DOI:
10.1039/C4RA00985A
(Review Article)
RSC Adv., 2014,
4, 21379-21404
The importance of various anchoring groups attached on porphyrins as potential dyes for DSSC applications
Received
4th February 2014
, Accepted 29th April 2014
First published on 30th April 2014
Abstract
In the design of new chromophores, with high efficiency, chemical stability and low cost materials for dye sensitized solar cells, porphyrin macrocycles could play a very important role. Their successful use in nature during photosynthesis must be the inspiration for new artificial antenna systems. Porphyrins offer an excellent platform for building such multi-chromophoric systems to self-assemble because of the availability of several substituent sites and their intrinsic spectroscopic properties. Specifically, their high absorption ability in the visible region can be extended, and electron donor and anchoring groups with high chemical affinity to the cell should be part of the new design. This review provides a summary of some of the most important developments and approaches that are used in order to improve the light collection efficiency of DSSCs based on porphyrin hybrid derivatives. For this reason we have attempted to describe the developments in the DSSCs of various porphyrin dyes with different anchoring groups linked through either meso or β-positions. Also, the influence of the anchoring groups in the cell performance is discussed. Studies containing chromophores other than porphyrin derivatives are not included in this work.
 K. Ladomenou | Kalliopi Ladomenou in 1998 obtained her B. Sc. in chemistry at University of Crete (Greece) and in 2002 obtained her Ph. D. at University of Liverpool (UK) with Dr Richard P. Bonar-Law. Her main work was the design, synthesis and binding studies of podand-type porphyrin molecules for carbohydrate recognition. She held a postdoctoral position at University of Edinburgh (UK) with Prof. David A. Leigh, where she synthesized novel chiral macrocycles with possible applications in catalysis. Then, she moved to University of Crete in the laboratory of Bioinorganic chemistry with Prof. Athanassios G. Coutsolelos. She is currently a researcher at the Chemistry Department of University of Crete. Her main research interest is the synthesis and physical-chemical characterization of mainly porphyrin based chromophores with potential use in dye-sensitized solar cells. Also, her research includes the design, synthesis and study of model compounds for artificial photosynthesis. |
 G. D. Sharma | G. D. Sharma was born in 1959. He received his Ph. D. degree in Physics from Indian Institute of Technology, New Delhi, India. in 1985. After that he joined as Assistant professor in November 1985 at Department of Physics, JNV University, Jodhpur and promoted to the post of Professor in same Department in 2004. During 1990–1991, he worked as post doctoral fellow in Department of Electrical Engineering, The state University of New Jersey, USA and worked on organic photovoltaic devices. At present, he is working as Director (Academic and Research) JEC group of Colleges, Jaipur Engineering College campus, Kukas, Jaipur. His research area includes organic solar cells based on conjugated polymers and small molecules dye sensitized solar cells. He had research national and international collaboration, published more than 170 research papers in international journals with high impact factor. 14 students have completed their Ph. D degree under his supervision. |
 A. G. Coutsolelos | Professor Dr Athanassios G. Coutsolelos received his B. Sc. in Chemistry in 1979 from the University of Thessaloniki, Greece and his Ph. D. from the University of Dijon in France in 1985, after completing his thesis in the Laboratory of Heterocyclic Chemistry in the field of new materials with gallium porphyrins. In the period 1985–1986 he worked as a postdoctoral fellow in the Department of Chemistry, University of Texas at Houston, USA. In 1988 joined the Department of Chemistry, University of Crete, as assistant professor, where he is currently Professor in Bioinorganic Chemistry. In the mid time, in summer of 1992 and September 1996 he spent two months as Visiting Researcher Laboratory of Coordination Chemistry (LCC), Centre National pour la Recherche Scientifique (CNRS), Toulouse, France. In spring of 1994 and fall of 2000 he was invited as Visiting Professor Institut de la Chimie Moleculaire d'Orsay (ICMO), Laboratoire de Chimie Bioinorganique, Université Paris-Sud, Orsay, France and from July 1995 to June 1996 he spend a year as Visiting Professor (Sabbatical Year), University of Notre Dame, Department of Chemistry and Biochemistry, Notre Dame, USA. His research interests include metalloporphyrins as very close proximity π-systems, metal–metal bonding and, one-dimensional materials with porphyrinic units, metalloporphyrins in biocompatible catalytic conversion of polyene-polymers and modeling of active centers of biological processes, new metalloporphyrinic complexes with DNA affinity (synthesis, physical-chemical characterization and applications), preparation of new chelated agents for cations with therapeutic interest as well hybrid derivatives porphyrin–nanohorns and self assembled materials as models for artificial photosynthesis. He has participated in many projects funded by EU and other national and international organizations. He is the author of more than 95 journal publications and more than 200 oral presentations. |
1. Introduction
The increasing demand for energy and the continuous depletion of fossil fuels creates an urgent need to discover alternative energy supplies. Hence, further research to explore alternative, safe and renewable sources of energy is extremely important. Among all renewable energy technologies, photovoltaics, which utilize solar energy, are believed to be the most promising ones. Solar energy, one of the most abundant and renewable sources of energy, plays an important role, as the amount of energy emitted from the sun to earth is several thousand times higher than the global requirement. At present most of the solar cells are fabricated from silicon. However, this type of solar cells has certain limitations due to the difficulties related to large scale production and high fabrication cost.
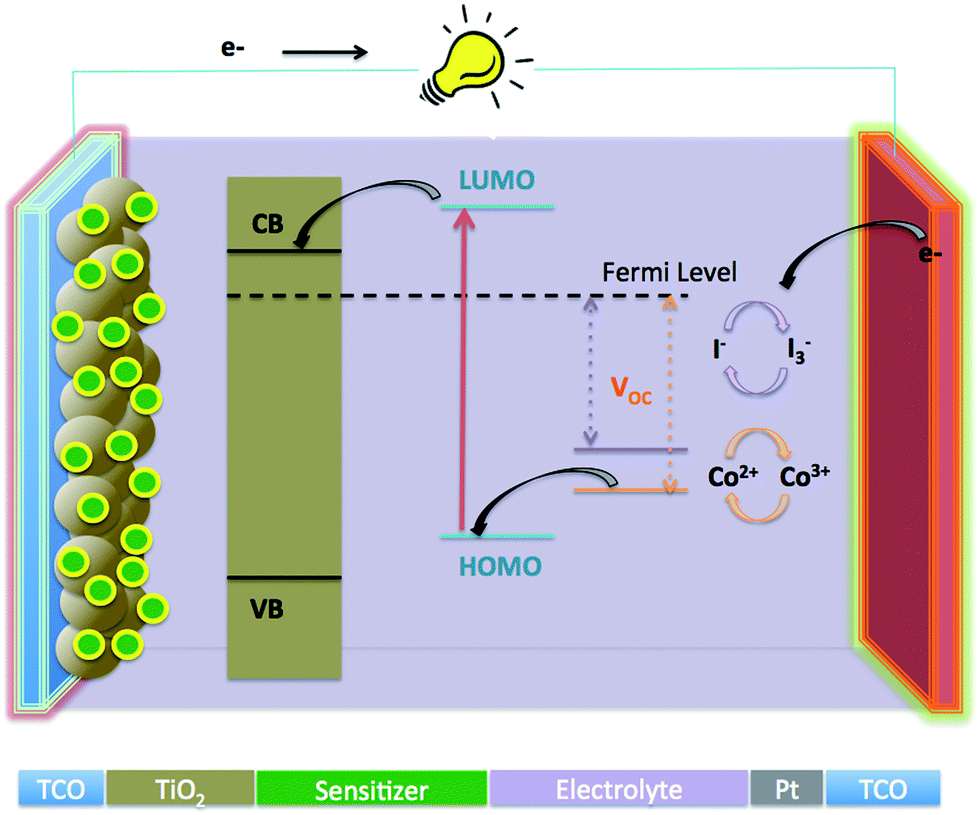
Since their first report of Grätzel research group in 1991,1,2 dye sensitized solar cells (DSSCs) have attracted lots of attention globally because of their low cost and environmentally friendly properties. These solar cells have the potential to replace the present conventional solar cells based on silicon.3–5 The operation of the typical solar cell is illustrated in Fig. 1, which consists of a dye sensitized mesoporous working electrode (generally wide energy band gap metal oxide semiconductor TiO2 or ZnO anode), a counter electrode (Pt coated cathode) and an electrolyte (iodide based or cobalt complex, redox mediator), filling the space between the anode and cathode. When the DSSC is illuminated, electrons of the sensitizer are excited to the lowest unoccupied molecular orbital (LUMO), then are rapidly injected into the conduction band of metal oxide semiconductor (TiO2), and subsequently transferred to the platinized counter electrode through the external circuit. The holes are reduced by the redox couple, either I3−/I− or Co2+/Co3+, and the oxidized dye thereby regenerated by the I− spices or Co2+ complex to produce I3− spices or a Co3+ complex. Finally, the diffusion of oxidized spices i.e. I3− or a Co3+ complex, to the surface of the cathode complete the circuit.
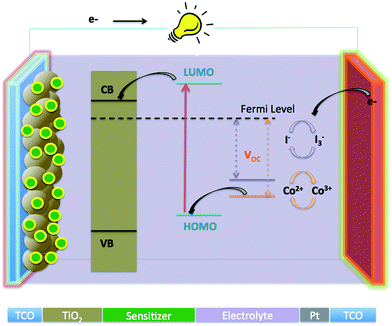 |
| | Fig. 1 Systematic illustration of components and operation of dye sensitized solar cell. | |
A plethora of photophysical sequences occur in a DSSC, all of which are dependent on the three main components of the DSSC: the light absorber (dye), the electron transport agent (wide band gap nanocrystalline semiconductor, mostly TiO2 or ZnO), a hole transport agent (redox couple in electrolyte). For an efficient DSSC, all three components require fine tuning.6 In particular, concerning the dye, there are certain features required for efficient performance such as suitable HOMO and LUMO levels for charge injection into TiO2, dye regeneration from electrolyte, an anchoring group to attach to the semiconducting material, and a broad absorption in the visible and NIR region for light harvesting, as well as photostability and solubility.7
The DSSCs based on nanocrystalline TiO2 photoanode sensitized with polypyridyl ruthenium complexes have yielded power conversion efficiency (PCE) over 11%.8–13 However, high cost, scarcity and environmental issues concerning the ruthenium complex limits their affordable commercial applications, and has prompted the exploration of lower cost and safer sensitizers. Consequently, scientists from all over the world have put a significant amount of effort into the design of new efficient DSSCs, using either no-metal or inexpensive-metals, and sensitizers, with high molar extinction coefficients.5,14–20 Moreover, this new DSSCs, are prepared using facile synthesis, flexible structural modification and ideal photovoltaic performances. The PCE's of the DSSCs sensitized with metal free dyes were reported to be 9–10% during last five years. The best performing DSSC sensitizer is one with an organic dye (C219) that achieved a PCE of 10.3%.21 This value however, remains lower than the state of the art ruthenium complex dyes, because of its weak light absorption in the near infrared region.
Therefore, it is significant to develop efficient sensitizers over a very wide spectral range for DSSCs. Inspired by the efficient energy and electron transfer behavior in the light harvesting antenna of natural systems,22 porphyrin derivatives23,24 and analogues25,26 have been widely used in photovoltaic devices. Porphyrin based compounds have several intrinsic advantages such as good photostability and rigid molecular structure with large absorption coefficients in visible region.16,27,28 Their photophysical and electrochemical properties can be tuned by peripheral substituents and inner metal complexations, by taking advantage of their multiple reactive sites, including four meso and eight β-positions.29–35 A drawback of porphyrins is the poor light-harvesting properties at wavelengths around 450 nm and above 600 nm. Elongation of strong electron-donating and electron-withdrawing groups can shift and broad the absorption of porphyrins making it possible to increase the light-harvesting property and the resulting DSSC efficiency. Therefore, a push–pull type porphyrin dye with diarylamine (donor) and ethynyl-benzoic acid (acceptor) substituents at meso positions has been reported to achieve a PCE of 12.3%,36 which remains the highest value for DSSCs so far and superior than Ru based DSSCs. This encouraging achievement stimulates the development of further porphyrin sensitizers to promote the performance of the DSSCs.
In DSSCs the presence of an anchoring group is essential to graft the dye on a TiO2 surface, in order to achieve fast electron injection into the TiO2 semiconductor.37 The efficiency of the electron-transfer step at the dye–semiconductor interface is highly dependent, among numerous other factors, on the way the chromophore is attached to the surface.38–40 Therefore, different porphyrin anchoring groups have been used and researchers have studied their effect on the TiO2 surface.41–43
This review provides a summary of some of the most important developments and approaches that were used in order to improve the light collection efficiency of DSSCs based on porphyrin hybrid derivatives. Studies concerning various synthetic approaches of porphyrins are not described. This manuscript is mainly focused on the influence of the various anchoring groups used for the attachment of the porphyrin derivatives to the TiO2 electrode.
2. Carboxylic acids as anchoring groups
Carboxylic acid present in the meso and β-positions of a porphyrin ring is an anchoring group that has been extensively studied. At the meso-position, two main classes of linking moieties have been employed most often, the classic meso-substituted benzoic acid linking and the direct attachment of a meso-alkynylbenzoic acid moiety. For DSSCs employing a β-position linking group the conjugated ones have been mostly studied in the literature.
Carboxylic acid groups can form ester linkages with the surface of the metal oxide to provide a strongly bound dye and good electronic communication between the two parts. However, the link can be hydrolyzed through the presence of water, an important factor in terms of the stability of the cell. For carboxylate groups, bidentate connections have been shown to be the preferred binding mode on TiO2.44–46
2.1 Meso-substituted benzoic acids
The concept for the design of meso ethynyl linked porphyrins was first given by Anderson47 and Therien.48 Before the discovery of DSSC, the ability of porphyrin to sensitize TiO2 semiconductor was first demonstrated by Grätzel in 1987.49 However, Kay and Grätzel first introduced a porphyrin as a sensitizer in DSSC in 1993. A substituted porphyrin with propionic acid as anchoring group achieved an overall efficiency of 2.6% (Jsc = 9.4 mA cm−2, Voc = 0.52 V).50
Cherian and Wamser51 in 2000 reported the first meso-substituted DSSC with 3.5% (Jsc = 0.133 mA cm−2, Voc = 0.36 V, and FF = 0.62) efficiency based on a free-base porphyrin 1 (Fig. 2). That report also analyzed the adsorption properties of 1 on TiO2 using XPS and resonance Raman spectroscopy to elucidate the binding characteristics of 1 and reported that low concentrations of dye in the adsorption solution (∼0.1 mM) are important to avoid dye aggregation.
 |
| | Fig. 2 Molecular structures of porphyrins 1–3. | |
One key issue to consider in designing porphyrin dyes relates to the position of the anchoring group in relation to the plane of macrocycle π-system. The presence of carboxyl anchoring groups attached to the meso-phenyl ring of the porphyrin has little influence on the ground-state electronic nature of the dye, because of the orthogonal orientation of the meso-phenyl ring.52–54 However, a few studies have shown that the positioning of carboxyl group vary the performance of the solar cells.42,51,55–57 For example, Campbell et al. have reported a 5-fold increase in the short circuit photocurrent (Jsc) and a significantly higher open circuit photovoltage (Voc) on changing the position of the COOH groups from the para (in p-ZnTCPP 2) to the meta position (in m-ZnTCPP 3) (Fig. 2).15 This effect was attributed to a more efficient electron injection from the meta porphyrin, which binds flat on the TiO2 surface, compared to that of the para, which binds vertically (Fig. 3).15
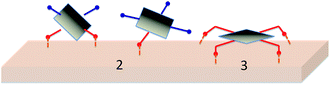 |
| | Fig. 3 Different binding modes of porphyrins 2 and 3 onto TiO2 surface. | |
In 2007 Galoppini and coworkers55 studied tetrachelated zinc porphyrins with four meta-substituted linkers at four meso positions of the porphyrin to give inside on how the distance and attachment of the sensitizing chromophore on colloidal TiO2 and ZnO films influence interfacial processes and solar cell efficiencies. The meta substitution favored a planar binding mode to the metal oxide surfaces, as determined by a combination of studies that included IR, UV, and solar cell efficiencies on TiO2. All studies indicated that only p-ZnTCPP 2 aggregated, suggesting close packing of the dye molecules on the semiconductor surface. Aggregation effects were not observed for the meta porphyrins. The greater efficiency of the rigid planar meta-substituted systems was explained in terms of a greater charge injection into the TiO2 semiconductor from rings that lie flat, and closer, to the surface. In the same study it was observed that the main binding modes for this class of molecules are a chelating and/or a bidentate bridging of the carboxylate on the TiO2 surface (Fig. 4).45,58–60
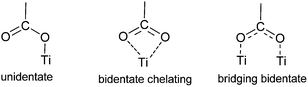 |
| | Fig. 4 Main binding modes of the carboxylate group to TiO2. | |
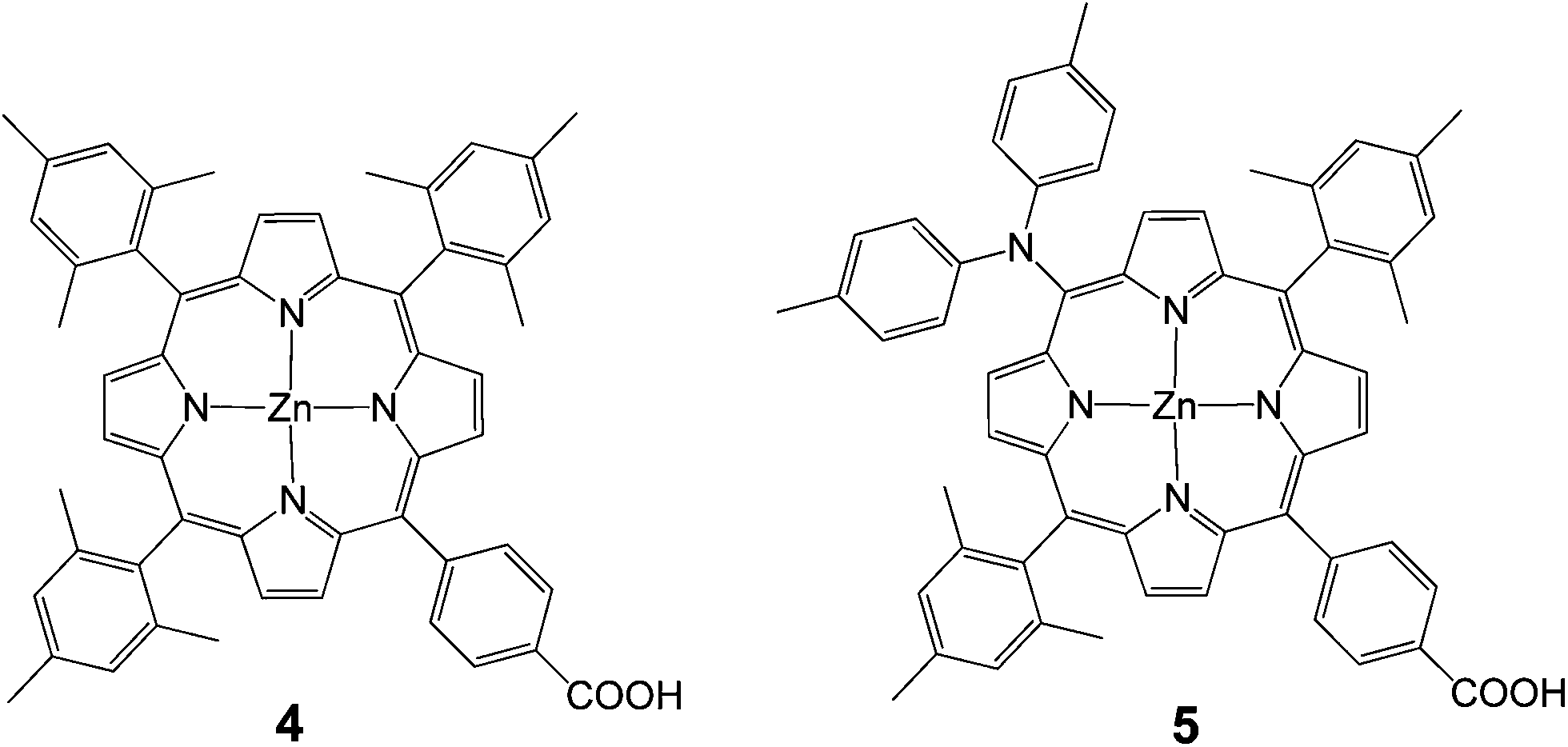
In 2009, Imahori et al. studied a series of monocarboxyphenyl triaryl porphyrins in DSSCs.61 Zn porphyrin 4 (Fig. 5) yielded the highest efficiency of 4.6% (Jsc = 9.4 mA cm−2, Voc = 0.76 V, and FF = 0.64) in a standard DSSC. As a part of their study, they optimized the adsorption time and adsorption solvent for dye loading on TiO2 films and determined that lower adsorption times and protic solvents (methanol) led to the best efficiencies. In an another publication from the same group integration of a push–pull structure with a trans-diphenylamino substituent 5 gave a very high efficiency of 6.5% (Jsc = 13.1 mA cm−2, Voc = 0.72 V, and FF = 0.69) (Fig. 5).62
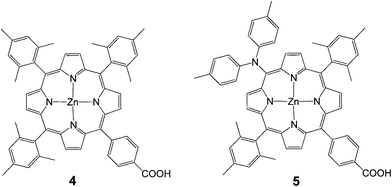 |
| | Fig. 5 Molecular structures of porphyrins 4 and 5. | |
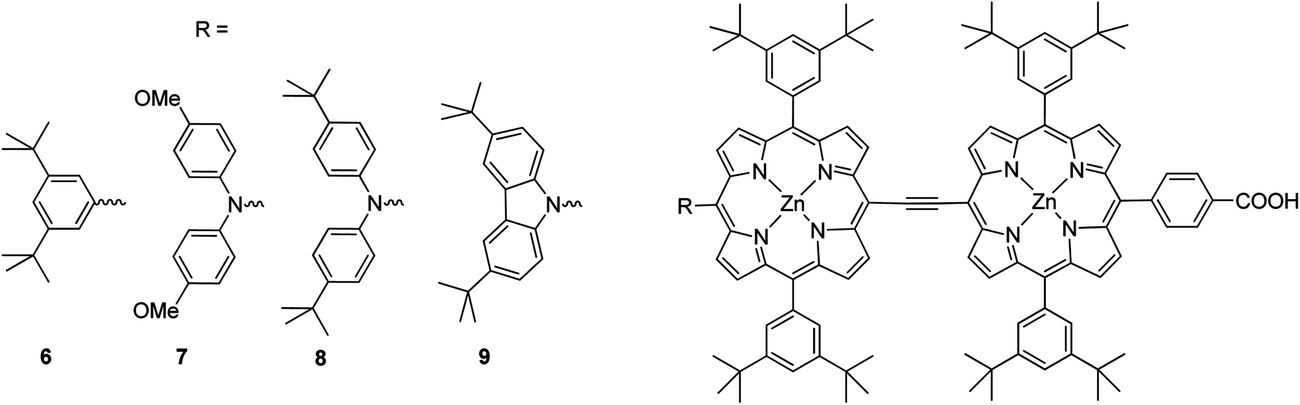
Moreover, in order to synthesize a dye with a broad spectral coverage two porphyrin units can be linked at the meso position either directly63 or by suitable π-conjugated linker.64 Furthermore, unsymmetrical porphyrin dyads can lead to enhancement of the solar cell efficiency.65 Combining the strategy of donor substation and the “built-in” energy gradient of the hetero-dimer taking advantage of the different energy levels of zinc coordinated and non-coordinated porphyrins,65 Segawa et al. synthesized a N-fused carbazole zinc porphyrin-free base porphyrin triad and obtained highly efficient light to current conversion throughout the visible to NIR region.66 The same research group synthesized ethynyl-linked porphyrin hetero-dimers substituted by a series of electron donors, namely, 3,6-di-tert-butylphenyl 6, bis(4-methoxyphenyl)amino 7, bis (4-tert-butylphenyl)amino 8 and 3,6-di-tert-butylcarbazol-9-yl 9 (Fig. 6).58 Despite the increased electron injection driving forces due to the bathochromic shift, the intensification of long-wavelength absorption bands and the elevated LUMO levels, the substitution of diphenylamino groups (7 and 8) with stronger electron-donating abilities gave rise to surprising mediocrity in the short-circuit photocurrent densities (Jsc). For that reason the overall energy conversion efficiencies followed the order 7 (3.94%) (Jsc = 10.55 mA cm−2, Voc = 0.543 V, and FF = 0.69) < 6 (4.57%) (Jsc = 12.51 mA cm−2, Voc = 0.541 V, FF = 0.68) < 8 (4.83%) (Jsc = 13.15 mA cm−2, Voc = 0.541 V, FF = 0.68) < 9 (5.21%) (Jsc = 14.26 mA cm−2, Voc = 0.547 V and FF = 0.67) (Table 1).
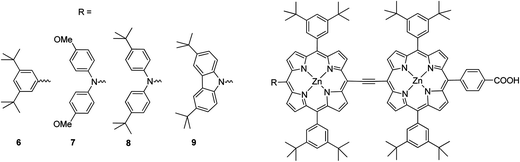 |
| | Fig. 6 Molecular structures of the dyes studied 6–9. | |
Table 1 Photovoltaic performances of DSSCs using 6–9 as sensitizers
| Dye |
Voc, mV |
Jsc, mA cm−2 |
FF |
n, % |
| 6 |
541 |
12.5 |
0.68 |
4.6 |
| 7 |
543 |
10.5 |
0.69 |
3.9 |
| 8 |
541 |
13.1 |
0.68 |
4.8 |
| 9 |
547 |
14.3 |
0.67 |
5.2 |
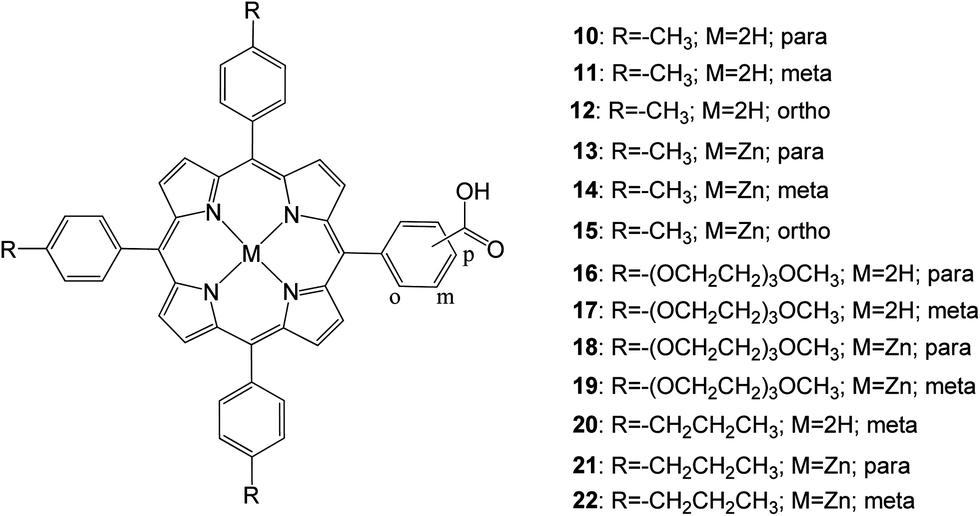
In 2013 D'Souza and coworkers67 studied, 13 porphyrins functionalized with a carboxylic acid anchoring group at the para, meta, or ortho positions of one of the phenyl rings of meso-tetra aryl porphyrin in addition to varying functional groups of the remaining meso positions of the porphyrin ring (Fig. 7). It was found that porphyrins anchored to the mesoporous TiO2 in the para and meta positions with respect to the porphyrin ring had improved photoelectrochemical properties compared to their ortho counterparts. This was supported by femtosecond transient absorption spectroscopy data collected on zinc-metalated porphyrin derivatives, showing a fast charge recombination time for the ortho derivative adsorbed electrode. Similarly, placing tolyl groups in the three remaining meso positions showed improved efficiency compared to the long alkyl chain and ethylene oxide groups mentioned in this study. Additionally, the zinc porphyrin derivatives outperformed their free-base porphyrin counterparts.
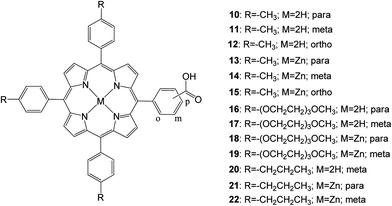 |
| | Fig. 7 Molecular structures of dyes 10–22. | |
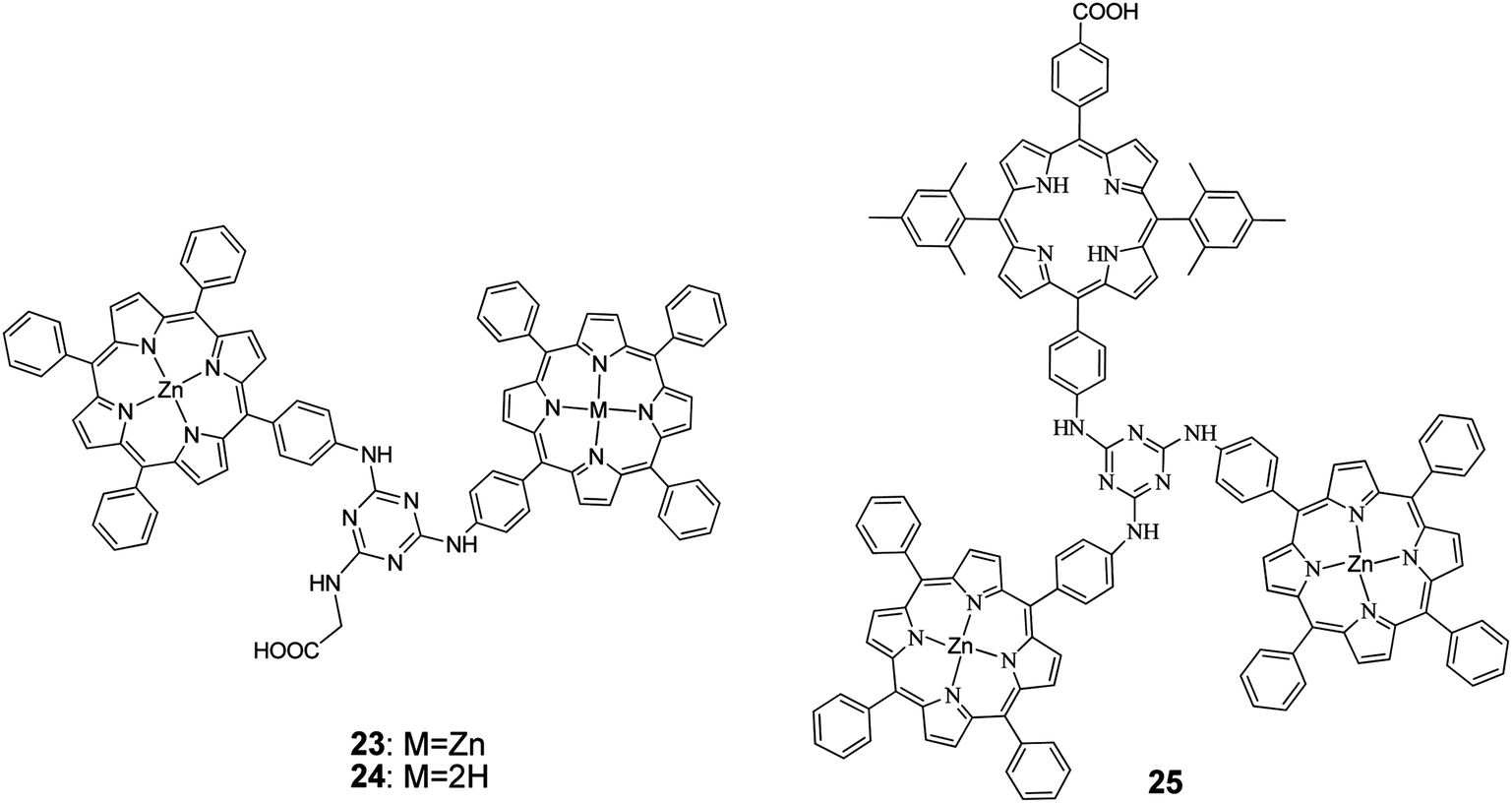
1,3,5-Triazine, a known mediator of intramolecular photoinduced electron transfer between chromophores, tethered two porphyrin units.68 The synthesis was performed by taking advantage of the stepwise and temperature-dependent reactivity of cyanuric chloride. The dyads were also functionalized by a terminal carboxylic acid group of a glycine moiety attached to the triazine group (Fig. 8). The study of the photophysical and electronic properties of the two compounds revealed no significant electronic interaction in their ground states but broadened spectral absorptions and suitable frontier orbital energy levels for use in DSSCs. Porphyrins 23 and 24 were fabricated for DSSC, demonstrating efficient intramolecular electron-transfer and charge-separation phenomena mediated by the triazine group. The PCE values were of 3.61% (Jsc = 8.82 mA cm−2, Voc = 0.63 V, and FF = 0.65) and 4.46% (Jsc = 9.94 mA cm−2, Voc = 0.66 V, and FF = 0.68), respectively. The higher PCE value of the 24-based DSSC is attributed to its larger dye loading and higher Jsc, Voc, and FF values.
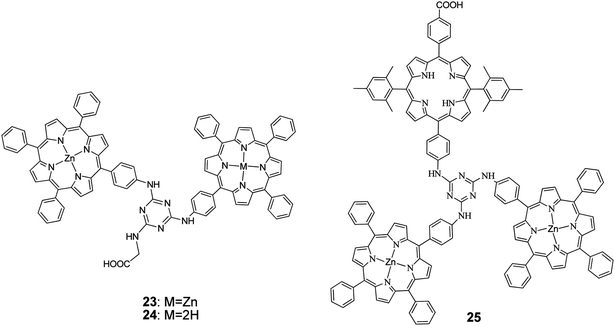 |
| | Fig. 8 Molecular structures of porphyrins 23–25. | |
Coutsolelos and coworkers,69 synthesized via stepwise amination reactions of cyanuric chloride a propeller shaped, triazine bridged, and unsymmetrical porphyrin triad 25. Photophysical and electrochemical studies of the triad revealed no significant electronic interactions between the porphyrin units in the triad ground state, but the frontier orbital energy levels are suitable for use as sensitizer in DSSCs, as they favour the electron injection and dye regeneration processes. These results were corroborated by DFT calculations which suggested that the triad 25 could also be described as a “push–pull” 2D-π-A system, with the A residing on the anchoring (carboxylic acid) group, that has the potential to promote triazine-mediated electron transfer and injection into the TiO2 electrode. Solar cells sensitized by triad 25 exhibited PCE of 4.91% (Jsc = 10.9 mA cm−2, Voc = 0.66 V, and FF = 0.68). The PCE of the latter solar cell was further improved up to 5.56% (Jsc = 12.4 mA cm−2, Voc = 0.64 V, and FF = 0.70) by co-sensitization by incorporation of chenodeoxychlolic acid (CDCA) in the dye solution (Table 2).
Table 2 Photovoltaic performances of DSSCs using 23–25 as sensitizers
| Dye |
Voc, mV |
Jsc, mA cm−2 |
FF |
n, % |
| Co-sensitization with CDCA. |
| 23 |
630 |
8.8 |
0.65 |
3.6 |
| 24 |
660 |
9.9 |
0.68 |
4.5 |
| 25 |
660 |
10.9 |
0.68 |
4.9 |
| 25a |
640 |
12.4 |
0.70 |
5.6 |
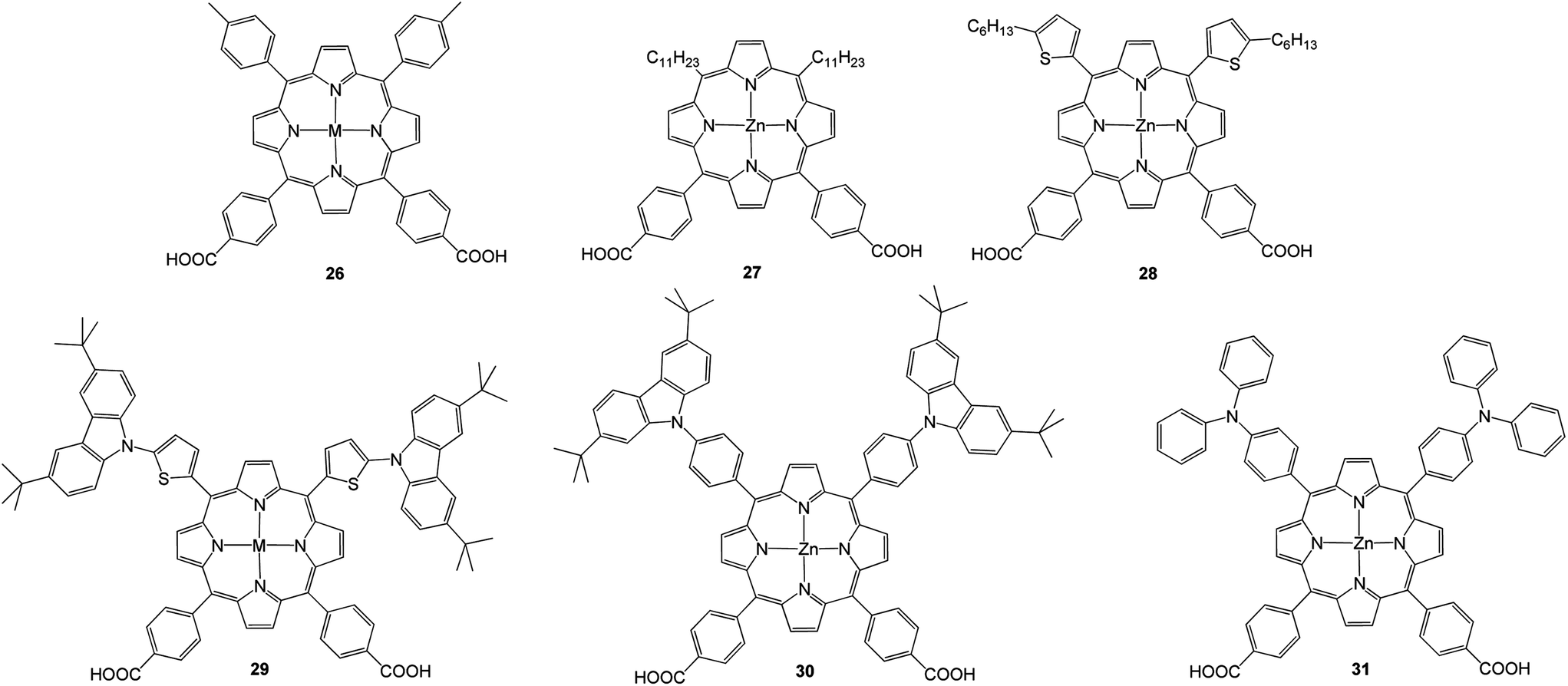
Recently, Hung et al.70 synthesized a series of porphyrins 26–31 (Fig. 9) having two electron donating groups and dual anchoring units that were connected through a porphyrin π-bridging unit and applied as sensitizers for DSSCs. The highest PCE of about 4.07% with Jsc = 9.81 mA cm−2, Voc = 0.63 V and FF = 0.66 was observed for 30 due to its high molar extinction coefficient (Table 3).
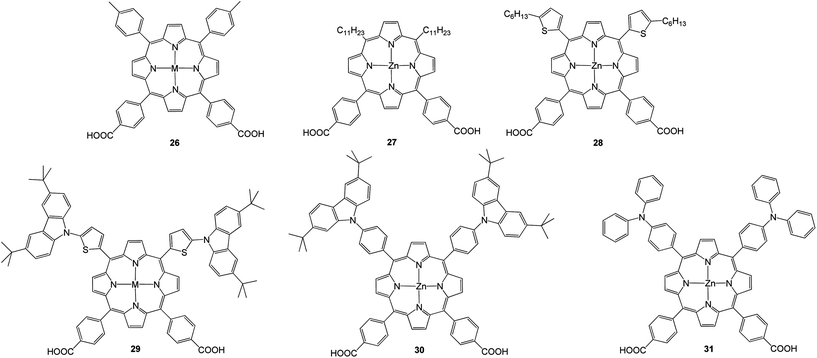 |
| | Fig. 9 Molecular structures of 26–31. | |
Table 3 Photovoltaic performances of DSSCs using 26–31 as sensitizers
| Dye |
Voc, mV |
Jsc, mA cm−2 |
FF |
n, % |
| 26 |
570 |
7.52 |
0.65 |
2.78 |
| 27 |
580 |
8.50 |
0.58 |
2.85 |
| 28 |
580 |
7.92 |
0.69 |
3.18 |
| 29 |
580 |
6.87 |
0.68 |
2.72 |
| 30 |
630 |
9.81 |
0.66 |
4.07 |
| 31 |
590 |
9.18 |
0.67 |
3.64 |
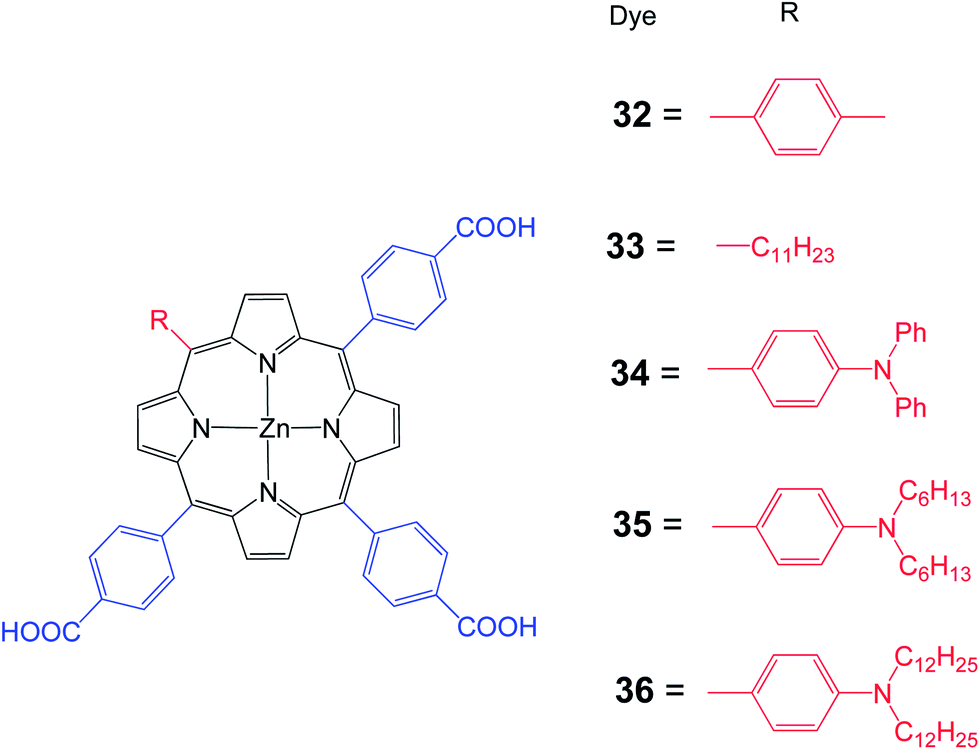
Moreover, Hung et al. synthesized a series of 1D-π-3A zinc porphyrin dyes 32–36 with three p-carboxyphenyl anchoring groups with various substituents at the meso position (Fig. 10).71 Porphyrin 34 is slightly red shifted compared to the other porphyrins. The Soret band of 34–36 is broadened because of the presence of an electron donating amino unit. Higher than 5% PCE has been achieved for a DSSC sensitized with either 34–36. The strong electron donating character, better electron injection, intrinsic anti-aggregation and suppressed charge recombination capacity contributed to the highest PCE of 6.10% (Jsc = 12.66 mA cm−2, Voc = 0.70 V and FF = 0.69) for the DSSC sensitized with 35 (Table 4). Moreover, it was demonstrated that the DSSC based on multi-anchoring unit porphyrin dye is more stable than their mono-anchoring analogs.
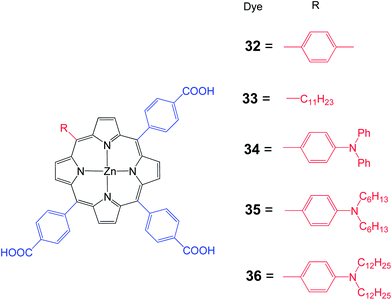 |
| | Fig. 10 Molecular structures of dyes 32–36. | |
Table 4 Photovoltaic performances of DSSCs using 32–36 as sensitizers
| Dye |
Voc, mV |
Jsc, mA cm−2 |
FF |
n, % |
| 32 |
630 |
10.55 |
0.68 |
4.51 |
| 33 |
590 |
9.36 |
0.69 |
3.80 |
| 34 |
660 |
11.75 |
0.69 |
5.36 |
| 35 |
700 |
12.66 |
0.69 |
6.10 |
| 36 |
650 |
13.61 |
0.66 |
5.80 |
Although meso-carboxyphenyl tethered porphyrins have been widely studied in DSSCs, there remains a concern about the torsional angle decoupling of the phenyl rings from the conjugated system of the macrocycle, which potentially limits the electronic communication between the excited dye and the linkage to the semiconductor. Therefore more attention has been paid to other structural designs such as porphyrins with meso-alkynylbenzoic acid or β-linked conjugated tethers and devices using these designs have reached remarkably high efficiencies.
2.2 Conjugated bridge carboxylic acids
The type of linkage between the porphyrin and the binding group is very important. It has been shown that conjugated linkers make a significant difference in the overall cell performance.
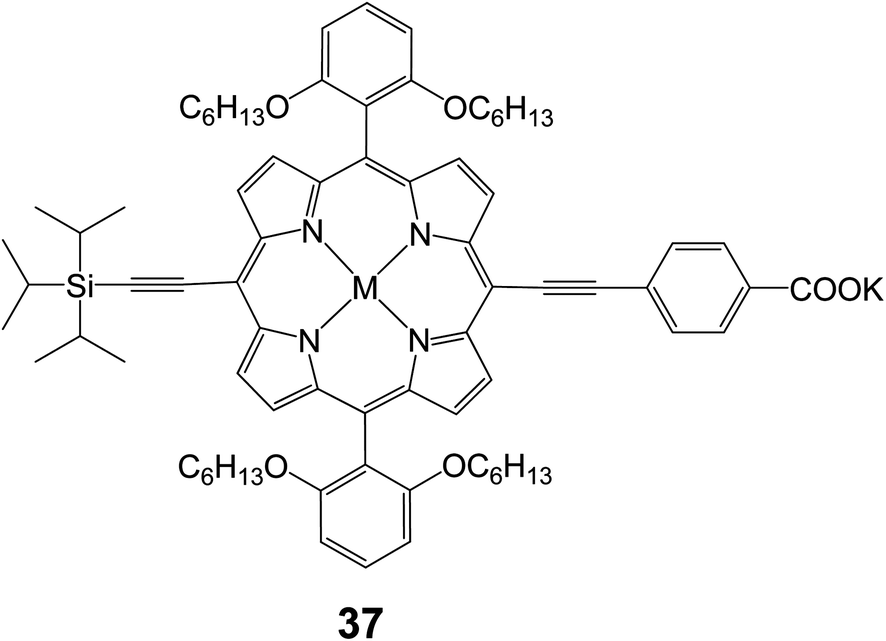
2.2.1 Meso-position linking. The use of meso-linked alkynylbenzoic acid tethers is a relatively new design strategy in porphyrin DSSC research. The first report of alkynylbenzoic acid tethered porphyrins was by Stromberg et al. in 200772 in which the reported photocurrent of the device was only 0.035 mA cm−2. Since then two main groups of Joseph Hupp and Eric Diau have studied this novel tether style. The use of the meso-alkynylbenzoic acid tether has led to an increase in solar cell efficiency for porphyrin DSSCs.73,74 In a paper by Hupp and coworkers, dye 37 (Fig. 11) was reported to give efficiencies of 2.5% (Jsc = 5.9 mA cm−2, Voc = 0.65 V and FF = 0.66) in standard TiO2 DSSC.75 In the report on 37, ZnO was also tested as a semiconductor, and it was determined that the more acidic dyes that tend to slightly corrode the ZnO surface showed better electron injection dynamics.
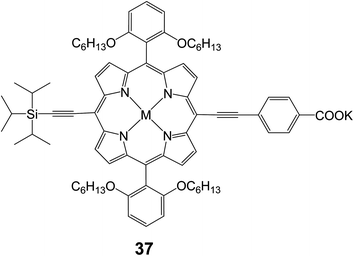 |
| | Fig. 11 Molecular structure of compound 37. | |
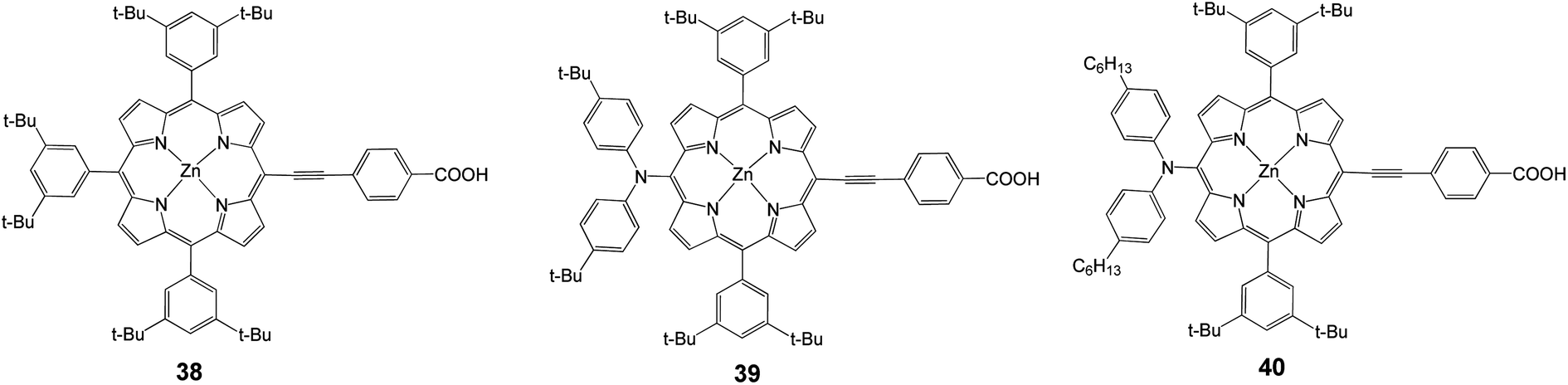
To solve the problem of porphyrin aggregation, 3,5-di-tertbutylphenyl groups were introduced at the meso-positions of the porphyrin ring. Based on this molecular design, Yeh and coworkers35 reported meso-substituted zinc porphyrin derivatives, for which 38 (Fig. 12) served as a reference compound. With the diarylamino group attached at the meso-position of the porphyrin core, 39 attained 6.0% efficiency (Jsc = 13.60 mA cm−2, Voc = 0.701 V and FF = 0.629). The appearance of porphyrins with a push–pull framework such as 39 and 40 indicated the arrival of a new period of DSSC.74 Porphyrin 40 had two long alkyl chains in order to improve the thermal and photochemical stability of a device with an efficiency of 6.8% (Jsc = 13.68 mA cm−2, Voc = 0.711 V and FF = 0.695), (Table 5).
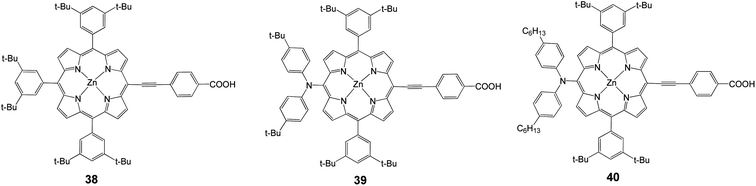 |
| | Fig. 12 Molecular structures of dyes 38–40. | |
Table 5 Photovoltaic performances of DSSCs using 38–40 as sensitizers
| Dye |
Voc, mV |
Jsc, mA cm−2 |
FF |
n, % |
| 38 |
617 |
5.79 |
0.667 |
2.4 |
| 39 |
701 |
13.60 |
0.629 |
6.0 |
| 40 |
711 |
13.68 |
0.695 |
6.8 |
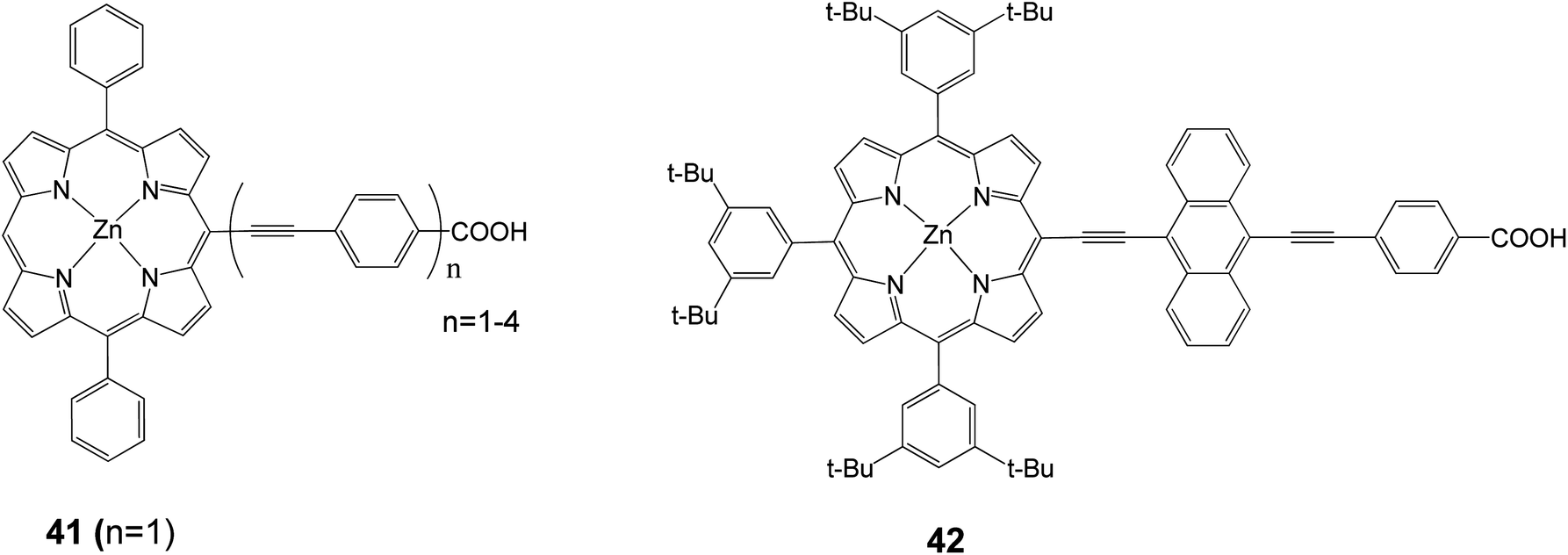
In 2010, the device performance of 40 was further improved by Grätzel and coworkers,30 giving an efficiency of 10.9%, (Jsc = 18.6 mA cm−2, Voc = 0.77 V and FF = 0.76) which lasted as a record for DSSC until 2011. Diau and coworkers synthesized a series of dyes in order to probe the effects of the bridging moiety length and the size of the intermediate acene.76–79 A series of porphyrin dyes with differing numbers of ethynylphenyl groups (1–4) were produced and tested; the optimal dye 41 (with n = 1) is shown in Fig. 13 and gave an efficiency of 2.7% (Jsc = 7.3 mA cm−2, Voc = 0.57 V, and FF = 0.65).77 In a different study in order to determine how the number of aromatic rings in the bridging group affects solar cell performance, a series of porphyrin dyes with benzene through pentacene moieties were synthesized and tested as DSSCs. The optimal dye 42 is shown in Fig. 13 and gave an efficiency of 5.4% (Jsc = 12.67 mA cm−2, Voc = 0.67 V, and FF = 0.64).78
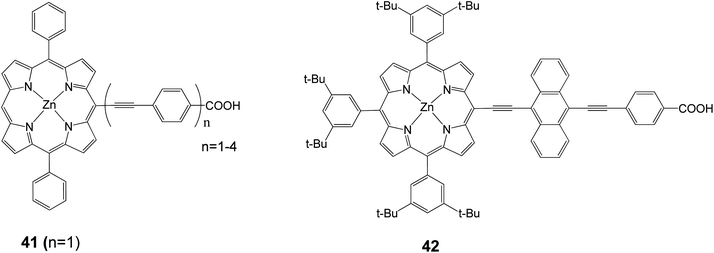 |
| | Fig. 13 Molecular structures of compounds 41 and 42. | |
The electronic structure of porphyrins is considerably influenced by introducing electron donors at the meso positions.29,30 In principle, the tuning of HOMO and LUMO levels could be achieved with the choice of suitable groups. Xie et al. synthesized porphyrins (43–47) with various number of triphenylamine and trimethoxyphenyl groups as electron donors, in order to modulate the HOMO and LUMO levels (Fig. 14).80 Among these porphyrin dyes, 47 showed the highest PCE of 5.45% (Jsc = 15.36 mA cm−2, Voc = 0.665 V, and FF = 0.53), associated with an efficient electron injection and dye regeneration, resulting from the suitable arrangements of the HOMO and LUMO energy levels, in combination of longer electron lifetime. It was demonstrated that by introducing appropriate electron donors might significantly enhance the performance by modulating the HOMO and LUMO levels of the porphyrin sensitizers (Table 6).
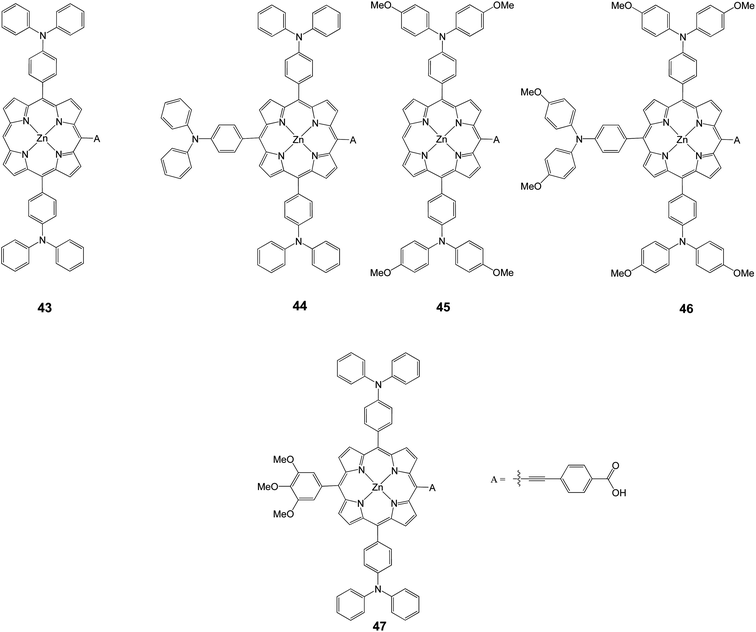 |
| | Fig. 14 Molecular structures of 43–47. | |
Table 6 Photovoltaic performances of DSSCs using 43–47 as sensitizers
| Dye |
Voc, mV |
Jsc, mA cm−2 |
FF |
n, % |
| 43 |
624 |
13.04 |
0.54 |
4.36 |
| 44 |
573 |
12.38 |
0.58 |
4.10 |
| 45 |
560 |
10.19 |
0.62 |
3.53 |
| 46 |
533 |
7.86 |
0.64 |
2.70 |
| 47 |
665 |
15.36 |
0.53 |
5.45 |
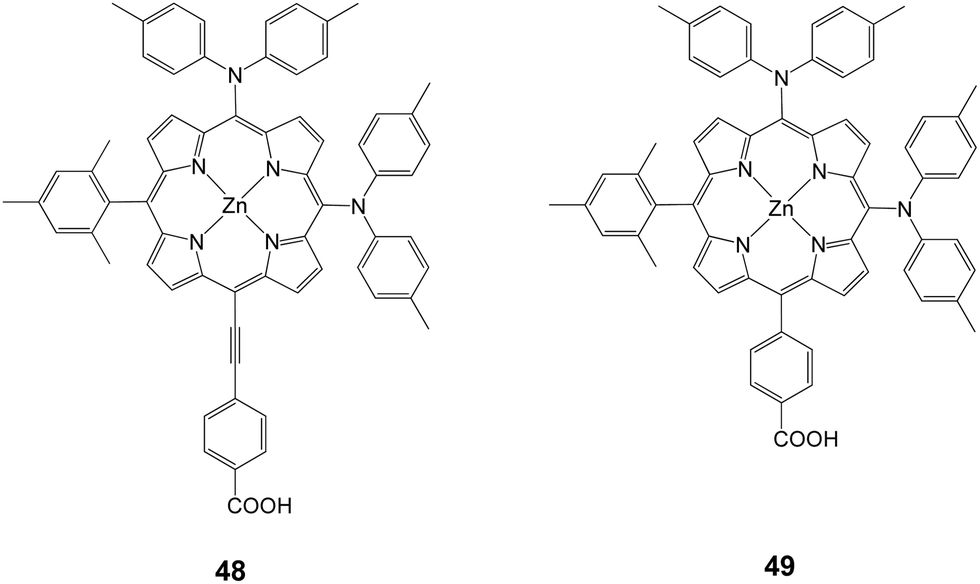
Following their research work on porphyrin dyes in order to improve the light harvesting properties, Imahori and coworkers demonstrated PCE of 10.1% (Jsc = 19.33 mA cm−2, Voc = 0.72 V and FF = 0.724) and 8.3% (Jsc = 16.26 mA cm−2, Voc = 0.71 V and FF = 0.72) for 48 and 49, respectively (Fig. 15).81 Compound 48 is an asymmetrically enhanced push–pull porphyrin bearing two dyarilamino groups at the meso position with a cis configuration.
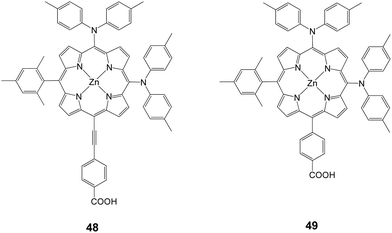 |
| | Fig. 15 Molecular structure of dyes 48 and 49. | |
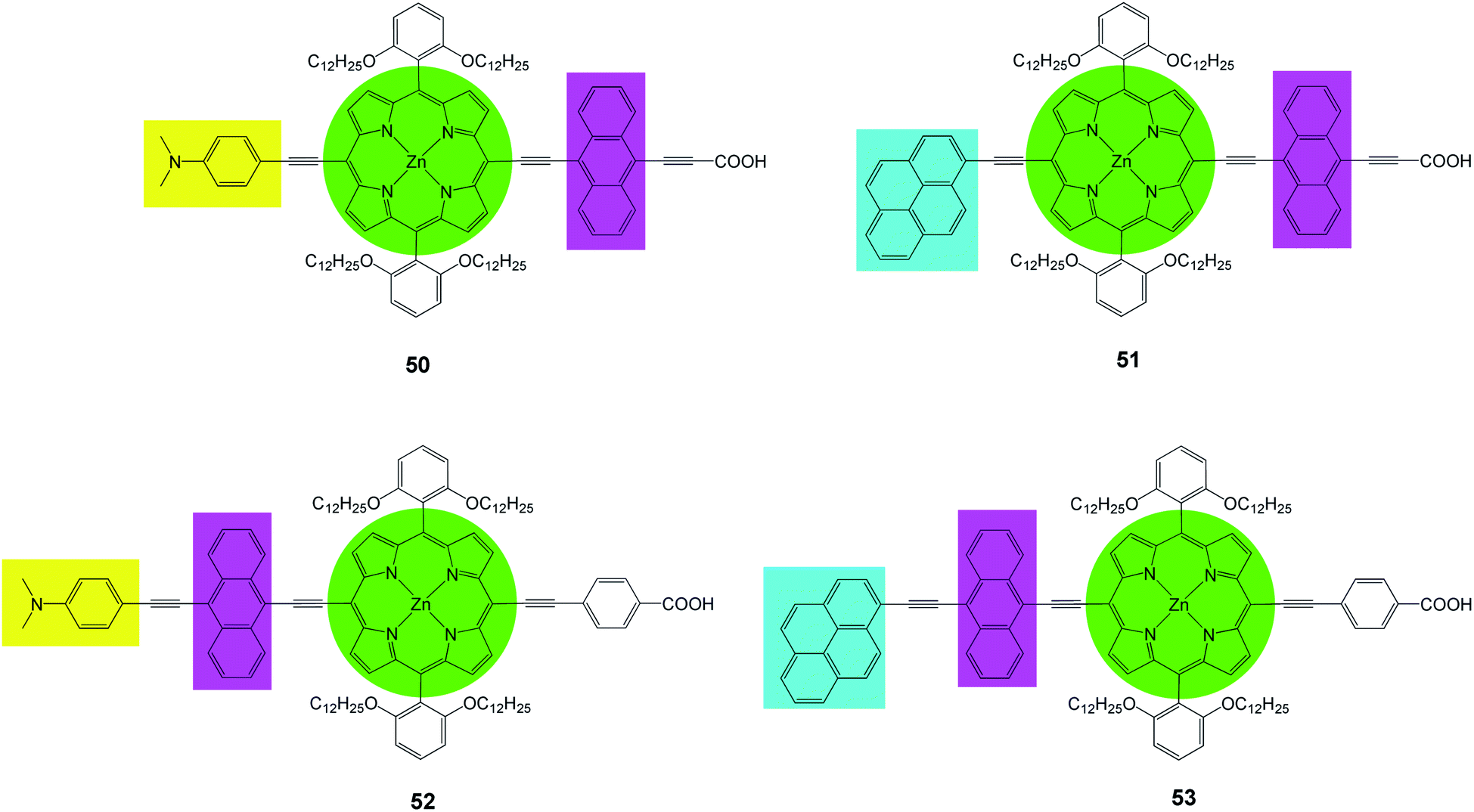
Recently Lin et al.82 prepared a series of new porphyrins (50–53) with near-IR light harvesting properties, for DSSCs, by attaching a pyrene or a 4-dimethylaminophenyl group in combination with anthracene to modify the porphyrin core (Fig. 16). It was shown that the incorporation of these moieties renders feasible tuning of spectral and redox properties of the porphyrins. The PCE values of the dyes was in the range of 6.7–9.73% and 50 sensitized DSSC showed a maximum PCE of 9.73% (Jsc = 17.77 mA cm−2, Voc = 0.73 V and FF = 0.75), (Table 7). These high performances are due to the following factors (a) expansion of the porphyrin spectral range, (b) reduction of the dye aggregation bye suitable dye soaking processes, (c) addition of polar electron releasing groups.
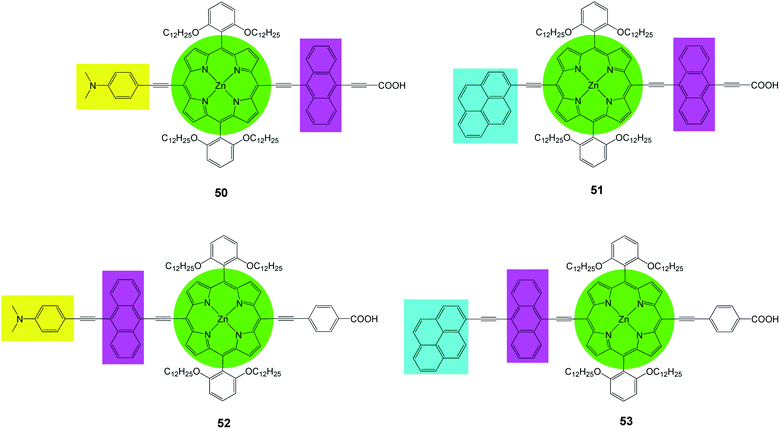 |
| | Fig. 16 Molecular structures of dyes 50–53. | |
Table 7 Photovoltaic performancesa of DSSCs using 50–53 as sensitizers
| Dye |
Voc, mV |
Jsc, mA cm−2 |
FF |
n, % |
| Under AM1.5 illumination (power 100 mW cm−2) with an active area of 0.096 cm2. |
| 50 |
730 |
17.77 |
0.75 |
9.73 |
| 51 |
680 |
14.17 |
0.72 |
6.97 |
| 52 |
720 |
17.76 |
0.74 |
9.51 |
| 53 |
680 |
14.20 |
0.69 |
6.70 |
2.2.2 β-Position linking. The use of conjugated β-substituted anchoring groups was first demonstrated in 2004 by Grätzel et al. when they introduced a dye 54 with an energy conversion efficiency of 4.1% (Jsc = 8.86 mA cm−2, Voc = 0.65 V and FF = 0.71).46 Also, for porphyrins with carboxylic binding groups, the Zn containing diamagnetic metalloporphyrins 63 and 55 had very high incident monochromatic photon-to-current conversion efficiencies compared to those observed for the Cu containing paramagnetic metalloporphyrin 56 (Fig. 17, Table 8).
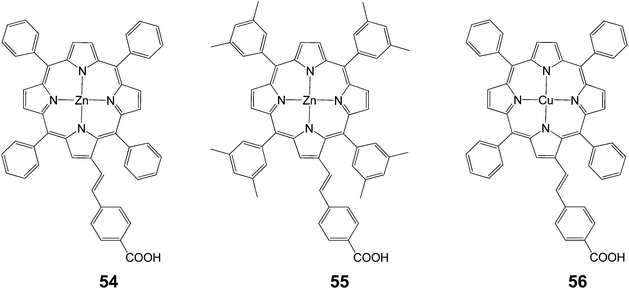 |
| | Fig. 17 Molecular structures of porphyrins 54–56. | |
Table 8 Photovoltaic performances of DSSCs using 54–56 as sensitizers
| Dye |
Voc, mV |
Jsc, mA cm−2 |
FF |
n, % |
| 54 |
654 |
8.86 |
0.71 |
4.11 |
| 55 |
660 |
9.70 |
0.75 |
4.80 |
| 56 |
490 |
1.35 |
0.69 |
0.45 |
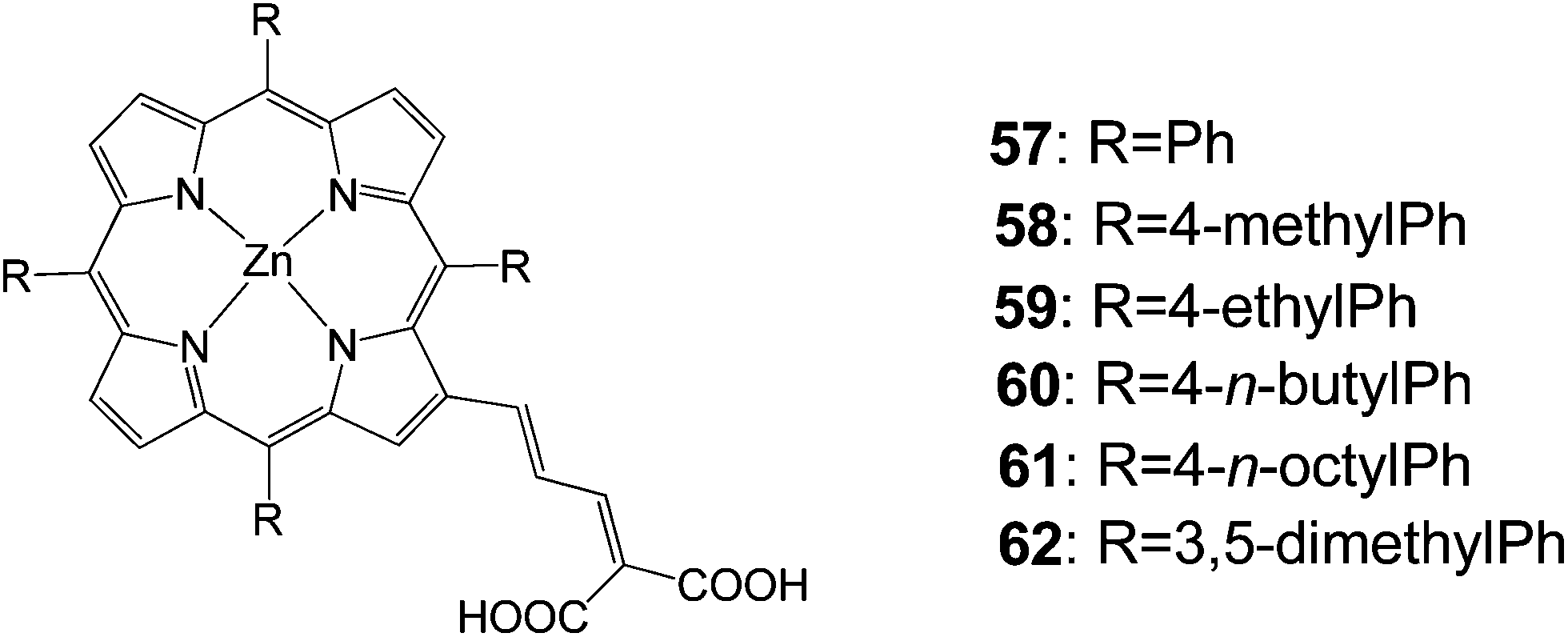
In 2007, Officer and coworkers reported a series of porphyrins 57–62 with a conjugated bridge to a dicarboxylic acid acceptor moiety (Fig. 18).83 Porphyrin 58 gave the best efficiency 7.1% and the rest of the dyes featured efficiencies over 5% (Table 9).
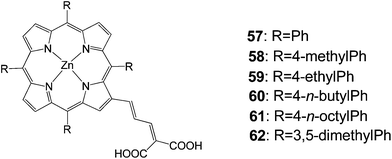 |
| | Fig. 18 Molecular structures of dyes 57–62. | |
Table 9 Photovoltaic performancesa of DSSCs using zinc tetraarylporphyrin malonic acids as sensitizers
| Dye |
Voc, mV |
Jsc, mA cm−2 |
FF |
n, % |
| Data obtained under cell illumination AM1.5 100 mW cm−2. |
| 57 |
638 |
12.1 |
0.66 |
5.1 |
| 58 |
680 |
14.0 |
0.74 |
7.1 |
| 59 |
642 |
14.8 |
0.63 |
5.8 |
| 60 |
701 |
13.4 |
0.68 |
6.4 |
| 61 |
649 |
13.4 |
0.61 |
5.3 |
| 62 |
685 |
13.3 |
0.68 |
6.1 |
Since the 2007 report on β-linked porphyrins, Officer and coworkers used β-linked derivatives to study several aspects of solar cell design and performance. The aspects of design under study included the use of a post adsorbed phosphinic acid blocking moiety to improve efficiencies by minimizing recombination events at the electrode surface.84 They also studied the use of ionic liquid electrolytes as a replacement for volatile organic solvents in typical liquid electrolytes.85 The Officer group had also studied the open-circuit voltage and electron injection dynamics using β-substituted porphyrins and suggested that the reason for limitations to the Voc and Jsc were related to reduced electron lifetime and less favorable electron injection dynamics.86,87
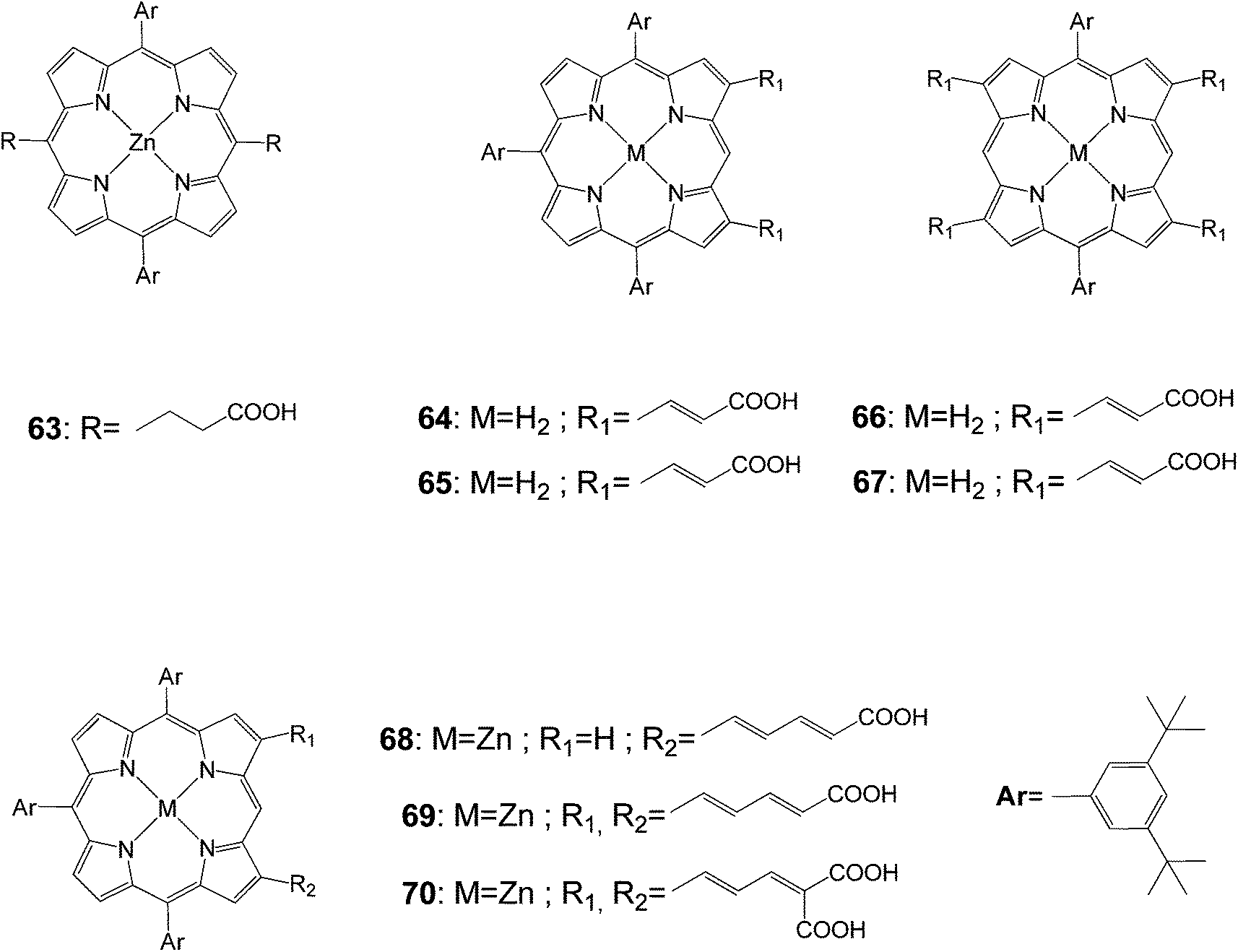
Porphyrins (63–70) functionalized at meso- and β-positions with different carboxylic acid groups were prepared to investigate the photovoltaic properties as dye sensitized nanocrystalline-TiO2 solar cells (Fig. 19).88 The electronic structures of the porphyrin macrocyclic core were strongly coupled with olefinic side chains so that the absorption spectrum exhibited largely broad and red-shifted Soret and Q-bands. In the case of the doubly functionalized porphyrin with malonic di-acid groups, the Soret band appeared at 475 nm. Among porphyrin derivatives prepared in this study, compound 70 exhibited the maximum overall conversion efficiency of 3.03% (Table 10). From such photovoltaic performances, it was suggested that multiple pathways through olefinic side chains at two β-positions enhanced the overall electron injection efficiency and the moderate distance between the porphyrin sensitizer and the TiO2 semiconductor layer was important, retarding the charge recombination processes. As a consequence, these combined effects gave rise to higher photovoltaic efficiency in photovoltaic regenerative solar cells.
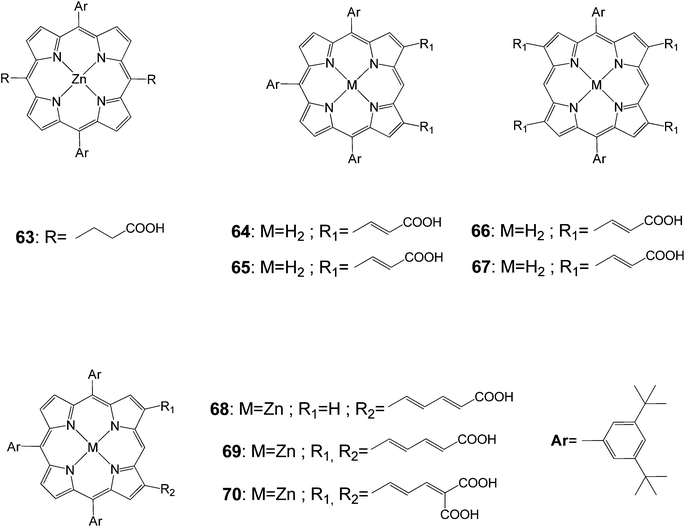 |
| | Fig. 19 Molecular structures of porphyrins 63–70. | |
Table 10 Photovoltaic performancesa of DSSCs using 63–70 as sensitizers
| Dye |
Voc, mV |
Jsc, mA cm−2 |
FF |
n, % |
| Overall conversion efficiency parameters were measured in the light condition of 100 mW cm−2 illumination. Not measured. |
| 63 |
450 |
0.51 |
0.60 |
0.14 |
| 64 |
510 |
1.84 |
0.67 |
0.63 |
| 65 |
540 |
4.45 |
0.61 |
1.48 |
| 66 |
b |
b |
b |
b |
| 67 |
b |
b |
b |
b |
| 68 |
540 |
6.20 |
0.62 |
2.08 |
| 69 |
560 |
6.74 |
0.62 |
2.37 |
| 70 |
590 |
8.38 |
0.62 |
3.03 |
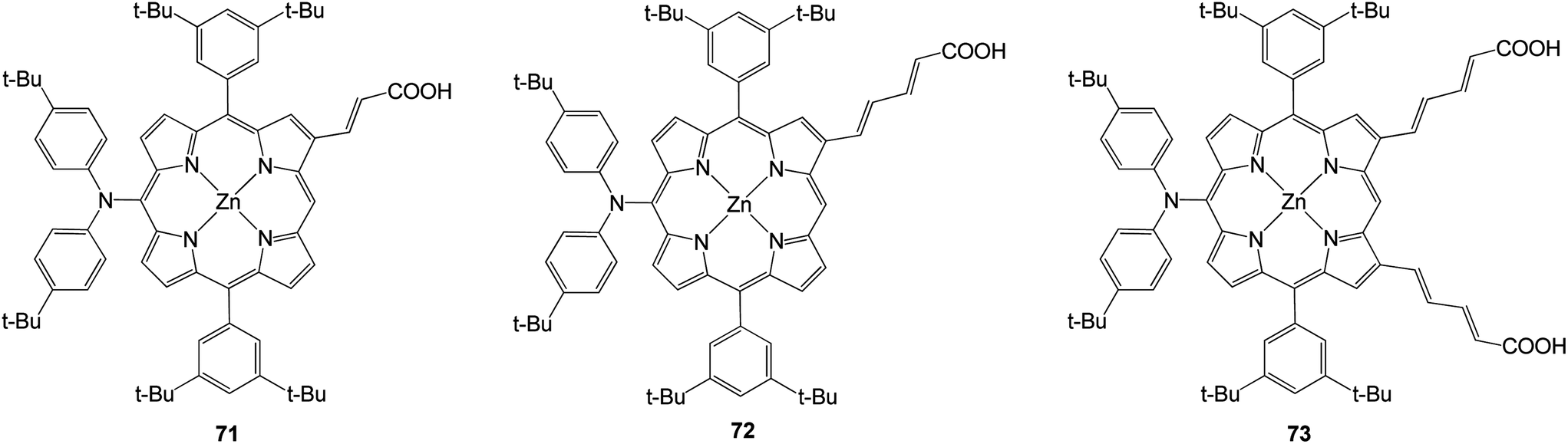
In 2011 three porphyrins with structures based on a molecular design that relies on donor–π–acceptor interactions (Fig. 20).89 At the meso position of the porphyrin was present a bis (4-tert-butylphenyl) amino group and at the opposite side were 2-propenoic or 2,4-pentadienoic acid anchoring groups at the β-pyrrolic positions. The efficiencies of all compounds are present in Table 11 and among the dyes prepared, the doubly functionalized carboxylic acid derivative 73 gave rise to the highest power conversion with an efficiency of 7.47%.
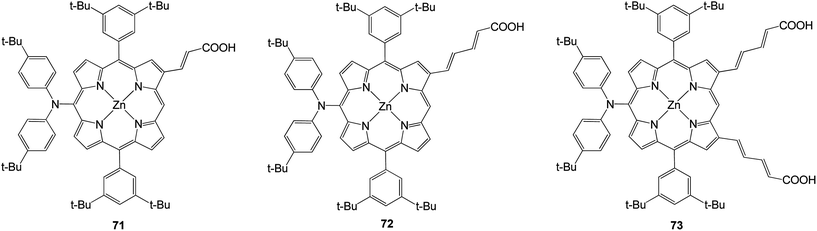 |
| | Fig. 20 Molecular structures of porphyrins 71–73. | |
Table 11 Photovoltaic performance of DSSCs using porphyrins 71–73 as sensitizers
| Dye |
Jsc, mA cm−2 |
Voc, mV |
FF |
n, % |
| 71 |
12.1 |
660 |
0.62 |
4.95 |
| 72 |
13.9 |
670 |
0.64 |
5.91 |
| 73 |
18.4 |
710 |
0.57 |
7.47 |
In another study following the same concept of push–pull, two meso-di-substituted porphyrinic dyes 74 and 75 (Fig. 21) were synthesized followed a facile route in order to compare the efficiency between them.90 It was found that the performance of DSSCs based on β-di-substituted push–pull dye 74 was measured to be slightly superior 4.1% (Jsc = 11.6 mA cm−2, Voc = 0.56 V and FF = 0.62) to this of DSSCs based on meso-bi-substituted dye 75 3.9% (Jsc = 11.1 mA cm−2, Voc = 0.60 V and FF = 0.59).
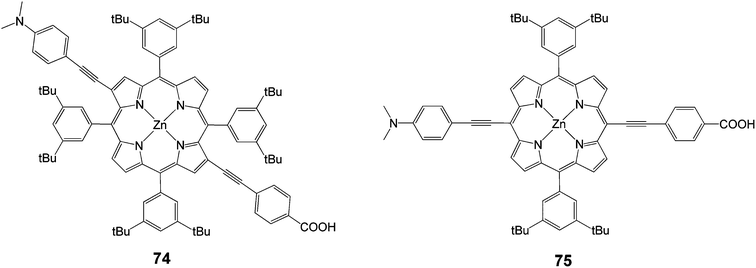 |
| | Fig. 21 Molecular structures of compounds 74 and 75. | |
Moreover, a very interesting study compared silatrane, phosphonic acid, and carboxylic acid functional groups (Fig. 22) for attachment of porphyrin sensitizers to TiO2 in photoelectrochemical cells. Porphyrin sensitizers bearing β-vinyl substituents with carboxylic acid 76, phosphonic acid 77, or silatrane moieties 78 had similar photophysical properties.91 All three molecules could be attached to nano-particulate anatase TiO2 by soaking in an appropriate solvent, but about twice the surface coverage was obtained with the carboxylic acid, relative to the other two linker groups. Maybe this was due to differences in the geometry of the anchoring groups on the oxide surface. In DSSCs, Voc and FF were comparable for all three linkers, but the short-circuit current and solar conversion efficiency of the cell with the carboxylic acid linked sensitizer were about twice those observed with the phosphonic acid and silatrane linkers. Thus the photoelectrochemical parameters for all three sensitizers were comparable, but the carboxylic acid bearing sensitizer gave about twice the solar conversion efficiency because of the increased surface coverage (Table 12). This was due to increased light absorption, absorption closer to the electrode FTO conducting surface and increased surface passivation. On the other hand, the carboxylate linkage was readily hydrolyzed, especially under basic conditions, resulting in loss of the sensitizer from the surface. Phosphonate linkages were much less labile, and the siloxyl linkages derived from the silatrane treatment were the most resistive to hydrolysis under the conditions examined. Such resistance to cleavage was important for long term stability of DSSCs, and especially for photoelectrochemical cells with aqueous electrolytes that were used for light-powered water oxidation and fuel production.
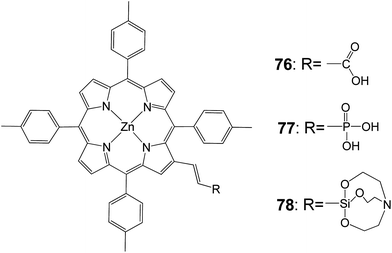 |
| | Fig. 22 Molecular structures of dyes 76–78. | |
Table 12 Photovoltaic performancea of DSSCs using porphyrins 76–78 as sensitizers
| Dye |
Jsc, mA cm−2 |
Voc, mV |
FF |
n, % |
| Illuminated using simulated AM1.5G sunlight at 1000 W m−2. |
| 76 |
2.49 |
444 |
0.52 |
0.57 |
| 77 |
1.11 |
426 |
0.53 |
0.25 |
| 78 |
1.10 |
423 |
0.53 |
0.25 |
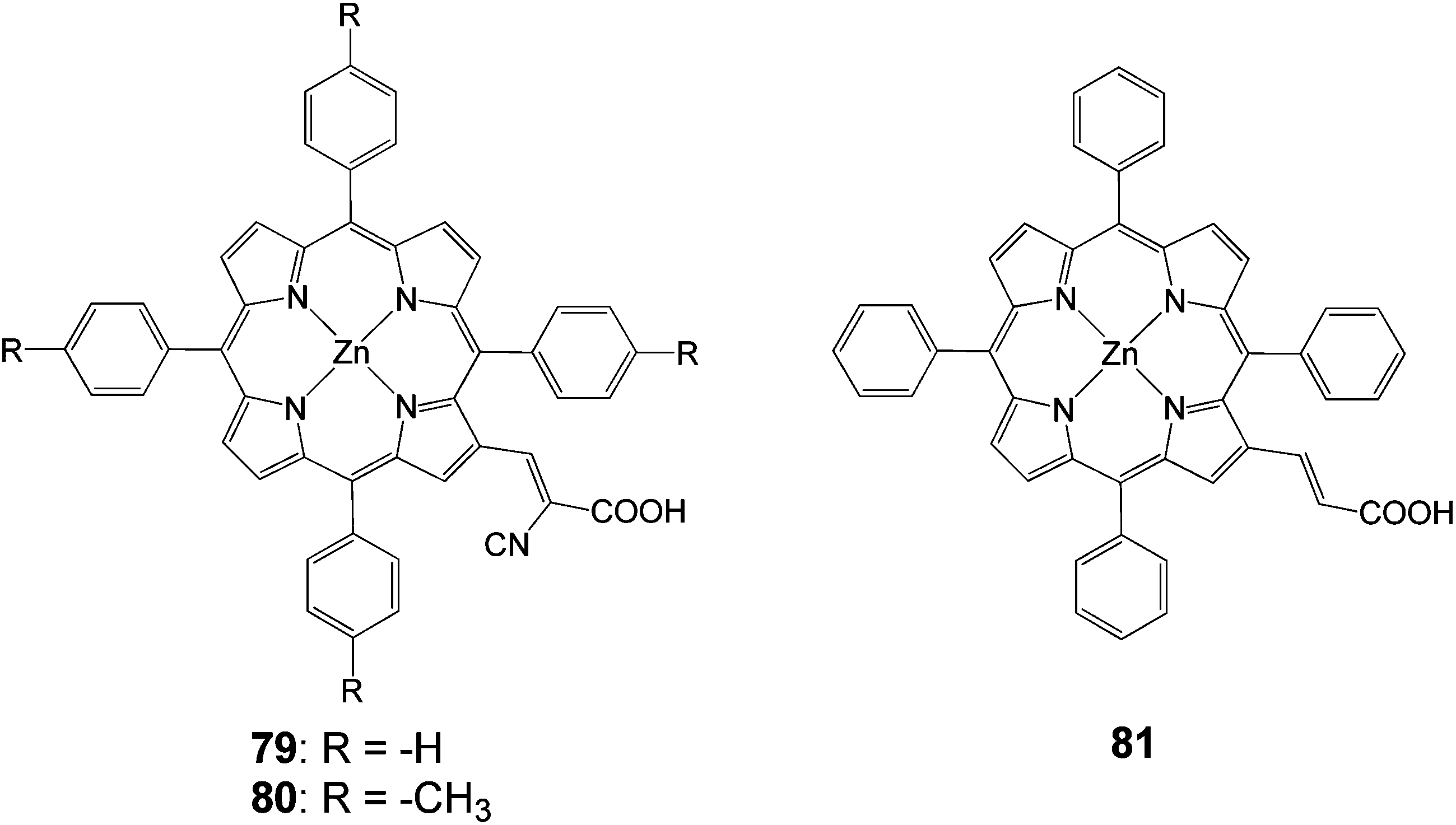
2.2.3 Cyanoacrylic acids. Another anchoring group that has been extensively used is the cyanoacrylic acid. In 2005, tetraphenylporphyrin with a β-substituted cyanoacrylic acid (Fig. 23), exhibited an efficiency of 5.2% (Jsc = 13.5 mA cm−2, Voc = 0.56 V).44 Dye 79 had a better efficiency compared to the carboxylic acid porphyrin 81 4.0% (Jsc = 10.9 mA cm−2, Voc = 0.55 V), synthesized from the same group.
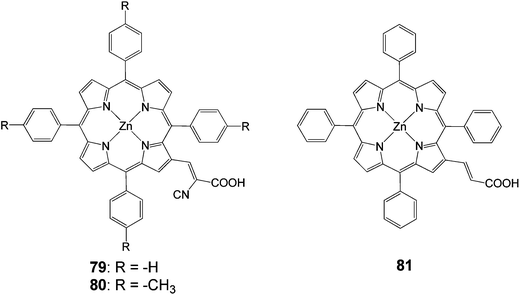 |
| | Fig. 23 Molecular structures of dyes 79–81. | |
In another study β-substituted zinc porphyrin analogues 79 and 80 (Fig. 23) were synthesized, where 80 had an added methyl group at the phenyl ring attached to the meso-position of the porphyrin macrocycle.92 These results showed that the different solvents used in the sensitization process affected how the dyes were aggregated and how much of the dyes were adsorbed on the semiconductor surface. The highest efficiency of 4.2% (Jsc = 13.0 mA cm−2, Voc = 0.48 V and FF = 0.67), 30 min soaking time and 4.0% (Jsc = 12.8 mA cm−2, Voc = 0.46 V and FF = 0.67), 1 hour soaking time was achieved for 79 and 80 analogues, respectively. The difference in the photovoltaic properties between 79 and 80 was caused by the amount of tilting of the molecular dye plane toward the dye surface. The 80 molecular planes were closer to the TiO2 surface due to the increased steric effect caused by the addition of a methyl group (Fig. 24). This increased the surface area of the dye adsorbed in the TiO2, resulting a much lower dye concentration. The soaking condition and the use of co-adsorbents giving much higher photovoltaic performances could control the amount of aggregation of the dye.
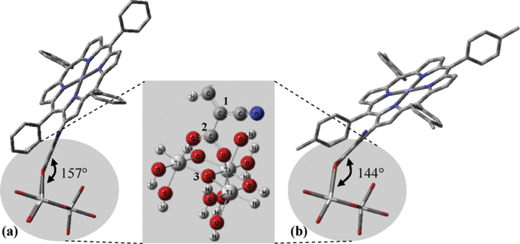 |
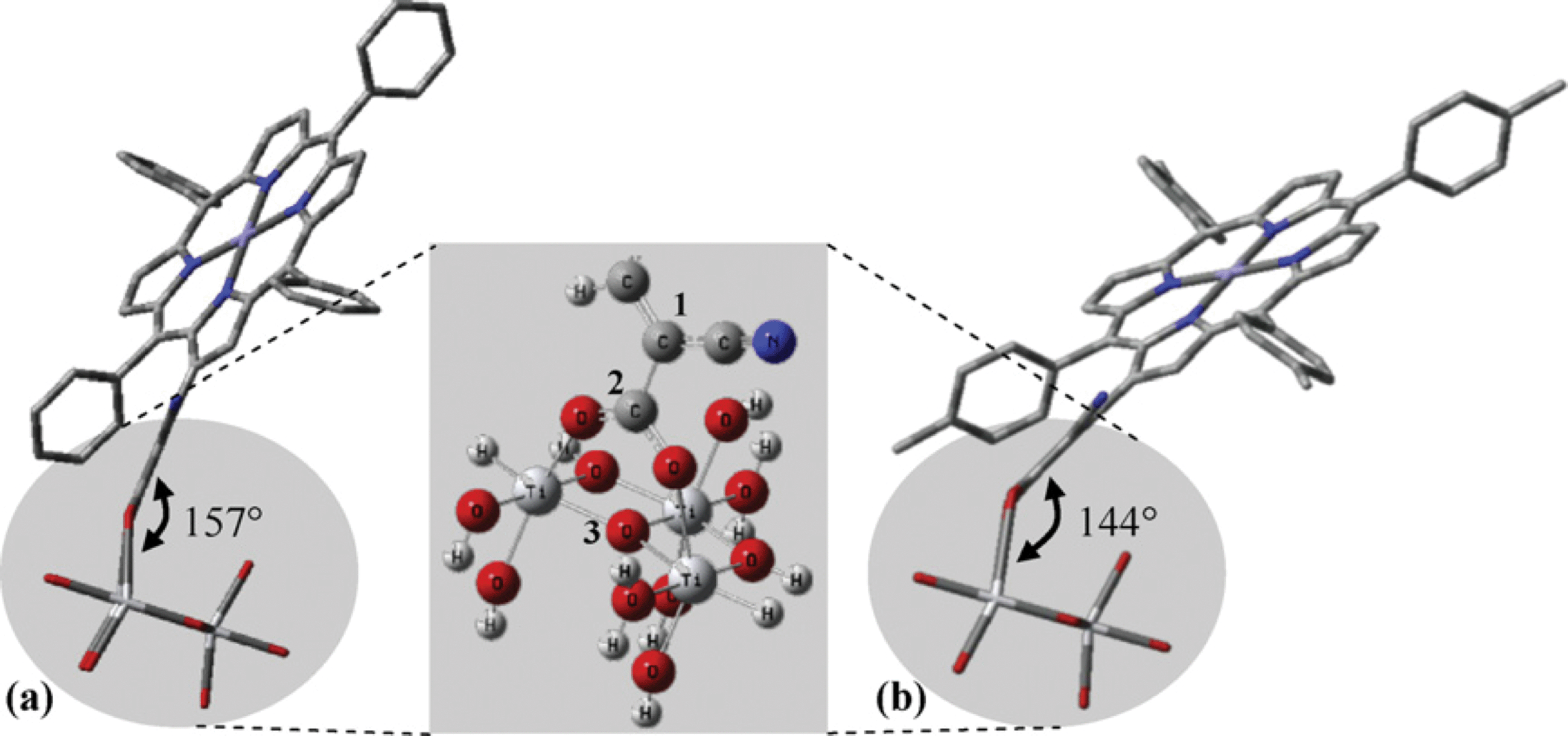
| | Fig. 24 Possible adsorption configurations of (a) 79 and (b) 80. The inset shows the bidentate adsorption of the dye. C1–C2–O3 determines the dye molecular angle against the TiO2. Adapted from ref. 92. | |
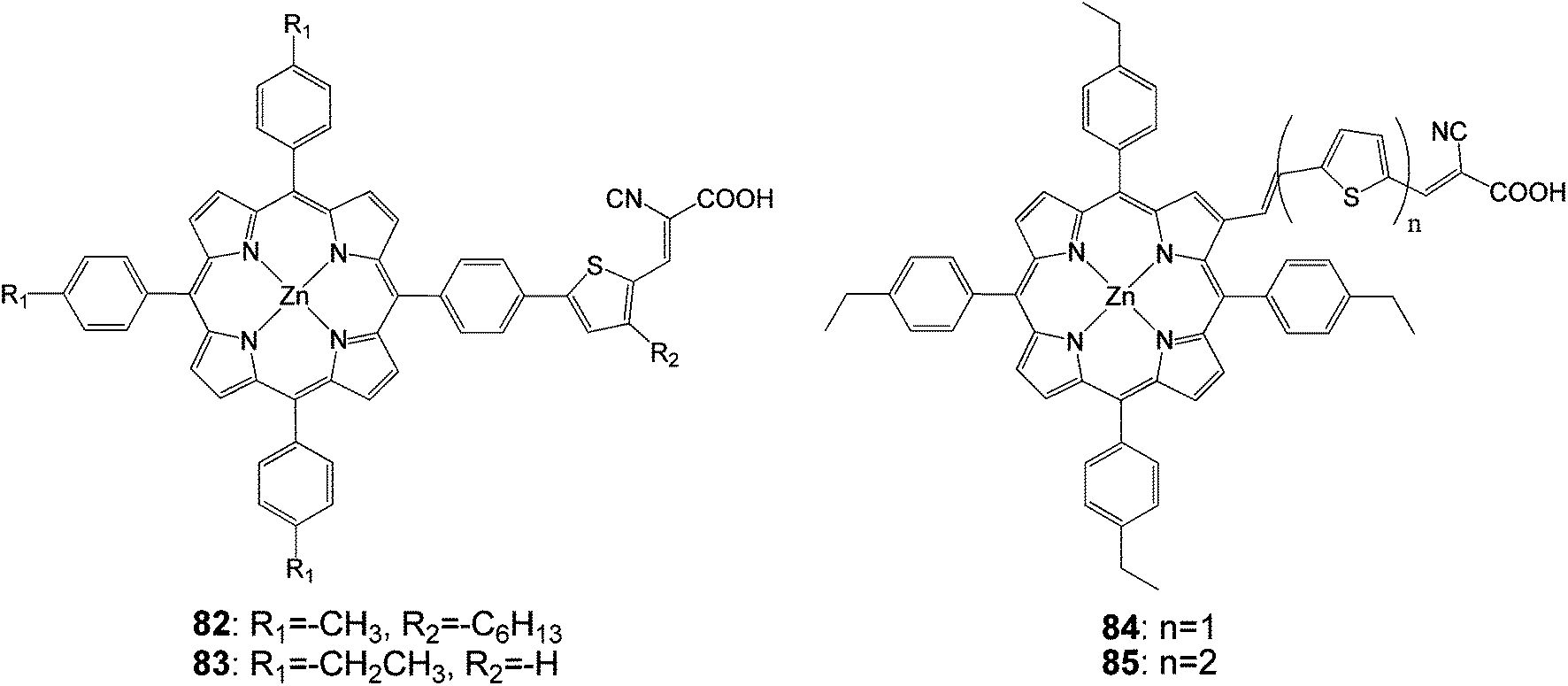
Research by Liu et al. was focused on adding thiophenyl groups to the para-position of meso-phenyl groups with and without a specific binding functionality (i.e. –COOH). Dye 82 (Fig. 25) gave an efficiency of 5.1% (Jsc = 12.9 mA cm−2, Voc = 0.60 V and FF = 0.66) when DMF was used as the adsorption solvent.93
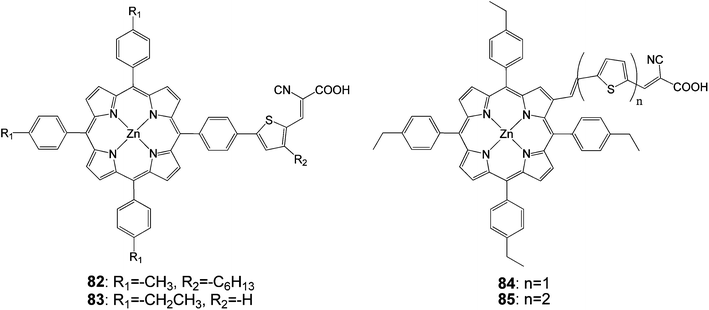 |
| | Fig. 25 Molecular structures of porphyrins 82–85. | |
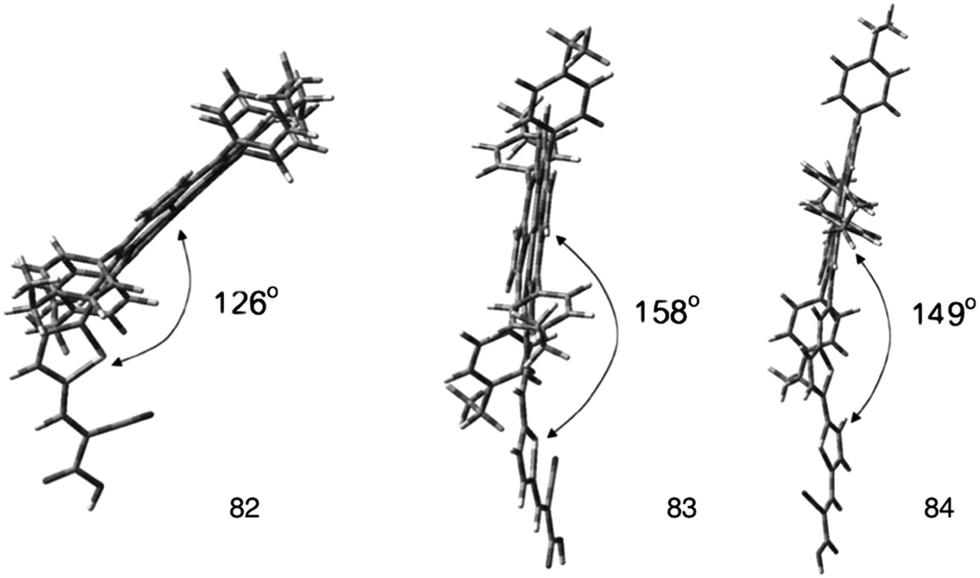
In order to explore the nature of the linker between the porphyrin and the anchoring group, β-substituted porphyrins 83–85 (Fig. 25) containing thiophene linker and ethyl group on meso-phenyl group were synthesized and studied their photoelectronic properties (Table 13).94 The results of this study showed that 84 and 85 had a tendency to undergo aggregation due to their almost planar structure (Fig. 26), which resulted to the reduced cell efficiency. However, the bent structure of 83 was able to suppress the aggregation. The low binding ability of 83 could be explained due to the shorter length of linker and bent structure. Porphyrin ring plane of 83 was largely bent from the linker group, leading to steric hindrance between porphyrin moiety and surface of TiO2.
Table 13 Photovoltaic performances of DSSC using 83–85
| Dye |
Jsc, mA cm−2 |
Voc, V |
FF |
n, % |
| 83 |
1.6 |
0.49 |
0.67 |
0.5 |
| 84 |
11.8 |
0.60 |
0.64 |
3.2 |
| 85 |
13.0 |
0.61 |
0.66 |
4.0 |
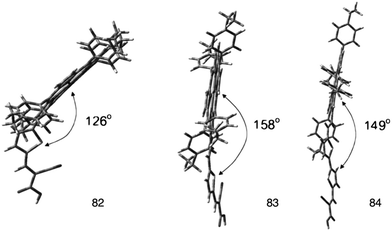 |
| | Fig. 26 Optimized structures of dyes 82–84. Adapted from ref. 94. | |
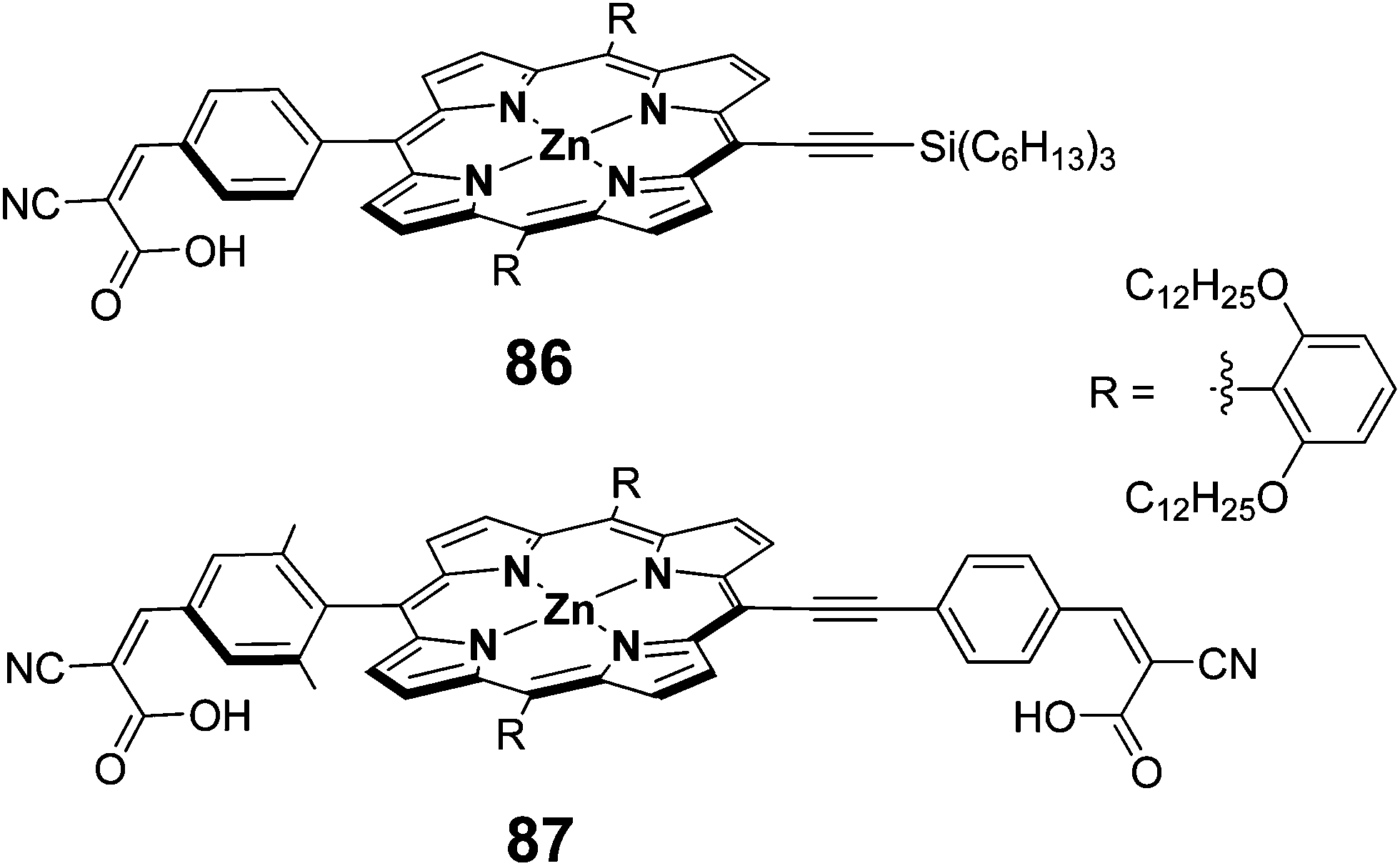
To improve the absorption profile of the porphyrin dyes towards longer wavelength region, it was reported that π-extended porphyrins27 and accessory pigment attached porphyrin95 that enhances their absorption profile in red region of the solar spectrum. Additionally, using four anchoring groups instead of one anchoring unit, improves the performance of the DSSCs.55 Hupp et al. have employed a π-extended porphyrin possessing two anchoring units 87 as sensitizer and achieved a superior PCE of 5.5% (Jsc = 11.3 mA cm−2, Voc = 0.68 V and FF = 0.70) compared to 86 (3.5%, Jsc = 7.5 mA cm−2, Voc = 0.66 V and FF = 0.69) sensitized solar cell (Fig. 27).96
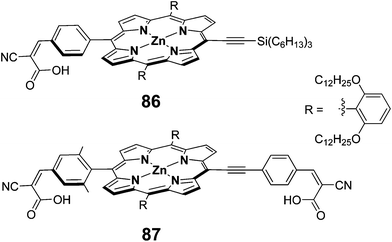 |
| | Fig. 27 Molecular structures of compounds 86 and 87. | |
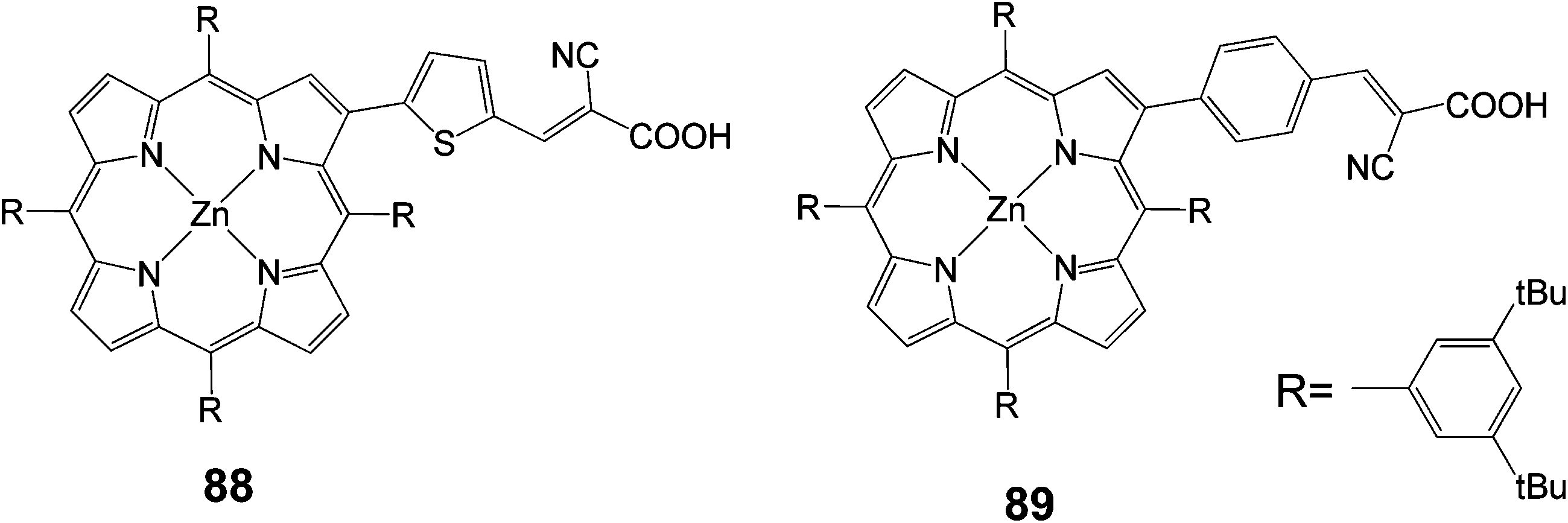
New β-pyrrolic functionalized porphyrins with donor–p–acceptor character were synthesized and characterized as dye sensitizers for DSSCs (Fig. 28).97 Two types of p-conjugated spacers, namely benzene and thiophene rings, with cyanoacrylic acid as an acceptor were linked to the porphyrin ring at the β-pyrrolic position. These porphyrins showed high thermal and electrochemical stability. As sensitizers, the porphyrin dye bearing a thiophene ring as the p-conjugated spacer 88 gave better cell performance with an overall conversion efficiency of 1.94% (Jsc = 4.57 mA cm−2, Voc = 0.59 V and FF = 0.59), compared to a 1.13% (Jsc = 2.62 mA cm−2, Voc = 0.59 V and FF = 0.57) for dye 89.
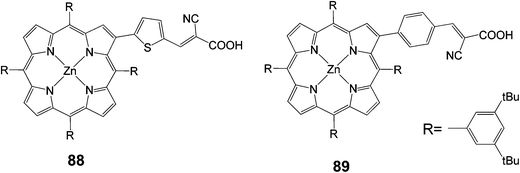 |
| | Fig. 28 Molecular structures of porphyrins 88 and 89. | |
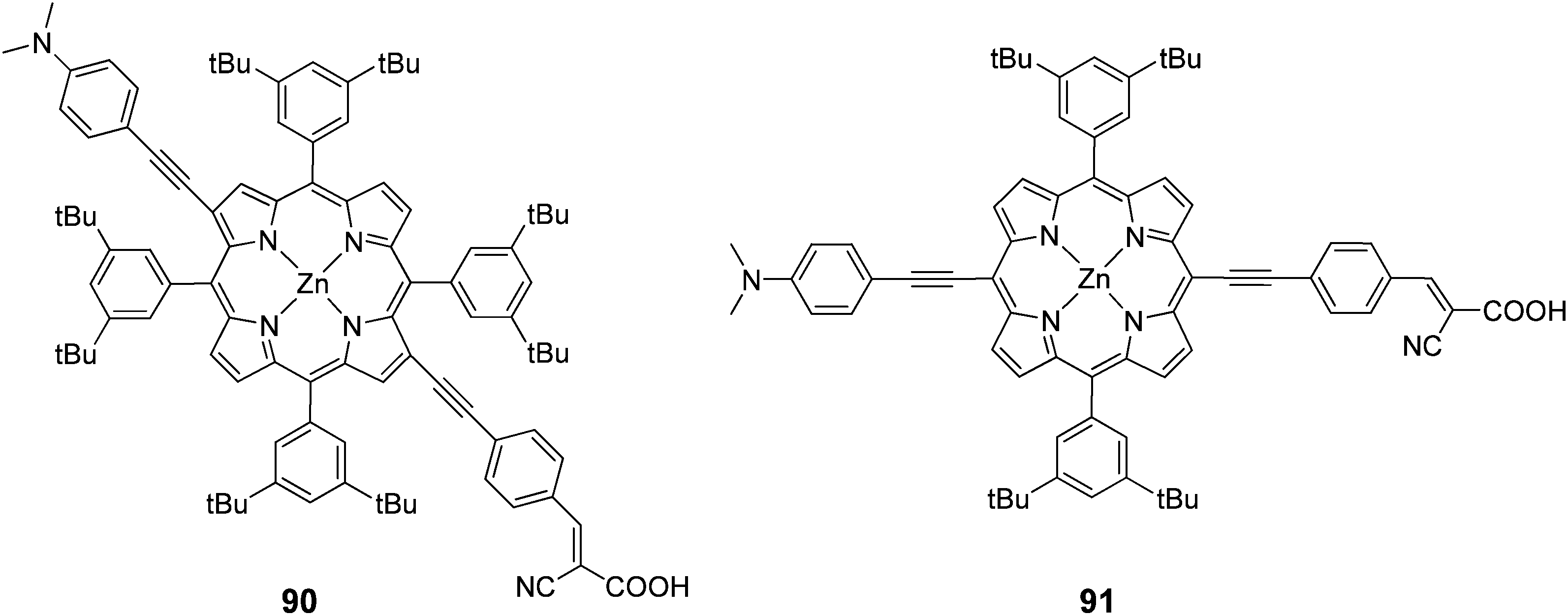
In another study porphyrins were synthesized with a push–pull orientation bearing cyanoacrylic acid in the β 90 and meso positions 91 of the porphyrin ring (Fig. 29).90 The “pull” ethynylphenyl substituent bearing the cyanoacrylic acid group in the β position had a higher efficiency 4.7% (Jsc = 14.0 mA cm−2, Voc = 0.59 V and FF = 0.58) over 4.2% (Jsc = 13.3 mA cm−2, Voc = 0.52 V and FF = 0.61). In the same study it was observed that the critical factor in determining injection efficiency in β-substituted DSSCs was the chemical nature of the anchoring group, with the cyanoacrylic acid group exhibiting a better performance than the carboxylic group of a benzoic acid moiety, confirming the findings of another report.98
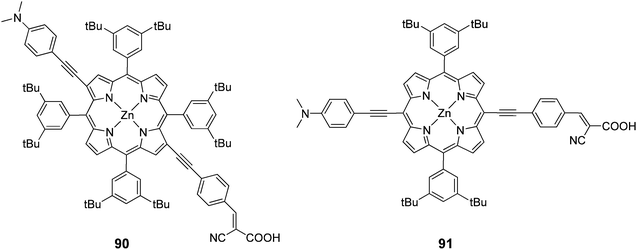 |
| | Fig. 29 Molecular structures of dyes 90 and 91. | |
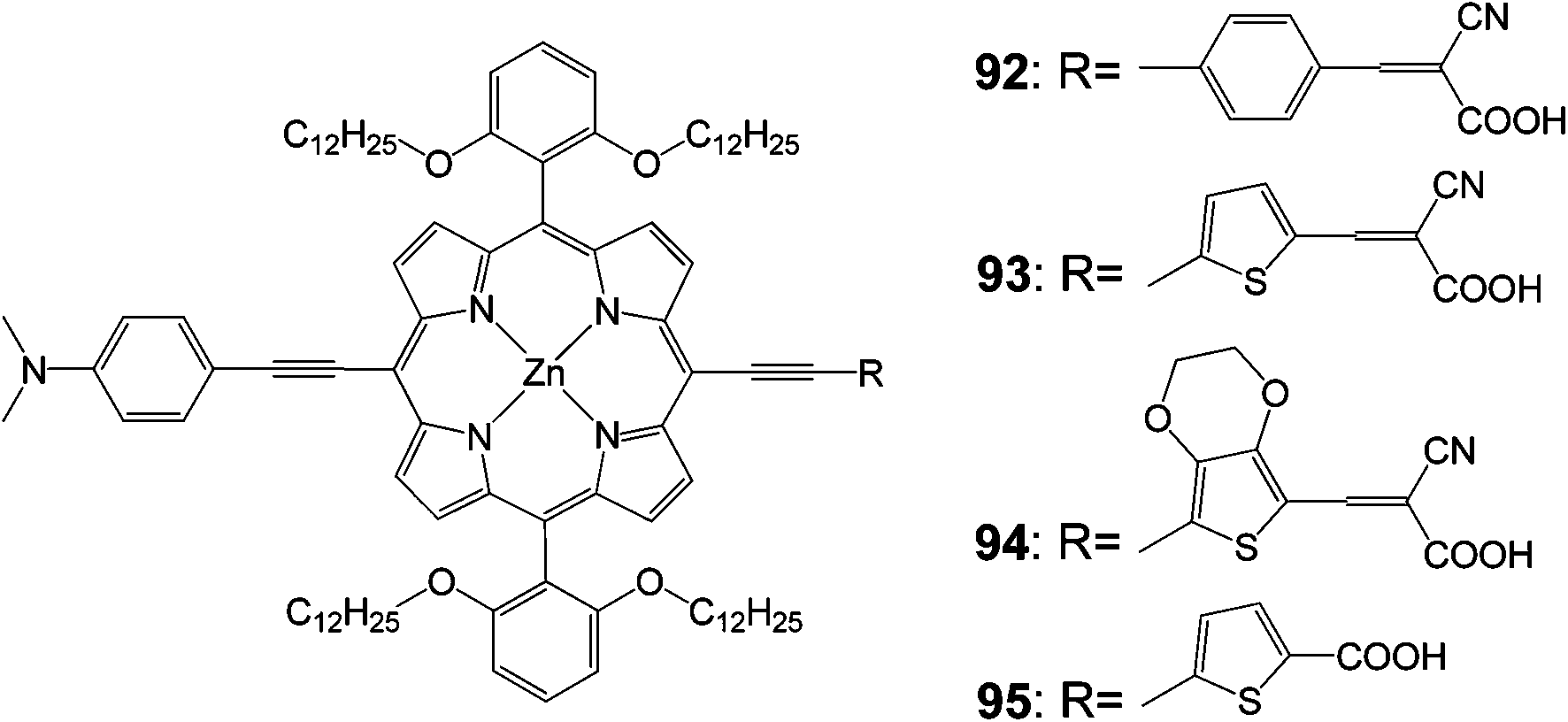
In another work, three push–pull structured new porphyrin dyes with various linkers (the 92 dye with 4-ethynylbenzene, the 93 dye with 2-ethynylthiophene, and the 94 dye with 5-ethynyl-2,3-dihydrothieno[3,4-b][1,4]dioxine) were synthesized in order to understand the influence of substitution of the p-linker with various hetero-aromatic groups (Fig. 30) on the light-harvesting ability of the sensitizers.99 These push–pull structured porphyrin sensitizers worked efficiently in DSSC devices, a detailed examination with impedance and transient photovoltage decay measurements revealed that a fast recombination process took place at the dye-sensitizer TiO2–electrolyte interface due to the introduction of cyanoacrylic acid as an anchoring group. Based on these studies the 95 porphyrin with a rigid π-linker feature structure (5-ethynylthiophene-2-carboxylic acid) was further designed and synthesized, which attained an improved PCE of 9.53% (Table 14).
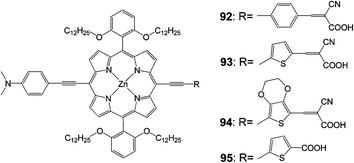 |
| | Fig. 30 Molecular structures of compounds 92–95. | |
Table 14 Photovoltaic performances of DSSC using 92–95
| Dye |
Jsc, mA cm−2 |
Voc, V |
FF |
n, % |
| 92 |
15.70 |
0.648 |
0.70 |
7.12 |
| 93 |
15.36 |
0.685 |
0.70 |
7.37 |
| 94 |
16.35 |
0.657 |
0.71 |
7.63 |
| 95 |
17.65 |
0.75 |
0.72 |
9.53 |
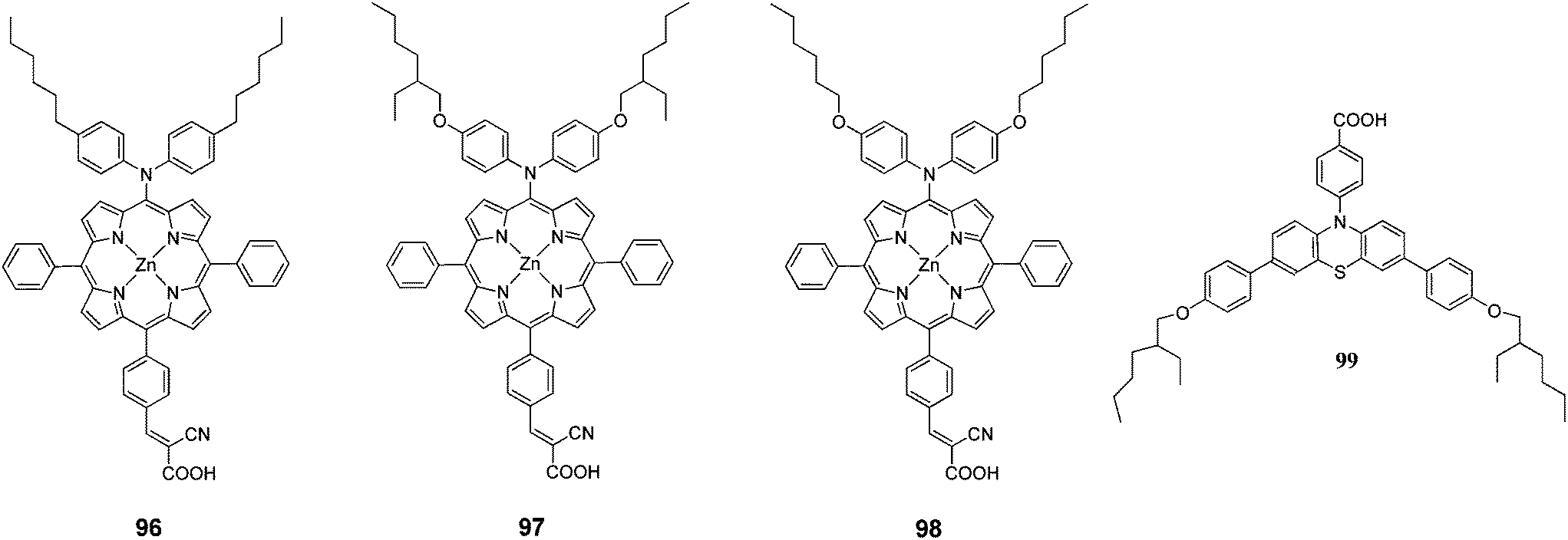
A series of push–pull structured (D–π–A) porphyrin dyes with an electron donating group attached at the meso position (96–98) were designed by Kim et al.100 for the use of sensitizers for DSSCs (Fig. 31). The absorption bands of these porphyrin dyes were broadened and red shifted upon introduction of alkoxy chains to the electron donating groups at meso position opposite to the anchoring cyanoacrylic acid group. Among the sensitizers, the highest PCE of DSSC fabricated with 98 was 4.7% (Jsc = 11.8 mA cm−2, Voc = 0.593 V and FF = 0.651) and further improved up to 7.6% (Jsc = 15.9 mA cm−2, Voc = 0.68 V and FF = 0.703) when 99 used as coadsorbent (Table 15).
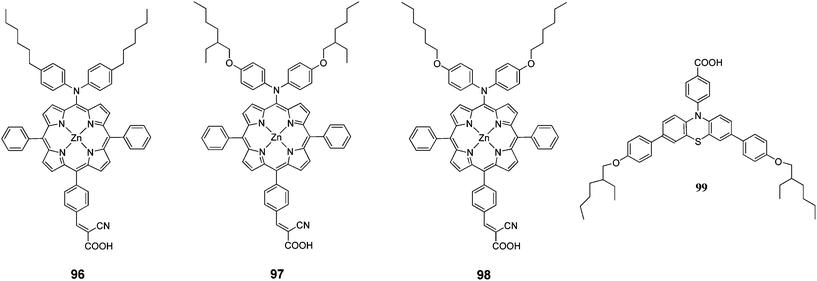 |
| | Fig. 31 Molecular structure of compounds 96–98 and coadsorbent 99. | |
Table 15 Photovoltaic performances of DSSC using 96–98
| Dye |
Jsc, mA cm−2 |
Voc, V |
FF |
n, % |
| Compound 99 was used as the co-adsorbent. |
| 96 |
4.9 (10.9)a |
0.546 (0.593) |
0.74 (0.77)a |
2.0 (5.0)a |
| 97 |
9.6 (13.4)a |
0.548 (0.632)a |
0.69 (0.72)a |
3.6 (6.1)a |
| 98 |
11.8 (15.9)a |
0.617 (0.678)a |
0.65 (0.70) |
4.7 (7.6)a |
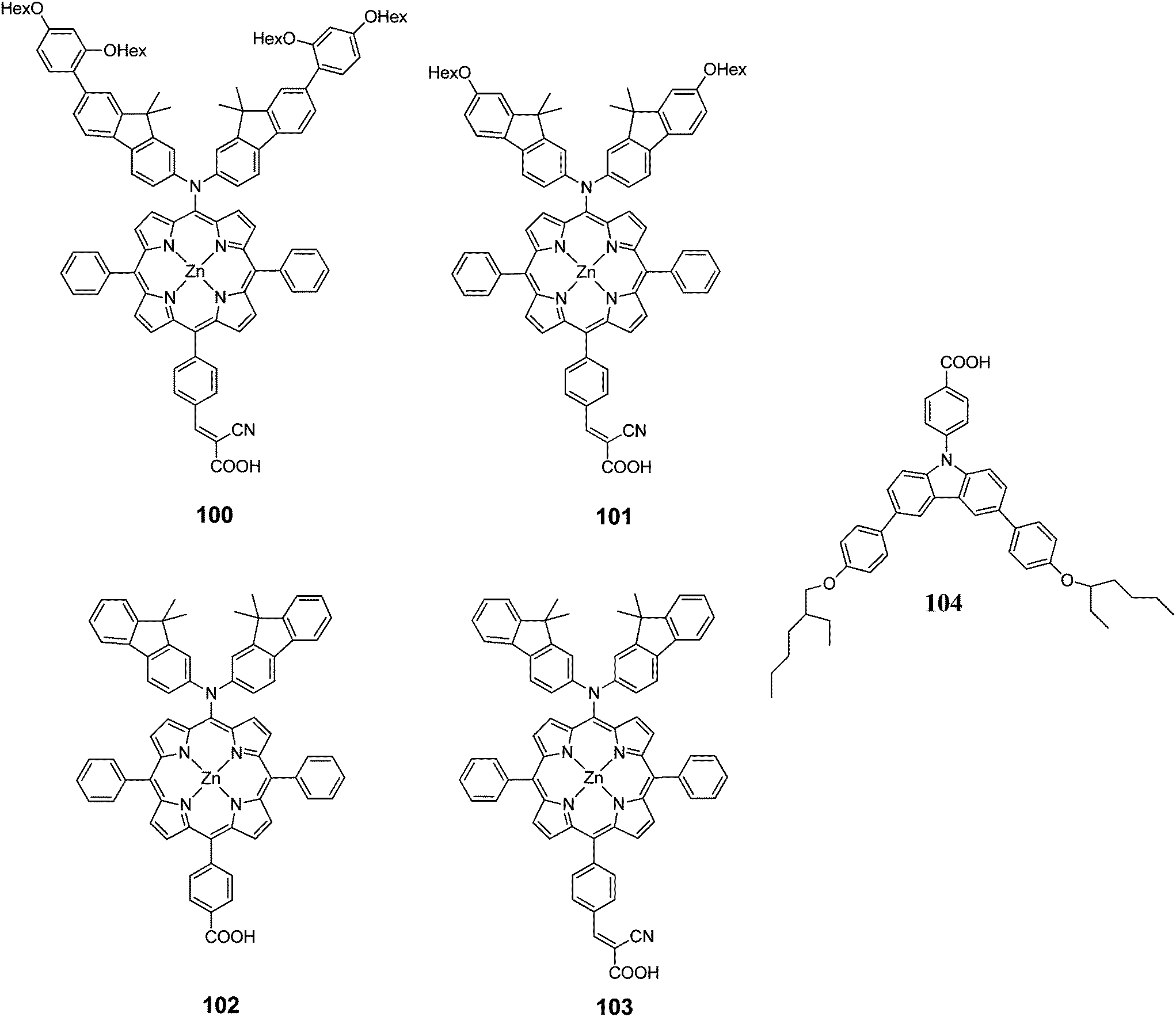
Kim et al. designed two Zn(II)-porphyrin sensitizers (100 and 101 with a bulky alkoxy group in order to retard the charge recombination (Fig. 32).101 The DSSCs sensitized 100 and 101 showed a PCE of 4.88% (Jsc = 10.92%, Voc = 0.63% and FF = 0.71) and 4.53% (Jsc = 10.36%, Voc = 0.60% and FF = 0.73), respectively. Moreover, when 104 incorporated into the dye solution as coadsorbent, the PCE has been enhanced significantly up to 8.46% (Jsc = 15.39 mA cm−2, Voc = 0.74 V, FF = 0.74) for 100 and 7.02% (Jsc = 13.72 mA cm−2, Voc = 0.686 V, FF = 0.75) for 101 (Table 16).
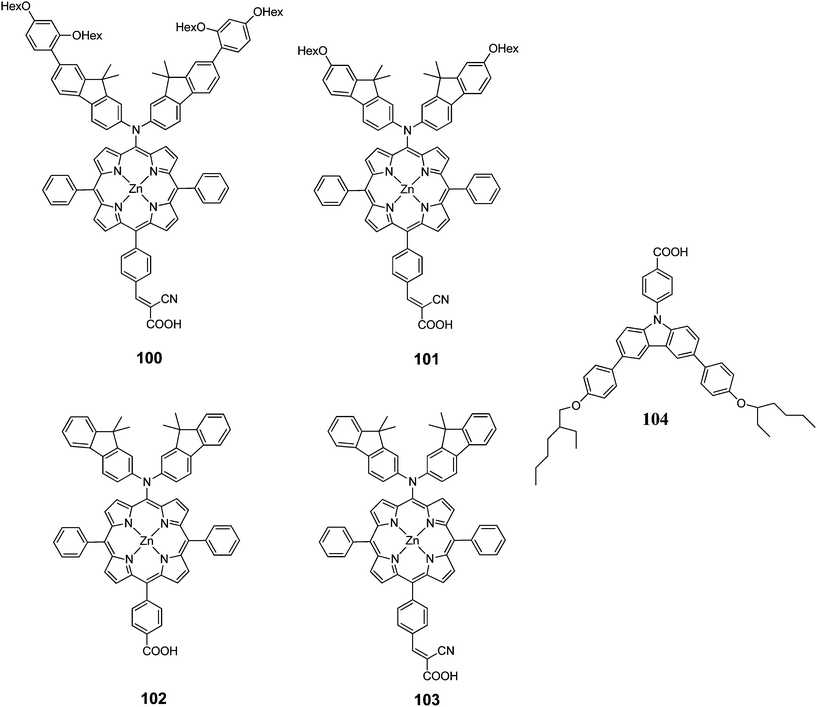 |
| | Fig. 32 Molecular structure of dyes 100–103 and coadsorbent 104. | |
Table 16 Photovoltaic performances of DSSC using 100–103
| Dye |
Jsc, mA cm−2 |
Voc, V |
FF |
n, % |
| Compound 104 was used as the co-adsorbent. |
| 100 |
10.92 (15.39)a |
0.63 (0.74) |
0.71 (0.74)a |
4.88 (8.46)a |
| 101 |
10.36 (13.72)a |
0.60 (0.686)a |
0.73 (0.75)a |
4.53 (7.02)a |
| 102 |
7.37 (15.30)a |
0.56 (0.67)a |
0.77 (0.71) |
3.16 (7.22)a |
| 103 |
10.04 (14.59)a |
0.58 (0.63)a |
0.75 (0.72) |
4.37 (6.64)a |
The same research group also synthesized two D–π–A structured Zn(II) porphyrin dyes 102 and 103 containing an electron donating bis(3,3-dimethylfluorenyl)amine moiety at the meso position and different carboxylic acid and cyanoacrylic acid at the opposite meso position linked through a phenylene π-conjugation bridge.33 The DSSCs sensitized with 102 and 103 showed PCE of 3.16% (Jsc = 7.37 mA cm−2, Voc = 0.56 and FF = 0.77) and 4.37% (Jsc = 10.04 mA cm−2, Voc = 0.58 V and FF = 0.75), respectively. The PCE of 102 sensitized was improved up to 7.22% (Jsc = 15.30 mA cm−2, Voc = 0.67 V and FF = 0.71) when the 0.1 mM 104 was used as coadsorbent (Table 16). These results clearly demonstrate that efficient intra-molecular charge separation of D–π–A Zn(II) porphyrin can by induced by inserting a strongly electron accepting cynanoacrylic acid substituents, resulting an relatively high overall PCE.
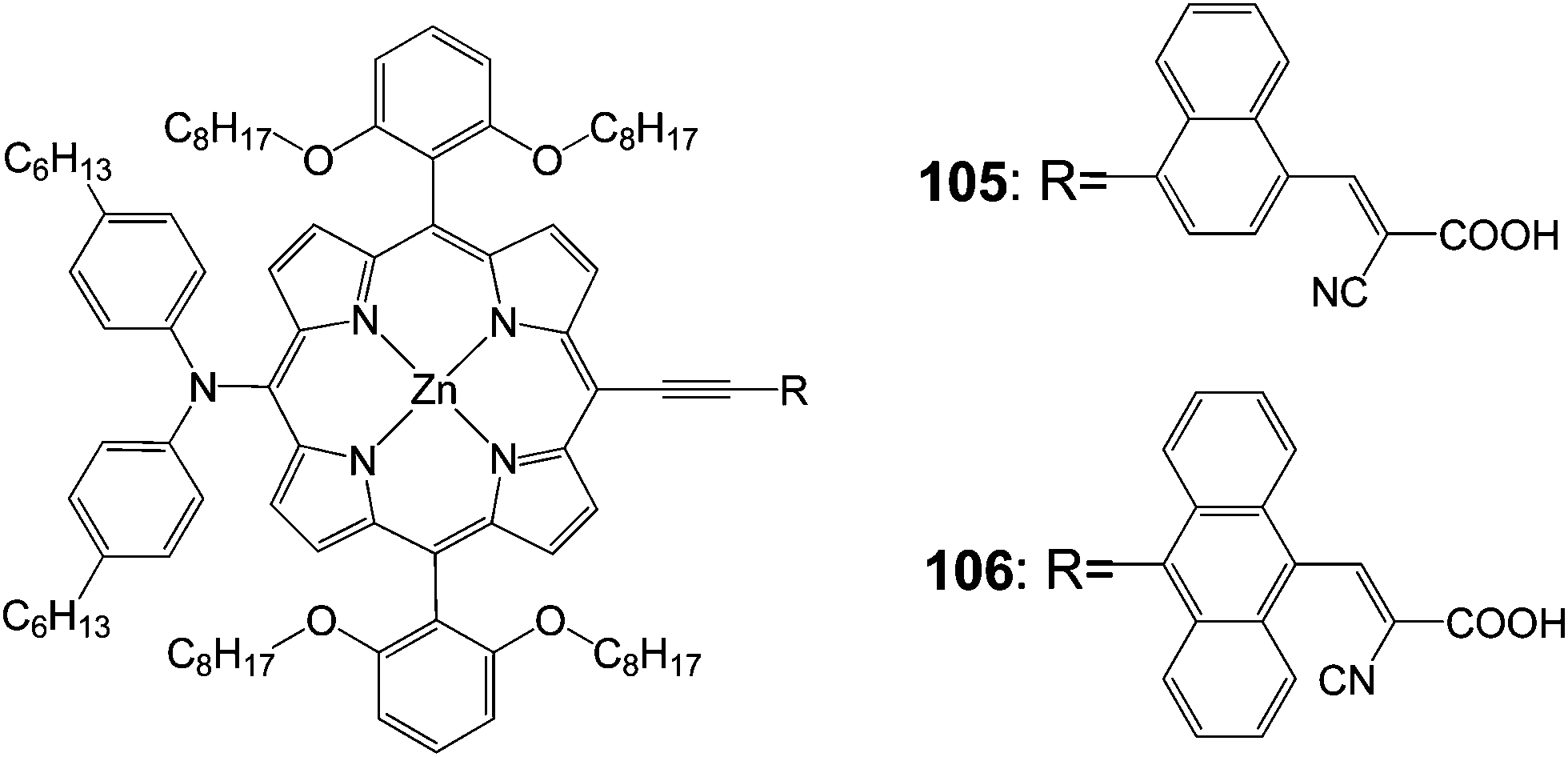
Diau and Yeh102 synthesized p-extended porphyrin dye105 and 106 with long alkoxyl chains at the ortho positions and cyanoacrylic acid as an anchoring group (Fig. 33). The efficiencies of both compounds were quite poor 5.8% (Jsc = 11.30 mA cm−2, Voc = 0.69 V and FF = 0.74) for 105 and 3.6% (Jsc = 7.17 mA cm−2, Voc = 0.67 V and FF = 0.75) for 106. This was due to the floppy structural nature and limited molecular rigidity of the cyanoacrylic acid anchor, which might lead the porphyrins to tilt down the TiO2 surface for an efficient charge recombination to occur.103,104
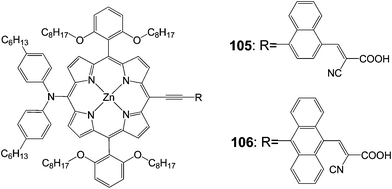 |
| | Fig. 33 Molecular structure of dyes 105 and 106. | |
2.3 Fused porphyrins
One issue with porphyrins as sensitizers in DSSCs is their limited absorptivity in the red and near infrared regions of the solar spectrum. With meso-tetraphenylporphyrins the phenyl moieties are typically considered perpendicular to the plane of the macrocycle and therefore are not in conjugation with the porphyrin system. One design strategy for increasing the light absorption of porphyrin sensitizers is to lock the meso-phenyl ring into the plane of the macrocycle in order to improve the conjugation between the porphyrin and the phenyl rings. This strategy has the added benefit of increasing the molecular asymmetry of the molecule. The combination of added conjugation and increased molecular asymmetry manifests itself as a red shift in the absorption spectrum of the molecule. The following examples synthesized towards this strategy.
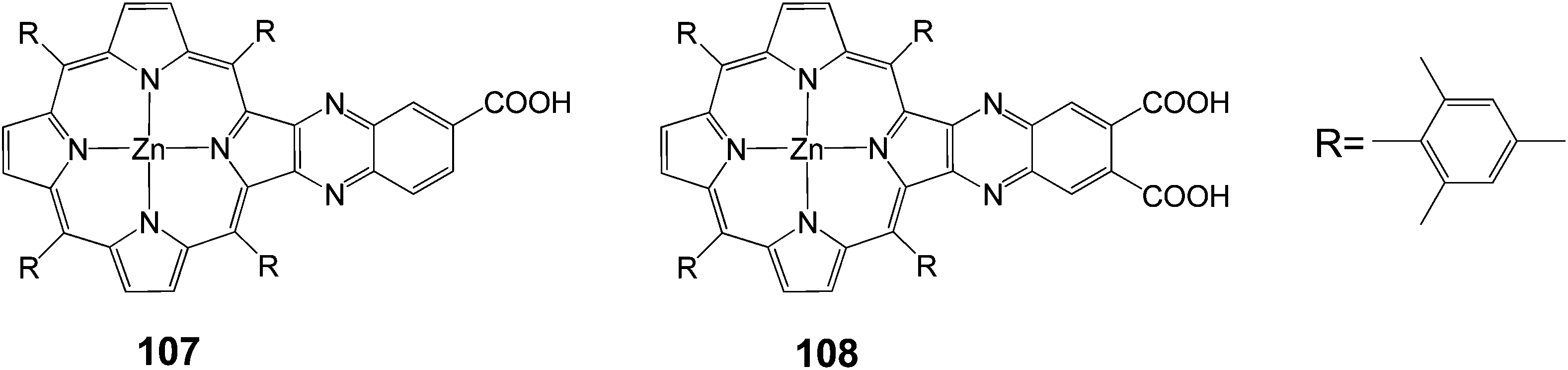
One way to improve the light harvesting property of porphyrin sensitizers is to introduce the unsymmetrical π-elongation in the porphyrins.27,62,105–110 Imahori and coworkers reported novel β-linked porphyrins 107 and 108 (Fig. 34) with carboxyquinoxalino or dicarboxyquinoxalino moieties respectively.106 Compound 107 gave a solar energy conversion efficiency of 5.2% (Jsc = 11.2 mA cm−2, Voc = 0.72 V and FF = 0.68) in a standard DSSC, while the dicarboxy derivative 108 gave 4% (Jsc = 9.3 mA cm−2, Voc = 0.67 V and FF = 0.64) efficiency. The superior performance of the 107-sensitized solar cell to the 108-sensitized one was originated from both the more favorable electron injection and charge collection efficiency.
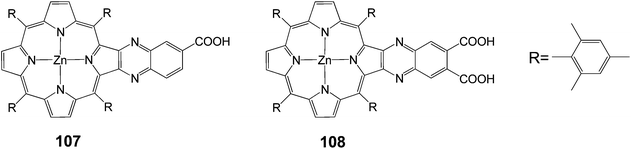 |
| | Fig. 34 Molecular structure of dyes 107 and 108. | |
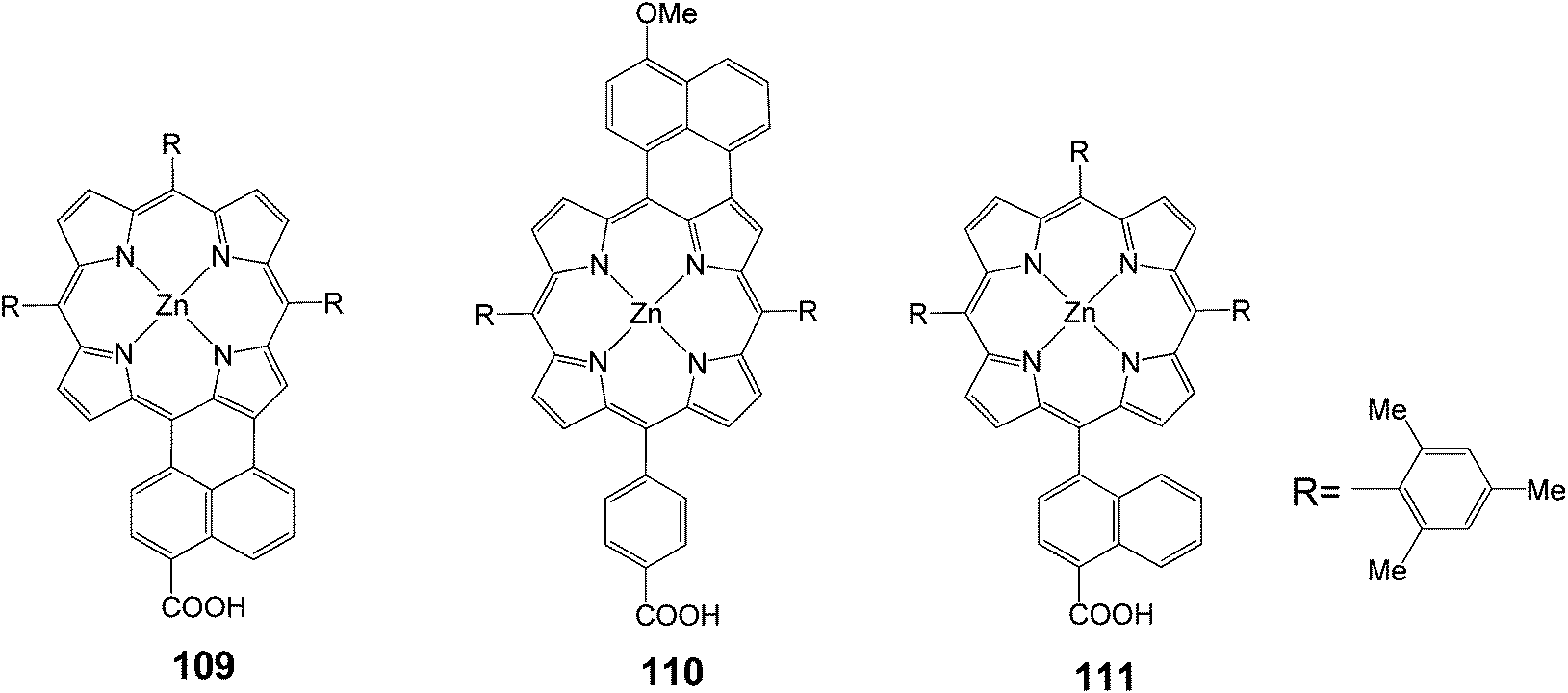
The same group synthesized unsymmetrical π-elongated porphyrins (Fig. 35), in which the naphthyl moiety was fused to the porphyrin core at the naphthyl bridge with a carboxyl group 109 or at the opposite side of the phenyl bridge with a carboxyl group 110.109 The 109 and 110-sensitized TiO2 solar cells showed the power conversion efficiencies (n) of 4.1% (Jsc = 10.6 mA cm−2, Voc = 0.62 V and FF = 0.62) and 1.1% (Jsc = 3.6 mA cm−2, Voc = 0.53 V and FF = 0.58), respectively, under standard AM1.5 conditions. The n value of the 109 cell was improved by 50% compared to the reference cell using unfused porphyrin 111. The 109-sensitized cell revealed high IPCE values of up to 55%, extending the response of photocurrent generation close to 800 nm. Thus, the improved photocurrent generation of the 109-sensitized cell relative to the 111-sensitized reference cell was responsible for the remarkable difference in the n values. The n value of the 110 cell was much lower than that of the 109 cell. DFT calculations disclosed that there were significant electron densities on the carboxyl group in the LUMO of 109, whereas there were little electron densities on the carboxyl group in the LUMO of 110. Accordingly, the larger electronic coupling between the porphyrin and the TiO2 surface in the 109-sensitized cell may be responsible for the high cell performance, due to the efficient electron injection from the porphyrin excited singlet state to the conduction band of the TiO2 electrodes. To further improve the cell performance, porphyrin 4, possessing different light-harvesting properties, was co-adsorbed with 109 onto a TiO2 electrode. Under the optimized conditions, the cosensitized cell yielded IPCE value of 86%, short circuit photocurrent density of 11.7 mA cm−2, open-circuit voltage of 0.67 V, fill factor (FF) of 0.64, and n of 5.0% under standard AM1.5 conditions (Table 17).
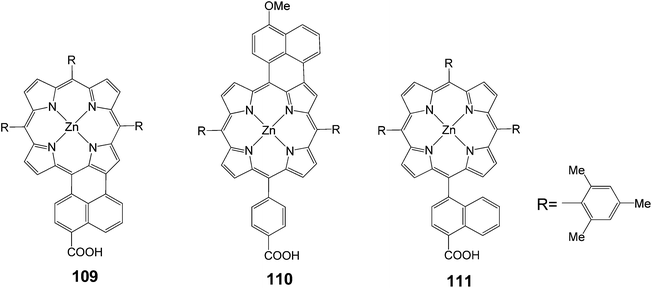 |
| | Fig. 35 Molecular structure of porphyrins 109–111. | |
Table 17 Photovoltaic performances of DSSC using 109–111
| Dye |
Jsc, mA cm−2 |
Voc, mV |
FF |
n, % |
| 109 |
10.6 |
620 |
0.62 |
4.1 |
| 110 |
3.6 |
530 |
0.58 |
1.1 |
| 111 |
6.7 |
610 |
0.68 |
2.8 |
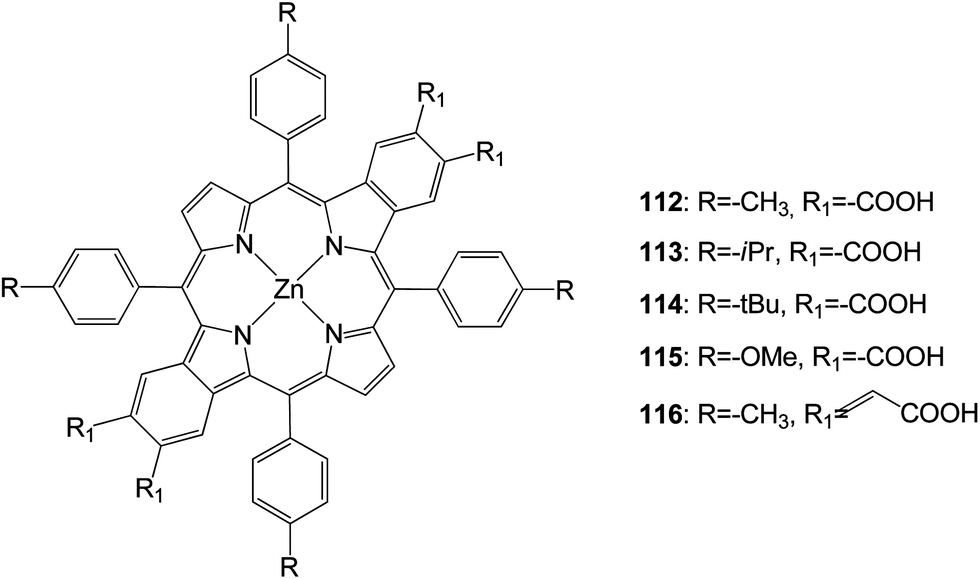
New opp-dibenzoporphyrins (Fig. 36) were synthesized through a Pd0-catalyzed cascade reaction.111 These opp-dibenzoporphyrins (112–116) were evaluated for the first time as light harvesters for dye-sensitized solar cells. These porphyrins displayed moderate solar energy-to-electricity conversion efficiencies of 1.54–3.14%. The installation of a more bulky group on the meso-phenyl rings of the porphyrin did not help to enhance the solar-energy conversion, presumably owing to lower adsorption of the porphyrin dye on the TiO2 surface. The incorporation of conjugated carboxylic acid linkers onto the porphyrin considerably broadened and red-shifted the absorption bands of the porphyrin, thereby led to a very high Jsc value (12.89), (Table 18).
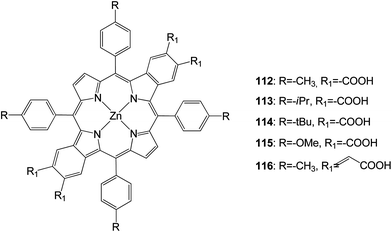 |
| | Fig. 36 Molecular structure of dyes 112–116. | |
Table 18 Photovoltaic performances of DSSC using 112–116
| Dye |
Jsc, mA cm−2 |
Voc, V |
FF |
n, % |
| 112 |
8.86 |
0.57 |
0.58 |
2.93 |
| 113 |
5.05 |
0.54 |
0.56 |
1.54 |
| 114 |
5.52 |
0.58 |
0.57 |
1.81 |
| 115 |
7.48 |
0.57 |
0.53 |
2.22 |
| 116 |
12.87 |
530 |
0.46 |
3.14 |
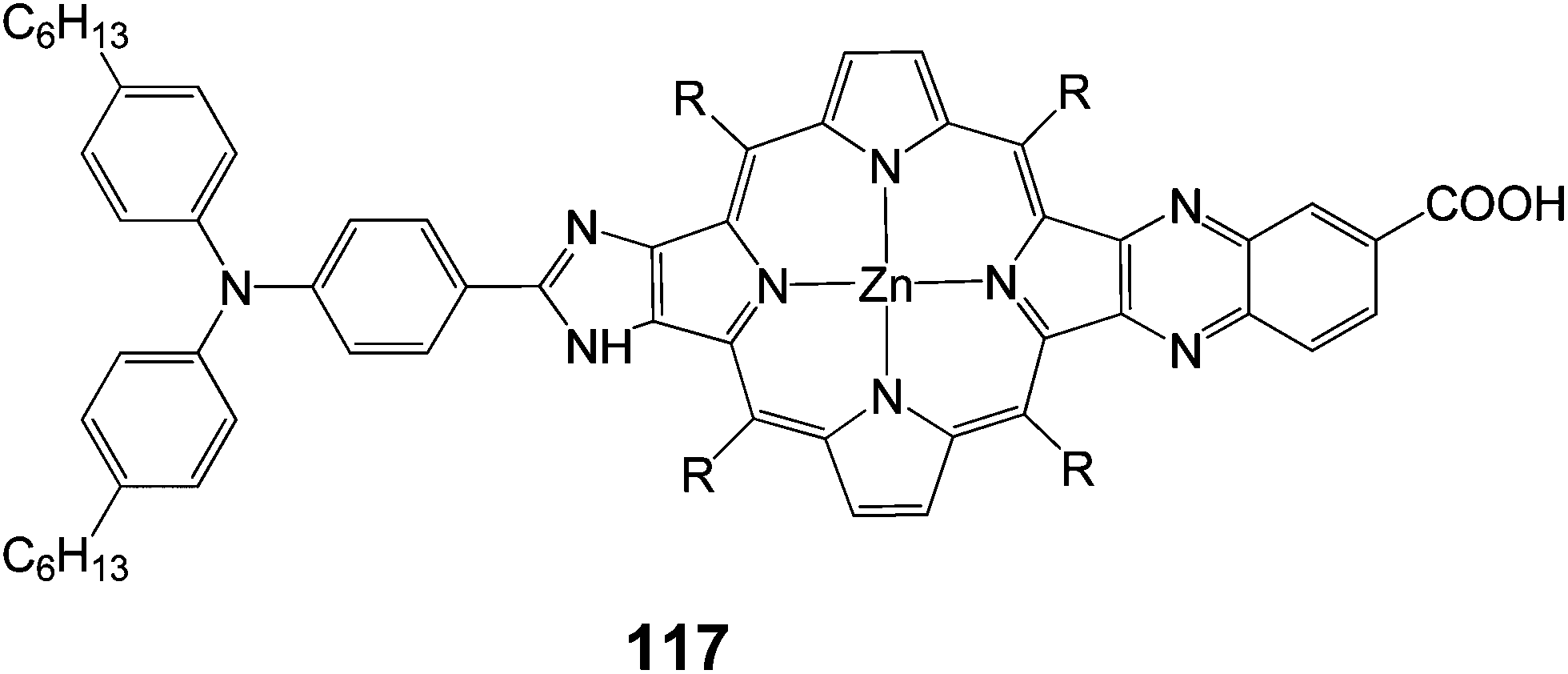
A push–pull porphyrin 117 was synthesized and studied with an electron donating triarylamino group at the β,β′-edge through a fused imidazole group and an electron withdrawing carboxyquinoxalino anchoring group at the opposite β,β′-edge (Fig. 37) and employed as sensitizer for DSSC.112 The 117-sensitized solar cell exhibited a relatively high PCE of 6.8% (Jsc = 13.9 mA cm−2, Voc = 0.68 V and FF = 0.71), which was larger than that of the 107-sensitized solar cell 6.3% (Jsc = 13.2 mA cm−2, Voc = 0.71 V and FF = 0.67) with chenodeoxycholic acid as co-absorbent, to reduce porphyrin aggregation on TiO2. The short-circuit current and FF of the 117-sensitized solar cell were larger than those of the 107-sensitized solar cell, whereas the open circuit potential of the 117-sensitized cell was smaller than that of the 107-sensitized cell, leading to an overall improved cell performance of 117.
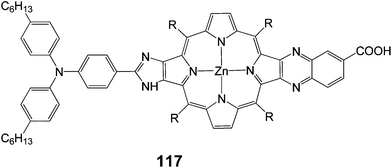 |
| | Fig. 37 Molecular structure of dye 117. | |
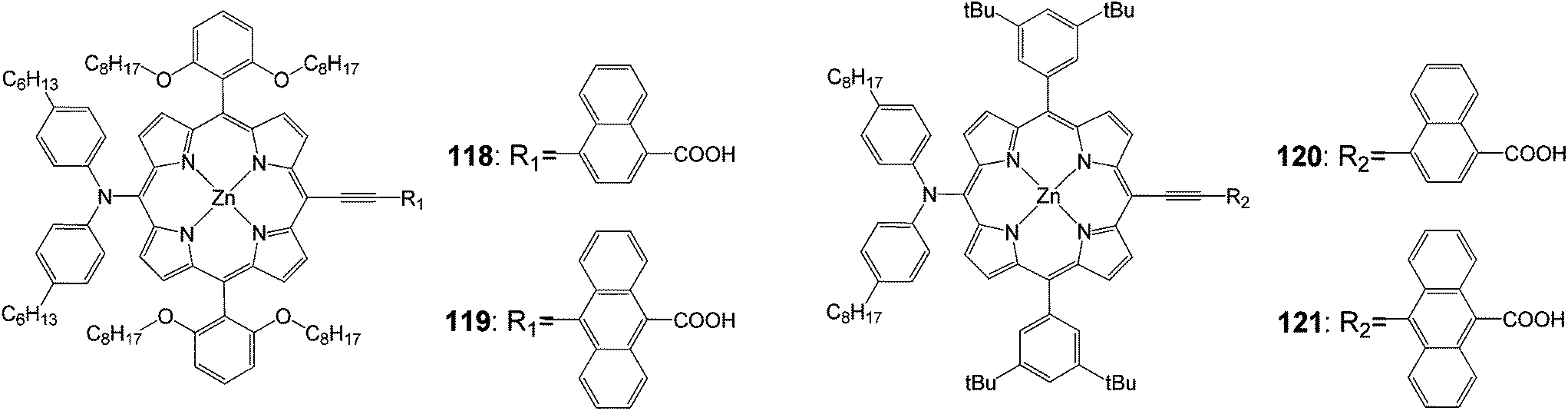
Yeh and coworkers102 synthesized p-extended porphyrin dyes 118 and 119 with long alkoxyl chains at the ortho positions of the meso phenyls, and meta di-tert-butylphenyl-substituted porphyrins for DSSCs; their optical, electrochemical and photovoltaic properties were investigated and compared with those of 120 and 121 (Fig. 38).
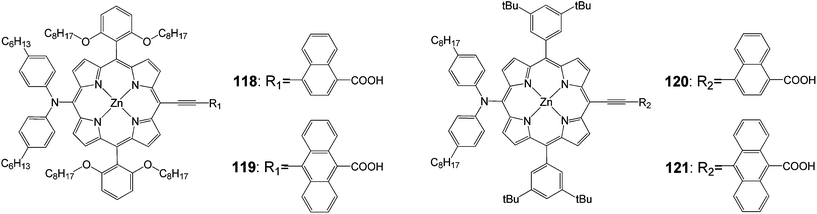 |
| | Fig. 38 Molecular structure of dye 118–121. | |
For the naphthalene-bridged porphyrins, the ortho-substituted long alkoxyl chains in the two meso-phenyls of 118 played a key role to prevent dye aggregation so that both Jsc and Voc were significantly enhanced for 118 than for 120. As a result, the PCE of 118 reached 8.04%, which was ∼11% higher than that of 120 (7.23%). The devices made of 121 were found to perform extremely poorly due to a serious problem of dye aggregation on the TiO2 surface that significantly reduced the efficiency of electron injection from the excited state of the dye molecule to the conduction band of TiO2.73 However, the ortho-substituted long alkoxyl chains in 119 exhibited dramatic effects to decrease the degree of dye aggregation so that the IPCE values of 119 were significantly improved by a factor of three over those of 121. As a result, Jsc of the 119 device was improved by more than a factor of two over that of the 121 device. Therefore, the device performance of 119 reached efficiency of 6.03%, which was remarkably improved by 135% compared with that of 121 (Table 19).
Table 19 Photocatalytic parameters under global AM1.5 irradiation for DSSCs absorbed on nanocrystalline TiO2 anodes
| Dye |
Jsc, mA cm−2 |
Voc, V |
FF |
n, % |
| 105 |
11.30 |
0.696 |
0.737 |
5.80 |
| 106 |
7.17 |
0.673 |
0.755 |
3.64 |
| 118 |
15.37 |
0.745 |
0.702 |
8.04 |
| 119 |
11.59 |
0.720 |
0.723 |
6.03 |
| 120 |
14.6 |
0.713 |
0.721 |
7.23 |
| 121 |
5.09 |
0.679 |
0.743 |
2.57 |
3. Phosphonic acids as anchoring groups
Another group that has been used as an anchoring group is the phosphonic acid. An interesting study investigated the effects of the anchoring group (carboxylate vs. phosphonate) in ruthenium-bipyridyl-based complexes on the photoelectrochemical behaviors when they were used as sensitizers in DSSCs.113 The surface binding on TiO2 and the overall cell performance were highly dependent on both the number and the kind of anchoring groups. The cell performance (or sensitization efficiency) of carboxylate-TiO2 systems was primarily governed by the surface binding mode and stability of carboxylate-complexes on TiO2, which was strongly influenced by the number of the carboxylate groups. As a result, the tetra-carboxylate TiO2 system showed the best cell performance despite the lowest visible light absorption because the most effective surface binding mode was allowed with this structure. On the contrary, the photoelectrochemical behaviors of phosponate-TiO2 systems were not influenced much by the surface binding properties. This could be attributed to the intrinsically stronger binding capability of the phosphonate group than the carboxylate linkage. Since the surface binding of phosphonate-complexes was strong enough even with only one phosphonate group, the presence of additional anchoring groups did not make a significant difference in the sensitizer adsorption and the electronic coupling between the sensitizer and TiO2. Therefore, the overall cell performance followed the order of the visible light absorptivity of phosphonate-complexes, which changed with the number of phosphonate groups.
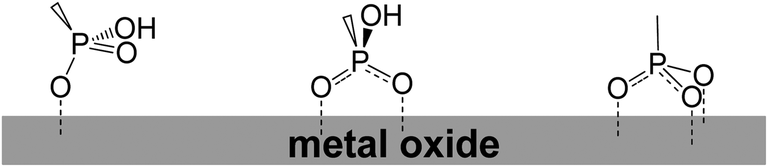
Phosphonic acid was placed at both meso and β-position of the porphyrin ring. For phosphonates there was evidence for both bi- and tridentate linkages, with bidentate theoretically determined to be the most stable on crystalline TiO2 lattices (Fig. 39).114–118
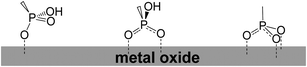 |
| | Fig. 39 Different binding modes of phosphonates on TiO2. | |
In an interesting study various porphyrins sensitizers were synthesized (Fig. 40) and the type and the position of the anchoring group were studied.43
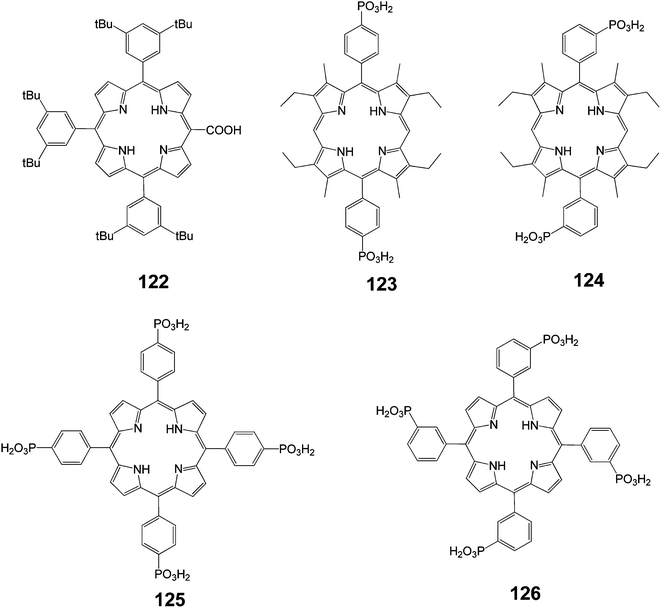 |
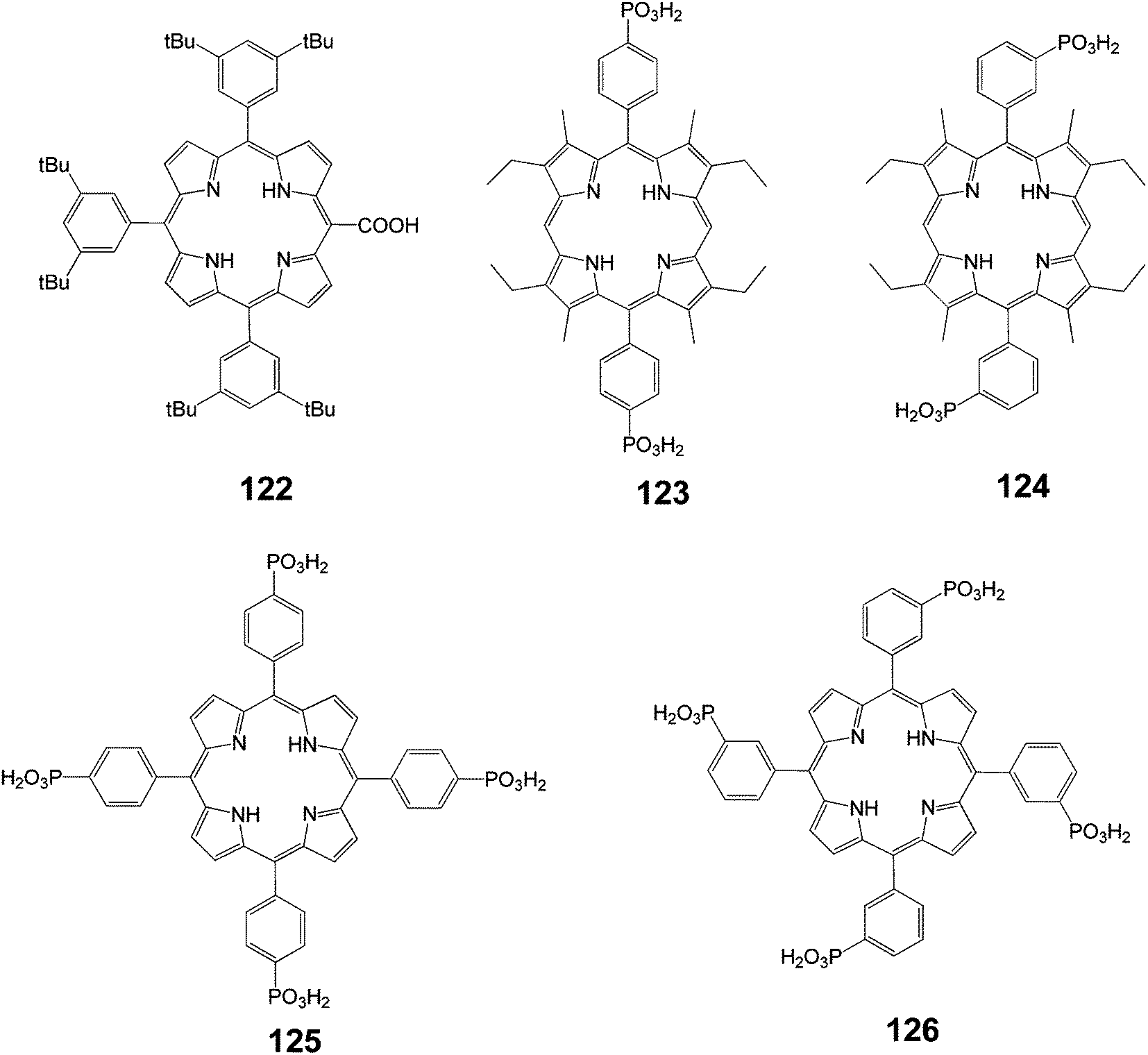
| | Fig. 40 Molecular structure of dyes 122–126. | |
In this study, it was shown that the nature of the anchoring group (phosphonic or carboxylic acids) had little impact on the photo-electrochemical performance of the cell. However, the substitution position of the anchoring group on the porphyrin strongly influenced the IPCE of the resulting cell. The results indicated that the electronic coupling of this type of dye with the d-band of the semiconductor was one of the key parameters in the design of efficient sensitizers.
Porphyrin 122 bears the COOH anchoring group directly on the p-aromatic core. This feature certainly allowed for a stronger electronic interaction with TiO2 compared to porphyrin 1, in which the remote COOH groups were electronically decoupled from the porphyrin macrocycle.
The sensitivity of the IPCE to the substitution position of the phosphonate anchoring groups in porphyrins 123 and 124 was also remarkable (Fig. 40). This can be interpreted as due to differences in the orientation and distance of the dye with respect to the TiO2 surface imposed by the directionality of the anchoring groups. The porphyrin that beard phosphonic acid groups in meta positions on the phenyl ring (dye 124) may lie closer to the TiO2 surface than that with para substituents (dye 123). Consequently, these variations may bring about significant differences in the magnitude of the electronic coupling. Activated electron transfer for dyes 124 and 125 can explain why the IPCE values of these sensitizers were the same and did not parallel the differences observed between dyes 123 and 124. The injection quantum yield of the former sensitizers might be limited by the lower excited state potentials. Finally, it was noted that replacement of the phosphonic with the carboxylic functions, as in the case of 125 and 1, did not seem to modify the dye photo-electrochemical performances. This could be explained by the fact that both dyes were electronically decoupled from TiO2 because the meso-aryl groups orient perpendicularly to the porphyrin macrocycle.
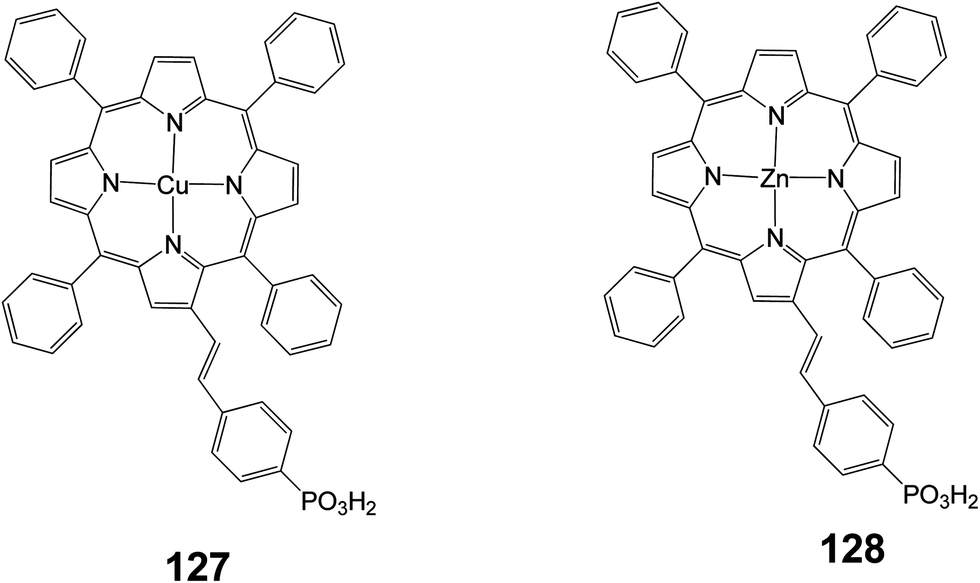
In another study two porphyrins (127 and 128) were synthesized bearing a phosphonic acid at the beta position of the porphyrin ring (Fig. 41) and were compared with the same porphyrins with carboxylic acid as anchoring groups.46 It was reported that porphyrin 128 showed much lower efficiency (0.89%) compared to the same porphyrin with a carboxylate anchoring group (4.11%), (Table 20).
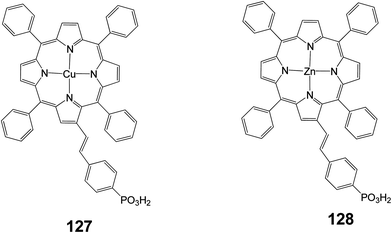 |
| | Fig. 41 Molecular structure of dyes 127 and 128. | |
Table 20 Photovoltaic performances of DSSC using 127 and 128
| Dye |
Jsc, mA cm−2 |
Voc, mV |
FF |
n, % |
| 127 |
1.1 |
561 |
0.67 |
0.41 |
| 128 |
2.05 |
580 |
0.75 |
0.89 |
4. Sulfonic acids as anchoring groups
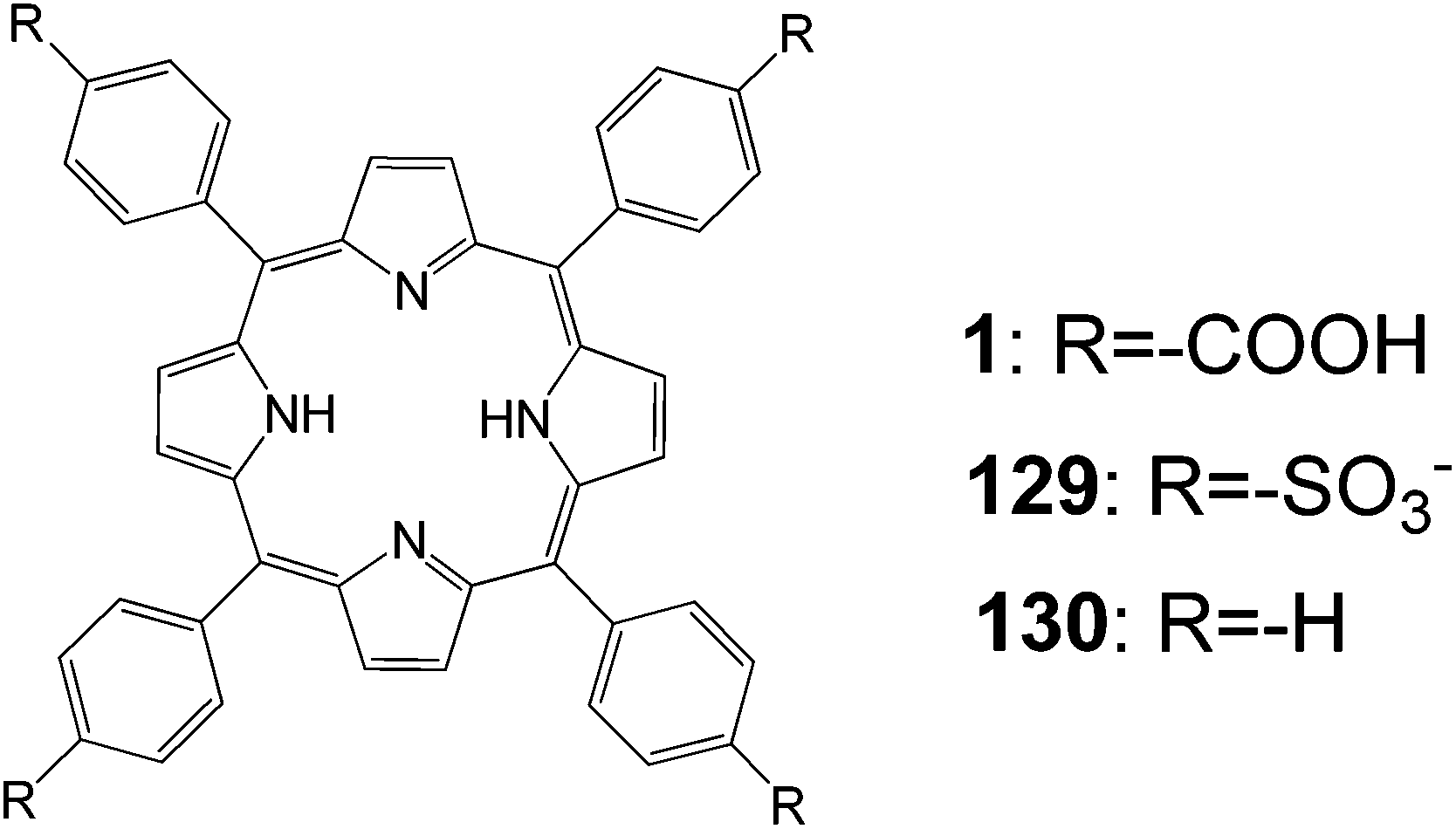
, A systematic study on the functional groups (including sulfonic acids) of porphyrin derivatives was carried out by various spectroscopic measurements to reveal the effects of these groups on the photochemical properties and conversion efficiency (Fig. 42).42 The adsorption behaviors of three kinds of porphyrins were different on the surface of the TiO2 electrode. An interaction between the porphyrin and the TiO2 surface was observed, and their strengths were quite different and decreased in the order 1 > 129 > 130. The ionization potentials of the porphyrins in the bulk and adsorbed onto the TiO2 electrode were determined, and a shift in the ionization potential between the two states was found. This approach provided a method to determine the energy level for a semiconductor sensitized by dye under the working conditions. The type of the functional groups on the dye significantly influenced the photocurrent and the conversion efficiency. These results indicated that the binding state between the dye and semiconductor was an important factor affecting the properties of the dye-sensitized solar cell.
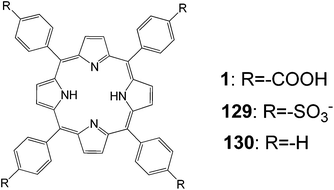 |
| | Fig. 42 Molecular structure of dyes 1, 129 and 130. | |
5. Pyridines as anchoring groups
A new type of push–pull dye sensitizer with a pyridine ring as an electron withdrawing anchoring group was developed.119,120 These dyes exhibited a higher short circuit photocurrent (Jsc) and PCE compared to their counterpart dyes having carboxyl groups. It was reported that the formation of coordinate bonds between the pyridine ring of the dye and the Lewis acid sites of the TiO2 surface led to efficient electron injection owing to good electron communication between them rather than the formation of an ester linkage between the dyes having a carboxyl anchoring group.
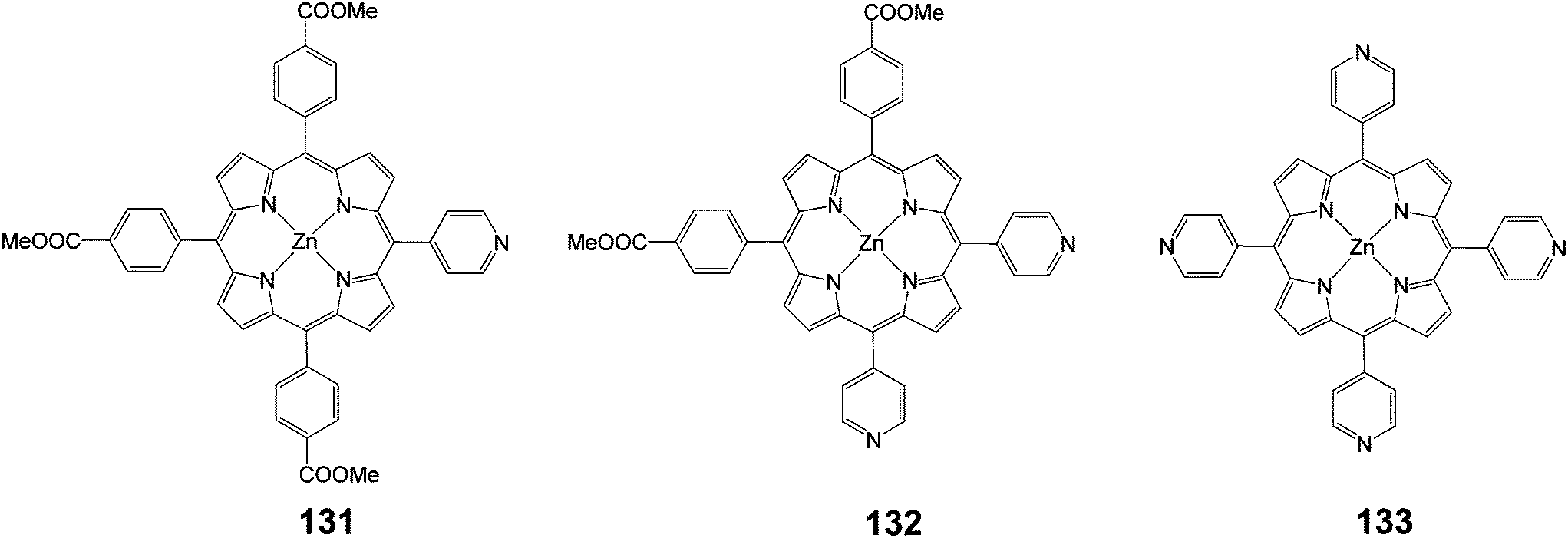
Three porphyrin dyes, 131–133 (Fig. 43), bearing one, two and four pyridyl groups, respectively, in the meso positions, acting as electron acceptor anchoring groups, were synthesized, characterized and investigated as sensitizers for the fabrication of DSSCs.121 The overall PCEs of DSSCs based on these dyes laid in the range of 2.46–3.9% using a 12 mm thick TiO2 photoanode. Porphyrin 132 achieved the maximum performance, which could be rationalized by the high dye loading, efficient electron injection, dye regeneration process and longer electron lifetime, as demonstrated by the electrochemical impedance spectroscopy (EIS) measurements (Table 21). The PCE of the DSSC of sensitizer 132 when the photoanode was treated with formic acid, showed an enhanced efficiency of 5.23% (Jsc = 12.04 mA cm−2, Voc = 0.68 V and FF = 0.64). This improvement, attributed to multifunctional properties such as higher dye uptake, reduced recombination process and enhanced charge collection efficiency.
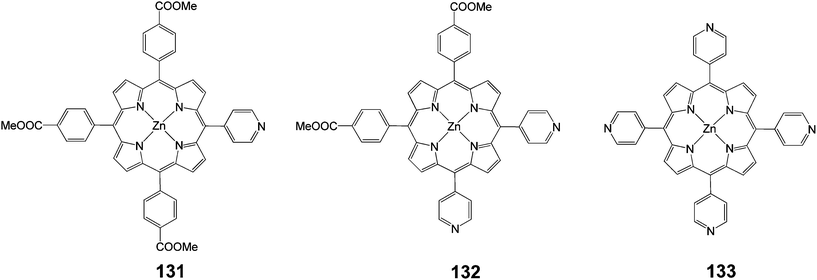 |
| | Fig. 43 Molecular structure of dyes 131–133. | |
Table 21 Photovoltaic performances of DSSC using 131–133
| Dye |
Jsc, mA cm−2 |
Voc, V |
FF |
n, % |
| 131 |
9.2 |
0.60 |
0.56 |
3.10 |
| 132 |
10.56 |
0.64 |
0.60 |
3.90 |
| 133 |
7.6 |
0.60 |
0.54 |
2.46 |
Deoxycholic acid (DCA) was also used as a co-adsorbent in order to prevent dye aggregation and it was found that the PCE improved up to 6.12% (Jsc = 12.86 mA cm−2, Voc = 0.70 V and FF = 0.68) for sensitizer 132 on the modified TiO2 photoanode, which could be attributed to further improvement in the electron injection efficiency and charge collection efficiency.
A novel porphyrin dye extended at one meso-position via a pyridinylethynyl group acting as anchoring group and three phenyl COOMe groups was synthesized and used as sensitizer for the fabrication DSSC (Fig. 44).122 The overall PCE of DSSCs based on porphyrin 134 with and without chenodeoxychlolic acid (CDCA) as coadsorbent were 3.36% (Jsc = 9.38 mA cm−2, Voc = 0.64 V and FF = 0.56) and 4.56% (Jsc = 11.56 mA cm−2, Voc = 0.72 V and FF = 0.68), respectively. In order to improve the PCE of DSSC, Ag nanoparticles were incorporated into the nano-porous TiO2 photoanode i.e. FTO/TiO2/Ag-NPs and found an enhancement up to 5.66% (Jsc = 11.56 mA cm−2, Voc = 0.72 V and FF = 0.68). The improved photovoltaic performance of the DSSCs with modified photoanode was attributed to the (i) the increased light harvesting efficiency due to the plasmon enhanced optical absorption induced by Ag nanoparticles, (ii) reduced back recombination process at TiO2/dye/electrolyte interface, (iii) improved electron lifetime, and (iv) formation of Schottky barrier at TiO2/NPs-Ag.
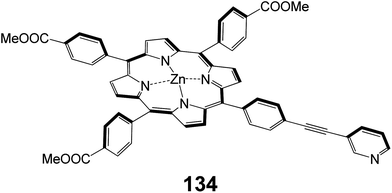 |
| | Fig. 44 Molecular structure of dye 134. | |
In another study,123 it was investigated the effect of a thiourea containing electrolyte on the photovoltaic response of a DSSC based on an A3B-type meso substituted porphyrin 134. The porphyrin structure contained three methyl-benzoate and a phenylethynyl-pyridine group. The overall PCE of the DSSC based on thiourea containing electrolyte was 5.34% (Jsc = 11.93 mA cm−2, Voc = 0.70 V and FF = 0.64), which was higher than the corresponding DSSC without thiourea electrolyte (3.36%, Jsc = 9.38 mA cm−2, Voc = 0.64 V and FF = 0.56), under identical conditions.
Since the anchoring groups were extremely important in controlling the performance of dye-sensitized solar cells (DSSCs), the design of sensitizers with new anchoring groups became quite popular.124 Therefore, donor–p–acceptor zinc-porphyrin dyes with a pyridine ring as an anchoring group designed and synthesized for DSSC applications (Fig. 45). Photophysical and electrochemical investigations demonstrated that the pyridine ring worked effectively as an anchoring group for the porphyrin sensitizers. DSSCs that were based on these new porphyrins showed an overall power conversion efficiency of about 4.0% for compound 135 under full sunlight (AM1.5G, 100 mW cm−2). Moreover, compounds 136–138 showed efficiencies of about 1, 4 and 9%, respectively (Table 22).
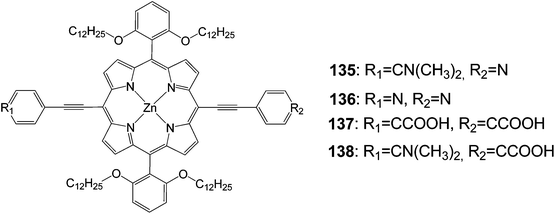 |
| | Fig. 45 Molecular structure of dyes 135–138. | |
Table 22 Photovoltaic performances of DSSC using 135–138
| Dye |
Jsc, mA cm−2 |
Voc, V |
FF |
n, % |
| 135 |
8.2 |
0.63 |
0.77 |
3.96 |
| 136 |
2.84 |
0.54 |
0.74 |
1.24 |
| 137 |
9.83 |
0.63 |
0.74 |
4.59 |
| 138 |
17.38 |
0.73 |
0.71 |
9.01 |
6. Conclusions
In this review, we have covered porphyrin based systems for solar cells bearing various anchoring groups as an effective binding site onto TiO2 electrode. The one that has been mostly used is the carboxylic acid, being meso-substituted or conjugated to the porphyrin ring at the trans or β position. Also, the most recent examples of phosphonic, sulfonic acids and pyridine anchoring groups are presented. The influence of various anchoring groups at the efficiency of the dyes is extensively discussed. In all the above examples, various donor groups have been added onto the porphyrinic macrocycle, with the diphenyl amide as a leading group. Since the best efficiency was present in a dye bearing a donor–acceptor group, much effort has been made towards this motif.
We strongly believe that the most promising approach is to develop panchromatic light harvesting single molecules that can absorb light over the entire visible/NIR spectrum and to achieve effective integration in photovoltaics. Innovative design strategies are necessary to avoid this problem. Along this path, a perfect combination of multiple sensitizers having complimentary absorption properties over the whole visible and NIR region (400–920 nm) is particularly promising. In addition, energy cascading by relay dyes exhibiting complimentary overlap between emission of one dye and absorption of another are also very interesting. Moreover, broad absorption by self-assembled or orderly aggregated dyes also poses encouraging aspects for photovoltaics if the side reactions or efficiency retarding interactions could be minimized inside the cell. Toward this aim, we believe that porphyrins offer an excellent platform for building such multi-chromophoric systems to self-assembly because the availability of several substituent sites and their intrinsic spectroscopic properties. Much deeper understanding of structural and functional features of natural light-harvesting systems is required for judicious design of more efficient single porphyrin molecules and multiporphyrin systems. Finally, appropriate physical requirements and advanced strategies are needed to assimilate these developments into the device that can utilize and transform light into other forms of storable energy.
Acknowledgements
Financial support from the European Commission (FP7-REGPOT-2008-1, Project BIOSOLENUTI no. 229927) is greatly acknowledged. This research has been also co-financed by the European Union (European Social Fund-ESF) and Greek national funds through the Operational Program "Education and Lifelong Learning" of the National Strategic Reference Framework (NSRF)-Research Funding Program: Heraklitos II and THALIS-UOA-MIS 377252. Finally Special Research account of the University of Crete is also acknowledged.
References
- G. W. Crabtree and N. S. Lewis, Phys. Today, 2007, 60, 37 CrossRef CAS PubMed.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef CAS.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382 CrossRef CAS.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788 CrossRef PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- B. E. Hardin, H. J. Snaith and M. D. McGehee, Nat. Photonics, 2012, 6, 162 CrossRef CAS.
- L. Giribabu, R. K. Kanaparthi and V. Velkannan, Chem. Rec., 2012, 12, 306 CrossRef CAS PubMed.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835 CrossRef CAS PubMed.
- Q. Wang, S. Ito, M. Grätzel, F. Fabregat-Santiago, I. Mora-Seró, J. Bisquert, T. Bessho and H. Imai, J. Phys. Chem. B, 2006, 110, 25210 CrossRef CAS PubMed.
- C.-Y. Chen, M. Wang, J.-Y. Li, N. Pootrakulchote, L. Alibabaei, C.-h. Ngoc-le, J.-D. Decoppet, J.-H. Tsai, C. Grätzel, C.-G. Wu, S. M. Zakeeruddin and M. Grätzel, ACS Nano, 2009, 3, 3103 CrossRef CAS PubMed.
- Y. Cao, Y. Bai, Q. Yu, Y. Cheng, S. Liu, D. Shi, F. Gao and P. Wang, J. Phys. Chem. C, 2009, 113, 6290 CAS.
- Q. Yu, Y. Wang, Z. Yi, N. Zu, J. Zhang, M. Zhang and P. Wang, ACS Nano, 2010, 4, 6032 CrossRef CAS PubMed.
- L. Han, A. Islam, H. Chen, C. Malapaka, B. Chiranjeevi, S. Zhang, X. Yang and M. Yanagida, Energy Environ. Sci., 2012, 5, 6057 CAS.
- M. K. Nazeeruddin, E. Baranoff and M. Grätzel, Sol. Energy, 2011, 85, 1172 CrossRef CAS PubMed.
- W. M. Campbell, A. K. Burrell, D. L. Officer and K. W. Jolley, Coord. Chem. Rev., 2004, 248, 1363 CrossRef CAS PubMed.
- M. G. Walter, A. B. Rudine and C. C. Wamser, J. Porphyrins Phthalocyanines, 2010, 14, 759 CrossRef CAS.
- L. L. Li and E. W. G. Diau, Chem. Soc. Rev., 2013, 42, 291 RSC.
- L. Giribabu, C. V. Kumar and P. Y. Reddy, J. Porphyrins Phthalocyanines, 2006, 10, 1007 CrossRef CAS.
- H. C. Mandalia, V. K. Jain and B. N. Pattanaik, J. Chem. Sci., 2012, 2, 89 CAS.
- L. Giribabu and R. K. Kanaparthi, Curr. Sci., 2013, 104, 847 CAS.
- W. Zeng, Y. Cao, Y. Bai, Y. Wang, Y. Shi, M. Zhang, F. Wang, C. Pan and P. Wang, Chem. Mater., 2010, 22, 1915 CrossRef CAS.
- J. Deisenhofer and J. R. Norris, The Photosynthetic Reaction, Academic Press, New York, 1993 Search PubMed.
- L. Luo, C.-F. Lo, C.-Y. Lin, I. J. Chang and E. W.-G. Diau, J. Phys. Chem. B, 2005, 110, 410 CrossRef PubMed.
- C.-Y. Lin, C.-F. Lo, L. Luo, H.-P. Lu, C.-S. Hung and E. W.-G. Diau, J. Phys. Chem. C, 2008, 113, 755 Search PubMed.
- B. C. O'Regan, I. López-Duarte, M. V. Martínez-Díaz, A. Forneli, J. Albero, A. Morandeira, E. Palomares, T. Torres and J. R. Durrant, J. Am. Chem. Soc., 2008, 130, 2906 CrossRef CAS PubMed.
- J.-J. Cid, J.-H. Yum, S.-R. Jang, M. K. Nazeeruddin, E. Martínez-Ferrero, E. Palomares, J. Ko, M. Grätzel and T. Torres, Angew. Chem., Int. Ed., 2007, 46, 8358 CrossRef CAS PubMed.
- H. Imahori, T. Umeyama and S. Ito, Acc. Chem. Res., 2009, 42, 1809 CrossRef CAS PubMed.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138 CrossRef CAS.
- C.-P. Hsieh, H.-P. Lu, C.-L. Chiu, C.-W. Lee, S.-H. Chuang, C.-L. Mai, W.-N. Yen, S.-J. Hsu, E. W.-G. Diau and C.-Y. Yeh, J. Mater. Chem., 2010, 20, 1127 RSC.
- T. Bessho, S. M. Zakeeruddin, C.-Y. Yeh, E. W.-G. Diau and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6646 CrossRef CAS PubMed.
- H. He, A. Gurung and L. Si, Chem. Commun., 2012, 48, 5910 RSC.
- W. Zhou, Z. Cao, S. Jiang, H. Huang, L. Deng, Y. Liu, P. Shen, B. Zhao, S. Tan and X. Zhang, Org. Electron., 2012, 13, 560 CrossRef CAS PubMed.
- M. S. Kang, S. H. Kang, S. G. Kim, I. T. Choi, J. H. Ryu, M. J. Ju, D. Cho, J. Y. Lee and H. K. Kim, Chem. Commun., 2012, 48, 9349 RSC.
- C. Jiao, N. Zu, K.-W. Huang, P. Wang and J. Wu, Org. Lett., 2011, 13, 3652 CrossRef CAS PubMed.
- C.-W. Lee, H.-P. Lu, C.-M. Lan, Y.-L. Huang, Y.-R. Liang, W.-N. Yen, Y.-C. Liu, Y.-S. Lin, E. W.-G. Diau and C.-Y. Yeh, Chem.–Eur. J., 2009, 15, 1403 CrossRef CAS PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed.
- K. Murakoshi, G. Kano, Y. Wada, S. Yanagida, H. Miyazaki, M. Matsumoto and S. Murasawa, J. Electroanal. Chem., 1995, 396, 27 CrossRef.
- J. N. Clifford, E. Palomares, M. K. Nazeeruddin, M. Grätzel, J. Nelson, X. Li, N. J. Long and J. R. Durrant, J. Am. Chem. Soc., 2004, 126, 5225 CrossRef CAS PubMed.
- E. Palomares, M. V. Martinez-Diaz, S. A. Haque, T. Torres and J. R. Durrant, Chem. Commun., 2004, 2112 RSC.
- J. R. Durrant, S. A. Haque and E. Palomares, Coord. Chem. Rev., 2004, 248, 1247 CrossRef CAS PubMed.
- T. Ma, K. Inoue, K. Yao, H. Noma, T. Shuji, E. Abe, J. Yu, X. Wang and B. Zhang, J. Electroanal. Chem., 2002, 537, 31 CrossRef CAS.
- T. Ma, K. Inoue, H. Noma, K. Yao and E. Abe, J. Photochem. Photobiol., A, 2002, 152, 207 CrossRef CAS.
- F. Odobel, E. Blart, M. Lagree, M. Villieras, H. Boujtita, N. El Murr, S. Caramori and C. Alberto Bignozzi, J. Mater. Chem., 2003, 13, 502 RSC.
- Q. Wang, W. M. Campbell, E. E. Bonfantani, K. W. Jolley, D. L. Officer, P. J. Walsh, K. Gordon, R. Humphry-Baker, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. B, 2005, 109, 15397 CrossRef CAS PubMed.
- M. K. Nazeeruddin, R. Humphry-Baker, P. Liska and M. Grätzel, J. Phys. Chem. B, 2003, 107, 8981 CrossRef CAS.
- M. K. Nazeeruddin, R. Humphry-Baker, D. L. Officer, W. M. Campbell, A. K. Burrell and M. Grätzel, Langmuir, 2004, 20, 6514 CrossRef CAS PubMed.
- T. E. O. Screen, K. B. Lawton, G. S. Wilson, N. Dolney, R. Ispasoiu, T. Goodson Iii, S. J. Martin, D. D. C. Bradley and H. L. Anderson, J. Mater. Chem., 2001, 11, 312 RSC.
- V. S. Lin, S. G. DiMangno and M. J. Therien, Science, 1994, 264, 1105 CAS.
- K. Kalyanasundaram, N. Vlachopoulos, V. Krishnan, A. Monnier and M. Grätzel, J. Phys. Chem., 1987, 91, 2342 CrossRef CAS.
- A. Kay and M. Grätzel, J. Phys. Chem., 1993, 97, 6272 CrossRef CAS.
- S. Cherian and C. C. Wamser, J. Phys. Chem. B, 2000, 104, 3624 CrossRef CAS.
- E. S. Schmidt, T. S. Calderwood and T. C. Bruice, Inorg. Chem., 1986, 25, 3718 CrossRef CAS.
- J. Seth, V. Palaniappan, T. E. Johnson, S. Prathapan, J. S. Lindsey and D. F. Bocian, J. Am. Chem. Soc., 1994, 116, 10578 CrossRef CAS.
- C. Bruckner, P. C. D. Foss, J. O. Sullivan, R. Pelto, M. Zeller, R. R. Birge and G. Crundwell, Phys. Chem. Chem. Phys., 2006, 8, 2402 RSC.
- J. Rochford, D. Chu, A. Hagfeldt and E. Galoppini, J. Am. Chem. Soc., 2007, 129, 4655 CrossRef CAS PubMed.
- S. Rangan, S. Katalinic, R. Thorpe, R. A. Bartynski, J. Rochford and E. Galoppini, J. Phys. Chem. C, 2009, 114, 1139 Search PubMed.
- J. Rochford and E. Galoppini, Langmuir, 2008, 24, 5366 CrossRef CAS PubMed.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS.
- K. S. Finnie, J. R. Bartlett and J. L. Woolfrey, Langmuir, 1998, 14, 2744 CrossRef CAS.
- A. Vittadini, A. Selloni, F. P. Rotzinger and M. Grätzel, J. Phys. Chem. B, 2000, 104, 1300 CrossRef CAS.
- H. Imahori, S. Hayashi, H. Hayashi, A. Oguro, S. Eu, T. Umeyama and Y. Matano, J. Phys. Chem. C, 2009, 113, 18406 CAS.
- H. Imahori, Y. Matsubara, H. Iijima, T. Umeyama, Y. Matano, S. Ito, M. Niemi, N. V. Tkachenko and H. Lemmetyinen, J. Phys. Chem. C, 2010, 114, 10656 CAS.
- J. K. Park, J. Chen, H. R. Lee, S. W. Park, H. Shinokubo, A. Osuka and D. Kim, J. Phys. Chem. C, 2009, 113, 21956 CAS.
- A. J. Mozer, M. J. Griffith, G. Tsekouras, P. Wagner, G. G. Wallace, S. Mori, K. Sunahara, M. Miyashita, J. C. Earles, K. C. Gordon, L. Du, R. Katoh, A. Furube and D. L. Officer, J. Am. Chem. Soc., 2009, 131, 15621 CrossRef CAS PubMed.
- J. T. Dy, K. Tamaki, Y. Sanehira, J. Nakazaki, S. Uchida, T. Kubo and H. Segawa, Electrochemistry, 2009, 77, 206 CrossRef CAS PubMed.
- Y. Liu, H. Lin, J. T. Dy, K. Tamaki, J. Nakazaki, D. Nakayama, S. Uchida, T. Kubo and H. Segawa, Chem. Commun., 2011, 47, 4010 RSC.
- A. S. Hart, C. B. Kc, H. B. Gobeze, L. R. Sequeira and F. D'Souza, ACS Appl. Mater. Interfaces, 2013, 5, 5314 CAS.
- G. E. Zervaki, M. S. Roy, M. K. Panda, P. A. Angaridis, E. Chrissos, G. D. Sharma and A. G. Coutsolelos, Inorg. Chem., 2013, 52, 9813 CrossRef CAS PubMed.
- G. E. Zervaki, E. Papastamatakis, P. A. Angaridis, V. Nikolaou, M. Singh, R. Kurchania, T. N. Kitsopoulos, G. D. Sharma and A. G. Coutsolelos, Eur. J. Inorg. Chem., 2014, 6, 1020 CrossRef.
- R. B. Ambre, G.-F. Chang, M. R. Zanwar, C.-F. Yao, E. W.-G. Diau and C.-H. Hung, Chem.–Asian. J., 2013, 8, 2144 CrossRef CAS PubMed.
- R. B. Ambre, G.-F. Chang and C.-H. Hung, Chem. Commun., 2014, 50, 725 RSC.
- J. R. Stromberg, A. Marton, H. L. Kee, C. Kirmaier, J. R. Diers, C. Muthiah, M. Taniguchi, J. S. Lindsey, D. F. Bocian, G. J. Meyer and D. Holten, J. Phys. Chem. C, 2007, 111, 15464 CAS.
- H.-P. Lu, C.-L. Mai, C.-Y. Tsia, S.-J. Hsu, C.-P. Hsieh, C.-L. Chiu, C.-Y. Yeh and E. W.-G. Diau, Phys. Chem. Chem. Phys., 2009, 11, 10270 RSC.
- H.-P. Lu, C.-Y. Tsai, W.-N. Yen, C.-P. Hsieh, C.-W. Lee, C.-Y. Yeh and E. W.-G. Diau, J. Phys. Chem. C, 2009, 113, 20990 CAS.
- R. A. Jensen, H. Van Ryswyk, C. She, J. M. Szarko, L. X. Chen and J. T. Hupp, Langmuir, 2009, 26, 1401 CrossRef PubMed.
- C.-W. Chang, L. Luo, C.-K. Chou, C.-F. Lo, C.-Y. Lin, C.-S. Hung, Y.-P. Lee and E. W.-G. Diau, J. Phys. Chem. C, 2009, 113, 11524 CAS.
- C.-Y. Lin, C.-F. Lo, L. Luo, H.-P. Lu, C.-S. Hung and E. W.-G. Diau, J. Phys. Chem. C, 2009, 113, 755 CAS.
- C.-Y. Lin, Y.-C. Wang, S.-J. Hsu, C.-F. Lo and E. W.-G. Diau, J. Phys. Chem. C, 2010, 114, 687 CAS.
- L. Luo, C.-J. Lin, C.-Y. Tsai, H.-P. Wu, L.-L. Li, C.-F. Lo, C.-Y. Lin and E. W.-G. Diau, Phys. Chem. Chem. Phys., 2010, 12, 1064 RSC.
- B. Liu, W. Zhu, Y. Wang, W. Wu, X. Li, B. Chen, Y.-T. Long and Y. Xie, J. Mater. Chem., 2012, 22, 7434 RSC.
- K. Kurotobi, Y. Toude, K. Kawamoto, Y. Fujimori, S. Ito, P. Chabera, V. Sundström and H. Imahori, Chem.–Eur. J., 2013, 19, 17075 CrossRef CAS PubMed.
- C.-H. Wu, M.-C. Chen, P.-C. Su, H.-H. Kuo, C.-L. Wang, C.-Y. Lu, C.-H. Tsai, C.-C. Wu and C.-Y. Lin, J. Mater. Chem. A, 2014, 2, 991 CAS.
- W. M. Campbell, K. W. Jolley, P. Wagner, K. Wagner, P. J. Walsh, K. C. Gordon, L. Schmidt-Mende, M. K. Nazeeruddin, Q. Wang, M. Grätzel and D. L. Officer, J. Phys. Chem. C, 2007, 111, 11760 CAS.
- A. Allegrucci, N. A. Lewcenko, A. J. Mozer, L. Dennany, P. Wagner, D. L. Officer, K. Sunahara, S. Mori and L. Spiccia, Energy Environ. Sci., 2009, 2, 1069 CAS.
- V. Armel, J. M. Pringle, M. Forsyth, D. R. MacFarlane, D. L. Officer and P. Wagner, Chem. Commun., 2010, 46, 3146 RSC.
- A. J. Mozer, P. Wagner, D. L. Officer, G. G. Wallace, W. M. Campbell, M. Miyashita, K. Sunahara and S. Mori, Chem. Commun., 2008, 4741 RSC.
- T. D. Santos, A. Morandeira, S. Koops, A. J. Mozer, G. Tsekouras, Y. Dong, P. Wagner, G. Wallace, J. C. Earles, K. C. Gordon, D. Officer and J. R. Durrant, J. Phys. Chem. C, 2010, 114, 3276 Search PubMed.
- J. K. Park, H. R. Lee, J. Chen, H. Shinokubo, A. Osuka and D. Kim, J. Phys. Chem. C, 2008, 112, 16691 CAS.
- M. Ishida, S. W. Park, D. Hwang, Y. B. Koo, J. L. Sessler, D. Y. Kim and D. Kim, J. Phys. Chem. C, 2011, 115, 19343 CAS.
- C. G. Di, B. A. Orbelli, M. Pizzotti, F. Tessore, V. Trifiletti, R. Ruffo, A. Abbotto, A. Amat, A. F. De and P. R. Mussini, Chem.–Eur. J., 2013, 19, 10723 CrossRef PubMed.
- B. J. Brennan, M. J. Llansola Portoles, P. A. Liddell, T. A. Moore, A. L. Moore and D. Gust, Phys. Chem. Chem. Phys., 2013, 15, 16605 RSC.
- M. P. Balanay, K. H. Kim, S. H. Lee and D. H. Kim, J. Photochem. Photobiol., A, 2012, 248, 63 CrossRef CAS PubMed.
- Y. Liu, N. Xiang, X. Feng, P. Shen, W. Zhou, C. Weng, B. Zhao and S. Tan, Chem. Commun., 2009, 2499 RSC.
- V. A. K. Nuay, D. H. Kim, S.-H. Lee and J.-J. Ko, Bull. Korean Chem. Soc., 2009, 2009, 2871 CrossRef.
- C. Y. Lee and J. T. Hupp, Langmuir, 2009, 26, 3760 CrossRef PubMed.
- C. Y. Lee, C. She, N. C. Jeong and J. T. Hupp, Chem. Commun., 2010, 46, 6090 RSC.
- K. Sirithip, S. Morada, S. Namuangruk, T. Keawin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, Tetrahedron Lett., 2013, 54, 2435 CrossRef CAS PubMed.
- M. J. Griffith, K. Sunahara, P. Wagner, K. Wagner, G. G. Wallace, D. L. Officer, A. Furube, R. Katoh, S. Mori and A. J. Mozer, Chem. Commun., 2012, 48, 4145 RSC.
- J. Lu, X. Xu, K. Cao, J. Cui, Y. Zhang, Y. Shen, X. Shi, L. Liao, Y. Cheng and M. Wang, J. Mater. Chem. A, 2013, 1, 10008 CAS.
- S. H. Kang, I. T. Choi, M. S. Kang, Y. K. Eom, M. J. Ju, J. Y. Hong, H. S. Kang and H. K. Kim, J. Mater. Chem. A, 2013, 1, 3977 CAS.
- M. S. Kang, I. T. Choi, Y. W. Kim, B. S. You, S. H. Kang, J. Y. Hong, M. J. Ju and H. K. Kim, J. Mater. Chem. A, 2013, 1, 9848 CAS.
- N. M. Reddy, T. Y. Pan, Y. C. Rajan, B. C. Guo, C. M. Lan, E. W. Diau and C. Y. Yeh, Phys. Chem. Chem. Phys., 2013, 15, 8409 RSC.
- T. Ripolles-Sanchis, B. C. Guo, H. P. Wu, T. Y. Pan, H. W. Lee, S. R. Raga, F. Fabregat-Santiago, J. Bisquert, C. Y. Yeh and E. W. G. Diau, Chem. Commun., 2012, 48, 4368 RSC.
- H. Imahori, S. Kang, H. Hayashi, M. Haruta, H. Kurata, S. Isoda, S. E. Canton, Y. Infahsaeng, A. Kathiravan, T. Pascher, P. Chabera, A. P. Yartsev and V. Sundstrom, J. Phys. Chem. A, 2011, 115, 3679 CrossRef CAS PubMed.
- S. Eu, S. Hayashi, T. Umeyama, A. Oguro, M. Kawasaki, N. Kadota, Y. Matano and H. Imahori, J. Phys. Chem. C, 2007, 111, 3528 CAS.
- S. Eu, S. Hayashi, T. Umeyama, Y. Matano, Y. Araki and H. Imahori, J. Phys. Chem. C, 2008, 112, 4396 CAS.
- M. Tanaka, S. Hayashi, S. Eu, T. Umeyama, Y. Matano and H. Imahori, Chem. Commun., 2007, 2069 RSC.
- S. Hayashi, Y. Matsubara, S. Eu, H. Hayashi, T. Umeyama, Y. Matano and H. Imahori, Chem. Lett., 2008, 37, 846 CrossRef CAS.
- S. Hayashi, M. Tanaka, H. Hayashi, S. Eu, T. Umeyama, Y. Matano, Y. Araki and H. Imahori, J. Phys. Chem. C, 2008, 112, 15576 CAS.
- A. Kira, Y. Matsubara, H. Iijima, T. Umeyama, Y. Matano, S. Ito, M. Niemi, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, J. Phys. Chem. C, 2010, 114, 11293 CAS.
- R. Deshpande, B. Wang, L. Dai, L. Jiang, C. S. Hartley, S. Zou, H. Wang and L. Kerr, Chem.–Asian J., 2012, 7, 2662 CrossRef CAS PubMed.
- H. Hayashi, A. S. Touchy, Y. Kinjo, K. Kurotobi, Y. Toude, S. Ito, H. Saarenpaa, N. V. Tkachenko, H. Lemmetyinen and H. Imahori, ChemSusChem, 2013, 6, 508 CrossRef CAS PubMed.
- H. Park, E. Bae, J.-J. Lee, J. Park and W. Choi, J. Phys. Chem. B, 2006, 110, 8740 CrossRef CAS PubMed.
- E. Bae, W. Choi, J. Park, H. S. Shin, S. B. Kim and J. S. Lee, J. Phys. Chem. B, 2004, 108, 14093 CrossRef CAS.
- C. N. Rusu and J. T. Yates, J. Phys. Chem. B, 2000, 104, 12292 CrossRef CAS.
- R. Luschtinetz, J. Frenzel, T. Milek and G. Seifert, J. Phys. Chem. C, 2009, 113, 5730 CAS.
- G. Guerrero, P. H. Mutin and A. Vioux, Chem. Mater., 2001, 13, 4367 CrossRef CAS.
- P. Wang, C. Klein, J.-E. Moser, R. Humphry-Baker, N.-L. Cevey-Ha, R. Charvet, P. Comte, S. M. Zakeeruddin and M. Grätzel, J. Phys. Chem. B, 2004, 108, 17553 CrossRef CAS.
- Y. Ooyama, S. Inoue, T. Nagano, K. Kushimoto, J. Ohshita, I. Imae, K. Komaguchi and Y. Harima, Angew. Chem., Int. Ed., 2011, 50, 7429 CrossRef CAS PubMed.
- Y. Ooyama, T. Nagano, S. Inoue, I. Imae, K. Komaguchi, J. Ohshita and Y. Harima, Chem.–Eur. J., 2011, 17, 14837 CrossRef CAS PubMed.
- D. Daphnomili, G. Landrou, S. Prakash Singh, A. Thomas, K. Yesudas, K. Bhanuprakash, G. D. Sharma and A. G. Coutsolelos, RSC Adv., 2012, 2, 12899 RSC.
- D. Daphnomili, G. D. Sharma, S. Biswas, T. K. R. Justin and A. G. Coutsolelos, J. Photochem. Photobiol., A, 2013, 253, 88 CrossRef CAS PubMed.
- G. D. Sharma, D. Daphnomili, P. A. Angaridis, S. Biswas and A. G. Coutsolelos, Electrochim. Acta, 2013, 102, 459 CrossRef CAS PubMed.
- J. Lu, X. Xu, Z. Li, K. Cao, J. Cui, Y. Zhang, Y. Shen, Y. Li, J. Zhu, S. Dai, W. Chen, Y. Cheng and M. Wang, Chem.–Asian J., 2013, 8, 956 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |