
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Remote functionalization of hydrocarbons with reversibility enhanced stereocontrol†
Alexandre
Vasseur
a,
Lionel
Perrin
*b,
Odile
Eisenstein
c and
Ilan
Marek
*a
aThe Mallat Family Laboratory of Organic Chemistry, Schulich Faculty of Chemistry and Lise Meitner-Minerva Center for Computational Quantum Chemistry, Technion-Israel Institute of Technology, Technion City, Haifa 32000, Israel. E-mail: chilanm@tx.technion.ac.il; Fax: +972-4-829-37-09; Tel: +972-4-829-37-09
bICBMS UMR 5246, Université de Lyon, Bat Curien, 43 Bd du 11 Novembre 1918, 69622 Villeurbanne cedex, France. E-mail: lionel.perrin@univ-lyon1.fr
cInstitut Charles Gerhardt, CNRS UMR 5253, Université de Montpellier, cc 1501, Place E. Bataillon, 34095 Montpellier, France
First published on 3rd March 2015
Abstract
Remote functionalization of hydrocarbons could be achieved through successive zirconocene-mediated allylic C–H bond activations followed by a selective C–C bond cleavage. Determination of the reaction mechanism by density functional theory (DFT) calculations shows that the high stereocontrol observed in this process results from a large number of energetically accessible equilibria feeding a preferred reactive channel that leads to the major product. A distinctive consequence of this pattern is that stereoselectivity is enhanced upon heating.
Introduction
Manipulation of functionality at a specific position of a hydrocarbon that would generate a reaction at a different location represents a major challenge in synthetic organic chemistry. The difficulty of such remote functionalization is even more pronounced for acyclic systems where flexible alkyl chains are present between the initiating and the final reactive centers. Since the pioneering work on remote functionalization of Breslow's in the 70's,1 numerous studies have appeared for the relay or transmission of stereochemical information along alkyl chains.2In this context, a particularly impressive example in the field of asymmetric induction is the foldamer-mediated 1,61-asymmetric remote induction reported by Clayden.3 On the other hand, as transition-metal complexes have the ability to isomerize double bonds along a carbon-skeleton,4–6 one could design a system where the isomerization would generate a selective remote transformation. However, due to the natural propensity for statistical isomerization, the selective migration of a metal complex along a hydrocarbon chain can only be directed if associated with a strongly thermodynamically favoured termination step. In this context, we have reported the transformation of unsaturated fatty alcohol derivatives as a source of substituted allylmetal species (Path A, Scheme 1)7 and the stereoselective preparation of conjugated dienyl metal complexes from non-conjugated enol ethers (Path B, Scheme 1)8 where the irreversible termination step is an elimination reaction. Initiating reversible allylic C–H bond activations triggered these reactions and up to 6 carbon atoms separated the initial double bond from the terminating center. In contrast, a very appealing unidirectional palladium-chain walking for the enantioselective redox-relay Heck-type arylation of alkenyl alcohols was reported where an oxidative deprotonation of the final α-alkoxy palladium–alkyl intermediate closes the catalytic cycle (Path C, Scheme 1).9
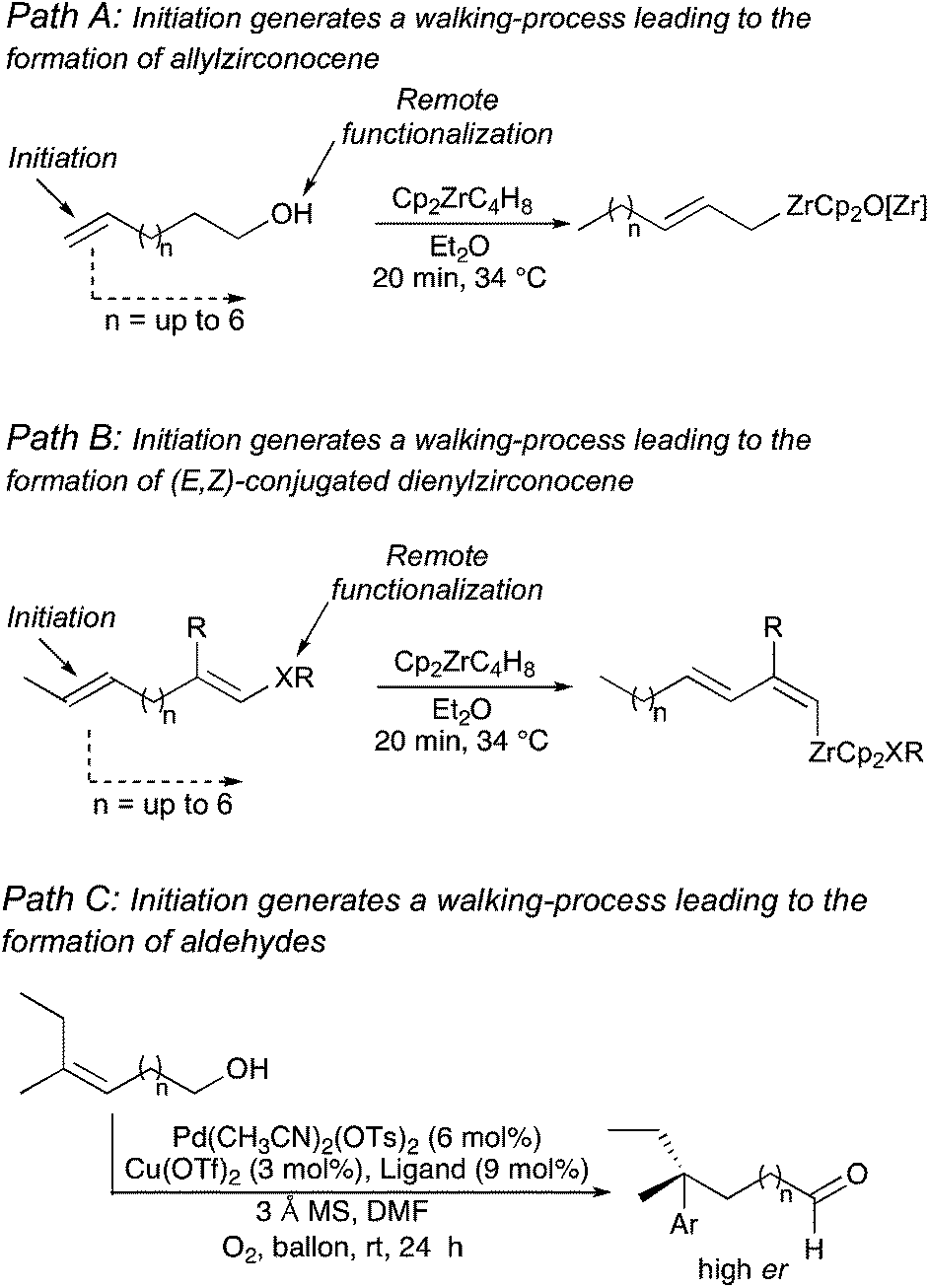 | ||
| Scheme 1 General approach for remote functionalization. | ||
However, reaction of a metal complex with a functionality that would generate a final unidirectional “walking-process” over an alkyl chain of an hydrocarbon (only composed of H and C atoms)4 producing a chemical reaction at a defined terminus position is a very promising, but still in its infancy, approach to functionalize molecules.5,6 To answer this challenging remote functionalization of hydrocarbons, we were interested to investigate the case of ω-ene cyclopropanes. If the trigger promotes the walking-process of a double bond to finally lead to a selective C–C bond carbon cleavage (Scheme 2), two cutting-edge methods of activation (C–H and C–C bond cleavage) would be unified into a single method through the use of a unique organometallic species.10
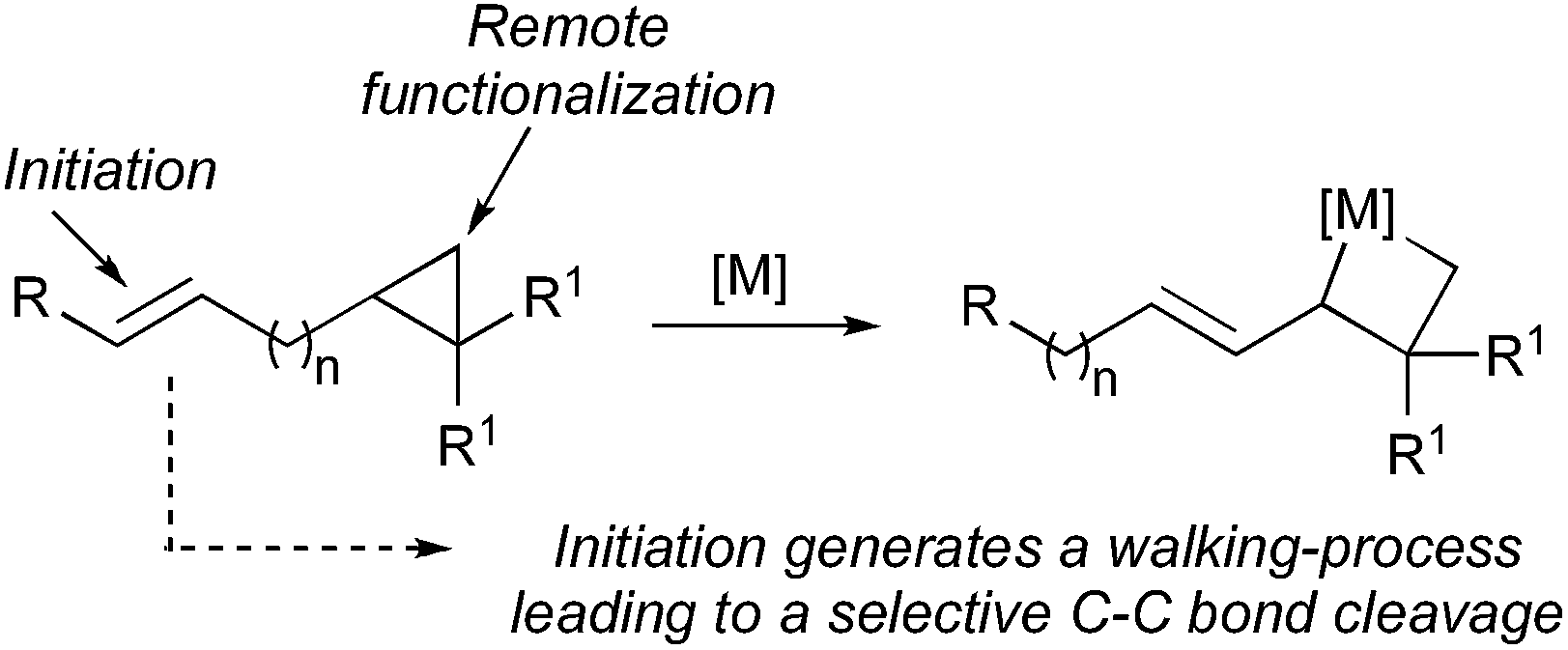 | ||
| Scheme 2 Proposed approach for remote functionalization of hydrocarbons. | ||
Results and discussion
We have shown that ω-ene cyclopropane 1a (Scheme 3) as well as alkylidenecyclopropanes (not described) can easily undergo a zirconocene-mediated allylic C–H bond activation followed by highly selective C–C bond cleavage.10 It should be noted that under the same experimental conditions, saturated cyclopropane (not possessing the remote double bond) does not lead to the carbon–carbon bond cleavage of the three-membered ring, confirming our hypothesis that the initiating step on the alkenyl moiety is required for the reaction to proceed. The allylic C–H bond activation most probably proceeds through the formation of an η3-allyl intermediate7,8 and the unique selectivity of the ring-opening results from the kinetically and thermodynamically preferred formation of primary organometallic species 2 over tertiary organometallic species in the ring cleavage (C1–C2 cleavage preferred over C1–C3, Scheme 3).11 Moreover, the reactivity of the bismetalated species (also called metallacycle) 2 can be fully controlled as the reactivity of the allylzirconocene moiety (C1–Zr) is higher than the reactivity of the alkylzirconocene (C2–Zr) moieties towards electrophiles.12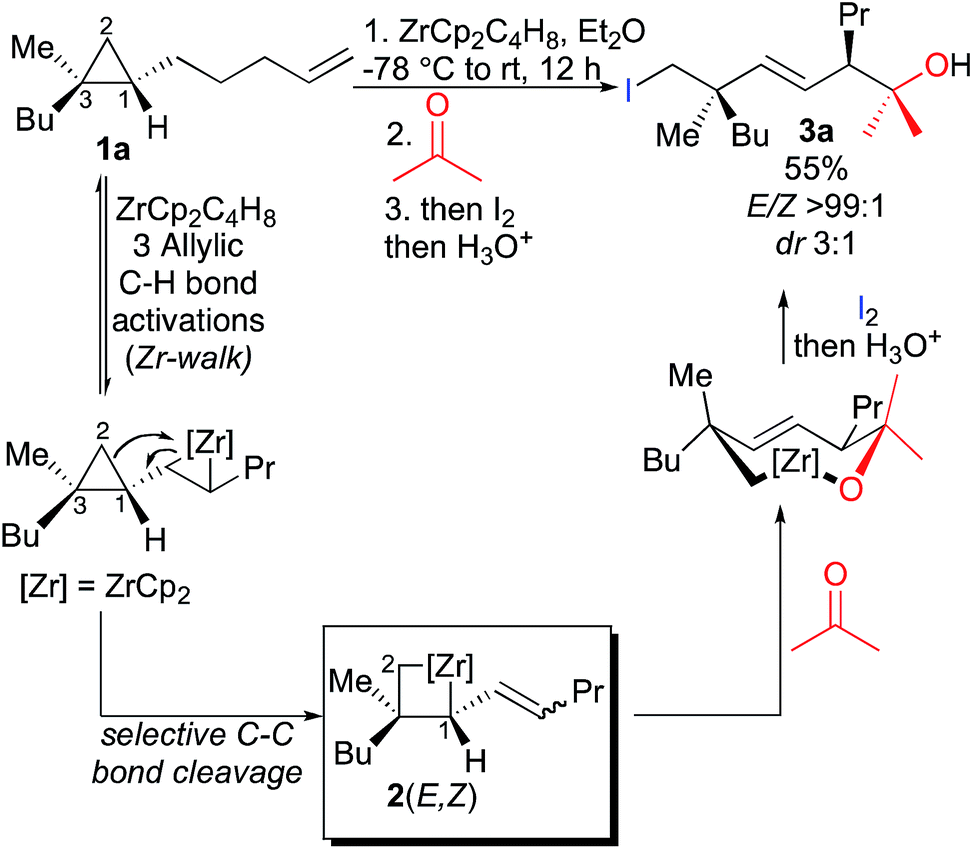 | ||
| Scheme 3 1,4-Diastereoselectivity in the zirconocene-promoted allylic C–H bond activation and C–C bond cleavage. | ||
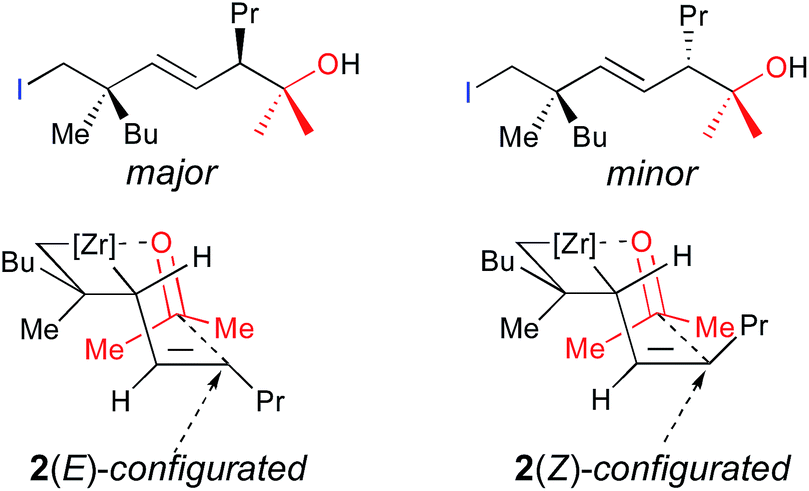
Therefore, addition of a carbonyl group to the first electrophile such as acetone leads to the unique formation of functionalized adducts at the allylic position, that could be also eventually represented as the cyclic mono-metallated cyclooctenolate derivative, and the second electrophile (i.e. I2) gives the final E-adduct 3a in 55% yield (Scheme 3).10 Although this strategy holds potential for 1,4-induction of diastereoselectivity with formation of the highly valued quaternary carbon and tertiary stereocenters in 1,4-relationship in an acyclic system,13,14 we could never reach a decent diastereoselectivity from ω-ene cyclopropanes (dr 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Scheme 3). This low 1,4-diastereoselectivity in the formation of the linear product was rationalized by the presence of a mixture of two geometrical E- and Z-isomers of the substituted allylzirconocene 2,10 both reacting with acetone through a chair-like transition state15 as described in Fig. 1 leading to the E-isomer 3a as a mixture of two sp3 centered diastereoisomers. In this communication, we report how we could improve the diastereoselectivity of the reaction and rationalize the results.
:1, Scheme 3). This low 1,4-diastereoselectivity in the formation of the linear product was rationalized by the presence of a mixture of two geometrical E- and Z-isomers of the substituted allylzirconocene 2,10 both reacting with acetone through a chair-like transition state15 as described in Fig. 1 leading to the E-isomer 3a as a mixture of two sp3 centered diastereoisomers. In this communication, we report how we could improve the diastereoselectivity of the reaction and rationalize the results.
 | ||
| Fig. 1 Proposed Zimmermann–Traxler transition state. | ||
Following our previous work on the zirconocene-mediated ring-opening of alkylidenecyclopropanes,10 we hypothesized that the diastereoselectivity could be improved if one could isomerize the (Z)-configured substituted allylzirconocene 2 into the thermodynamically more stable 2(E)-isomer. When the diastereomerically pure ω-ene cyclopropanes 1b,e were treated with the Negishi reagent16 at room temperature overnight in Et2O, the intermediate metallacycles 2b,e were initially obtained as two geometrical (E,Z)-isomers. We were delighted to observe that the isomerization of the substituted allylzirconocene into the single E-allylzirconocene species 2 could be promoted by addition of THF as a co-solvent and heating to 55 °C for 3 h. Then, the addition of the carbonyl groups followed by the second electrophile gave diastereoisomerically enriched 3b–m as described in Table 1. It should be noted that neither the addition of THF alone without heating nor heating (in Et2O) without addition of the cosolvent THF was sufficient to fully isomerize the (Z)-allylzirconocenes 2 into the (E)-isomer. We hypothesized that the addition of this co-solvent is needed to reach a temperature high enough to promote the isomerization of the Z- into E-allylzirconocene. In all cases, the combined C–H allylic bond activation followed by the selective C–C bond cleavage leads, after isomerization of the substituted allylzirconocene and reaction with two different electrophiles, to acyclic E-alkenes possessing two stereogenic centers in a 1,4-relationship, including the quaternary carbon stereocenter with very high diastereoselectivity. It should be stressed that the selectivity of the ring-cleavage is complete as no trace of activation along the C1–C3 bond was detected in the crude reaction mixture. This tandem reaction is not limited to a one-carbon tether (Table 1, entries 1–5) as it could be extended similarly to longer alkyl chains (Table 1, entry 6, n = 2, entries 7–11, n = 3 and entry 12, n = 4) with similar selectivities. When the migrating double bond is 1,2-disubstituted such as in 1c (Table 1, entry 6), our tandem sequence of allylic C–H isomerization/carbon–carbon cleavage still proceeds very efficiently as 3g is obtained in 62% yield with a 1,4-diastereoisomeric ratio of 98:2. It should be noted that although 1c was present as two geometrical (E)/(Z)-isomers in a 1:1 ratio, only the (E)-isomer of 3g is obtained.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | n | E1 | E2 | dra | Yieldb (%) |
| a Determined by 1H NMR analysis of crude reaction mixture. b Determined after purification by column chromatography on silica gel. | ||||||||
| 1 | Pr | Bu | H | 1(1b) | MeCOMe | H3O+ | 98:2 |
83 (3b) |
| 2 | Pr | Bu | H | 1(1b) | [CH2]4CO | H3O+ | 98:2 |
62 (3c) |
| 3 | Pr | Bu | H | 1(1b) | [CH2]5CO | H3O+ | 98:2 |
52 (3d) |
| 4 | Pr | Bu | H | 1(1b) | EtCOEt | I2 | 98:2 |
56 (3e) |
| 5 | Pr | Bu | H | 1(1b) | MeCOMe | I2 | 98:2 |
51 (3f) |
| 6 | Et | Bu | Me | 2(1c) | MeCOMe | H3O+ | 98:2 |
62 (3g) |
| 7 | Et | Bu | H | 3(1d) | MeCOMe | H3O+ | 98:2 |
61 (3h) |
| 8 | Et | Bu | H | 3(1d) | [CH2]4CO | H3O+ | 97:3 |
61 (3i) |
| 9 | Et | Bu | H | 3(1d) | [CH2]5CO | H3O+ | 98:2 |
62 (3j) |
| 10 | Et | Bu | H | 3(1d) | MeCOMe | I2 | 96:4 |
55 (3k) |
| 11 | Et | Bu | H | 3(1d) | EtCOEt | H3O+ | 98:2 |
59 (3l) |
| 12 | Et | Bu | H | 4(1e) | MeCOMe | H3O+ | 98:2 |
50 (3m) |
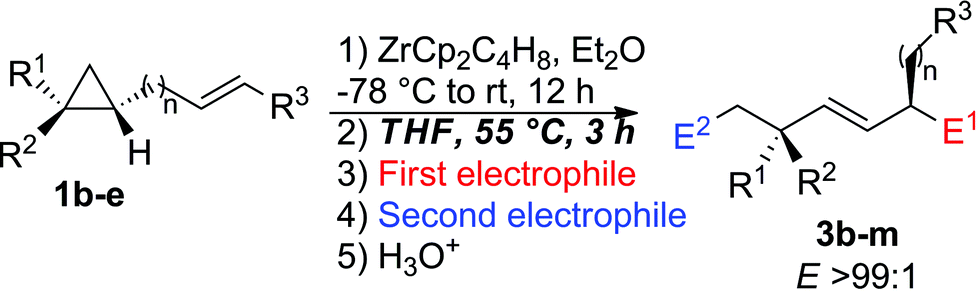
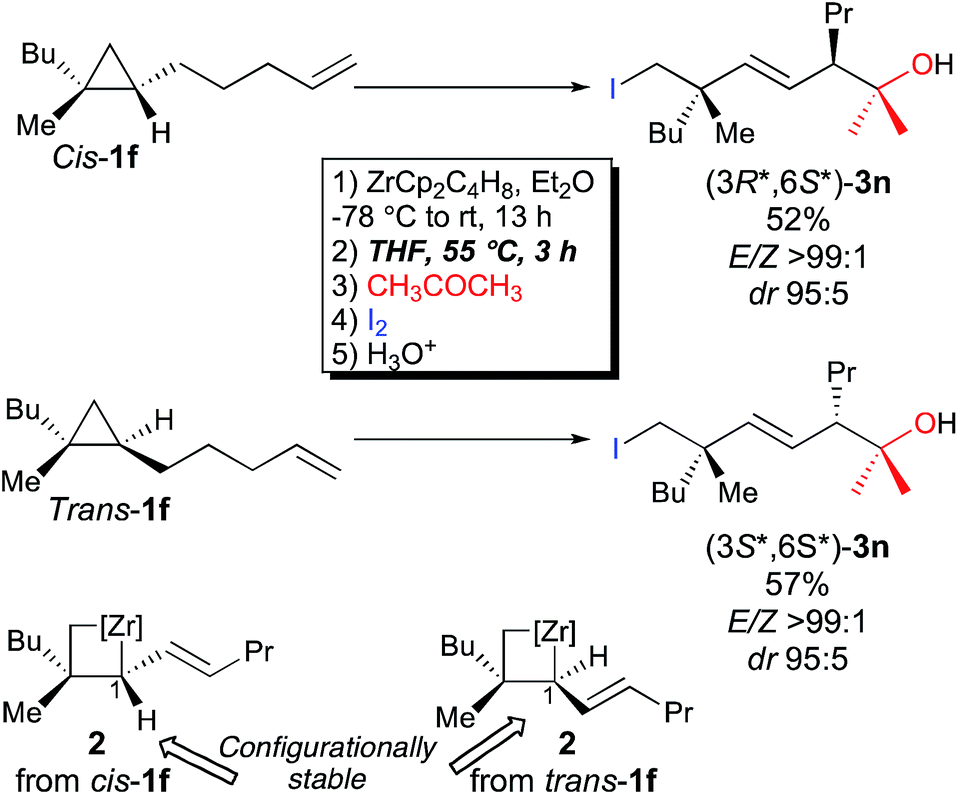
Importantly, when the two diastereoisomers of ω-ene cyclopropanes 1f are subjected to our experimental conditions, the two opposite diastereoisomers of 3n are obtained with comparable diastereomeric ratios and yields (Scheme 4). In addition to the synthetic importance of this transformation, this result clearly shows that the C1–[Zr] bond of zirconacyclobutane intermediates 2 is configurationally stable. In other words, the two independently formed intermediate alkyl–allyl zirconacyclobutanes cis-2c and trans-2c respectively do not interconvert at the metalated center C1 (Path A, Fig. 2) despite the isomerization of the (Z)-2c into (E)-2c allylzirconocene fragment at C5 by heating at 55 °C for 3 h (Path B, Fig. 2).
 | ||
| Scheme 4 Tandem reactions on the two opposite diastereoisomers of 1f. | ||
 | ||
| Fig. 2 Configurational stability at C1 and isomerization at C5. | ||
To gain a better understanding of the mechanism of this surprising Z- to E-isomerization of the allylzirconocene fragment on C5 without eroding the configurational stability at C1 of the zirconacyclobutane 2c, this reaction was studied with density functional theory (DFT) calculations using the M06 functional that includes dispersion contributions.17 An implicit solvent model was used for modeling bulk solvation by Et2O according to the SMD method. Gibbs free energies calculated at T = 298.15 K and P = 1 atm are used to discuss the reaction pathways. More details are given in the ESI.†
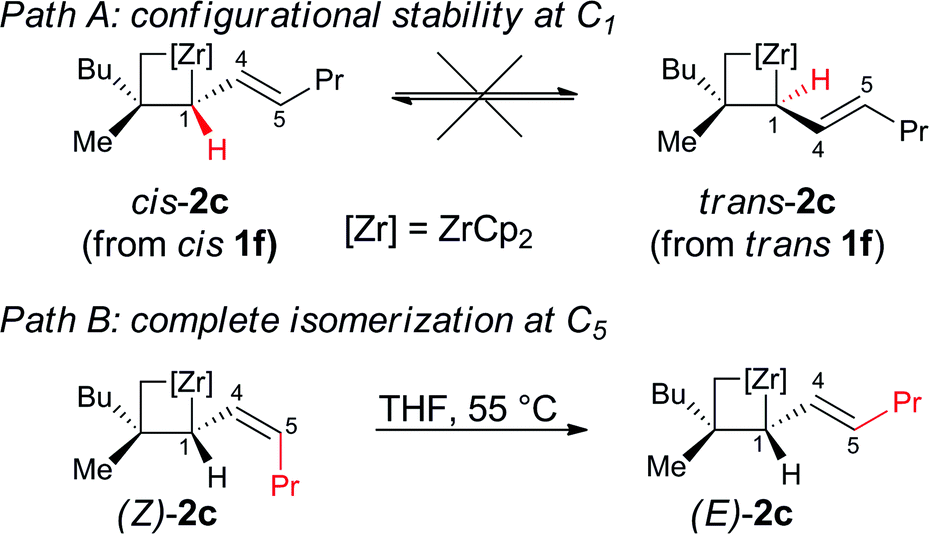
Experimentally, the chain walking mechanism has been established by deuterium labeling. It was shown that successive migration of the double bond is promoted by allylic C–H bond activations to yield Zr–hydride–allyl intermediate complexes as depicted in Fig. 3I.7a,8a,10a Calculations show that this mechanism is energetically preferred for the chain walking along the alkenyl chain of a cyclopropane substrate. When moving the double bond of one carbon unit, the rate-determining step involves the rotation along the Zr–C bond of the σ-allyl ligand. The highest transition state is 6 kcal mol−1 above the initial reactants taken as energy reference hereafter. Calculations also reveal the existence of a complex set of equilibria built on rotations within σ-allyl ligands at the transient zirconocene–hydride–allyl complexes (Fig. 3I–III) that was characterized for a related Zr–allyl complex.18 These equilibria are kinetically and thermodynamically comparable leading to kinetically and thermodynamically unselective chain walking (zirconocene can switch between the alkene faces (Fig. 3II) and isomerize the Z- into the E-olefin, Fig. 3III). A fully detailed computational mechanistic of these isomerizations and others will be given in a forthcoming report.
 | ||
| Fig. 3 Reaction pathways showing the key extrema for zirconocene (I) chain walking; (II) face switching; (III) E to Z-olefin isomerization; (IV) Newman projection for complexes B and C to illustrate the anti- and syn-periplanar configurations between the 3-membered rings; (V) reaction pathways involved in the formation of cis-2c and trans-2c. Extrema are labeled in bold black. Gibbs free energies are in kcal mol−1 relative to separated [Zr]Bu2 and substrate taken as energy reference with values for minima and transition states in pink and teal, respectively. In multistep sequences, max refers to the Gibbs free energy of the highest transition state. The sign * refers to values obtained for pent-2-ene as substrate. | ||
The ability to transform the olefin complex into a hydride allylic complex where many isomerizations can be achieved at a low energy cost is thus an important feature of this system. Indeed, the Zr-promoted walking process potentially leads to a mixture of four cyclopropyl diastereoisomeric zircona–cyclopropane complexes (A to D, Fig. 3V).17 These four intermediates are in equilibrium via the isomerization aforementioned and share I as the most stable common intermediate, which is 7 kcal mol−1 below Cp2ZrBu2 and free substrate and more stable than A to D. Therefore, species I is the most abundant intermediate before the C–C bond cleavage. Importantly, the relative ratios of these four complexes have little to no consequence on the stereochemical outcome of the reaction as described below.
The four complexes A to D differ strongly in their ability to undergo the ring opening of the cyclopropyl ring since they are determined by the position of the Zr relative to the ring. An anti-periplanar relationship between the zirconacyclopropane and the cyclopropyl ring in C and D prevents the C1–C2 ring opening, (activation barrier of ca. 40 kcal mol−1, Fig. 3IV) whereas a syn-periplanar relationship between the reactive function in A and B leads to activation barriers for the C–C bond cleavage of around 5 kcal mol−1. Furthermore, the ring opening of the cyclopropyl ring always occurs at the least substituted carbon–carbon bond as shown in Scheme 3 and Fig. 3V. No transition state for C1–C3 bond cleavage could be located presumably due to the steric hindrance.
Cyclopropane ring opening of A and B respectively yields Imin and IMaj that are the calculated structures for (Z)-2c and (E)-2c, respectively (Fig. 2 and 3). Whereas, isomers A and B are isoenergetic, IMaj is energetically preferred over Imin by 4 kcal mol−1, an energy difference that is also present in the corresponding transition states. The transition state for the cyclopropane ring opening occurs by approaching the methylene C2 to the Zr while maintaining the Zr–β-ene interaction. In both TSB–IMaj and TSA–Imin, the Zr–C2 bond distance is 2.66 Å and the C2–C1 is 1.72 Å. Thus, the difference in energy between the two transition states TSB–IMaj and TSA–Imin appears to be due to nonbonded interactions between the ethyl chain and the Cp2Zr fragment. As illustrated in Fig. 4, these interactions are present in IMaj and Imin and account for similarity in the kinetic and thermodynamic preferences. Thus, the kinetic preference for going via the transition state that forms the thermodynamically preferred IMaj, accounts for the experimental 3:1 diastereoisomeric ratio of 3 (Scheme 3) when the reaction is performed at room temperature. Importantly, intermediates C and D, which cannot perform the cyclopropane ring opening, isomerize to either A or Bvia the Zr–allyl hydride intermediate I and thus also contribute to product formation.
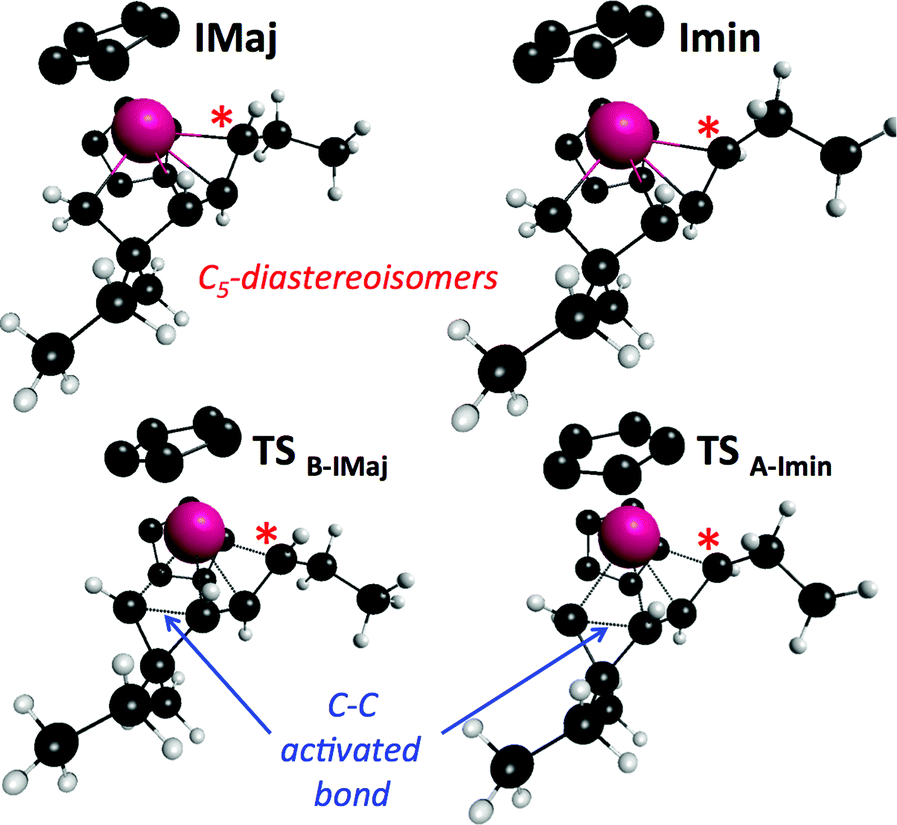 | ||
| Fig. 4 Structures of Imin, IMaj and their associated transition states of formation. | ||
Calculations indicate that direct isomerization between (Z)- and (E)-2c, (i.e. between Imin to IMaj) is not allowed. This originates from the restricted rotation around C4–C5 caused by either the presence of a π-bond or the constraint of the 6-membered ring in the two σ-allyl complexes. Alternatively, a mechanism that connects Imin to I and thus to IMajvia allylic C–H activation and formation of a zirconium–hydride–diene complex has been computed; the highest transition state of this pathway is located at 22 kcal mol−1 above energy reference.19 Consequently, the most energetically preferred pathway from Imin to IMaj is viaA, I and B with the highest transition state being 10 kcal mol−1 above the energy reference. This isomerization between A, B, C and DviaI controls the stereoselectivity of the reaction and highlights the importance of the reversibility of the allylic C–H bond activation and C–C bond cleavage by heating the reaction mixture at 55 °C for 3 h.
These computations show that the high diastereoselectivity of this reaction is not due to the existence of a single preferred path but to a large number of energetically accessible equilibria that enables the isomerization of all intermediates into a single one, responsible for the formation of the major product. This mechanism of dynamic thermodynamic resolution has already been reported in the literature mainly for the equilibration of sp3 alkyllithium species,20 but also for the formation of substituted allyllithium species with sparteine.21 Having now a good understanding of the reaction mechanism, we turned our attention to the last synthetic challenge, namely the functionalization of the second Csp3–[Zr] bond (second electrophile) through the creation of a new C–C bond.
To perform such C–C bond forming events, transmetalation reactions usually provide a unique and powerful means of expanding the synthetic scope of organozirconium chemistry.22 Once the tandem allylic C–H and selective C–C activations is performed on 1b, THF is added and the solution is heated at 55 °C for 3 h. Then acetone was first added and the resulting alkyl zirconium species was transmetalated into alkyl copper species by addition of catalytic amount of copper salt.23 The in situ generated alkyl copper can then react with classical electrophiles of copper chemistry such as allyl bromide and aromatic as well as heteroaromatic acyl chloride to give functionalized adducts (Scheme 5, formation of 3o–q, respectively). The corresponding alkyl copper species could also be transmetalated into palladium by addition of 10 mol% of Pd(PPh3)4 and coupled with aromatic iodide (formation of 3r, Scheme 5).
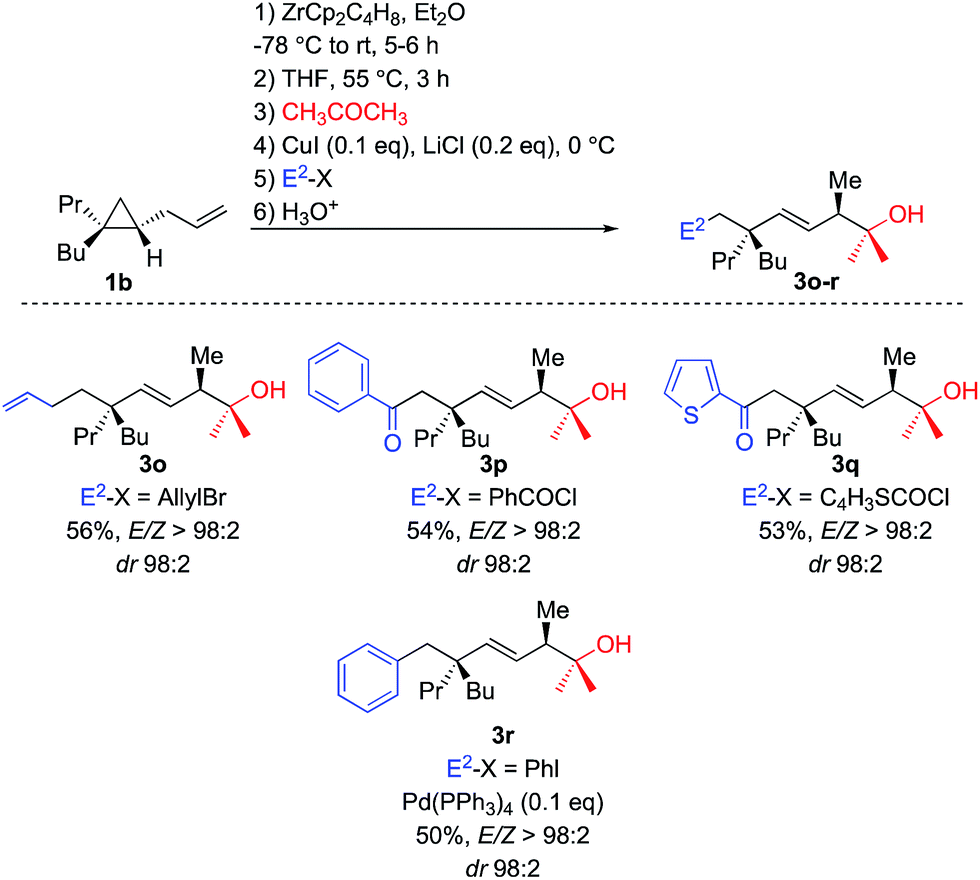 | ||
| Scheme 5 Transmetalation of Csp3–[Zr] bond for further C–C bond forming processes. | ||
Conclusions
In conclusion, a diastereoselective remote functionalization of ω-ene cyclopropane species is reported that could lead, in addition, to the formation of functionalized adducts with high 1,4-diastereocontrol. As highly enantiomerically enriched cyclopropane derivatives are easily accessible,24 this reaction could represent an interesting entry to the formation of enantiomerically enriched quaternary and tertiary carbon stereocenters in acyclic systems. Computational studies reveal an original way to achieve high stereocontrol by having a plethora of equilibria feeding a preferred reactive channel leading to the major isomer through thermodynamic control. This accounts for the counter-intuitive result that stereocontrol is enhanced by thermal treatment.Acknowledgements
This research is supported by the European Research Council under the European Community's Seventh Framework Program (ERC grant agreement no. 338912). LP thanks CCIR of ICBMS and CALMIP for providing computational resources and technical support. OE thanks the Technion for an invited Schulich Professorship position during which this collaboration was initiated.Notes and references
- (a) R. Breslow, Chem. Soc. Rev., 2003, 1, 553 RSC; (b) R. Breslow, Acc. Chem. Res., 1980, 13, 170 CrossRef CAS.
- For reviews, see: (a) I. Franzoni and C. Mazet, Org. Biomol. Chem., 2014, 12, 233 RSC; (b) H. Jiang, L. Albrecht and K. A. Jørgensen, Chem. Sci., 2013, 4, 2287 RSC; (c) H. Schwarz, Acc. Chem. Res., 1989, 22, 282 CrossRef CAS.
- L. Byrne, J. Solà, T. Boddaert, T. Marcelli, R. W. Adams, G. A. Morris and J. Clayden, Angew. Chem., Int. Ed., 2014, 53, 151 CrossRef CAS PubMed.
- G. P. Moss, P. A. S. Smith and D. Tavernier, Pure Appl. Chem., 1995, 67, 1307 Search PubMed.
- For metal-walking over electron-rich π-system, see: (a) D. Strawser, A. Karton, A. V. Zenkina, A. A. Iron, L. J. W. Shimon, J. M. L. Martin and M. E. van der Boom, J. Am. Chem. Soc., 2005, 127, 9322 CrossRef CAS PubMed; (b) O. Zenkina, M. Altman, G. Leitus, L. J. W. Shimon, R. Cohen and M. E. van der Boom, Organometallics, 2007, 26, 4528 CrossRef CAS. For metal-walking on hydrocarbons, see: (c) C. R. Larsen, G. Erdogan and D. B. Grotjahn, J. Am. Chem. Soc., 2014, 136, 1226 CrossRef CAS PubMed; (d) G. Hilt, ChemCatChem, 2014, 6, 2484 CrossRef CAS; (e) J. Tao, F. Sun and T. Fang, J. Organomet. Chem., 2012, 698, 1 CrossRef CAS; (f) F. Ding, Y. Sun and F. Verpoort, Eur. J. Inorg. Chem., 2010, 1536 CrossRef CAS; (g) G. Erdogan and D. B. Grotjahn, Top. Catal., 2010, 53, 1055 CrossRef CAS; (h) G. Erdogan and D. B. Grotjahn, J. Am. Chem. Soc., 2009, 131, 10354 CrossRef CAS PubMed; (i) T. O. Northcutt, D. D. Wick, A. J. Vetter and W. D. Jones, J. Am. Chem. Soc., 2001, 123, 7257 CrossRef CAS PubMed; (j) R. A. Periana and R. G. Bergman, J. Am. Chem. Soc., 1986, 108, 7332 CrossRef CAS.
- (a) W.-C. Lee, C.-H. Wang, Y.-H. Lin, W.-C. Shih and T.-G. Ong, Org. Lett., 2013, 15, 5358 CrossRef CAS PubMed; (b) L. Wu, I. Fleicher, R. Jackstell, I. Profir, R. Franke and M. Beller, J. Am. Chem. Soc., 2013, 135, 14306 CrossRef CAS PubMed; (c) M. Vilches-Herrera, L. Domke and A. Börner, ACS Catal., 2014, 4, 1706 CrossRef CAS; (d) J. S. Bair, Y. Schramm, A. G. Sergeev, E. Clot, O. Eisenstein and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 13098 CrossRef CAS PubMed.
- (a) N. Chinkov, A. Levin and I. Marek, Angew. Chem., Int. Ed., 2006, 45, 465 CrossRef CAS PubMed; (b) I. Marek, N. Chinkov and A. Levin, Synlett, 2006, 4, 501 CrossRef.
- (a) N. Chinkov, S. Majumdar and I. Marek, J. Am. Chem. Soc., 2003, 125, 13258 CrossRef CAS PubMed; (b) N. Chinkov, S. Majumdar and I. Marek, J. Am. Chem. Soc., 2002, 124, 10282 CrossRef CAS PubMed.
- (a) T.-S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340 CrossRef CAS PubMed; (b) T.-S. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830 CrossRef CAS PubMed; (c) E. W. Werner, T.-S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455 CrossRef CAS PubMed; (d) M. J. Hilton, L.-P. Xu, P.-O. Norrby, Y.-D. Wu, O. Wiest and M. S. Sigman, J. Org. Chem., 2014, 79, 11841 CrossRef CAS PubMed.
- (a) A. Masarwa, D. Didier, T. Zabrodski, M. Schinkel, L. Ackermann and I. Marek, Nature, 2014, 505, 199 CrossRef CAS PubMed; (b) S. Harada, H. Kiyono, R. Nishio, T. Taguchi and Y. Hanzawa, J. Org. Chem., 1997, 62, 3994 CrossRef CAS; (c) P. W. Dimmock and R. J. Whitby, J. Chem. Soc., Chem. Commun., 1994, 2323 RSC; (d) J.-L. Vasse and J. Szymoniak, Tetrahedron Lett., 2004, 45, 6449 CrossRef CAS.
- A. Masarwa and I. Marek, Chem.–Eur. J., 2010, 16, 9712 CrossRef CAS PubMed.
- L. T. Kiman, S. N. Mlynarski, G. E. Ferris and J. P. Morken, Angew. Chem., Int. Ed., 2012, 51, 521 CrossRef PubMed.
- For reviews; see: (a) Y. Minko and I. Marek, Chem. Commun., 2014, 50, 12597 RSC; (b) A. Y. Hong and B. M. Stoltz, Eur. J. Org. Chem., 2013, 2745 CrossRef CAS PubMed; (c) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593 RSC; (d) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295 RSC; (e) M. Bella and T. Gasperi, Synthesis, 2009, 1583 CrossRef CAS; (f) P. G. Cozzi, R. Hilgraf and N. Zimmermann, Eur. J. Org. Chem., 2007, 5969 CrossRef CAS; (g) I. Marek and G. Sklute, Chem. Commun., 2007, 1683 RSC; (h) B. M. Trost and C. Jiang, Synthesis, 2006, 369 CrossRef CAS; (i) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363 CrossRef CAS PubMed; (j) I. Denissova and L. Barriault, Tetrahedron, 2003, 59, 10105 CrossRef CAS; (k) Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis, ed. J. Christoffers and A. Baro, Wiley-VCH, Weinheim, 2005 Search PubMed; (l) E. J. Corey and A. Guzman-Perez, Angew. Chem., Int. Ed., 1998, 37, 388 CrossRef.
- (a) I. Marek, Y. Minko, M. Pasco, T. Mejuch, N. Gilboa, H. Chechik and J. D. Das, J. Am. Chem. Soc., 2014, 136, 2682 CrossRef CAS PubMed; (b) D. Didier, P.-O. Delaye, M. Simaan, B. Island, G. Eppe, H. Eijsberg, A. Kleiner, P. Knochel and I. Marek, Chem.–Eur. J., 2014, 20, 1038 CrossRef CAS PubMed; (c) M. Pasco, N. Gilboa, T. Mejuch and I. Marek, Organometallics, 2013, 92, 942 CrossRef; (d) P.-O. Delaye, D. Didier and I. Marek, Angew. Chem., Int. Ed., 2013, 52, 5333 CrossRef CAS PubMed; (e) Y. Minko, M. Pasco, L. Lercher, M. Botoshansky and I. Marek, Nature, 2012, 490, 522 CrossRef CAS PubMed; (f) T. Mejuch, B. Dutta, M. Botoshansky and I. Marek, Org. Biomol. Chem., 2012, 10, 5803 RSC; (g) N. Gilboa, H. Wang, K. N. Houk and I. Marek, Chem.–Eur. J., 2011, 17, 8000 CrossRef CAS PubMed; (h) B. Dutta, N. Gilboa and I. Marek, J. Am. Chem. Soc., 2010, 132, 5588 CrossRef CAS PubMed; (i) S. Simaan and I. Marek, J. Am. Chem. Soc., 2010, 132, 4066 CrossRef CAS PubMed; (j) S. Simaan, A. F. G. Goldberg, S. Rosset and I. Marek, Chem.–Eur. J., 2010, 16, 774 CrossRef CAS PubMed; (k) J. P. Das, H. Chechik and I. Marek, Nat. Chem., 2009, 1, 128 CrossRef CAS PubMed; (l) G. Kolodney, G. Sklute, S. Perrone, P. Knochel and I. Marek, Angew. Chem., Int. Ed., 2007, 46, 9291 CrossRef CAS PubMed; (m) G. Sklute and I. Marek, J. Am. Chem. Soc., 2006, 128, 4642 CrossRef CAS PubMed.
- (a) T. Luker and R. J. Whitby, Tetrahedron Lett., 1996, 37, 7661 CrossRef CAS; (b) G. J. Gordon and R. J. Whitby, Synlett, 1995, 77 CrossRef CAS; (c) T. Luker and R. J. Whitby, Tetrahedron Lett., 1994, 35, 9465 CrossRef CAS.
- E. Negishi and T. Takahashi, Synthesis, 1988, 1 CrossRef CAS.
- As a model, the Bu and ω-ene chains in 1c were respectively simplified by Et and δ-ene groups. Calculations were performed using the M06 hybrid functional. Zr was described by a Stuttgart–Dresden–Köln pseudopotential with its valence extended basis set, other atoms were represented by 6-311G(d,p) basis sets. Optimizations without any constraint and frequency calculations were carried out with implicit SMD solvent (Et2O). See ESI† for full description of the computational procedure.
- M. Vatamanu, Organometallics, 2014, 33, 3683 CrossRef CAS.
- This pathway involves an energetically demanding transition state to form a cis-butadiene complex via allyl C–H at C4′, followed by the ring closing at the C1–C2 bond. Participation of a butadiene complex in a related chemistry was suggested. See Scheme 6 in H. Yasuda and A. Nakamura, Angew. Chem., Int. Ed., 1987, 26, 723 CrossRef.
- (a) P. Beak, D. R. Anderson, M. D. Curtis, J. M. Laumer, D. J. Pippel and G. A. Weisenburger, Acc. Chem. Res., 2000, 33, 715 CrossRef CAS PubMed; (b) W. K. Lee, Y. S. Park and P. Beak, Acc. Chem. Res., 2009, 42, 224 CrossRef CAS PubMed; (c) E. Vedejs and M. Jure, Angew. Chem., Int. Ed., 2005, 44, 3974 CrossRef CAS PubMed; (d) J. Andraos, J. Phys. Chem. A, 2003, 107, 2374 CrossRef CAS.
- D. Hoppe and O. Zschage, Angew. Chem., Int. Ed. Engl., 1989, 28, 69 CrossRef.
- P. Wipf and H. Jahn, Tetrahedron, 1996, 52, 12853 CrossRef CAS.
- (a) S. Farhat, I. Zouev and I. Marek, Tetrahedron, 2004, 60, 1329 CrossRef CAS; (b) N. Chinkov, H. Chechik, S. Majumbar, A. Liard and I. Marek, Synthesis, 2002, 2473 CAS; (c) S. Farhat and I. Marek, Angew. Chem., Int. Ed., 2002, 114, 361 CrossRef; (d) A. Liard and I. Marek, J. Org. Chem., 2000, 65, 7218 CrossRef CAS PubMed.
- (a) A. B. Charette and A. Beauchemin, Org. React., 2001, 58, 1 CAS; (b) H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS PubMed; (c) H. Y. Kim and P. J. Walsh, Acc. Chem. Res., 2012, 45, 1533 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, computational details, list of coordinates and energies (E, H and G) of optimized structures, spectroscopic data and copies of 1H and 13C NMR spectra. See DOI: 10.1039/c5sc00445d |
| This journal is © The Royal Society of Chemistry 2015 |