
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic analysis of aliphatic β-lactones in Vibrio harveyi reveals a quorum sensing independent mode of action†
Weining
Zhao
a,
Nicola
Lorenz
b,
Kirsten
Jung
b and
Stephan A.
Sieber
*a
aDepartment of Chemistry, Center for Integrated Protein Science Munich (CIPSM), Technische Universität München, Garching, Germany. E-mail: stephan.sieber@tum.de
bCenter for Integrated Protein Science Munich (CIPSM) at the Department of Biology I, Ludwig-Maximilians-Universität München, Germany
First published on 31st August 2016
Abstract
N-Acylhomoserine lactones are autoinducers of quorum sensing (QS) in Gram-negative bacteria. We exploit here the role of structurally related β-lactones in the inhibition of Vibrio harveyi bioluminescence and identify a derivative with nanomolar potency. Surprisingly, QS was not affected and combined proteomic/biochemical studies revealed insights into the cellular mode of action.
Quorum sensing (QS) represents a common bacterial communication strategy controlling diverse cellular mechanisms including biofilm formation,1–3 virulence4,5 and bioluminescence.6 A growing population of bacteria secretes small autoinducer molecules (AIs) that are sensed by cytoplasmic or transmembrane receptors. Upon exceeding the threshold concentration of AIs an intracellular signaling cascade activates the transcription of QS-regulated genes.7 Bioluminescent strains such as Vibrio harveyi ATCC BAA-1116, recently reclassified as Vibrio campbellii ATCC BAA-1116,8 served as a model organism in the mechanistic analysis of QS9,10 as well as for inhibitor screens.11–13V. harveyi recognizes three different classes of AIs including species-specific HAI-1, the interspecies molecule AI-2 and Vibrio genus specific CAI-1. The binding of these AIs to their cognate receptors LuxN, LuxQ/P and CqsS induces a phosphorelay involving LuxU and LuxO to regulate the production of the QS master regulator LuxR. LuxR in turn induces transcription of genes required for bioluminescence, biofilm formation, or exoproteolytic activity, and represses the expression of the type III secretion system or siderophores.6 The inhibition of QS represents an attractive strategy for combating bacterial infections by attenuating virulence and biofilm formation.4,5,14,15 Several targets were exploited in the past that play crucial roles as key QS regulators, such as the QS-receptor itself or the master regulator LuxR.16
Interestingly, most compounds active in QS inhibition exploit certain structural motifs derived from the N-acylhomoserine lactone (AHL) scaffold.7 Here, among other features, the length of the acyl chain as well as the homoserine lactone core was identified as essential to promote QS. Interestingly, structurally related fimbrolide natural products are potent irreversible inhibitors of QS and bioluminescence by binding to LuxS, an enzyme required for AI-2 biosynthesis, as well as LuxE, an enzyme required for luciferase activity.17 Similarly, the structural composition of natural product β-lactones18 resembles some features of AHL, raising the question of whether a direct interference with QS pathways is also possible.
We investigate here the effects of diverse β-lactones on V. harveyi bioluminescence and identify a β-lactone equipped with a long aliphatic chain that even exceeds the potency of fimbrolides. Target identification via activity based protein profiling (ABPP) revealed an outer membrane protein, enzymes involved in fatty acid biosynthesis as well as a cyclic-di-GMP synthase as specific binders. Proteomic studies showed that β-lactones affect pathways relevant for fatty acid synthesis as well as electron transfer required for bioluminescence.
To unravel if the electrophilic β-lactone scaffold could substitute the larger and unreactive γ-lactone core of AHLs, we tested a panel of previously established β-lactones in bioluminescence assays (Fig. S1, ESI†).19 For benchmarking of their potencies, we included fimbrolides as the gold standard QS and bioluminescence inhibitors.17 All the compounds (50 μM final concentration) were added to the growing culture of Vibrio harveyi NBRC 15634, used here for initial screens, and bioluminescence was monitored in a plate reader. Out of the lactones tested, R1 decorated with a nonenyl-chain at the 2-position and a methyl group at the 3-position revealed a strong reduction of luminescence by 90%, comparable to those of fimbrolides that ranged from 75 to 97% inhibition (Fig. S2, ESI†). A closer inspection of the β-lactone structures revealed that although some of these compounds exhibited an identical nonenyl-substituent at the 2-position, larger decorations at the 3-position such as decenyl (T1), pentynyl (D3) or phenylethyl (U1) significantly decreased the potency down to 20%. Thus small substituents at the 2-position seem to be a key structural aspect of bioluminescence inhibition by β-lactones.
Based on the R1 substitution pattern, we expanded the investigation of structural analogs to exploit the inhibitory effect on bioluminescence of various long-chain aliphatic substituents at either 2- or 3-position. Three additional derivatives were synthesized (LP1, BL3 and LP3) and the collection was further complemented by the use of one previously established lactone (AV17).19 The derivatives were prepared by the alkylation of dimethyl malonate with alkyl iodide, followed by partial hydrolysis of the diester, reduction of the remaining ester to a hydroxy group with LiBH4 and cyclization by HBTU (Scheme 1 and Fig. 1A).
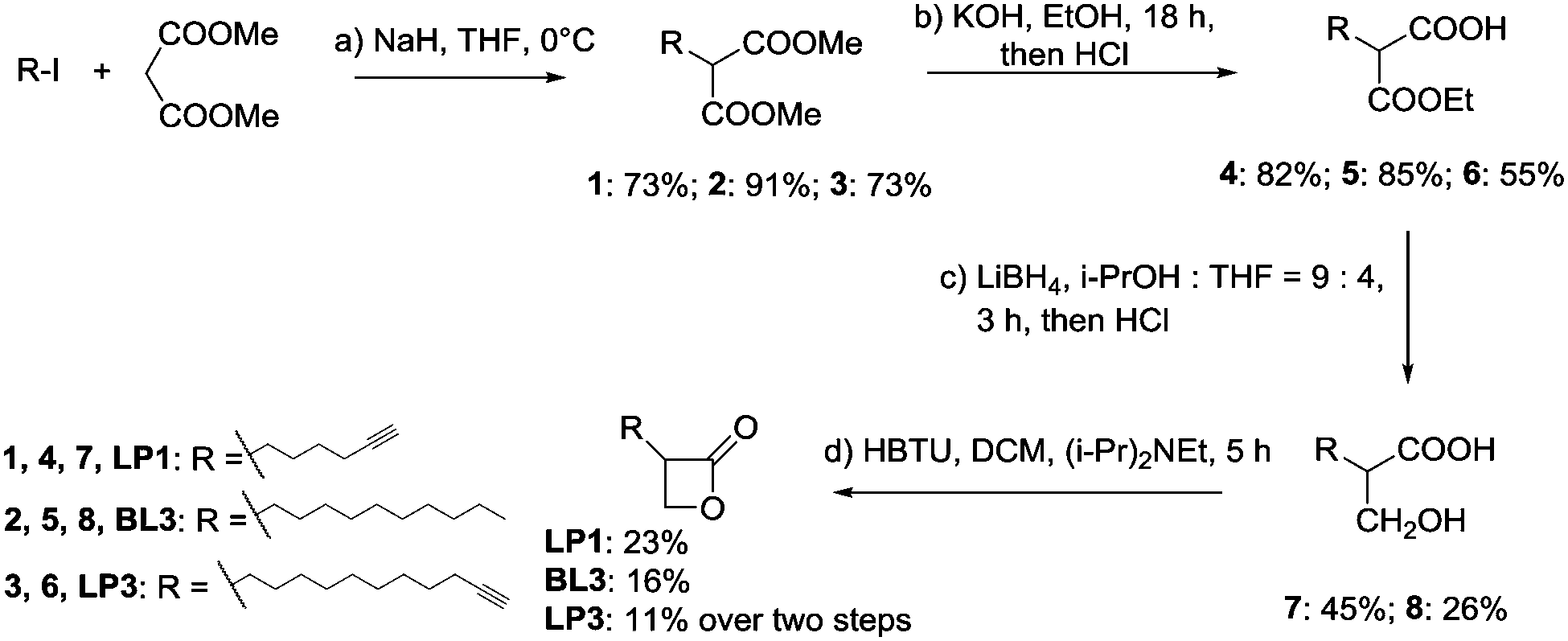 | ||
Scheme 1 Synthesis of β-lactones. (a) NaH, THF, 0 °C, then add R-I; (b) KOH, EtOH, rt, 18 h, then add HCl; (c) LiBH4, i-PrOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF = 9:4, 3 h, then add HCl; (d) HBTU, DCM, (i-Pr)2NEt, 5 h. THF = tetrahydrofuran, DCM = dichloromethane, HBTU = 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate. :THF = 9:4, 3 h, then add HCl; (d) HBTU, DCM, (i-Pr)2NEt, 5 h. THF = tetrahydrofuran, DCM = dichloromethane, HBTU = 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate. | ||
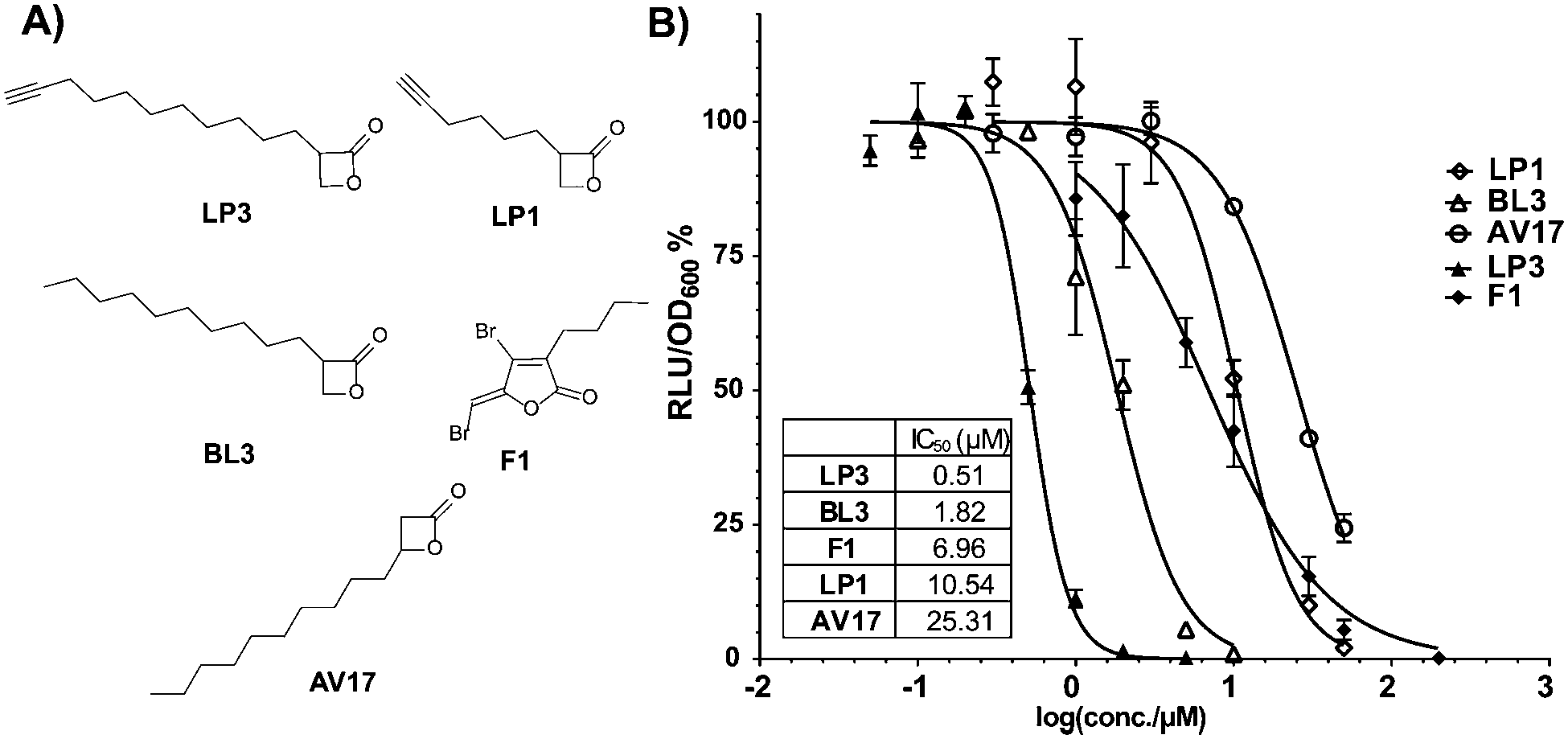 | ||
| Fig. 1 (A) Structures of compounds used in V. harveyi bioluminescence assay. (B) Residual bioluminescence production of V. harveyi after 30 min incubation with the compounds. Relative luminescence units (RLU) were normalized to cell density (OD600) and to the DMSO control. F1 is used as the benchmark. The data were based on three biological experiments with technical triplicates. | ||
For the determination of bioluminescence IC50 values, we switched to V. harveyi BAA-1116 as a commonly applied reference strain. As a general trend an increase of chain length at the 2-position dramatically enhanced the potency, while a substitution at the 3-position was largely inactive (Fig. 1B). Importantly, compounds BL3 and LP3 equipped with the longest aliphatic chains of this series, decyl and undecynyl, exhibited the best potencies with IC50s of 1.8 and 0.5 μM, respectively. LP3 as the most potent derivative exhibits a terminal alkyne, and was subsequently utilized for target identification via ABPP. For the identification of optimal labeling conditions, intact V. harveyi cells were incubated with various concentrations of LP3. Subsequently, the cells were lysed, separated in soluble and insoluble fractions and clicked to rhodamine azide under copper catalysis to append a fluorescent dye for target visualization.20 SDS-PAGE analysis of the labeled proteomes and subsequent fluorescence scanning revealed a concentration dependent labeling of distinct proteins (Fig. S3, ESI†). A concentration of 10 μM LP3 showed the optimal signal intensity and was selected for competition experiments. Here the cells were treated with various excesses of BL3 (no alkyne tag) and subsequently incubated with the LP3 probe. Interestingly, several protein bands in soluble and insoluble fractions vanished suggesting specific binding of both molecules into the same active sites (Fig. S3, ESI†).
As fluorescence labeling does not directly reveal protein identities, we utilized a quantitative proteomic labeling strategy via direct mass-spectrometric target identification. In brief, intact cells were treated with the probe, lysed, clicked to biotin azide and labeled proteins were enriched on avidin beads (Fig. 2).21 To account for unspecific avidin background binders, an identical experiment was carried out with DMSO instead of the probe in parallel. Avidin bound proteins were released by tryptic digestion and the resulting peptides were appended to different isotopes via dimethyl labeling.22 The samples were pooled and analyzed via LC-MS/MS to determine isotope ratios. These were ranked in the corresponding volcano plots according to their enrichment and significance (proteins with an enrichment ratio of 8 and p-value < 0.00001 for insoluble samples and a ratio of 4 with p-value < 0.003 for soluble proteins were regarded as hits). Interestingly, insoluble and soluble proteome fractions showed distinct target proteins including a 3-oxoacyl-[acyl-carrier-protein] synthase (KASII), the outer membrane protein A (OmpA) and an acetyl-CoA-acetyltransferase (ACAT). Surprisingly, a putative diguanylate cyclase with a GGDEF-domain (DGC) was not detected in the soluble fraction but emerged as the best target from the insoluble samples (Fig. 3A and Fig. S4A, ESI†). To confirm that these proteins are specific β-lactone targets, we performed a competition experiment with a 20-fold excess of BL3 and subsequent probe labeling in parallel (Fig. 2, 3B and Fig. S4B, ESI†). Interestingly, with the exception of KASII, all other targets were efficiently outcompeted. A recombinant expression of the remaining three proteins in Escherichia coli confirmed their specific interaction with LP3 upon fluorescence labeling of SDS-gels (Fig. S5, ESI†).
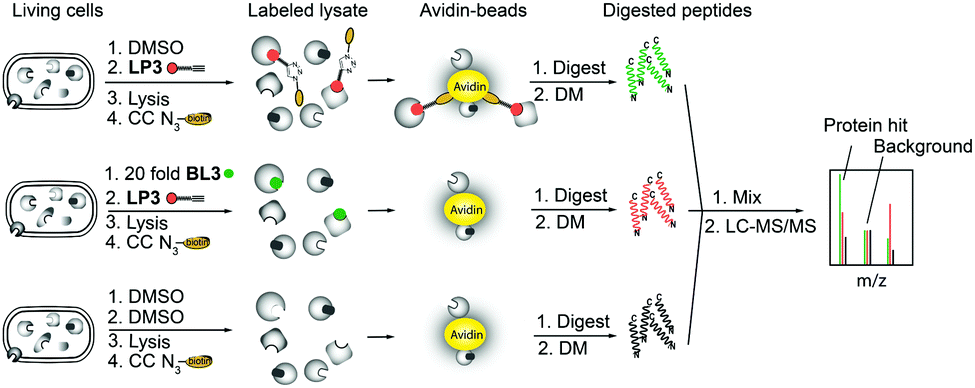 | ||
| Fig. 2 Principle of gel-free activity based protein profiling (ABPP) employing isotope labeling of amino acids. CC denotes click chemistry and DM denotes stable isotope dimethyl labeling. | ||
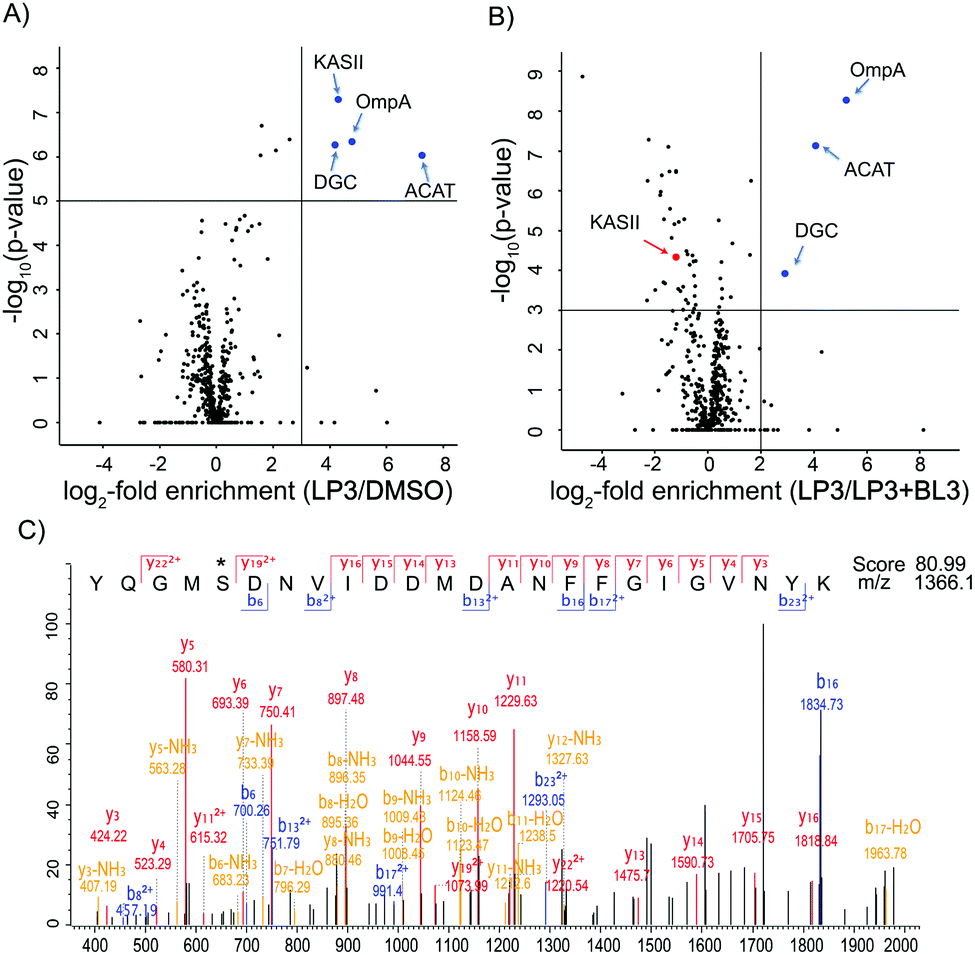 | ||
| Fig. 3 (A) Volcano plot of gel-free quantitative ABPP experiments with 10 μM LP3vs. DMSO in V. harveyi insoluble fraction. The blue dots depict enriched targets (criteria: log2-fold enrichment ≥ 3 and −log10(p-value) ≥ 5). (B) Volcano plot of V. harveyi insoluble fraction treated with 10 μM LP3vs. a 20-fold excess of BL3 (LP3 + BL3). The blue dots depict selected targets that are outcompeted by BL3 (criteria: log2-fold enrichment ≥ 2 and −log10(p-value) ≥ 3). The red dots denote those targets enriched by LP3 but not outcompeted by BL3. Both results were derived from three biological replicates with technical triplicates and −log10(p-values) were calculated using a two-sided one sample Student's t-test based on Z scores with Perseus. (C) MS/MS sequencing shows the binding of LP3 to Ser167 of OmpA. * denotes the modified amino acid. | ||
A closer inspection of the three proteins revealed that, with the exception of ACAT, the functional roles of DGC and OmpA are less explored.23 DGC is a predicted cyclic-di-GMP cyclase. Cyclic-di-GMP acts as a second messenger in bacteria regulating motility and biofilm production.24 In V. harveyi there are 31 proteins, which have a GGDEF-domain and are putative DGCs. This suggests that the production of cyclic-di-GMP is an important trait and that multiple proteins could substitute each other. Due to the predicted effect of cyclic-di-GMP on motility, we performed swimming plate assays in the presence and absence of LP3 and BL3 (Fig. S6, ESI†).25 Interestingly, in the presence of the compounds, the cells consistently swim a slightly larger distance compared to the DMSO treated samples. In addition, the lactone treated bacteria formed a diffuse outer ring on the plates which was much sharper in the case of DMSO treated samples, suggesting that cells are not fully chemotactic.26 Although the exact role of the identified DGC in V. harveyi is elusive and it is unclear if other homologs could substitute its inhibition, the effects observed in the swimming plate assays point towards an impairment of the cyclic-di-GMP mediated motility.
OmpA is a β-barrel protein of the outer membrane. In E. coli this protein was found to be important for membrane stability.27 β-Lactones preferentially react with serine or cysteine residues of elevated nucleophilicity, e.g., in enzyme active sites. As OmpA lacks enzymatic activity we were interested to unravel the lactone binding site. Recombinant OmpA was incubated with LP3, digested and analyzed by LC-MS/MS sequencing and serine167 was identified as the modification site (Fig. 3C). To further analyze if OmpA was relevant for the observed bioluminescence phenotype, we constructed a V. harveyi ompA deletion mutant. Labeling with LP3 confirmed a lack of OmpA in the mutant via fluorescence SDS-gel analysis (Fig. S7, ESI†). However, the mutant strain exhibited almost the same behavior as the wild type with respect to growth, motility and bioluminescence, suggesting that OmpA is not responsible for the lactone mediated phenotype (Fig. S8, ESI†).
QS in V. harveyi is controlled by a phosphorylation cascade which allows the determination of the interaction with LP3 at various stages.28 We thus first tested the effect of LP3 and BL3 on the LuxN (AHL receptor) mediated phosphotransfer to LuxU in vitro. Neither β-lactone revealed a change in phosphorylation, suggesting that there is no direct interaction with the receptor (Fig. S9, ESI†). In addition, bioluminescence produced from a luxO deletion strain (constitutively QS-ON) was still inhibited by LP3 and BL3, suggesting a downstream target (Fig. S10, ESI†). To clarify if QS is addressed at all by β-lactones we additionally analyzed the exoproteolytic activity, another QS-dependent phenotype, which should be reduced in the case of QS inhibition.6 However, both lactones did not show any effect, highlighting a QS independent mechanism (Fig. S11, ESI†).
To further dissect the β-lactone mode of action, we performed a global proteome analysis of β-lactone and DMSO treated cells. V. harveyi was incubated with 25 μM BL3 or DMSO for 2 h, lysed, and the proteomes were digested by trypsin treatment. Isotope labeling of each sample followed by consolidated LC-MS/MS analysis revealed several significantly up- and down-regulated proteins under β-lactone treatment (Fig. 4). Interestingly, among the most down-regulated proteins are the α and β subunits of the electron transfer flavoprotein (ETF) under β-lactone treatment. ETF is involved in the β-oxidation of fatty acids and thereby linked to acyl- and acetyl-CoA production.29,30 The down-regulation of ETF may thus stall the myristic acid biosynthesis required for luciferase activity establishing a putative link to the observed reduction in bioluminescence with the β-lactone.31 In addition, we utilized the MS-based method for a comparison of the ompA deletion and wild type to compare profiles with the β-lactone treated samples (Fig. S12, ESI†). Malonyl CoA-acyl carrier protein transacylase (FabD), an enzyme also involved in the fatty acid metabolism, was among the most up-regulated proteins in the mutant.32 Interestingly, the same protein also exhibited significantly elevated levels in the case of β-lactone treatment connecting OmpA, aliphatic lactone binding and fatty acid biosynthesis. Although this connection is intriguing, future studies are needed to further elucidate the function and putative role of OmpA in fatty acid biosynthesis.
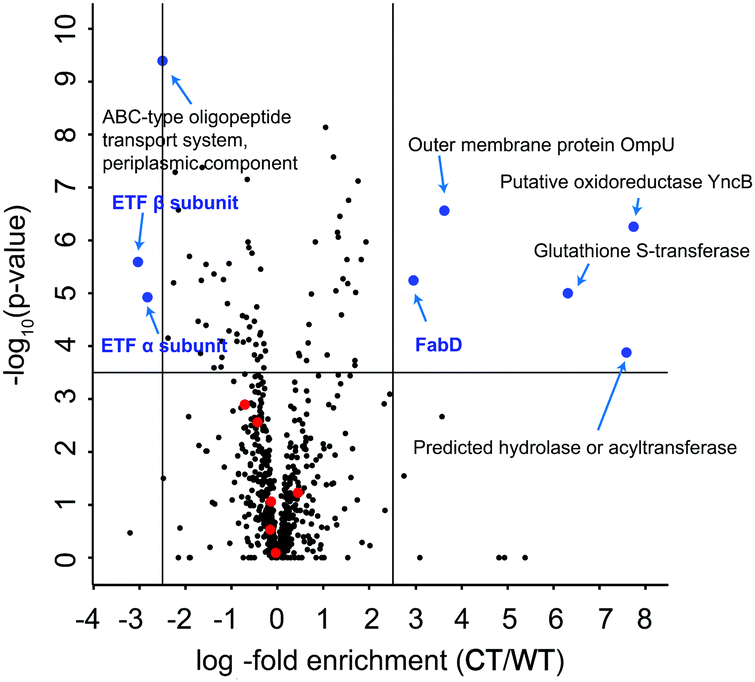 | ||
| Fig. 4 Volcano plot of whole proteome comparison between 25 μM BL3 treated V. harveyi ATCC BAA-1116 (CT) and non-treated samples (WT). The blue dots depict selected targets that are significantly up-regulated or down-regulated (criteria: log2-fold enrichment ≥ 2.5 or ≤ −2.5 and −log10(p-value) ≥ 3.5). Red dots represent proteins expressed from luxCDABEG operon. The targets discussed in the text are highlighted in blue. The data were derived from three biological replicates with technical triplicates and −log10(p-value) were calculated using a two-sided one sample Student's t-test based on Z scores with Perseus. | ||
Due to the structural similarity between AHLs produced by Gram-negative bacteria and β-lactones, we analyzed their potential effects on QS-regulated bioluminescence in V. harveyi. Although β-lactone LP3 was identified as a nanomolar bioluminescence inhibitor, a detailed mode of action analysis suggested no direct link to QS. Instead the compound likely interfered with the fatty acid metabolism, which is largely attributed to its long aliphatic chain. None of the identified LP3 targets were directly linked to bioluminescence; however, the down-regulation of ETF α and β subunits pointed towards luciferase activity, which needs to be further evaluated in future studies, e.g. by the use of photoprobes to analyze reversible binding target proteins. In conclusion, although AHLs and long chain aliphatic β-lactones share similar structural motifs the identity of the lactone core scaffold (4 vs. 5 membered ring) remains a crucial switch between QS regulation and QS-independent inhibition of bioluminescence.
S. A. S. acknowledges the funding by the Deutsche Forschungs-gemeinschaft: FOR1406 and the ERC starting grant (250924-antibacterials) (to S. A. S.), and Exc114/2 and Ju270/13-1 (to K. J.). W. Z. acknowledges the support from the China Scholarship Council.
Notes and references
- J. S. Dickschat, Nat. Prod. Rep., 2010, 27, 343 RSC.
- C. Solano, M. Echeverz and I. Lasa, Curr. Opin. Microbiol., 2014, 18, 96 CrossRef CAS PubMed.
- I. Olsen, Eur. J. Clin. Microbiol. Infect. Dis., 2015, 34, 877 CrossRef CAS PubMed.
- B. Wang and T. W. Muir, Cell Chem. Biol., 2016, 23, 214 CrossRef CAS PubMed.
- I. Castillo-Juarez, T. Maeda, E. A. Mandujano-Tinoco, M. Tomas, B. Perez-Eretza, S. J. Garcia-Contreras, T. K. Wood and R. Garcia-Contreras, World J. Clin. Cases, 2015, 3, 575 Search PubMed.
- C. Anetzberger, M. Reiger, A. Fekete, U. Schell, N. Stambrau, L. Plener, J. Kopka, P. Schmitt-Kopplin, H. Hilbi and K. Jung, PLoS One, 2012, 7, e48310 CAS.
- W. R. Galloway, J. T. Hodgkinson, S. D. Bowden, M. Welch and D. R. Spring, Chem. Rev., 2011, 111, 28 CrossRef CAS PubMed.
- B. Lin, Z. Wang, A. P. Malanoski, E. A. O'Grady, C. F. Wimpee, V. Vuddhakul, N. Alves Jr, F. L. Thompson, B. Gomez-Gil and G. J. Vora, Environ. Microbiol. Rep., 2010, 2, 81 CrossRef CAS PubMed.
- M. G. Surette, M. B. Miller and B. L. Bassler, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1639 CrossRef CAS.
- T. Defoirdt, C. M. Miyamoto, T. K. Wood, E. A. Meighen, P. Sorgeloos, W. Verstraete and P. Bossier, Environ. Microbiol., 2007, 9, 2486 CrossRef CAS PubMed.
- J. A. Smith, J. Wang, S. M. Nguyen-Mau, V. Lee and H. O. Sintim, Chem. Commun., 2009, 7033 RSC.
- T. Jiang and M. Li, Expert Opin. Ther. Pat., 2013, 23, 867 CrossRef CAS PubMed.
- M. Guo, S. Gamby, Y. Zheng and H. O. Sintim, Int. J. Mol. Sci., 2013, 14, 17694 CrossRef CAS PubMed.
- G. Brackman and T. Coenye, Curr. Pharm. Des., 2015, 21, 5 CrossRef CAS PubMed.
- W. R. Abraham, Antibiotics, 2016, 5, 3 CrossRef PubMed.
- S. T. Rutherford and B. L. Bassler, Cold Spring Harbor Perspect. Med., 2012, 2, a012427 Search PubMed.
- W. Zhao, N. Lorenz, K. Jung and S. A. Sieber, Angew. Chem., Int. Ed., 2016, 55, 1187 CrossRef CAS PubMed.
- T. Bottcher and S. A. Sieber, Angew. Chem., Int. Ed., 2008, 47, 4600 CrossRef PubMed.
- M. Gersch, F. Gut, V. S. Korotkov, J. Lehmann, T. Bottcher, M. Rusch, C. Hedberg, H. Waldmann, G. Klebe and S. A. Sieber, Angew. Chem., Int. Ed., 2013, 52, 3009 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- M. H. Wright and S. A. Sieber, Nat. Prod. Rep., 2016, 33, 681 RSC.
- P. J. Boersema, R. Raijmakers, S. Lemeer, S. Mohammed and A. J. Heck, Nat. Protoc., 2009, 4, 484 CrossRef CAS PubMed.
- S. Thompson, F. Mayerl, O. P. Peoples, S. Masamune, A. J. Sinskey and C. T. Walsh, Biochemistry, 1989, 28, 5735 CrossRef CAS PubMed.
- D. A. Ryjenkov, M. Tarutina, O. V. Moskvin and M. Gomelsky, J. Bacteriol., 2005, 187, 1792 CrossRef CAS PubMed.
- D. Srivastava, M. L. Hsieh, A. Khataokar, M. B. Neiditch and C. M. Waters, Mol. Microbiol., 2013, 90, 1262 CrossRef CAS PubMed.
- G. L. Hazelbauer, R. E. Mesibov and J. Adler, Proc. Natl. Acad. Sci. U. S. A., 1969, 64, 1300 CrossRef CAS.
- Y. Wang, Biochem. Biophys. Res. Commun., 2002, 292, 396 CrossRef CAS PubMed.
- K. Jung, Nat. Chem. Biol., 2011, 7, 502 CrossRef CAS PubMed.
- A. E. Kazakov, D. A. Rodionov, E. Alm, A. P. Arkin, I. Dubchak and M. S. Gelfand, J. Bacteriol., 2009, 191, 52 CrossRef CAS PubMed.
- Y. Fujita, H. Matsuoka and K. Hirooka, Mol. Microbiol., 2007, 66, 829 CrossRef CAS PubMed.
- E. A. Meighen, Microbiol. Rev., 1991, 55, 123 CAS.
- J. W. Campbell and J. E. Cronan, Annu. Rev. Microbiol., 2001, 55, 305 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc05807h |
| This journal is © The Royal Society of Chemistry 2016 |