
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoselective reductive alkynylation of tertiary amides by Ir and Cu(I) bis-metal sequential catalysis†
Pei-Qiang
Huang
 *ab,
Wei
Ou
a and
Feng
Han
a
*ab,
Wei
Ou
a and
Feng
Han
a
aDepartment of Chemistry and The Key Laboratory for Chemical Biology of Fujian Province, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, P. R. China. E-mail: pqhuang@xmu.edu.cn
bState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China
First published on 5th September 2016
Abstract
We report herein a convenient and versatile method for the direct reductive alkynylation of tertiary amides to give propargylic amines through sequential Ir-catalysed hydrosilylation–Cu(I)-catalysed alkynylation. The reactions proceed chemoselectively at the amide group in the presence of several sensitive functional groups including the very reactive aldehyde group on either the amide or the alkyne coupling partner. The method is general for tert-amides with or without α-hydrogen.
Reductive alkylation of amides is an essential transformation in the synthesis of alkaloids and nitrogen-containing medicinal agents.1,2a,b However, due to the poor electrophilicity of the amide carbonyl group, those transformations frequently require the use of highly reactive organometallic reagents (e.g. RLi and RMgX) and harsh conditions, which lead to low chemoselectivity and poor functional group tolerance. Consequently, multi-step methods are being routinely employed instead.1b,f,g
Within the context of developing efficient synthetic methods, the step-economical direct transformation of amides has attracted considerable attention in recent years, leading to the accumulation of a host of synthetically useful methods.2,3 Despite the advances, catalytic C–C bond forming reactions that employ common amides as substrates remain rare and challenging. Recently, based on Nagashima's iridium-catalysed transformation of amides into enamines (Scheme 1a),4 Dixon and Chida/Sato have independently developed a catalytic intramolecular reductive nitro-Mannich-type reaction,5 and a chemoselective reductive nucleophilic addition to N-methoxyamides.6 Very recently, Chida and Sata have reported an iridium-catalysed reductive transformation of N-hydroxyamides into nitrones.7 However, only tert-amides with α-hydrogens have been employed as substrates in all these methods.4–7
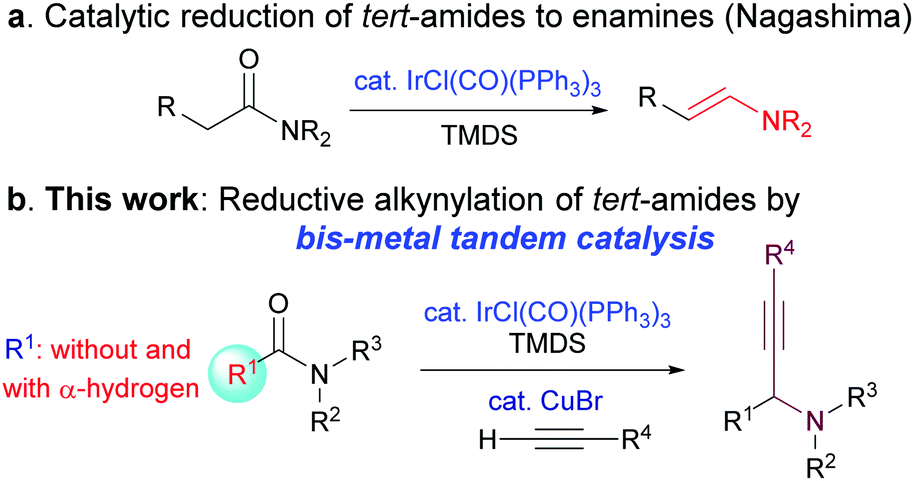 | ||
| Scheme 1 (a) Nagashima's catalytic reduction of amides to enamines and (b) bis-metal sequential catalytic transformation of amides with C–C bond formation. | ||
Propargylic amines are a class of versatile building blocks in organic synthesis and medicinal chemistry.1f,g,8 The catalytic synthesis of these important structures has been extensively investigated in the last 15 years.8,10 These efforts have resulted in several highly efficient and powerful methodologies.8–11 However, to the best of our knowledge, the access to propargylic amines through the direct catalytic reductive alkynylation of amides and lactams has hitherto been unknown. Considering the widespread use of amides as versatile synthetic intermediates in the synthesis of alkaloids and medicinal agents,1,2a and the fact that amides are products of numerous powerful synthetic methodologies,12 the catalytic transformation of amides leading to C–C bond formation is highly desirable. Except for one report,13 the known methods for the reductive alkynylation of amides are stepwise and require additional steps to convert amides/lactams to more reactive thioamides/lactams,1f,g,14 and selenoamides.15 As a continuation of our research program on the direct transformation of amides,3b,e,g–i we report herein a bis-metallic Ir- and Cu(I)-catalysed reductive alkynylation of common tertiary amides with or without an α-hydrogen to give propargylic amines (Scheme 1b).
On the basis of the abovementioned literature precedents, our goal was to develop a direct, sequential catalytic hydrosilylation–alkynylation of common tert-amides. Although Nagashima's method (Scheme 1a)4 has been developed and applied only for tert-amides containing α-hydrogens,5–7 we wondered if this method could be extended to include common tert-amides without an α-hydrogen as substrates. Thus, a toluene solution of N,N-dimethylbenzamide (1a) was treated with a catalytic amount of Vaska's complex [IrCl(CO)(PPh3)2, 1 mol%], 1.2 equiv. of TMDS, 1.2 equiv. of phenylacetylene, and 5 mol% of CuBr (Table 1).9b To our delight, the desired propargylamine 3a was obtained in 89% yield after being stirred at room temperature for 12 h. Similarly, subjecting of amide 1b (the structures of all amides and alkynes used are listed in the ESI†) to the two-step, one-pot reductive alkynylation reaction afforded propargylic amine 3b in 90% yield. Reactions of aliphatic amides 1c and 1d produced the corresponding propargylic amines 3c and 3d in 60% and 80% yield, respectively. The relatively low yield of 3c could be attributed to the steric hindrance of the amide substrate. The reaction of sterically hindered N,N-diisopropylbenzamide 1e also provided the corresponding amine (3e) in a moderate yield (67%), much lower than those of sterically unencumbered N,N-dimethylbenzamide (1a) and N,N-diallylbenzamide (1f). Functionalized alkyne 2b participated in the reaction to afford amine 3g in an excellent yield (91%). On the other hand, amides bearing either electron-rich groups such as furan-2-yl (1h) and benzo[b]thiophen-2-yl (1i) or electron-poor groups such as para-nitrophenyl (1j) all reacted similarly to give the corresponding amines 3h, 3i, and 3j in 80%, 81%, and 77% yield, respectively.
After examining the steric and electronic effects, we explored the chemoselectivity and functional group tolerance.4,6,9e The mild reductive alkynylation reaction showed remarkable functional group tolerance and chemoselectivity as demonstrated by the formation of products 3j–3p. The reaction tolerated redox-sensitive functional groups such as nitro (3j), cyano (3k), and aldehyde (3l), and common nitrogen protecting groups such as Boc (3m) and Cbz (3n). It is well known that amides are far less reactive than aldehydes. However, under the current conditions, the reaction of an aldehyde-containing substrate took place chemoselectively at the carbonyl of the amide instead of that of the aldehyde, leading to the isolation of amino aldehyde 3l in 64% yield, along with the (aldehyde group) concomitantly reduced product 3l′ in 8% yield. Moreover, the reductive alkynylation reaction is fully compatible with a Boc-protected amine that contains an acidic proton (NHBoc, 1o), affording the desired product 3o in excellent yield (89%). Interestingly, the reaction of an amide bearing a free hydroxyl group (1p) yielded the O-silylated product 3p in 52% yield when 2.0 equiv. of TMDS was used. It is worth mentioning that o-cyanobenzamide 1k is a product of metal-catalysed C–H cyanation.12e
We next investigated the stereoselectivity of the reductive alkynylation reaction. For this purpose, benzamide derivatives derived from methyl (S)-prolinate 1q–1s were selected as substrates. These functionalized substrates also provided opportunities for the further examination of the functional group tolerance and chemoselectivity of the reaction. The reductive phenylacetylenation of amido ester 1q proceeded chemoselectively at the amido group to give a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture from which the major diastereomer 3q was isolated in 78% yield (Table 1).
:1 diastereomeric mixture from which the major diastereomer 3q was isolated in 78% yield (Table 1).
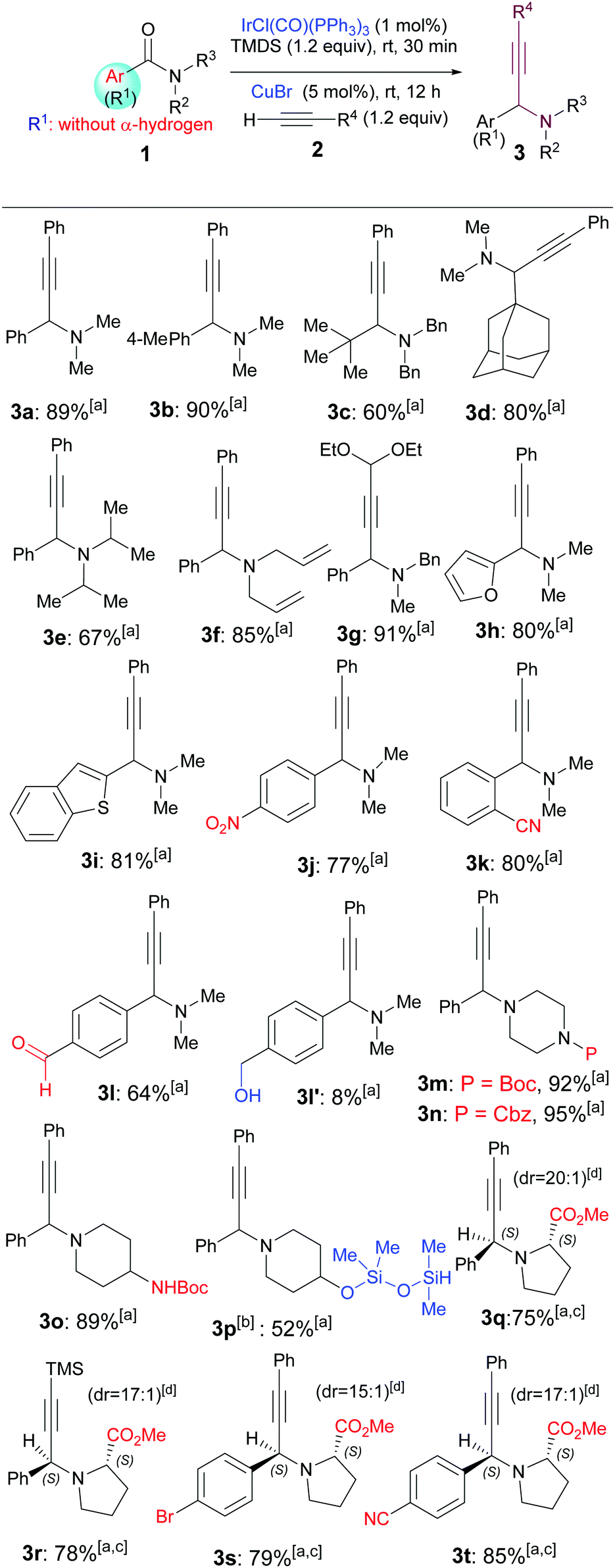
Similarly, excellent chemoselectivities and diastereoselectivities (dr = 17:1, 15:1, and 17:1) were also observed for the reactions of trimethylsilylacetylene with 1q, and phenylacetylene with the bromo-, cyano-substituted prolinates 1r and 1s, which afforded the major diastereomers 3r, 3s and 3t in 78%, 79%, and 85% yield, respectively. Note that in the latter two reactions, the bromo, cyano, and ester groups remained intact during the reductive alkynylation process. The relative stereochemistry of the major diastereomer 3q was assigned to S,S by comparing the NMR data with those reported by Tu,9d and Che9e (two diastereomers of ethyl ester of 3q, cf. ESI†), and those of 3r–3t were assigned accordingly.
After the successful development of catalytic reductive alkynylation of amides without α-hydrogen, we turned our attention to extend the reaction to amides bearing α-hydrogen atoms. Moreover, to further examine the scope of the alkynes, functionalized ones were employed. Thus, the reductive alkynylation of amide 1t with propiolaldehyde diethylketal 2b furnished the propargylic amine 3u in 94% yield (Scheme 2). This gram-scale reaction used 2 equiv. of TMDS instead of the usual 1.2 equiv. Subsequent reduction of 3u with H2 and Pd/C could be combined with the reductive alkynylation in one-pot to give the alkaloid (±)-bgugaine (4)16 (Scheme 2). Bgugaine is an alkaloid isolated from the tubers of Arisarum vulgare and has been shown to be a strong hepatotoxin with antibacterial and antimycotic activities.16 Methyl propiolate could also be used for the direct reductive alkynylation to afford propargylic amine 3v in 88% yield even on a 5 mmol scale. Remarkably, the direct reductive alkynylation with an acetylene bearing an unprotected aldehyde group underwent smooth reaction to afford the desired amino aldehyde 3w in 67% yield.
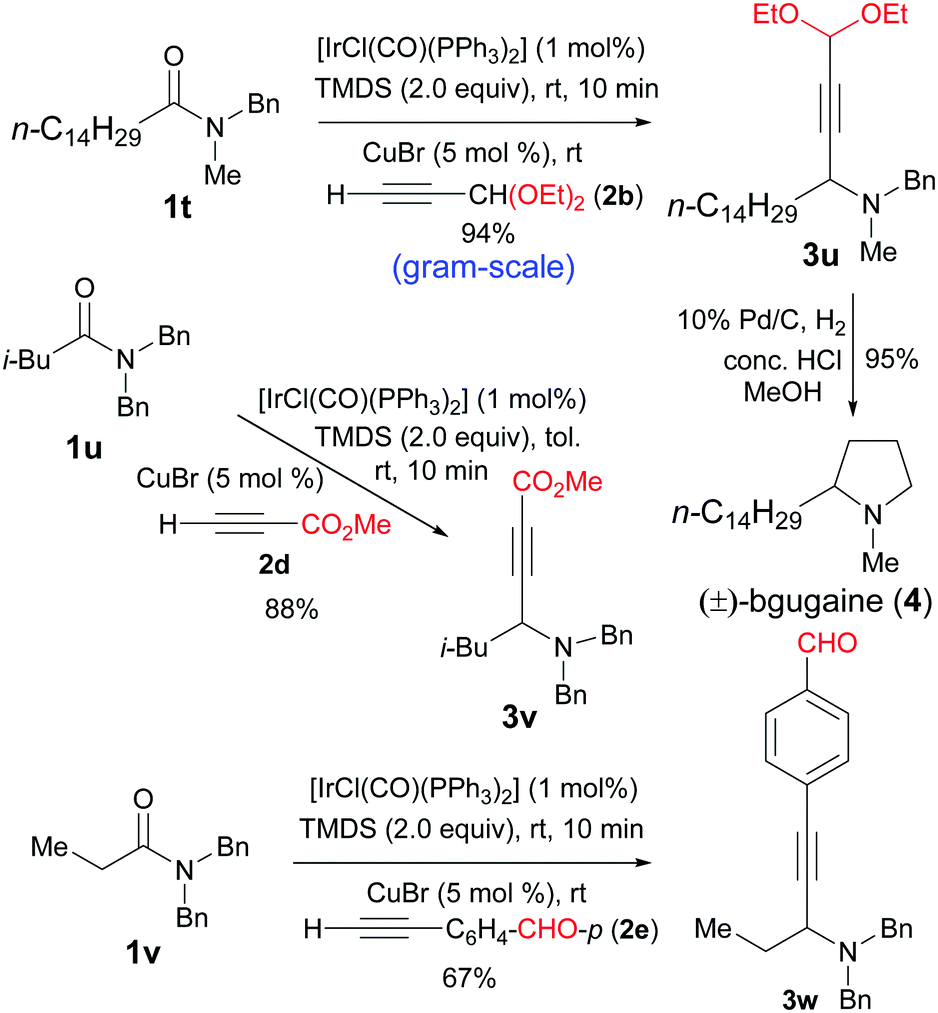 | ||
| Scheme 2 Direct catalytic reductive alkynylation of tert-amides containing α-hydrogens. | ||
We next undertook a preliminary mechanistic study. For this purpose, 1H NMR spectra of starting amide 1v (spectrum A, cf. ESI†), its mixture with TMDS (spectrum B, cf. ESI†), and the mixture formed 10 min after addition of the Ir catalyst (spectrum C, Fig. S1, ESI†) were recorded. Spectrum C showed that the starting amide was completely consumed (δα-H = 2.06, q) with the clean formation of enamine A-v (δenamine-H = 6.07, d). On the other hand, the 1H NMR spectra of starting benzamide 1b (spectrum A′, cf. ESI†), an amide without an α-hydrogen, its mixture with TMDS (spectrum B′, cf. ESI†), and the mixture formed 30 min after the addition of the Ir-catalyst (spectrum C′, Fig. S2, ESI†) indicated that the amide (δNMe–H = 2.64, s) was transformed cleanly to give hemiaminal silyl ether B-b (δNCHO = 5.61, s). On the basis of the above-mentioned information, a plausible mechanism for the reaction is depicted in Scheme 3. Upon coordination with CuBr, terminal hydrogen in alkyne C became more acidic and was deprotonated with either enamine A or hemiaminal silyl ether intermediate B to generate an alkynylide ion D and an iminium intermediate E or E′. The scenario of the reaction between C and A is in analogy with that proposed by Knochel.10a,c The addition of the alkynylide ion D to the iminium ions E/E′ then gives F/F′, which, after work-up, affords the propargylic amines 3.
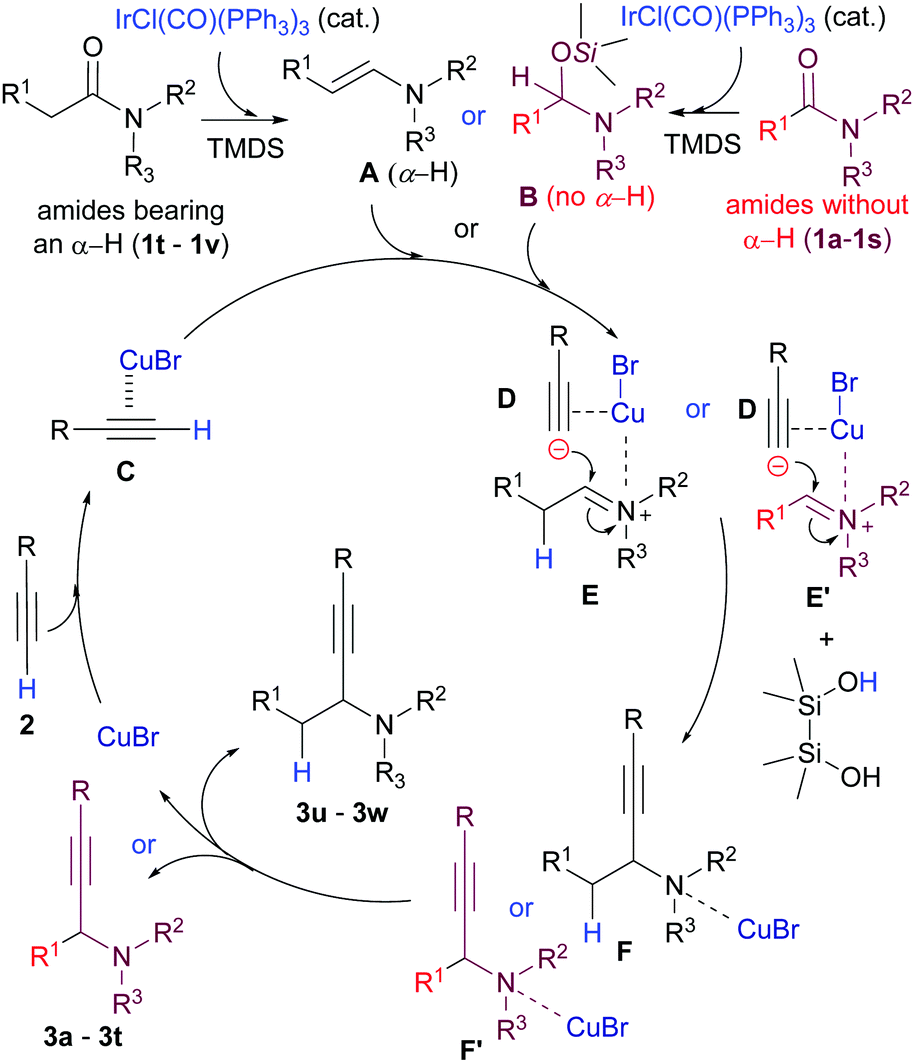 | ||
| Scheme 3 Plausible mechanism for the bis-metallic Ir- and Cu(I)-catalysed reductive alkynylation of tert-amides. | ||
In summary, we have demonstrated that the Ir-catalysed hydrosilylation of tert-amides is compatible with the Cu(I)-catalysed alkynylation reaction. On the basis of this finding, we have developed a mild, sequential catalysis method for the direct reductive alkynylation of tert-amides to give propargylic amines.
The method is broad in scope, allowing the employment of aromatic as well as aliphatic tert-amides with or without α-hydrogen and bearing functional groups with diverse electronic and steric properties. The method also shows excellent 1,3-asymmetric induction. The reaction exhibits remarkable chemoselectivity at the least reactive amide group and is compatible with several sensitive functional groups such as aldehyde, cyano, ester, and nitro groups on either the amide or alkyne partners.
General procedure for the reductive alkynylation of amides. To a dried 10 mL round-bottom flask containing IrCl(CO)(PPh3)2 (1 mol%, weighted in a glove box) were added a toluene (5 mL) solution, a tert-amide (1.0 mmol) and TMDS (1.2 mmol) or TMDS (2.0 mmol) (for amides with α-hydrogen) at rt. After being stirred for 30 min (for amides without α-hydrogen) or 10 min (for amides with α-hydrogen), the resulting solution and an alkyne (1.2 mmol) were added to a suspension of CuBr (5 mol%) in toluene (3 mL). The mixture was stirred for 12 h at rt and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel to afford the corresponding propargylic amine 3.
The authors are grateful for financial support from the National Natural Science Foundation of China (21332007), and the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) of the Ministry of Education, China.
Notes and references
- For selected examples, see: (a) C. Lindemann and C. Schneider, Synthesis, 2016, 828–844 CAS; (b) A. S. Lee, B. B. Liau and M. D. Shair, J. Am. Chem. Soc., 2014, 136, 13442–13452 CrossRef CAS PubMed; (c) K. Shirokane, T. Wada, M. Yoritate, R. Minamikawa, N. Takayama, T. Sato and N. Chida, Angew. Chem., Int. Ed., 2014, 53, 512–516 CrossRef CAS PubMed; (d) P. Jakubec, A. Hawkins, W. Felzmann and D. J. Dixon, J. Am. Chem. Soc., 2012, 134, 17482–17485 CrossRef CAS PubMed; (e) J. W. Medley and M. Movassaghi, Angew. Chem., Int. Ed., 2012, 51, 4572–4576 CrossRef CAS PubMed; (f) T. Murai and F. Asai, J. Am. Chem. Soc., 2007, 129, 780–781 CrossRef CAS PubMed; (g) P. Schär and P. Renaud, Org. Lett., 2006, 8, 1569–1571 CrossRef PubMed.
- For reviews and perspectives on nucleophilic addition to amides, see: (a) V. Pace, W. Holzer and B. Olofsson, Adv. Synth. Catal., 2014, 356, 3697–3736 CrossRef CAS; (b) T. Sato and N. Chida, Org. Biomol. Chem., 2014, 12, 3147–3150 RSC; (c) D. Kaiser and N. Maulide, J. Org. Chem., 2016, 81, 4421–4428 CrossRef CAS PubMed.
- For selected examples, see: (a) C. Madelaine, V. Valerio and N. Maulide, Angew. Chem., Int. Ed., 2010, 49, 1583–1586 CrossRef CAS PubMed; (b) K.-J. Xiao, J.-M. Luo, K.-Y. Ye, Y. Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2010, 49, 3037–3040 CrossRef CAS PubMed; (c) K. Shirokane, Y. Kurosaki, T. Sato and N. Chida, Angew. Chem., Int. Ed., 2010, 49, 6369–6372 CrossRef CAS PubMed; (d) W. S. Bechara, G. Pelletier and A. B. Charette, Nat. Chem., 2012, 4, 228–234 CrossRef CAS PubMed; (e) K.-J. Xiao, A.-E Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2012, 51, 8314–8317 CrossRef CAS PubMed; (f) A. Romanens and G. Bélanger, Org. Lett., 2015, 17, 322–325 CrossRef CAS PubMed; (g) J.-F. Zheng, Z.-Q. Xie, X.-J. Chen and P.-Q. Huang, Acta Chim. Sin., 2015, 73, 705–715 CrossRef CAS; (h) P.-Q. Huang, Y.-H. Huang, K.-J. Xiao, Y. Wang and X.-E. Xia, J. Org. Chem., 2015, 80, 2861–2868 CrossRef CAS PubMed; (i) P.-Q. Huang, Y.-H. Huang, H. Geng and J.-L. Ye, Sci. Rep., 2016, 6, 28801, DOI:10.1038/srep28801.
- Y. Motoyama, M. Aoki, N. Takaoka, R. Aoto and H. Nagashima, Chem. Commun., 2009, 1574–1576 RSC.
- A. W. Gregory, A. Chambers, A. Hawkins, P. Jakubec and D. J. Dixon, Chem. – Eur. J., 2015, 21, 111–114 CrossRef CAS PubMed.
- M. Nakajima, T. Sato and N. Chida, Org. Lett., 2015, 17, 1696–1699 CrossRef CAS PubMed.
- S. Katahara, S. Kobayashi, K. Fujita, T. Matsumoto, T. Sato and N. Chida, J. Am. Chem. Soc., 2016, 138, 5246–5249 CrossRef CAS PubMed.
- For selected examples, see: (a) S. Diethelm and E. M. Carreira, J. Am. Chem. Soc., 2015, 137, 6084–6096 CrossRef CAS PubMed; (b) P. B. Huleatt, M. L. Khoo, Y. Y. Chua, T. W. Tan, R. S. Liew, B. Balogh, R. Deme, F. Gölöncsér, K. Magyar, D. P. Sheela, H. K. Ho, B. Sperlágh, P. Mátyus and C. L. L. Chai, J. Med. Chem., 2015, 58, 1400–1419 CrossRef CAS PubMed; (c) W. Lin and S. M. Ma, Org. Chem. Front., 2014, 1, 338–346 RSC; (d) Z.-H. Chen, Y.-Q. Zhang, Z.-M. Chen, Y.-Q. Tu and F.-M. Zhang, Chem. Commun., 2011, 47, 1836–1838 RSC; (e) K. A. Miller, C. S. Shanahan and S. F. Martin, Tetrahedron, 2008, 64, 6884–6900 CrossRef CAS PubMed; (f) B. Jiang and M. Xu, Angew. Chem., Int. Ed., 2004, 43, 2543–2546 CrossRef CAS PubMed; (g) B. Jiang and Y.-G. Si, Angew. Chem., Int. Ed., 2004, 43, 216–218 CrossRef CAS PubMed.
- (a) C. Fischer and E. M. Carreira, Org. Lett., 2001, 3, 4319–4321 CrossRef CAS PubMed; (b) C.-J. Li and C. Wei, Chem. Commun., 2002, 268–269 RSC; (c) C. Wei, Z. Li and C.-J. Li, Org. Lett., 2003, 5, 4473–4476 CrossRef CAS PubMed; (d) L. Shi, Y.-Q. Tu, M. Wang, F.-M. Zhang and C.-A. Fan, Org. Lett., 2004, 6, 1001–1003 CrossRef CAS PubMed; (e) V. K.-Y. Lo, Y. Liu, M.-K. Wong and C.-M. Che, Org. Lett., 2006, 8, 1529–1532 CrossRef CAS PubMed.
- For enantioselective alkynylation reactions, see: (a) C. Koradin, K. Polborn and P. Knochel, Angew. Chem., Int. Ed., 2002, 41, 2535–2538 CrossRef CAS; (b) C. Wei and C.-J. Li, J. Am. Chem. Soc., 2002, 124, 5638–5639 CrossRef CAS PubMed; (c) C. Koradin, N. Gommermann, K. Polborn and P. Knochel, Chem. – Eur. J., 2003, 9, 2797–2811 CrossRef CAS PubMed; (d) N. Gommermann, C. Koradin, K. Polborn and P. Knochel, Angew. Chem., Int. Ed., 2003, 42, 5763–5766 CrossRef CAS PubMed; (e) T. F. Knöpfel, P. Aschwanden, T. Ichikawa, T. Watanabe and E. M. Carreira, Angew. Chem., Int. Ed., 2004, 43, 5971–5973 CrossRef PubMed; (f) P. Aschwanden, C. R. J. Stephenson and E. M. Carreira, Org. Lett., 2006, 8, 2437–2440 CrossRef CAS PubMed; (g) N. Gommermann and P. Knochel, Chem. – Eur. J., 2006, 12, 4380–4392 CrossRef CAS PubMed; (h) W. Fan and S.-M. Ma, Chem. Commun., 2013, 49, 10175–10177 RSC; (i) W. Lin, T. Cao, W. Fan, Y. Han, J. Kuang, H. Luo, B. Miao, X. Tang, Q. Yu, W. Yuan, J. Zhang, C. Zhu and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 277–281 CrossRef CAS PubMed; (j) C. Zhao and D. Seidel, J. Am. Chem. Soc., 2015, 137, 4650–4653 CrossRef CAS PubMed.
- A3-coupling reaction: (a) J. Li, M. Rudolph, F. Rominger, J. Xie and A. S. K. Hashmi, Adv. Synth. Catal., 2016, 358, 207–211 CrossRef CAS; (b) V. A. Peshkov, O. P. Pereshivko and E. V. Van der Eycken, Chem. Soc. Rev., 2012, 41, 3790–3807 RSC; (c) W.-J. Yoo, L. Zhao and C.-J. Li, Aldrichimica Acta, 2011, 44, 43–51 CAS; (d) C.-J. Li, Acc. Chem. Res., 2010, 43, 581–590 CrossRef CAS PubMed . CDC reaction: ; (e) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed . Redox-A3 reaction: ; (f) D. Seidel, Org. Chem. Front., 2014, 1, 426–429 RSC . See also: ; (g) H.-P. Bi, Q. Teng, M. Guan, W.-W. Chen, Y.-M. Liang, X. Yao and C.-J. Li, J. Org. Chem., 2010, 75, 783–788 CrossRef CAS PubMed; (h) J. Xie, S. Shi, T. Zhang, N. Mehrkens, M. Rudolph, A. Stephen and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2015, 54, 6046–6050 CrossRef CAS PubMed.
- (a) L. Xu, C. Zhang, Y. He, L. Tan and D.-W. Ma, Angew. Chem., Int. Ed., 2016, 55, 321–325 CrossRef CAS PubMed; (b) J. R. Cabrero-Antonino, I. Sorribes, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 387–391 CrossRef CAS PubMed; (c) J.-X. Yan, H. Li, X.-W. Liu, J.-L. Shi, X. Wang and Z.-J. Shi, Angew. Chem., Int. Ed., 2014, 53, 4945–4949 CrossRef CAS PubMed; (d) J. Hara, M. Ishida, M. Kobayashi, K. Noguchi and K. Tanaka, Angew. Chem., Int. Ed., 2014, 53, 2956–2959 CrossRef CAS PubMed; (e) W. Liu and L. Ackermann, Chem. Commun., 2014, 50, 1878–1881 RSC; (f) P. A. Donets and N. Cramer, J. Am. Chem. Soc., 2013, 135, 11772–11775 CrossRef CAS PubMed; (g) S. L. Zultanski and G. C. Fu, J. Am. Chem. Soc., 2011, 133, 15362–15364 CrossRef CAS PubMed.
- M. Yamaguchi and I. Hirao, Tetrahedron Lett., 1983, 24, 1719–1722 CrossRef CAS.
- (a) H. Takahata, K. Takahashi, E.-C. Wang and T. Yamazaki, J. Chem. Soc., Perkin Trans. 1, 1989, 1211–1214 RSC; (b) T. Murai, Y. Mutoh, Y. Ohta and M. Murakami, J. Am. Chem. Soc., 2004, 126, 5968–5969 CrossRef CAS PubMed; (c) T. Murai, R. Toshio and Y. Mutoh, Tetrahedron, 2006, 62, 6312–6320 CrossRef CAS; (d) T. Murai and F. Asai, J. Org. Chem., 2008, 73, 9518–9521 CrossRef CAS PubMed.
- (a) T. Murai, S. Nogawa and Y. Mutoh, Bull. Chem. Soc. Jpn., 2007, 80, 2220–2225 CrossRef CAS; (b) T. Mitamura and A. Ogawa, Org. Lett., 2009, 11, 2045–2048 CrossRef CAS PubMed.
- (a) A. Melhaoui, A. Jossang and B. Bodo, Nat. Prod. Lett., 1993, 2, 237–242 CrossRef CAS; (b) N. Rakba, A. Melhaoui, P. Loyer, J. G. Delcros, I. Morel and G. Lescoat, Toxicol. Lett., 1999, 104, 239–248 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc05318a |
| This journal is © The Royal Society of Chemistry 2016 |