) G. Granucci and M. Persico, J. Chem. Phys., 2007, 126, 134114 CrossRef PubMed; (g) G. Granucci, M. Persico and A. Zoccante, J. Chem. Phys., 2010, 133, 134111 CrossRef PubMed; (h) O. V. Prezhdo and P. J. Rossky, J. Chem. Phys., 1997, 107, 5863 CrossRef CAS; (i) B. J. Schwartz, E. R. Bittner, O. V. Prezhdo and P. J. Rossky, J. Chem. Phys., 1996, 104, 5942 CrossRef CAS; (j) J. E. Subotnik and N. Shenvi, J. Chem. Phys., 2011, 134, 024105 CrossRef PubMed; (k) J. E. Subotnik, J. Phys. Chem. A, 2011, 115, 1208 CrossRef PubMed; (l) N. Shenvi, J. E. Subotnik and W. Yang, J. Chem. Phys., 2011, 134, 144102 CrossRef PubMed; (m) C. Zhu, S. Nangia, A. W. Jasper and D. G. Truhlar, J. Chem. Phys., 2004, 121, 7658 CrossRef CAS PubMed; (n) M. Thachuk, M. Y. Ivanov and D. M. Wardlaw, J. Chem. Phys., 1998, 109, 5747 CrossRef CAS.
- G. A. Worth, H.-D. Meyer, H. Köppel, L. S. Cederbaum and I. Burghardt, Using the MCTDH wavepacket propagation method to describe multimode non-diabatic dynamics, Int. Rev. Phys. Chem., 2008, 27, 569–606 CrossRef CAS.
- H. Tamura and I. Burghardt, Ultrafast charge separation in organic photovoltaics enhanced by charge delocalization and vibronically hot exciton dissociation, J. Am. Chem. Soc., 2013, 135, 16364–16367 CrossRef CAS PubMed.
- (a) G. A. Worth, M. A. Robb and B. Lasorne, Solving the time-dependent Schrödinger equation for nuclear motion in one step: direct dynamics of non-adiabatic systems, Mol. Phys., 2008, 106(16), 2077–2091 CrossRef CAS; (b) R. Car and M. Parrinello, Unified Approach for Molecular Dynamics and Density-Functional Theory, Phys. Rev. Lett., 1985, 55, 2471 CrossRef CAS PubMed; (c) M. E. Tuckerman, P. J. Ungar, T. von Rosenvinge and M. L. Klein, Ab initio molecular dynamics simulations, J. Phys. Chem., 1996, 100, 12878 CrossRef CAS; (d) M. Ben-Nun and T. J. Martínez, Ab Initio Quantum Molecular Dynamics, Adv. Chem. Phys., 2002, 121, 439 CrossRef CAS.
- (a) G. A. Worth and I. Burghardt, Full Quantum Mechanical Molecular Dynamics Using Gaussian Wavepackets, Chem. Phys. Lett., 2003, 368, 502 CrossRef CAS; (b) G. W. Richings, I. Polyak, K. E. Spinlove, G. A. Worth, I. Burghardt and B. Lasorne, Quantum dynamics simulations using Gaussian wavepackets: the vMCG method, Int. Rev. Phys. Chem., 2015, 35, 269 CrossRef.
- (a) T. J. Martinez, M. Ben-Nun and G. Ashkenazi, Classical/quantal method for multistate dynamics: a computational study, J. Chem. Phys., 1996, 104, 2847 CrossRef CAS; (b) D. V. Shalashilin, Quantum mechanics with the basis set guided by Ehrenfest trajectories: theory and application to spin-boson model, J. Chem. Phys., 2009, 130(24), 244101 CrossRef PubMed; (c) S. L. Fiedler and J. Eloranta, Nonadiabatic dynamics by mean-field and surface hopping approaches: energy conservation considerations, Mol. Phys., 2010, 108(11), 1471–1479 CrossRef CAS; (d) D. V. Shalashilin, Nonadiabatic dynamics with the help of multiconfigurational Ehrenfest method: improved theory and fully quantum 24D simulation of pyrazine, J. Chem. Phys., 2010, 132(24), 244111 CrossRef PubMed; (e) D. V. Shalashilin, Multiconfigurational Ehrenfest approach to quantum coherent dynamics in large molecular systems, Faraday Discuss., 2011, 153, 105 RSC; (f) K. Saita and D. V. Shalashilin, On-the-fly ab initio molecular dynamics with multiconfigurational Ehrenfest method, J. Chem. Phys., 2012, 137, 8 CrossRef PubMed; (g) D. V. Makhov, W. J. Glover, T. J. Martinez and D. V. Shalashilin, Ab initio multiple cloning algorithm for quantum nonadiabatic molecular dynamics, J. Chem. Phys., 2014, 141(5), 054110 CrossRef PubMed; (h) D. V. Makhov, K. Saita, T. J. Martinez and D. V. Shalashilin, Ab initio multiple cloning simulations of pyrrole photodissociation: TKER spectra and velocity map imaging, Phys. Chem. Chem. Phys., 2015, 17, 3316 RSC.
- (a) H.-D. Meyer and W. H. Miller, A classical analog for electronic degrees of freedom in nonadiabatic collision processes, J. Chem. Phys., 1979, 70, 3214 CrossRef CAS; (b) G. D. Billing, On the use of Ehrenfest's theorem in molecular scattering, Chem. Phys. Lett., 1983, 100, 535 CrossRef CAS; (c) J. W. Negele, The mean-field theory of nuclear structure and dynamics, Rev. Mod. Phys., 1982, 54, 913 CrossRef CAS.
- S. Fernandez-Alberti, A. E. Roitberg, T. Nelson and S. Tretiak, Identification of unavoided crossings in nonadiabatic photoexcited dynamics involving multiple electronic states in polyatomic conjugated molecules, J. Chem. Phys., 2012, 137(1), 014512 CrossRef PubMed.
- (a) S. F. Swallen, et al., Dendrimer Photoantenna Supermolecules: Energetic Funnels, Exciton Hopping and Correlated Excimer Formation, J. Mol. Struct., 1999, 485, 585–597 CrossRef; (b) C. Devadoss, P. Bharathi and J. S. Moore, Energy Transfer in Dendritic Macromolecules: Molecular Size Effects and the Role of an Energy Gradient, J. Am. Chem. Soc., 1996, 118, 9635–9644 CrossRef CAS; (c) Z. F. Xu, et al., Phenylacetylene Dendrimers by the Divergent, Convergent, and Double-Stage Convergent Methods, J. Am. Chem. Soc., 1994, 116, 4537–4550 CrossRef CAS; (d) M. R. Shortreed, et al., Directed Energy Transfer Funnels in Dendrimeric Antenna Supermolecules, J. Phys. Chem. B, 1997, 101, 6318–6322 CrossRef.
- D. V. Shalashilin and M. S. Child, Basis set sampling in the method of coupled coherent states: coherent state swarms, trains, and pancakes, J. Chem. Phys., 2008, 128, 054102 CrossRef PubMed.
- (a) V. Chernyak, M. F. Schulz, S. Mukamel, S. Tretiak and E. V. Tsiper, J. Chem. Phys., 2000, 113, 36 CrossRef CAS; (b) S. Tretiak, C. Isborn, A. Niklasson and M. Challacombe, J. Chem. Phys., 2009, 130, 054111 CrossRef PubMed.
- (a) T. Nelson, S. Fernandez-Alberti, V. Chernyak, A. E. Roitberg and S. Tretiak, Nonadiabatic Excited-State Molecular Dynamics Modeling of Photoinduced Dynamics in Conjugated Molecules, J. Phys. Chem. B, 2011, 115, 5402–5414 CrossRef CAS PubMed; (b) F. Furche and R. Ahlrichs, J. Chem. Phys., 2002, 117, 7433 CrossRef CAS; (c) S. Tretiak and V. Chernyak, J. Chem. Phys., 2003, 119, 8809 CrossRef CAS.
- (a) M. Tommasini, V. Chernyak and S. Mukamel, Int. J. Quantum Chem., 2001, 85, 225 CrossRef CAS; (b) V. Chernyak and S. Mukamel, J. Chem. Phys., 2000, 112, 3572 CrossRef CAS; (c) R. Send and F. Furche, J. Chem. Phys., 2010, 132, 044107 CrossRef PubMed.
- (a) S. Tretiak, V. Chernyak and S. Mukamel, J. Chem. Phys., 1996, 105, 8914 CrossRef CAS; (b) S. Tretiak, W. M. Zhang, V. Chernyak and S. Mukamel, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13003 CrossRef CAS PubMed; (c) S. Mukamel, S. Tretiak, T. Wagersreiter and V. Chernyak, Science, 1997, 277, 781 CrossRef CAS.
- T. Nelson, S. Fernandez-Alberti, A. Roitberg and S. Tretiak, Nonadiabatic Excited-State Molecular Dynamics: Modeling Photophysics in Organic Conjugated Materials, Acc. Chem. Res., 2014, 47(4), 1155–1164 CrossRef CAS PubMed.
- (a) S. Hammes-Schiffer and J. C. Tully, Proton Transfer in Solution: Molecular Dynamics with Quantum Transitions, J. Chem. Phys., 1994, 101, 4657–4667 CrossRef CAS; (b) J. C. Tully, Molecular Dynamics with Electronic Transitions, J. Chem. Phys., 1990, 93, 1061–1071 CrossRef CAS; (c) J. C. Tully, Nonadiabatic Molecular Dynamics, Int. J. Quantum Chem., 1991, 40, 299–309 CrossRef.
- (a) S. Fernandez-Alberti, V. D. Kleiman, S. Tretiak and A. E. Roitberg, Nonadiabatic molecular dynamics simulations of the energy transfer between building blocks in a phenylene ethynylene dendrimer, J. Phys. Chem. A, 2009, 113, 7535–7542 CrossRef CAS PubMed; (b) S. Fernandez-Alberti, V. D. Kleiman, S. Tretiak and A. E. Roitberg, Unidirectional energy transfer in conjugated molecules: the crucial role of high frequency C(triple)C bonds, J. Phys. Chem. Lett., 2010, 1, 2699–2704 CrossRef CAS; (c) S. Fernandez-Alberti, A. E. Roitberg, V. D. Kleiman, T. Nelson and S. Tretiak, Shishiodoshi unidirectional energy transfer mechanism in phenylene ethynylene dendrimers, J. Chem. Phys., 2012, 137, 22A526 CAS.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, The Development and Use of Quantum-mechanical molecular models. AM1 – A New General Purpose Quantum-mechanical Molecular-model, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- S. Tretiak, et al., CEO/Semiempirical Calculations of UVVisible Spectra in Conjugated Molecules, Chem. Phys. Lett., 2000, 331, 561–568 CrossRef CAS.
- S. Tretiak, Conformational Dynamics of Photoexcited Conjugated Molecules, Phys. Rev. Lett., 2002, 89, 097402 CrossRef CAS PubMed.
- I. Franco and S. Tretiak, Electron-Vibrational Dynamics of Photoexcited Polyfluorenes, J. Am. Chem. Soc., 2004, 126, 12130–12140 CrossRef CAS PubMed.
- (a) V. D. Kleiman, J. S. Melinger and D. McMorrow, J. Phys. Chem. B, 2001, 105, 5595 CrossRef CAS; (b) E. Atas, Ultrafast Time Resolved Excitation Dynamics in Conjugated Dendrimers, PhD thesis, University of Florida, Gainesville, FL, 2006 Search PubMed.
- J. Huang, L. Du, D. Hu and Z. Lan, Theoretical analysis of excited states and energy transfer mechanism in conjugated dendrimers, J. Comput. Chem., 2015, 36, 151–163 CrossRef CAS PubMed.
- J. F. Galindo, E. Atas, A. Altan, D. G. Kuroda, S. Fernandez-Alberti, S. Tretiak, V. Kleiman and A. E. Roitberg, Dynamics of Energy Transfer in a Conjugated Dendrimer Driven by Ultrafast Localization of Excitations, J. Am. Chem. Soc., 2015, 137, 11637–11644 CrossRef CAS PubMed.
- (a) M. A. Soler, A. E. Roitberg, T. Nelson, S. Tretiak and S. Fernandez-Alberti, Analysis of state-specific vibrations coupled to the unidirectional energy transfer in conjugated dendrimers, J. Phys. Chem. A, 2012, 116, 9802–9810 CrossRef CAS PubMed; (b) M. Soler, T. Nelson, A. Roitberg, S. Tretiak and S. Fernandez-Alberti, Signature of Nonadiabatic Coupling in Excited-State Vibrational Modes, J. Phys. Chem. A, 2014, 118, 10372–10379 CrossRef CAS PubMed.
- W. C. Swope, H. C. Andersen, P. H. Berens and K. R. Wilson, J. Chem. Phys., 1982, 76, 637 CrossRef CAS.
- (a) T. E. Hull, W. Enright, K. Jackson, User's guide for DVERK – A subroutine for solving non-stiff ODEs, Technical Report 100, Department of Computer Science, University of Toronto, Canada, 1976; (b) IMSL MATH/LIBRARY Special Functions, Visual Numerics, Inc., Houston, TX 77042, USA.
- A. L. Thompson, C. Punwong and T. J. Martinez, Optimization of width parameters for quantum dynamics with frozen Gaussian basis set, Chem. Phys., 2010, 370, 70–77 CrossRef CAS.
- (a) D. V. Makhov, C. Symonds and D. Shalashilin, Chem. Phys. Search PubMed , in preparation; (b) C. Symonds, Development and Applications of New Basis Set Sampling and Basis Set Handling Procedures for the Coupled Coherent States Family of Methods, PhD thesis, University of Leeds, 2015 Search PubMed.
- (a) S. Tretiak, V. Chernyak and S. Mukamel, Chem. Phys. Lett., 1996, 259, 55 CrossRef CAS; (b) S. Tretiak, V. Chernyak and S. Mukamel, J. Am. Chem. Soc., 1997, 119, 11408 CrossRef CAS.
- (a) F. Plasser and H. Lischka, Analysis of Excitonic and Charge Transfer Interactions from Quantum Chemical Calculations, J. Chem. Theory Comput., 2012, 8, 2777–2789 CrossRef CAS PubMed; (b) F. Plasser, M. Wormit and A. Dreuw, New tools for the systematic analysis and visualization of electronic excitations. I. Formalism, J. Chem. Phys., 2014, 141, 024106 CrossRef PubMed; (c) F. Plasser, S. A. Bäppler, M. Wormit and A. Dreuw, New tools for the systematic analysis and visualization of electronic excitations. I. Applications, J. Chem. Phys., 2014, 141, 024107 CrossRef PubMed; (d) A. A. Voityuk, Fragment transition density method to calculate electronic coupling for excitation energy transfer, J. Chem. Phys., 2014, 140, 244117 CrossRef PubMed; (e) W. Liu, B. Lunkenheimer, V. Settels, B. Engels, R. F. Fink and A. Köhn, J. Chem. Phys., 2015, 143, 084106 CrossRef PubMed.
- W. Chao, S. V. Malinin, S. Tretiak and V. Chernyak, Nat. Phys., 2006, 2, 631 CrossRef.
- T. Tada, D. Nozaki, M. Kondo and K. Yoshizawa, J. Phys. Chem. B, 2003, 107, 14204 CrossRef CAS.
| This journal is © the Owner Societies 2016 |