
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEt3B-mediated two- and three-component coupling reactions via radical decarbonylation of α-alkoxyacyl tellurides: single-step construction of densely oxygenated carboskeletons†
Masanori
Nagatomo
,
Daigo
Kamimura
,
Yuki
Matsui
,
Keisuke
Masuda
and
Masayuki
Inoue
*
Graduate School of Pharmaceutical Sciences, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: inoue@mol.f.u-tokyo.ac.jp
First published on 6th March 2015
Abstract
The single-step construction of various densely oxygenated carboskeletons was achieved by radical-based two- and three-component coupling reactions of sugar derivatives, without the need for light or heat. Et3B/O2-mediated decarbonylation readily converted α-alkoxyacyl tellurides to α-alkoxy carbon radicals, which intermolecularly added to glyoxylic oxime ether or enones to provide the two-component adducts. Furthermore, the three-component adducts were produced by an intermolecular aldol reaction between the aldehyde and the boron enolates generated by capture of the two-component radical intermediates by Et3B. This powerful coupling method serves as a novel strategy for the convergent synthesis of polyol natural products.
Introduction
Contiguously substituted polyol structures are found not only in numerous saccharides, but also in diverse secondary metabolites. For example, highly oxygenated terpenoids often accommodate such substructures (e.g., sororianolide B,1 klysimplexin U,2Scheme 1). As the oxygen-based functional groups potentially form hydrogen bonds with biological polymers, polyol natural products often exhibit potent bioactivities. Therefore, these complex terpenoids are expected to function as selective cellular probes and pharmacologically useful compounds.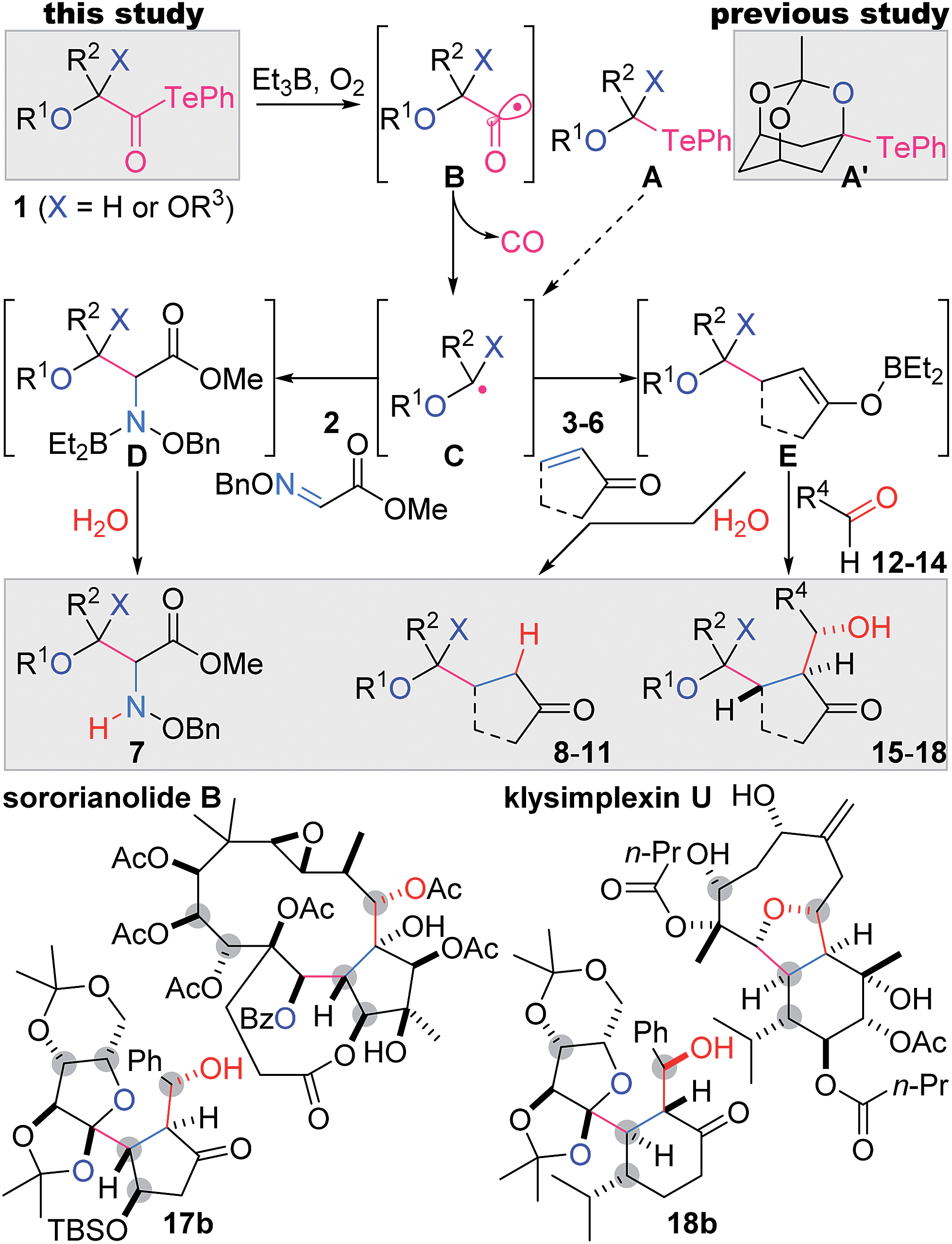 | ||
| Scheme 1 The present two- and three-component coupling reactions via radical decarbonylation of α-alkoxyacyl tellurides, and representative polyol natural products. | ||
The presence of a greater number of oxygen-attached stereocenters with increased density in bioactive polyol natural products heightens the challenge of their chemical synthesis.3 In designing shorter and simpler synthetic routes to these complex targets, the convergent assembly of oxygenated fragments is most desirable, as this would minimize post-coupling manipulations of the functional groups. Radical-based reactions enable the highly chemoselective formation of carbon–carbon (C–C) bonds without affecting most oxygen-based functional groups and thus are particularly suitable as powerful convergent methodologies.4 Here we report mild and robust two- and three-component radical coupling reactions using multiply oxygenated α-alkoxyacyl tellurides 1 and Et3B as the key reagents. The versatility of this process was demonstrated by the single-step preparation of variously adorned structures (7–11, 15–18), including the polyol fragments of sororianolide B (17b) and klysimplexin U (18b).
Results and discussion
We recently demonstrated facile bridgehead radical formation from a mono-telluro acetal A′ by the action of Et3B and O2 (Scheme 1).5,6 However, expansion of the substrate scope of this reaction was unsuccessful because of the chemical instability of the mono-telluro acetal (X = H) and the orthoester A (X = OR3).7 Thus, the C–Te bond of A typically undergoes heterolytic cleavage by electron donation from the oxygen lone pair, while such electron donation is uniquely impeded in A′ due to the anti-Bredt character of the resultant oxocarbenium ion. In this context, we alternatively designed more stable α-alkoxyacyl tellurides 1 as precursors to α-alkoxy radical intermediates C.8 An Et radical generated from Et3B/O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 would induce the C–Te homolysis of 1, leading to the acyl radical B, the decarbonylation of which would give rise to the requisite radical C.10,11 According to our presumed scenario, the nucleophilic radical C would then react intermolecularly with the electrophilic C
9 would induce the C–Te homolysis of 1, leading to the acyl radical B, the decarbonylation of which would give rise to the requisite radical C.10,11 According to our presumed scenario, the nucleophilic radical C would then react intermolecularly with the electrophilic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of 2 and the CC bond of 3–6 to generate the corresponding N- and C-radicals, respectively. Et3B in turn would transform these radicals into the polar intermediates D and E through ejection of an Et radical,12 and subsequent protonation would afford the two-component adducts 7 and 8–11, respectively. The boron enolate E would further react with aldehydes 12–14via the aldol reaction, resulting in the formation of the three-component products 15–18. Most importantly, this intricate yet attractive plan would be realized only when all of the reactants properly follow the series of depicted radical and polar reactions in a single flask.
N bond of 2 and the CC bond of 3–6 to generate the corresponding N- and C-radicals, respectively. Et3B in turn would transform these radicals into the polar intermediates D and E through ejection of an Et radical,12 and subsequent protonation would afford the two-component adducts 7 and 8–11, respectively. The boron enolate E would further react with aldehydes 12–14via the aldol reaction, resulting in the formation of the three-component products 15–18. Most importantly, this intricate yet attractive plan would be realized only when all of the reactants properly follow the series of depicted radical and polar reactions in a single flask.
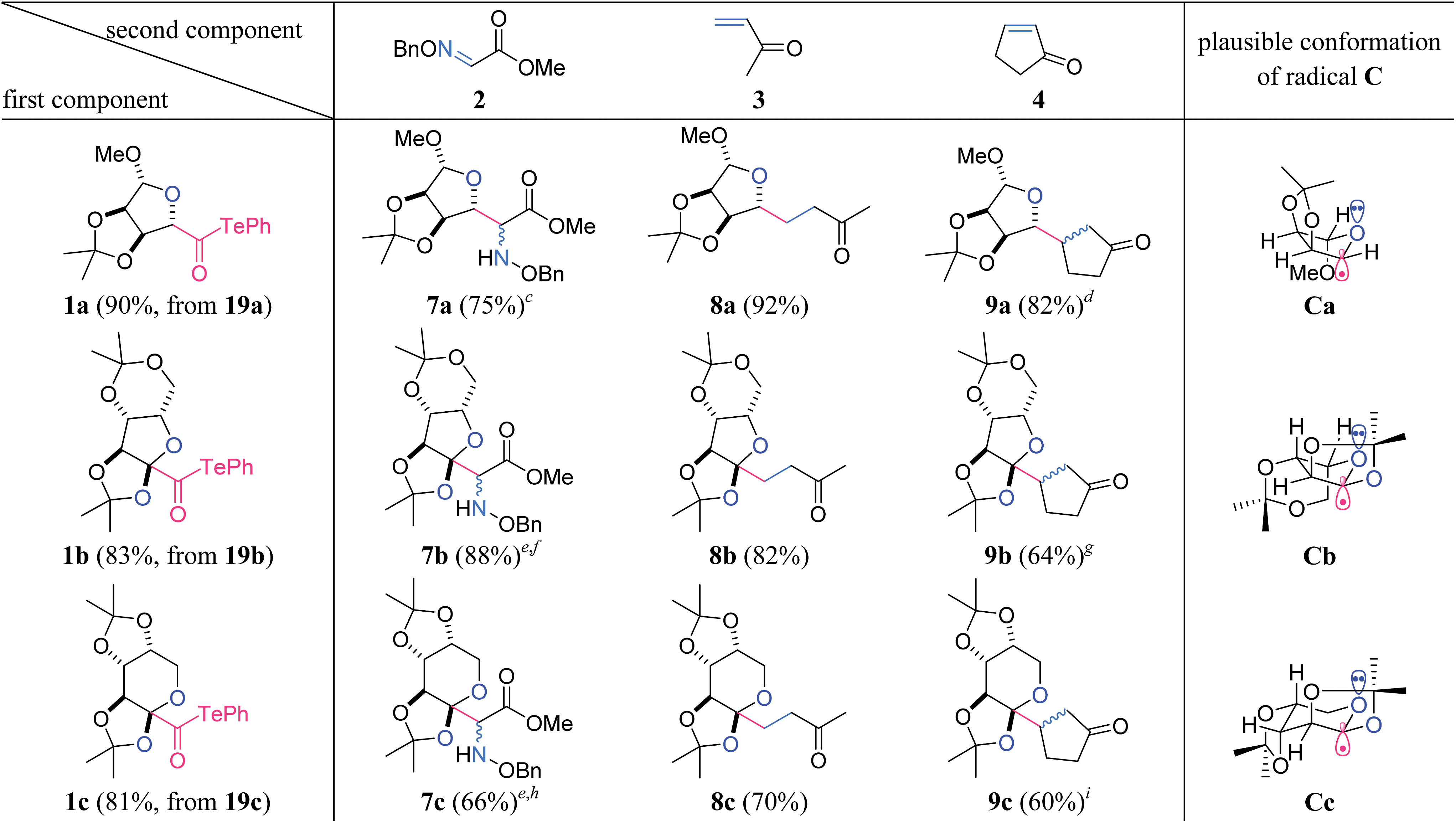
β-D-Ribofuranosiduronic acid derivative 19a, 2-keto-L-gulonic acid derivative 19b, and β-D-arabino-2-hexulopyranosonic acid derivative 19c were selected as readily available carboxylic acids with four contiguous oxygen-attached stereocenters (Table 1). α-Alkoxyacyl tellurides 1a–c were then obtained from the sugar-derived acids 19a–c in one pot. Namely, 19 was first transformed into the activated ester via condensation with i-butyl chloroformate, and was then treated in situ with PhTeNa, prepared from (PhTe)2 and NaBH4, providing 1. The structurally distinct alkoxyacyl tellurides 1a–c were found to be stable to air, light and silica gel.
|
|
|---|
|
a Reagents and conditions: 19 (1 equiv.), i-butyl chloroformate (1.2 equiv.), N-methylmorpholine (1.2 equiv.), THF (0.2 M), 0 °C, 30 min; (PhTe)2 (1 equiv.), NaBH4 (3 equiv.), THF/MeOH (10:1, 0.2 M), 0 °C, 30 min; rt, 30 min.
b
1 (1 equiv.), 2–4 (2 equiv.), Et3B (3 equiv.), CH2Cl2 (0.02 M), rt, under air.
c dr = 2:1.
d dr = 3:1.
e The reaction was carried out in CH2Cl2 (0.1 M).
f dr = 1:1.
g dr = 1.3:1.
h dr = 1.5:1.
i dr = 1.4:1.
|
|
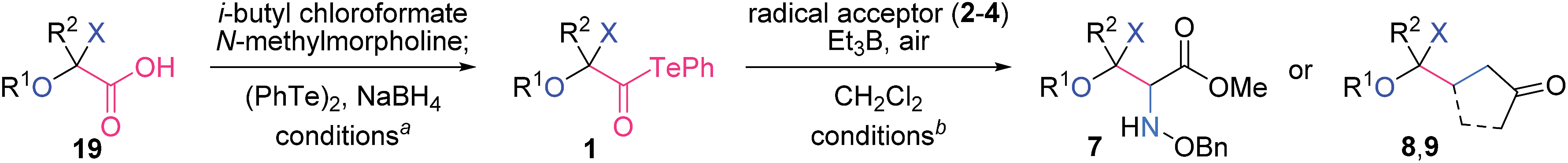
Significantly, the two-component radical reactions between the three radical donors 1a–c and three radical acceptors 2–4 all efficiently proceeded to generate nine complex adducts (Table 1).13,14 In these reactions, α-alkoxyacyl telluride (1a, b or c) was simply treated with 2 equiv. of the acceptor (2, 3 or 4) and 3 equiv. of Et3B under air in CH2Cl2 at room temperature.15 As a result, three highly oxygenated α-amino acid derivatives 7a–c were directly constructed using the O-benzyl-protected methyl glyoxylate oxime 2 as the acceptor.16 An alternative application for α,β-unsaturated ketones 3 and 4 permitted the attachment to the linear carboskeleton of 8a–c and the branched carboskeleton of 9a–c, respectively. The absence of the formation of any potential by-products derived from the addition of the acyl radicals proved that radical decarbonylation consistently occurred prior to the intermolecular addition to the CN or CC bond. C–C homolysis of the acyl radicals is likely facilitated by the stabilizing orbital interaction between the alkyl radical and the adjacent oxygen lone pair in Ca–c.17
The resultant compounds Ca–c exhibited potent reactivity as nucleophilic radicals, and stereoselectively constructed the trisubstituted (7a–9a) and tetrasubstituted carbons (7b–9b, 7c–9c) on the ether rings (Table 1). The observed stereochemical outcome could be explained by a combination of stereoelectronic and steric effects. The three-dimensional structures of Ca–c represent their assumed conformations, where the interaction of the σ-radical orbital and the non-bonding O-electron pair increase both the stability and nucleophilicity of the axial σ-radical.18,19 The approach of the acceptor only occurs from the α-face of the molecule because of the concave nature of the β-face.20 In the case of 7b–9b and 7c–9c, the favorable formation of the cis-fused bicycle in comparison to the strained trans-fused counterpart contributes to the excellent stereoselectivity at the ring junction.21 Overall, the results in Table 1 proved the high utility of the contiguously substituted polyol radical donors 1a–c.
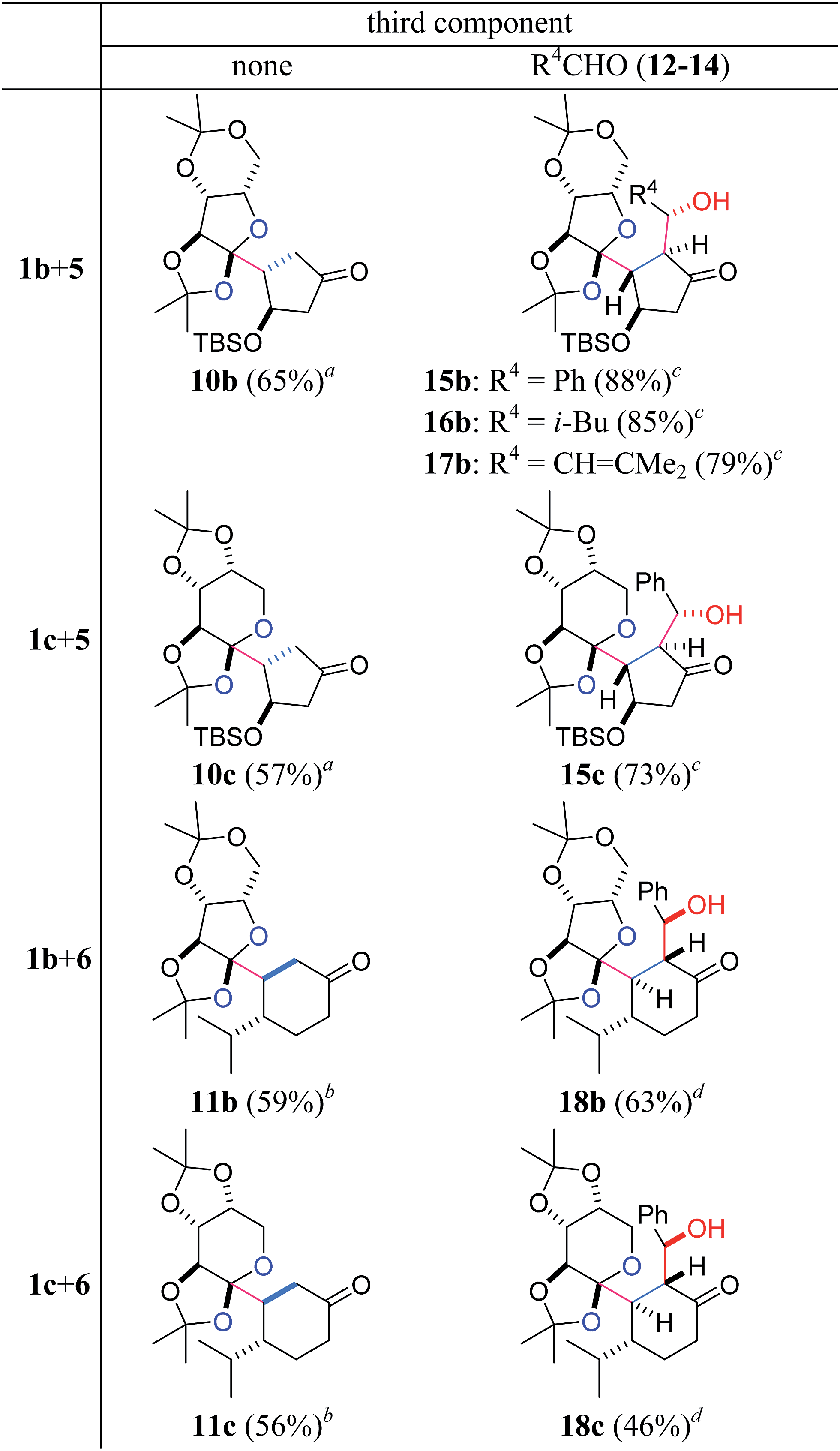
Although the stereoselectivity at the radical accepting position in 7a–c and 9a–c was not high (dr = 3:1–1:1), the pre-existing stereocenters of acceptors 5 and 6 perfectly governed the stereochemical outcome of the two-component reactions (Table 2). Specifically, the β-oriented TBS-oxy group of 5 induced the selective α-face addition of the radical that was generated from 1b or 1c in the presence of Et3B/O2, giving rise to 10b or 10c as the sole stereoisomer. On the other hand, the radical addition to 6 occurred trans to its α-configured isopropyl group, predominantly leading to 11b or 11c.
|
|
|---|
| a Reagents and conditions: 1b,c (1 equiv.), 5 (2 equiv.), Et3B (3 equiv.), CH2Cl2 (0.02 M), rt, under air. b 1b,c (1 equiv.), 6 (5 equiv.), Et3B (3 equiv.), CH2Cl2 (0.1 M), rt, under air. c 1b,c (1 equiv.), 5 (2 equiv.), 12–14 (3 equiv.), Et3B (3 equiv.), CH2Cl2 (0.1 M), rt, under air. d 1b,c (1 equiv.), 6 (3 equiv.), 12 (5 equiv.), Et3B (3 equiv.), CH2Cl2 (0.1 M), rt, under air. |
|
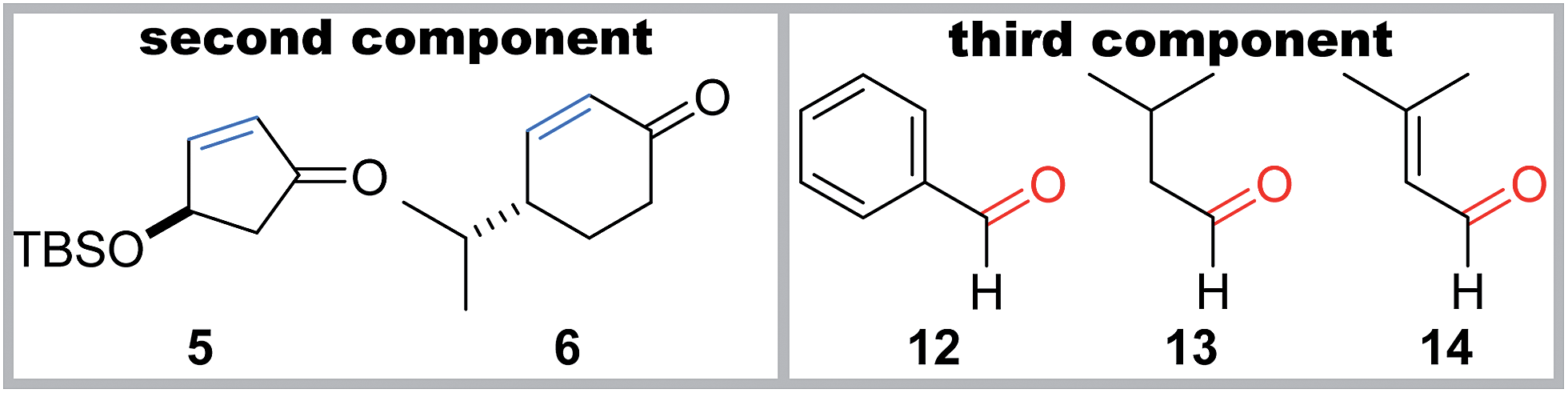
Furthermore, the three-component coupling reactions were realized with complete control of the four stereocenters (Table 2). When a mixture of acyl telluride 1b, 5 (2 equiv.) and benzaldehyde 12 (3 equiv.) was treated with Et3B/O2 at room temperature, only 15b was obtained in a high yield. The aliphatic aldehyde 13 and the even α,β-unsaturated aldehyde 14 also served as the third components without decreasing the yields to afford 16b and 17b, respectively. Application of the different radical donor 1c with 5 and 12 also resulted in the smooth formation of the adduct 15c. The 6-membered enone 6 participated in the three-component reactions upon use of 1b and 1c in the presence of 12, delivering 18b and 18c, respectively. It is noteworthy that the one-step construction of the complex products with eight contiguous stereocenters was achieved under neutral conditions, and the five stereocenters of 15b–17b and the four stereocenters of 18b directly matched those of sororianolide B and klysimplexin U, respectively (highlighted as gray circles in Scheme 1). These data together proved the power of the present convergent methodology for the synthesis of densely oxidized carboskeletons.
The radical-polar crossover mechanism of the two- and three-component reactions was exemplified by the synthesis of 10b and 15b (Scheme 2).22 The Et3B-mediated radical reaction between 1b and 5 stereoselectively generates the radical intermediate Fb,23 which is transformed to the boron enolate Eb by capture with Et3B. Protonation of the polar intermediate Eb affords the two-component adduct 10b. Alternatively, aldehyde 12 approaches Eb from the face opposite to the bulky fused tricycle and forms the chair-like six-membered transition state Gb,24 subsequently establishing the stereocenters of the α- and β-positions of the ketone of 15b. Three of the four newly introduced stereocenters of 15–17 and 18 are opposite because the configuration of the isopropyl group of 6 is the inverse of the TBS-oxy group of 5 (Table 2). Hence the stereochemical consistency in both the radical and aldol reactions is one of the most remarkable features of the present methodology.
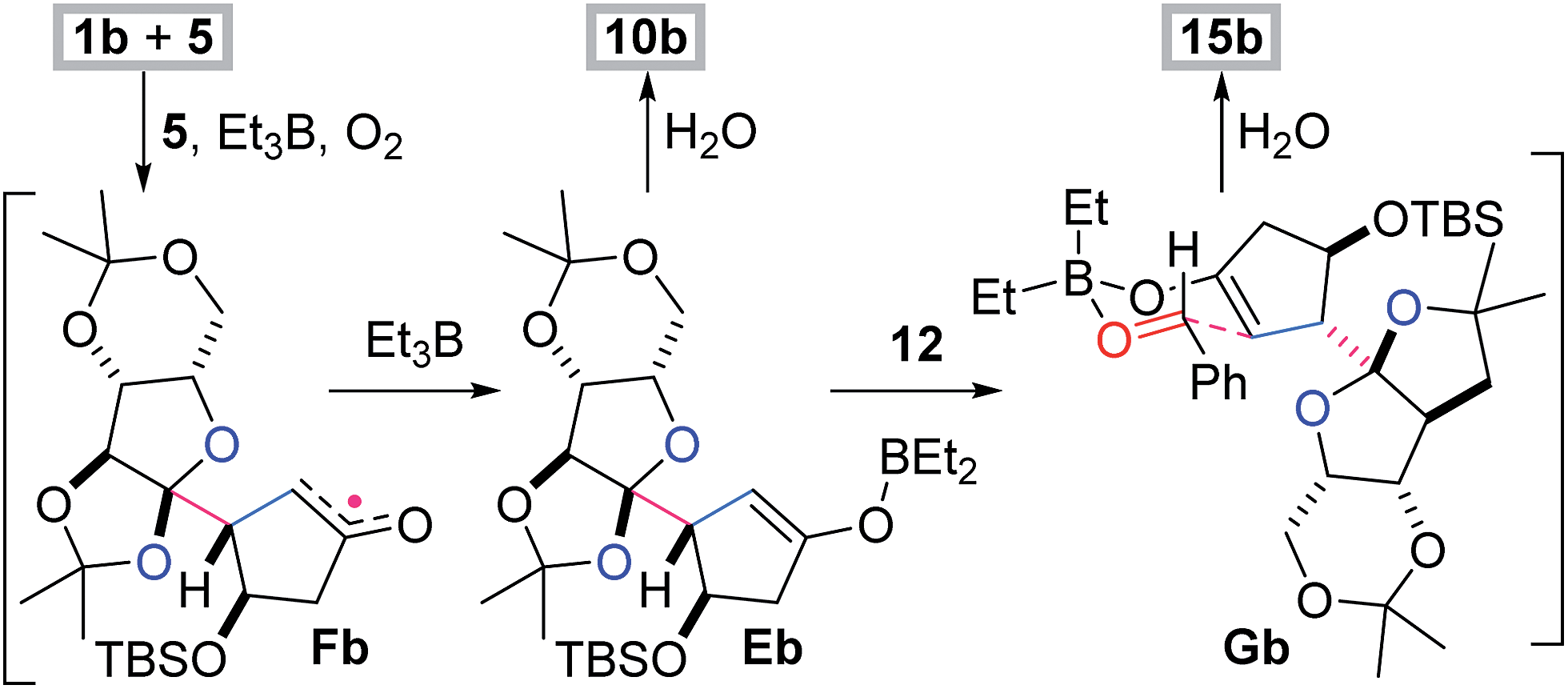 | ||
| Scheme 2 Rationale of the stereochemical outcome. | ||
Conclusions
In summary, we devised new radical-based two- and three-component coupling reactions, and realized the one-step construction of contiguously substituted polyol structures, which are significantly difficult to access by other methods. Upon activation with Et3B/O2, α-alkoxyacyl tellurides were rapidly converted to α-alkoxy radicals through expulsion of CO, thereby serving as the surrogate of the unstable mono-telluro acetals or orthoesters. Despite the sterically hindered nature, the resultant secondary or tertiary radical possessed potent reactivity toward the oxime and enone to stereoselectively furnish a variety of two-component adducts. Furthermore, the boron enolate, the intermediate in the two-component reaction, effectively participated in the aldol reaction with the third component, leading to another series of products. The three component coupling reactions simultaneously installed four contiguous stereocenters, and thus are particularly useful in increasing the complexity of the molecule in a single step. Because of the simplicity of the reagent system and the mildness of the conditions, this powerful radical technology introduces a novel convergent strategy for the expedited total synthesis of densely oxidized natural products.Acknowledgements
This research was financially supported by the Funding Program for a Grant-in-Aid for Scientific Research (A) to M. I., and a Grant-in-Aid for Young Scientists (B) (JSPS) to M. N.Notes and references
- Y. Huang and H. A. Aisa, Helv. Chim. Acta, 2010, 93, 1156 CrossRef CAS
.
- B.-W. Chen, C.-Y. Huang, Z.-H. Wen, J.-H. Su, W.-H. Wang, P.-J. Sung, Y.-C. Wu and J.-H. Sheu, Bull. Chem. Soc. Jpn., 2011, 84, 1237 CrossRef CAS
- For selected reviews on the total synthesis of terpenoids, see:
(a) K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44 CrossRef CAS
- For recent reviews on radical reactions, see:
(a) G. S. C. Srikanth and S. L. Castle, Tetrahedron, 2005, 61, 10377 CrossRef CAS PubMed
- D. Kamimura, D. Urabe, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 5122 CrossRef CAS PubMed
- For reviews on the radical reactions of organotellurides, see:
(a) S. Yamago, Synlett, 2004, 1875 CrossRef CAS PubMed
- Although mono-telluro acetals have been utilized as radical donors, the synthesis of a mono-telluro orthoester has not been reported.
(a) D. H. R. Barton and M. Ramesh, J. Am. Chem. Soc., 1990, 112, 891 CrossRef CAS
- Most recently, we developed decarbonylative two-component coupling reactions of α-aminoacyl tellurides. M. Nagatomo, H. Nishiyama, H. Fujino and M. Inoue, Angew. Chem., Int. Ed., 2015, 54, 1537 CrossRef CAS PubMed
- K. Nozaki, K. Oshima and K. Utimoto, J. Am. Chem. Soc., 1987, 109, 2547 CrossRef CAS
- For a review on acyl radical reactions and decarbonylation rates, see:
(a) D. L. Boger, Isr. J. Chem., 1997, 37, 119 CrossRef CAS
- For selected examples of recent decarboxylative (–CO2) two-component radical coupling, see:
(a) Y. Yoshimi, K. Kobayashi, H. Kamakura, K. Nishikawa, Y. Haga, K. Maeda, T. Morita, T. Itou, Y. Okada and M. Hatanaka, Tetrahedron Lett., 2010, 51, 2332 CrossRef CAS PubMed
-
(a) N. Miyaura, M. Harada, M. Itoh and A. Suzuki, Chem. Lett., 1973, 1145 CrossRef CAS
- The stereochemistries of the newly synthesized compounds were established from NMR experiments on derivatized compounds. See ESI† for details.
- We confirmed from a separate experiment that the selenide version of 1a was inert toward our reagent system (Et3B/O2/rt), corroborating the superior reactivity of the acyl telluride in comparison to the acyl selenide. See also ref. 5.
- Acyl tellurides were previously converted to the acyl radicals either by thermolysis (>100 °C) or UV photolysis.
(a) C. Chen, D. Crich and A. Papadatos, J. Am. Chem. Soc., 1992, 114, 8313 CrossRef CAS
-
(a) H. Miyabe, C. Ushiro, M. Ueda, K. Yamakawa and T. Naito, J. Org. Chem., 2000, 65, 176 CrossRef CAS PubMed
- Stabilization of the alkyl radical was known to accelerate decarbonylation of the acyl radical.
(a) H. Fischer and H. Paul, Acc. Chem. Res., 1987, 20, 200 CrossRef CAS
- Interaction between the singly occupied orbital and the σ* orbital of the coplanar β-C–OR bond could further stabilize the radical. For structures of α-alkoxy radicals, see:
(a) B. Giese and J. Dupuis, Tetrahedron Lett., 1984, 25, 1349 CrossRef CAS
- For reviews on stereoselectivity on reactions of α-alkoxy radicals, see:
(a) B. Giese, Angew. Chem., Int. Ed. Engl., 1989, 28, 969 CrossRef
-
(a) D. H. R. Barton, S. D. Géro, B. Quiclet-Sire and M. Samadi, J. Chem. Soc., Chem. Commun., 1988, 1372 RSC
- D. Crich and L. B. L. Lim, J. Chem. Soc., Perkin Trans. 1, 1991, 2209 RSC
- For reviews on the radical-polar crossover reactions, see:
(a)
J. A. Murphy, The Radical-Polar Crossover Reaction, in Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, 2001, vol. 1, pp. 298–315 Search PubMed
- Formation of PhTeEt was observed.
-
(a) W. Fenzl and R. Köster, Liebigs Ann. Chem., 1975, 1322 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, characterization data, and NMR spectra of all new compounds. See DOI: 10.1039/c5sc00457h |
| This journal is © The Royal Society of Chemistry 2015 |