Ultrasmall and monodisperse colloidal amorphous Nd–Fe–B–Na magnetic nanoparticles with high TC†
Q.
Zhang
a,
Z.
Jiang
b and
B.
Yan
*a
aDepartment of Chemistry, Tongji University, Shanghai 200092, PR China. E-mail: byan@tongji.edu.cn; Fax: (+86) 21-65982287
bShanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201204, China
First published on 22nd April 2014
Abstract
Ultrasmall and monodisperse air-stable colloidal Nd–Fe–B–Na nanoparticles (NPs) have been synthesized by the solution phase colloidal method and characterized by TEM, EXAFS, Mössbauer spectroscopy, and SQUID. Their size can be controlled in the sub-5 nm regime. The critical temperature (TC) of Nd–Fe–B–Na nanoparticles with 3 nm diameter is surprisingly high, higher than 650 K. EXAFS gives the chemical environmental information of the Fe center, and Mössbauer spectroscopy confirms that the 3 nm Nd–Fe–B–Na nanoparticles contain the Fe nanocluster. A possible magnetic coupling mechanism is proposed.
Colloidal magnetic nanoparticles (NPs) have attracted intensive research attention in the past few decades from synthesis to various applications, ranging from ultrahigh-density magnetic recording,1 single-electron tunneling magnetic devices,2 magnetic energy storage,3 biomedical applications,4–6 and magnetic separable catalysis7 to magnetically responsive photonic crystals.8 As a potential candidate to realize ultrahigh-density recording at the single particle level, an essential prerequisite for practical one particle one bit ultrahigh-density magnetic recording is to synthesize ultrasmall colloidal magnetic nanoparticles with enough high transition temperature. However, the blocking temperatures (TB) of colloidally synthesized freestanding monodisperse magnetic nanoparticles and surfactant-assisted ball milled Nd2Fe14B nanoparticles with about 5 nm diameter based on long-range ferromagnetic interactions are far below room temperature, such as 20–30 K for 4 nm chemically disordered fcc FePt,1 80 K for 10 nm ε-Co,2b 160 K for 5.2 nm CoFe2O4,4a 25 K for 4 nm γ-Fe2O3,4b 20 K for 1.8 nm CoPt3,4c 40 K for 5 nm Fe3O4,5a and 65 K for 2.7 nm Nd2Fe14B.9a At the same time, amorphous phase RE–TM and RE–TM–B alloys based on short-range ferromagnetic interactions (RE = rare-earth metal, TM = transition metals) with a high magnetic anisotropy constant (Ku usually larger than 107 erg cm−3) have drawn much attention for their commercial applications in a magnetooptic sensor, a high-density magnetooptic disk and high TC.9b,c,10–12 The short-range ordered structure and random magnetic anisotropy (RMA) of the above mentioned amorphous ferromagnetic alloys suggest that the undesirable effects of decreasing size on TB do not behave like those of the crystalline phase. However, controlled synthesis of colloidal magnetic nanoparticles of amorphous phase metal alloys remains a challenge. It is necessary to develop a colloidal method to synthesize amorphous magnetic nanoparticles and investigate their magnetic properties at the nanometer scale.
In this communication, we report the controllable colloidal synthesis and magnetic properties of air-stable monodisperse amorphous Nd–Fe–B–Na nanoparticles, one of them with 3 nm diameter and TC higher than 650 K.
To increase the ratio of magnetic centers, we try to eliminate sodium from the final product by selecting tetrabutylammonium borohydride (TBAB) and triethylamine borane complex. For the former, the reaction system produces a large amount of bubbles when the reducing agent is added in at 100 °C and no product is separable by centrifugation; for the latter, triethylamine borane is sublimated on the cool part of glassware and no product can be obtained. We also tested KBH4 and LiBH4: no reaction has been found for KBH4, but the reaction is very fast for LiBH4 and no product is separable by centrifugation. The hydride, with low melting point (LiBH4, 275 °C) or compatible with the organic phase (TBAB), reacts too fast to form stable nuclei before the dissociation of Nd(acac)3 and Fe(acac)3. The triethylamine borane complex is stable at the reflux temperature of the solvent and surfactants for its three B–C covalent chemical bonds. This could be the reason why superhydride (LiBEt3H) has been selected to synthesize colloidal ε-Co nanoparticles: to form a homogeneous solution in the organic phase (dioctylether) and to avoid possible incorporation of B atoms into ε-Co. In the series of LiBH4, NaBH4 (m.p., 400 °C), and KBH4 (m.p, 500 °C), only NaBH4 can react steadily with the precursors and surfactants (OA and OM) in organic solution near 300 °C to form stable nuclei and its solid state ensures the following growth process. Thus, the suitable chemical reactivity of NaBH4 and its solid state at the selected aging temperature realize a reasonable separation between nucleation and growth. When the temperature increases to 275 °C, there are small bubbles in the reaction, which are the bubbling effects taking place through absorbing local latent heat released from the exothermic reactions involved in the nucleation and growth of magnetic nanoparticles. The bubbling effects facilitate the formation of a huge number of nuclei.13 The reactions in the system are rather complex. At least, the amidation reaction between OA and OM, the dissociation of Nd(acac)3 and Fe(acac)3 (facilitated by surfactants, such as OA and OM, and/or by hydrides), and the reduction of Fe(III) by borohydride are involved.14 Because of the very high positive redox potential of the metal Nd, NaBH4 cannot reduce Nd(III) to Nd, supported by the XPS peak of Nd 3d5/2 at 982 eV, being the same value as that of Nd2O3 (see ESI, Fig. S3†). Even if this reduction reaction occurs, the water molecules from the above-mentioned amidation reaction and OA can oxidize it to Nd(III). Considering the intrinsic oxophilic properties of Nd(III) and the presence of water and other oxygen-containing molecular species, the Nd–Fe–B–Na nanoparticles could contain some oxygen. These factors can contribute to the chemical stability to air, evaluated by the magnetic properties in the following part. These chemical reactions are different from the sputter deposition and melt spinning techniques on chemical composition control. We speculate that the reasons for formation of amorphous colloidal nanoparticles instead of crystalline ones are as follows. Firstly, the chemical potentials of the monomers (reactive atomic or molecular species) are usually high, and according to Ostwald's rule, the kinetically favoured metastable amorphous phase forms first.15 Secondly, the presence of Nd(III), sodium, some oxygen, and high content of boron could suppress the formation of the crystalline phase. Thirdly, the temperature allowing us to synthesize ultrasmall and monodisperse nanoparticles (300–320 °C) is not high enough to rearrange and anneal the constituent atoms during growth. This rearrangement is usually associated with significantly high thermal barriers.16 It is worth noting that it is a likely trend that ultrasmall nanoparticles adopt disordered structure compared to their bulk crystalline phases, such as chemically disordered face-centered cubic (fcc) for FePt colloidal nanocrystals,1 (γ-Fe2O3)0.8(Fe3O4)0.2 for Fe3O4 nanocrystals with a diameter of 5 nm,10 and non-periodic and lacking long-range atomic ordered structure for the Co and FeCo nanoparticles.17
The magnetic properties of Nd–Fe–B–Na colloidal nanoparticles with size being 1 nm, 2 nm, and 3 nm corresponding to Fig. 1 are measured with a SQUID–VSM system (MPMS, Quantum) in the temperature range lower than 400 K. The M–H magnetic hysteresis curves are depicted in Fig. 2. The linear paramagnetic characterization is for the colloidal nanoparticle with 1 nm diameter. When the diameter increases to 2 nm, the ferromagnetic magnetic hysteresis loop is observed, with the coercive field (Hc) being 120 Oe at 300 K (Fig. 2a). The coercive fields (Hc) of nanoparticles with 3 nm diameter are 262 Oe at 300 K and 245 Oe at 400 K (Fig. 2b). These Hc values are higher than that of bulk amorphous alloy measured at 300 K, about 10 Oe for Nd8Fe72B20.12d The saturation magnetization (Ms) at 6 T is 14 emu g−1, comparable with that of Fe3O4 nanoparticles with the same size,5c which can exclude the possibility of the ferromagnetic properties originating from defect magnetism. In Fig. 2, the time for the M–H curve measured at 400 K is twenty days later than the M–H curve measured at 300 K. From the M–H curve at 300 K in Fig. 2b inset, we can see that the magnetic nanoparticles with 3 nm diameter cannot be spontaneously magnetized or induced by the magnetic bar during colloidal synthesis. However, after the magnetic measurement at 300 K, the nanoparticle maintains its magnetic remanence at least for twenty days, as shown in Fig. 2b inset for the M–H curve at 400 K. The stability in air of the Nd–Fe–B–Na colloidal nanoparticles is interrogated by the change of the magnetic properties after being exposed to air for two months. The coercive field (Hc) and magnetic remanence (Mr) decrease only by 0.5 percent, while the saturation magnetization (Ms) at 6 T decreases by 2.5 percent (see ESI, Fig. S4†). These indicate that the 3 nm nanoparticles are stable in air. The magnetic properties decreasing after exposing to air for several days is due to the oxidation of the trace surface unstable Fe (e.g., Fe(I) and Fe(II)) or Fe(0) nanocluster.
 | ||
| Fig. 1 TEM characterizations of colloidal Nd–Fe–B–Na nanoparticles with different sizes. (a) 1 nm; (b) 2 nm; (c) 3 nm; (d) 5 nm. | ||
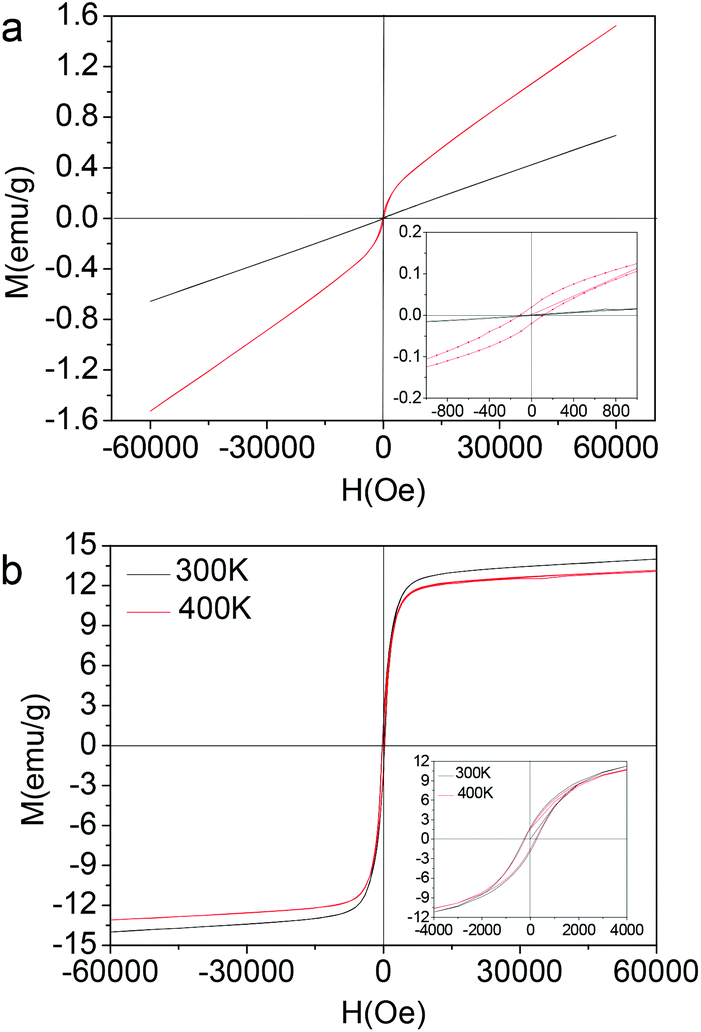 | ||
| Fig. 2 Magnetic properties of colloidal Nd–Fe–B–Na nanoparticles with different sizes. (a) Magnetic hysteresis of nanoparticles with 1 nm diameter (black curve) and 2 nm (red curve). (b) Magnetic hysteresis of nanoparticles with 3 nm diameter at 300 K (black curve) and 400 K (red curve). | ||
To shed more light on the structure, the magnetic properties of the 3 nm nanoparticles under temperatures higher than 400 K are determined using a LakeShore 7407 vibrating sample magnetometer (VSM) equipped with a Hall probe. The M–H magnetic hysteresis curves and the magnetic parameters are given in Fig. 3. As the temperature of the sample increases to 650 K, the saturation magnetization and magnetic remanence decrease. However, the coercive field (Hc) decreases first and then increases a little, from 308 Oe to 270 Oe and then to 293 Oe (see ESI, Fig. S5†). We do not measure the magnetic properties at temperatures higher than 650 K for the following considerations: higher temperature would decompose the organic surfactants (OA and OM) and result in the aggregation by annealing and sintering. The thermal analysis of the colloidal magnetic nanoparticles under a nitrogen atmosphere is conducted (see ESI, Fig. S6†). There are an endothermic peak of the DSC curve and a weight loss peak of the TGA curve near 723 K, and these peaks can be tentatively assigned to the corresponding process of decomposition of the organic surfactants of OA and OM and the fusion and aggregation of the nanoparticles. The magnetic properties at temperatures higher than 650 K would arise from the fusion and aggregation of the ‘naked’ nanoparticles, not at the single nanoparticle level. When the size increases to 5 nm (Fig. 1d), the magnetic nanoparticles can be spontaneously magnetized or induced by the magnetic bar during colloidal synthesis, shown in Fig. S7.† The coercive field (Hc) and magnetic remanence (Mr) are 612 Oe and 11.2 emu g−1 measured by a LakeShore 7407 VSM at 300 K, while the saturation magnetization (Ms) at 1.8 T is 29.6 emu g−1.
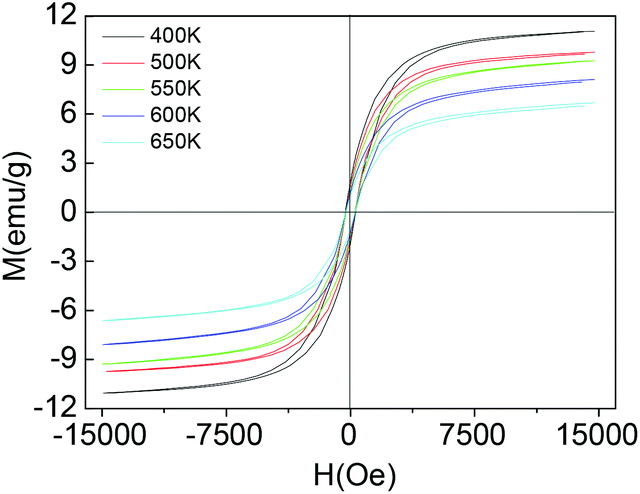 | ||
| Fig. 3 Magnetic properties of colloidal Nd–Fe–B–Na nanoparticles with 3 nm diameter at high temperatures between 400 K and 650 K. | ||
To study the magnetic properties of the 3 nm Nd–Fe–B–Na nanoparticles, we performed extended X-ray absorption fine structure (EXAFS) and Mössbauer spectroscopy characterization. The radical distribution functions (RDF) of the 3 nm Nd–Fe–B–Na nanoparticle and Fe foil, obtained from their k3χ(k) by Fourier transform in Fig. S8,† are depicted in Fig. 4. There are several distinct peaks of the sample. Compared with the peaks of Fe foil, the peaks at 2.58 Å, 3.9 Å, and 4.6 Å of the sample may partially result from the Fe0 nanocluster. The peak at 1.86 Å can be assigned to the amorphous phase. Structural parameters obtained by fitting the Fe K-edge data shown in Fig. 4 are listed in Table S1.† There are about 3.5 boron atoms and 1.2 Fe atoms around each Fe center. The nearest Fe–B and Fe–Fe distances are 1.99 Å and 2.91 Å. Although Nd2Fe14B nanomagnets and nanocomposites as crystlline sample are characterized by EXAFS, the data on the fitted results for chemical bonds of Fe–B and Fe–Fe are still lacking. It is difficult to directly compare the crystalline phase with the amorphous phase. The distance of Fe–B is shorter while the distance of Fe–Fe is larger than those of the amorphous Fe–Si–B alloy, 2.27 Å and 2.54 Å, respectively.11b The first peak in the RDF of the 3 nm nanoparticle is in a slightly higher position than those of standard Fe2O3 and Fe3O4 in Fig. S9,† indicating the Fe–B bonds being slightly larger than the Fe–O chemical bonds.
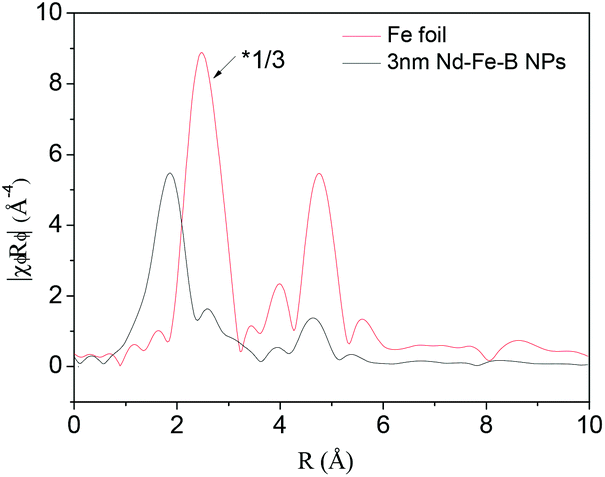 | ||
| Fig. 4 RDFs of the 3 nm Nd–Fe–B–Na nanoparticles and the standard Fe foil. | ||
57Fe Mössbauer spectroscopy of the sample measured at 300 K in Fig. 5 and Table S2† confirms that there is a sextet subspectrum of subarea of 24% (subspectrum 2) with the same isomer shift (δ) as that of Fe foil and a hyperfine field of 33.3 kOe attributed to unoxidized Fe0 nanoparticles in a blocked state, assigned to Fe0 nanocluster(s).18 To exclude the possibility of the sextet subspectrum resulting from very large Fe nanoparticles not observed by TEM, we characterize the dynamic light scattering (DLS) of the 3 nm Nd–Fe–B–Na nanoparticles dispersed in hexane; there is no sign of particles with hydrodynamic diameter larger than 7 nm (Fig. S10†). For freestanding colloidal Fe nanoparticles, the blocking temperature of unoxidized 4.5 nm Fe nanoparticles is just 110 K.19 We speculate that the sextet subspectrum of the Fe0 nanocluster(s) could originate from their exchange-coupling with the amorphous parts surrounding them inside the 3 nm Nd–Fe–B–Na nanoparticles. This structural feature is in accordance with the RDF of the nanoparticles in Fig. 4. Although the temperature to measure the Mössbauer effect is far below TC, there is still a doublet peak with 40% subarea. This phenomenon is different from the magnetic behavior of CoFe2O4 nanocrystals and Fe nanocube assembly with only one sextet peak under the blocking temperature, probably due to structural disorder and surface effect.4a,18 Because of the surface effect and complex chemical composition, the Mössbauer spectra are also different from those of bulk amorphous Fe–B–Si, the bulk without any doublet peak.11b However, there is no sign of Fe0 nanoclusters in the 5 nm nanoparticles shown in Fig. S11 and Table S3,† probably due to higher synthetic temperature being able to destroy the Fe nanocluster by forming Fe–B chemical bonds. As the size increases to 5 nm, the subarea of the doublet peak from Fe nuclei with unblocked state decreases slightly to 37%. For the nanoparticles with 1 nm or 2 nm diameter, there are only two doublet peaks, although the Hc is 120 Oe for the 2 nm one (Fig. S11 and Table S3†).
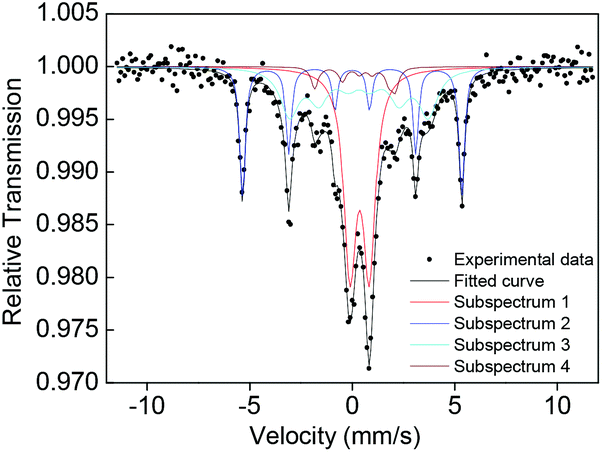 | ||
| Fig. 5 57Fe Mössbauer spectrum of the 3 nm Nd–Fe–B–Na nanoparticles measured at 300 K. | ||
For the present colloidal Nd–Fe–B–Na nanoparticles, when the size decreases to 1 nm, the zero-field-cooling (ZFC) magnetization (Fig. S12†) under 1000 Oe magnetic field indicates that there is no peak magnetization between 2 K and 305 K, unlike those of ε-Co, CoFe2O4, and Fe3O4 magnetic nanoparticles.1,4a,5a These results probably arise from finite size effects suppressing the magnetic ordering. Considering the ferromagnetic properties of magnetic nanoparticles larger than 2 nm (Fig. 2a, red curve), the size is a crucial factor to determine the magnetic exchange model. The critical size of the present colloidal Nd–Fe–B–Na nanoparticles from the experimental results should be between 1 nm and 2 nm, which is close to the estimated grain size of Gd–Co amorphous films (smaller than 25 Å) and the diameter of Fe-enriched clusters simulated in the inhomogeneous Fe67Y33 alloys (about 15–20 Å).10a,11a
Conclusions
In summary, the controlled colloidal synthesis of amorphous Nd–Fe–B–Na nanoparticles and the size dependence of the magnetic properties have been demonstrated in the sub-5 nm regime. The magnetic nanoparticles with 3 nm diameter exhibit ferromagnetic magnetic properties even at temperatures as high as 650 K. The structure of the 3 nm Nd–Fe–B–Na nanoparticles has been characterized by EXAFS and Mössbauer spectroscopy. These characteristics clearly demonstrate that such Nd–Fe–B–Na nanoparticles offer opportunities for fundamental studies of nanomagnetics, as well as for potential ultrahigh-density magnetic recording, permanent magnetic nanocomposites, catalysis, and biological applications. The present colloidal method can also be used to grow colloidal nanoparticles of other amorphous metal alloys with nanometer size, with potential importance for magnetic, magnetooptic, and electronic applications.Acknowledgements
This work was supported by the National Natural Science Foundation of China (91122003) and the Developing Science Funds of Tongji University.Notes and references
- S. H. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989 CrossRef CAS.
- (a) S. H. Sun and C. B. Murray, J. Appl. Phys., 1999, 85, 4325 CrossRef CAS PubMed; (b) C. T. Black, C. B. Murray, R. L. Sandstrom and S. H. Sun, Science, 2000, 290, 1131 CrossRef CAS; (c) V. F. Puntes, K. M. Krishnan and A. P. Alivisatos, Science, 2001, 291, 2115 CrossRef CAS PubMed.
- H. Zeng, J. Li, J. P. Liu, Z. L. Wang and S. H. Sun, Nature, 2002, 420, 395 CrossRef CAS PubMed.
- (a) C. Liu, B. S. Zou, A. J. Rondinone and J. Zhang, J. Am. Chem. Soc., 2000, 122, 6263 CrossRef CAS; (b) T. Hyeon, S. S. Lee, J. Park, Y. Chung and H. Bin Na, J. Am. Chem. Soc., 2001, 123, 12798 CrossRef CAS PubMed; (c) J. I. Park and J. Cheon, J. Am. Chem. Soc., 2001, 123, 5743 CrossRef CAS PubMed; (d) S. H. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204 CrossRef CAS PubMed.
- (a) J. Park, K. J. An, Y. S. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891 CrossRef CAS PubMed; (b) J. Park, E. Lee, N. M. Hwang, M. S. Kang, S. C. Kim, Y. Hwang, J. G. Park, H. J. Noh, J. Y. Kini, J. H. Park and T. Hyeon, Angew. Chem., Int. Ed., 2005, 44, 2872 CrossRef CAS PubMed; (c) B. H. Kim, N. Lee, K. An, Y. I. Park, Y. Choi, K. Shin, Y. Lee, S. G. Kwon, H. B. Na, J. G. Park, T. Y. Ahn, Y. W. Kim, W. K. Moon, S. H. Choi and T. Hyeon, J. Am. Chem. Soc., 2011, 133, 12624 CrossRef CAS PubMed; (d) J. Dobson, Nat. Nanotechnol., 2008, 3, 139 CrossRef CAS PubMed.
- (a) C. Tassa, S. Y. Shaw and R. Weissleder, Acc. Chem. Res., 2011, 44, 842 CrossRef CAS PubMed; (b) D. Yoo, J. H. Lee, T. H. Shin and J. Cheon, Acc. Chem. Res., 2011, 44, 863 CrossRef CAS PubMed; (c) D. Ho, X. L. Sun and S. H. Sun, Acc. Chem. Res., 2011, 44, 875 CrossRef CAS PubMed; (d) J. H. Lee, J. T. Jang, J. S. Choi, S. H. Moon, S. H. Noh, J. W. Kim, J. G. Kim, I. S. Kim, K. I. Park and J. Cheon, Nat. Nanotechnol., 2011, 6, 418 CrossRef CAS PubMed.
- (a) A. H. Lu, W. Schmidt, N. Matoussevitch, H. Bonnemann, B. Spliethoff, B. Tesche, E. Bill, W. Kiefer and F. Schuth, Angew. Chem., Int. Ed., 2004, 43, 4303 CrossRef CAS PubMed; (b) M. Shokouhimehr, Y. Z. Piao, J. Kim, Y. J. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 7039 CrossRef CAS PubMed; (c) V. Mazumder, M. F. Chi, K. L. More and S. H. Sun, J. Am. Chem. Soc., 2010, 132, 7848 CrossRef CAS PubMed; (d) Y. M. Lee, M. A. Garcia, N. A. F. Huls and S. H. Sun, Angew. Chem., Int. Ed., 2010, 49, 1271 CrossRef CAS PubMed; (e) S. J. Guo, S. Zhang, X. L. Sun and S. H. Sun, J. Am. Chem. Soc., 2011, 133, 15354 CrossRef CAS PubMed.
- (a) J. P. Ge, Y. X. Hu and Y. D. Yin, Angew. Chem., Int. Ed., 2007, 46, 7428 CrossRef CAS PubMed; (b) J. P. Ge, L. He, J. Goebl and Y. D. Yin, J. Am. Chem. Soc., 2009, 131, 3484 CrossRef CAS PubMed; (c) J. P. Ge and Y. D. Yin, Angew. Chem., Int. Ed., 2011, 50, 1492 CrossRef CAS PubMed; (d) Y. X. Hu, L. He and Y. D. Yin, Angew. Chem., Int. Ed., 2011, 50, 3747 CrossRef CAS PubMed.
- (a) N. G. Akdogan, W. F. Li and G. C. Hadjipanayis, J. Appl. Phys., 2011, 109, 07A759 CrossRef PubMed; (b) C. W. Kim, Y. H. Kim, U. Pal and Y. S. Kang, J. Mater. Chem. C, 2013, 1, 275 RSC; (c) A. Hussain, A. P. Jadhav, Y. K. Baek, H. J. Choi, J. Lee and Y. S. Kang, J. Nanosci. Nanotechnol., 2013, 13, 7717 CrossRef CAS PubMed.
- (a) P. Chaudhari, J. J. Cuomo and R. J. Gambino, Appl. Phys. Lett., 1973, 22, 337 CrossRef CAS PubMed; (b) Y. Togami, Appl. Phys. Lett., 1978, 32, 673 CrossRef CAS PubMed; (c) R. Harris, M. Plischke and M. J. Zuckermann, Phys. Rev. Lett., 1973, 31, 160 CrossRef CAS.
- (a) D. Spisak, J. Hafner and R. Lorenz, J. Magn. Magn. Mater., 1997, 166, 303 CrossRef CAS; (b) Y. Takahara and N. Narita, Mater. Sci. Eng., A, 2001, 315, 153 CrossRef; (c) N. Hassanain, H. Lassri, R. Krishnan and A. Berrada, J. Magn. Magn. Mater., 1995, 146, 37 CrossRef CAS; (d) N. Hassanain, H. Lassri, R. Krishnan and A. Berrada, J. Magn. Magn. Mater., 1995, 146, 315 CrossRef CAS.
- (a) M. Slimani, M. Hamedoun, H. Lassri, S. Sayouri and R. Krishnan, J. Magn. Magn. Mater., 1996, 153, 132 CrossRef CAS; (b) M. Slimani, M. Hamdoun, M. Tlemcani, H. Arhchoui and S. Sayouri, Physica B, 1997, 240, 372 CrossRef CAS; (c) E. H. Sayouty, F. Annouar, H. Lassri, N. Randrianantoandro and J. M. Greneche, J. Alloys Compd., 2005, 397, 47 CrossRef CAS PubMed; (d) N. Hassanain, H. Lassri, A. Berrada and R. Krishnan, Phys. Status Solidi C, 2006, 3, 3239 CrossRef CAS.
- J. Lynch, J. Q. Zhuang, T. Wang, D. LaMontagne, H. M. Wu and Y. C. Cao, J. Am. Chem. Soc., 2011, 133, 12664 CrossRef CAS PubMed.
- (a) N. R. Jana, Y. F. Chen and X. G. Peng, Chem. Mater., 2004, 16, 3931 CrossRef CAS; (b) J. Joo, S. G. Kwon, J. H. Yu and T. Hyeon, Adv. Mater., 2005, 17, 1873 CrossRef CAS; (c) H. M. Wu, Y. G. Yang and Y. C. Cao, J. Am. Chem. Soc., 2006, 128, 16522 CrossRef CAS PubMed; (d) H. T. Zhang, D. H. Ha, R. Hovden, L. F. Kourkoutis and R. D. Robinson, Nano Lett., 2011, 11, 188 CrossRef CAS PubMed; (e) Q. Zhang, B. Yan, F. Lei and H. H. Chen, Nanoscale, 2012, 4, 7646 RSC.
- W. Z. Ostwald, Phys. Chem., 1897, 22, 289 CAS.
- L. E. M. Howard, H. L. Nguyen, S. R. Giblin, B. K. Tanner, I. Terry, A. K. Hughes and J. S. O. Evans, J. Am. Chem. Soc., 2005, 127, 10140 CrossRef CAS PubMed.
- (a) D. P. Dinega and M. G. Bawendi, Angew. Chem., Int. Ed., 1999, 38, 1788 CrossRef CAS; (b) B. Chaudret, C. Desvaux, C. Amiens, P. Fejes, P. Renaud, M. Respaud, P. Lecante and E. Snoeck, Nat. Mater., 2005, 4, 750 CrossRef PubMed.
- F. Dumestre, B. Chaudret, C. Amiens, P. Renaud and P. Fejes, Science, 2004, 303, 821 CrossRef CAS PubMed.
- T. C. Monson, E. L. Venturini, V. Petkov, Y. Ren, J. M. Lavin and D. L. Huber, J. Magn. Magn. Mater., 2013, 331, 156 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qi00049h |
| This journal is © the Partner Organisations 2014 |