DOI:
10.1039/C5RA01315A
(Paper)
RSC Adv., 2015,
5, 17606-17614
Synthesis and anion binding studies of tris(3-aminopropyl)amine-based tripodal urea and thiourea receptors: proton transfer-induced selectivity for hydrogen sulfate over sulfate†
Received
22nd January 2015
, Accepted 29th January 2015
First published on 29th January 2015
Abstract
Tris(3-aminopropyl)amine-based tripodal urea and thiourea receptors, tris([(4-cyanophenyl)amino]propyl)urea (L1) and tris([(4-cyanophenyl)amino]propyl)thiourea (L2), have been synthesized and their anion binding properties have been investigated for halides and oxoanions. As investigated by 1H NMR titrations, each receptor binds an anion with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry via hydrogen-bonding interactions (NH⋯anion), showing the binding trend in the order of F− > H2PO4− > HCO3− > HSO4− > CH3COO− > SO42− > Cl− > Br− > I in DMSO-d6. The interactions of the receptors were further studied by 2D NOESY, showing the loss of NOESY contacts of two NH resonances for the complexes of F−, H2PO4−, HCO3−, HSO4− or CH3COO− due to the strong NH⋯anion interactions. The observed higher binding affinity for HSO4− than SO42− is attributed to the proton transfer from HSO4− to the central nitrogen of L1 or L2 which was also supported by the DFT calculations, leading to the secondary acid–base interactions. The thiourea receptor L2 has a general trend to show a higher affinity for an anion as compared to the urea receptor L1 for the corresponding anion in DMSO-d6. In addition, the compound L2 has been exploited for its extraction properties for fluoride in water using a liquid–liquid extraction technique, and the results indicate that the receptor effectively extracts fluoride from water showing ca. 99% efficiency (based on L2).
:1 stoichiometry via hydrogen-bonding interactions (NH⋯anion), showing the binding trend in the order of F− > H2PO4− > HCO3− > HSO4− > CH3COO− > SO42− > Cl− > Br− > I in DMSO-d6. The interactions of the receptors were further studied by 2D NOESY, showing the loss of NOESY contacts of two NH resonances for the complexes of F−, H2PO4−, HCO3−, HSO4− or CH3COO− due to the strong NH⋯anion interactions. The observed higher binding affinity for HSO4− than SO42− is attributed to the proton transfer from HSO4− to the central nitrogen of L1 or L2 which was also supported by the DFT calculations, leading to the secondary acid–base interactions. The thiourea receptor L2 has a general trend to show a higher affinity for an anion as compared to the urea receptor L1 for the corresponding anion in DMSO-d6. In addition, the compound L2 has been exploited for its extraction properties for fluoride in water using a liquid–liquid extraction technique, and the results indicate that the receptor effectively extracts fluoride from water showing ca. 99% efficiency (based on L2).
Introduction
Anion coordination chemistry is a major area of research in supramolecular chemistry, since anions play critical roles in many biological, chemical and environmental applications.1–7 As learned from nature, hydrogen-bonding interactions are key factors in controlling many important functions of biomolecules, e.g. information storage, signal transfer, replication and catalysis.8 In order to understand and mimic the natural interactions involved in complex living systems, several types of neutral synthetic molecules including amides,9 thioamides,10 ureas,11 thioureas,12 pyrroles,13 and indoles14 have been broadly employed as effective receptors for a variety of anions in solution and solid state.
Among these various receptors that possess hydrogen bonding capabilities in anion binding via NH⋯anion interactions, urea-based receptors have received much attention recently, due to the acidic nature and directional properties of NH groups for anionic guests.11a,15 An early example reported by Hamilton et al. demonstrated that a simple acyclic urea containing a single urea functionality showed an affinity for acetate (K = 45 M−1) in DMSO.16 Fabbrizzi et al. synthesized a bis(4-nitrophenyl) urea receptor that formed a strong complex with fluoride (K = 2.40 × 107 M−1) in CH3CN.17 Gale et al. developed a urea-based receptor linked with indole groups that formed a carbonate complex stabilized by NH donor groups from both indole and urea functional groups.18 Johnson et al. reported a rigid dipodal urea linked with acetylene groups, which was shown to form a five-coordinate chloride complex.19
Recently, a number of urea- and thiourea-based receptors have been developed based on the use of tris(2-aminoethyl)amine (tren) as a framework appended with different aromatic groups.20,21 For example, a m-cyanophenyl-based tripodal urea reported by Custelcean et al. was shown to form a silver-based MOF that encapsulated sulfate by a total of twelve hydrogen bonds.20a Wu et al. reported a 3-pyridyl-based tripodal urea that also showed strong affinity for sulfate.20b Ghosh et al. reported a pentafluorophenyl-based tripodal urea for the selective binding of phosphate.20c A m-nitrophenyl substituted tripodal urea synthesized by Das et al. was found to form capsular complexes with carbonate and sulfate.20h The progression from urea to thiourea leads to an enhanced acidity of a NH group in the later, thereby a thiourea could have a stronger affinity for an anion than its urea analogue.22 Gale et al. reported a phenyl-based thiourea tripodal receptor that formed a carbonate complex from a mixture of the host with [Et4N][HCO3].21a The compound was able to transport bicarbonate across lipid membranes. While fluorinated tripodal ureas and thioureas were shown to transport chloride anions through a lipid bilayer.21b In the case of p-fluorophenyl tripodal thiourea, an encapsulated chloride complex and a sulfate capsular complex were structurally characterized.21b A tren-based tris(thiourea) receptor substituted with p-nitrophenyl groups was shown to form a rigid dimeric capsule with trivalent phosphate.21c Our group has recently reported a p-cyanophenyl tripodal urea for sulfate forming a seven coordinate sulfate complex.23a Further work on this receptor for halides has demonstrated the binding trend in the order of fluoride > chloride > bromide > iodide in solution.23b Ghosh et al. has recently reported that the thiourea analogue p-cyanophenyl tripodal receptor is capable of forming a 1:1 complex with fluoride and 2:1 complex with sulfate, showing moderate extraction efficiencies for fluoride and sulfate from aqueous solutions.21d
Our continued interests in the development of urea/thiourea-based anion receptors24 have led us to use a slightly larger tripodal framework as tris(3-aminopropyl)amine linked with three p-cyanophenyl groups. Because of the longer chain in the propylene group as compared to the ethylene chain analogue, such receptors are expected to provide larger and flexible cavities; which could affect their selectivity patterns for an anion. The choice of cyanophenyl-substituted spacers was derived from their ability to act as electron-withdrawing groups, which was further supported by DFT calculations, showing the highest electron potential on cyano-groups. In particular, recent studies showed that the structural manipulation of simple receptors with variable lengths, sizes, functional groups and spacers can lead to selective binding of a particular anion.15 Herein, we report the synthesis of two propylene-linked new receptors L1 and L2 (Scheme 1), and their comparative anion binding studies by 1H NMR titrations and 2D NOESY experiments in DMSO-d6, showing the unusual selectivity for hydrogen sulfate than sulfate. In addition, L2 was further used for the extraction of fluoride in water using a liquid–liquid extraction technique.
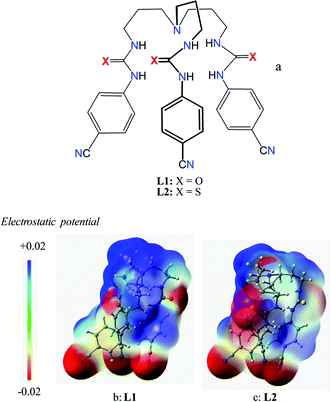 |
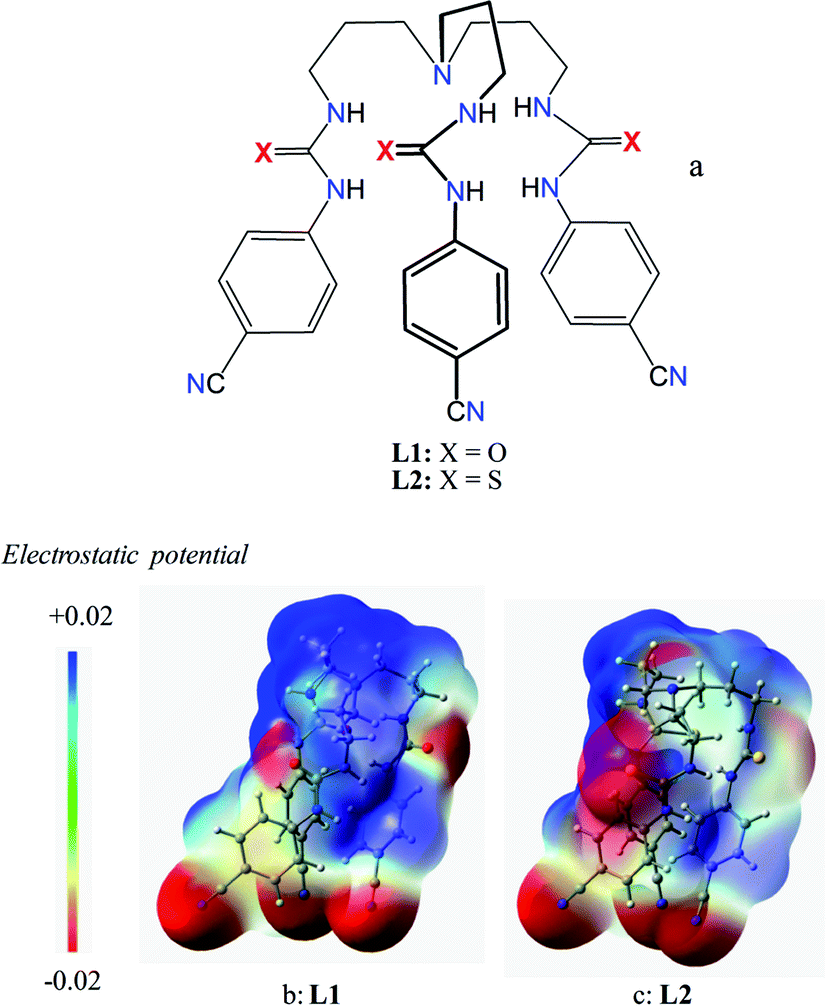
| | Scheme 1 Schematic representation of chemical structures of L1 and L2 (a), and electrostatic potential map for L1 (b) and L2 (c) calculated at M06-2X/6-31G(d,p) level theory (red is negative potential and blue is positive potential). | |
Results and discussion
Synthesis
The synthesis of L1 (urea) and L2 (thiourea) was accomplished from the reaction of tris(3-aminopropyl)amine (1) with three equivalents of 4-cyanophenyl isocyanate/isothiocyanate (2) in CH2Cl2 (Scheme 2), following the similar method as reported before for ethylene chain analogues.23,24 In general, a higher yield was achieved for urea-based receptor (90%) than the thiourea-based receptor (73%). Attempts to obtain X-ray quality crystals of free receptors or anion complexes were unsuccessful.
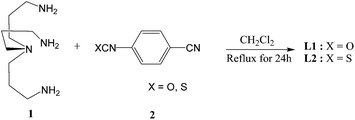 |
| | Scheme 2 Synthetic pathway of L1 and L2. | |
NMR titration studies
The binding properties of the new receptors (L1 and L2) for a number of anions including F−, Cl−, Br−, I−, ClO4−, NO3−, HSO4−, H2PO4−, CH3COO−, HCO3− and SO42− were investigated by 1H NMR studies in DMSO-d6. Initially, the anion binding abilities of L1 and L2 were screened by the addition of one equivalent of the respective anion to a host solution.
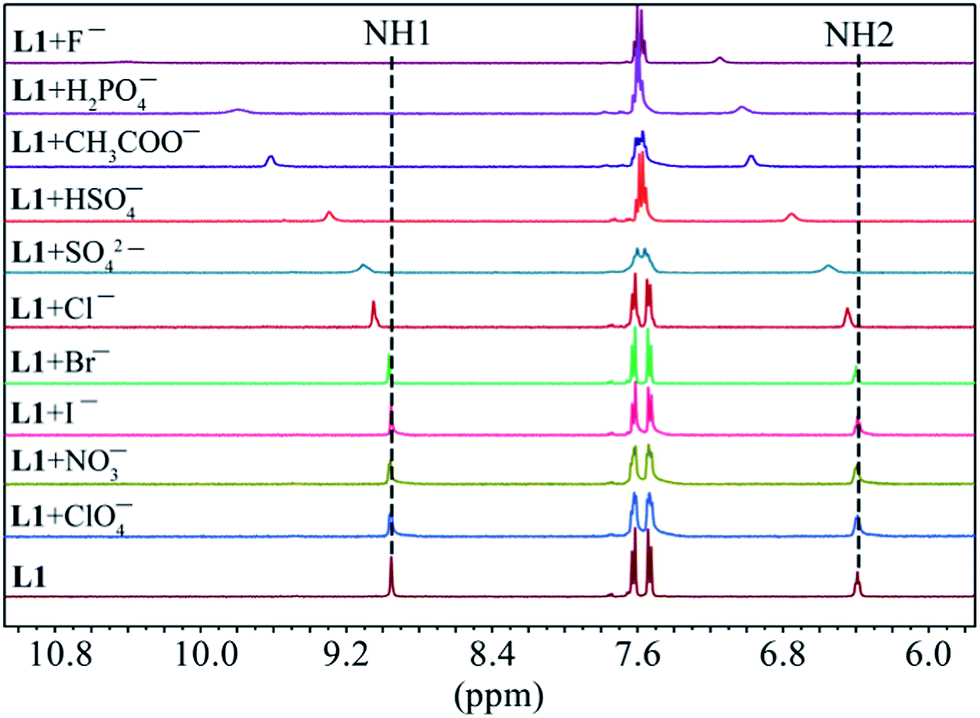
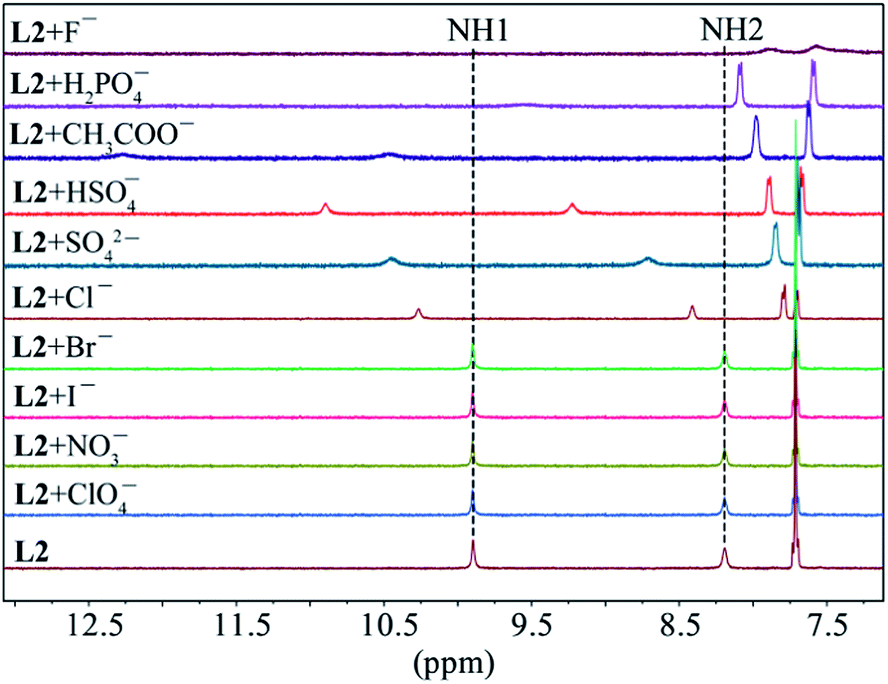
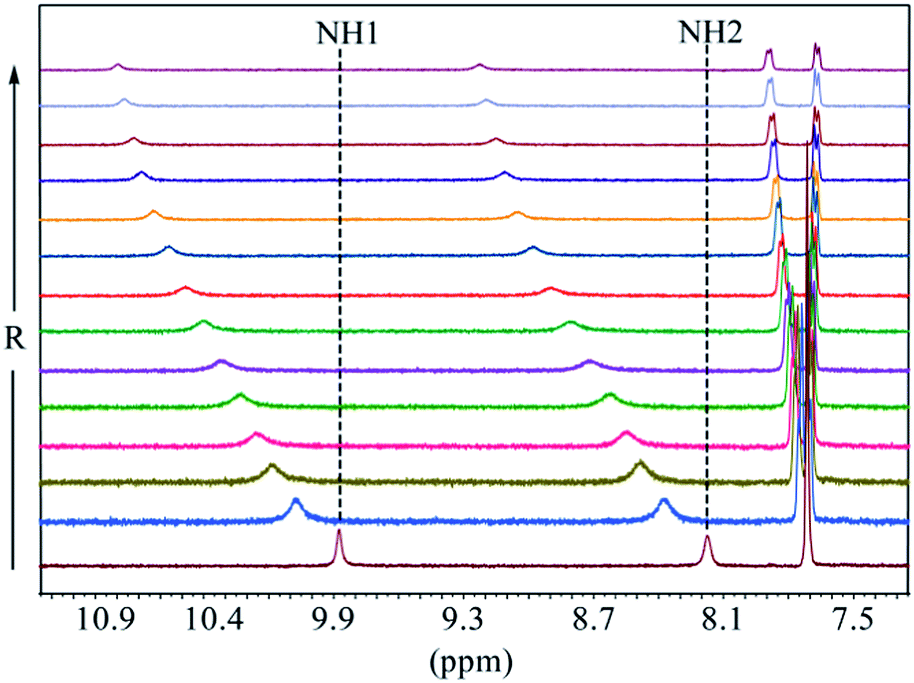
As shown in Fig. 1, two NH protons of urea group of L1 appeared at 8.94 ppm (H1) and 6.37 ppm (H2). These protons shifted downfield after the addition of oxoanions including HSO4−, H2PO4−, CH3COO−, HCO3− and SO42−. However, no appreciable shift was observed in the presence of ClO4−, NO3−, Br− and I−. Among the all anions, the highest shift of NH's was observed for fluoride followed by H2PO4− and CH3COO−. The addition of F− or H2PO4− to L1 resulted in the broadening of NH peaks.25 Such a significant downfield shift of both NH resonances for an anion is attributed to the direct involvement of the NH groups in anion binding via NH⋯anion interactions. For the thiourea-based receptor L2, two corresponding NH protons that appeared at 9.86 ppm (H1) and 8.17 ppm (H2) were also found to respond with different anions exhibiting the similar trend (Fig. 2) as observed for L1 (Fig. 1). However, a higher downfield shift was observed for L2 with oxoanions and halides as compared to L1 with the corresponding anions. In the case of F− and H2PO4− and HCO3− with L2, peak broadening of NHs occurred similar to that observed for L1.
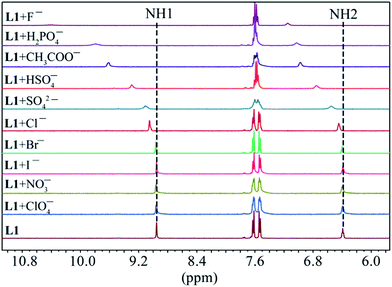 |
| | Fig. 1 Partial 1H NMR spectra of L1 (2 mM) in the presence of one equivalent of different anions in DMSO-d6 (H1 = CONHAr, H2 = CH2NHCO). | |
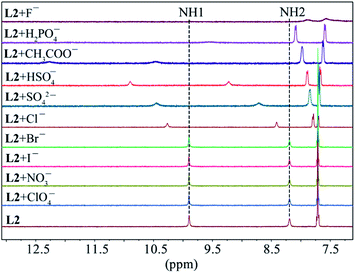 |
| | Fig. 2 Partial 1H NMR spectra of L2 (2 mM) in the presence of one equivalent of different anions in DMSO-d6 (H1 = CSNHAr, H2 = CH2NHCS). | |
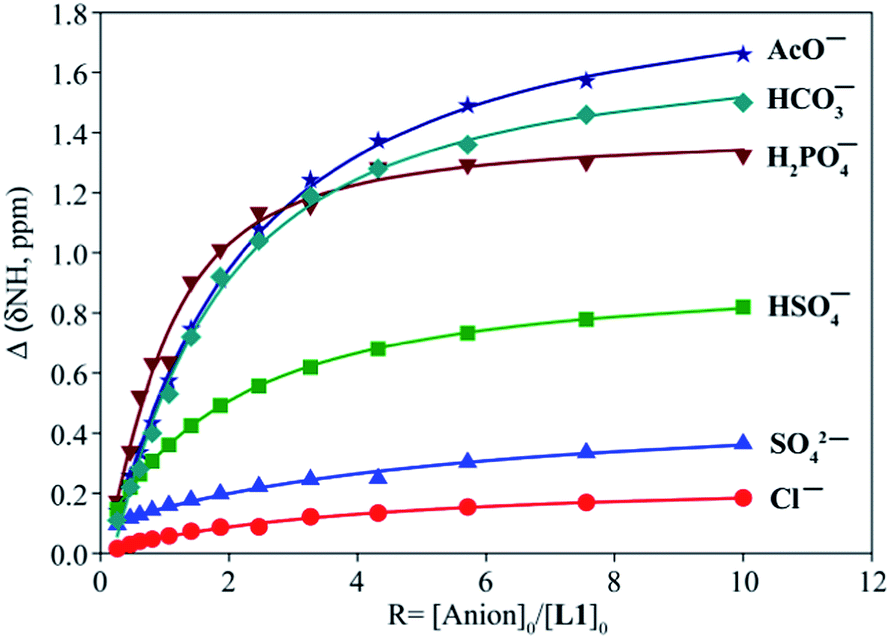
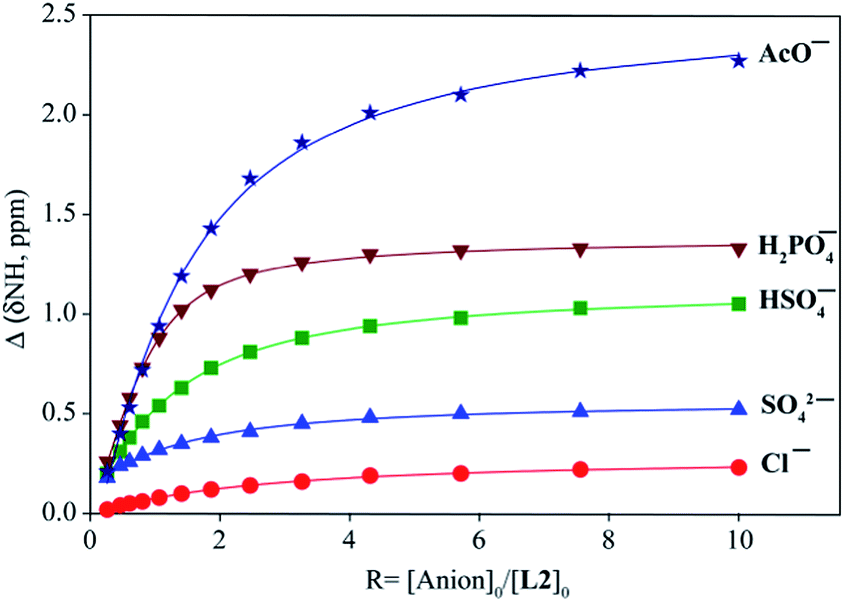
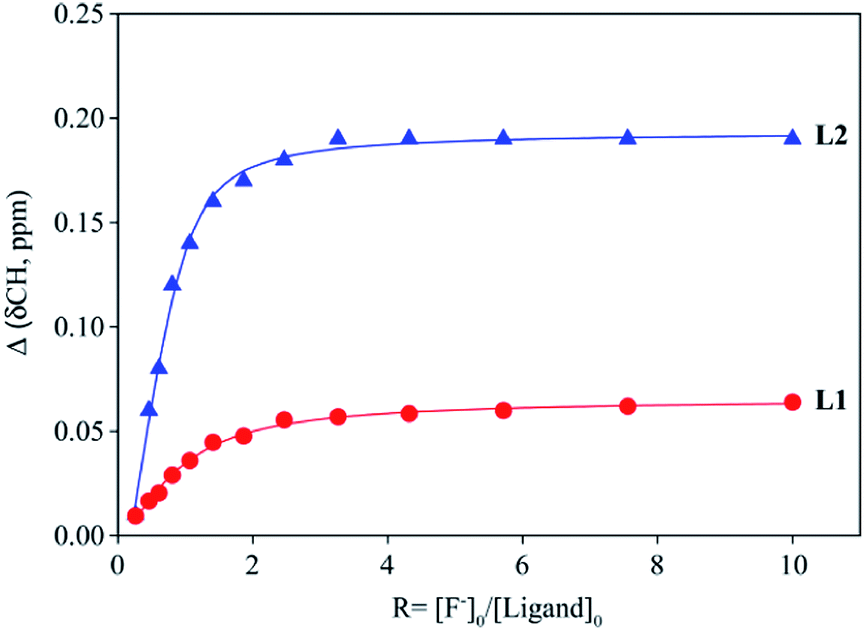
The binding constants of L1 and L2 for different anions were measured by 1H NMR titration experiments in DMSO-d6. Fig. 3 shows a representative example of 1H NMR titration spectra obtained from the incremental addition of hydrogen sulfate to L2, displaying a gradual shift change in both NH's resonances. The changes in the chemical shifts of NH's of L1 or L2 were plotted with an increasing amount of an anion, providing the best fit for a 1:1 binding model for the anions,26 as shown in Fig. 4 for L1 and Fig. 5 for L2. The 1:1 stoichiometry was further verified by a Job plot, showing a maximum at a 0.5 mole fraction for each anion (Fig. S30–35 in ESI†). Because of the peak broadening of NH's after the addition of F− to both receptors, the binding constants for fluoride were determined from shift changes of aromatic CH protons (Fig. 6).
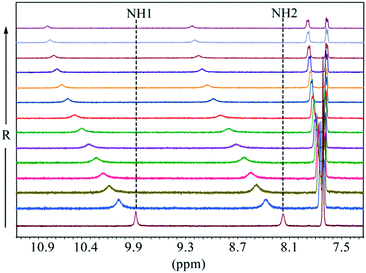 |
| | Fig. 3 Partial 1H NMR titration of L2 showing changes in the NH chemical shifts of the receptor with an increasing amount of HSO4− in DMSO-d6. (H1 = CSNHAr and H2 = CH2NHCS). | |
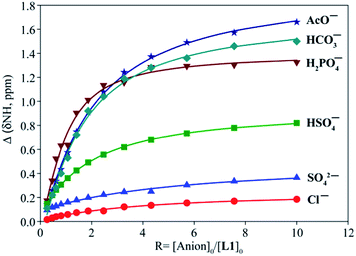 |
| | Fig. 4 1H NMR titration plot of changes in the NH (CH2NHCO) chemical shifts of L1 with an increasing amount of different anions in DMSO-d6. | |
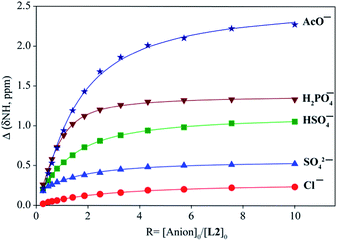 |
| | Fig. 5 1H NMR titration plot of changes in the NH (CH2NHCS) chemical shifts of L2 with an increasing amount of different anions in DMSO-d6. | |
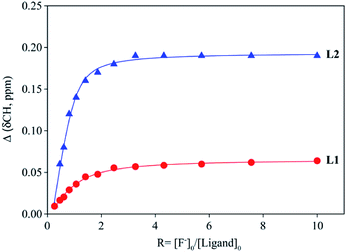 |
| | Fig. 6 1H NMR titration plot of changes in the aromatic CH chemical shifts (CHCNH) of L1 and L2 with an increasing amount of F− in DMSO-d6. | |
The binding constants of L1 and L2 for different anions determined from nonlinear regression analyses of chemical shift changes are listed in Table 1. An inspection of the binding data suggests that both receptors show a similar trend of binding for the investigated anions exhibiting the highest affinity for F−. In general, the thiourea-based receptor L2 exhibits higher affinity for an anion as compared to L1, which is due to the enhanced acidity of NHs in L2 incorporated with thiourea groups, as expected.12b Both receptors, however, show negligible affinity for other halides. For oxoanions, the highest binding was achieved for H2PO4−, followed by HSO4−, HCO3−, CH3COO− and SO42−. The observed binding constants broadly reflect the influence of relative basicity of the anions.27 However, the higher binding constants of both receptors for HSO4− as compared to the corresponding values for SO42− were somewhat unanticipated, although SO42− is more basic than HSO4− and has a higher charge. Such a discrepancy could be attributed to acid–base interactions of the central amine group of L1 or L2 with the acidic HO group of HSO4−,28 providing a secondary interaction of N+⋯H–O that was also verified by DFT calculations (discussed in later). Previously reported urea-based receptors linked with ethylene chains showed stronger binding for SO42− than HSO4−, in DMSO-d6.20h,23a Thus, the expansion of the tripodal cavity with propylene chains leads to the change of the selectivity patterns for HSO4− and SO42−, showing greater selectivity for HSO4−. As compared to ethylene-chain analogues,20h,23a,b the propylene chains in L1 and L2 might result in the higher basicity of the central nitrogen, which could be due to the weaker inductive effect29 of urea/thiourea groups through the longer propylene chains. Thus the central nitrogen can act as a base to transfer a proton from HSO4−. Both receptors showed higher binding for HCO3− as well, supporting this assumption. For highly basic acetate anion, the non-compliment shape of CH3COO− with the tripodal binding pocket might be a probable reason lowering the binding constant than that of H2PO4−. In general, the propylene-based receptors showed lower binding affinity for anions as compared to ethylene-based analogues, which could be due to the flexible nature of the cavity and enhanced basicity of the central nitrogen in L1 or L2.
Table 1 Binding constants of L1 and L2 in DMSO-d6
| Anion |
L1 (logK) |
L2 (logK) |
| Chemical shift changes were too small to calculate the K. |
| F− |
3.16 |
3.81 |
| Cl− |
1.96 |
2.34 |
| Br− |
1.75 |
a |
| I− |
<1 |
<1 |
| H2PO4− |
3.02 |
3.35 |
| HSO4− |
2.56 |
2.82 |
| SO42− |
1.61 |
1.89 |
| CH3COO− |
2.50 |
2.75 |
| HCO3− |
2.55 |
3.24 |
| ClO4− |
<1 |
<1 |
| NO3− |
<1 |
<1 |
NOESY NMR experiments
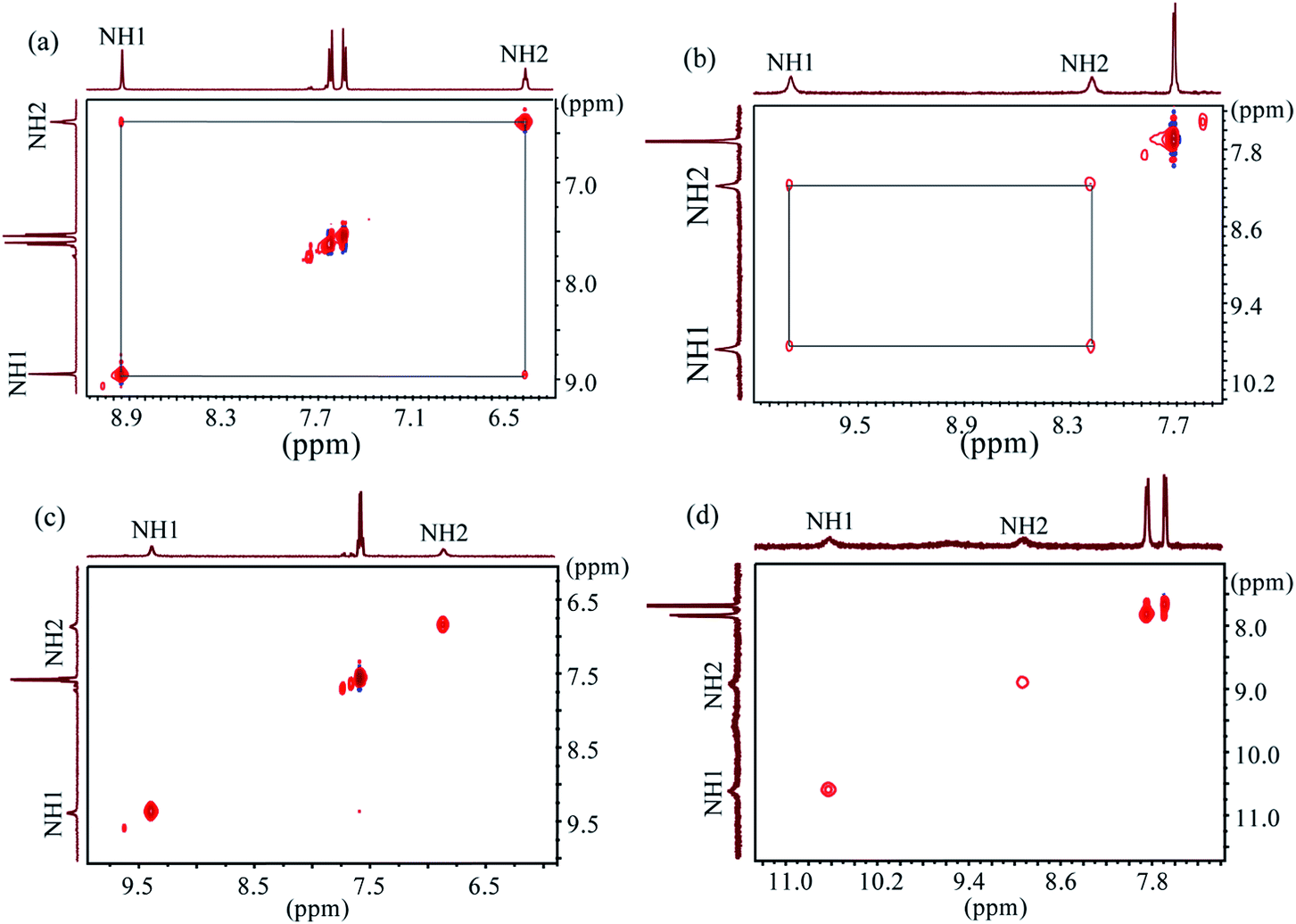
2D NOESY NMR experiments were performed to characterize the structures and conformational changes of the complexes in solution. Previous studies by us23a,b and others20h suggested that 2D NOESY NMR can effectively be used to evaluate the binding strength. In order to corroborate the data from NMR titrations, all 2D NOESY spectra were recorded for free L1 and L2 and their spectra were compared after the addition of one equivalent of the respective anions in DMSO-d6 at room temperature (Fig. 7 and Fig. S36–55 in ESI†). The Fig. 7a and b show the NOESY NMR spectra of free L1 and L2, respectively, each displaying a strong NH1⋯NH2 NOESY contact. After the addition of one equivalent of hydrogen sulfate, the NOESY contacts for both receptors completely disappeared (Fig. 7c and d), indicating the interactions of NHs with the added anion and a possible anion-induced conformational change of the receptors.23a,30 Similar spectral changes in NOESY were previously reported for anion complexes with tren-based receptors by us,23a,b Schneider30 and Das.20h,21c Indeed, both receptors show appreciable affinities for HSO4− as measured from 1H NMR titrations in DMSO-d6 (Table 1). We also observed a similar loss of NOESY signals for L1 in the presence of certain anions including F−, H2PO4−, CH3COO− and SO42−, and for L2 in the presence of SO42− (ESI†). However, the spotting of NH1⋯NH2 NOESY signals was hampered for L2 in the presence of F−, H2PO4− and CH3COO− due to the broadening of NH resonances of the receptor (ESI†). The addition of chloride or bromide to the receptors results in the weakening of NH1⋯NH2 NOESY signals. In contrast, the corresponding signals for both receptors were almost unchanged after the addition of one equivalent of I−, NO3− and ClO4−. This observation suggests the absence of interactions of the NHs with added anions, which is in agreement with the results obtained from NMR titrations (Table 1).
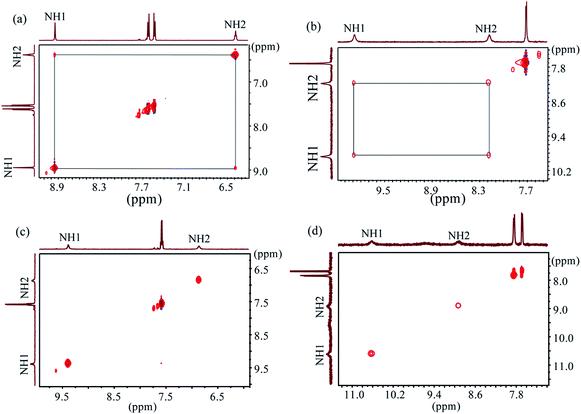 |
| | Fig. 7 2D NOESY NMR of (a) free L1, (b) free L2, (c) L1 + HSO4− (1 eq.) and (d) L2 + HSO4− (1 eq.) (H1 = ArNH and H2 = CH2NH). | |
DFT calculations
In order to evaluate the binding discrepancies of the receptors for SO42− and HSO4−, theoretical calculations were performed by density functional theory (DFT) with hybrid meta exchange-correlation functional M06-2X,31 using the Gaussian 09 package of programs.32 Molecular geometries were fully optimized without symmetry constraints at the M06-2X/6-31G(d,p) level of theory33 in gas phase and also in a polarizable continuum model (PCM) solvent model to approximate a DMSO environment (dielectric constant = 46.8). The binding energies (ΔE) of L1 and L2 were calculated for SO42− and HSO4−, using the equation: ΔE = E(complex) − E(receptor) − E(anion). The results show that the binding energies ΔE of [L1(SO4)]2− and [L1(HSO4)]− are −173.0 and −74.4 kcal mol−1, respectively in gas phase; while, as expected, the corresponding values are much lower in solvent phase, which are −42.1 and −37.8 kcal mol−1, respectively. The higher binding energies for SO42− is the effect of two charges on this anion as compared to one charge on HSO4−. On the other hand, the binding energies of [L2(SO4)]2− and [L2(HSO4)]− are −200.0 and −94.5 kcal mol−1, respectively in gas phase. In solvent phase the ΔE of [L2(SO4)]2− and [L2(HSO4)]− are −55.5 and −47.4 kcal mol−1. It is obvious that the binding energies of L2 are higher for both anions than those of L1, agreeing with the trend of experimental binding constants obtained from 1H NMR titrations (Table 1).
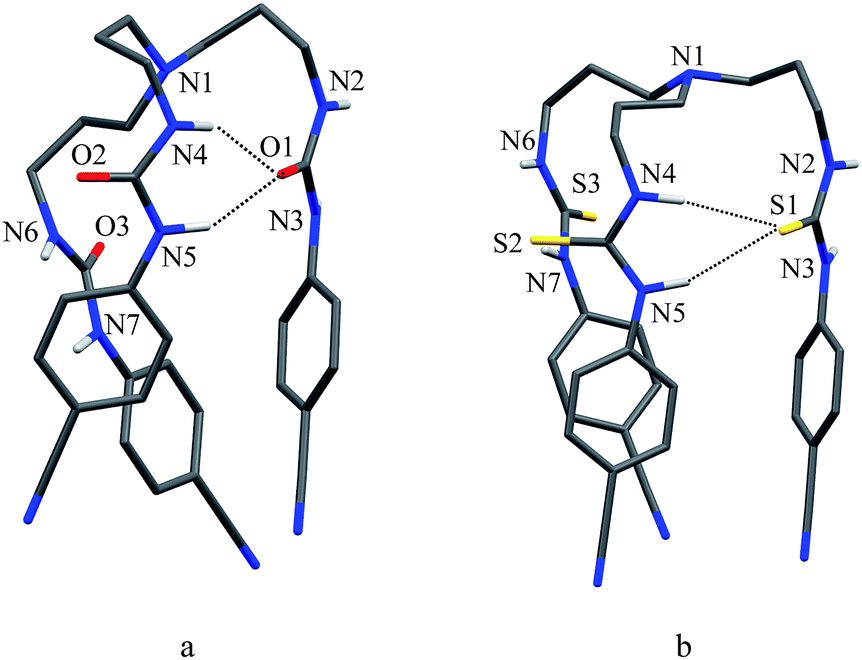
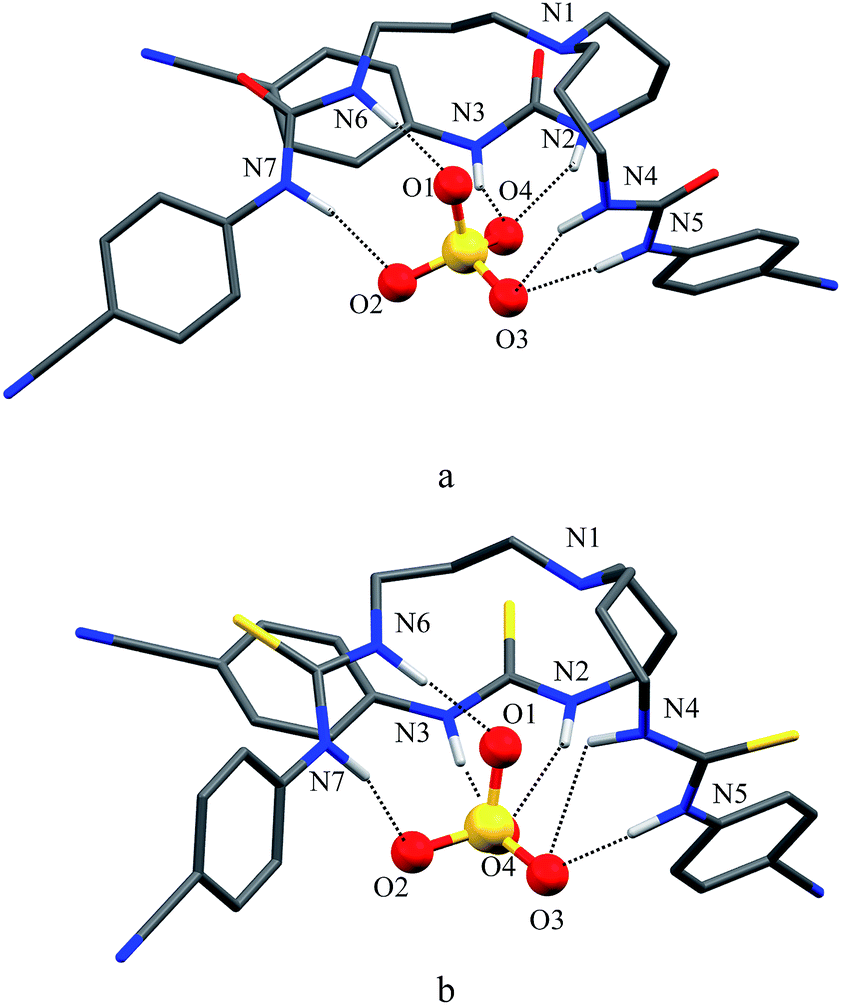
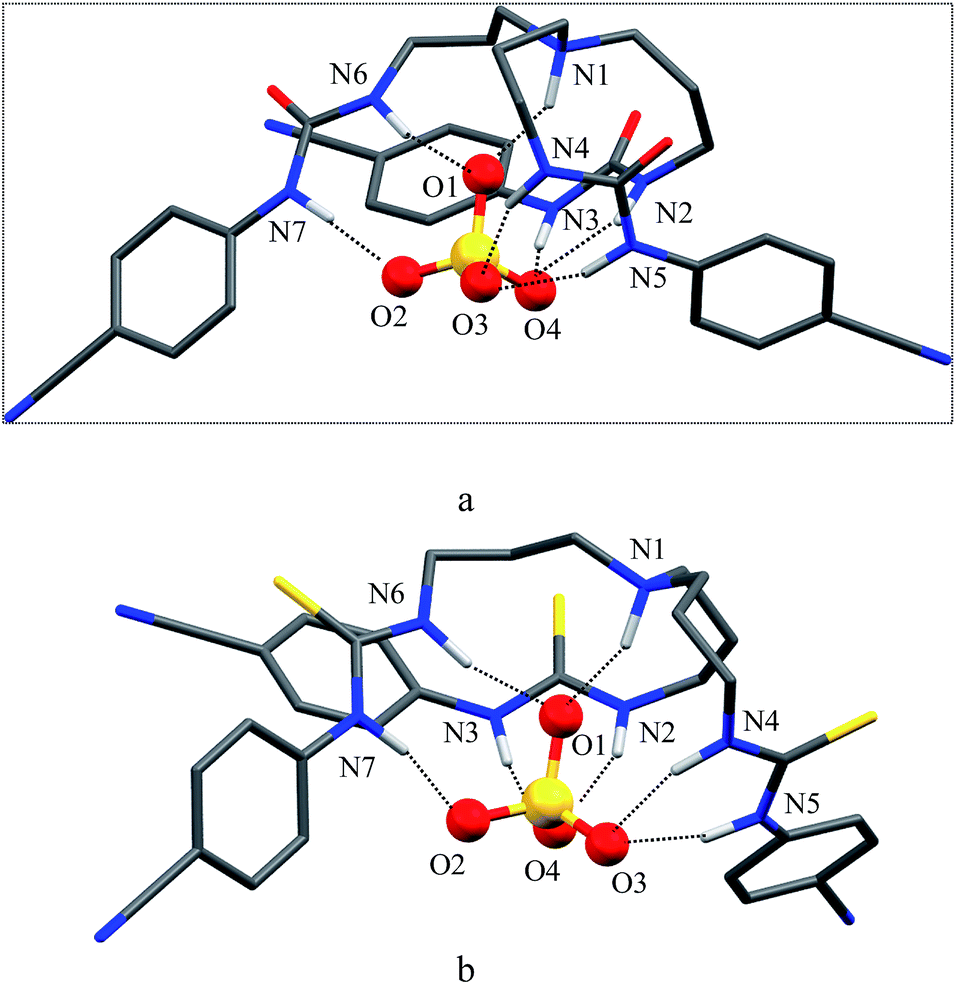
As shown in Scheme 1b and c, a strong electrostatic positive potential is created inside the cavities due to the presence of cyano-groups on aromatic rings, making them potential to host an anion. Fig. 8a and b show the optimized structures of the free receptors L1 and L2 in the solvent phase. For both cases, one NH group of an arm is hydrogen-bonded to oxygen/sulfate of another arm via NH⋯O/S interactions, thus creating a suitable cavity for guest. We previously observed similar hydrogen bonding interactions in a free p-cyanophenyl tripodal urea.23a The optimized structures of L1 and L2 complexes with SO42− are shown in Fig. 9, while those with HSO4− are displayed in Fig. 10. The corresponding hydrogen bonding distances are listed in Table 2. It is noteworthy to mention that both receptors are deformed in order to interact with SO42− or HSO4− through NH binding sites. In the sulfate complexes of L1 and L2, one sulfate is encapsulated within the cavity via a total six NH⋯O bonds, exhibiting a 1:1 binding for each case. Such a binding mode is in consistence with that observed in solution binding studies in DMSO-d6. Interestingly, in the optimized complexes with HSO4− as shown in Fig. 10, one proton from HSO4− is transferred to the bridgehead nitrogen of L1 or L2, providing an additional binding site as NH+ to the receptor. Thus the anion is held via a total of seven NH⋯O bonds, supporting the higher binding for HSO4− determined in solution by 1H NMR titrations. Such a proton transfer was previously observed experimentally23a as well as theoretically.34
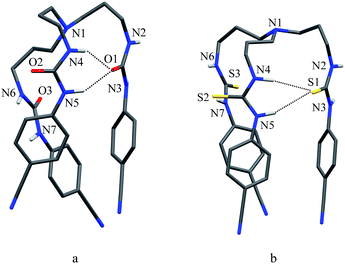 |
| | Fig. 8 Optimized structures of (a) L1 and (b) L2 calculated at the M06-2X/6-31G(d,p) level of theory. | |
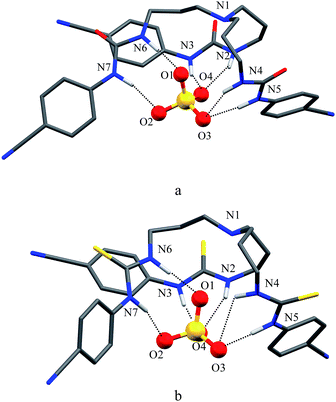 |
| | Fig. 9 Optimized structures of (a) [L1(SO4)]2− and (b) [L2(SO4)]2− calculated at the M06-2X/6-31G(d,p) level of theory. | |
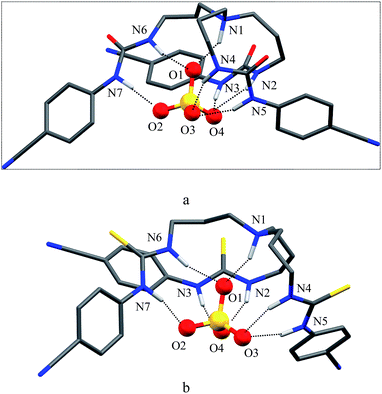 |
| | Fig. 10 Optimized structures of (a) [L1(HSO4)]− and (b) [L2(HSO4)]− calculated at the M06-2X/6-31G(d,p) level of theory. | |
Table 2 Hydrogen bonding interactions (Å, °) for the complexes of L1 and L2 with sulfate and hydrogen sulfate calculated with DFT at M06-2X/6-31G(d,p)
| Complex |
L1 |
L2 |
| D–H⋯A |
D⋯A (Å, °) |
D–H⋯A |
D⋯A (Å, °) |
| SO42− |
N2–H⋯O4 |
2.936 |
N2–H⋯O4 |
2.994 |
| N3–H⋯O4 |
2.758 |
N3–H⋯O4 |
2.742 |
| N4–H⋯O3 |
2.946 |
N4–H⋯O3 |
3.280 |
| N5–H⋯O3 |
2.962 |
N5–H⋯O3 |
2.785 |
| N6–H⋯O1 |
2.792 |
N6–H⋯O1 |
2.875 |
| N7–H⋯O2 |
2.937 |
N7–H⋯O2 |
2.827 |
| HSO4− |
N1–H⋯O1 |
2.738 |
N1–H⋯O1 |
2.705 |
| N2–H⋯O4 |
2.902 |
N2–H⋯O4 |
2.913 |
| N3–H⋯O4 |
2.835 |
N3–H⋯O4 |
2.813 |
| N4–H⋯O3 |
2.934 |
N4–H⋯O3 |
2.853 |
| N5–H⋯O3 |
2.902 |
N5–H⋯O3 |
2.854 |
| N6–H⋯O1 |
2.945 |
N6–H⋯O1 |
2.909 |
| N7–H⋯O2 |
2.812 |
N7–H⋯O2 |
2.819 |
Fluoride extraction studies
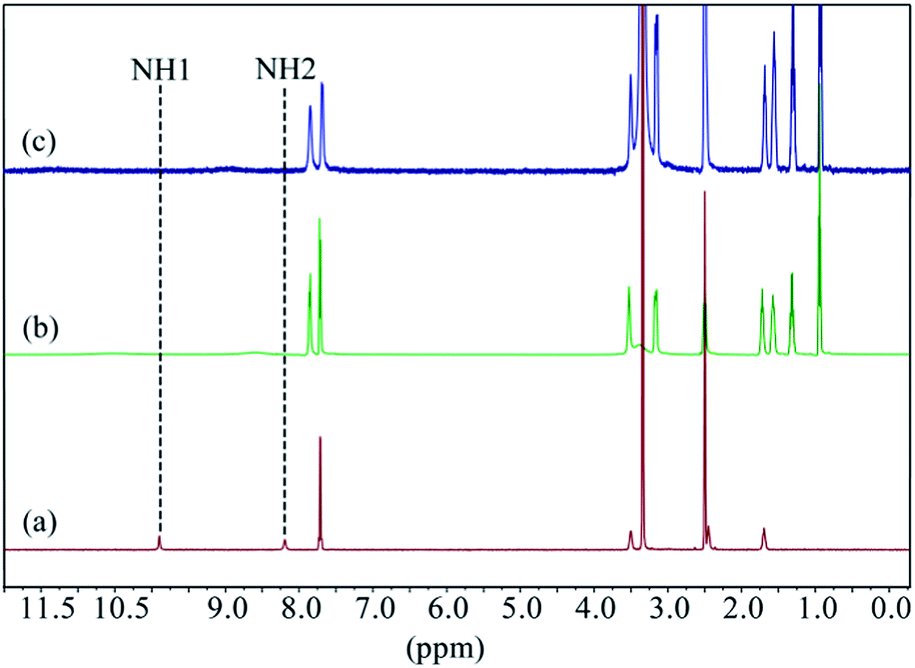
The fluoride extraction studies of L2 were successfully performed by liquid–liquid extraction technique using tetrabutylammonium iodide as the anion exchanger and the phase transfer agent, following the methods reported previously.21d,35 For a typical extraction experiment, distilled water solution (5 mL) of sodium fluoride (44.9 mg, 1 mmol) was added to the mixture of L2 (66.89 mg, 0.1 mmol) and tetrabutylammonium iodide (36.94 mg, 0.1 mmol) in chloroform (5 mL). The biphasic solution was mixed for 3 hours, and the two layers formed were separated. After the evaporation of the organic phase, the white solid product was washed with diethyl ether to remove the remaining tetrabutylammonium iodide, and collected after drying. The extraction efficiency was calculated gravimetrically as 99%. Fig. 11 represents the comparative 1H NMR spectra of the free receptor, extracted fluoride complex and L2 in presence of one equivalent of [n-Bu4N]+F− in DMSO-d6. The 1H NMR spectra of the extracted fluoride complex shows broadening and significant downfield shifting of NH peaks (Δδ = 0.67 and 0.43 ppm) with respect to receptor L2, which is very similar to the one obtained after adding one equivalent of [nBu4N]+F− to the receptor. This result clearly indicates the formation of fluoride complex after performing the liquid–liquid extraction by L2.
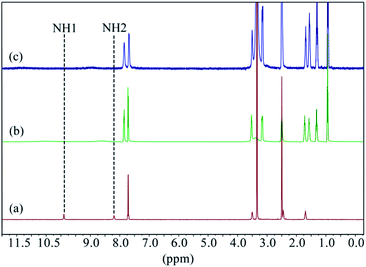 |
| | Fig. 11 Comparative 1H NMR spectra of (a) L2, (b) extracted fluoride–L2 complex, (c) L2 in the presence of one equivalent of [n-Bu4N]+F− in DMSO-d6. (H1 = CSNHAr and H2 = CH2NHCS). | |
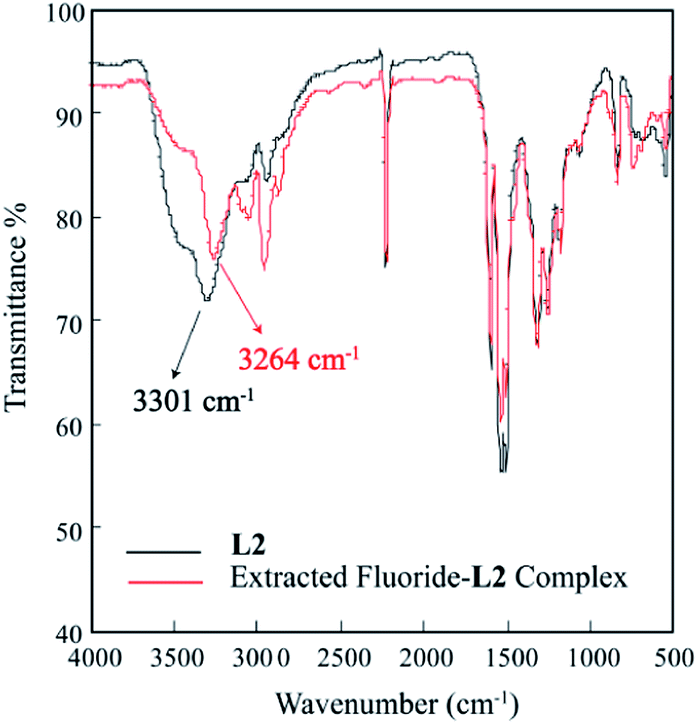
The solid state FT-IR analysis was also performed to examine the interactions of the receptor with fluoride in the extracted complex. The significant downward shift (Δν(N–H) = 37 cm−1) of broad NH's stretching frequency from 3301 cm−1 (L2) to 3264 cm−1 (extracted fluoride complex) was observed,36 suggesting the strong N–H⋯F− interactions between NH groups and the fluoride and ultimately deprotonation of the receptor by highly basic fluoride anion (Fig. 12).
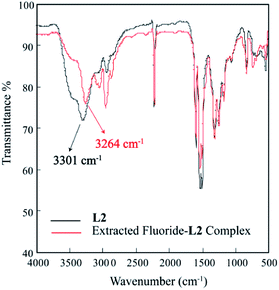 |
| | Fig. 12 Comparative FT-IR spectra of L2 (black) and extracted fluoride–L2 complex (red). | |
Conclusions
In summary, we report two simple acyclic tripodal urea/thiourea-based receptors containing propylene chain-induced cavity, showing strong selectivity for fluoride and dihydrogen phosphate in DMSO-d6. 1H NMR titrations suggest that both receptors show a similar binding trend for investigated anions following the order of: F− > H2PO4− > HCO3− > HSO4− > CH3COO− > SO42− > Cl−. Further 2D NOESY was used as a probe showing an obvious encapsulation of certain anions by the receptors via NH⋯anion interactions. Because of the enhanced acidity of NH's, the thiourea receptor showed higher binding affinity for anions as compared to the corresponding urea receptor. As opposed to the commonly observed binding trend for ethylene chain analogues20h,23a for HSO4− and SO42−, the present binding data suggests that the selectivity patterns of new tripodal receptors can be influenced by the chain length and cavity size, showing the higher binding constant for singly charged HSO4− than that for doubly charged SO42−. We assume that the higher binding affinity for HSO4− than SO42− is due to the acid–base interactions18 between the acidic HSO4− and the basic tertiary amine of urea/thiourea. This assumption was further supported by DFT calculations of the complexes with HSO4−, revealing that a proton from HSO4− is transferred to the tertiary nitrogen of each receptor, providing an additional binding site to a receptor. Further, the thiourea-based receptor has successfully been used for liquid–liquid extraction of biologically and environmentally important fluoride anion from aqueous phase with high efficiency.
Experimental
General
All reagents and solvents were purchased as reagent grade and were used without further purification. Nuclear magnetic resonance (NMR) spectra were recorded on a Varian Unity INOVA 500 FT-NMR. Chemical shifts for samples were measured in DMSO-d6 and calibrated against sodium salt of 3-(trimethylsilyl) propionic-2,2,3,3-d4 acid (TSP) as an external reference in a sealed capillary tube. NMR data were processed and analyzed with MestReNova Version 6.1.1-6384. The IR spectra was recorded on a Perkin Elmer-Spectrum One FT-IR spectrometer with KBr disks in the range of 4000–400 cm−1. The melting point was determined on a Mel-Temp (Electrothermal 120 VAC 50/60 Hz) melting point apparatus and was uncorrected. Mass spectral data were obtained at ESI-MS positive mode on a TSQ Quantum GC (Thermo Scientific). Elemental analysis was carried out by Columbia Analytical Services (Tucson, AZ 85714).
Synthesis
L1. Tris(3-aminopropyl)amine (526 μL, 2.52 mmol) was added to p-cyanophenyl isocyanate (1.12 g, 7.57 mmol) in dichloromethane (400 mL) at room temperature under constant stirring. The mixture was refluxed for 24 hours. A white precipitate formed and was collected by filtration. The residue was washed with dichloromethane and dried under vacuum for overnight to give the tripodal host (L1). Yield: 1.40 g, 90%. 1H NMR (500 MHz, DMSO-d6, TSP): δ 8.94 (s, 3H, Ar-NH), 7.62 (d, J = 8.50 Hz, 6H, ArH), 7.53 (d, J = 8.55 Hz, 6H, ArH), 6.37 (s, 3H, CH2NH), 3.10 (m, J = 6.20 Hz, 6H, NHCH2), 2.38 (t, J = 6.68 Hz, 6H, NCH2), 1.56 (m, J = 6.68 Hz, 6H, CH2CH2CH2). 13C NMR (125 MHz, DMSO-d6): δ 155.32 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 145.38 (Ar-C), 133.77 (Ar-CH), 119.62 (Ar-CN), 117.88 (Ar-CH), 102.96 (ArC-CN), 51.38 (NHCH2), 37.92 (NCH2), 27.68 (CH2CH2CH2). ESI-MS (+ve): m/z 620.4 [M]+. Mp: 210–211 °C. Anal. calcd for C33H36N10O3: C, 63.86; H, 5.85; N, 22.57. Found: C, 63.91; H, 5.96; N, 22.59. IR frequencies (KBr): ν(N–H) 3315 cm−1; ν(CN) 2207 cm−1; ν(CO) 1225 cm−1.
O), 145.38 (Ar-C), 133.77 (Ar-CH), 119.62 (Ar-CN), 117.88 (Ar-CH), 102.96 (ArC-CN), 51.38 (NHCH2), 37.92 (NCH2), 27.68 (CH2CH2CH2). ESI-MS (+ve): m/z 620.4 [M]+. Mp: 210–211 °C. Anal. calcd for C33H36N10O3: C, 63.86; H, 5.85; N, 22.57. Found: C, 63.91; H, 5.96; N, 22.59. IR frequencies (KBr): ν(N–H) 3315 cm−1; ν(CN) 2207 cm−1; ν(CO) 1225 cm−1.
L2. Tris(3-aminopropyl)amine 1 (526 μL, 2.52 mmol) was added to p-cyanophenyl isothiocyanate (1.24 g, 7.57 mmol) in dichloromethane (400 mL) at room temperature under constant stirring. The mixture was refluxed for 24 hours. A white precipitate formed and was collected by filtration. The residue was washed with dichloromethane and dried under vacuum for overnight to give the tripodal host (L2). Yield: 1.24 g, 73%. 1H NMR (500 MHz, DMSO-d6, TSP): δ 9.86 (s, 3H, Ar-NH), 8.17 (s, 3H, CH2NH), 7.71 (s, 12H, ArH), 3.51 (broad s, 6H, NHCH2), 2.45 (t, J = 6.97 Hz, 6H, NCH2), 1.70 (m, J1 = 6.90 Hz, J2 = 7.15 Hz, 6H, CH2CH2CH2). 13C NMR (125 MHz, DMSO-d6): δ 179.87 (CS), 143.99 (Ar-C), 132.80 (Ar-CH), 121.21 (Ar-CN), 119.10 (Ar-CH), 104.58 (ArC-CN), 51.06 (NHCH2), 42.57 (NCH2), 25.71 (CH2CH2CH2). ESI-MS (+ve): m/z 668.7 [M]+. Mp: 120 °C. Anal. calcd for C33H36N10S3: C, 59.25; H, 5.42; N, 20.94. Found: C, 59.31; H, 5.56; N, 20.98. IR frequencies (KBr): ν(N–H) 3301 cm−1; ν(CN) 2231 cm−1; ν(CS) 1176 cm−1.
NMR binding studies
Binding constants were obtained by 1H NMR titrations of L1 and L2 using[n-Bu4N]+A (F−, Cl−, Br−, I−, ClO4−, NO3−, HSO4−, H2PO4−, CH3COO−, HCO3− and SO42−) in DMSO-d6. Initial concentrations were [host]0 = 2 mM, and [anion]0 = 20 mM. Sodium salt of 3-(trimethylsilyl)-propionic-2,2,3,3-d4 acid (TSP) in DMSO-d6 was used as an external reference in a capillary tube. Each titration was performed by 13 measurements at room temperature. The association constant K was calculated by fitting of several independent NMR signals with a 1:1 association model using Sigma Plot software, from the following equations: Δδ = ([A]0 + [L]0 + 1/K − (([A]0 + [L]0 + 1/K)2 − 4[L]0[A]0)1/2)Δδmax/2[L]0 (where, L = receptor and A = anion). Error limit in K was less that 10%.
DFT calculations
DFT calculations were performed using the M06-2X hybrid functional which incorporates an improved description of dispersion energies. From the equilibrium geometry, anion was added at the center of the receptor's cavity. The geometries of the anion–receptor complexes were then optimized at the M06-2X/6-31g(d,p) level of theory in gas phase and also in DMSO solvent (dielectric constant = 46.8). All the calculations were carried out using Gaussian 09 package of programs.32
Fluoride extraction studies
Distilled water solution (5 mL) of sodium fluoride (44.9 mg, 1 mmol) was added to the mixture of L2 (66.89 mg, 0.1 mmol) and tetrabutylammonium iodide (36.94 mg, 0.1 mmol) in chloroform (5 mL). The biphasic solution was mixed for 3 hours. Then the two layers were separated. After solvent evaporation of the organic phase, the white solid product was washed with diethyl ether to remove the remaining tetrabutylammonium iodide, and collected after drying. Yield: 92.3 mg, 99%. 1H NMR (500 MHz, DMSO-d6, TSP): δ 10.53 (broad s, 3H, Ar-NH), 8.60 (broad s, 3H, CH2NH), 7.85 (d, J = 8.10 Hz, 6H, ArH), 7.71 (d, J = 8.65 Hz, 6H, ArH), 3.53 (broad s, 6H, NHCH2), 3.17 (t, J = 8.32 Hz, 8H, NCH2CH2CH2CH3), 2.50 (broad s, 6H, NCH2), 1.72 (m, J = 6.65 Hz, 6H, CH2CH2CH2), 1.57 (m, 8H, NCH2CH2CH2CH3), 1.32 (m, 8H, NCH2CH2CH2CH3), 0.94 (t, J = 7.32 Hz, 12H, NCH2CH2CH2CH3). IR frequencies (KBr): ν(N–H) 3264 cm−1; ν(CN) 2231 cm−1; ν(CS) 1176 cm−1.
Acknowledgements
The National Science Foundation is acknowledged for a CAREER award (CHE-1056927) to M.A.H. Analytical core facility at Jackson State University was supported by the National Institutes of Health (G12RR013459). Ismet Basaran acknowledges the Scientific and Technological Research Council of Turkey (TUBITAK) for financial support during his stay at Jackson State University. The authors thank for support of the NSF CREST Interdisciplinary Nanotoxicity Center NSF-CREST - Grant # HRD-0833178 for the computational work described in this paper.
Notes and references
- F. P. Schmidtchen and M. Berger, Chem. Rev., 1997, 97, 1609 CrossRef CAS PubMed.
- C. R. Bondy and S. J. Loeb, Coord. Chem. Rev., 2003, 240, 77 CrossRef CAS.
- P. A. Gale, Chem. Commun., 2005, 3761 RSC.
- C. Caltagirone and P. A. Gale, Chem. Soc. Rev., 2009, 38, 520 RSC.
- K. Bowman-James, A. Bianchi and E. Garcia-Espana, Anion Coordination Chemistry, Wiley-VCH, New York, 2011 Search PubMed.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486 CrossRef CAS.
- M. A. Hossain, Curr. Org. Chem., 2008, 12, 1231 CrossRef CAS.
-
(a) G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Structures, Springer-Verlag, Berlin, 1991 Search PubMed;
(b) J. L. Sessler and J. Jayawickramarajah, Chem. Commun., 2005, 1939 RSC;
(c) M. A. Mateos-Timoneda, M. Crego-Calama and D. N. Reinhoudt, Chem. Soc. Rev., 2004, 33, 363 RSC.
-
(a) C. R. Bondy and S. J. Loeb, Coord. Chem. Rev., 2003, 240, 77 CrossRef CAS;
(b) M. A. Hossain, J. M. Llinares, D. Powell and K. Bowman-James, Inorg. Chem., 2001, 40, 2936 CrossRef CAS;
(c) S. O. Kang, V. W. Day and K. Bowman-James, Org. Lett., 2009, 11, 3654 CrossRef CAS PubMed;
(d) R. Begum, S. O. Kang and K. Bowman-James, Angew. Chem., Int. Ed., 2006, 45, 7882 CrossRef PubMed.
-
(a) M. A. Hossain, S. K. Kang, L. M. Llinares, D. Powell and K. Bowman-James, Inorg. Chem., 2003, 42, 5043 CrossRef CAS PubMed;
(b) Y. Inoue, T. Kanbara and T. Yamamoto, Tetrahedron Lett., 2003, 44, 5167 CrossRef CAS.
-
(a) V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889 RSC;
(b) R. Custelcean, Chem. Commun., 2008, 295 RSC;
(c) C. N. Carroll, O. B. Berryman, C. A. Johnson II, L. N. Zakharov, M. M. Haley and D. W. Johnson, Chem. Commun., 2009, 2520 RSC;
(d) D. R. Turner, M. J. Paterson and J. W. Steed, J. Org. Chem., 2006, 71, 1598 CrossRef CAS PubMed;
(e) M. Olivari, C. Caltagirone, A. Garau, F. Isaia, M. E. Light, V. Lippolis, R. Montis and M. Andrea Scorciapino, New J. Chem., 2013, 37, 663 RSC;
(f) R. Dutta and P. Ghosh, Chem. Commun., 2014, 50, 10538 RSC.
-
(a) Z. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187 RSC;
(b) A.-F. Li, J.-H. Wang, F. Wang and Y.-B. Jiang, Chem. Soc. Rev., 2010, 39, 3729 RSC.
-
(a) J. L. Sessler, S. Camiolo and P. A. Gale, Coord. Chem. Rev., 2003, 240, 17 CrossRef CAS;
(b) R. Custelcean, L. H. Delmau, B. A. Moyer, J. L. Sessler, W.-S. Cho, D. Gross, G. W. Bates, S. J. Brooks, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2005, 44, 2537 CrossRef CAS PubMed;
(c) J. L. Sessler, D. E. Gross, W.-S. Cho, V. M. Lynch, F. P. Schmidtchen, G. W. Bates, M. E. Light and P. A. Gale, J. Am. Chem. Soc., 2006, 128, 12281 CrossRef CAS PubMed;
(d) G. W. Bates, L. M. E. Triyanti, M. Albrecht and P. A. Gale, J. Org. Chem., 2007, 72, 8921 CrossRef CAS PubMed.
-
(a) K.-J. Chang, D. Moon, M. S. Lah and K.-S. Jeong, Angew. Chem., Int. Ed., 2005, 44, 7926 CrossRef CAS PubMed;
(b) K.-J. Chang, M.-K. Chae, C. Lee, J.-Y. Lee and K.-S. Jeong, Tetrahedron Lett., 2006, 47, 6385 CrossRef CAS PubMed;
(c) C. Caltagirone, P. A. Gale, J. R. Hiscock, S. J. Brooks, M. B. Hursthouse and M. E. Light, Chem. Commun., 2008, 3007 RSC.
- M. A. Hossain, R. A. Begum, V. W. Day and K. Bowman-James, Amide and Urea-Based Receptors, Supramolecular Chemistry: From Molecules to Nanomaterials, John Wiley & Sons, Ltd, 2012 Search PubMed.
- E. Fan, S. A. Van Arman, S. Kincaid and A. D. Hamilton, J. Am. Chem. Soc., 1993, 115, 369 CrossRef CAS.
- M. Boiocchi, L. Del Boca, D. Esteban-Gomez, L. Fabbrizzi, M. Licchelli and E. Monzani, J. Am. Chem. Soc., 2004, 126, 16507 CrossRef CAS PubMed.
- G. W. Bates, P. A. Gale and M. E. Light, Chem. Commun., 2007, 2121 RSC.
- C. A. Johnson, O. B. Berryman, A. C. Sather, L. N. Zakharov, M. M. Haley and D. W. Johnson, Cryst. Growth Des., 2009, 9, 4247 CAS.
-
(a) R. Custelcean, B. A. Moyer and B. P. Hay, Chem. Commun., 2005, 5971 RSC;
(b) B. Wu, J. Liang, J. Yang, C. Jia, X.-J. Yang, H. Zhang, N. Tang and C. Janiak, Chem. Commun., 2008, 1762 RSC;
(c) I. Ravikumar, P. S. Lakshminarayanan, M. Arunachalam, E. Suresh and P. Ghosh, Dalton Trans., 2009, 4160 RSC;
(d) K. J. Bell, A. N. Westra, R. J. Warr, J. Chartres, R. Ellis, C. C. Tong, A. J. Blake, P. A. Tasker and M. Schroder, Angew. Chem., Int. Ed., 2008, 47, 1745 CrossRef CAS PubMed;
(e) R. Custelcean, A. Bock and B. A. Moyer, J. Am. Chem. Soc., 2010, 132, 7177 CrossRef CAS PubMed;
(f) M. Li, B. Wu, F. Cui, Y. Hao, X. Huang and X.-J. Yang, Z. Anorg. Allg. Chem., 2011, 637, 2306 CrossRef CAS;
(g) N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernandez, R. Perez-Tomas and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136 CrossRef CAS PubMed;
(h) S. K. Dey, R. Chutia and G. Das, Inorg. Chem., 2012, 51, 1727 CrossRef CAS PubMed;
(i) P. Bose, R. Dutta and P. Ghosh, Org. Biomol. Chem., 2013, 11, 4581 RSC;
(j) A. Basu and G. Das, J. Org. Chem., 2014, 79, 2647 CrossRef CAS PubMed.
-
(a) N. Busschaert, P. A. Gale, C. J. E. Haynes, M. E. Light, S. J. Moore, C. C. Tong, J. T. Davis Jr and W. A. Harrell, Chem. Commun., 2010, 46, 6252 RSC;
(b) N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernández, R. Pérez-Tomás and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136 CrossRef CAS PubMed;
(c) S. K. Dey and G. Das, Dalton Trans., 2012, 41, 8960 RSC;
(d) P. Bose, R. Dutta, S. Santra, B. Chowdhury and P. Ghosh, Eur. J. Inorg. Chem., 2012, 5791 CrossRef CAS;
(e) I. Marques, A. R. Colaço, P. J. Costa, N. Busschaert, P. A. Gale and V. Félix, Soft Matter, 2014, 10, 3608 RSC;
(f) D. Horvat, M. E. Khansari, A. Pramanik, M. R. Beeram, T. J. Kuehl, M. A. Hossain and M. N. Uddin, Int. J. Environ. Res. Public Health, 2014, 11, 7456 CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS.
-
(a) A. Pramanik, B. Thompson, T. Hayes, K. Tucker, D. R. Powell, P. V. Bonnesen, E. D. Ellis, K. S. Lee, H. Yu and M. A. Hossain, Org. Biomol. Chem., 2011, 9, 4444 RSC;
(b) A. Pramanik, D. R. Powell, B. M. Wong and M. A. Hossain, Inorg. Chem., 2012, 51, 4274 CrossRef CAS PubMed.
-
(a) T. H. Russ, A. Pramanik, M. E. Khansari, B. M. Wong and M. A. Hossain, Nat. Prod. Commun., 2012, 7, 301 CAS;
(b) M. Emami Khansari, K. D. Wallace and M. A. Hossain, Tetrahedron Lett., 2014, 55, 438 CrossRef CAS PubMed;
(c) A. Pramanik, M. Emami Khansari, D. R. Powell, F. R. Fronczek and M. A. Hossain, Org. Lett., 2014, 16, 366 CrossRef CAS PubMed.
- M. Boiocchi, L. D. Boca, D. E. Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, J. Am. Chem. Soc., 2004, 126, 16507 CrossRef CAS PubMed.
- H. J. Schneider, R. Kramer, S. Simova and U. Schneider, J. Am. Chem. Soc., 1988, 110, 6442 CrossRef CAS.
- K. R. Dey, B. M. Wong and M. A. Hossain, Tetrahedron Lett., 2010, 51, 1329 CrossRef CAS PubMed.
- M. A. Hossain, J. M. Llinares, D. Powell and K. Bowman-James, Inorg. Chem., 2001, 40, 2936 CrossRef CAS.
- N. E. Schore and P. C. Vollhardt, Organic Chemistry, Structure and Function, W.H. Freeman & Company, New York, 5th edn, 2007 Search PubMed.
- F. Werner and H. J. Schneider, Helv. Chim. Acta, 2000, 83, 465 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Chem. Phys. Lett., 2011, 502, 1 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, version D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327 CrossRef CAS.
- M. A. Rhaman, L. Ahmed, J. Wang, D. Powell, J. Leszczynski and M. A. Hossain, Org. Biomol. Chem., 2014, 12, 2045 CAS.
-
(a) C. J. Fowler, T. J. Haverlock, B. A. Moyer, J. A. Shriver, D. E. Gross, M. Marquez, J. L. Sessler, M. A. Hossain and K. Bowman-James, J. Am. Chem. Soc., 2008, 130, 14386 CrossRef CAS PubMed;
(b) B. A. Moyer, F. V. Sloop Jr, C. J. Fowler, T. J. Haverlock, H.-A. Kang, L. H. Delmau, D. M. Bau, M. A. Hossain, K. Bowman-James, J. A. Shriver, N. L. Bill, D. E. Gross, M. Marquez, V. M. Lynch and J. L. Sessler, Supramol. Chem., 2010, 22, 653 CrossRef CAS;
(c) C. Jia, B. Wu, S. Li, X. Huang, Q. Zhao, Q.-S. Li and X.-J. Yang, Angew. Chem., 2011, 123, 506 CrossRef;
(d) C. Jia, B. Wu, S. Li, X. Huang, Q. Zhao, Q.-S. Li and X.-J. Yang, Angew. Chem., Int. Ed., 2011, 50, 486 CrossRef CAS PubMed.
- S. Edrah, J. Appl. Sci. Res., 2010, 4, 1014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of the receptors, 1H NMR titration spectra and binding isotherms, Job's Plots, additional 2D NOESY NMR experiments, 1H NMR spectra for fluoride extraction studies. See DOI: 10.1039/c5ra01315a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Aemal Nafisa,
Jing Wanga,
Jerzy Leszczynski*a and
Md. Alamgir Hossain*a
ab,
Aemal Nafisa,
Jing Wanga,
Jerzy Leszczynski*a and
Md. Alamgir Hossain*a