Self-assembling peptide scaffolds for regenerative medicine
John B.
Matson
a and
Samuel I.
Stupp
*abcd
aInstitute for BioNanotechnology in Medicine, Northwestern University, Chicago, IL 60611, USA. E-mail: s-stupp@northwestern.edu; Fax: +312 503-2482; Tel: +312 503-6713
bDepartment of Chemistry, Northwestern University, Evanston, IL 60208, USA
cDepartment of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, USA
dFeinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA
First published on 14th November 2011
Abstract
Biomaterials made from self-assembling, short peptides and peptide derivatives have great potential to generate powerful new therapies in regenerative medicine. The high signaling capacity and therapeutic efficacy of peptidic scaffolds has been established in several animal models, and the development of more complex, hierarchical structures based on peptide materials is underway. This highlight discusses several classes of self-assembling peptide-based materials, including peptide amphiphiles, Fmoc-peptides, self-complementary ionic peptides, hairpin peptides, and others. The self-assembly designs, bioactive signalling strategies, and cell signalling capabilities of these bioactive materials are reported. The future challenges of the field are also discussed, including short-term goals such as integration with biopolymers and traditional implants, and long term goals, such as immune system programming, subcellular targeting, and the development of highly integrated scaffold systems.
 John B. Matson | John Matson received his AB degree in Chemistry at Washington University in St. Louis in 2004. He completed his PhD in Chemistry in 2009 at the California Institute of Technology under the guidance of Robert H. Grubbs. John is currently a postdoctoral scholar in the group of Sam Stupp at Northwestern University. His research interests include hydrogel drug delivery, dynamic self-assembling peptides, and the development of new materials for regenerative medicine. |
 Samuel I. Stupp | Samuel I. Stupp received his BS in Chemistry in 1972 from the University of California at Los Angeles and his PhD in Materials Science in 1977 from Northwestern University. He remained at Northwestern as a faculty member until 1980 before moving to the University of Illinois at Urbana-Champaign. In 1999 he returned to Northwestern University as a Board of Trustees Professor of Chemistry, Materials Science, and Medicine. He is also the Director of the Institute for BioNanotechnology in Medicine at Northwestern. His research is focused on self-assembly, supramolecular chemistry, and the development of functional systems of interest in medicine and solar energy. |
Biocompatible and bioactive small molecules capable of self-assembly and degradation over time into predictable metabolites are ideal building blocks for scaffolds to regenerate tissues and organs. Of the major biological building blocks—sugars, amino acids and nucleic acids—amino acids offer the widest variety of functionality and cell signalling capacity with rapid and facile synthesis of complex molecules. Using peptide secondary structure motifs provided by proteins, small peptides can be rationally designed to self-assemble into a variety of supramolecular nanostructures, such as spheres, cylinders, tubes, and many other morphologies.1–4
Research on implantable materials for tissue regeneration has been primarily focused on biodegradable polymers and more recently synthetic proteins.5 Currently various types of hydrogels are of great interest in this field.6 While simple to produce, chemically crosslinked hydrogels have several shortcomings, such as limited biodegradation, potentially toxic monomers and crosslinking agents, and shrinkage of the hydrogels after crosslinking. The alternative approach that has emerged in the last decade has been to focus on supramolecular nanostructures using fully degradable small molecules.7 These systems can form implantable gels or can be injected as supramolecular nanostructures into diseased or damaged tissue to promote regeneration. Another approach with these systems is to create shear-thinning gels that can be effectively injected as liquids and gel again in vivo.8 Injectable systems facilitate simple procedures for the minimally invasive syringe delivery of molecular precursors that can assemble at the desired tissue site, making such materials ideal for cell encapsulation and drug or protein delivery. Furthermore, with suitable chemical design, materials constructed through supramolecular interactions offer the potential for complete biodegradation. It should also be noted that problems commonly encountered with peptidic drugs (e.g., rapid bloodstream clearance) are not seen with peptide-based injectable materials due to their supramolecular structure and direct tissue delivery. The field of self-assembled peptide scaffolds is growing rapidly with increasing potential to provide unique therapeutic strategies for disease and regeneration.9 Here we describe some of the most important contributions to the field of peptide-based scaffolds for regenerative medicine over the past decade, with a discussion of upcoming goals and challenges in the future of these versatile materials.
Classes of filamentous supramolecular scaffolds
Scaffolds for regenerative medicine have been prepared from a number of different peptide self-assembly motifs. Our group has extensively studied peptide amphiphiles (PAs), which are short peptide sequences attached to a hydrophobic tail (Fig. 1A).2,10–12 A key component of the PAs developed in our laboratory is a β-sheet forming region of 4–8 amino acids directly adjacent to a palmitic acid tail. This component induces cylindrical nanofiber formation, which depends heavily on hydrogen-bonding in the first few residues neighbouring the hydrophobic tail.13,14 The β-sheet region also has a large effect on the ultimate mechanical properties of PA gels, with less twisted β-sheet structures generating stiffer materials.15 The PA structure is completed by the addition of one or more charged residues to aid in solubility and gelation.7,16Self-assembly of PA molecules into cylindrical nanofibers occurs through hydrophobic collapse in concert with the formation of a hydrogen-bonding network down the long axis of the nanofiber. This process of self-assembly has been studied by simulations using a mixed Monte Carlo-stochastic dynamics method.17Gelation of the nanofibers into networks can be triggered by charge screening through the addition of electrolytes or a change in pH.18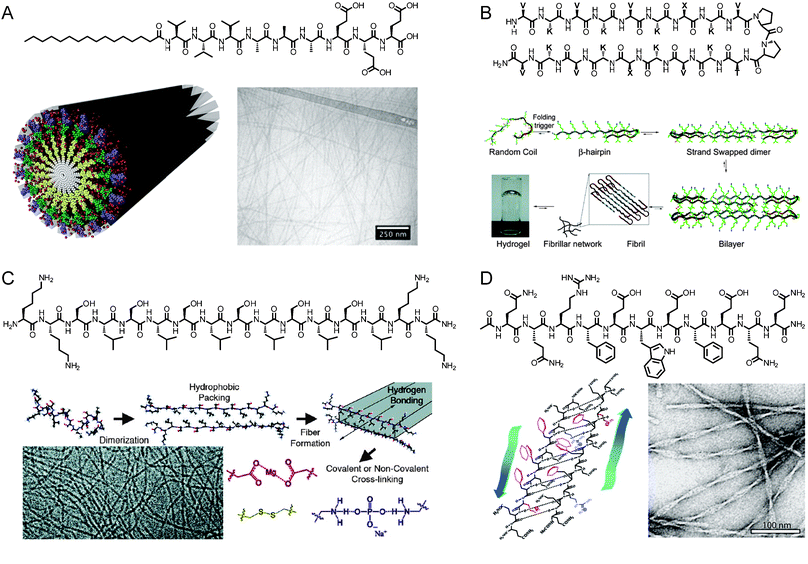 | ||
| Fig. 1 (A) Chemical structure of a peptide amphiphile, schematic representation of PA self-assembly into cylindrical nanofibers, and cryogenic transmission electron microscopy (cryoTEM) of assembled aggregates. Reprinted with permission from ref. 15 (Copyright 2010) and ref. 9 (Copyright 2011) (B) Chemical structure of a β-hairpin peptide and mechanism of folding and gelation. Reprinted with permission from ref. 21 (Copyright 2009 American Chemical Society). (C) Chemical structure of a multidomain peptide, mechanism of folding and gelation, and cryoTEM image of assembled peptides. Reprinted with permission from ref. 22 (Copyright 2009 American Chemical Society). (D) Chemical structure of self-assembling peptide P11-II, its β-sheet-driven assembly into tape-like aggregates, and negatively stained TEM image of the nanostructures. Reprinted with permission from ref. 23 (Copyright 2003 American Chemical Society). | ||
Several other groups have developed peptide-based scaffolds that rely on self-assembly of short peptides into one-dimensional nanostructures. For example, Zhang has developed a class of self-complementary ionic peptides inspired by the repeating AX sequence of the Z-DNA binding protein zuotin, where X is a charged amino acid.19 These materials have hydrophilic and hydrophobic faces that stack to form long, entangled nanofibers. Schneider and Pochan have worked extensively with β-hairpin peptides, which include a Pro-DPro hairpin to induce folding and gelation (Fig. 1B).8,20,21 Other peptide gelators based on a variety of supramolecular interactions have also been developed by Hartgerink (Fig. 1C),22,24 Aggeli (Fig. 1D),23,25,26 Ulijn,27,28 Xu,29 and others.30–34
Early studies on self-assembling peptide-based nanostructures showed that this new class of compounds was not only structurally interesting, but might also generate scaffolds for templating the formation of chemical reactions. In work from our group, the hydrophobic core of PA nanofibers was used as a template to facilitate the topochemical polymerization of a set of diacetylene-containing PAs (Fig. 2).35 The highly-ordered hydrophobic core of the PA nanofibers oriented the diacetylene units to form a conjugated polydiacetylene backbone, covalently bonding the entire PA nanofiber and generating the characteristic blue color of polydiacetylenes. The increased mechanical robustness of the material after polymerization permitted micropatterning of the PA gel surface.36Diacetylene crosslinking was also used by Tirrell to generate RGDS-functionalized PA surfaces that could be reused multiple times as robust, cell-adherent surfaces.37
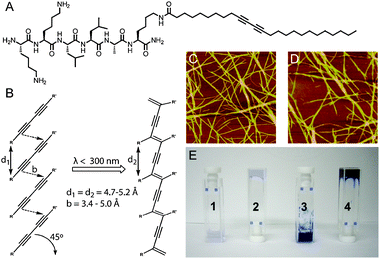 | ||
| Fig. 2 (A) Chemical structure of diacetylene-containing PA. (B) Photoinitiated topochemical polymerization of diacetylene units. (C and D) AFM image of unirradiated (C) and irradiated (D) PA nanofibers. (E) Digital images of PA samples showing (1) PA solution without irradiation, (2) gelled PA solution without irradiation, (3) PA solution after irradiation, and (4) gelled PA solution after irradiation. Reprinted with permission from ref. 35 (Copyright 2008 American Chemical Society). | ||
The surface of PA nanofibers was also used to template the formation of aligned structures, most notably hydroxyapatite (HAP), the inorganic component of bone derived from calcium and phosphate.7 Using a PA that contained a phosphoserine residue, the crystallographic c-axis of the mineralized HAP was found to be aligned with the long axis of the PA nanofiber. This was a significant finding, indicating that one-dimensional, peptide-based materials could template the growth of the mineral component of bone in a way that emulates biomineralization of collagen. We later found that three-dimensional PA nanofiber gels can also nucleate HAP formation using alkaline phosphatase and an organic phosphate source to regulate phosphate concentration.38X-ray diffraction (XRD) experiments indicated that the PA nanofibers played an essential role in templating HAP mineralization. In recent in vivo experiments on bone growth, we tested a mixture of a phosphoserine-containing PA and a PA containing the integrin binding sequence RGDS in a critical sized bone defect rat model.39 The PA mixture itself, with no additional growth factors or cells, was found to facilitate bone growth as effectively as demineralized bone matrix, the current clinical standard.
Epitope-presenting scaffolds
The extracellular matrix (ECM) is a complex mixture of proteins and polysaccharides that provides structural support and domains for cell adhesion, among many other roles. Major components of the ECM include fibronectin, laminin, collagen, chondroitin sulfate, and others. Fibrous materials that display common cell-binding epitopes, including RGDS, YIGSR, and IKVAV, are currently under intense study as ECM mimetics due to their dual roles as structural and adhesive frameworks.40Our group has explored the use of cell-binding epitopes including RGDS and IKVAV as bioactive sequences on PAs for a variety of regenerative applications.41–47 Two or more PAs can be coassembled to generate nanofibers with multiple epitopes or to optimize epitope spacing by addition of a non-bioactive diluent PA.48 Atomistic molecular dynamics simulations show that even hydrophobic sequences such as IKVAV are located on the surface of PA nanofibers, making epitopes available for interaction with cell surface receptors.49
The fibronectin-derived RGDS (or RGD) sequence is the most commonly-employed integrin-biding motif in biomaterials. We studied the adhesion of bone marrow derived stem cells onto RGDS PA-containing surfaces and found the optimal matrix for these cells to contain a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 RGDS PA to diluent PA.43 Encapsulation of the stem cells into the coassembled PA gel and subcutaneous injection into mice showed that RGDS-containing gels supported viability of the transplanted cells significantly better than PA gels without the epitope. Other studies from our group showed that multiple RGDS epitopes can be placed within a single PA molecule using orthogonal protecting group chemistry to generate branches in the PA sequence.41 The branches were found to increase rotational freedom and spacing of the epitopes in fluorescence anisotropy experiments, potentially leading to increased availability for binding.
:9 RGDS PA to diluent PA.43 Encapsulation of the stem cells into the coassembled PA gel and subcutaneous injection into mice showed that RGDS-containing gels supported viability of the transplanted cells significantly better than PA gels without the epitope. Other studies from our group showed that multiple RGDS epitopes can be placed within a single PA molecule using orthogonal protecting group chemistry to generate branches in the PA sequence.41 The branches were found to increase rotational freedom and spacing of the epitopes in fluorescence anisotropy experiments, potentially leading to increased availability for binding.
Ulijn recently engineered a self-assembling system for generating cell-adhesive nanofibers (Fig. 3).50 Aqueous solutions of two short Fmoc-terminated peptides, Fmoc-RGD and Fmoc-FF, were mixed to yield a self-supporting gel upon addition of cell media. Encapsulated fibroblasts within gels of a 2:1 to 3:1 ratio of Fmoc-FF to Fmoc-RGD were well spread after 48 h, suggesting integrin binding to the scaffold. Scaffolds made with an Fmoc-RGE peptide did not show significant spreading. Gazit examined cell viability on similar self-assembled Fmoc-oligopeptides, including gels that contained non-natural amino acids.51 These simple Fmoc di- and tri-peptides are especially intriguing due to their minimal structural components and ease of synthesis.
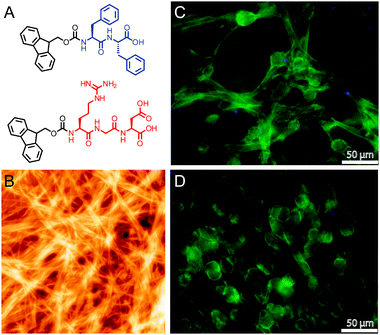 | ||
| Fig. 3 (A) Chemical structure of Fmoc-FF and Fmoc-RGD peptides. (B) AFM height image of self-assembled Fmoc-peptides. (C and D) Fluorescence image of human adult dermal fibroblasts in Fmoc-FF/RGD gel (C) and Fmoc-FF/RGE gel (D) with nuclear staining (DAPI, blue) and actin staining (phalloidin, green). Reprinted from ref. 50 with permission from Elsevier. | ||
Collier recently published a study on RGD-bearing β-sheet fibrillizing hydrogels where the RGD ligand could be non-uniformly distributed throughout the scaffold. Dramatic changes in the mechanical properties of the material and in cell growth within the gel were observed compared with uniformly-distributed ligands. The added degree of complexity and control of gel properties without additional materials likely foreshadows the future of the field.52
The power of synthetic scaffolds that present epitopes can be extended beyond cell encapsulation to stem cell differentiation and tissue regeneration. Controlled differentiation of stem cells remains an important problem in regenerative medicine, and differentiation within three-dimensional matrices presents a new route to potential therapies. Our group has recently reported new methodologies to differentiate stem cells for hard tissue replacement using micropatterned PA-coated surfaces containing the RGDS PA.36 Surfaces bearing shallow holes either 20 or 40 μm in diameter were found to increase expression of the late osteogenic marker osteopontin compared with a smooth surface. The patterned surfaces are thought to enclose individual cells, inducing differentiation by simulating a high cell density environment. Increased expression of osteogenic markers has also been observed in self-complementary ionic peptide scaffolds.53 In this report, gels bearing a cell-adhesion sequence derived from osteopontin showed increased expression of osteogenic genes compared to controls, similar to effects noted for the RGDS sequence.54
Differentiation of neural progenitor cells into mature neurons is a major goal of regenerative therapies for the central nervous system. Ionic self-complementary gels were observed in 2000 to support neural cell attachment and facilitate neurite outgrowth in uncommitted P12 neural progenitor cells.55 This platform was later tested in experiments to restore vision in hamsters where the optic tract had been severed.56 The PAs designed in our laboratory bearing the laminin-derived IKVAV epitope were found to induce for the first time rapid and most importantly selective differentiation of encapsulated neural progenitor cells into neurons, supressing differentiation into astrocytes.44 Remarkably, differentiation into neurons was greater in cells exposed to bioactive PA nanofibers than those exposed to laminin itself. Injection of the material in vivo in spinal cord injury models has shown that significant behavioral improvements are observed in animals treated with the IKVAV-bearing PA nanofibers.45,46 We attribute the success of this example and other epitope-bearing peptide scaffolds to the high density of signals presented on the fiber surfaces as well as signal dynamics and spatial orientation. In one calculation, PA nanofibers were estimated to display IKVAV epitopes in densities that could be up to 1000 times greater than those expected from ordered arrays of native laminin.44
In a very recent report, we showed that epitope-bearing PA gels could be used not only for cell adhesion, but also to generate a pronounced therapeutic effect by mimicking a natural protein. A PA that presented a sequence known to activate the receptor of the angiogenic protein vascular endothelial growth factor (VEGF)57 was designed and tested in an in vivoangiogenesis assay and also in an animal model of ischemic tissue disease.58 The PA was found to exhibit a therapeutic effect in an ischemic hind-limb study as a model of human peripheral arterial disease. The therapeutic effect of the PA was substantially better than free VEGF mimetic peptide and similar to a high dose of recombinant VEGF protein. These results demonstrate the potential of peptide-based materials as immobilized protein mimetics in regenerative medicine.
Protein and small molecule delivery
Peptide-based scaffolds are well suited for designing therapeutic materials that combine cell adhesion functionality with controlled release of proteins and small molecules. In the simplest method of protein encapsulation, unmodified proteins can be sequestered inside peptide-based gels. Physical entrapment of proteins within β-hairpin gels has been studied by Schneider, where charge and size were both found to affect release kinetics.59 Similar effects were noted by Zhang in self-complementary ionic peptide gels.60,61Our laboratory has extensively studied growth factor delivery from PA nanofiber gels based on peptide sequences that bind heparin and sequences that directly bind growth factors of interest. In 2006 we reported a heparin-binding PA (HBPA),62 which contains a heparin-binding domain inspired by the consensus sequence proposed by Cardin and Weintraub.63Heparin is a natural biopolymer known to interact with spcific binding domains on several growth factors. We have since used HBPA in combination with heparin to release growth factors for in vivo and in vitro studies on angiogenesis,62,64,65 islet transplantation,66,67 and cardiovascular disease.68 In a different strategy, transforming growth factor-β1 (TGF-β1) was directly bound to a PA nanofiber using a binding sequence derived from phage display.69 The PA gel presenting this growth factor was shown to promote cartilage regeneration in a rabbit model.
Delivery of small molecules from peptide-based gels has been explored to a lesser extent than delivery of proteins.70,71 Physical encapsulation is the simplest strategy, whereby hydrophobic small molecules can be sequestered in peptide gels, presumably occupying hydrophobic pockets in the nanostructures. Pochan recently reported on the use of a β-hairpin gel to physically encapsulate the hydrophobic polyphenol curcumin.72 Sustained release was observed over two weeks, and the release rate could be controlled by varying the concentration of the peptide gelator. Our group employed the physical encapsulation strategy to sequester the nitric oxide prodrug PROLI/NO into a PA nanofiber gel.73PROLI/NO is a diazeniumdiolated proline derivative, which spontaneously releases two equivalents of NO in physiological conditions with a t1/2 of 2 s. Sequestration of PROLI/NO in a PA nanofiber gel extended the release of NO to over four days. In a rat carotid artery injury model, the NO-releasing PA gels were shown to reduce vascular smooth muscle cell proliferation and stimulate regeneration of the damaged arteries. In a more recent paper on release of chemotherapeutics, doxorubicin (Dox) was physically sequestered inside PA nanofibers.74 The drug was released upon PA nanofiber disassembly triggered by enzymatic phosphorylation of a serine residue. In order to achieve greater control over small molecule release from PA gels, we recently reported the use of hydrazones to covalently attach the anti-inflammatory drug nabumetone to PA nanofibers (Fig. 4).75 The addition of the drug did not disrupt assembly of the PAs into robust gels, and the t1/2 for drug release was found to be over 30 d with minimal burst release. Efforts to further understand and control small molecule release from PA gels are currently underway in our laboratory.
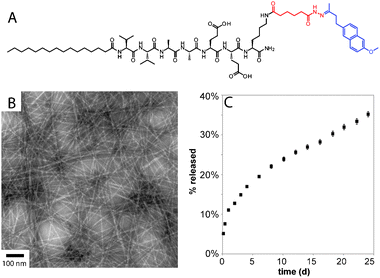 | ||
| Fig. 4 (A) Chemical structure of drug releasing PA, with the hydrazone linker shown in red and the drug (nabumetone) shown in blue. (B) conventional TEM of nabumetone-containing PA containing a hydrolysable hydrazone. (C) Profile of nabumetone release from PA gel. Reprinted with permission from ref. 75. | ||
Hierarchically ordered structures
Peptide-based materials for regenerative medicine have become increasingly complex over the past decade. Scaffolds for biomineralization have evolved from nanofibers that template mineral to materials that regenerate bone, and cell encapsulating constructs continue to inspire new therapeutic strategies. The next generation of self-assembled scaffolds will organize molecular building blocks into larger structures in a hierarchical fashion. Hierarchical structures would more closely resemble natural structures such as collagen, which is assembled over several length scales beginning from 1.5 nm triple helical protein bundles to create micron-scale fibers. A more complex architecture that was recently reported from our laboratory is the development of a lyotropic liquid crystalline PA nanofiber network that forms aligned gels of arbitrary length (Fig. 5).76 The monodomain gels are formed by first heating a solution of PA, inducing a thermally triggered dehydration into lamellar, two-dimensional sheets or plaques. Upon cooling, the plaques break into large bundles of aligned PA nanofibers, which can be formed into long, highly-aligned constructs by gently shearing the solution into salt-containing media using a pipette. The resulting gels, which are capable of encapsulating and aligning cells, are many hundreds of micrometres in diameter and can be up to several cm long. Aligned PA nanofiber scaffolds have already been studied as materials for cavernous nerve regeneration to release sonic hedgehog (SHH), a potent morphogen thought to play an important role in nerve regeneration.77 In this work directional guidance of the aligned scaffold was proposed to facilitate axon regeneration across the damaged nerve. Such a macroscopically aligned construct may also be a good template for highly aligned tissue, such as muscle fibers, the spinal cord, and parts of the brain. | ||
| Fig. 5 Aligned monodomain PA gels. (A) Birefringence images of two aligned PA noodles, showing light extinction at the crosspoint. (B) Scanning electron microscopy (SEM) image of aligned bundles. (C) Phase image of human mesenchymal stem cells (hMSCs) preferentially aligned along the string axis. (D) Fluorescence image of calcein-stained hMSCs cultured in the aligned PA gel. Reproduced with permission from ref. 76 | ||
Another family of hierarchically ordered macroscopic materials recently reported by our laboratory are PA-biopolymer hybrid membranes or liquid-filled sacs.78 These hierarchically ordered constructs, which were discovered upon mixing of two oppositely charged aqueous solutions of PA and polyelectrolyte, assemble according to a unique, multistep mechanism. First, a diffusion barrier instantaneously forms as a result of electrostatic complexation at the interface of the two solutions. Over a period of hours to days, osmotic pressure-driven diffusion of HA into the PA solution leads to dynamic assembly and growth of aligned nanofiber bundles perpendicular to the interface. The resulting membranes or enclosed sacs are on the order of 2–20 μm in thickness and have two chemically and morphologically distinct faces—one containing amorphous polyelectrolyte and the other displaying highly aligned PA nanofiber bundles.79 The assembly of ordered membranes made from oppositely charged polymers and amphiphiles has recently been shown to be possible for simple (non-peptidic) surfactants as well.80 Miniaturization of the enclosed sacs for biological applications has recently been reported by our group using a spray technique, and these microsacs were shown to release proteins and other macromolecules.81 Such cell-sized constructs may be useful in biomaterials as protein reservoirs or microbioreactors encapsulated in three-dimensional scaffolds, or as cell-isolating containers for cell co-culture.
Future outlook
In addition to its role as the scaffolding for cells and tissues, native ECM has many diverse functions, including modulating signalling pathways that control cell growth, proliferation, differentiation, morphogenesis, and survival. Signalling pathways are regulated by complex changes in epitope presentation, mechanical properties, growth factor delivery, and other complex phenomena. The synthetic cell scaffolds described here exhibit only extremely simple capacity for cell signalling relative to native ECM. The future of peptide-based scaffolds for regenerative medicine will require increasingly complex capacity for cell regulation, ideally using rationally designed small molecules and polymers.Fig. 6 shows our vision for the future of the field over the next decade and beyond. Early targets include simple hierarchically structured materials that display order parameters over several length scales, similar to those described here. Such structures begin to approach some of the organizational features in native ECM and will improve with advances in supramolecular self-assembly. Also on the horizon are new materials that interface peptide-based supramolecular structures with naturally occurring biopolymers or specifically designed synthetic polymers.82 Peptide-polymer hybrid structures have already generated self-assembled bioactive materials,65 and these bioactive composites are poised to enhance the capabilities of ECM mimetics.
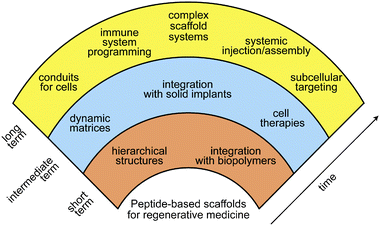 | ||
| Fig. 6 Vision for the future of peptide-based materials in regenerative medicine. | ||
We foresee several advances likely to have a major impact on regenerative medicine in the intermediate term. One is the development of dynamic scaffolds that can respond to their environment through internal or external stimuli. Materials that assemble or degrade in response to specific enzymes have been extensively studied,83–85 though few reports on cellular interactions with these scaffolds have been published.29,74,86 One can expect further developments on enzyme-responsive dynamic scaffolds in cell culture and regenerative medicine. Additionally, light-mediated changes in epitope display have been reported and are likely to play a significant role in future ECM mimetics.14,87 Another area that needs to grow is the integration of bioactive peptide materials with solid implants (e.g., titanium rods and bulk polymers).88,89 Such hybrid constructs could become a useful strategy to afford better outcomes at tissue-implant interfaces. The last intermediate term application is clinically relevant materials that will transform stem cell therapies. Induced pluripotent stem cells (iPS cells) have enormous potential to cure diseases without the attendant political and ethical concerns of embryonic stem cells. Scaffolds that can facilitate both the reprogramming and differentiation of somatic cells into the desired cell population would have a dramatic impact in organ and tissue regeneration.
Long-term contributions of chemistry to the field of regenerative medicine could follow many important directions over the next few decades. For example, molecularly sophisticated materials could serve as paths or conduits to direct migration of endogenous stem cell populations to damaged or diseased areas. The conduits could also contain the molecular triggers to expand these cell populations at the desired locations. This would be particularly exciting in the human brain to reverse neurodegenerative diseases. These complex conduits would eliminate the steps of harvesting, expanding, and implanting exogenous stem cells for regenerative therapies. Another goal would be the systemic injection of targeted molecular precursors that assemble as a cell scaffold at a desired site in vivo. Such an advance would provide a minimally invasive method for generating bioactive matrices in difficult to access areas of the body. In the area of cell signalling, the use of scaffolds for intracellular targets remains a largely unexplored area. Some peptide nanostructures programmed for intracellular delivery have been reported,90–92 but the vast majority of bioactive peptide nanostructures act on extracellular targets. Future developments in this arena will include the design of materials that can deliver a specific signal to the cytosol or a desired organelle. Another possible avenue that requires both systemic targeting and intracellular bioactivity is the use of nanostructures in programming the immune system. Recent work indicates that peptide nanostructures can be effective immune adjuvants, leading to strong antibody responses.93 Lastly, it will be important for future regenerative strategies to design complex scaffold systems that comprise heterogeneous cell populations, multiplexed signalling, as well as capacity to dynamically change their composition and physical properties. In fact, this last feature of an ECM is found in non-mammalian species capable of complete organ and limb regeneration.
Regenerative medicine is a critical need for this century and beyond in order to raise quality of life and reduce the cost of health care in the context of an increasing human lifespan. It is clear that this field offers an extraordinary opportunity for chemists to design the covalent and supramolecular structures that can promote regeneration in the human body.
Research in the authors' laboratory described in this contribution was supported with grants from the National Institutes of Health (grant numbers 2R01DE015920-06, 2R01EB003806-06A2, 1U54CA151880-01), the Department of Energy (grant number DE-FG02-00ER45810), DARPA, and the National Science Foundation (DMR-1006713). JBM was supported by a Baxter Early Career Award in Bioengineering and an NIH postdoctoral fellowship. The authors are also grateful for the use of experimental facilities at the Peptide Synthesis Core at the Institute for BioNanotechnology in Medicine (IBNAM), the Biological Imaging Facility (BIF), the Integrated Molecular Structure Education and Research Center (IMSERC), the Northwestern University Atomic- and Nanoscale Characterization Experimental Center (NUANCE, EPIC, NIFTI, Keck-II) and Keck Biophysics Facilities, all at Northwestern University.
Notes and references
- S. I. Stupp, M. U. Pralle, G. N. Tew, L. M. Li, M. Sayar and E. R. Zubarev, MRS Bull., 2000, 25, 42–48 CrossRef CAS.
- H. G. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS.
- H. Cui, T. Muraoka, A. G. Cheetham and S. I. Stupp, Nano Lett., 2009, 9, 945–951 CrossRef CAS.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487–492 CrossRef CAS.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS.
- J. B. Matson, R. H. Zha and S. I. Stupp, Curr. Opin. Solid State Mater. Sci., 2011, 15, 225–235 CrossRef CAS.
- Y. C. Yu, P. Berndt, M. Tirrell and G. B. Fields, J. Am. Chem. Soc., 1996, 118, 12515–12520 CrossRef CAS.
- M. J. Webber, J. A. Kessler and S. I. Stupp, J. Intern. Med., 2010, 267, 71–88 CrossRef CAS.
- S. I. Stupp, Nano Lett., 2010, 10, 4783–4786 CrossRef CAS.
- S. E. Paramonov, H. W. Jun and J. D. Hartgerink, J. Am. Chem. Soc., 2006, 128, 7291–7298 CrossRef CAS.
- T. Muraoka, C. Y. Koh, H. G. Cui and S. I. Stupp, Angew. Chem., Int. Ed., 2009, 48, 5946–5949 CrossRef CAS.
- E. T. Pashuck, H. G. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041–6046 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5133–5138 CrossRef CAS.
- Y. S. Velichko, S. I. Stupp and M. O. de la Cruz, J. Phys. Chem. B, 2008, 112, 2326–2334 CrossRef CAS.
- M. A. Greenfield, J. R. Hoffman, M. O. de la Cruz and S. I. Stupp, Langmuir, 2010, 26, 3641–3647 CrossRef CAS.
- S. G. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3334–3338 CrossRef CAS.
- D. J. Pochan, J. P. Schneider, J. Kretsinger, B. Ozbas, K. Rajagopal and L. Haines, J. Am. Chem. Soc., 2003, 125, 11802–11803 CrossRef CAS.
- R. A. Hule, R. P. Nagarkar, B. Hammouda, J. P. Schneider and D. J. Pochan, Macromolecules, 2009, 42, 7137–7145 CrossRef CAS.
- L. Aulisa, H. Dong and J. D. Hartgerink, Biomacromolecules, 2009, 10, 2694–2698 CrossRef CAS.
- C. W. G. Fishwick, A. J. Beevers, L. M. Carrick, C. D. Whitehouse, A. Aggeli and N. Boden, Nano Lett., 2003, 3, 1475–1479 CrossRef CAS.
- H. Dong, S. E. Paramonov, L. Aulisa, E. L. Bakota and J. D. Hartgerink, J. Am. Chem. Soc., 2007, 129, 12468–12472 CrossRef CAS.
- A. Aggeli, M. Bell, N. Boden, J. N. Keen, P. F. Knowles, T. C. B. McLeish, M. Pitkeathly and S. E. Radford, Nature, 1997, 386, 259–262 CrossRef CAS.
- A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. B. McLeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11857–11862 CrossRef CAS.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. E. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- Z. M. Yang, G. L. Liang, L. Wang and B. Xu, J. Am. Chem. Soc., 2006, 128, 3038–3043 CrossRef CAS.
- M. M. Pires, D. E. Przybyla and J. Chmielewski, Angew. Chem., Int. Ed., 2009, 48, 7813–7817 CrossRef CAS.
- J. H. Collier and P. B. Messersmith, Adv. Mater., 2004, 16, 907–910 CrossRef CAS.
- J. P. Jung, A. K. Nagaraj, E. K. Fox, J. S. Rudra, J. M. Devgun and J. H. Collier, Biomaterials, 2009, 30, 2400–2410 CrossRef CAS.
- E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596–600 CrossRef CAS.
- C. J. Bowerman and B. L. Nilsson, J. Am. Chem. Soc., 2010, 132, 9526–9527 CrossRef CAS.
- L. Hsu, G. L. Cvetanovich and S. I. Stupp, J. Am. Chem. Soc., 2008, 130, 3892–3899 CrossRef CAS.
- A. Mata, L. Hsu, R. Capito, C. Aparicio, K. Henrikson and S. I. Stupp, Soft Matter, 2009, 5, 1228–1236 RSC.
- M. A. Biesalski, A. Knaebel, R. Tu and M. Tirrell, Biomaterials, 2006, 27, 1259–1269 CrossRef CAS.
- E. D. Spoerke, S. G. Anthony and S. I. Stupp, Adv. Mater., 2009, 21, 425–430 CrossRef CAS.
- A. Mata, Y. B. Geng, K. J. Henrikson, C. Aparicio, S. R. Stock, R. L. Satcher and S. I. Stupp, Biomaterials, 2010, 31, 6004–6012 CrossRef CAS.
- J. P. Jung, J. V. Moyano and J. H. Collier, Integr. Biol., 2011, 3, 185–196 RSC.
- M. O. Guler, L. Hsu, S. Soukasene, D. A. Harrington, J. F. Hulvat and S. I. Stupp, Biomacromolecules, 2006, 7, 1855–1863 CrossRef CAS.
- H. Storrie, M. O. Guler, S. N. Abu-Amara, T. Volberg, M. Rao, B. Geiger and S. I. Stupp, Biomaterials, 2007, 28, 4608–4618 CrossRef CAS.
- M. J. Webber, J. Tongers, M. A. Renault, J. G. Roncalli, D. W. Losordo and S. I. Stupp, Acta Biomater., 2010, 6, 3–11 CrossRef CAS.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS.
- V. M. Tysseling-Mattiace, V. Sahni, K. L. Niece, D. Birch, C. Czeisler, M. G. Fehlings, S. I. Stupp and J. A. Kessler, J. Neurosci., 2008, 28, 3814–3823 CrossRef CAS.
- V. M. Tysseling, V. Sahni, E. T. Pashuck, D. Birch, A. Hebert, C. Czeisler, S. I. Stupp and J. A. Kessler, J. Neurosci. Res., 2010, 88, 3161–3170 CrossRef CAS.
- J. E. Goldberger, E. J. Berns, R. Bitton, C. J. Newcomb and S. I. Stupp, Angew. Chem., Int. Ed., 2011, 50, 6292–6295 CrossRef CAS.
- H. A. Behanna, J. Donners, A. C. Gordon and S. I. Stupp, J. Am. Chem. Soc., 2005, 127, 1193–1200 CrossRef CAS.
- O. S. Lee, S. I. Stupp and G. C. Schatz, J. Am. Chem. Soc., 2011, 133, 3677–3683 CrossRef CAS.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS.
- R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646–2651 CrossRef CAS.
- J. Z. Gasiorowski and J. H. Collier, Biomacromolecules, 2011, 12, 3549–3558 CrossRef CAS.
- A. Horii, X. M. Wang, F. Gelain and S. G. Zhang, PLoS One, 2007, 2, e190 Search PubMed.
- H. Shin, J. S. Temenoff, G. C. Bowden, K. Zygourakis, M. C. Farach-Carson, M. J. Yaszemski and A. G. Mikos, Biomaterials, 2005, 26, 3645–3654 CrossRef CAS.
- T. C. Holmes, S. de Lacalle, X. Su, G. S. Liu, A. Rich and S. G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6728–6733 CrossRef CAS.
- R. G. Ellis-Behnke, Y. X. Liang, S. W. You, D. K. C. Tay, S. G. Zhang, K. F. So and G. E. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5054–5059 CrossRef CAS.
- L. D. D'Andrea, G. Iaccarino, R. Fattorusso, D. Sorriento, C. Carannante, D. Capasso, B. Trimarco and C. Pedone, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 14215–14220 CrossRef CAS.
- M. J. Webber, J. Tongers, C. J. Newcomb, K. T. Marquardt, J. Bauersachs, D. W. Losordo and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13438–13443 CrossRef CAS.
- M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, Biomaterials, 2010, 31, 9527–9534 CrossRef CAS.
- S. Koutsopoulos, L. D. Unsworth, Y. Nagaia and S. G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4623–4628 CrossRef CAS.
- F. Gelain, L. D. Unsworth and S. G. Zhang, J. Controlled Release, 2010, 145, 231–239 CrossRef CAS.
- K. Rajangam, H. A. Behanna, M. J. Hui, X. Q. Han, J. F. Hulvat, J. W. Lomasney and S. I. Stupp, Nano Lett., 2006, 6, 2086–2090 CrossRef CAS.
- A. D. Cardin and H. J. R. Weintraub, Arterioscler., Thromb., Vasc. Biol., 1989, 9, 21–32 CrossRef CAS.
- S. Ghanaati, M. J. Webber, R. E. Unger, C. Orth, J. F. Hulvat, S. E. Kiehna, M. Barbeck, A. Rasic, S. I. Stupp and C. J. Kirkpatrick, Biomaterials, 2009, 30, 6202–6212 CrossRef CAS.
- L. W. Chow, R. Bitton, M. J. Webber, D. Carvajal, K. R. Shull, A. K. Sharma and S. I. Stupp, Biomaterials, 2011, 32, 1574–1582 CrossRef CAS.
- J. C. Stendahl, L. J. Wang, L. W. Chow, D. B. Kaufman and S. I. Stupp, Transplantation, 2008, 86, 478–481 CrossRef CAS.
- L. W. Chow, L. J. Wang, D. B. Kaufman and S. I. Stupp, Biomaterials, 2010, 31, 6154–6161 CrossRef CAS.
- M. J. Webber, X. Q. Han, S. N. P. Murthy, K. Rajangam, S. I. Stupp and J. W. Lomasney, J. Tissue Eng. Regener. Med., 2010, 4, 600–610 CrossRef CAS.
- R. N. Shah, N. A. Shah, M. M. D. Lim, C. Hsieh, G. Nuber and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3293–3298 CrossRef CAS.
- Y. Nagai, L. D. Unsworth, S. Koutsopoulos and S. G. Zhang, J. Controlled Release, 2006, 115, 18–25 CrossRef CAS.
- G. L. Liang, Z. M. Yang, R. J. Zhang, L. H. Li, Y. J. Fan, Y. Kuang, Y. Gao, T. Wang, W. W. Lu and B. Xu, Langmuir, 2009, 25, 8419–8422 CrossRef CAS.
- A. Altunbas, S. J. Lee, S. A. Rajasekaran, J. P. Schneider and D. J. Pochan, Biomaterials, 2011, 32, 5906–5914 CrossRef CAS.
- M. R. Kapadia, L. W. Chow, N. D. Tsihlis, S. S. Ahanchi, J. A. Hrabie, J. Murar, J. Martinez, D. A. Popowich, Q. Jiang, J. E. Saavedra, L. K. Keefer, J. F. Hulvat, S. I. Stupp and M. P. Kibbe, J. Vasc. Surg., 2008, 47, 173–182 CrossRef.
- M. J. Webber, C. J. Newcomb, R. Bitton and S. I. Stupp, Soft Matter, 2011, 7, 9665–9672 RSC.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2011, 47, 7962–7964 RSC.
- S. M. Zhang, M. A. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio, M. O. de la Cruz and S. I. Stupp, Nat. Mater., 2010, 9, 594–601 CrossRef CAS.
- N. L. Angeloni, C. W. Bond, Y. Tang, D. A. Harrington, S. M. Zhang, S. I. Stupp, K. E. McKenna and C. A. Podlasek, Biomaterials, 2011, 32, 1091–1101 CrossRef CAS.
- R. M. Capito, H. S. Azevedo, Y. S. Velichko, A. Mata and S. I. Stupp, Science, 2008, 319, 1812–1816 CrossRef CAS.
- D. Carvajal, R. Bitton, J. R. Mantei, Y. S. Velichko, S. I. Stupp and K. R. Shull, Soft Matter, 2010, 6, 1816–1823 RSC.
- D. B. Carew, K. J. Channon, I. Manners and D. N. Woolfson, Soft Matter, 2011, 7, 3475–3481 RSC.
- D. I. Rozkiewicz, B. D. Myers and S. I. Stupp, Angew. Chem., Int. Ed., 2011, 50, 6324–6327 CrossRef CAS.
- H. A. Klok, Macromolecules, 2009, 42, 7990–8000 CrossRef CAS.
- R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19–24 CrossRef CAS.
- Z. M. Yang, H. W. Gu, D. G. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS.
- Y. Gao, Z. M. Yang, Y. Kuang, M. L. Ma, J. Y. Li, F. Zhao and B. Xu, Biopolymers, 2010, 94, 19–31 CrossRef CAS.
- K. M. Galler, L. Aulisa, K. R. Regan, R. N. D'Souza and J. D. Hartgerink, J. Am. Chem. Soc., 2010, 132, 3217–3223 CrossRef CAS.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS.
- T. D. Sargeant, S. M. Oppenheimer, D. C. Dunand and S. I. Stupp, J. Tissue Eng. Regener. Med., 2008, 2, 455–462 CrossRef CAS.
- T. D. Sargeant, M. O. Guler, S. M. Oppenheimer, A. Mata, R. L. Satcher, D. C. Dunand and S. I. Stupp, Biomaterials, 2008, 29, 161–171 CrossRef CAS.
- D. Missirlis, D. V. Krogstad and M. Tirrell, Mol. Pharmaceutics, 2010, 7, 2173–2184 CrossRef CAS.
- L. Aulisa, N. Forraz, C. McGuckin and J. D. Hartgerink, Acta Biomater., 2009, 5, 842–853 CrossRef CAS.
- D. Easterhoff, J. T. M. DiMaio, T. M. Doran, S. Dewhurst and B. L. Nilsson, Biophys. J., 2011, 100, 1325–1334 CrossRef CAS.
- J. S. Rudra, Y. F. Tian, J. P. Jung and J. H. Collier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 622–627 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |