Metal phosphonate hybrid materials: from densely layered to hierarchically nanoporous structures
Yun-Pei
Zhu
a,
Tian-Yi
Ma
a,
Ya-Lu
Liu
a,
Tie-Zhen
Ren
b and
Zhong-Yong
Yuan
*a
aKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: zyyuan@nankai.edu.cn
bSchool of Chemical Engineering and Technology, Hebei University of Technology, Tianjin 300130, China
First published on 9th April 2014
Abstract
Metal phosphonate materials are promising non-siliceous inorganic–organic hybrids that are synthesized by combining metal joints and organophosphonic linkages at the molecular scale. The mild conditions for metal phosphonate synthesis, their homogeneous composition and the combined merits of inorganic units and organic groups have permitted the rational design and incorporation of various functionalities through constituent building units. In this critical review, we present the development and recent advances related to the field of metal phosphonates and the relevant nanocomposites. The possibility to integrate the functionalities from both inorganic and organic moieties is discussed. The incorporation of well-defined porosity and capacity for post-modification have extended the application potential to the area of adsorption, separation, catalysis, environmental intervention, energy storage and biology. Metal phosphonates thus present an unprecedented opportunity for the rational and precise design of sophisticated materials with multifunctionality.
 Yun-Pei Zhu | Yun-Pei Zhu received his BSc degree in 2011 at Henan Polytechnic University. He is currently a PhD candidate under the supervision of Prof. Zhong-Yong Yuan at Nankai University. His current research relates to the rational design and practical applications of porous inorganic–organic hybrid materials. |
 Tian-Yi Ma | Tian-Yi Ma received his BSc and PhD degrees in Chemistry at Nankai University in 2008 and 2013, under the supervision of Prof. Zhong-Yong Yuan. He is currently a postdoctoral research fellow in the School of Chemical Engineering, University of Adelaide, Australia. |
 Ya-Lu Liu | Ya-Lu Liu received her BSc degree in 2012 at Langfang Teachers University. She is currently a graduate student under the supervision of Prof. Zhong-Yong Yuan. Her current research is focused on the synthesis and application of inorganic–organic hybrid materials. |
 Tie-Zhen Ren | Tie-Zhen Ren obtained her PhD degree from the University of Namur, Belgium in 2005. After two-year postdoctoral research at Stockholm University, Sweden, she joined the faculty of the Hebei University of Technology in 2007, where she is currently Professor in Chemical Technology. Her group's research areas include nanoporous photoelectrochemical materials and metal–organic frameworks. |
 Zhong-Yong Yuan | Zhong-Yong Yuan received his PhD degree from Nankai University in 1999. After his postdoctoral research at the Institute of Physics, Chinese Academy of Sciences, he joined the Laboratory of Inorganic Materials Chemistry at the University of Namur, Belgium in 2001. In 2005, he was appointed Professor in Nankai University. In 2006, he was awarded the “Program for New Century Excellent Talents in University” by the Ministry of Education. His research interests are mainly focused on the self-assembly of hierarchically nanoporous and nanostructured materials for energy and environmental applications. |
1 Introduction
Deliberate efforts to combine the favourable properties of inorganic units and organic moieties in a single composite material represent an old challenge which started at the beginning of the industrial era. With the establishment of “chimie douce”, Livage opened the gates towards a new galaxy of materials, inorganic–organic hybrid materials.1–5 Noticeably, the concept of “inorganic–organic hybrid materials” has more to do with chemistry than with simple physical mixtures. In general, inorganic–organic hybrid materials are nanocomposites with the inorganic and organic components interacting intimately at the molecular scale.6–10Nowadays the field of organic–inorganic materials has been extended to other fields as diverse as molecular and supramolecular materials or polymer chemistry.11,12 Furthermore, due to the combined physicochemical merits of organic and inorganic components, a very significant trend has been the growing research interest towards functional hybrids, which extends the field even broader and further. In addition to structural hybrid materials, there is a quickly expanding area of research on functional materials that focus on chemical, electrochemical, and biochemical activities, as well as on magnetic, electronic, and optical properties, or a combination of them.13 The particular nanostructures, the degree of organization and the properties for the hybrid materials certainly depend on the chemical nature of the building components, but they are also affected by the interactions between these components.
The key point for designing new hybrids is to adjust the nature, extent and accessibility of the inner interfaces. As a result, the nature of the interface and the interactions exchanged by the organic and inorganic units can be employed to categorize the hybrids into two main classes.14–16 Class I corresponds to the hybrid systems that involve no covalent or weak chemical bonding. In this class, only hydrogen bonding, van der Waals or electrostatic forces are usually present. Conversely, Class II hybrid materials show strong chemical interactions between the components, which are formed when the discrete inorganic building blocks are covalently bonded to the organic polymer or inorganic and organic polymers are covalently connected with each other.17,18 On the other hand, hybrids can also be characterized by the type and size of the organic or the inorganic precursors.14,15 Precursors can be two separate monomers or polymers and even covalently linked ones. Because of the mutual insolubility between inorganic and organic components, phase separation will occur. However, homogeneous or single-phased hybrids can be obtained through judiciously choosing bifunctional monomers that contain organic and inorganic components, or by combining both types of components in phases where one of them is in large excess.19
The chemical strategies to construct Class II hybrid frameworks rely on the relative stability of the interactions between the components and the chemical linkages that connect different components. Periodic mesoporous organosilicas (PMOs) containing organic siloxane groups in the silica network have received much attention since 1999.20–22 The stable Si–C bonds under hydrolytic conditions allow for the easy incorporation of a large variety of organic bridges in the silica network during the sol–gel process. Nevertheless, besides the limited choice and high cost of the precursors of organosilicane, functionalization or modification of organosilicas is confined to the physical properties concerned with adsorption, ion-exchange and catalysis, and thereby the rational design of hybrid materials has been extended to non-siliceous organic–inorganic hybrid materials including metal sulfonates, carboxylates and phosphonates.23 In comparison with their sulfonate and carboxylate counterparts, metal phosphonates exhibit much higher thermal and chemical stability due to the strong affinity of organophosphonic linkers to metal ions, making them promising in the fields of energy conversion, adsorption/separation, catalysis, biotechnology and so forth.23–25 Furthermore, various organophosphonic acids and corresponding derivatives (i.e. salts and esters) have been discovered in nature. Judicious design of the phosphonic bridging groups can introduce desirable properties into the hybrid frameworks. The different reactivity of phosphonic coupling molecules leads to structural diversity and physicochemical peculiarities of the resultant hybrid materials and may provide decisive advantages in the sol–gel synthesis of homogeneous hybrids. The chemical and thermal stability against hydrolysis of phosphorus–carbon bonds in phosphonates may be considerably high. By using organically bridged phosphonic acids as coupling molecules, the homogeneous and efficient incorporation of organic functional groups into the framework of the materials can be realized, allowing for uniform physicochemical properties from the external surface to the internal skeleton.
Porosity endows materials with higher surface area, larger pore volume and lower density as compared with dense materials, thus allowing atoms, ions, molecules and even large guest molecules to interact with the host materials not only at the surface but also throughout the bulk.26,27 According to the IUPAC convention,28 porous materials are divided into three types depending on the pore size: microporous materials with pores smaller than 2 nm, mesoporous materials with pores ranging from 2 to 50 nm, and macroporous materials, where the pores are larger than 50 nm. The distribution of shapes and sizes of the void spaces in nanoporous materials is intimately related to their capability to perform a desired function in a particular area. The demand to create uniformity in pore size and shape and greater volumes has been steadily increasing over the past decades, which can lead to superior physicochemical properties. For instance, zeolites with uniform micropores can be employed to separate molecules on the basis of the pore size by selectively adsorbing smaller molecules from a mixture containing molecules too large to enter into the pores. Noticeably, incorporation of well-structured porosity into the metal phosphonate frameworks can enhance the accessibility to the interior pore system. Therefore, functions due to inorganic units and organic moieties and their combined properties are valuable and interesting for the further exploration of porous metal phosphonate hybrid materials.
Recently, Kimura briefly reviewed the development of mesoporous aluminium organophosphonates prepared by surfactant-assisted reaction between metal sources and bisphosphonates.29 Clearfield et al. summarized the porous pillared zirconium and tin(IV) diphosphonates.30 Indeed, mesoporous non-siliceous hybrids including metal sulfonates, carboxylates and phosphonates have been considered as a promising platform for designing multifunctional materials.23 The synthesis of mesostructured titanium phosphonate hybrids has attracted considerable interest due to their extended functions for clean energy and other applications.31 Complicated porosity (from microporosity to macroporosity and even porous hierarchy) and various compositions of hybrid frameworks (inorganic metal centers and organophosphonic linkages) can lead to remarkable physicochemical properties. The present critical review provides a comprehensive understanding of metal phosphonate hybrids from densely layered to porous networks, and also covers surface-phosphonated oxides. The key elements in the rational design and synthesis of phosphonate-based hybrids are discussed. Moreover, typical and emerging applications in fields ranging from conventional catalysis and adsorption to the burgeoning biotechnology and energy conversion and storage are elaborated, in an attempt to attract more interest in the exploration of these innovative hybrid materials.
2 Crystalline metal phosphonates
Crystalline metal phosphonate hybrids, usually known as phosphonate-based metal–organic frameworks (MOFs), represent a particularly versatile field. Nonetheless, phosphonate-based MOFs are considerably rarer than their carboxylate-based counterparts. This is mainly due to the less predictable coordination chemistry of phosphonate linkers and the greater diversity of possible ligating modes,32 leading to the formation of densely layered structures that are poorly porous. This has prohibited their application as adsorbents. Thus various tactics have been employed to create porosity in metal phosphonates, such as utilizing multidimensional polyphosphonate ligands with spatially divergent phosphonate groups to generate open frameworks. In this section, the development of crystalline metal phosphonate materials from layered structures to microporous or open-framework structures is discussed.2.1 Dense layered metal phosphonate materials
Metal phosphates have been studied as layered inorganic networks, and later evolved into inorganic–organic hybrids by having organic pillars appended off the rigid inorganic layers.13,33,34 Layered structures are predominant for most metals with the organic groups being oriented perpendicularly into the interlamellar region (Fig. 1).35,36 Thus the interlayer distance can be easily tuned by changing the pillar groups and it is even possible to exfoliate the layers into film.37 Pendant groups can impart other functionalities including chirality38 and photoactivity.39 Some initial layered metal phosphonate materials with very simple structures emerged decades ago. Alberti et al. reported the first layered zirconium phenylphosphonates and alkylphosphonates.40 The structure of the phenylphosphonate was solved from the powder XRD patterns, which yielded only 35–40 reflections rather than the hundred or more expected for a complex structure, even after a long reflux time in HF. Zirconium alkylphosphonates have structures similar to that of the phenylphosphonate, which is to say they possess α-type layers. Layered metal phosphonates with monophosphonates are polymeric species that contain alternating hydrophilic and hydrophobic regions. In these compounds, the oxygen-bridged metal atoms form the central two-dimensional layers that are separated on either side by the pendant organic moieties of the phosphonate group. Many derivatives based on this initial work were reported later. For example, zirconium phenylphosphonate was sulfonated by treatment with fuming sulfuric acid to generate metal sulfophosphonates,41 demonstrating great potential in solid-acid catalysis and proton conductivity. It was found that layered polyether phosphates and phosphonates of zirconium could swell in water and complete exfoliation occurred when the value of n for the polyether chain (CH2CH2O)n was greater than nine.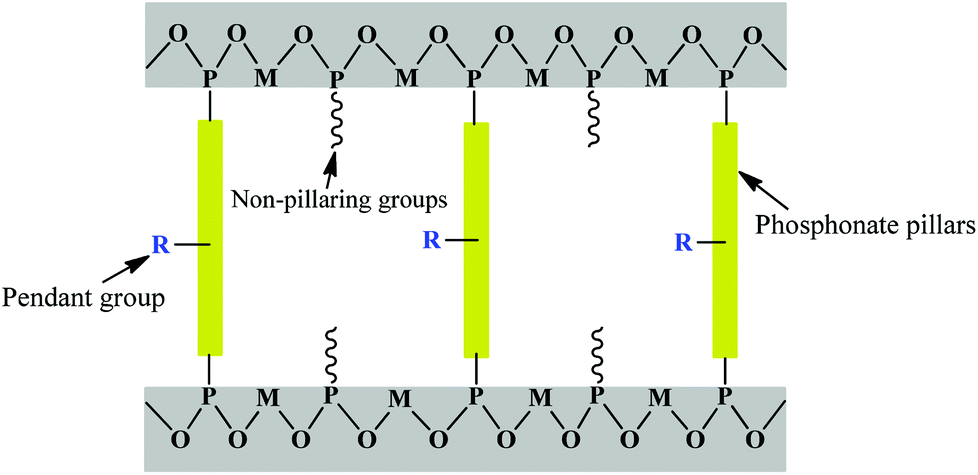 | ||
| Fig. 1 Schematic representation of layered metal phosphonates. To improve the porosity, one way is to insert small non-pillaring “spacer” groups, including metal oxide clusters, phosphoric, phosphors, methylphosphonic acids, and crown ethers, between the phosphonate pillars. Another route is to attach pendant functional groups, such as imino, pyridine, hydroxyl, carboxylic and sulfonic acid, on the organophosphonic linkages. | ||
Noticeably, because of their low solubility, metal phosphonates tend to be less crystalline than carboxylates. Also, phosphonate ligands do not form the types of secondary building units with metal ion joints that carboxylates are known to form, so the rational design of desired structures is considerably difficult. Toxic hydrofluoric acid is usually used to enhance the crystallization of the resultant frameworks.42–44 Alternatively, additional functional groups such as crown ether, carboxylate, hydroxyl, and amine groups have been attached to the phosphonic acid ligand to improve the solubility and crystallinity of phosphonates.45 Several lanthanide phosphonates with a crown ether or calixarene moiety have been reported.46–49 A series of N-(phosphonomethyl) aza-crown ethers were prepared and reacted with M(IV) and M(II) ions.48 With respect to zirconium phosphonates, the layered compounds have structures similar to N-(phosphonomethyl)iminodiacetic acid. These compounds have been described as macroscopic leaflets since the crown ether portions resemble leaves bound to twigs. The interlayer spacing is 20 Å, indicating that there is no interdigitation of crown ether groups. Sulfonate groups were also incorporated into the metal phosphonate frameworks. Mao et al. synthesized a series of novel layered lanthanide sulfonate-phosphonates through the hydrothermal reaction of lanthanide metal salts with MeN(CH2CO2H)(CH2PO3H2) and 5-sulfoisophthalic acid monosodium salt (NaH2BTS).45 This revealed that by using a second metal linker, such as carboxylic or sulfonic acid, whose lanthanide complexes have better solubility in water or other solvents, the solubility and crystallinity of lanthanide phosphonates can be greatly improved, which allowed us to determine their structures accurately and easily.
The elucidation of the structures of lanthanide phosphonates is quite significant and valuable due to their meaningful luminescent properties. Rare-earth elements as the metallic nodes in the construction of multidimensional phosphonate-based coordination polymers have been widely investigated, with the ultimate objective of isolating novel photoluminescent materials.50,51 The use of the polyfunctional tetraphosphonic acid ligand (H2O3PCH2)2NCH2CH2CH2CH2N(CH2PO3H2)2, in combination with oxalic acid, gave rise to two different types of lanthanide-based luminescent frameworks.52 Moreover, magnetism has been another focus for metal phosphonates, of which cobalt phosphonates are typical examples. In the framework of a layered cobalt carboxylate–phosphonate, Co[HO2C(CH2)3NH(CH2PO3H)2]2,53 the Co(II) ion in the compound was octahedrally coordinated by six phosphonate oxygen atoms from four carboxylate phosphonate ligands. While Bujoli et al. synthesized a 3D cobalt phosphonate,54 the compound was isostructural with the pillared layered metal phosphonates M3(O3PC2H4CO2)2, with M = Zn or Mn. In this structure, the magnetic transition metal ions are arranged within layers formed by the interconnection of CoO4 tetrahedra and CoO6 octahedra.
The strong binding ability of phosphonic acids usually leads to dense layered architectures of metal phosphonates. Incorporating other functional groups onto phosphonic linkages improved the resulting crystallization. Different metal nodes introduced distinct physicochemical properties into the metal phosphonate hybrid frameworks. For example, rare-earth elements led to photoluminescence, while magnetism was formed when cobalt was incorporated. Many other metallic precursors could also be utilized as the central ions to coordinate with phosphonic acids to obtain layered structures.55,56 However, it is noteworthy that layered phosphonate hybrids often show up in the form of dense motifs. The pillars are too crowded to leave sufficient free space in the interlayer region, and thus poor or no porosity is expected. A further step should be taken to generate well-defined porosity in the phosphonate networks to make them fit the qualifications for research and practical applications.
2.2 Metal phosphonate materials with open framework
A layered motif is the most common observation for a simple metal phosphonate, however an important exception is the family of complexes observed with methylphosphonic acid. The first 3D phosphonate framework with open channels was reported in 1994 by Bujoli and co-workers.57 The synthesized β-Cu(O3PCH3) contains 24-membered rings lined by pendant methyl groups. Because the distance between the opposite methyl carbons is 5.97 Å, the effective pore size is estimated at about 3 Å, which is too narrow for small adsorbate molecules such as nitrogen to enter. Thereafter, Maeda and co-workers reported the synthesis of polymorphs α- and β-Al2(CH3PO3)3 with zeolite-type frameworks,58,59 which contained trivalent aluminum cations in both tetrahedral and octahedral geometries with bridging phosphonate groups, leaving hexagonal channels lined with methyl groups, and thus giving ∼5 Å free diameter channels for both of these compounds. Then, the preparation of 3D open frameworks of metal phosphonates was fully expanded. Not only methylphosphonic acid but also methylenediphosphonic acid was used in the synthesis of open frameworks. The open-framework Co2(O3PCH2PO3)·H2O has 1D inorganic channels including 20-membered rings lined with methylene bridges.60Phosphonate bridging moieties are almost limited to those with small organic groups such as methylphosphonate and methylenediphosphonate.33 Varying the chain length in metal alkylphosphonates can alter the structure considerably. Organophosphonic linkers with –CH2– chain lengths of n = 2–4 typically resulted in pillared layered structures.61 However, when the alkyl chain length was increased to n = 8 with Co2+, a low-dimensional structure was obtained, composed of cationic [Co(H2O)4(H4L)]2+ (H4L = 1,8-octylenediphosphonic acid) chains with charge-balancing 1,8-octylenediphosphonate clathrated in the structure.62 On the other hand, many attempts have been made to create micropores in open framework compounds by introducing metal oxide clusters as interlayer pillars.63,64 The synthesized open framework of [NH3(CH2)4NH3]Cu3(HEDP)2·2H2O and [NH2(C2H4)2NH2]Cu3(HEDP)2 (HEDP = 1-hydroxy ethylidene-1,1-diphosphonic acid)63 adopts a two-dimensional layered structure with four- and eight-membered rings assembled from vertex-sharing {CuO4} units and {CPO3} tetrahedra. Co3(O3PCH2-NC4H7-COO)2·5H2O containing a proline-derived phosphonate anion has ordered micropores composed of 12 polyhedral units,64 and straight 2-D channels are formed by stacking of layers. Similarly, substitution of aryl biphosphonic acid by some non-pillaring groups, such as phosphoric, phosphors, and methylphosphonic acids, could lead to the creation of interlayer pores and the increase of surface area (Fig. 1).30 This method was employed to overcome the “close-pillar disposition” and resulted in a “dilution” of the phosphonate moieties. Although a porous phosphonate network can be obtained, the problem of this approach is that replacement is random and uncontrollable, and the structural characterization and narrow pore size distribution still constitute challenges. The water solubility of metal phosphonates generally decreases as the metal ion valences increase.32,65 The inclusion of a second smaller “spacer” has been shown to produce porosity in tetravalent metal phosphonates,66 while it often results in the generation of two phases when coming to divalent metal ions.
The study of novel open-framework or microporous metal phosphonates was aided by anchoring functional groups to the phosphonic linkages. A new functional group on the organophosphonate ligand would perturb the layered structure in metal phosphonates, which was deemed desirable for the formation of a new 3D open framework.23,31 The typical functional groups attached to the phosphonic acid includes imino, pyridine, hydroxyl, carboxylic and sulfonic acid (Fig. 1).13 Herein the use of bifunctional phosphonate anions and metal cations that can adopt different coordination environments is proposed as a strategy for synthesizing microporous metal phosphonates. For example, Férey and co-workers reported the open-framework lanthanide-carboxyphosphonates, M4(H2O)7[O2CC5H10-NCH2PO3]4·(H2O)5 (M = Pr, Y).67 These isostructural lanthanide compounds had a three-dimensional open-framework structure and possessed a reversible hydration–dehydration capacity, demonstrating significant thermal stability up to 523 K. But the sorption data were not available. The use of amino acid derived phosphonates to form porous architectures led to a homochiral metal-phosphonate solid.68 The chirally pure (S)-N-(phosphonomethyl)proline was reacted with a series of lanthanides to form isostructural solids, and the resultant 1D tubular channels were of 4.32 × 3.81 Å free diameter. Since the introduction of pendant functional groups on the phosphonic linkers, the porosity and crystallization of crystalline metal phosphonates have been significantly perfected. From another point of view, favourable physicochemical properties can be incorporated in the hybrid framework, thus showing a promising methodology for crystalline growth and deep application investigation.
A large and multidimensional polyphosphonic bridging molecule would disfavour the formation of the layered motif, thereby necessitating an open framework (Fig. 2). The organophosphonic ligand 1,3,5,7-tetrakis(4-phenylphosphonic acid)-adamantane is such a molecule as it possesses four phosphonic acid moieties spaced by rigid phenyl groups from the adamantane core. A number of literature reports of metal complexes with this ligand have appeared. A vanadium phosphonate material was prepared through the non-hydrolytic condensation of vanadium(V) alkoxide with this complicated ligand, and the corresponding surface area value was 118 m2 g−1.69 The resultant porous vanadium phosphonate showed favorable catalytic activity for the aerobic oxidation or benzylic alcohols to aldehydes. Hybrid frameworks with titanium(IV) and this complicated and rigid ligand could be prepared as well,70 resulting in a paracrystalline material with 22 × 9 Å pores and a high specific surface area of 557 m2 g−1.
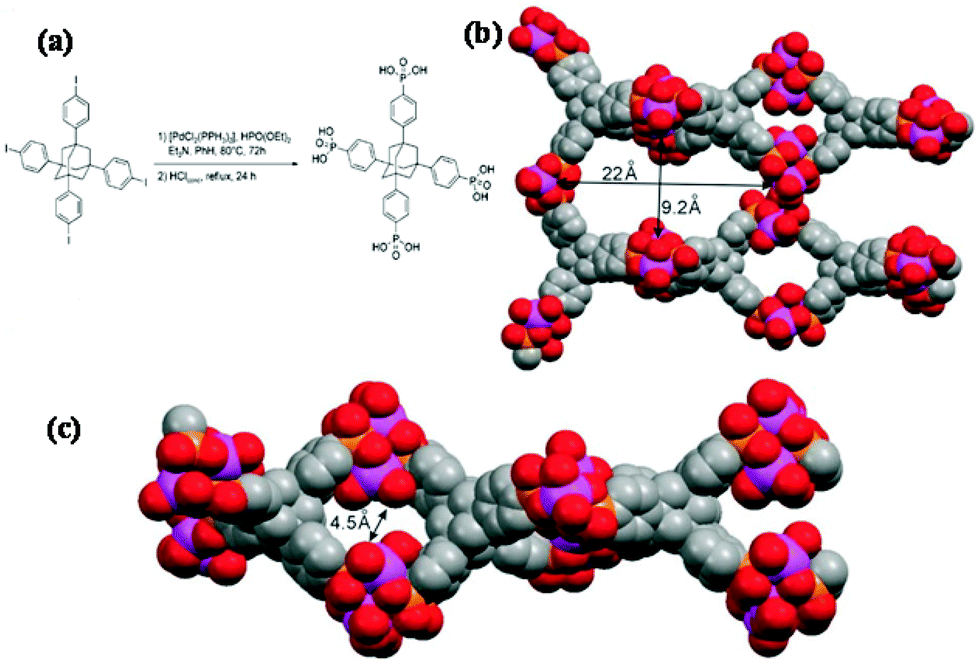 | ||
| Fig. 2 Synthesis of tetrakis-1,3,5,7-(4-phosphonatophenyl)adamantine (a), and computer-stimulated model of titanium tetraphosphonate material (b, c).70 | ||
Hitherto, a number of strategies have been employed to improve the porosity of crystalline metal phosphonate frameworks. The main strategies can be assigned to three types, namely, using non-pillaring groups as interlayer spacers, attaching a functional group on the ligand and extending the geometry of the organic core in a polyphosphonate to direct the structure away from layered motif. Phosphonate-based MOFs have distinct differences from the carboxylate-based counterparts, such as the relatively high thermal and chemical stability and extremely low solubility, making metal phosphonates attractive candidates for porous materials. Conversely, this typically renders it difficult to obtain crystalline phases with determined structures. Developments of high-throughput hydrothermal or solvothermal techniques and advances in powder XRD modeling and refinement will significantly increase the number of structurally characterized phosphonate-based MOFs, and it will be exciting to watch this field as it develops.
3 Mesoporous metal phosphonate materials
The dimensions and accessibility of dense or microporous crystalline metal phosphonates are restrained to the sub-nanometer scale, which confines their applications for small molecules. In parallel with the above work on dense metal phosphonates and related open frameworks, recent progress has been made towards mesoporous materials with uniform channel dimensions and pore distributions which can be adjusted over a wide range of length scales. Because of the larger pore size and pore volume of mesoporous materials in comparison with those of micropores, they display superior physicochemical properties and application potential. Indeed, the adaptability of sol–gel chemistry allows the mixing of inorganic and organic components at the nanometer scale, resulting in mesoporous organic–inorganic nanocomposites with homogeneous composition.71 This process can be accomplished in the absence or presence of surfactants, and the pore wall of the resultant solids is the assembly of hybrid nanoparticles, which is different from the covalent bonding networks of crystalline metal phosphonates.Template-free self-assembly usually initiates the assembly for the interactions among the precursor molecules, and the ordered attachment tends to form porous nanostructures.23 This method does not involve the use of pre-formed templates or structure-directing agents, simplifying the preparation process. Synthesis parameters, such as the ratio of the precursors, are of significance in influencing the structural properties of the synthesized mesoporous metal phosphonates. By simply changing the ratios and the concentrations of the reagents, a series of α-pillared zirconium phosphite-phosphonates with large surface area (230 to 400 m2 g−1) and great pore volume (0.3 to 0.7 cm3 g−1) were obtained.72 These materials showed a narrow pore size distribution that was tunable over the range of 4–14 nm by varying the preparation conditions, especially the concentration of the reagents. The formation of interparticle mesoporosity was attributable to edge–edge interactions between rigid packets of a few pillared α-layers giving rise to stable aggregates with a house of cards structure.
Self-assembly in the absence of structure-directing agents often involves weak interactions between the nano-building units, including atoms, molecules and related secondary blocks, to direct the assembly process. Mesoporous metal phosphonates with well-defined porosity and nanostructures have been obtained through self-assembly strategy in the past decades. Nevertheless, this process is mainly dependent on the synthesis conditions. A more controllable method is thus required from the research and practical applications point of view.
Surfactant-assisted synthesis has been thoroughly and widely developed for the design of periodic mesoporous materials since 1992.73,74 Rather than the individual molecule as void filler in the ordering of the reagents to form microporous hybrid frameworks, assemblies of molecules dictated by solution energetic are indispensable for the formation of the periodic pore systems.75 During the synthesis process, the pre-formed surfactant micelle scaffold performs as a lyotropic liquid crystalline phase, and subsequently the oligomers from the condensation of inorganic/organic precursors grow around the arranged micelles, leading to the assembly of an ordered mesostructured composite (Fig. 3). Removal of the surfactant by extraction or calcination can leave mesovoids in the framework. Hard-templating routes have been shown to be sufficiently efficient in synthesizing solid inorganic frameworks including silicas, oxides, cyanides and carbons.76 However, the prepared materials often have a wider pore size distribution than the pristine replicas; and multiple preparation procedures at the sacrifice of the costly hard templates render this approach expensive, complicated, and consequently unsuitable for large-scale production and industrial applications. Meanwhile, the template removal always involves strong acids, bases, and high-temperature calcination, which make it favorable for the synthesis of special mesoporous materials with solid networks including metal sulfides and oxides, carbons, and silicon carbides.23,76 As to hybrid frameworks containing organic components, the soft-templating approach is much more appropriate due to the modest preparation conditions to protect the hybrid frameworks, relative simplicity and environmental friendliness. Some typical examples of ordered mesoporous metal phosphonates are summarized in Table 1, which contains the experimental parameters, textual properties and mesophases.
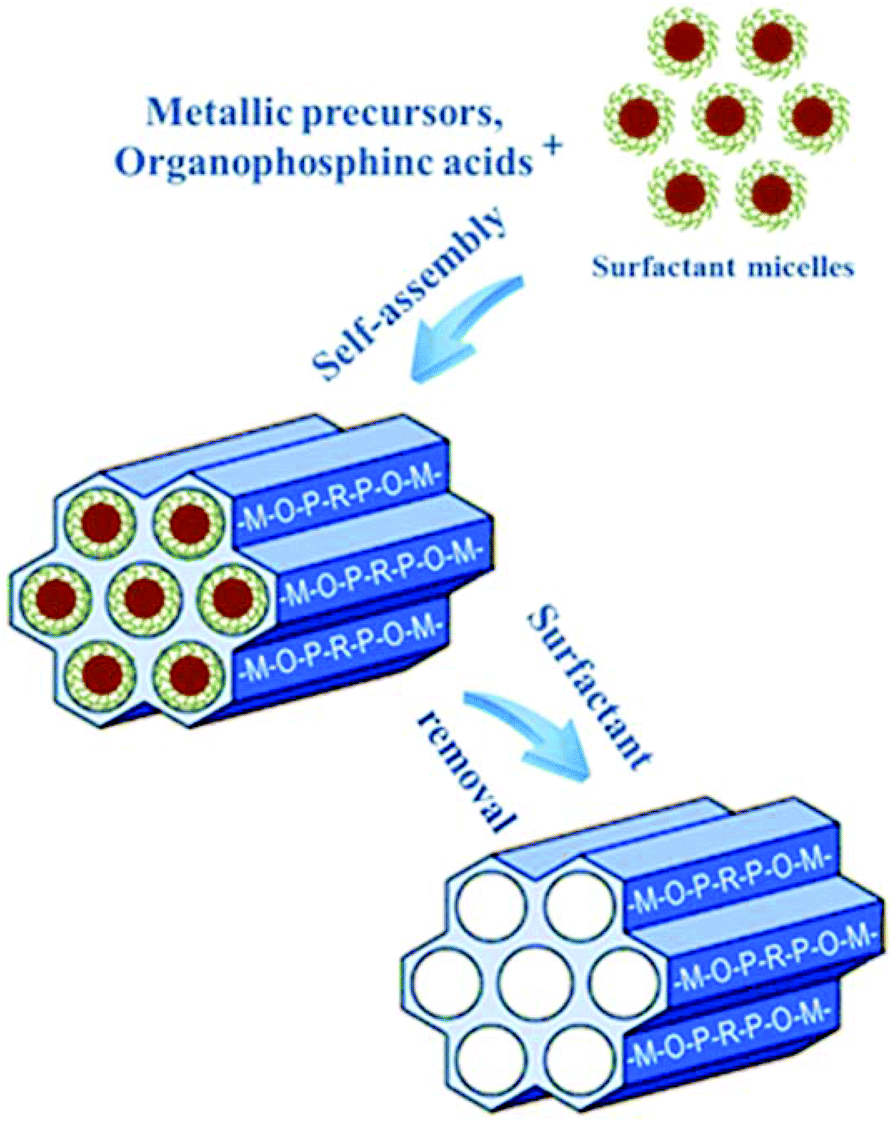 | ||
| Fig. 3 Schematic model for the formation of mesoporous metal phosphonates through soft-templating strategy. | ||
| Metallic precursor | Phosphonic acida | Synthesis strategy | Surface area/m2 g−1 | Pore volume/cm3 g−1 | Pore sizeb/nm | Surfactant | Mesophase | Microstructural phasec | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a PDP: propylene diphosphonic acids, HEDP: 1-hydroxyethane-1,1-diphosphonic acid, EDTMP: ethylenediamine tetra(methylene phosphonic acid). b The microporosity was resulted from the intraparticular aggregation. c Crystal structures have still not been defined due to the low crystallization. | |||||||||
| Al(OBus)3 | PDP, n = 2 | Atrane route | 675 | 0.63 | 3.3 | C16TABr | Hexagonal | — | 77,78 |
| Al(OiC3H7)3 | PDP, n = 1 | — | 738 | 0.32 | 1.8 | C18TACl | Hexagonal | — | 79 |
| AlCl3 | PDP, n = 1 | EISA | 788 | 0.44 | 2.2 | C16TACl | Hexagonal | — | 80 |
| AlCl3 | PDP, n = 2 | EISA | 708 | 0.32 | 1.9 | C16TACl | Hexagonal | — | 80 |
| AlCl3 | PDP, n = 3 | EISA | 217 | 0.27 | 3.0 | Brij 58 | Hexagonal | — | 81 |
| AlCl3 | PDP, n = 3 | EISA | 172 | 0.59 | 7.3 | F68 | Hexagonal | — | 81 |
| AlCl3 | PDP, n = 2 | EISA | 309 | 0.71 | 9.4 | P123 | Hexagonal | — | 81 |
| AlCl3 | PDP, n = 2 | EISA | 337 | 0.79 | 11.6 | F127 | Hexagonal | — | 81 |
| TiCl4 | EDTMP | Hydrothermal and EISA | 1066 | 0.83 | 2.8 | Brij 56 | Hexagonal | Amorphous | 82 |
| TiCl4 | HEDP | Hydrothermal and EISA | 1052 | 0.58 | 2.6 | C16ATBr | Cubic | Amorphous | 86 |
| TiCl4 | HEDP | Hydrothermal and EISA | 1034 | 0.57 | 2.4 | Brij 56 | Hexagonal | Amorphous | 143 |
| TiCl4 | EDTMP | Hydrothermal and EISA | 606 | 0.44 | 2.2 | Brij 56 | Hexagonal | Amorphous | 144 |
| AlCl3 | EDTMP | Microwave-assisted hydrothermal | 498 | 0.61 | 1.7, 7.5 | F127 | Hexagonal | Crystalline aluminum phosphonate | 130 |
| TiCl4 | EDTMP | Microwave-assisted hydrothermal | 522 | 0.63 | 1.4, 7.2 | F127 | Hexagonal | Crystalline titanium phosphonate | 130 |
| ZrCl4 | EDTMP | Microwave-assisted hydrothermal | 513 | 0.64 | 1.5, 7.1 | F127 | Hexagonal | Crystalline zirconium phosphonate | 130 |
| VCl4 | EDTMP | Microwave-assisted hydrothermal | 538 | 0.68 | 1.3, 7.1 | F127 | Hexagonal | Crystalline vanadium phosphonate | 130 |
| Al(OiC3H7)3 | PDP, n = 1 or 2 | EISA | — | — | — | C16TACl | Lamellar | Crystalline aluminum phosphonate | 83 |
| ZrOCl2 | HEDP | Hydrothermal | 702 | 0.86 | 3.6 | C16TABr | Wormhole-like mesopores | Amorphous | 133 |
Pure mesoporous aluminum phosphonates and diphosphonates (UVM-9) were reported by Haskouri et al. with organophosphorus moieties incorporated into the framework.77 The resulting solids were prepared through a one-pot surfactant-assisted procedure that was based on the use of cationic surfactant (CTAB), a complexing polyalcohol (2,2′,2′′-nitrilotriethanol), ethylenephosphonic acid, and methylphosphonic acid or mixed phosphate–phosphonate. All materials displayed XRD patterns with at least one strong peak in the low 2θ range (associated with the [100] reflection if a hexagonal cell is assumed), characteristic of periodic mesoporous materials. TEM showed ordered mesoporous channels as well. The BET surface area of the mesoporous hybrid could reach up to 793 cm2 g−1 accompanied with a narrow pore width distribution around 2.7 nm. An S+I− surfactant-assisted cooperative mechanism through a one-pot preparative procedure from aqueous solution was assumed. The “atrane” complexes from the aluminum and the complexing polyalcohol were confirmed to mediate the hydrolytic rate of aluminum precursors,78 thus effectively enhancing the mesoporous periodicity.
Alternatively, ordered mesoporous aluminum phosphonates could be synthesized with alkyltrimethylammonium as surfactant by acidic strategy79,80 and also with oligomeric surfactant and triblock copolymer through evaporation-induced self-assembly (EISA).81 By using octadecyltrimethylammonium (C18TMA) based on S+I− mechanism, hexagonal mesostructured aluminum phosphonates with organically bridged diphosphonic acids ((HO)2OPCnH2nPO(OH)2, n = 1, 2 and 3) homogeneously distributed in the network.79,80 The removal of surfactant from the as-synthesized hybrid framework has always been a difficult problem. Extraction of as-synthesized materials by acid treatment could not be achieved due to the less condensed and easily hydrolyzed frameworks. On the basis of the difference of thermal stabilities between the methylene groups of phosphonic acid and the C18TMA molecules, surfactant removal could be achieved by low-temperature calcination without decomposition of the bridged methylene groups. Oligomeric surfactants (e.g., Brij 56 and 58) or triblock copolymers (e.g., P123, F68 and F127) depending on (S0H+)X−I+ mesostructure formation mechanism were also employed for the preparation of mesoporous aluminum phosphonates.81 However, only simple alkyl-bridged diphosphonic acids were incorporated into ordered mesoporous phosphonate materials in these studies and the metal sources are restricted to main group metals such as aluminum.
Since different metal joints and organophosphonate linkages in the network would result in distinct physicochemical and mechanical performances, Ma et al. reported a periodic mesoporous titanium phosphonate (PMTP-1) material synthesized using the sodium salt of ethylene diamine tetra(methylene phosphonic acid) (EDTMP) as the claw molecule with the assistance of the oligomeric surfactant Brij 56 in the acidic solution through (S0H+)X−I+ surfactant-induced mechanism.82 During the synthesis, a cryosel bath was used to create low-temperature conditions for slowing down the hydrolysis and condensation speeds of the reactants, and the autoclaving process was utilized and combined with the so-called EISA method to obtain the periodic porous structure. After autoclaving at 120 °C, titanium phosphonate solution was fully condensed containing oligomeric Brij 56 micelles, followed by the EISA process under the stable temperature to give a flaxen gel (Fig. 4). The hexagonal arrangement of the mesopores can be clearly seen from the TEM image with an average pore size of around 2.8 nm and a pore wall thickness of 1.8 nm. The specific surface area and pore volume of PMTP-1 were 1066 m2 g−1 and 0.83 cm3 g−1, respectively.
 | ||
| Fig. 4 Photographs of (a) EDTMP solution, (b) as-synthesized gel before surfactant extracted, (c) mesostructured monoliths after extraction, and (d) final mashed powder. TEM images of the hexagonal mesoporous titanium phosphonate hybrid material (e, f).82 | ||
Unlike mesoporous silicas and carbons, the effective preparation of mesoporous phosphonate hybrids remains challenging, due to unmanageable reaction rates between the precursors, and the intricate interactions between the organic and inorganic components and the surfactant micelles. However, it should be kept in mind that the key factors for the efficient assemblies of periodic mesophases include the control of the aggregation of precursors, sufficient interactions between oligomers/precursors and surfactants, and a suitable size and charge of the building blocks.
Most of the reported mesoporous metal phosphonates are of hexagonal or lamellar phases. As to lamellar phases, the removal of surfactants leads to the irreversible collapse of the mesostructures,83,84 revealing a limited scope from the viewpoint of practical applications. As compared with hexagonal phases, cubic mesostructures possess interconnected pore systems, showing great potential in the areas of adsorption, catalysis and electronics.85 Periodic cubic and hexagonal mesoporous titanium phosphonates were first prepared using organophosphonic acid HEDP in the presence of cationic surfactant C16TAB, and the different phases of the mesopores could be transformed into each other by simply adjusting the molar ratio of the adding amount of surfactant CTAB and inorganic precursor TiCl4.86 This coincided with the previously reported molecular surfactant packing parameter theory that the hexagonal phase is formed at a low surfactant/inorganic species ratio and the cubic phase formed at a high ratio.87 Interestingly, the hexagonal pore structure was well preserved after calcination, leaving a purely inorganic framework, offering a new procedure to synthesize ordered mesoporous titanium phosphate materials.86 However, periodic mesoporous metal phosphonates usually tend to form hexagonal mesophases. This may be due to the fact that the incompletely condensed hybrid network, the complex coordination chemistry between metal ions and organic bridges and the weak interactions among the organic components may converge to form relatively stable hexagonal phases.
On the other hand, the pore size is mainly dependent on the hydrophobic portions of the soft-templating molecules (Table 1). The pore size increased with the hydrophobic volume, admitting the adjustment of pore width at the nanometer scale. In general, the resultant pore size distributes in the region of 2–4 nm for ionic and oligomeric surfactants with low molecular weight,77–81 6–10 nm for some Pluronic block-copolymers,81 even large mesopores (30 nm) when using polystyrene-block-poly(oxyethylene) colloidal template.88,89
Each synthesis pathway has its own advantages and disadvantages. EISA method can fit wide preparation conditions owing to the alleviated hydrolysis speed of metallic precursors, though this strategy needs relatively rigorous conditions including temperature and humidity. Hydrothermal autoclaving is a quick and efficient route, though energy consumption is required for the high temperature and pressure. One should choose the most suitable pathway depending on the practical situation. Exploring low-cost, environmentally friendly and reproducible method is still an urgent requirement.
The syntheses of disordered wormhole-like mesoporous metal phosphonates templated by supramolecular surfactants have also contributed a great deal to the exploiting of the organization principles in the surfactant-assisted strategy. Disordered mesostructures have no unit cell, symmetry or space group. Although the resultant mesostructures are disordered, uniformly sized mesopores, high surface area and easy modulation can usually be achieved, offering them potential in catalysis, adsorption, separation and immobilization. Due to the complexity, it is still difficult to pass a definitive verdict as to which kind of mesostructures, disordered or ordered, are more beneficial in applications. The exploration of the territory of preparing mesoporous phosphonate-based hybrids is not as mature as the silicas and carbonaceous materials owing to the complex interconnected factors, coordination and sol–gel chemistry. Thus further investigation should be of significance and justifies investment.
4 Metal phosphonates with hierarchical porosity
Hierarchical materials containing interconnected porous structures have enhanced properties compared with single-sized pore materials due to increased mass transport through the material and maintenance of a specific surface area on the level of fine pore systems.90 Thus, an elevated performance from adsorption/separation to shape-selective catalysis and biology can be achieved.91 Metal phosphonates with hierarchical porosity (micro-/mesoporosity and meso-/macroporosity) are summarized and tabulated in Table 2.| Porous hierarchy | Metal sources | Phosphonic acidsa | Pore sizeb/nm | Surface area/m2 g−1 | Pore volume/cm3 g−1 | Surfactant | Crystallization | Ref. |
|---|---|---|---|---|---|---|---|---|
| a HDTMP: hexamethylenediamine-N,N,N′,N′-tetrakis-(methylphosphonic acid), BTPA: benzene-1,3,5-triphosphonic acid, BDC: butylene-1,4-diphosphonic acid, PEHMP: pentaethylenehexamine-octakis-(methylphosphonic acid) hexadecasodium salt, ATMP: amino tri(methylene phosphonic acid). b Micro- and mesoporous size are calculated from the N2 sorption isotherm, and the macropore size distributions are determined from the observation of electron microscopy. | ||||||||
| Micro-/mesoporosity | SnCl4 | BTPA | 1.3, 3.5 | 380 | 0.16 | — | Hexagonal crystal phase | 92 |
| NiCl2 | HDTMP | 1.3, — | 241 | 0.29 | — | Tetragonal crystal phase | 93 | |
| SnCl4 | Phytic acid | 1.5, 2.5 | 347 | — | — | Low crystallization | 131 | |
| FeCl3 | BTPA | 1.1, 2.6 | 556 | 0.029 | — | Triclinic crystal phase | 132 | |
| SnCl4 | BDC | 1.2, 2.6, 6.8 | 338 | 0.54 | — | Tetragonal crystal phase | 135 | |
| SnCl4 | PEHMP | 1.4, 2.9, 4.8 | 723 | 0.87 | C16TABr | — | 136 | |
| Meso-/macroporosity | Ti(OnBu)4 | EDTMP | 2.5–10, 100–300 | 86 | 0.074 | — | Amorphous | 101 |
| Ti(OnBu)4 | HEDP | 2.0, 90–400 | 257 | 0.263 | — | Titania-phosphonate | 102 | |
| Ti(OnBu)4 | ATMP | 4.9, 50–100 | 323 | 0.22 | — | Amorphous | 103 | |
| Ti(OnBu)4 | BHMTPMP | 4.8, 400–600 | 307 | 0.21 | — | Amorphous | 107 | |
| Ti(OnBu)4 | DTPMP | 5.1, 500–1000 | 269 | 0.22 | — | Amorphous | 134 | |
| Ti(OnBu)4 | HEDP | 2–3, 800–1200 | 511 | 0.67 | P123 | Amorphous | 106 | |
| Al(OiC3H7)3 | ATMP | 5.4, 500–2000 | 154 | 0.44 | F127 | Amorphous | 108 | |
| BHMTPMP | 5.0, 500–2000 | 128 | 0.42 | F127 | Amorphous | 108 | ||
Self-assembled crystalline metal phosphonates can lead to bimodal porosity, intracrystalline micropores and interparticular mesovoids.92,93 Furthermore, hybrids with self-assembled nanostructures and hierarchical porosity are highly desirable in the context of heterogeneous catalysis. Macropores or macrovoids can be created by using either hard or soft templates, such as colloidal crystals,94 emulsions,95 and polymers,96 or through a spontaneous process in the absence of any surfactants.97 For example, an inverse opals method was applied to prepare a three-dimensionally ordered macroporous titanium phosphonate material.98 The synthesized titanium phosphonate presented a 3D-ordered arrangement of interconnected macropores with a mean pore diameter of 400 nm, which was ca. 10–15% smaller than the size range of the original templated PS spheres, suggesting significant shrinkage during latex sphere extraction. Although the templating method is efficient and general, the further removal of the templates not only perplexes the fabrication procedures, but may also result in the collapse of the structures, and even detrimentally introduce some impurities. Thus a hierarchical architecture through a simple approach with both mesopores and macropores is desirable, which could combine the advantages of the porosity at two scales.
The microemulsion-mediated method has been effectively used to prepare meso-/macroporous materials,99,100 as well as metal phosphonates.101 Uniformly arranged macroporous channels (100–300 nm) were composed of mesostructured porous walls. Wormhole-like mesopores of 2.5–5.8 nm in size were present in the vicinity of the surface, and were accompanied by a new mesocellular foam structure (8–10 nm) in the core region. Since the titanium tetrabutoxide rapidly hydrolyzed in the EDTMP solution, nanometer-sized titanium phosphonate formed immediately with butanol molecules as byproducts. Thus, microemulsion drops formed in the multicomponent system composed of alkoxide–organophosphonate–alcohol–water. The phosphonate sols aggregated along with the microemulsions, evolving into a mesocellular foam structure. The interactions between the sols caused the formation of mesostructured nanoclusters several nanometers in size. If HEDP was chosen as the organic precursor, according to the microemulsion methodology, the interfacial polymerization of titanium phosphonate sols and titanium-oxo clusters resulted in the formation of mesostructured hybrid nanorods with a length of 80–105 nm and a thickness of 18–38 nm.102
Interestingly, organic additives could be utilized to create macropores in the mesoporous hybrid substrates. By using β-cyclodextrin as an organic additive, well-structured mesopores were produced from worm-like cyclodextrin aggregates.103,104 Additionally, large spherical pores with sizes between 50–100 nm were found between the wormhole-like mesopores, which could constitute the inverse spaces of spherical cyclodextrin aggregates.105 It is noteworthy that the pH conditions of the reaction system influence the hydrolysis rate of inorganic metallic precursors. Too high pH values would cause a rapid hydrolysis rate, which would then disrupt the microphase separation process and lead to poor porosity. However, when the pH was lowered to strongly acidic values, the newly formed metal phosphonate clusters were partially dissolved, resulting in poor structured porosity and incompletely condensed frameworks. Consequently, moderate pH conditions were found to be vital in obtaining hierarchical porous phosphonates with stable hybrid networks.
A mild solvent evaporation strategy was used to synthesize titanium phosphonate hybrid materials with a hierarchically meso-/macroporous structure in which copolymers F127 and P123 were used as structure-directing agents and HEDP as phosphonic linker.106 All the samples possessed a macroporous morphology of the mesoporous framework, resulting in high surface areas (>370 m2 g−1). Noticeably, mesostructured titanium phosphonates with unusual uniform lines of macropores were synthesized by using bis(hexamethylenetriamine) penta(methylenephosphonic acid) (BHMTPMP) as the coupling molecule, through a one-pot hydrothermal process without any surfactant assistance (Fig. 5).107 A wormhole-like mesostructure and many uniform parallel lines of macropores were divided by solid ridges in the same direction. This novel macropore architecture has never been observed in other metal phosphonate materials, which may be directly related to the structural nature of BHMTPMP with extra-long alkyl chains. The hydrophobic interaction between the hexamethylenetriamine bridges in the hybrid phosphonate clusters would promote the ordered arrangement of the clusters with a consistent orientation. This two-dimensional polymeric structure might be described as layers built up of alternate inorganic polymers (–Ti⋯O–P–) and alkyl groups, followed by directional extending along the layers. But it was not expected as a molecule-scaled ordered arrangement, and thus the material framework was still amorphous. The microphase separation progressed during the entire process along with the layered stacking of titanium phosphonate nanoparticles. By increasing the content of BHMTPMP, the hydrophobic interaction was further enhanced, causing an improvement in the regularity of the layers, which led to the formation of an ordered arrangement of the macroscopic layers and thicker ridges. Other metal phosphonate networks with hierarchical meso-/macroporosity could also be prepared. For example, hybrid aluminum phosphonate materials with a hierarchical meso-/macroporous structure were synthesized with the assistance of amino tri(methylene phosphonic acid) (ATMP).108 The preparation was accomplished both with and without the assistance of surfactant F127. The product possessed a uniform macroporous (500–2000 nm) framework of mesopores (4–5 nm). Although various surfactants have been used in the construction of hierarchical porosity, the surfactants played no role in the formation of micro- and macropores other than to influence the hydrodynamic conditions during synthesis,109 which could influence the texture of the final materials.
 | ||
| Fig. 5 Time-dependent SEM images of hierarchical porous titanium phosphonates with P/Ti = 1/4 (a–d) and 1/2 (e–h), scale bar: 2 μm. Bottom: a proposed formation mechanism for the obtained macrostructures.107 | ||
A great deal of progress has recently been made in the field of hierarchical porous metal phosphonates. Incorporation of porous hierarchy in metal phosphonate hybrids combines benefits from microporous through mesoporous to macroporous structures. In comparison with single-sized metal phosphonates, hierarchical ones exhibit enhanced properties due to increased mass transport and accessible surface. By adjusting the synthetic conditions and using organic additives and surfactants, the resulting textual properties could be effectively controlled. To obtain ordered macroporous architectures, some supermolecular copolymers are preferred. This is due to the fact that these copolymer templates can be removed under relatively mild conditions while keeping the preformed macroporous channels. However, the general syntheses involve multiple steps, wherein the template removal may result in the collapse of the structures, and even detrimentally introduce some impurities. The use of special metallic precursors, typically metal alkoxides, would favour spontaneous formation of hierarchical pores, during which the phase separation occurred. The uncontrollable hydrolysis rate of metal alkoxides usually leads to poor porous hierarchy with modest yields. The combination of templating method and self-generated mechanism provides an alternative way to construct well-structured hierarchical porous metal phosphonates.
5 Surface modification of oxides with phosphonate layers
Initially, phosphonate ligands were used to construct metal phosphonates with a variety of structures and compositions. In other words, the phosphonate groups were homogeneously incorporated into the hybrid framework. Surface modification of metal oxides with phosphonic acid monolayers to form phosphonate-oxides and/or phosphonated oxides has also attracted extensive research interest. The absence of condensation between the phosphonic ligands, as well as the tendency to form P–O⋯M bonds with good hydrolytic stability, allow the straightforward and reproducible formation of robust phosphonate layers on an extremely wide range of substrates (Fig. 6). Surface phosphonation has raised increasing interest in interfacial chemistry.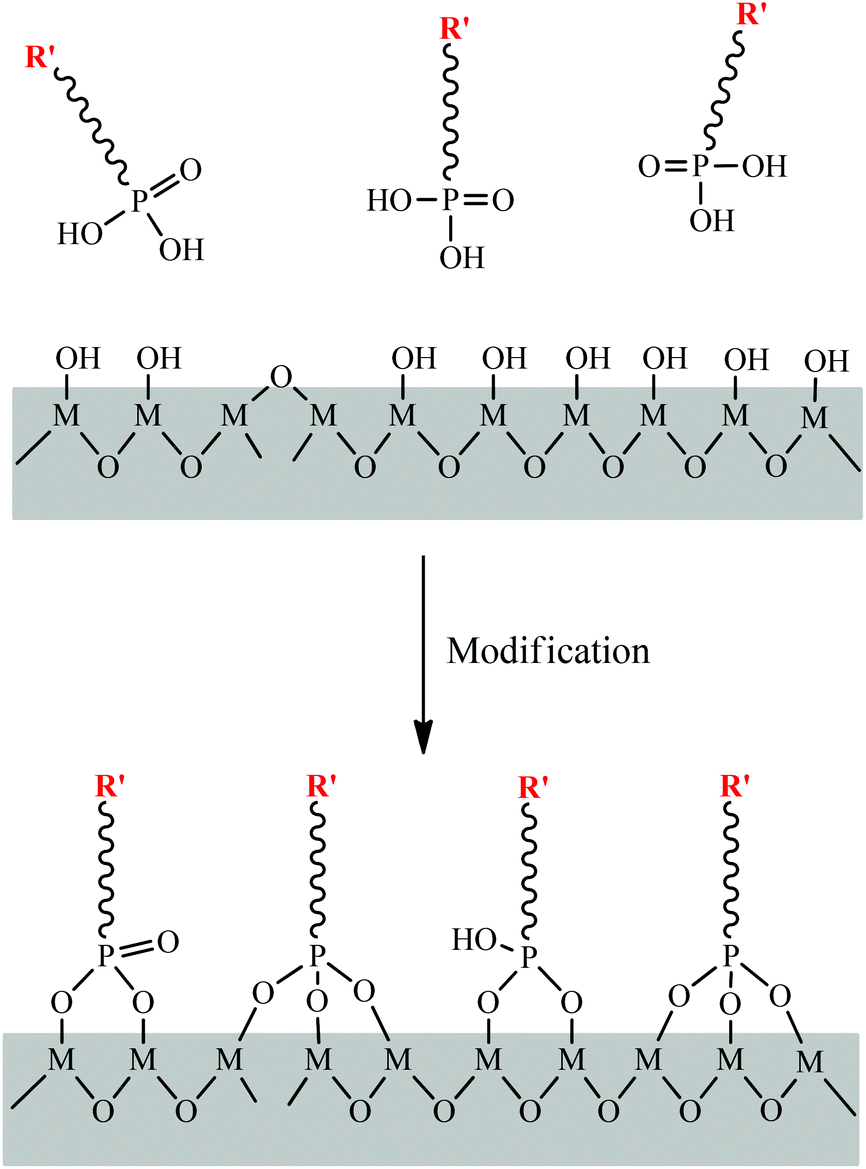 | ||
| Fig. 6 Schematic illustration of surface modification of phosphonic acids on an oxide surface, R′ represents the introduced functional groups. | ||
Mallouk et al. proposed to employ bisphosphonic acids for the formation of metal phosphonate multilayers on surfaces.110 Later on, Guerrero et al. reported the anchoring of phosphonate and phosphinate coupling molecules on titania particles,111 with the use of six organophosphorous compounds: phenylphosphonic and diphenylphosphonic acids, their ethyl esters, and their trimethylsilyl esters. In the case of organophosphorus coupling molecules, reaction with the surface involves not only the condensation with surface hydroxyl groups but also the coordination of the phosphoryl on Lewis acid sites, and the cleavage of the M–O–M bonds depending on the anchoring conditions. The hydrolytic stability of organic monolayers supported on metal oxides was also investigated.112 It was found that the monolayers of C18H37P(O)(OH)2 demonstrated a better hydrolytic stability than other octadecyl organosilane modifiers. The high stability of these phosphonate monolayers is explained by the strong specific interactions of the phosphonic acid group with the surfaces of metal oxides. On the basis of the above mentioned literature reports, the feasibility of grafting phosphonic acids onto metal oxides is fully confirmed. Soler-Illia et al. prepared organic modified transition-metal oxide mesoporous thin films and xerogels by using dihexadecyl phosphate (DHDP), monododecyl phosphate (MDP) and phenyl phosphate (PPA).113 Dramatic differences were observed for the incorporation of organophosphonates in mesoporous versus nonmesoporous solids, demonstrating that the organic functions were incorporated inside the pore system. Incorporation behaviors were also observed depending on the mesostructure; cubic 3D mesostructures are more accessible than their 2D hexagonal counterparts.114 Furthermore, the functionalized pores were found to be further accessible to other molecules (solvent, fluorescent probes) or ions (i.e., Hg2+), opening the way for sensor or sorption applications.
Besides the monophosphonic acids mentioned above, Yuan and co-workers reported the use of a series of amine-based organophosphonic acids and their salts as organophosphorus coupling molecules in the one-step synthesis and the application exploration of oxide-phosphonates and metal organophosphonate hybrid materials with mesopores and hierarchical meso-/macroporous architectures.115,116 Claw molecules of ethylene diamine tetra(methylene phosphonic acid) and diethylene triamine penta(methylene phosphonic acid) were anchored to the titania network homogeneously. The synthesized titania–phosphonate hybrids showed irregular mesoporosity formed by the assembly of nanoparticles in a crystalline anatase phase. The synthesis process is quite simple in comparison with the previously reported two-step sol–gel processing involving first the formation of P–O⋯M bonds by non-hydrolytic condensation of a metal alkoxide with a phosphonic acid and then the formation of the M–O–M bonds of the metal oxide network by hydrolysis/condensation of the remaining alkoxide group.8 The burdensome work to remove the residual organic solvent was not needed.
Phosphonic acids can bind strongly to a wide variety of metal oxides, metals, sulfides and nitrides. On one hand, the original physicochemical properties of the bulk materials can be preserved. On the other hand, diverse novel phosphonates with special properties including bioactivity, electronic conductivity and photochemical properties can be incorporated, showing the capacity to be further modified as well. Furthermore, the phosphonic ligands are not sensitive to moisture and do not polymerize, thus presenting facile and robust procedures for deposition of dense and homogeneous phosphonate layers.
6 Potential applications
Applications for inorganic–organic metal phosphonate hybrid materials are emerging. Owing to their extensive porosity, adjustable composition and controllable structures, metal phosphonates have been developed as multifunctional materials to display versatile excellent performances beyond the traditional use as catalysts and adsorbents, even contributing to the developments in fields ranging from energy storage and conversion to medical diagnosis and therapy. In this section, the typical and emerging applications of metal phosphonates are highlighted and discussed.6.1 Multiphase adsorption and separation
The capture of greenhouse gases such as carbon dioxide (CO2) under practical conditions is quite significant because of the implications for global warming, and the removal of CO2 from industrial flue gas has become an important issue. Among a number of CO2 capture solids including porous carbons,117,118 amine-modified mesoporous silicas,119 carbon–CaO nanocomposites,120 and carboxylate-based MOFs,121 phosphonate-based MOFs exhibit certain advantages such as high surface area, large pore volume, uniform pore width, low cost and relatively high stability and are therefore promising for CO2 capture over a wide range of operating conditions. Cu(1,4-benzenediphosphonate bis(monoalkyl ester)) isomorphous framework with an ethyl tether could capture CO2 with a high isosteric heat of adsorption of 45 kJ mol−1,122 and a modest uptake of CO2 at ambient temperature was observed, 6.06 wt% at 273 K and 1200 mbar. The phosphonate monoesters balanced the crystallinity and the permanent porosity rendered the material accessible to CO2. Using a triazole modified phosphonate ligand, 4-(1,2,4-triazol-4-yl)phenylphosphonic acid, resulted in high selectivity for CO2 over N2 (114![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1),123 which might be due to the higher polarizability and quadrupole moment of CO2 compared with N2 which causes a higher affinity of the pending polar functionalities (–PO3H−1) within the channels. The resultant CO2 uptake amount was 15.8 wt%.
:1),123 which might be due to the higher polarizability and quadrupole moment of CO2 compared with N2 which causes a higher affinity of the pending polar functionalities (–PO3H−1) within the channels. The resultant CO2 uptake amount was 15.8 wt%.
Recent theoretical and experimental studies have revealed the correlation between the amount of CO2 adsorbed and the surface area or pore volume, as well as the increase of adsorption enthalpy for the host materials with open metal sites and active organic functionalities.117,124 In particular, the incorporation of mesoporosity and even hierarchical porosity can optimize the adsorption capacity and kinetics. The CO2 adsorption equilibrium for meso-/macroporous titanium phosphonates could be reached within 50 min.107 The CO2 uptake was 0.89 mmol g−1 at 40 °C, which was higher than some pure silica adsorbents (0.52 mmol g−1) with specific surface area up to 900 m2 g−1.125
It is noteworthy that the adsorption capacity of porous metal phosphonates remained stable even after multiple cycles, making them promising in practical applications.106 Increasing the CO2–framework interactions and improving the porosity of the phosphonate hybrid frameworks presents the major challenge and bottleneck in capturing CO2. Creating open metal sites and introducing basic sites has been shown to effectively increase the interactions. Moreover, in comparison with the carboxylate-based MOFs, the storage of fuel gases such as hydrogen and methane on porous metal phosphonates is scarcely reported. Rigorous investigation towards highly stable and affordable metal phosphonates for theoretical research and practical applications is urgently needed.
Adsorption from liquid phase is much more complicated compared with gas phase adsorption due to the competitive behaviors between solute and solvent for the solid surface. The adsorption of a solute is mainly dependent on the molecular sizes and physicochemical properties, and on the textual properties and surface chemistry of an adsorbent. It can be envisioned that metal phosphonates can perform as good adsorbents for the removal of heavy metal ions from waste waters, due to the fact that the organic functionalities aid the formation of complexes with metal ions through acid–base interactions, and an easy separation of the loaded solid adsorbent from the liquid phase is preferable. For instance, organophosphonic molecules were homogeneously incorporated into the mesoporous walls of the titanium phosphonate PMTP-1 materials, and the specific structure of ethylenediamine could chelate metal ions.82 The maximum adsorption capacity based on the Langmuir isotherm model was determined to be 36.49, 29.03, and 26.87 mmol g−1 adsorbent for Cu2+, Pb2+, and Cd2+, respectively. Further experiments confirmed that, besides the ethylenediamine groups, other electronegative groups or atoms including –OH, –SH, and N could contribute to metal ion adsorption. Meanwhile, the adsorption characteristics of organic pollutants on mesoporous metal phosphonate materials were also identified. It was revealed that mesoporous titanium phosphonate PMTP-1 exhibited excellent adsorption performance for the cationic dye methylene blue (MB) as target pollutant from aqueous solution.126 The adsorption equilibrium was achieved after 30 min of contact time, and the adsorption of MB on PMTP-1 was best fitted to the Langmuir isotherm model with the maximum monolayer adsorption capacity of 617.28 mg g−1, indicating that the PMTP-1 could be used as an efficient adsorbent for the removal of textile dyes from effluents.
The adsorption of biomacromolecules such as proteins from solution onto solid surfaces is also of great scientific importance in many areas, such as biology, medicine, biotechnology and food processing.127 Under pH conditions close to the isoelectric point, the adsorption of lysozyme on aluminum phosphonate hybrid materials was dominated by host–guest hydrophobicity–hydrophobicity interactions.108 Interestingly, unlike inorganic framework adsorbents used for the adsorption of proteins,128 the porous phosphonate hybrid adsorbents had an organic–inorganic framework, which contains plenty of hydrophobic alkyl groups inside the framework.108 The hydrophobicity/hydrophilicity could be controlled at a chemical dimension. So the hydrophobic interactions between the organic groups inside the channel walls and the nonpolar side chains of the amino acids on the surface of lysozyme was greatly enhanced, leading to an increased monolayer adsorption capacity. When extra-long hydrophobic alkyl chains (–[CH2]6–) was incorporated, the resultant adsorption ability was higher than for organophosphonates with fewer hydrophobic –CH2– groups. Since various biomolecules exhibit distinct isoelectric points and spatial sizes, the molecules can be effectively adsorbed and separated by changing the pH and the porosity and pore structures of metal phosphonates.
Chromatography (e.g., gas- and liquid-phase) is one of the most powerful separative methods in analytical chemistry. PMOs and organically modified mesoporous silicas have been employed as the stationary phases in reverse-phase high-performance liquid chromatography (HPLC) in the form of packed columns, for the separation of only neutral compounds.90,129 However, packed columns usually result in low resolution as a result of peak broadening, which impairs the separation efficiency of these hybrid materials. Recently, a series of hexagonal periodic mesoporous metal phosphonates with semi-crystalline pore walls synthesized by microwave irradiation were first employed as the stationary phase in the open-tubular capillary electrochromatography (OTCEC) separation technique,130 which combines the efficiency of capillary electrophoresis and the selectivity of HPLC. The presence of functional groups inside the framework and the excellent properties of the obtained metal phosphonates, including well-ordered pores with large surface area and pore volume and high thermal stability, improved selectivity and so encouraged their use as the stationary phase in wall-coated OTCEC separation for various substances including acidic, basic, and neutral compounds. In the case of neutral compounds including benzene, nitrobenzene, naphthalene, and anthracene (Fig. 7), the elution order was benzene < nitrobenzene < naphthalene < anthracene on the mesoporous titanium phosphonate coated capillary, suggesting a hydrophobicity mechanism for the separation of the species. In contrast, for the ordered mesoporous titanium phosphonate constructed from the same phosphonate linkers (EDTMP) but with an amorphous framework, benzene, naphthalene, and anthracene could be well separated, while benzene and nitrobenzene could not be completely separated. This indicated that, in addition to hydrophobic interactions, suitable polarization of the crystalline mesoporous phosphonates is responsible for the effective separation of benzene and nitrobenzene. Since the separation efficiency mainly depends on the host–guest interactions between stationary and mobile phases, such as hydrophobicity–hydrophobicity interaction, polarity, and intermolecular forces, phosphonate hybrid materials, constructed by long alkyl chains,108 multidimensional linkages,69,70 or phosphonic linkers with polar pendant groups,30,32 can also be used in chromatography techniques, and this is worth exploring.
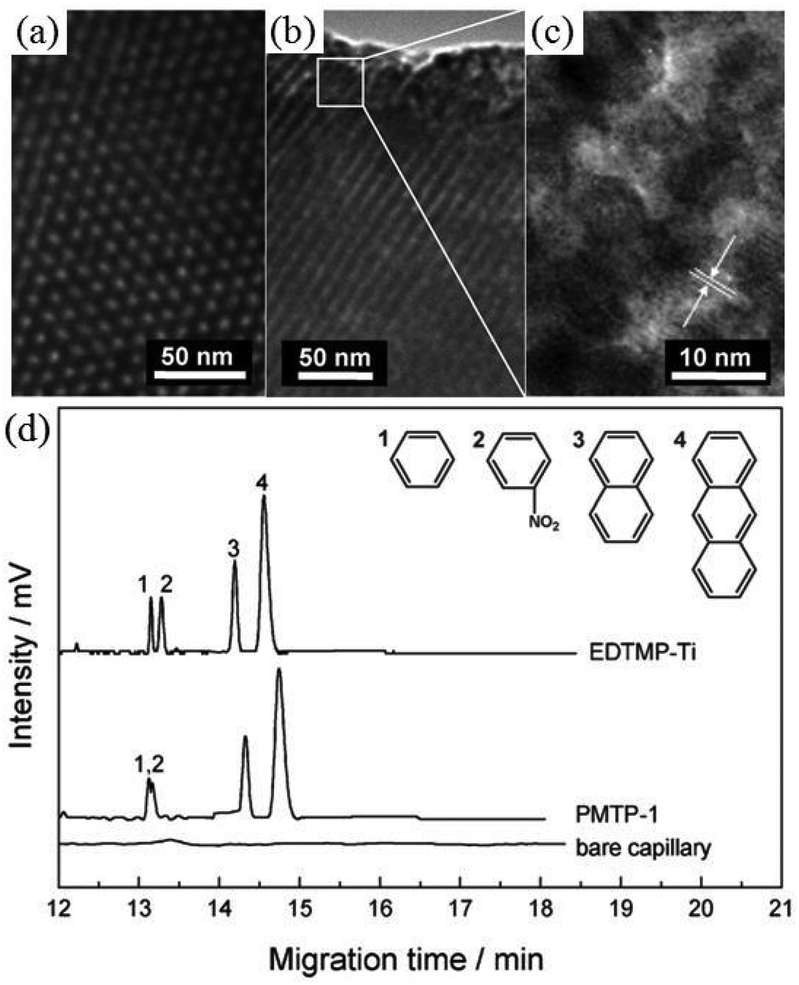 | ||
| Fig. 7 TEM images of hexagonal mesoporous titanium phosphonate (a, b, c), and OTCEC separation of neutral compounds at pH = 3.0 (d). PMTP-1 and EDTMP-Ti stand for amorphous82 and crystalline130 ordered mesoporous titanium phosphonates, respectively. | ||
Although porous metal phosphonates have been successfully applied to adsorbing CO2, toxic metal ions and organic contaminants, and separating organic compounds, some further studies are still needed to illustrate the host–guest interactions, such as model fitting and calculation analysis, supplying theoretical basis to direct the synthesis and to optimize the adsorption/separation capabilities. Thus it is believed that metal phosphonates will find their real value in adsorption and separation in the near future.
6.2 Catalysis
The exploration of porous metal phosphonates as heterogeneous catalysts is of considerable interest due to their porosity which is favorable for the diffusion of reactants and products. It is possible to tailor the porous structures and functionality to yield chemo-, regio-, stereo- and/or enantioselectivity by creating an appropriate environment around the catalytic center in the restricted space available. Furthermore, owing to the homogeneous compositions of hybrid frameworks, a homogeneous distribution of active sites can thus be envisioned.Acid content is one of the key elements to determine catalytic activity and efficiency. Mesostructured zirconium organophosphonate, possessing a specific surface area of 702 m2 g−1 and a uniform pore size of 3.6 nm, was synthesized with the assistance of C16TAB using HEDP as coupling molecule.133 The existence of defective P–OH was confirmed, showing an ion exchange capacity of 1.65 mmol g−1. The resultant hydroxyethylidene bridged mesoporous zirconium phosphonate served as acid catalyst for the synthesis of methyl-2,3-O-isopropylidene-β-D-ribofuranoside from D-ribose, exhibiting high catalytic activity with rapid reaction rate (a product yield of 35.6% after reaction at 70 °C for 3 h), which were comparable to the catalytic performance of liquid HCl (yield of 26.2%) or commercial ion-exchange resin (yield of 33.0%). The excellent catalytic activity could be attributable to the high surface area of the synthesized mesoporous zirconium phosphonates. The high specific surface area is beneficial for the distribution of the active sites and for their exposure so that they can readily be attached by the reactants, and the mesopores aid in the acceleration of mass transfer.
Nonetheless, it was difficult to achieve a high concentration of the desired metal phosphonates in the conventional synthesis methods, since the condensation between P–OH and metal ions during the preparation process often results in the extensive formation of P–O–M (M = Ti, Zr, V, Al, etc.) bonds. In order thus to increase the defective P–OH concentration in metal phosphates and phosphonates for the improvement of the H+ exchange capacity, a series of alkyl amines were used as protecting groups during the condensation process, based on the reversible reaction between alkyl amines and P–OH groups in the phosphonic bridging molecules (Fig. 8).134 The alkyl amines first partially occupied the P–OH sites by acid–base reactions, followed by the condensation between the added alkoxides and residual P–OH and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups. Extraction with HCl finally released the P–OH defects of the resultant solids, leading to a high H+ exchange capacity and acid content. In the absence of amines, the P/Ti ratio reached a plateau of 1.35–1.51 when the added P/Ti ratio was larger than 1.75, due to the limit of coordination ability of Ti4+ ions with phosphonic acids. In the presence of amines, the P/Ti ratio of obtained solids exhibited a sharp initial rise, and finally reached a plateau of 1.59–1.80, which was higher than without amines added. Correspondingly, a similar tendency was observed for the H+ exchange capacity of the synthesized materials. The highest H+ exchange capacities were confirmed as 2.44–2.79 and 5.51–5.80 mmol g−1 for the samples synthesized without and with amines assistance, respectively. A high yield of 48.7% for methyl-2,3-O-isopropylidene-β-D-ribofuranoside was achieved. The product yield did not decrease even after 10 reuses.
O groups. Extraction with HCl finally released the P–OH defects of the resultant solids, leading to a high H+ exchange capacity and acid content. In the absence of amines, the P/Ti ratio reached a plateau of 1.35–1.51 when the added P/Ti ratio was larger than 1.75, due to the limit of coordination ability of Ti4+ ions with phosphonic acids. In the presence of amines, the P/Ti ratio of obtained solids exhibited a sharp initial rise, and finally reached a plateau of 1.59–1.80, which was higher than without amines added. Correspondingly, a similar tendency was observed for the H+ exchange capacity of the synthesized materials. The highest H+ exchange capacities were confirmed as 2.44–2.79 and 5.51–5.80 mmol g−1 for the samples synthesized without and with amines assistance, respectively. A high yield of 48.7% for methyl-2,3-O-isopropylidene-β-D-ribofuranoside was achieved. The product yield did not decrease even after 10 reuses.
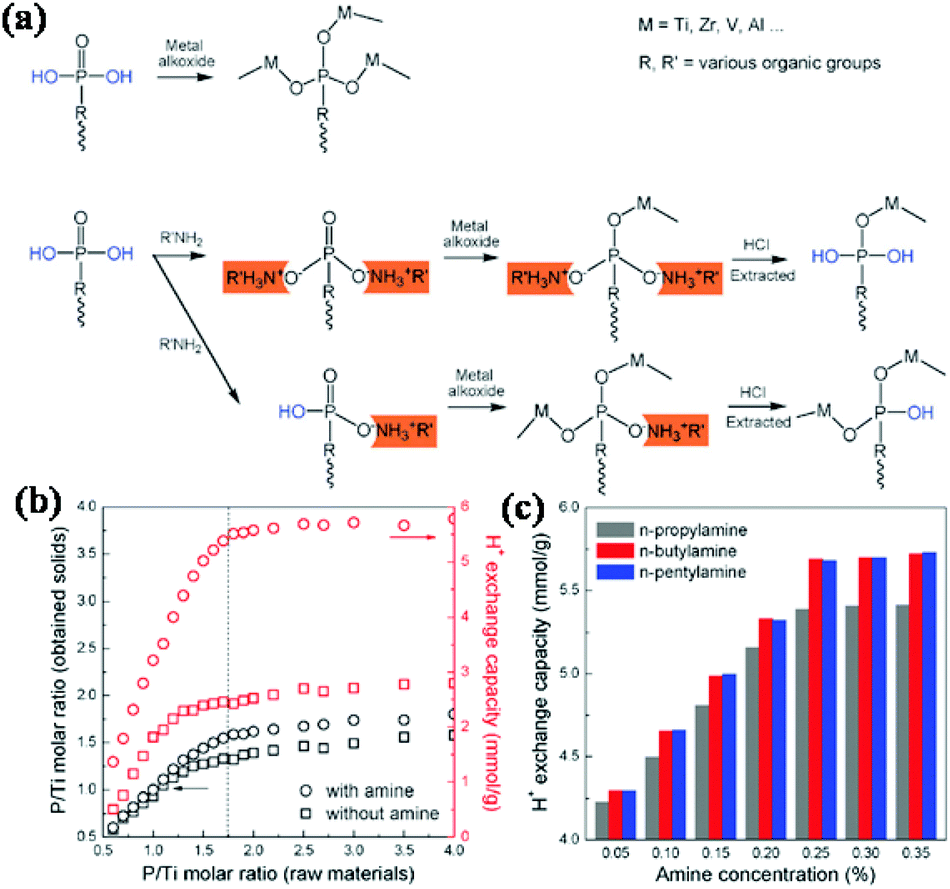 | ||
| Fig. 8 Alkyl amine-assisted preparation of titanium phosphonates (a), the P/Ti molar ratios (b) and ion-exchange capacity (c) of the resultant titanium phosphonates.134 | ||
Tetragonal tin phosphonate hybrid with mesoscopic voids, synthesized by using diphosphonic acid as spacers, were employed as catalyst for the polymerization of styrene to polystyrene in the absence of solvent, and for the partial oxidation of styrene to phenylacetaldehyde and acetophenone in the presence of various aprotic solvents with dilute aqueous H2O2 as an initiator/oxidant.135 The polymerization was completed at room temperature within 2–3 h, but the BET surface area of tetragonal tin phosphonate was relatively low (338 m2 g−1). By using CTAB as structure-directing agent, the surface area of tin phosphonate could be increased to 723 m2 g−1, with the formation of micropores due to crosslinking of the ligand.136 This hybrid demonstrated excellent catalytic activity in the direct one-pot oxidation of cyclohexanone to adipic acid using molecular oxygen under liquid phase conditions. The tin in the framework activated the molecular oxygen, helping to form the cyclic six-membered transition state, which further rearranged into a cyclic ester.
CO2 can serve as C1 building block for various organic chemicals. One of the most promising reaction schemes currently seems to be the formation of cyclic carbonate via coupling of CO2 and epoxides, which are useful as monomers, solvents, and pharmaceutical/fine chemical intermediates, and in biomedical applications.137 Carboxylate-based MOFs have been extensively studied in this area.138–142 Song et al. reported the coupling reaction of CO2 with propylene oxide to produce propylene carbonate catalyzed by MOF-5 in the presence of quaternary ammonium salts.138 The synergetic effect between MOF-5 and quaternary ammonium salts had excellent effect in promoting the reaction. The cycloaddition of CO2 with epoxides is considered to be catalyzed by basicity and promoted by the Lewis acidic sites. On the basis of the intrinsic catalytic sites of the metal-connecting points (weak Lewis acid), the introduction of basic amino groups could lead to an enhanced catalytic performance.142 Bifunctional hybrid catalysts containing moderate Lewis acidic and basic sites are preferred in the cycloaddition reactions. The bifunctionality can not only enhance the conversion and selectivity, but can also simplify the reaction conditions. The attempted bifunctionality can be obtained through judicious selection of precursors or through pre- and post-modification of the organic linkers. Up to now, reports concerning metal phosphonates for the coupling of CO2 and epoxides have been relatively scarce. Since metal phosphonates show similar characteristics of composition and structure and higher stability compared to their carboxylate counterparts, it is meaningful to explore the catalytic activity of metal phosphonates in this burgeoning area.
Inspired by the Langmuir adsorption behavior of metal ions onto porous phosphonates,82,86 a step further would transform the metal ions into active components, which could have potential application in some catalytic reactions. To achieve a high dispersion of CuO nanoparticles, the Cu2+ ions were firstly coordinated on the metal phosphonate material in the form of monolayers, and a subsequent calcination at 450 °C generated the highly dispersed CuO active components.144 Since the PMTP-1 microspheres were thermally stable to 450 °C, the high temperature calcination could achieve a high dispersion of CuO while maintaining the mesoporous hybrid framework. The density and distribution of the surface organic functional groups could be tuned, allowing for an indirect adjustment of the dispersion of the Cu2+ and the final CuO loading amounts. A main reduction peak at 217 °C was observed in the temperature-programmed reduction (H2-TPR) analysis of the synthesized catalysts, which was lower than for pure CuO and CuO catalysts supported on inorganic metal phosphates without organic ligands (252 °C). It is commonly accepted that a high dispersion of active components on the supports can contribute towards improving the catalytic oxidation performance.145 The oxidation of toxic CO was selected as the probe reaction, and the catalytic activity of the synthesized supported catalyst was higher than for those materials with the same CuO content but prepared by the conventional impregnation methods. Moreover, the synthesized catalyst showed a significant stability for low-temperature CO catalytic oxidation.
Noble metals, such as Au, Ag, Pt and Pd, have been known for their catalytic performances. Encapsulation of noble metal nanoparticles inside the metal phosphonate frameworks may extend their applications in catalysis and energy conversion by catalytic spillover.146,147 Interestingly, noble metal nanoparticles with different size regimes could be made through different reduction methods, reduction in ethanol (10–15 nm) and at elevated temperature under hydrogen (2–4 nm).146 This provides a simple way to control the size of the loaded active components.
Catalytic reactions are indeed surface interaction processes, where the metal joints and organophosphonate bridging groups perform as active sites. The special compositions and a variety of structural features can engender catalytic activity in metal phosphonates (Fig. 9). Post-modification can mainly be based on the organic motifs inside the framework, which can create novel physicochemical properties, such as acidity/alkalinity, hydrophobicity/hydrophilicity, chirality. However, there are still issues concerning thermal stability and chemical robustness, due to the nature of the components of the materials. Catalysis supported by metal phosphonates is still in its infancy.
 | ||
| Fig. 9 Summary of some emerging and potential catalytic applications of metal phosphonates. | ||
6.3 Energy conversion and storage
With the rapid development of modern society, energy issues have received increasing attention. On the basis of the ever-increasing demand for clean energy, environmentally sustainable energy resources are urgently needed, and renewable solar energy is regarded as an alternative to conventional fossil fuels. Moreover, there has been significant interest in the development of alternative electronic devices.Mesoporous phosphonated titania hybrid materials were prepared with the use of ATMP as the coupling molecule and triblock copolymer F127 as the template,115 in which the phosphonate groups homogeneously anchored on the mesoporous titania, allowing monolayer adsorption of Zn2+ by extensive coordination with the organic bridging groups. The highly dispersed photoactive ZnO nanoparticles were then formed through low-temperature annealing (180 °C) of the Zn2+ adsorbed mesoporous phosphonated titania, and the resultant ZnO coupled mesoporous phosphonated titanium oxide photocatalysts exhibited excellent photocatalytic activity and stability in the photo-degradation of Rhodamine B under both UV and visible light irradiation.148 In comparison with the pristine mesoporous phosphonate titania, the commercial titania P25, and the ZnO/mesoporous titania prepared by conventional impregnation, the superior photocatalytic performance and stability of the coupled catalyst of ZnO nanoparticles highly dispersed on the mesoporous phosphonated titania might be due to the coupling effect, the well-defined mesoporosity and the incorporation of phosphonic moieties into the TiO2 network, presenting potential applications in the fields of environmental remediation and solar cells.
Solar sensitized cells (SSCs) have been intensively investigated since 1991.149,150 Typical SSCs consist of three parts: work electrodes, counter electrodes, and liquid or polymeric electrolytes (Fig. 10). As regards the work electrode, TiO2 is the most commonly used semiconductor, performing as an electron selective layer between the photo-sensitizers (organic dyes and quantum dots) and electron-collecting conducting glasses. Important aspects for optimization of the cell performance are selection of the photo-sensitizers and its attachment motif to the semiconductor surface. Several of the most efficient dye-sensitized SCs (DSSCs) contain ruthenium–polypyridyl complexes as dyes.151,152 Though efficient in terms of good interfacial electronic coupling in DSSCs, dyes bearing carboxylate anchors have shown limited stability in aqueous and highly oxidizing conditions,153 and bifunctional long-chain carboxylic acids tend to form undesirable looping structures. Very recently, phosphonic acids were found to offer a promising alternative owing to their high affinity toward the surfaces of metal oxides and the relatively stronger binding than carboxylic acids,154,155 and they would thereby give better long-term stability of DSSCs. Mulhern et al. have analyzed the influence of the surface-attachment functions of the dyes on electron transfer at the dye–TiO2 interface and long-term stability.156 Chalcogenorhodamine dyes were attached to the surface of nanocrystalline TiO2 through phosphonic or carboxylic acid functions. No significant changes in the photoelectrochemical performances of DSSCs were observed. H aggregation (i.e. plane-to-plane π-stacking, which broadens the absorbance and causes a blue shift) and electron transfer reactivity were observed when varying the nature of the anchoring group. However, phosphonic linkers were found to enhance the dye–TiO2 bond stability, particularly upon immersion of the material in acidified acetonitrile. Carboxylic-functionalized dyes desorbed completely from TiO2 within 30 min, while no more than 20% desorption occurred with phosphonic-functionalized dyes after 2 days of immersion under the same conditions. By varying the solvent, pH, electrolyte, semiconductor, and presence of oxygen, Hanson et al. carried out a series of investigations of the relative desorption of –PO3H2versus –COOH substituted [RuII(bpy)3]2+ under different conditions.153 Carboxylic-based dyes were found to detach between 5 and 1000 times faster than their phosphonic counterparts in all tested media.
 | ||
| Fig. 10 Schematic illustration of sensitized solar cells. A typical work electrode (WE) is composed of transparent conductive glass (TCG), TiO2 and sensitizers (organic dyes and quantum dots). The sensitized nanostructures are immersed in redox electrolyte, and the circuit is closed by a counter electrode (CE). The latter is usually illuminated through a counter electrode (CE). Energy-band diagram showing the conduction- (CB) and valence-band (VB) edges of the wide-bandgap semiconductor (e.g., TiO2), the ground and excited level of the sensitizers and the redox potential Eredox. Upon solar light illumination, electrons are injected from the excited state into the TiO2, while the oxidized QD is recharged by the redox electrolyte. | ||
Nonetheless, in addition to the fact that phosphonic acids present more stable alternatives, the charge-injection rates can be prohibited to some extent due to the tetrahedral phosphorus center and loss of conjugation.156,157 Combining the superior binding stability of phosphonate and the good electron injection efficiency of carboxylate has resulted in a feasible method (Fig. 11).158 A bis(tridentate)-ruthenium complex containing phosphonic and carboxylic acids was elaborated.157 The underlying basis of this strategy was that the carboxylate moiety only needed to be positioned on the ligand that was involved in the charge transfer to the TiO2, while the phosphonate moieties could be installed on the opposing ligand that did not need to participate directly in the injection process. This led to interesting electron injection properties from the dye into the semiconductor, and a good stability in aqueous media.
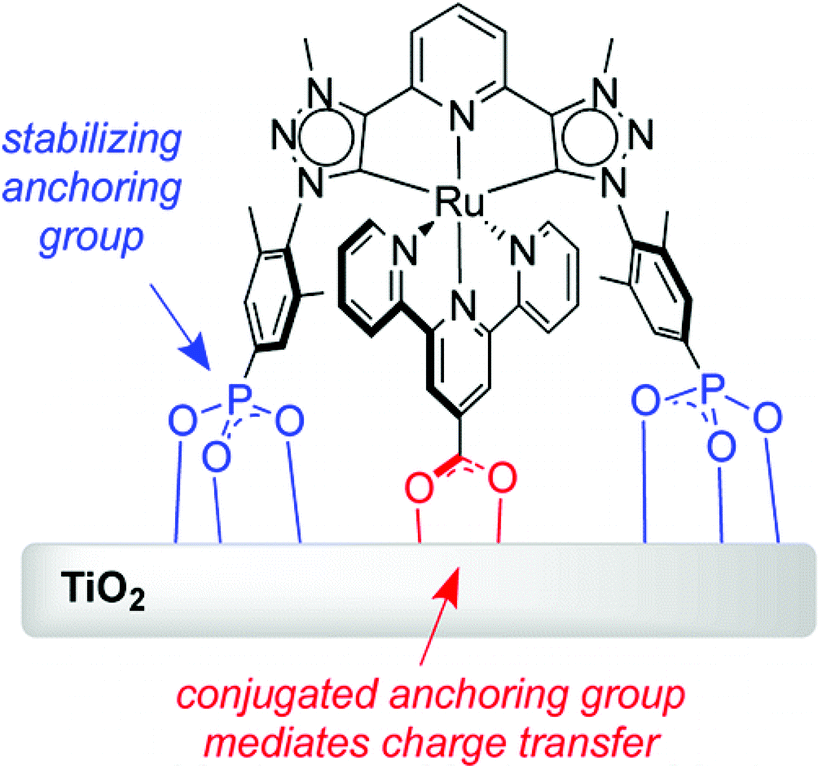 | ||
| Fig. 11 Ruthenium dye functionalized by both phosphonic and carboxylic acids were anchored on TiO2 substrate.158 | ||
The conventional preparation of the dye-sensitized electrodes of solar cells is accomplished by the adsorption of dye molecules onto the pre-synthesized semiconductor membranes, which usually leads to a very low loading amount of the photosensitive molecules.149,150,153 Overloading would result in dye aggregation. This is a waste of the high-cost dyes, because the poor contact between the aggregated dye molecules and the semiconductor could block the transmission of photoelectrons. The sol–gel method allows the molecular-level penetration of large π-aromatic photosensitive groups into the mesoporous semiconductor network homogeneously,159 leading to the achievement of an unprecedented high loading amount of organic dyes. Meanwhile, the aggregation of dye molecules was efficiently suppressed. Thus the resultant mesoporous hybrids were proved to be efficient photocurrent conductors and better electrode materials than the traditional dye-modified titania materials under the same experimental conditions.159 This model supplies us with an alternative strategy for the construction of new dye-sensitized solar cells from organic–inorganic hybrid mesoporous materials.
In the context of solar energy conversion, quantum-dot-sensitized solar cells (QDSSCs) are a promising alternative to existing photovoltaic technologies due to the tunable band gap and promise of stable, low-cost performance.160 In addition, the QDs open up a way to utilize hot electrons and to generate multiple electron–hole pairs with a single photon through impact ionization. The use of organic linkers between the QDs and titania provides a means of eliminating recombination and leads to an increased conversion efficiency and improved stability.160,161 Ardalan et al. investigated the effects of self-assembled monolayers with phosphonic acid head-groups on the bonding and the performance of cadmium sulfide (CdS) SSCs.162 Several organophosphonic acids with different tail-groups (–NH2, –COOH and –CH3) were taken as the linkers. It was demonstrated that the nature of the tail-group does not significantly affect the uptake of CdS quantum dots on TiO2 nanocrystallites nor their optical properties, but the presence of the phosphonic-based linkers had a significant effect on the photovoltaic device performance. The power conversion efficiencies in devices made with phosphonic acids were up to ∼3 times higher than those without any anchoring agent, which might be due to the organic linkers acting as recombination barriers or the passivated defects at the TiO2 surface.162 Furthermore, the electron injection yield depends on the distance between QDs and TiO2, and it decreases with the increase of linkage chain length.163,164 This is a factor worth considering in understanding the functionality of phosphonic linkers and rational design of better photoelectrochemical materials.
Sensitized solar cells play an indispensable role in sustainable development and the exploration of clean energy. It is noteworthy that phosphonate-based DSSCs and QDSSCs show inadequate photo-light conversion efficiency, though the corresponding stability of the electrodes shows potential for long term use. Since QDs can effectively capture solar energy due to the size-dependent adsorbance, QD–dye co-sensitized solar cells can be worthy of investigation. This can not only make full use of sun light, but also combines the advantages of QDs and organic dyes.
Conductivity is a product of the magnitude of the charge, the number of charge carriers, and the mobility of the charges. Conductivity can be tuned by introducing acidic and hydrophilic units, such as carboxylate, phosphonate and sulfonate groups, due to the presence of hydrophilic oxygen atoms acting as hydrogen-bonding acceptors. After the C3-symmetric trisulfonate ligand in PCMOF2 (trisodium 2,4,6-trihydroxy-1,3,5-trisulfonate benzene) was isomorphously substituted with the C3-symmetric tris(hydrogen phosphonate) ligand, the resulting material PCMOF21/2 had its proton conduction raised 1.5 orders of magnitude compared to the parent material, to 2.1 × 10−2 S cm−1 at 90% RH and 85 °C, while maintaining the parent MOF structure.167 This was due to the pores being partially lined with the hydrogen phosphonate groups rather than exclusively nonprotonated sulfonate groups, which should augment proton conduction. A series of MOF–polymer composite membranes exhibited an enhanced low-humidity proton conductivity, compared with that of pure MOF submicrometer crystals, {[Ca(D-Hpmpc)(H2O)2]·2HO0.5}n, at 25 °C and ∼53% RH.168 It was found that the available proton carriers in the MOF structure provided a basis for the conductivity, and the large humidification effect of PVP with adsorbed water molecules greatly contributed to the proton transport in the composite membrane.
Phosphonate-based MOFs have gradually attracted scientists’ interest. As discussed above, the efficient preparation of porous crystalline metal phosphonates still presents many difficulties. If metal phosphonates are to serve as proton conductors for practical application, it would be preferable that they function under relatively mild conditions (e.g., at low temperatures and in anhydrous conditions). It is considered that this goal may be achieved through pre-protection or post-functionalization of the phosphonic bridging groups.
Notably, the considerable ion-exchange capability of metal phosphonates has been confirmed.133,134 Zirconium tetraphosphonates possess an open framework structure with 1D cavities decorated with polar and acidic PO and P–OH groups.169 In addition to the excellent proton conductivity, the hybrid was fully protonated by adding HCl and then subjected to several acid–base ion-exchange reactions with alkaline metal ions, such as Li+, Na+, and K+. Anionic MOF of Zn2.5(H)0.4–0.5(C6H3O9P3)(H2O)1.9–2(NH4)0.5–0.6 was synthesized with the use of urea and 1,3,5-benzenetriphosphonic acid,170 in which ammonium ions are exchangeable with Li+. Due to a certain degree of flexibility of the hybrid framework,13,32,34,170 a reversible insertion/desertion of Li+ through the pores and elastic network can be envisioned, showing potential for secondary batteries. Although this aspect is not extensively studied, the intrinsic porosity within the conductive hybrid materials (ions or protons) remain largely unknown but worthy of research effort.
6.4 Bioapplications
The capacity of metal phosphonates to be designed with specialized functionalities gives them great potential as a new category of materials for bioapplications. Immobilization of enzymes on solid supports can improve enzyme stability, facilitate separation and recycling, and maintain the catalytic activity and selectivity.171 Mesoporous zirconium organophosphonates using 1-phosphomethylproline (H3PMP) as the bridging molecule possess tunable mesopores, high surface area and large pore volume, exhibiting high adsorption capacity and adsorption rates for enzymes.172 For lysozyme (Lz) adsorption, the adsorption equilibrium was reached within 30 min. The adsorption capacity for Lz and papain was as high as 438 and 297 mg g−1, respectively. Furthermore, Lz loaded on mesoporous zirconium phosphonates retained a structural conformation similar to its free state, suggesting that no denaturation of Lz occurred during the adsorption process.163 No leaching of Lz from the solid was observed when shaking the Lz-loaded solid in a buffer solution. The loading of biomolecules into the porous phosphonate hybrid networks is directly correlated with the strength of host–guest interactions,108 surface area and pore size.171,172 Correspondingly, separation of biomolecules can be feasibly realized through utilizing the targeted phosphonic bridging groups and adjusting the porosity of the phosphonate materials.The incorporation of 1,4-bis(phosphomethyl)piperazine (BPMP) introduced pH-sensitivity into the metal phosphonate hybrid network.173 The pH-sensitivity was derived from the reversible protonation under acidic conditions and deprotonation with weakly basic piperazines under different pH conditions, thereby endowing mesoporous zirconium phosphonates with reversible cationic–neutral surface properties. The designed delivery systems of “molecular lock” are able to selectively release the entrapped guests. For instance, the negatively charged PDS (a photosensitizer of sulfonated phthalocyanine for photodynamic therapy of tumors) could then be adsorbed or released through strong electrostatic interaction according to the pH conditions. The integration of H3PMP and BPMP would lead to phosphonate hybrids with bifunctionality, pH-sensitivity and functionalizability.174 The reversible protonation–deprotonation of L-proline groups of H3PMP and piperazine groups of BPMP on the mesoporous walls under different pH values (pH sensitivity) as well as the further functionalization with cell-penetrating peptides via the carboxyl in L-proline group of H3PMP on outer surface (functionalizability) endowed the materials with pH-controllable release function and high cell penetration capability.174 Thus a time- and pH-controlled oral colon-targeted nucleic acid delivery system was developed. Using salmon sperm DNA as model nucleic acid, allowed it to remain intact during delivery. The penetration capability through biomembranes was enhanced through further functionalization with a cell-penetrating peptide of octaarginine.
Numerous studies have indicated that nanoparticle-based therapeutics and diagnostic agents show enhanced efficacy and reduced side effects, due to their unique physicochemical properties.175,176 The vast majority of nanocarriers can be classified into two categories: either purely inorganic (e.g., quantum dots) or purely organic (e.g., liposomes). Noticeably, nanosized metal phosphonate hybrids have the potential to combine attractive characteristics of both inorganic and organic nanocarriers including robust particle morphologies, compositional and structural diversity, biocompatibility and bioactivity, to provide a unique platform for delivering agents, therapeutics and biosensing.23,31,177 Surface modification of iron oxide nanoparticles by phosphonates have a wealth of applications including magnetic resonance imaging (MRI), drug delivery and hyperthermia for cancer treatment.178–180 Lartigue et al. reported the modification of iron oxide nanoparticles with carbohydrates derivatized by phosphonate groups.180 The magnetic, hyperthermal, and relaxometric properties of the phosphonated nanoparticles made them promising candidates for MRI imaging and hyperthermia. On the basis of poly(quaternary ammonium) brushes grown by atom transfer radical polymerization using an initiator grafted via a phosphonate group to the surface of magnetite nanoparticles,181 recyclable antibacterial magnetic nanoparticles were successfully synthesized. Given the convenience of separation of the nanoparticles from the bacterial culture tests using an external magnetic field, the resultant nanoparticles presented high antibacterial activity against E. coli even after eight exposure tests. When cyclodextrin groups were attached to magnetite nanoparticles by using a phosphonic linkage,182 the anchored cyclodextrin formed inclusion complexes with diclofenac sodium salt, a non-steroidal anti-inflammatory drug, demonstrating the potential for targeted drug delivery.
In the past few years, some implant semiconductor biomaterials functionalized by phosphonic acids, such as In2O3 and TiO2, have been investigated for biosensor applications. In2O3 nanowires were first grafted with 3-phosphonopropionic acid, and then the terminal carboxylic acid groups were activated by EDC–NHS aqueous solution,183 resulting in a nanowire surface reactive toward the amine groups present on antibodies. After passivation with an amphipathic polymer (Tween 20), the resultant sensors were found to be capable of performing rapid, label-free, electrical detection of cancer biomarkers directly from human whole blood collected by a finger prick. However, up to now, detection and treatment of organism diseases are two consecutive and inseparable processes in clinical diagnostics and medicine, but their academic studies are often isolated from each other. It is still challenging and significant to design a “diagnospy” carrier that combines the functions of biomolecule quantitative detection and bioresponsive drug controlled release.184,185 Li et al. pioneered a study to intentionally design a smart system on the basis of hybrid phosphonate–TiO2 mesoporous nanostructures capped with fluorescein labelled oligonucleotides, which could realize simultaneous and highly-efficient biomolecule sensing and controlled drug release (Fig. 12).186 The incorporation of phosphonate could shift the absorption edge of titania to the visible light range and introduce positively charged amino groups to interact with negatively charged fluorescein labelled oligonucleotides, resulting in the closing of the mesopores and the fluorescence quenching of fluorescein at the same time. The further addition of complementary single DNA strands or protein target led to the displacement of the capped DNA due to hybridization or protein–aptamer reactions. Correspondingly, the pores were opened, causing the release of entrapped drugs as well as the restoration of dye fluorescence. Moreover, target concentration-dependent fluorescent signal response could be used to monitor treatment effects in real time, thus providing proof for determining drug dose or adjusting the treatment program. This provides a novel perception to utilize non-siliceous hybrid materials as the supports in sensing and control release applications.
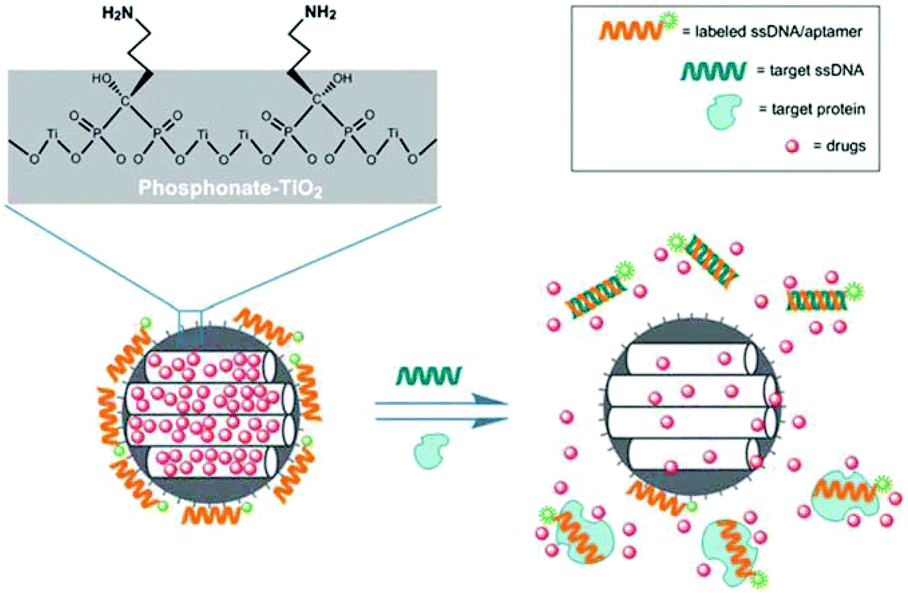 | ||
| Fig. 12 Schematic illustration of bioresponsive detection and drug controlled release system based on phosphonate–TiO2 hybrid material.186 | ||
Metal phosphonates are host materials in the fields of biosensing and biotechnology owing to their well-defined porosity, low biotoxicity and capacity of incorporation of biogroups. Introduction of specific organophosphonic linkers could result in biomimic performance. However, many practical detecting or sensing applications require extraordinarily high sensitivities. Optical sensors are molecular receptors whose optical properties can be changed upon binding to specific guests. Optical sensing and imaging systems have been intensively investigated for their capability of providing high sensitivity, fast and easy detection processing, biocompatibility, and adaptability to a wide variety of conditions.187 Since lanthanide phosphonates are photoluminescent hybrid materials with ease of functionalization,50–54 they are a promising class of materials for applications in sensing and optical imaging.177,188 These areas are of great significance for further exploration.
7 Summary and outlook
Inorganic–organic metal phosphonate materials and the related hybrid nanocomposites have received much attention in the past decades due to their unique physicochemical properties. Due to the less predictable coordination chemistry of phosphonate linkers, densely structured phosphonate motifs are formed. Through inserting non-pillaring spacers, complicating the organophosphonic linkages and attaching a functional group on the ligand could lead to the creation of porosity in the hybrid frameworks. To improve the accessibility of the porous systems, template-free and surfactant-assisted strategies on the basis of sol–gel chemistry have proven to be effective in obtaining porous phosphonate hybrids with adjustable porosity. Surface modification of the inorganic oxide substrates with phosphonates can not only preserve the nature of bulk, but also control the surface and interface properties of materials. The developments in modification of metal phosphonates demonstrate how modern physical techniques and chemical design can be applied to functionalize their internal surfaces. Metal phosphonate hybrids and their relevant composites have demonstrated significant potential ranging from adsorption/separation and catalysis to the burgeoning biotechnology and energy conversion and storage.Although metal phosphonates have been shown to outperform other porous materials in some applications, the relatively inferior stability of the hybrid materials compared with inorganic frameworks has impeded further application. The effort for the enhancement of the thermal and hydrothermal stability of metal phosphonate materials is of great importance, and the crystalline pore wall is first in line. However, for phosphonates, the unpredictable ligating mode would facilitate the formation of networks, while it could also result in precipitates with low degrees of order. Furthermore, it remains contradictory and challenging to achieve the high crystallization and well-structured hierarchical porosity (especially micro-/mesoporosity) simultaneously. The coordination rate between the inorganic units and organic moieties determines the nucleation kinetics and thus the crystal growth.189 “Crown-mediated controlled-release” methodology has recently exhibited the possibility to construct porous sulfonate- and carboxylate-based MOFs,189,190 which might provide a new route in the rational design of crystalline metal phosphonates with well-defined porous structures. Considering the intimate relation between phosphonates and phosphates, phosphonates may act as precursors in preparing phosphates with interesting structures and novel properties, thus improving the performances of phosphates in catalysis, photoelectricity and secondary batteries.191–194
Overall, the development in inorganic–organic metal phosphonate materials signifies a pivotal step towards exploring and finding new multifunctional hybrid materials and has significantly expanded the application ranges. Nowadays, the molecular approaches of solid state chemistry and organic synthesis have reached a high level of sophistication. As a consequence, original phosphonate hybrids can be designed through the synthesis of new hybrid nanosynthons, allowing for the coding of hybrid assemblies presenting a spatial ordering at different length scales. Particularly, the synthesis through the simultaneous use of self-assembly processes together with external factors, such as electrical or magnetic fields, or even through the use of strong compositional flux variations of the reagents during the synthesis are worthwhile areas to explore. The increasing interest in the field of functional hybrid metal phosphonate materials will be amplified in the future by the growing interest of materials scientists, chemists and biologists to fully exploit this opportunity for discovering materials and devices benefiting from the best of the two realms: inorganic and organic.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21076056 and 21073099), the Specialized Research Fund for the Doctoral Program of Higher Education (20110031110016), the Program for Innovative Research Team in University (IRT13022), and the 111 project (B12015). Z.Y.Y. also thanks the Royal Academy of Engineering for a Research Exchanges with China and India Award.Notes and references
- J. Livage and J. Lemerle, Annu. Rev. Mater. Sci., 1982, 12, 103 CrossRef CAS.
- J. Livage, Chem. Mater., 1991, 3, 578 CrossRef CAS.
- J. Livage, M. Henry and C. Sanchez, Prog. Solid State Chem., 1988, 18, 259 CrossRef CAS.
- J. Livage, New J. Chem., 2001, 25, 1 RSC.
- C. Sanchez and J. Livage, New J. Chem., 1990, 14, 503 Search PubMed.
- K. Nakanishi and K. Kanamori, J. Mater. Chem., 2005, 15, 3776 RSC.
- P. Innocenzia and B. Lebeau, J. Mater. Chem., 2005, 15, 3821 RSC.
- W. J. Hunks and G. A. Ozin, J. Mater. Chem., 2005, 15, 3716 RSC.
- S. Mann, Biomimetic Materials Chemistry, Wiley-VCH, Weinheim, 1997 Search PubMed.
- S. Mann, Angew. Chem., Int. Ed., 2000, 39, 3393 CrossRef.
- P. Gomez-Romero, Adv. Mater., 2001, 13, 163 CrossRef CAS.
- R. Gangopadhyay and A. De, Chem. Mater., 2000, 12, 608 CrossRef CAS.
- G. K. H. Shimizu, R. Vaidhyanathan and J. M. Taylor, Chem. Soc. Rev., 2009, 38, 1430 RSC.
- C. Sanchez and F. Ribot, New J. Chem., 1994, 18, 1007 CAS.
- P. Judeinstein and C. Sanchez, J. Mater. Chem., 1996, 6, 511 RSC.
- R. J. P. Corriu, New J. Chem., 2001, 25, 2 RSC.
- T. Z. Ren, Z. Y. Yuan and L. B. Su, Chem. Commun., 2004, 2730 RSC.
- E. G. Vrieling, T. P. M. Beelen, R. A. van Santen and W. W. C. Gieskes, Angew. Chem., Int. Ed., 2002, 41, 1543 CrossRef CAS.
- C. Sanchez, L. Rozes, F. Ribot, C. Laberty-Robert, D. Grosso, C. Sassoye, C. Boissiere and L. Nicole, C. R. Chim., 2010, 13, 3 CrossRef CAS PubMed.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611 CrossRef CAS.
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867 CAS.
- B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302 CrossRef CAS.
- Y. P. Zhu, T. Z. Ren and Z. Y. Yuan, New J. Chem., 2014, 38, 1905 RSC.
- A. Clearfield and Z. Wang, J. Chem. Soc., Dalton Trans., 2002, 2937 RSC.
- A. Clearfield, Curr. Opin. Solid State Mater. Sci., 1996, 1, 268 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed.
- K. S. W. Sing, D. H. Everett, R. H. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- T. Kimura, J. Nanosci. Nanotechnol., 2013, 13, 2461 CrossRef CAS PubMed.
- A. Clearfield, Dalton Trans., 2008, 6089 RSC.
- T. Y. Ma and Z. Y. Yuan, ChemSusChem, 2011, 4, 1407 CrossRef CAS PubMed.
- K. J. Gagnon, H. P. Perry and A. Clearfield, Chem. Rev., 2012, 112, 1034 CrossRef CAS PubMed.
- K. Maeda, Microporous Mesoporous Mater., 2004, 73, 47 CrossRef CAS PubMed.
- A. Clearfield, C. V. K. Sharma and B. L. Zhang, Chem. Mater., 2001, 13, 3099 CrossRef CAS.
- R. C. Finn, R. Lam, J. E. Greedan and J. Zubieta, Inorg. Chem., 2001, 40, 3745 CrossRef CAS PubMed.
- C. Herdes, A. Valente, Z. Lin, J. Rocha, A. A. P. Coutinho, F. Medina and L. F. Vega, Langmuir, 2007, 23, 7299 CrossRef CAS PubMed.
- D. B. Mitzi, Chem. Mater., 2001, 13, 3283 CrossRef CAS.
- F. Fredoueil, M. Evain, D. Massiot, M. Bujoli-Doeuff and B. Bujoli, J. Mater. Chem., 2001, 11, 1106 RSC.
- J. L. Snover, H. Byrd, E. P. Suponeva, E. Vicenzi and M. E. Thompson, Chem. Mater., 1996, 8, 1490 CrossRef CAS.
- G. Alberti, U. Costsntino, S. Allulli and N. Tomsssini, J. Inorg. Nucl. Chem., 1978, 4, 1113 CrossRef.
- G. Y. Yang and A. Clewfield, React. Polym., 1987, 5, 13 Search PubMed.
- C. Y. Ortiz-Avila and A. Clewfield, Inorg. Chem., 1985, 24, 1773 CrossRef CAS.
- H. Byrd, A. Clearfield, D. Poojary, K. P. Reis and M. E. Thompson, Chem. Mater., 1996, 8, 2239 CrossRef CAS.
- A. Clearfield, Z. K. Wang and P. Bellinghausen, J. Solid State Chem., 2002, 167, 376 CrossRef CAS.
- J. G. Mao, Coord. Chem. Rev., 2007, 251, 1493 CrossRef CAS PubMed.
- O. R. Evans, H. L. Ngo and W. B. Lin, J. Am. Chem. Soc., 2001, 123, 10395 CrossRef CAS.
- H. L. Ngo and W. B. Lin, J. Am. Chem. Soc., 2002, 124, 14298 CrossRef CAS PubMed.
- A. Clearfield, C. V. K. Sharma and B. L. Zhang, Chem. Mater., 2001, 13, 3099 CrossRef CAS.
- S. W. A. Bligh, N. Choi, C. F. G. C. Geraldes, S. Knoke, M. McPartlin, M. J. Sanganee and T. M. Woodroffe, J. Chem. Soc., Dalton Trans., 1997, 4119 RSC.
- F. N. Shi, L. Cunha-Silva, R. A. S. Ferreira, L. Mafra, T. Trindade, L. D. Carlos, F. A. A. Paz and J. Rocha, J. Am. Chem. Soc., 2008, 130, 150 CrossRef CAS PubMed.
- L. Cunha-Silva, L. Mafra, D. Ananias, L. D. Carlos, J. Rocha and F. A. A. Paz, Chem. Mater., 2007, 19, 3527 CrossRef CAS.
- S. M. Ying and J. G. Mao, Cryst. Growth Des., 2006, 6, 964 CAS.
- B. P. Yang, A. V. Prosvirin, Y. Q. Guo and J. G. Mao, Inorg. Chem., 2008, 47, 1453 CrossRef CAS PubMed.
- P. Rabu, P. Janvierb and B. Bujoli, J. Mater. Chem., 1999, 9, 1323 RSC.
- J. G. Mao, Z. K. Wang and A. Clearfield, Inorg. Chem., 2002, 41, 6106 CrossRef CAS PubMed.
- J. T. Li, L. R. Guo, Y. Shen and L. M. Zheng, CrystEngComm, 2009, 11, 1674 RSC.
- J. Bideau, C. L. Payen, P. Palvadeau and B. Bujoli, Inorg. Chem., 1994, 33, 4885 CrossRef.
- K. Maeda, Y. Kiyozumi and F. Mizukami, Angew. Chem., Int. Ed., 1994, 33, 2335 CrossRef.
- K. Maeda, J. Akimoto, Y. Kiyozumi and F. Mizukami, Angew. Chem., Int. Ed., 1995, 34, 1199 CrossRef CAS.
- D. L. Lohse and S. C. Sevov, Angew. Chem., Int. Ed., 1997, 36, 1619 CrossRef CAS.
- R. N. Devi, P. Wormald, P. A. Cox and P. A. Wright, Chem. Mater., 2004, 16, 2229 CrossRef CAS.
- E. V. Bakhmutova, X. Ouyang, D. G. Medvedev and A. Clearfield, Inorg. Chem., 2003, 42, 7046 CrossRef CAS PubMed.
- L. M. Zheng, H. H. Song, C. Y. Duan and X. Q. Xin, Inorg. Chem., 1999, 38, 5061 CrossRef CAS PubMed.
- A. Turner, B. P. A. Jaffres, E. J. MacLean, D. Villemin, V. McKee and G. B. Hix, Dalton Trans., 2003, 1314 RSC.
- G. Guerrero, J. G. Alauzun, M. Granier, D. Laurencin and P. H. Mutin, Dalton Trans., 2013, 42, 12569 RSC.
- H. P. Perry, J. Law, J. Zon and A. Clearfield, Microporous Mesoporous Mater., 2012, 149, 172 CrossRef CAS PubMed.
- C. Serre, N. Stock, T. Bein and G. Férey, Inorg. Chem., 2004, 43, 3159 CrossRef CAS PubMed.
- Q. Yue, J. Yang, G. H. Li, G. D. Li and J. S. Chen, Inorg. Chem., 2006, 45, 4431 CrossRef CAS PubMed.
- M. Vasylyev and R. Neumann, Chem. Mater., 2006, 18, 2781 CrossRef CAS.
- M. Vasylyev, E. J. Wachtel, R. Popovitz-Biro and R. Neumann, Chem. – Eur. J., 2006, 12, 3507 CrossRef CAS PubMed.
- L. Nicole, C. Boissière, D. Grosso, A. Quach and C. Sanchez, J. Mater. Chem., 2005, 15, 3598 RSC.
- G. Alberti, F. Marmottini, R. Vivani and P. Zappelli, J. Porous Mater., 1998, 5, 221 CrossRef CAS.
- Y. P. Zhu, Y. L. Liu, T. Z. Ren and Z. Y. Yuan, RSC Adv., 2014, 4, 16018 RSC.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- N. Pal and A. Bhaumik, Adv. Colloid Interface Sci., 2013, 189, 21 CrossRef PubMed.
- T. Y. Ma, L. Liu and Z. Y. Yuan, Chem. Soc. Rev., 2013, 42, 3977 RSC.
- J. El. Haskouri, C. Guillem, J. Latorre, A. Beltrán, D. Beltrán and P. Amorós, Eur. J. Inorg. Chem., 2004, 1804 CrossRef.
- J. El. Haskouri, C. Guillem, J. Latorre, A. Beltrán, D. Beltrán and P. Amorós, Chem. Mater., 2004, 16, 4359 CrossRef.
- T. Kimura, Chem. Mater., 2003, 15, 3742 CrossRef CAS.
- T. Kimura, Chem. Mater., 2005, 17, 337 CrossRef CAS.
- T. Kimura, Chem. Mater., 2005, 17, 5521 CrossRef CAS.
- T. Y. Ma, X. Z. Lin and Z. Y. Yuan, J. Mater. Chem., 2010, 20, 7406 RSC.
- T. Kimura, D. Nakashima and N. Miyamoto, Chem. Lett., 2009, 38, 916 CrossRef CAS.
- Y. H. Deng, J. Wei, Z. K. Sun and D. Y. Zhao, Chem. Soc. Rev., 2013, 42, 4054 RSC.
- Y. Wan and D. Y. Zhao, Chem. Rev., 2007, 107, 2821 CrossRef CAS PubMed.
- T. Y. Ma, X. Z. Lin and Z. Y. Yuan, Chem. – Eur. J., 2010, 16, 8487 CrossRef CAS PubMed.
- Q. S. Huo, D. I. Margolese and G. D. Stucky, Chem. Mater., 1996, 8, 1147 CrossRef CAS.
- T. Kimura and Y. Yamauchi, Chem. – Asian J., 2013, 8, 160 CrossRef CAS PubMed.
- T. Kimura and Y. Yamauchi, Langmuir, 2012, 28, 12901 CrossRef CAS PubMed.
- Z. Y. Yuan and B. L. Su, J. Mater. Chem., 2006, 16, 663 RSC.
- T. Z. Ren, Z. Y. Yuan and B. L. Su, Chem. Commun., 2004, 2730 RSC.
- M. Pramanik and A. Bhaumik, J. Mater. Chem. A, 2013, 1, 11210 CAS.
- A. Dutta, A. K. Patra and A. Bhaumik, Microporous Mesoporous Mater., 2012, 155, 208 CrossRef CAS PubMed.
- P. Yang, T. Deng, D. Zhao, P. Feng, D. Pine, B. F. Chmelka, G. M. Whitesides and G. D. Stucky, Science, 1998, 282, 2244 CrossRef CAS.
- A. Imhof and D. J. Pine, Nature, 1997, 389, 948 CrossRef CAS PubMed.
- K. J. Nakanishi, J. Porous Mater., 1997, 4, 67 CrossRef CAS.
- T. Amatani, K. Nakanishi, K. Hirao and T. Kodaira, Chem. Mater., 2005, 17, 2114 CrossRef CAS.
- T. Y. Ma, X. J. Zhang, G. S. Shao, J. L. Cao and Z. Y. Yuan, J. Phys. Chem. C, 2008, 112, 3090 CAS.
- X. Y. Yang, A. Leonard, A. Lemaire, G. Tian and B. L. Su, Chem. Commun., 2011, 47, 2763 RSC.
- Z. Y. Yuan, T. Z. Ren, A. Azioune, J. J. Pireaux and B. L. Su, Chem. Mater., 2006, 18, 1753 CrossRef CAS.
- T. Y. Ma, X. J. Zhang and Z. Y. Yuan, Microporous Mesoporous Mater., 2009, 123, 234 CrossRef CAS PubMed.
- X. J. Zhang, T. Y. Ma and Z. Y. Yuan, Eur. J. Inorg. Chem., 2008, 2721 CrossRef CAS.
- T. Y. Ma and Z. Y. Yuan, Eur. J. Inorg. Chem., 2010, 19, 2941 CrossRef.
- S. Polarz, B. Smarsly, L. Bronstein and M. Antonietti, Angew. Chem., Int. Ed., 2001, 40, 4417 CrossRef CAS.
- M. Bonini, S. Rossi, G. Karlsson, M. Almgren, P. L. Nostro and P. Baglioni, Langmuir, 2006, 22, 1478 CrossRef CAS PubMed.
- T. Y. Ma, X. Z. Lin, X. J. Zhang and Z. Y. Yuan, New J. Chem., 2010, 34, 1209 RSC.
- T. Y. Ma, X. Z. Lin, X. J. Zhang and Z. Y. Yuan, Nanoscale, 2011, 3, 1690 RSC.
- T. Y. Ma, X. J. Zhang and Z. Y. Yuan, J. Phys. Chem. C, 2009, 113, 12854 CAS.
- W. H. Deng and B. H. Shanks, Chem. Mater., 2005, 17, 3092 CrossRef CAS.
- H. Lee, L. J. Kepley, H. G. Hong and T. E. Mallouk, J. Am. Chem. Soc., 1988, 110, 618 CrossRef CAS.
- G. Guerrero, P. H. Mutin and A. Vioux, Chem. Mater., 2001, 13, 4367 CrossRef CAS.
- S. Marcinko and A. Y. Fadeev, Langmuir, 2004, 20, 2270 CrossRef CAS.
- P. C. Angelomé and G. J. de A. A. Soler-Illia, Chem. Mater., 2005, 17, 3223 CrossRef.
- P. C. Angelomé and G. J. de A. A. Soler-Illia, J. Mater. Chem., 2005, 15, 3903 RSC.
- X. J. Zhang, T. Y. Ma and Z. Y. Yuan, J. Mater. Chem., 2008, 18, 2003 RSC.
- T. Y. Ma, X. J. Zhang and Z. Y. Yuan, J. Mater. Sci., 2009, 44, 6775 CrossRef CAS.
- L. Liu, Q. F. Deng, X. X. Hou and Z. Y. Yuan, J. Mater. Chem., 2012, 22, 15540 RSC.
- L. Liu, Q. F. Deng, T. Y. Ma, X. Z. Lin, X. X. Hou, Y. P. Liu and Z. Y. Yuan, J. Mater. Chem., 2011, 21, 16001 RSC.
- R. Serna-Guerrero, E. Dána and A. Sayari, Ind. Eng. Chem. Res., 2008, 47, 9406 CrossRef CAS.
- Z. X. Wu, N. Hao, G. K. Xiao, L. Y. Liu, P. Webley and D. Y. Zhao, Phys. Chem. Chem. Phys., 2011, 13, 2495 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 CrossRef CAS PubMed.
- S. S. Iremonger, J. M. Liang, R. Vaidhyanathan, I. Martens, G. K. H. Shimizu, T. D. Daff, M. Z. Aghaji, S. Yeganegi and T. K. Woo, J. Am. Chem. Soc., 2011, 133, 20048 CrossRef CAS PubMed.
- F. P. Zhai, Q. S. Zheng, Z. X. Chen, Y. Ling, X. F. Liu, L. H. Weng and Y. M. Zhou, CrystEngComm, 2013, 15, 2040 RSC.
- L. F. Song, J. Zhang, L. X. Sun, F. Xu, F. Li, H. Z. Zhang, X. L. Si, C. L. Jiao, Z. B. Li, S. Liu, Y. L. Liu, H. Y. Zhou, D. L. Sun, Y. Du, Z. Cao and Z. Gabelica, Energy Environ. Sci., 2012, 5, 7508 CAS.
- G. P. Knowles, S. W. Delaney and A. L. Chaffee, Ind. Eng. Chem. Res., 2006, 45, 2626 CrossRef CAS.
- T.-Z. Ren, X.-H. Zhu, T.-Y. Ma and Z.-Y. Yuan, Adsorpt. Sci. Technol., 2013, 31, 535 CrossRef CAS PubMed.
- V. Lykourinou, Y. Chen, X. S. Wang, L. Meng, T. Hoang, L. J. Ming, R. L. Musselman and S. Q. Ma, J. Am. Chem. Soc., 2011, 133, 10382 CrossRef CAS PubMed.
- F. N. Shi, L. Cunha-Silva, R. A. S. Ferreira, L. Mafra, T. Trindade, L. D. Carlos, F. A. A. Paz and J. Rocha, J. Am. Chem. Soc., 2008, 130, 150 CrossRef CAS PubMed.
- T. Salesch, S. Bachmann, S. Brugger, R. Rabelo-Schaefer, K. Albert, S. Steinbrecher, E. Plies, A. Mehdi, C. Reyé, R. J. P. Corriu and E. Lindner, Adv. Funct. Mater., 2002, 13, 134 CrossRef.
- T. Y. Ma, H. Li, A. N. Tang and Z. Y. Yuan, Small, 2011, 4, 1407 CAS.
- A. Dutta, A. K. Patra, H. Uyama and A. Bhaumik, ACS Appl. Mater. Interfaces, 2013, 5, 9913 CAS.
- M. Pramanik and A. Bhaumik, Chem. – Eur. J., 2013, 19, 8507 CrossRef CAS PubMed.
- X. Z. Lin and Z. Y. Yuan, Eur. J. Inorg. Chem., 2012, 2661 CrossRef CAS.
- T. Y. Ma, L. Liu, Q. D. Deng, X. Z. Lin and Z. Y. Yuan, Chem. Commun., 2011, 47, 6015 RSC.
- M. Pramanik, M. Nandi, H. Uyama and A. Bhaumik, Catal. Sci. Technol., 2012, 2, 613 CAS.
- A. Dutta, M. Pramanik, A. K. Patra, M. Nandi, H. Uyama and A. Bhaumik, Chem. Commun., 2012, 48, 6738 RSC.
- A. A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951 CrossRef CAS PubMed.
- J. L. Song, Z. F. Zhang, S. Q. Hu, T. B. Wu, T. Jiang and B. X. Han, Green Chem., 2009, 11, 1031 RSC.
- D. A. Yang, H. Y. Cho, J. Kim, S. T. Yang and W. S. Ahn, Energy Environ. Sci., 2012, 5, 6465 CAS.
- Y. W. Ren, Y. C. Shi, J. X. Chen, S. R. Yang, C. R. Qi and H. F. Jiang, RSC Adv., 2013, 3, 2167 RSC.
- D. W. Feng, W. C. Chung, Z. W. Wei, Z. Y. Gu, H. L. Jiang, Y. P. Chen, D. J. Darensbourg and H. C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105 CrossRef CAS PubMed.
- J. Kim, S. N. Kim, H. G. Jang, G. Seo and W. S. Ahn, Appl. Catal., A, 2013, 453, 175 CrossRef CAS PubMed.
- T. Y. Ma and Z. Y. Yuan, Chem. Commun., 2010, 46, 2325 RSC.
- T. Y. Ma and Z. Y. Yuan, Dalton Trans., 2010, 39, 9570 RSC.
- J. L. Cao, Y. Wang, X. L. Yu, S. R. Wang, S. H. Wu and Z. Y. Yuan, Appl. Catal., B, 2008, 79, 26 CrossRef CAS PubMed.
- H. P. Perry, J. Law, J. Zon and A. Clearfield, Microporous Mesoporous Mater., 2012, 149, 172 CrossRef CAS PubMed.
- X. Y. Liu, A. Q. Wang, X. F. Yang, T. Zhang, C. Y. Mou, D. S. Su and J. Li, Chem. Mater., 2009, 21, 410 CrossRef CAS.
- Y. P. Zhu, T. Y. Ma, T. Z. Ren, J. Li, G. H. Du and Z. Y. Yuan, Appl. Catal., B, 2014, 156–157, 44 CrossRef CAS PubMed.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef CAS.
- J. Burschka, N. Pellet, S. J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316 CrossRef CAS PubMed.
- P. G. Bomben, K. C. D. Robson, B. D. Koivisto and C. P. Berlinguette, Coord. Chem. Rev., 2012, 256, 1438 CrossRef CAS PubMed.
- G. C. Vougioukalakis, A. I. Philippopoulos, T. Stergiopoulos and P. Falaras, Coord. Chem. Rev., 2011, 255, 2602 CrossRef CAS PubMed.
- K. Hanson, M. K. Brennaman, H. Luo, C. R. K. Glasson, J. J. Concepcion, W. Song and T. J. Meyer, ACS Appl. Mater. Interfaces, 2012, 4, 1462 CAS.
- T. P. Brewster, S. J. Konezny, S. W. Sheehan, L. A. Martini, C. A. Schmuttenmaer, V. S. Batista and R. H. Crabtree, Inorg. Chem., 2013, 52, 6752 CrossRef CAS PubMed.
- R. Luschtinetz, J. Frenzel, T. Milek and G. Seifert, J. Phys. Chem. C, 2009, 113, 5730 CAS.
- K. R. Mulhern, A. Orchard, D. F. Watson and M. R. Detty, Langmuir, 2012, 28, 7071 CrossRef CAS PubMed.
- K. Hanson, M. K. Brennaman, A. Ito, H. Luo, W. Song, K. A. Parker, R. Ghosh, M. R. Norris, C. R. K. Glasson, J. J. Concepcion, R. Lopez and T. J. Meyer, J. Phys. Chem. C, 2012, 116, 14837 CAS.
- D. G. Brown, P. A. Schauer, J. Borau-Garcia, B. R. Fancy and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 1692 CrossRef CAS PubMed.
- T. Y. Ma, Y. S. Wei, T. Z. Ren, L. Liu, Q. Guo and Z. Y. Yuan, ACS Appl. Mater. Interfaces, 2010, 2, 3563 CAS.
- S. Rühle, M. Shalom and A. Zaban, ChemPhysChem, 2010, 11, 2290 CrossRef PubMed.
- I. Robel, V. Subramanian, M. Kuno and P. V. Kamat, J. Am. Chem. Soc., 2006, 128, 2385 CrossRef CAS PubMed.
- P. Ardalan, T. P. Brennan, H. Lee, J. R. Bakke, I. K. Ding, M. D. McGehee and S. F. Bent, ACS Nano, 2011, 3, 1495 CrossRef PubMed.
- R. S. Dibbell, D. G. Youker and D. F. Watson, J. Phys. Chem. C, 2009, 113, 18643 CAS.
- R. S. Dibbell and D. F. Watson, J. Phys. Chem. C, 2009, 113, 3139 CAS.
- Q. Li, R. He, J. O. Jensen and N. J. Bjerrum, Chem. Mater., 2003, 15, 4896 CrossRef CAS.
- J. M. Taylor, R. K. Mah, I. L. Moudrakovski, C. I. Ratcliffe, R. Vaidhyanathan and G. K. H. Shimizu, J. Am. Chem. Soc., 2010, 132, 14055 CrossRef CAS PubMed.
- S. Kim, K. W. Dawson, B. S. Gelfand, J. M. Taylor and G. K. H. Shimizu, J. Am. Chem. Soc., 2013, 135, 963 CrossRef CAS PubMed.
- X. Q. Liang, F. Zhang, W. Feng, X. Q. Zou, C. J. Zhao, H. Na, C. Liu, F. X. Sun and G. S. Zhu, Chem. Sci., 2013, 4, 983 RSC.
- F. Costantino, A. Donnadio and M. Casciola, Inorg. Chem., 2012, 51, 6992 CrossRef CAS PubMed.
- T. L. Kinnibrugh, A. A. Ayi, V. I. Bakhmutov, J. Zoń and A. Clearfield, Cryst. Growth Des., 2013, 13, 2973 CAS.
- S. Hudson, J. Cooney and E. Magner, Angew. Chem., Int. Ed., 2008, 47, 8582 CrossRef CAS PubMed.
- X. Shi, J. Liu, C. M. Li and Q. H. Yang, Inorg. Chem., 2007, 46, 7944 CrossRef CAS PubMed.
- X. Shi, J. P. Li, Y. Tang and Q. H. Yang, J. Mater. Chem., 2010, 20, 6495 RSC.
- Y. Tang, Y. B. Ren and X. Shi, Inorg. Chem., 2013, 52, 1388 CrossRef CAS PubMed.
- W. T. Al-Jamal and K. Kostarelos, Acc. Chem. Res., 2011, 44, 1094 CrossRef CAS PubMed.
- Y. Namiki, T. Fuchigami, N. Tada, R. Kawamura, S. Matsunuma, Y. Kitamoto and M. Nakagawa, Acc. Chem. Res., 2011, 44, 1080 CrossRef CAS PubMed.
- C. Wang, D. M. Liu and W. B. Lin, J. Am. Chem. Soc., 2013, 135, 13222 CrossRef CAS PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995 CrossRef CAS PubMed.
- E. Duguet, S. Vasseur, S. Mornet and J.-M. Devoiselle, Nanomedicine, 2006, 1, 157 CrossRef CAS PubMed.
- L. Lartigue, C. Innocenti, T. Kalaivani, A. Awwad, D. M. Sanchez, Y. Guari, J. Larionova, C. Guerin, J. L. G. Montero, V. Barragan-Montero, P. Arosio, A. Lascialfari, D. Gatteschi and C. Sangregorio, J. Am. Chem. Soc., 2011, 133, 10459 CrossRef CAS PubMed.
- H. Dong, J. Huang, R. R. Koepsel, P. Ye, A. J. Russell and K. Matyjaszewski, Biomacromolecules, 2011, 12, 1305 CrossRef CAS PubMed.
- C. Tudisco, V. Oliveri, M. Cantarella, G. Vecchio and G. G. Condorelli, Eur. J. Inorg. Chem., 2012, 32, 5323 CrossRef.
- H. K. Chang, F. N. Ishikawa, R. Zhang, R. Datar, R. J. Cote, M. E. Thompson and C. W. Zhou, ACS Nano, 2011, 5, 9883 CrossRef CAS PubMed.
- K. Wang, Z. Tang, C. J. Yang, Y. Kim, X. Fang, W. Li, Y. Wu, C. D. Medley, Z. Cao, J. Li, P. Colon, H. Lin and W. Tan, Angew. Chem., Int. Ed., 2009, 48, 856 CrossRef CAS PubMed.
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181 CrossRef CAS PubMed.
- H. Li, T. Y. Ma, D. M. Kong and Z. Y. Yuan, Analyst, 2013, 4, 1084 RSC.
- E. Z. Lee, Y. S. Jun, W. H. Hong, A. Thomas and M. M. Jin, Angew. Chem., Int. Ed., 2010, 49, 9706 CrossRef CAS PubMed.
- M. Zhou and S. J. Dong, Acc. Chem. Res., 2011, 44, 1232 CrossRef CAS PubMed.
- T. Y. Ma, H. Li, Q. F. Deng, L. Liu, T. Z. Ren and Z. Y. Yuan, Chem. Mater., 2012, 24, 2253 CrossRef CAS.
- S. Cao, G. Gody, W. Zhao, S. Perrier, X. Y. Peng, C. Ducati, D. Y. Zhao and A. K. Cheetham, Chem. Sci., 2013, 4, 3573 RSC.
- Y. P. Bi, H. Y. Hu, S. X. Ouyang, G. X. Lu, J. Y. Cao and J. H. Ye, Chem. Commun., 2012, 48, 3748 RSC.
- G. Wang, B. B. Huang, X. C. Ma, Z. Y. Wang, X. Y. Qin, X. Y. Zhang, Y. Dai and M. H. Whangbo, Angew. Chem., Int. Ed., 2013, 52, 4810 CrossRef CAS PubMed.
- R. Cai, Y. P. Du, W. Y. Zhang, H. T. Tan, T. Zeng, X. Huang, H. F. Yang, C. P. Chen, H. Liu, J. X. Zhu, S. J. Peng, J. Chen, Y. L. Zhao, H. C. Wu, Y. Z. Huang, R. Xu, T. M. Lim, Q. C. Zhang, H. Zhang and Q. Y. Yan, Chem. – Eur. J., 2013, 19, 1568 CrossRef CAS PubMed.
- C. S. Pan, J. Xu, Y. J. Wang, D. Li and Y. F. Zhu, Adv. Funct. Mater., 2012, 22, 1518 CrossRef CAS.
| This journal is © the Partner Organisations 2014 |