
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formal total synthesis of (±)-strictamine – the [2,3]-Stevens rearrangement for construction of octahydro-2H-2,8-methanoquinolizines†
Ruben
Eckermann
,
Michael
Breunig
and
Tanja
Gaich
*
Department of Chemistry, University of Konstanz, Universitätsstrasse 10, 78457 Konstanz, Germany. E-mail: tanja.gaich@uni-konstanz.de
First published on 16th August 2016
Abstract
For decades, akuammiline alkaloids have attracted synthetic chemists due to their intriguing molecular architecture. Among the different structural elements embedded in their carboskeleton, the methanoquinolizidine system constitutes the signature structural element of this alkaloid family. Herein, we describe a novel synthetic access to this system which relies on a [2,3]-Stevens rearrangement and results in the formal synthesis of strictamine.
Akuammiline alkaloids belong to the group of monoterpenoid indole alkaloids. Akuamma, one of the native names of the tree Picralima nitida (family: Apocinaceae, synonyms: Picalima klaineana, Picralima macrocarpa), is a rainforest tree occurring in the African forest region from the Ivory Coast to Uganda. The plant is used for the treatment of a variety of diseases in traditional medicine in these regions of Africa.1 Extracts from its seeds, fruit rind and stem bark demonstrated antimalarial activity2 as well as antimicrobial3 and anti-inflammatory effects.3b,4
Its alcoholic extracts contain 3.5–4.8% of alkaloids.5 Early on, these alkaloids were studied as potential sources of pharmaceutical compounds. Henry and Sharp characterized four different alkaloids from the akuamma seeds in 19271a and named the first structure they elucidated akuammine (1). From these seeds, the second indole alkaloid akuammiline5 (2) served as an eponym of a whole natural product family – the akuammiline alkaloids (Fig. 1).
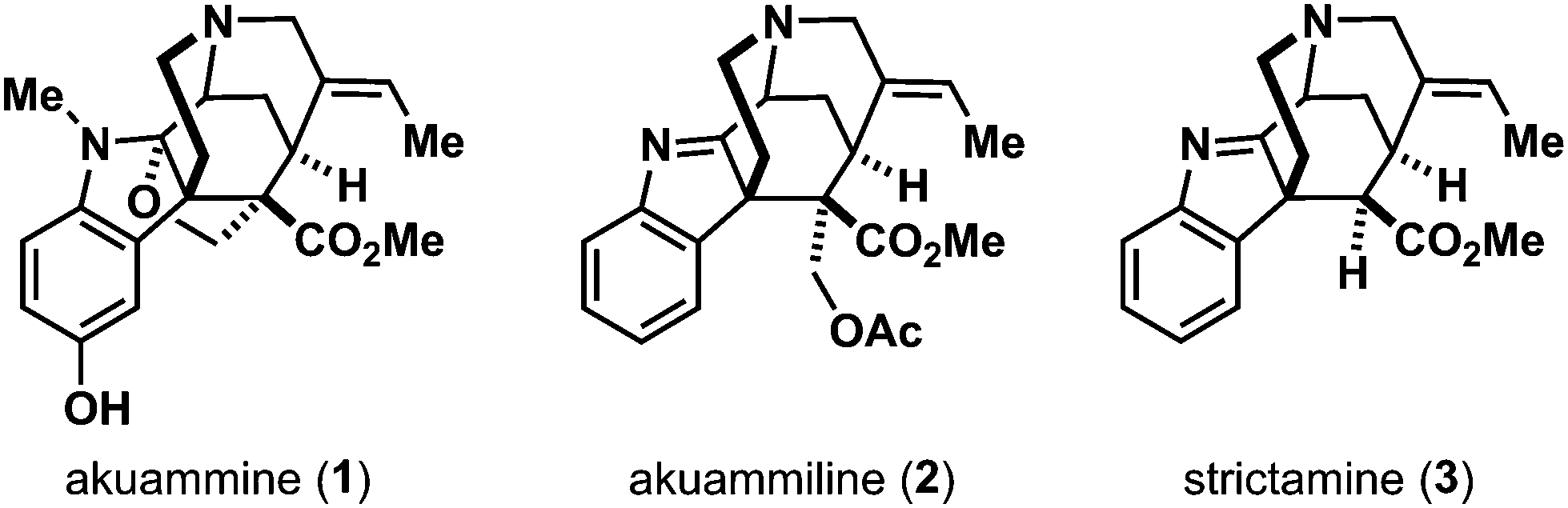 | ||
| Fig. 1 Prominent members of the akuammiline alkaloids. | ||
In 1966, strictamine (3) was isolated from the plant Rhazya Stricta (family: Apocinaceae).6 It shows inhibitory effects of the nuclear factor-κB (NF-κB),7 which plays an important role in the regulation of gene expression in immune and inflammatory responses.
The unique challenging chemical structure of akuammiline alkaloids together with their biological activity has sparked the interest of the synthetic community during the last decades.8 Among them, strictamine (3) has succumbed only recently to total synthesis by the groups of Garg (enantioselective),9 Zhu (racemic),10 and Ohno (formal synthesis).11
The architecture of strictamine (3) is characterized by the presence of methanoquinolizidine system 4 (highlighted in red, Fig. 2).1b This structure element represents a tricyclic system containing the C–D–E rings of strictamine (3). The methanoquinolizidine is assembled by the C7–C16 carbon carbon bond (biosynthetic numbering). This bond is crucial, since it accounts for the dearomatized indolenine system bearing a quaternary carbon center at C7. This leads to a very compact, cage like structure comparable to adamantane (5) (see Fig. 2).
 | ||
| Fig. 2 Cage-like methanoquinolizidine structure 4 of the akuammiline alkaloids. | ||
Our retrosynthetic analysis commences with the removal of the indolenine core – a very sensitive structure element. Intramolecular 1,4-addition disassembles the methanoquinolizidine motif, leading to ketone 6. This compound constitutes the product of [2,3]-Stevens rearrangement-II,12 obtained from bicycle 7 and iodide 8. In this rearrangement, the C–D ring system of 6 – an 2-azabicyclo[3.3.1]nonane – is assembled (Fig. 3).
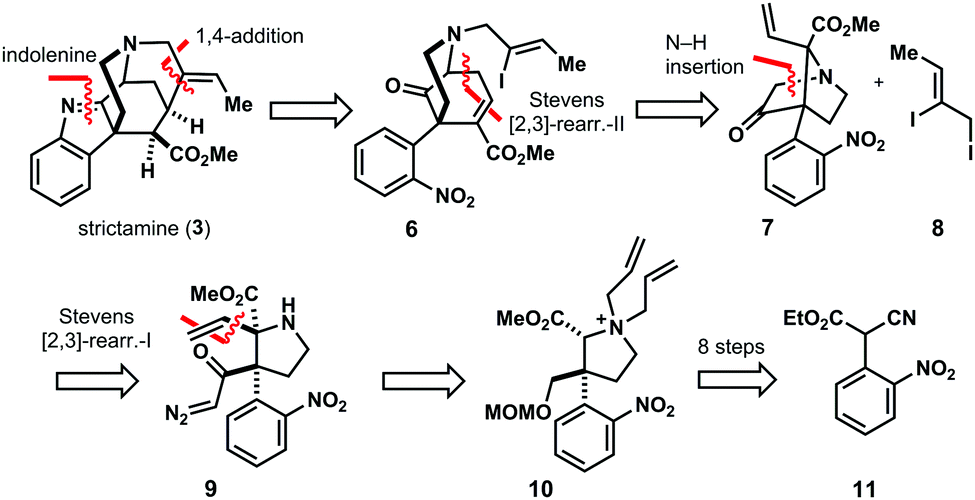 | ||
| Fig. 3 Retrosynthetic analysis of strictamine (3). | ||
Therefore, this Stevens rearrangement-II represents the key transformation of our synthesis. Its mechanism is outlined in Scheme 1 detailing the assembly of the C–D bicyclic system of methanoquinolizidine 4.
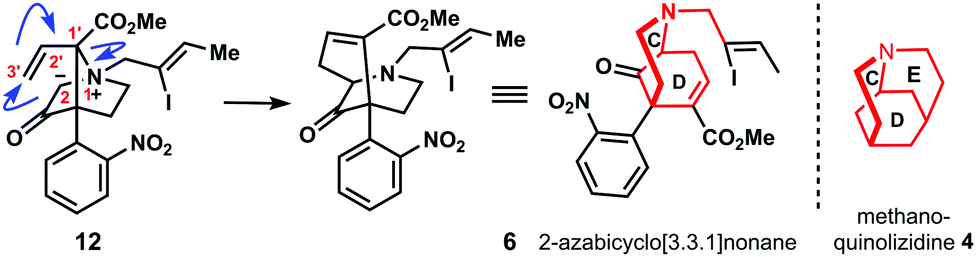 | ||
| Scheme 1 Mechanism of Stevens rearrangement-II. | ||
The bridged bicyclic core of rearrangement precursor 7 is generated via N–H insertion of diazoketone 9 (Fig. 3). This highly substituted pyrrolidine harbors two vicinal stereocenters, the β-center constituting a quaternary carbon center rendering its synthesis a formidable synthetic challenge. The α-stereocenter of 9 is introduced by [2,3]-Stevens rearrangement-I of ammonium salt 10 with complete control of diastereoselectivity. Pyrrolidine 10 is obtained in eight isolated steps from known nitrobenzene 11.13
Our synthesis started with α-cyanoacetate 11,13 which was alkylated with dibromoethane and K2CO3 in dimethylformamide at 90 °C to generate the quarternary carbon center of 12 after hydrolysis of the nitrile group with concentrated sulfuric acid to afford carboxylic amide 12 in 68% overall yield (Scheme 2). Lactam formation was affected with sodium hexamethyldisilazane at −78 °C, which was subsequently protected as tert-butyl carbamate to give hemiaminal 13 in 67% yield upon reduction with superhydride in the presence of catalytic amounts of boron trifluoroetherate.
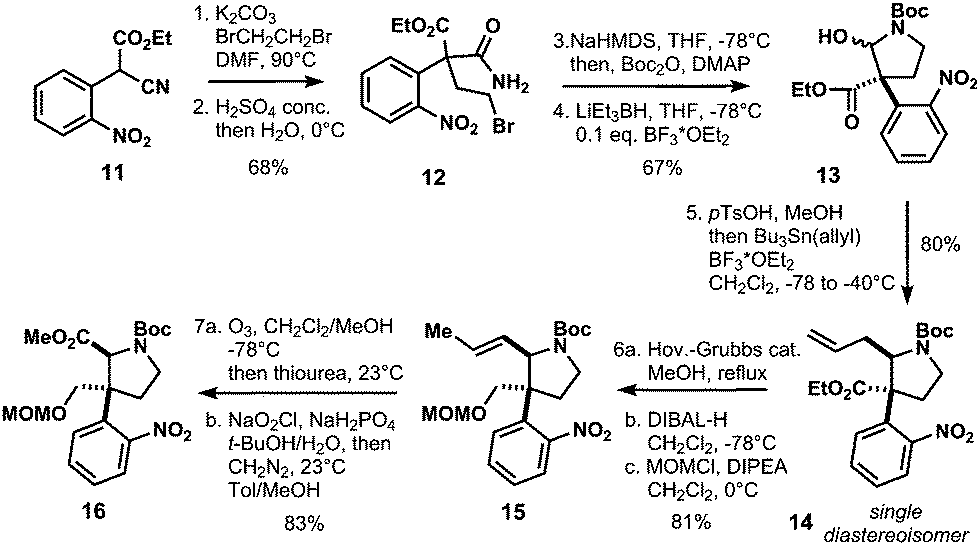 | ||
| Scheme 2 Establishment of quaternary carbon center in 12. | ||
Conversion of 13 to the methoxy aminal with p-toluenesulfonic acid and concomitant allylation via N-acyl iminium ion chemistry afforded compound 14 as a single diastereoisomer in 80% yield. The allyl-group in 14 was isomerized with Hoveyda–Grubbs II catalyst, then reduction of the ester with DIBAL-H and subsequent protection with chloromethyl methyl ether (MOMCl) gave 15 in 81% yield (Scheme 2). The isomerized double bond of 15 was thus converted to the methyl ester by ozonolysis, Pinnick oxidation and methylation with diazomethane to furnish 16 in 83% yield. Deprotection of the Boc-group with trifluoroacetic acid in dichloromethane followed by N-alkylation with allyl bromide gave access to allyl amine 17 (Scheme 3). Introduction of the tertiary α-stereocenter at the pyrrolidine ring proceeded via Stevens rearrangement-I with silver(I) triflate to give product 18 with complete diastereoselectivity (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in 53% yield (21% recovered starting material). The stereochemistry of 18 was determined by X-ray analysis. We surmise that this selectivity originates from steric hindrance exerted by the nitrophenyl group in the course of the rearrangement of 10. Therefore, the α-stereocenter is established in a cis fashion with respect to the MOM-protected alcohol (see 10 and 18 in Scheme 3).
:1) in 53% yield (21% recovered starting material). The stereochemistry of 18 was determined by X-ray analysis. We surmise that this selectivity originates from steric hindrance exerted by the nitrophenyl group in the course of the rearrangement of 10. Therefore, the α-stereocenter is established in a cis fashion with respect to the MOM-protected alcohol (see 10 and 18 in Scheme 3).
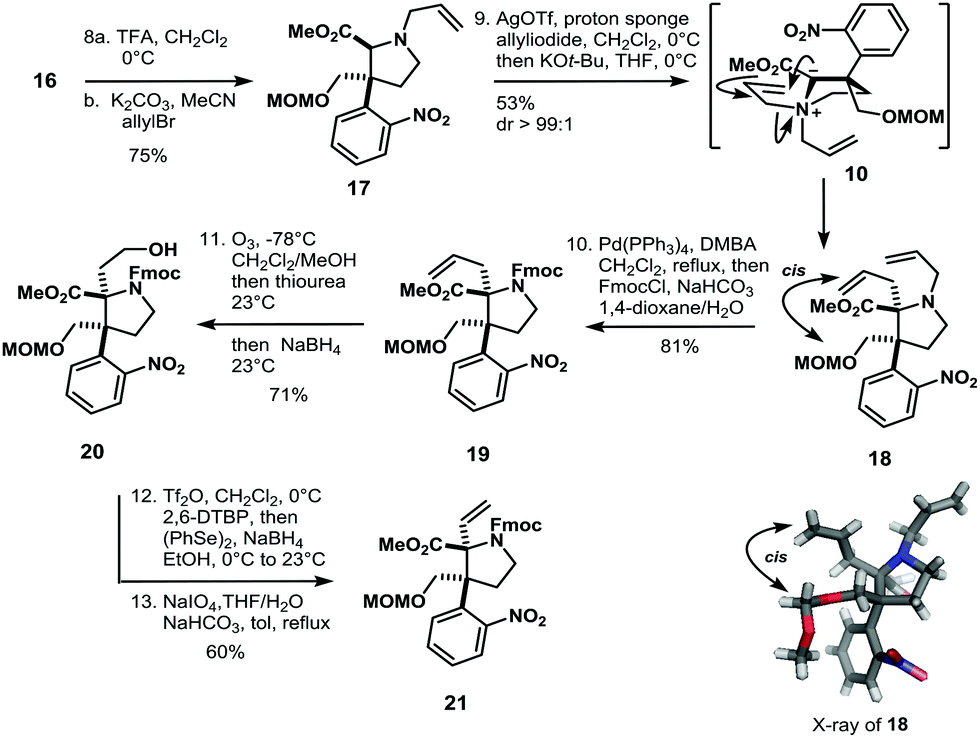 | ||
| Scheme 3 Establishment of the α-stereocenter in 18. | ||
The so obtained allyl amine 18 was converted to Fmoc-carbamate 19 in two transformations and 81% yield. In order to obtain envisioned Stevens rearrangement-II substrate 7, the next task was to transform the allylic side chain of 19 to the corresponding vinyl-chain (see compound 21 in Scheme 3). This was achieved by ozonolysis/reduction to furnish alcohol 20. Triflation of 20 and SN2-reaction with NaSePh afforded vinyl-compound 21 in 60% yield after oxidative workup with sodium periodate (Scheme 3). With the vinyl-chain established, the bridged bicyclic system of 7 was assembled in five isolated steps from 21 (Scheme 4).
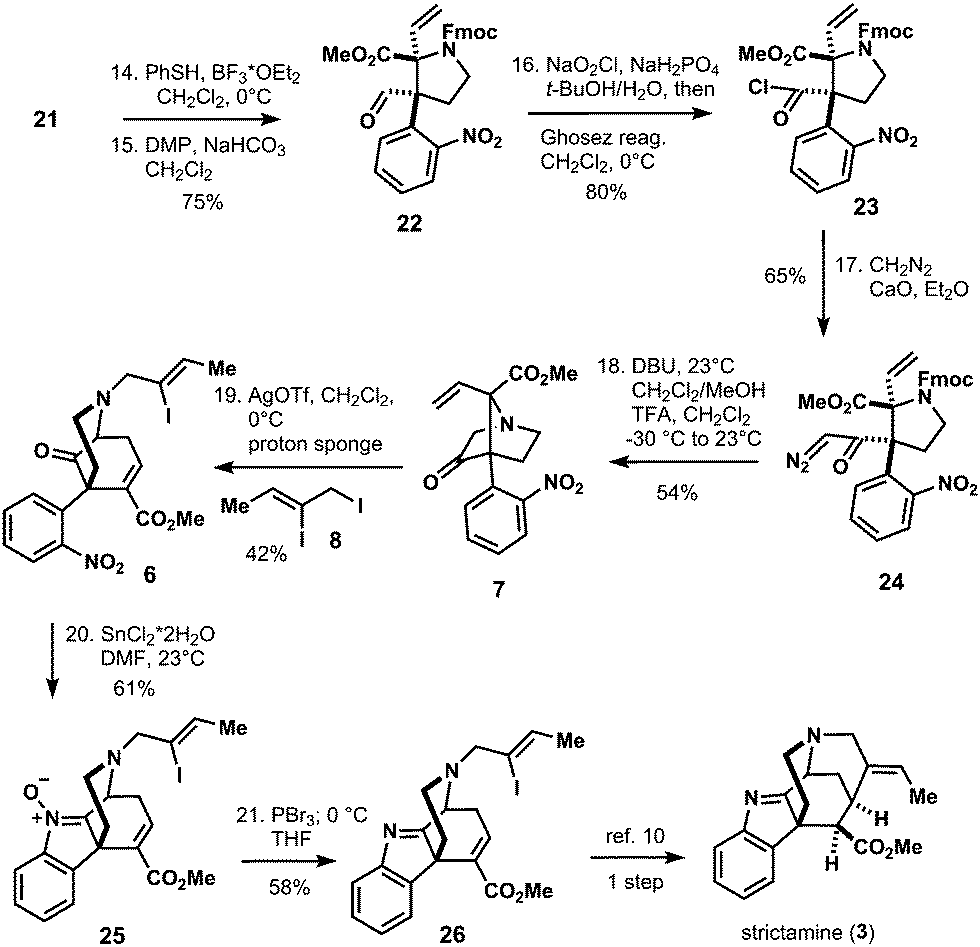 | ||
| Scheme 4 Endgame of the synthesis. | ||
Thus, deprotection of the MOM-ether of 21 and subsequent oxidation with Dess–Martin periodinane (DMP) afforded aldehyde 22. Pinnick oxidation to the corresponding carboxylic acid with concomitant acid chloride formation with Ghosez's reagent14 delivered 23. The latter was reacted with diazomethane in a sealed tube to give desired α-diazo ketone 24. With this compound in hands, bridged bicyclic system 7 was formed by Fmoc-deprotection of 24 and subsequent acid-triggered N–H insertion to yield Stevens rearrangement-II precursor 7 in 54% yield. N-Alkylation with iodide 8 and AgOTf in combination with proton sponge in dichloromethane provided the corresponding ylide, which directly underwent the envisioned [2,3]-sigmatropic Stevens rearrangement-II to give the crucial azabicyclo-[3.3.1]nonane skeleton 6 in 42% yield. With compound 6 in hands, reduction of the nitro group was attempted and resulted in decomposition under various conditions. However, treatment of 6 with tin(II)chloride dihydrate delivered 61% of incompletely reduced product 25 as a single product. After extensive experimentation we were able to reduce nitrone 25 to imine 26 with phosphorus tribromide in tetrahydrofuran and 58% yield.15 Compound 26 can be converted to strictamine (3) in one more step according to Zhu's protocol.10
In conclusion, we have accomplished a formal total synthesis of strictamine (3) based on 21 isolated steps (34 chemical transformations). Our synthetic route is highly diastereoselective, very robust and was performed on multigram-scale up to diazo-compound 24. We were able to show the versatility of a [2,3]-Stevens rearrangement for the construction of the 2-azabicyclo[3.3.1]nonane system, which is a very challenging structure motif to synthesize and represents the key compound for the total synthesis of strictamine (3).
Financial support was granted by the Fonds der Chemischen Industrie via a Liebigstipendium.
Notes and references
- (a) T. A. Henry and T. M. Sharp, J. Chem. Soc., 1927, 1950 RSC ; for reviews, see: ; (b) R. Eckermann and T. Gaich, Synthesis, 2013, 2813 CAS; (c) J. M. Smith, J. Moreno, B. W. Boal and N. K. Garg, Angew. Chem., Int. Ed., 2015, 54, 400 CAS.
- (a) I. C. Ezeamuzie, M. C. Ojinnaka, E. O. Uzogara and S. E. Oji, Afr. J. Med. Sci., 1994, 23, 85 CAS; (b) G. J. Kapadia, C. K. Angerhofer and R. Ansa-Asamoah, Planta Med., 1993, 59, 565 CrossRef CAS PubMed.
- (a) T. O. Fakeye, O. A. Itiola and H. A. Odelola, Phytother. Res., 2000, 14, 368 CrossRef CAS PubMed; (b) W. Wang, M.-H. Cheng and X.-H. Wang, Molecules, 2013, 18, 7309 CrossRef CAS PubMed.
- M. Duwiejua, E. Woode and D. D. Obiri, J. Ethnopharmacol., 2002, 81, 73 CrossRef CAS PubMed.
- T. A. Henry, J. Chem. Soc., 1932, 2759 RSC.
- H. K. Schnoes, K. Biemann, J. Mokry, I. Kompis, A. Chatterjee and G. Ganguli, J. Org. Chem., 1966, 31, 1641 CrossRef CAS.
- Y. Hou, X. Cao, L. Wang, B. Cheng, L. Dong, X. Luo, G. Bai and W. Gao, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2012, 908, 98 CrossRef CAS PubMed.
- (a) M. Zhang, X. Huang, L. Shen and Y. Qin, J. Am. Chem. Soc., 2009, 131, 6013 CrossRef CAS PubMed; (b) G. L. Adams, P. J. Carroll and A. B. Smith, J. Am. Chem. Soc., 2012, 134, 4037 CrossRef CAS PubMed; (c) B. D. Horning and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 6442 CrossRef CAS PubMed; (d) L. Zu, B. W. Boal and N. K. Garg, J. Am. Chem. Soc., 2011, 133, 8877 CrossRef CAS PubMed; (e) W. Ren, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2014, 53, 1818 CrossRef CAS PubMed; (f) M. Teng, W. Zi and D. Ma, Angew. Chem., Int. Ed., 2014, 53, 1814 CrossRef CAS PubMed; (g) J. M. Smith, J. Moreno, B. W. Boal and N. K. Garg, J. Am. Chem. Soc., 2014, 136, 4504 CrossRef CAS PubMed; (h) M. W. Smith and S. A Snyder, J. Am. Chem. Soc., 2013, 135, 12964 CrossRef CAS PubMed; (i) W. Zi, W. Xie and D. Ma, J. Am. Chem. Soc., 2012, 134, 9126 CrossRef CAS PubMed; (j) G. L. Adams, P. J. Carroll and A. B. Smith, J. Am. Chem. Soc., 2013, 135, 519 CrossRef CAS PubMed; (k) A. Ramirez and S. Garcia-Rubio, Curr. Med. Chem., 2003, 10, 1891 CrossRef CAS PubMed.
- J. Moreno, E. Picazo, L. A. Morrill, J. M. Smith and N. K. Garg, J. Am. Chem. Soc., 2016, 138, 1162 CrossRef CAS PubMed.
- W. Ren, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 3500 CrossRef CAS PubMed.
- D. Nishiyama, A. Ohara, H. Chiba, H. Kumagai, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2016, 18, 1670 CrossRef CAS PubMed.
- J. A. Vanecko, H. Wan and F. G. West, Tetrahedron, 2006, 62, 1043 CrossRef CAS.
- M. Makosza, R. Podraza and A. Kwast, J. Org. Chem., 1994, 59, 6796 CrossRef CAS.
- L. Ghosez and J. Marchand-Brynaert, in Advances in Organic Chemistry, ed. R. A. Raphael, E. C. Taylor and H. Wynberg, Interscience, New York, 1976, vol. 1, pp. 421–523 Search PubMed.
- T. Doi, K. Oikawa, J. Suzuki, M. Yoshida and N. Iki, Synlett, 2012, 306 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1474384. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc06125g |
| This journal is © The Royal Society of Chemistry 2016 |