Cation-induced molecular motion of spring-like [2]catenanes†
Alexandre V.
Leontiev
,
Christopher J.
Serpell
,
Nicholas G.
White
and
Paul D.
Beer
*
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: paul.beer@chem.ox.ac.uk; Fax: +44-(0)1865-272690; Tel: +44-(0)1865-285142
First published on 8th March 2011
Abstract
The syntheses and cation recognition studies of two novel heteroditopic [2]catenanes that are capable of reversible rotary motion are described. Prepared by chloride anion templation, both catenanes possess a calixdiquinone unit able to bind Na+, K+, NH4+ and Ba2+ cations. Following characterization of the chloride salts of both catenanes by NMR, mass spectrometry and crystal structure determination, hexafluorophosphate salts were investigated for their behaviour upon addition of cations. Ba2+ complexation caused a partial intra-ring rotation of the two macrocycles in both species, which was reversed upon addition of the sequestering SO42− anion.
Introduction
With the objective of controlling the dynamics of the mechanical bond for molecular switch and machine-like nanotechnological applications, the imaginative design and construction of interlocked molecules of increasing complexity and function is continuing to attract intense research interest in the chemical community.1 In addition the potential of their unique, topologically constrained three dimensional cavities in molecular recognition and sensing applications is only recently beginning to be realized.2Whilst interlocked host systems for cation,3 and to a limited extent, anion4 binding have been reported for recognition and dynamic applications, the challenge of fabricating heteroditopic5 interlocked systems remains underexploited. This is surprising given the number of potential mechanisms charged guest binding may induce in changes of co-conformation between interlocked components of such systems. Integrating the molecular design features of our recently reported heteroditopic calix[4]diquinone macrocyclic receptors,6 which bind group 1 metal and ammonium chlorides as contact ion-pair species, into an interlocked structural framework, we report herein the synthesis of novel heteroditopic [2]catenanes which undergo cation induced dynamic co-conformational intra-ring rotary behaviour (Fig. 1).
![Proposed switchable calixdiquinone [2]catenane driven by cation-cation electrostatic repulsion.](/image/article/2011/SC/c1sc00034a/c1sc00034a-f1.gif) | ||
| Fig. 1 Proposed switchable calixdiquinone [2]catenane driven by cation-cation electrostatic repulsion. | ||
Results and discussion
Design and synthetic strategy
Common to the two target heteroditopic catenanes is the calix[4]diquinone macrocyclic component 15a which contains a calix[4]diquinone ether cation recognition site covalently linked via hydroquinone groups to an isophthalamide anion binding motif designed to facilitate interpenetrated assembly via anion templation with 3,5-bis-vinyl amide substituted N-methyl- and N-benzyl-pyridinium-chloride threading components. Favourable π–π stacking interactions between the electron rich hydroquinone groups and the respective positively charged electron deficient pyridinium group, combined with cooperative association of the unsaturated chloride anion of the pyridinium ion-pair to the macrocycle isophthalamide binding site, stabilizes the pseudorotaxane intermediate which upon undergoing a ‘clipping’ Grubbs'7 catalysed ring closing metathesis (RCM) reaction affords the heteroditopic [2]catenane system. Mutual electrostatic repulsion8 between a cation bound at the calix[4]diquinone ether macrocycle recognition site and the positively charged pyridinium group of the other macrocycle is predicted to induce an intra-ring co-conformational rotary motion between the two macrocycles. (Fig. 1).Synthesis
The synthesis of the target catenanes is shown in Scheme 1. RCM reactions were carried out by the addition of Grubbs' 2nd generation catalyst to equimolar dichloromethane solutions of calixdiquinone macrocycle 1 with either threading derivative 2Cl or 3Cl. After purification by silica gel chromatography the target [2]catenanes 4Cl and 5Cl were isolated in 29% and 27% yields, respectively (see ESI†).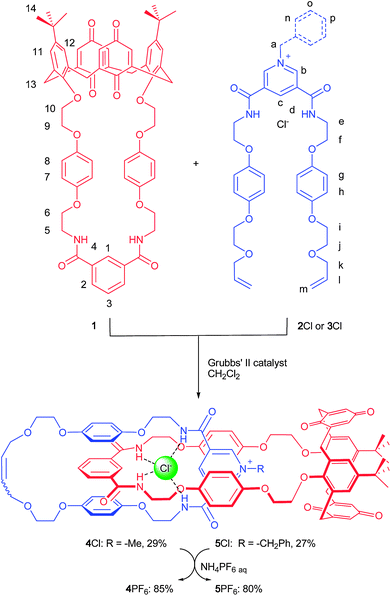 | ||
| Scheme 1 Synthesis of catenanes 4Cl and 5Cl. Catenanes 4PF6 and 5PF6 were prepared from corresponding chloride salts by anion exchange with NH4PF6. | ||
The catenanes were characterized by NMR spectroscopy and electrospray mass spectroscopy. The 1H NMR spectra of catenanes 4Cl and 5Cl compared to their individual components (see ESI†) provide evidence of a ‘stacked’ arrangement of the respective positively charged pyridinium ring between the hydroquinone groups of the calixdiquinone macrocycle as depicted in Scheme 1. Among the most diagnostic for catenane formation are the splitting and upfield shifts of hydroquinone protons 7 and 8, due to π–π stacking interactions and the large downfield shifts observed in the amide protons 4 and internal isophthalamide proton 1 indicating complexation of the templating chloride anion to the calixdiquinone macrocycle (Fig. 2). Further evidence for the interlocked nature of the macrocyclic rings in the catenanes was provided by 1H ROESY NMR spectra which revealed several characteristic through-space couplings between the interlocked components of the catenanes (a → 13, b → 7(8), d → 7(8)) (see ESI†).
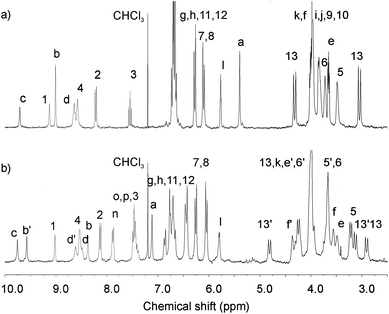 | ||
| Fig. 2 Partial 1H NMR spectra (CDCl3, 293 K) of a) 4Cl and b) 5Cl catenanes. | ||
The presence of a bulkier benzyl group in the catenane 5Cl leads to desymmetrization of the 1H NMR spectrum (Fig. 2b).
Use of 2D COSY, ROESY and HSQC experiments enabled complete assignment of all signals in the 1H NMR spectrum of 5Cl in CDCl3. Broken symmetry imposed by pyridinium PhCH2-N substitution resulted in splitting of most of the signals for the calixdiquinone and the pyridinium rings. However, the benzyl protons a and n are not split suggesting unrestricted rotation of the phenyl ring along the CH2–Ph bond.
The described orientation is consistent with that observed in the solid state structures of catenanes 4Cl and 5Cl (Fig. 3), single crystals of which were grown by slow evaporation of DCM–CH3CN solutions of the compounds. X-ray diffraction data were collected using synchrotron radiation at Diamond Light Source beamline I19 (details in ESI†).
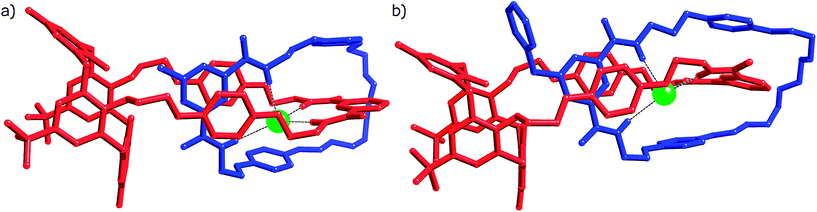 | ||
| Fig. 3 X-ray crystal structures of catenanes a) 4Cl and b) 5Cl. Solvent molecules and hydrogen atoms have been omitted for clarity. Chloride anion represented as a spacefill green sphere. | ||
As evident from Fig. 3, with respect to the supramolecular forces involved and ring co-conformations, both catenanes have similar solid-state structures. Comparison of the relevant features of the two catenated crystal structures reveals no significant differences between the supramolecular behaviour of the two systems, despite the presence of the greater steric bulk in the N-benzyl-pyridinium catenane. The distance between the chloride anion centroid of the anion binding cavity (defined by the four amide nitrogen atoms) differs by only approximately 0.1 Å (0.778(3) Å and 0.749(3) Å for the methyl system versus 0.657(1) Å for benzyl). Similarly, the distance between the sp3 methyl or benzyl pyridinium carbon and the centroid of the four oxygens at the bottom of the calixquinone varies by about 0.1 Å (2.298(14) Å and 2.406(14) Å versus 2.270(6) Å). These results indicate that the benzyl group neither causes a rotation of the pyridinium macrocycle within the calixquinone cavity, nor affects the nature of the anion binding site, compared with the N-methyl-pyridinium analogue.
The hexafluorophosphate salts 4PF6 and 5PF6 were prepared in 85% and 80% yields, respectively, by repetitive washing of the chloride salts with aqueous NH4PF6. While in the N-methyl pyridinium catenane, the 1H NMR spectra (recorded in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CD2Cl2:CD3CN) of the two salts are similar (see ESI†) apart from the expected upfield shifts of protons involved in the anion binding site (1, 4, d and c), dramatically different behaviour is observed with the N-benzyl pyridinium catenane.
:1 CD2Cl2:CD3CN) of the two salts are similar (see ESI†) apart from the expected upfield shifts of protons involved in the anion binding site (1, 4, d and c), dramatically different behaviour is observed with the N-benzyl pyridinium catenane.
Fig. 4a shows that, although somewhat broad, the spectrum of 5Cl in CD2Cl2:CD3CN (4:1) solvent mixture demonstrates characteristic splitting in most of the signals for the calixdiquinone and the pyridinium rings reflecting its ‘broken symmetry’ nature. However, after removal the chloride anion from the interpenetrated cavity, the 1H NMR spectrum of 5PF6 is simplified significantly suggesting symmetrisation of the interlocked structure (Fig. 4b). Presumably, without the templating Cl− anion, conformational flexibility allows the benzyl group to flip inside and out of the calixdiquinone macrocycle cavity, a process which is fast on the NMR time scale. Conversely, with 5Cl the observed inequivalence of Hb and Hd can be attributed to steric considerations where the interlocked complexed chloride anion results in a catenane co-conformation in which as a consequence of the pyridinium ring residing between the hydroquinone groups, it is sterically not possible for the N-benzyl pyridinium group to undergo the intramolecular flipping process.
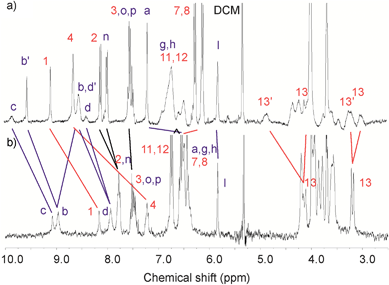 | ||
| Fig. 4 Partial 1H NMR spectra (CD2Cl2:CD3CN 4:1, 293 K) of a) catenane 5Cl and b) 5PF6. | ||
At the same time, 2D NMR experiments demonstrate (Fig. 5) the symmetrical arrangement of the positively charged pyridinium ring within the calixdiquinone macrocycle is preserved after anion exchange in 5PF6 as for the N-methyl pyridinium catenane 4PF6 (see ESI†).
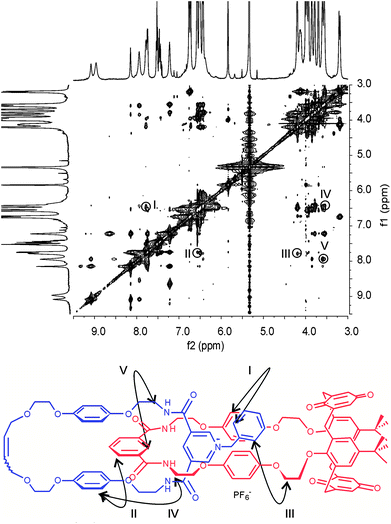 | ||
| Fig. 5 Partial 1H-1H ROESY NMR (500 MHz, CD2Cl2:CD3CN (4:1), 293 K) of catenane 5PF6. Cross couplings are shown in the schematic diagram below. See ESI for the corresponding spectrum of 4PF6. | ||
Cation binding studies
Having established the co-conformations in both catenanes, it was now of interest to investigate their cation binding properties and in particular dynamic behaviour. To elucidate evidence of cation complexation, 1H NMR and UV-vis spectroscopic titrations of catenanes 4PF6 and 5PF6 with sodium, potassium, ammonium and barium cations were undertaken.Association constants for binding of metal cations were determined by SPECFIT analysis9 of the UV-vis titration data using a 1:1 stoichiometric binding model (Table 1 and ESI†). The values obtained are in general agreement with the association constants derived from the 1H NMR spectroscopic titrations (see ESI†) and show that the relatively weaker cation binding properties of the catenanes compared to macrocycle 1 can be attributed to unfavourable electrostatic repulsion and steric effects between the complexed cation and the respective pyridinium motifs of the catenanes. Surprisingly, both catenanes display similar binding affinities toward the studied cations, which suggests the sterically bulkier benzyl group of 5PF6 does not significantly disfavour cation binding.
:1) at 293 K. (errors < 15%)
| Cation | 4PF6 log Ka | 5PF6 log Ka | Macrocycle 1 log Ka |
|---|---|---|---|
| Na+ | 3.84 | 4.17 | 4.48 |
| K+ | 4.51 | 3.96 | 4.59 |
| NH4+ | 3.46 | 3.83 | 4.48 |
| Ba2+ | >6 | >6 | >6 |
In order to gain more insight into the cation binding properties of the catenanes, 1H NMR titration experiments were undertaken. Contrasting behaviour was observed for the monocationic cations compared to the barium dication with both catenanes. Upon addition of the monovalent salts, the spectra typically sharpened but remained largely unchanged. As shown in Fig. 6, with 5PF6 downfield shifts in the position of methylene protons 13 are noted, which is attributed to conformational changes of the calixdiquinone rings upon binding of Na+. In addition, methylene protons a of NCH2Ph experience a 0.5 ppm upfield shift which is indicative of loss of hydrogen bonding with calixdiquinone and ethylene glycol oxygens. Noticeable splitting and further upfield shifts of hydroquinone protons 7 and 8 are presumably the result of enhanced π–π stacking interactions caused by the pyridinium group of the respective catenane residing in a relatively lower position experiencing a shielding effect from the hydroquinone groups of the second macrocycle.
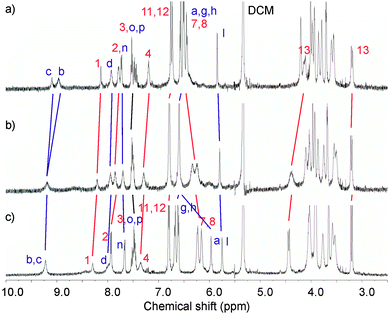 | ||
| Fig. 6 Partial 1H NMR spectra (500 MHz, CD2Cl2:CD3CN (4:1), 293 K) of a) 5PF6, b) 5PF6 + 1 eq of NaClO4 and c) 5PF6 + 5 eq of NaClO4. See ESI for corresponding spectra of 4PF6.† | ||
The analogous 1H NMR titrations carried out on both catenanes with Ba(ClO4)2 displayed slow exchange kinetics on the NMR time scale. Upon addition of excess equivalents of barium cations, significant splitting of the protons b and d (Fig. 7 and ESI†) were observed. These signals, representing pairs of –CH– and –NH– amide groups of the catenane pyridinium rings related by symmetry are indicative of the desymmetrisation of the catenane structure. Their chemical inequivalence suggests co-conformational partial rotation of the pyridinium ring component.
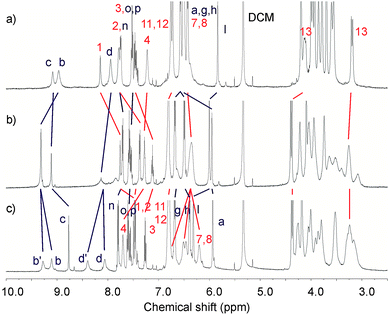 | ||
| Fig. 7 Partial 1H NMR spectra (500 MHz, CD2Cl2:CD3CN (4:1), 293 K) of a) 5PF6, b) 5PF6 + 1 eq of Ba(ClO4)2 and c) 5PF6 + 5 eq of Ba(ClO4)2. See ESI for corresponding spectra of 4PF6.† | ||
Comparison of 1H ROESY NMR spectra obtained before and after the addition of barium cations show contrasting through space interactions to exist between the pyridinium and the calixdiquinone macrocycle (Fig. 8 and ESI†). Importantly, with 5PF6 on complexation of Ba2+ cations, cross coupling interactions cannot be seen between calixdiquinone protons 7,8 or 9,10 and the benzyl protons n and a. The absence of these interactions along with other identified proton–proton through space couplings indicate the barium cation-induced expulsion of the pyridinium motif from the lower rim of the calixdiquinone cavity in a spring-like mode (Fig. 8).
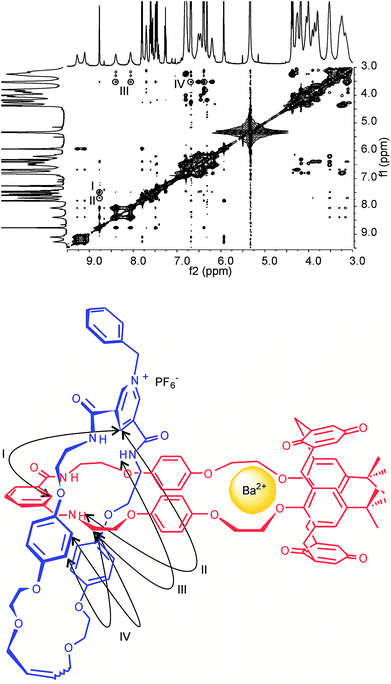 | ||
| Fig. 8 Partial 1H−1H ROESY NMR (500 MHz, CD2Cl2:CD3CN (4:1), 293 K) of N-benzyl catenane 5PF6 with 5 eq of Ba(ClO4)2. Cross couplings are shown in the schematic diagram below. See ESI for corresponding spectra of 4PF6.† | ||
Analogous 1H NMR observations were noted on addition of Ba(ClO4)2 to catenane 4PF6 (see ESI†).
The reversibility of barium-induced rotational motion was probed by 1H NMR titrations of the corresponding barium catenane complexes with tetrabutylammonium sulfate. Sequestration of barium cations by BaSO4 precipitation led to the restoration of the original co-conformation as evidenced by the 1H NMR spectra reverting to that of the hexafluorophosphate salts of the catenanes (Fig. 9).10
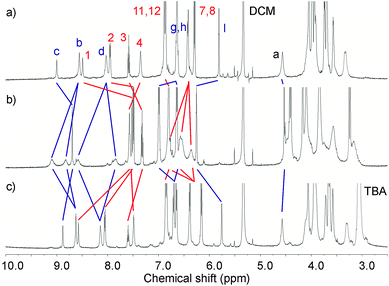 | ||
| Fig. 9 Partial 1H NMR spectra (500 MHz, CD2Cl2:CD3CN (4:1), 293 K) of a) 4PF6, b) 4PF6 + 5 eq of Ba(ClO4)2 and c) 4PF6 + 5 eq of Ba(ClO4)2 + 5 eq of (TBA)2SO4. | ||
Conclusion
Two novel heteroditopic [2]catenanes containing a calixdiquinone macrocycle interlocked with respectively N-methyl- and N-benzyl-pyridinium cyclic components have been prepared via chloride anion templation. The calixdiquinone motif of each catenane was demonstrated by 1H NMR and UV-vis spectroscopy to bind Na+, NH4+, and Ba2+ cations. Barium complexation caused partial intra-ring rotations of the catenanes, where well defined co-conformations dominate, which can be attributed to mutual electrostatic repulsion between the calixdiquinone bound dication and the positively charged pyridinium group of the other macrocycle. By contrast, the monovalent cations were bound without producing a significant co-conformational perturbation in the catenanes, thus indicating the increased charge of barium is responsible for effecting the partial rotatory molecular motion process.11 Addition of sulfate reversed the Ba2+ induced rotary motion of the two macrocycles.Acknowledgements
We thank the EPSRC for a postdoctoral fellowship (EP/E033962/1) (A.V.L.) and Johnson Matthey for a CASE studentship (C.J.S.). N.G.W. thanks Clarendon Fund and Trinity College for a studentship. We also thank Diamond Light Source for an award of beamtime on I19 (MT1880) and the beamline instrument scientists for their assistance.Notes and references
- For recent reviews, see: (a) K. Konstas, S. J. Langford and M. J. Latter, Int. J. Mol. Sci., 2010, 11, 2453–2472 Search PubMed; (b) J.-P. Sauvage, J.-P. Collin, S. Durot, J. Frey, V. Heitz, A. Sour and C. Tock, C. R. Chim., 2010, 13, 315–328 CrossRef CAS; (c) S. Silvi, M. Venturi and A. Credi, J. Mater. Chem., 2009, 19, 2279–2294 RSC; (d) D. Li, W. F. Paxton, R. H. Baughman, T. J. Huang, J. F. Stoddart and P. S. Weiss, MRS Bull., 2009, 34, 671–681 CrossRef CAS; (e) E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS.
- (a) J. J. Davis, G. A. Orlowski, H. Rahman and P. D. Beer, Chem. Commun., 2010, 46, 54–63 RSC; (b) M. J. Chmielewski, J. J. Davis and P. D. Beer, Org. Biomol. Chem., 2009, 7, 415–424 RSC.
- (a) G. J. E. Davidson, S. Sharma and S. J. Loeb, Angew. Chem., Int. Ed., 2010, 49, 4938–4942 CrossRef CAS; (b) J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC; (c) D. S. Marlin, D. G. Cabrera, D. A. Leigh and A. M. Z. Slawin, Angew. Chem., Int. Ed., 2006, 45, 77–83 CrossRef CAS; (d) D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, Chem. Commun., 2005, 4919–4921 RSC.
- (a) J. J. Gassensmith, S. Matthys, J.-J. Lee, A. Wojcik, P. V. Kamat and B. D. Smith, Chem.–Eur. J., 2010, 16, 2916–2921 CrossRef; (b) T.-C. Lin, C.-C. Lai and S.-H. Chiu, Org. Lett., 2009, 11, 613–616 CrossRef CAS; (c) K.-Y. Ng, V. Felix, S. M. Santos, N. H. Rees and P. D. Beer, Chem. Commun., 2008, 1281–1283 RSC; (d) Y.-L. Huang, W.-C. Hung, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Angew. Chem., Int. Ed., 2007, 46, 6629–6633 CrossRef CAS; (e) C.-F. Lin, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Chem.–Eur. J., 2007, 13, 4350–4355 CrossRef CAS; (f) C. M. Keaveney and D. A. Leigh, Angew. Chem., Int. Ed., 2004, 43, 1222–1224 CrossRef CAS; (g) B. W. Laursen, S. Nygaard, J. O. Jeppesen and J. F. Stoddart, Org. Lett., 2004, 6, 4167–4170 CrossRef CAS.
- (a) A. V. Leontiev, C. A. Jemmett and P. D. Beer, Chem.–Eur. J., 2011, 17, 816–825 CrossRef CAS; (b) X. Wang, J. Zhu and D. B. Smithrud, J. Org. Chem., 2010, 75, 3358–3370 CrossRef CAS; (c) M. J. Barrell, D. A. Leigh, P. J. Lusby and A. M. Z. Slawin, Angew. Chem., Int. Ed., 2008, 47, 8036–8039 CrossRef CAS; (d) M. J. Deetz, R. Shukla and B. D. Smith, Tetrahedron, 2002, 58, 799–805 CrossRef CAS; (e) R. Shukla, M. J. Deetz and B. D. Smith, Chem. Commun., 2000, 2397–2398 RSC.
- (a) M. D. Lankshear, I. M. Dudley, K.-M. Chan, A. R. Cowley, S. M. Santos, V. Felix and P. D. Beer, Chem.–Eur. J., 2008, 14, 2248–2263 CrossRef CAS; (b) M. D. Lankshear, I. M. Dudley, K.-M. Chan and P. D. Beer, New J. Chem., 2007, 31, 684–690 RSC; (c) M. D. Lankshear, A. R. Cowley and P. D. Beer, Chem. Commun., 2006, 612–614 RSC.
- For selected examples, see: (a) K.-Y. Ng, V. Felix, S. M. Santos, N. H. Rees and P. D. Beer, Chem. Commun., 2008, 1281–1283 RSC; (b) A.-M. L. Fuller, D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, J. Am. Chem. Soc., 2005, 127, 12612–12619 CrossRef CAS; (c) T. J. Kidd, D. A. Leigh and A. J. Wilson, J. Am. Chem. Soc., 1999, 121, 1599–1600 CrossRef CAS.
- For selected examples, see: (a) S. Nygaard, Y. Liu, P. C. Stein, A. H. Flood and J. O. Jeppesen, Adv. Funct. Mater., 2007, 17, 751–762 CrossRef CAS; (b) M. J. Gunter, Eur. J. Org. Chem., 2004, 1655–1673 CrossRef CAS; (c) M. Asakawa, P. R. Ashton, V. Balzani, S. E. Boyd, A. Credi, G. Mattersteig, S. Menzer, M. Montalti, F. M. Raymo, C. Ruffilli, J. F. Stoddart, M. Venturi and D. J. Williams, Eur. J. Org. Chem., 1999, 985–994 CrossRef CAS; (d) M. J. Gunter and M. R. Johnston, Chem. Commun., 1994, 829–830 RSC; (e) M. Asakawa, P. R. Ashton, V. Balzani, A. Credi, C. Hamers, G. Mattersteig, M. Montalti, A. N. Shipway, N. Spencer, J. F. Stoddart, M. S. Tolley, M. Venturi, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 333–337 CrossRef CAS; (f) J. D. Crowley, I. M. Steele and B. Bosnich, Chem.–Eur. J., 2006, 12, 8935–8951 CrossRef CAS; (g) X.-B. Wang, B. Dai, H.-K. Woo and L.-S. Wang, Angew. Chem., Int. Ed., 2005, 44, 6022–6024 CrossRef CAS; (h) V. Balzani, A. Credi, G. Mattersteig, O. A. Matthews, F. M. Raymo, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Org. Chem., 2000, 67, 1924–1936 CrossRef.
- SPECFIT ν. 2.02, Spectrum Software Associates, Chapel Hill, NC Search PubMed.
- The addition of metal cation ClO4− or PF6− salts to catenanes 4Cl and 5Cl in 4:1 CD2Cl2:CD3CN resulted in sequestration of the chloride anion accompanied by precipitation of metal chloride salts.
- As pointed out by a helpful referee, the 1H NMR spectral observations upon monocation catenane complexation could also be rationalized by fast exchange on the NMR timescale between two degenerate unsymmetrical co-conformations. VT 1H NMR experiments however led to precipitation problems at lower temperatures which unfortunately prevented any meaningful data being obtained.
Footnote |
| † Electronic supplementary information (ESI) available: compound characterization, full synthetic details and titration experiments. CCDC reference numbers 809243–809245. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00034a |
| This journal is © The Royal Society of Chemistry 2011 |