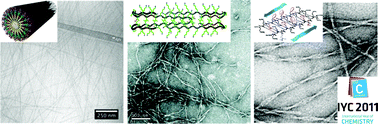
Biomaterials made from self-assembling, short peptides and peptide derivatives have great potential to generate powerful new therapies in regenerative medicine. The high signaling capacity and therapeutic efficacy of peptidic scaffolds has been established in several animal models, and the development of more complex, hierarchical structures based on peptide materials is underway. This highlight discusses several classes of self-assembling peptide-based materials, including peptide amphiphiles, Fmoc-peptides, self-complementary ionic peptides, hairpin peptides, and others. The self-assembly designs, bioactive signalling strategies, and cell signalling capabilities of these bioactive materials are reported. The future challenges of the field are also discussed, including short-term goals such as integration with biopolymers and traditional implants, and long term goals, such as immune system programming, subcellular targeting, and the development of highly integrated scaffold systems.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?